
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sonogashira cross-coupling as a key step in the synthesis of new glycoporphyrins†
Bartosz
Godlewski‡
a,
Dariusz
Baran‡
a,
Morgane
de Robichon
bc,
Angélique
Ferry
bc,
Stanisław
Ostrowski
a and
Maciej
Malinowski
 *a
*a
aFaculty of Chemistry, Warsaw University of Technology, ul. Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: maciej.malinowski@pw.edu.pl
bCY Cergy-Paris Université, BioCIS, CNRS, 5 mail Gay-Lussac, 95000 Cergy-Pontoise cedex, France
cUniversité Paris-Saclay, BioCIS, CNRS, 5, rue J-B Clément, 92296 Châtenay-Malabry cedex, France
First published on 15th March 2022
Abstract
Glycoporphyrins are an important subclass of the porphyrin family of compounds. They have been intensively studied because of their biological importance, i.e. they exhibit increased solubility in polar solvents and they might provide better selectivity towards biological targets. New methods for the efficient synthesis of these compounds are highly desired. Unfortunately, in most of the described methodologies, carbohydrate-linked porphyrins are obtained through hydrolyzable connections. Herein, the original route to new C–C conjugated glycoporphyrins via the Sonogashira cross-coupling is presented. The optimal conditions have been established and the most convenient synthetic strategy has been determined.
1. Introduction
Over the last decades, porphyrins and porphyrinoids have become well recognised as therapeutic agents in modern anticancer therapies such as photodynamic therapy (PDT).1 Porphyrins possess an interesting set of features that make them potential photosensitizers (PS) i.e. biocompatibility, high affinity towards cancer cells, absorption in the visible range of the electromagnetic spectrum, and efficient phototoxicity towards cancer cells. Despite the indisputable advantages of using porphyrins as PS, there are still challenges that need to be overcome to fully reveal their potential in PDT. The new generation of PS is expected to be more selective, with increased amphiphilicity and preferably with a red-shifted absorption maximum.2Nowadays, new synthetic strategies towards amphiphilic porphyrin systems are of great interest.3 Especially intriguing are the methods leading to glycoporphyrin hybrids, because these compounds may have a better solubility in polar solvents and they might exhibit an increased potential to aggregate nearby cancer cells.4,5 However, the synthesis of new porphyrin–sugar hybrids suffers from the absence of general methodologies.
The covalent binding of porphyrins with carbohydrates is mainly achieved via C–O,6 C–S,7 and C–N bonds,8 an amide or ester function,9 or a triazole linker.10 The C–C conjugated porphyrins with sugars might be considered particularly interesting as non-hydrolyzable hybrids. The well-recognized methodologies to obtain meso-glyco-substituted porphyrins are the Lindsey and the MacDonald strategy (Scheme 1A).11 However, the final products are often complex mixtures of atropoisomers that are difficult to interpret.
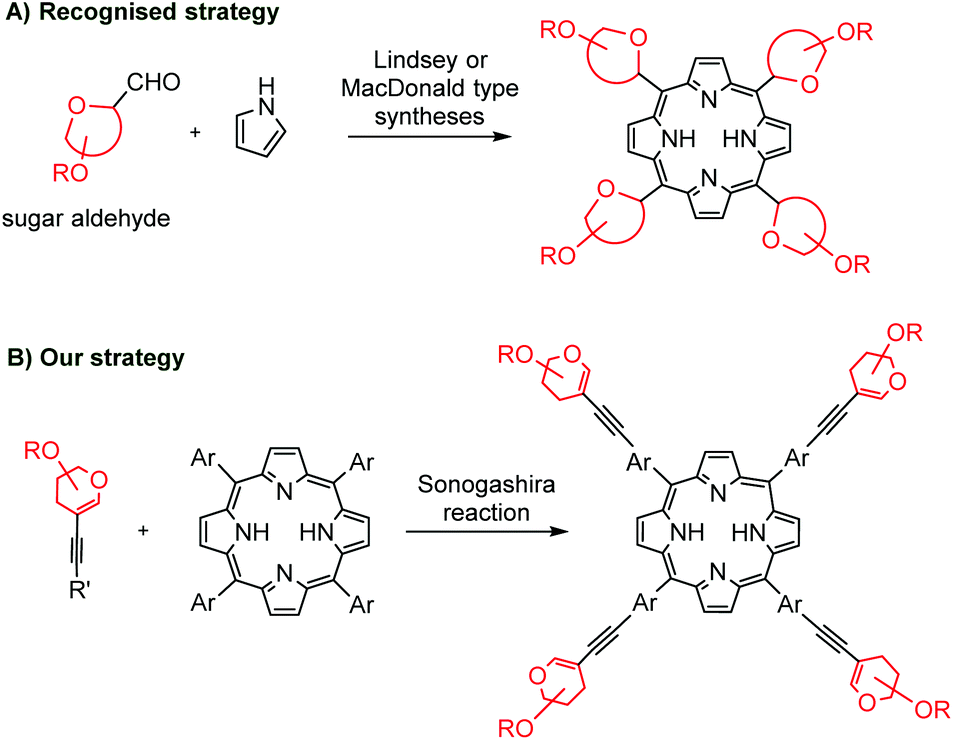 | ||
| Scheme 1 Strategies towards C–C linked glycoporphyrins. | ||
To the best of our knowledge, except for a singular example of a 12-step synthesis,12 the literature does not cover any other examples of methods to introduce carbohydrate units into meso-aryl rings via C–C bonds. What is more, palladium-catalysis has not yet been explored in the field of C–C linked glycoporphyrins.
On the other hand, in recent years, continuous progress has been made in the recognition of 2-haloglycals as convenient starting materials for metal-catalyzed reactions.13 New methodologies have been developed, focusing mostly on the creation of new C–C bonds via the Heck14 and Suzuki–Miyaura reactions,15 cyanation,16 or other transformations.17 More recently, carbonylative equivalents have been put in the spotlight,18 as well as reactions leading to new C-heteroatom bonds.19
Since the first report of palladium-catalyzed couplings between aryl halides and acetylenes in the presence of copper(I),20 the Sonogashira reaction has become one of the most reliable and default strategies to conjugate molecules via ethynyl linkers.21 The potential of this reaction has already been demonstrated for 2-iodoglycals, and conditions for the Sonogashira coupling have been developed for peracetylated sugar starting materials, however only relativily simple alkynes have been applied in these methodologies.22 On the other hand, the Sonogashira reaction has been widely exploited in the chemistry of porphyrins and other macroheterocycles.23 Despite this, no porphyrin–glycal hybrids have been developed till date using this strategy. This approach is particularly interesting since the possibility of using metal-catalysis opens a range of new routes towards C–C conjugated glycoporphyrins and therefore towards PS of a new generation.
Taking all of this into account, we focused our attention on the development of new suitable conditions for the Sonogashira cross-coupling to couple two groups of compounds that are quite different in terms of chemical origins: porphyrins and glycals (Scheme 1B). Herein, we present a new, short and efficient synthetic route towards non-hydrolyzable C–C conjugated glycoporphyrin hybrids.
2. Results and discussion
In the literature, there are two general approaches to introduce the carbohydrate moiety into macroheterocycles. The first one involves the prefunctionalization of the starting material by combining a simple scaffold with a sugar, followed by the formation of porphyrinoids. This approach gives the opportunity to easily obtain a diverse library of compounds. However, it is limited to acid-resistant functions and it is possible that porphyrin cyclisation step is a low-yield process. The second strategy is based on the creation of a macroheterocyclic system at first, followed by the introduction of a sugar unit in the second stage. In this approach, attention must be paid to the glycosylation step which should be performed very efficiently to avoid the separation of the complex, statistical mixtures resulting from low-yield reactions.A. Glycosylation prior to the porphyrin formation
We began our research with the exploration of the first strategy to conjugate perbenzylated 2-iodo-D-glucal (1) with the easily accessible 4-ethynylbenzaldehyde (2). The crucial transformation leading to the porphyrin was expected to be realized in the next stage via the Lindsey or the Adler–Longo method. Thus, the protective groups on the sugar moiety should be resistant to the acidid milieu. We chose the benzyl protecting groups as they were proven to be resistant to Lindsey's reaction conditions.11As the initial conditions for the conjugation of 1 with 2, we selected reagents inspired by the procedures developed for the Sonogashira cross-couplings of peracetylated glycals (Table 1, entries 1 and 2).22 However, in both cases, the procedures proved to be unsuitable for the chosen starting materials. We believe that this is due to the differences in the solubility of peracetylated glycals vs. perbenzylated ones and/or the stereoelectronic effects that make the oxidative addition step in the catalytic cycle more challenging.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (eq.) | CuI (eq.) | Other (eq.) | Yieldb |
| a General conditions: perbenzylated 2-iodo-D-glucal (0.110 mmol, 1.0 eq.), 4-ethynylbenzaldehyde (0.130 mmol, 1.2 eq.), catalyst (0.011 mmol, 0.1 eq.), copper(I) iodide (0.022 mmol, 0.2 eq.), toluene (0.5 mL), triethylamine (0.5 mL), 80 °C, 18 h, argon atmosphere. b Determined from the crude mixture by 1H NMR with the internal standard. c Reaction in 0.5 mL of solvent(s). d Corresponds to 85% of the isolated product. e Reaction at 60 °C. | |||||
| 1 | DMF | Pd(OAc)2 (0.1) | — | Cs2CO3 (1.5) | 0%c |
| 2 | Et3N | Pd(PPh3)2Cl2 (0.1) | 0.2 | — | 21%c |
| 3 | Toluene | Pd(PPh3)2Cl2 (0.1) | 0.2 | NEt3 (2.0) | 29%c |
| 4 | 1,4-Dioxane | Pd(PPh3)2Cl2 (0.1) | 0.2 | NEt3 (2.0) | 28%c |
| 5 | Toluene/NEt3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
Pd(PPh3)2Cl2 (0.1) | 0.2 | — | 44% c |
| 6 | Toluene/NEt3 (1:1) |
Pd(PPh3)2Cl2 (0.1) | 0.2 | — | 51% |
| 7 | Toluene/NEt3 (1:1) |
Pd(PPh3)4 (0.1) | 0.2 | — | 59% |
| 8 | Toluene/NEt3 (1:1) |
Pd(PPh3)4 (0.1) | — | — | 35% |
| 9 | Toluene/NEt3 (1:1) |
PdCl2 (0.1) | 0.2 | PPh3 (0.20) | 53% |
| 10 | Toluene/NEt3 (1:1) |
PdCl2 (0.1) | 0.2 | XPhos (0.20) | 63% |
| 11 |
Toluene/NEt
3
(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
Pd-XPhos-G3 (0.1) | 0.2 | XPhos (0.10) | 86% |
| 12 | Toluene/NEt3 (1:1) |
Pd-XPhos-G3 (0.1) | 0.1 | XPhos (0.10) | 85%e |
| 13 | Toluene/NEt3 (1:1) |
Pd-XPhos-G3 (0.05) | 0.05 | XPhos (0.05) | 75%e |
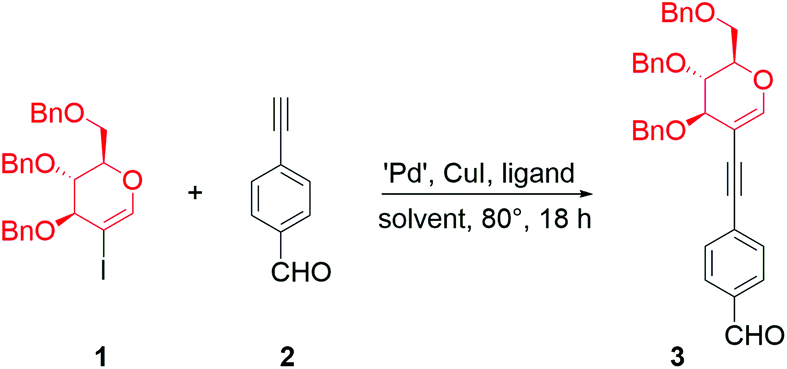
The lack of conversion of 1 and the decomposition of aldehyde 2 were observed when Cs2CO3 was used as a base (Table 1, entry 1). Changing the palladium source to Pd(PPh3)2Cl2 and the base to triethylamine (used herein as a solvent) slightly improved the conversion; however the product was obtained in a moderate yield (Table 1, entry 2).
Due to the lack of appropriate conditions for the coupling of 1 with 2, we decided to determine the optimized reaction conditions (for a detailed study, see the ESI†). The use of various solvents with 2 equivalents of triethylamine improved the yield only slightly (Table 1, entries 3 and 4). In Sonogashira cross-couplings, high yields are usually observed when triethylamine is used as a solvent, especially for copper-free variants. However, in our case, a modest yield was observed when triethylamine was used as the only solvent. We anticipated that using a mixture of solvents might limit the solubility issues while, on the other hand, providing quite high concentrations of triethylamine. Indeed, some noticeable improvement was observed when a mixture of toluene and triethylamine (1:1) was used as solvent (Table 1, entry 5), especially when the reaction was performed under diluted conditions (Table 1, entry 6).
Changing the catalytic system from Pd(PPh3)2Cl2 to palladium(0), i.e. Pd(PPh3)4, improved the yield to an acceptable level (Table 1, entry 7). The influence of copper(I) iodide was tested, proving its beneficial effect on the reaction (Table 1, entry 8); however, the reaction proceeded without the presence of a cocatalyst as well. Palladium(II) chloride with PPh3 as a ligand also catalysed the reaction, but an improvement in the yield was observed when the more sterically hindered ligand XPhos was used (Table 1, entry 9 vs. entry 10). Encouraged by these results, we decided to test Pd-XPhos-G3 as a well-recognized source of active palladium(0) species. The new catalytic system led to a very good yield (86%, Table 1, entry 11). Pd-XPhos-G3 enabled the lowering of the reaction temperature; however, halving the catalyst loading resulted in a slight decrease in the yield (Table 1 entry 12 vs. 13).24
With aldehyde 3 in hand, we next subjected it to porphyrin cyclocondensation conditions (Scheme 2). Despite several attempts, we were not able to detect porphyrin formation when Lindsey's procedure was applied to 3 (TFA or BF3·Et2O used as catalysts). Pyrrole polymerisation was mostly observed in reactions with a higher concentration of catalyst whereas lowering the amount of catalyst did not induce the condensation step and no conversion of 3 was observed. However, inspired by published results of porphyrin formation from 4-ethynylbenzaldehyde via the Adler–Longo strategy,25 we synthesized the expected product 5 in 10% yield. Since the reaction itself was hardly reproducible and sometimes led to a mixture of products that were difficult to separate, we decided to change the strategy and obtain the final glycoporphyrins using another approach.
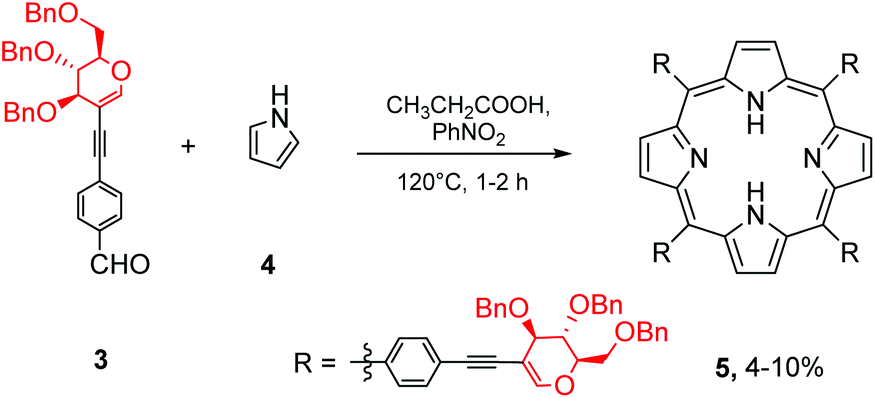 | ||
| Scheme 2 The Adler–Longo method in the synthesis of porphyrin 5. | ||
B. Glycosylation of porphyrins – benzyl group protection of the hydroxyl function
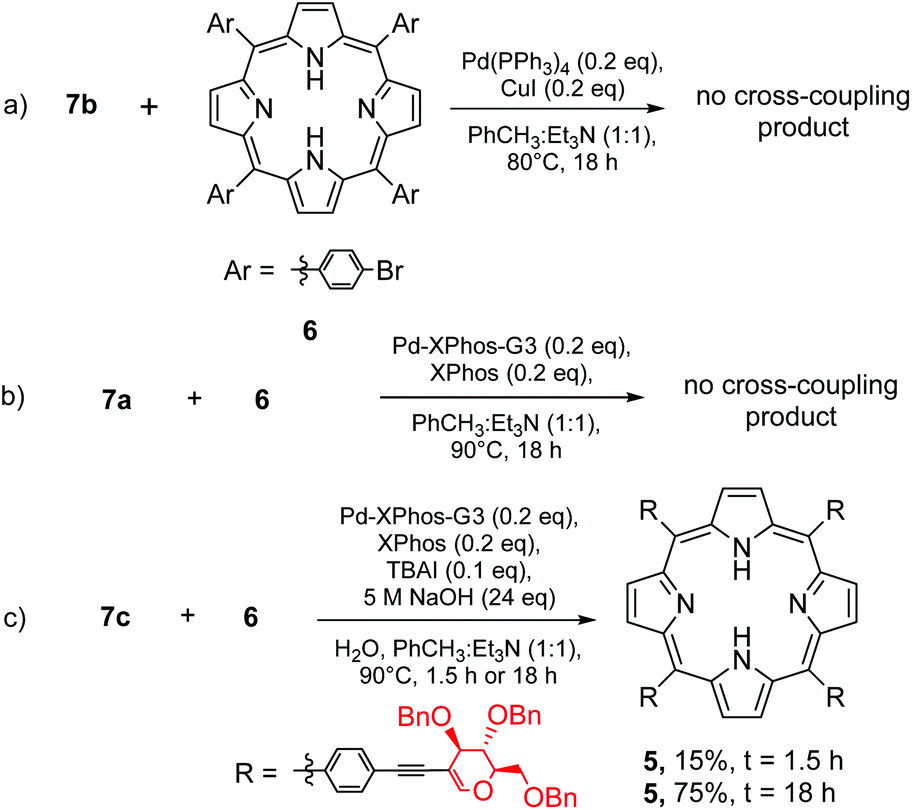
In this strategy we started with the synthesis of a porphyrin core, followed by the introduction of a glycal moiety via the Sonogashira reaction. As a model porphyrin we chose (5,10,15,20)-tetrakis(4-bromophenyl)porphyrin (6) because it is bench-stable and smoothly obtained from the reaction of 4-bromobenzaldehyde and pyrrole under Lindsey's conditions.26 The glycal starting materials (Scheme 3, 7a–7c) were obtained from perbenzylated 2-iodo-D-glucal under the optimised conditions described in Table 1.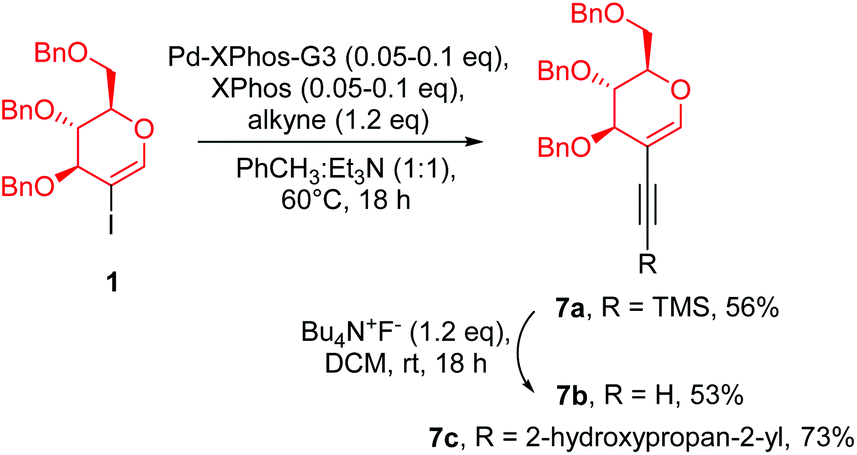 | ||
| Scheme 3 Synthesis of starting materials for the Sonogashira coupling. | ||
The route to synthesize porphyrin 5 from 6 required highly efficient conditions for the cross-coupling step. In the case of an incomplete process, a complicated mixture of products was obtained, making the separation process laborious.
At first, we allowed alkyne 7b to react with 6 under the standard conditions of Sonogashira cross-coupling (Scheme 4, path a). As a catalyst amount, we decided to use 0.20 equivalents of palladium(0) with regard to porphyrin which corresponded to 0.05 equivalents of palladium with regard to the aryl halide in the molecule. However, from the post-reaction mixture, unreacted 6 was isolated and a product resulting from the homocoupling of 7b was observed. This Glaser-type reactivity is a common side-reaction usually detected in unsuccessful Sonogashira transformations.21
 | ||
| Scheme 4 Variation of the Sonogashira cross-coupling reaction performed on porphyrin 6. | ||
To suppress this dimerization process, we decided to utilize 7a as the starting material and to apply the sila-Sonogashira reaction (Scheme 4, path b) to produce the desired alkyne 7bin situ. Despite that, this reaction also showed a similar result: porphyrin 6 was recovered and a product of homocoupling was observed. We concluded that a high concentration of compound 7b is undesirable and this was observed in both experiments, thus resulting in glycal homocoupling. To slow down the in situ production of 7b, another strategy to produce ethynyl compounds in a controlled manner was studied via β-alkynyl elimination.27 Indeed, the reaction of 7c with porphyrin 6 under conditions similar to those given in Table 1 led to the glycosylated product 5 in a very good overall yield of 76% (Scheme 4, path c). It seems that among all the tested variations under the Sonogashira conditions, the β-alkynyl elimination strategy provided the required amount of alkyne at the most optimal pace. Low concentration of alkyne 7b supressed the catalytic cycle path leading to homocoupling, and enabled a cross-coupling process. The desired product has been observed even after a shortening of reaction time to 90 min (Scheme 4c). Another crucial parameter for the porphyrin glycosylation strategy was using a suitable excess of glycal substrate. The change in the ratio between 6 and 7c from 1:8 to 1:6 resulted in a significant decrease in the reaction yield (32%) with the formation of a complex mixture of products.
With the discovery of an efficient route to glycoporphyrin 5, we faced the problem of removal of the benzyl group. Since unsaturated bonds were present in 5, the hydrogenolysis conditions were excluded. The first strategy involved the application of BCl3 in dichloromethane. However, the nature of this reagent limited the choice to a non-coordinating reaction solvent. Dichloromethane was an appropriate solvent to solubilize 5; however, the polarity of intermediates was increased significantly with progressive removal of the benzyl groups and their precipitation was observed in the reaction mixture. All of this resulted in the formation of a complex and inseparable mixture of semi-deprotected products. We tested several different solvents and their mixtures (CHCl3, THF, CH3CN, PhCH3, 1,4-dioxane, etc.) and higher temperatures; however, no change in the reaction outcome was observed. Polar solvents provided good solubility of intermediates and deactivated BCl3, while non-polar solvents led to the precipitation of intermediates. Acid hydrolysis did not bring about any improvement either (MsOH or 6 N HCl), similar to the strategies involving an exchange of the benzyl group with the acetyl one (AcCl with SnBr2 or TMSOTf in Ac2O).28 Since the persistence of the benzyl group terminated this synthetic pathway, we finally applied the glycosylation strategy to glycals with a group more prone to removal, namely the acetyl group.
C. Glycosylation of porphyrins –acetyl group protection of the hydroxyl function
The final approach towards glycoporphyrins was inspired by the experience gained from using perbenzylated glycals. As the most suitable model porphyrin, compound 6 was chosen. Glycal starting materials 9a and 9b were synthesized according to the described procedure,22b with a modification of solvent and products 9a and 9b were furnished in excellent yields (Scheme 5). Once again we observed the beneficial effect of a mixed solvent system on the reaction outcome, since the alkynylation process almost doubled the yield of 9b in comparison to its previously published yields.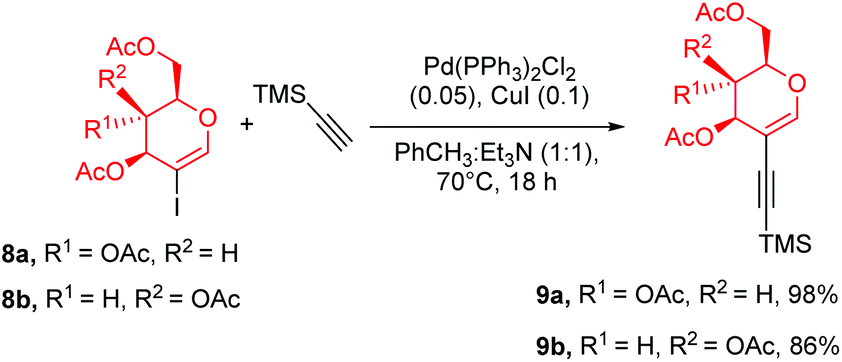 | ||
| Scheme 5 Synthesis of peracetylated starting materials. | ||
With peracetylated glycal as the starting material in hand, we started the optimization of the sila-Sonogashira cross-coupling with 6 (Table 2, for expanded studies, see the ESI†). Initially, the conditions, catalytic system, and reagent ratios described for 5 were applied (Scheme 4). The copper-free variant allowed the avoidance of the risk of porphyrin metalation that often occurs when copper(I) iodide is used in the reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (eq.) | Ligand (eq.) | Solvent | Yieldb |
| a General conditions: porphyrin 6 (0.024 mmol, 1.0 eq.), glycal 8a (0.190 mmol, 8.0 eq.), Bu4N+F− (0.194 mmol, 8.2 eq.), Pd-XPhos (0.005 mmol, 0.2 eq.), XPhos (0.005 mmol, 0.2 eq.), 1,4-dioxane (2.5 mL), triethylamine (2.5 mL), 18 h, 90 °C, argon atmosphere. b Yield of the isolated product. c The reaction was performed on peracetylated 2-ethynyl-D-glucal as a glycal substrate. d Reaction time was 1.5 h. | ||||
| 1 | Pd-XPhos-G3 (0.2) | XPhos (0.2) | Toluene/NEt3 (1:1) |
0%c |
| 2 | Pd-XPhos-G3 (0.2) | XPhos (0.2) | Toluene/NEt3 (1:1) |
56% |
| 3 | Pd-XPhos-G3 (0.2) | XPhos (0.2) | 1,4-dioxane/NEt3 (1:1) |
75% |
| 4 | Pd-XPhos-G3 (0.2) | XPhos (0.2) | 1,4-dioxane/NEt3 (1:1) |
71%d |
| 5 | Pd-XPhos-G3 (0.2) | XPhos (0.2) | 1,4-dioxane ( + NEt3, 8.0 eq.) | 42% |
| 6 | Pd-XPhos-G3 (0.2) | XPhos (0.2) | NEt3 | 13% |
| 7 | Pd-XPhos-G3 (0.15) | XPhos (0.15) |
1,4-dioxane/NEt
3
(1:1)
|
85% |
| 8 | Pd-XPhos-G3 (0.2) | — | 1,4-dioxane/NEt3 (1:1) |
60% |
| 9 | Pd-XPhos-G3 (0.2) | XPhos (0.4) | 1,4-dioxane/NEt3 (1:1) |
48% |
| 10 | PdCl2 (0.2) | XPhos (0.2) | 1,4-dioxane/NEt3 (1:1) |
45% |
| 11 | Pd-RuPhos-G3 (0.15) | RuPhos (0.15) | 1,4-dioxane/NEt3 (1:1) |
56% |
| 12 | Pd-XantPhos-G3 (0.15) | XantPhos (0.15) | 1,4-dioxane/NEt3 (1:1) |
42% |
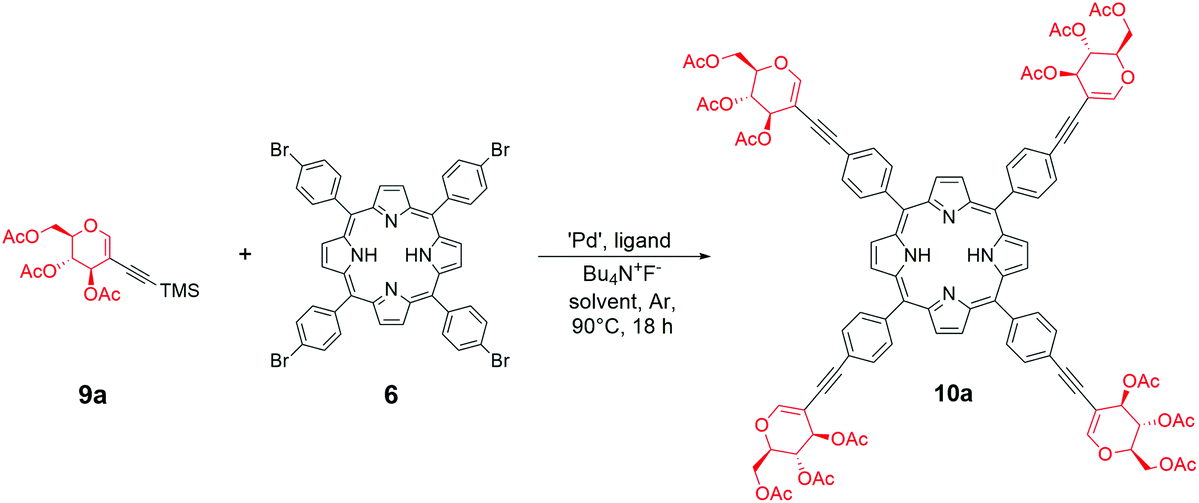
Similar to the results obtained with the perbenzylated analogue, the direct Sonogashira cross-coupling mainly resulted in the oxidative dimerization of the glycal (Table 2, entry 1). However, the sila-Sonogashira conditions in a mixture of solvents allowed glycoporphyrin 10a to be obtained in an acceptable yield (Table 2, entry 2). We observed the improvement in the reaction when 1,4-dioxane was used as a co-solvent (Table 2, entry 3). In contrast to the results obtained with the perbenzylated glycal 7a (Scheme 4, path a), probably, the catalytic pathway of the Sonogashira reaction was more efficient for compound 9a, and the reaction was almost complete after 90 min (Table 2, entry 4). It seems plausible that both starting materials differed in the electron density of their alkyne part, which has been proposed to be one of the crucial factors determining the final Sonogashira mechanism and the catalytic cycle rate.29 Using a mixture of solvents was beneficial for this reaction, providing porphyrin 6 with good solubility and a high concentration of triethylamine (Table 2, entry 3 vs. entries 5 and 6) and improving the conversion of the starting material significantly. Decreasing the amount of the catalyst to 0.15 equivalents furnished product 10a in a very good overall yield of 85% (Table 2, entry 7). The equimolar amount of ligand was optimal, since an increased amount resulted in a lower yield. This can be explained by the high stabilization of palladium(0). On the other hand, the lack of additional XPhos probably makes the catalyst insufficiently stable during the reaction (Table 2, entries 8 and 9). Other catalytic systems proved to be active in the reaction; however, their use resulted in a lower yield (Table 2, entries 10–12) and in the formation of a mixture of semi-glycosylated porphyrins.
To check the utility of the developed methodology towards glycoporphyrins, the optimal conditions used for the synthesis of 10a were then applied to another porphyrin ((5,10,15,20)-tetrakis(3-bromophenyl)porphyrin, 11) and tested on peracetylated galactal 9b as well (Scheme 6). The products of the sila-Sonogashira reaction (for 9a and 9b) were obtained in good to very good yields.
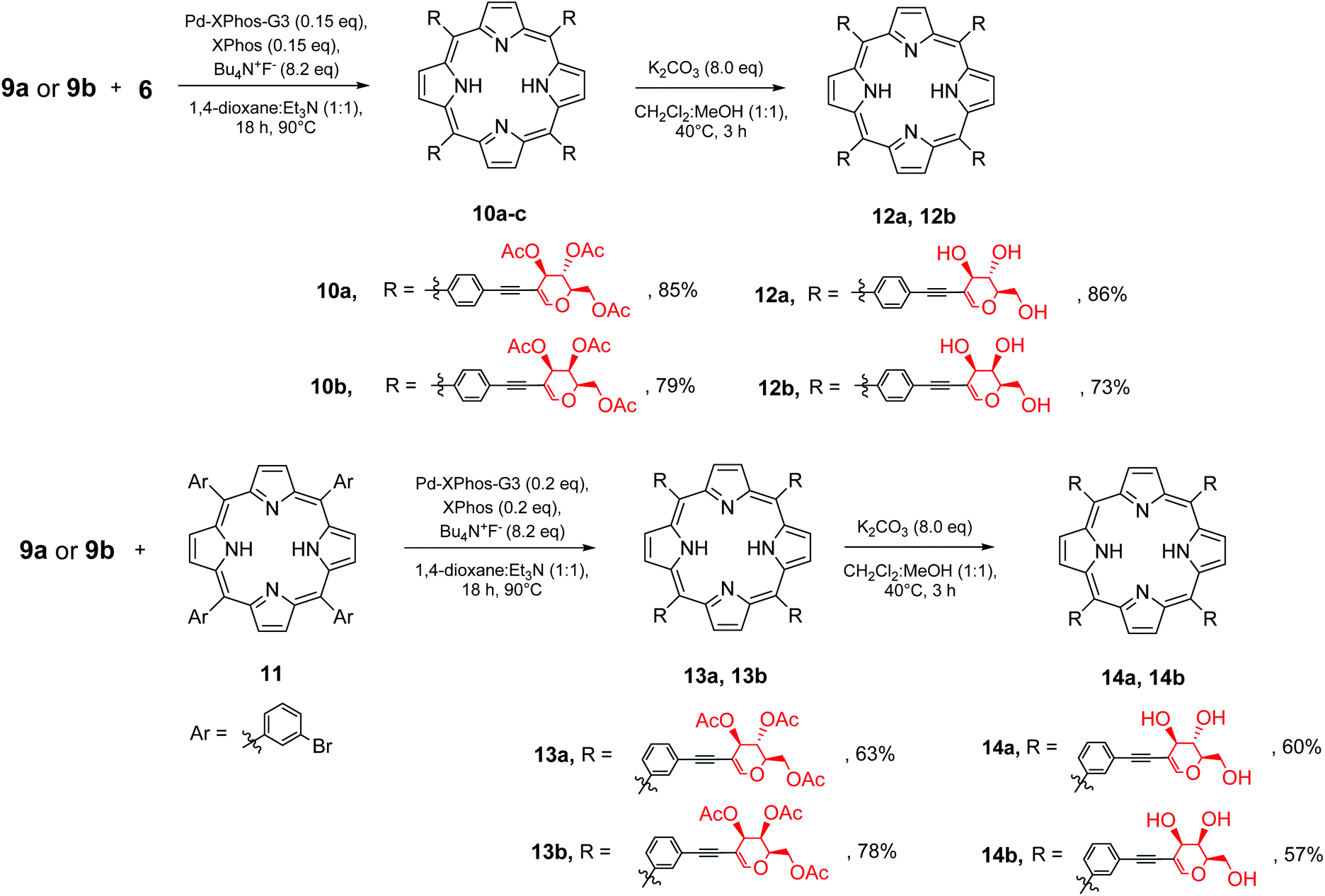 | ||
| Scheme 6 Synthesis of the final glycoporphyrin hybrids. | ||
The deprotection step required a small modification in comparison to the typical approach. Since porphyrins 10 and 13 were only slightly soluble in alcohol, in order to ensure efficient methanolysis, dichloromethane was used as the co-solvent, which significantly improved the solubility of the starting material.
The final products showed significantly improved solubility in polar solvents including DMSO and methanol. However, only a negligible solubility in water was observed for the final glycoporphyrins.
To further test our strategy, we decided to check if linker prolongation does not change the outcome of our methodology. In particular we became interested if more complex linkers, such as ethynyl–aryl–ethynyl substituents might be introduced into the meso-phenyl rings. As a linker we decided to use 4-ethynyl-(2-trimethylsilylethynyl)benzene. The installation of the linker on glycal 8a (Scheme 7) proceeded in a manner similar to previously discussed reactions, and product 9c was obtained in a very good yield. Then glycal 9c was reacted with porphyrin 6 under our developed conditions, affording product 10c in a good yield. This proved that this strategy could be used with the ethynyl–aryl–ethynyl linkers as well without any particular modifications.
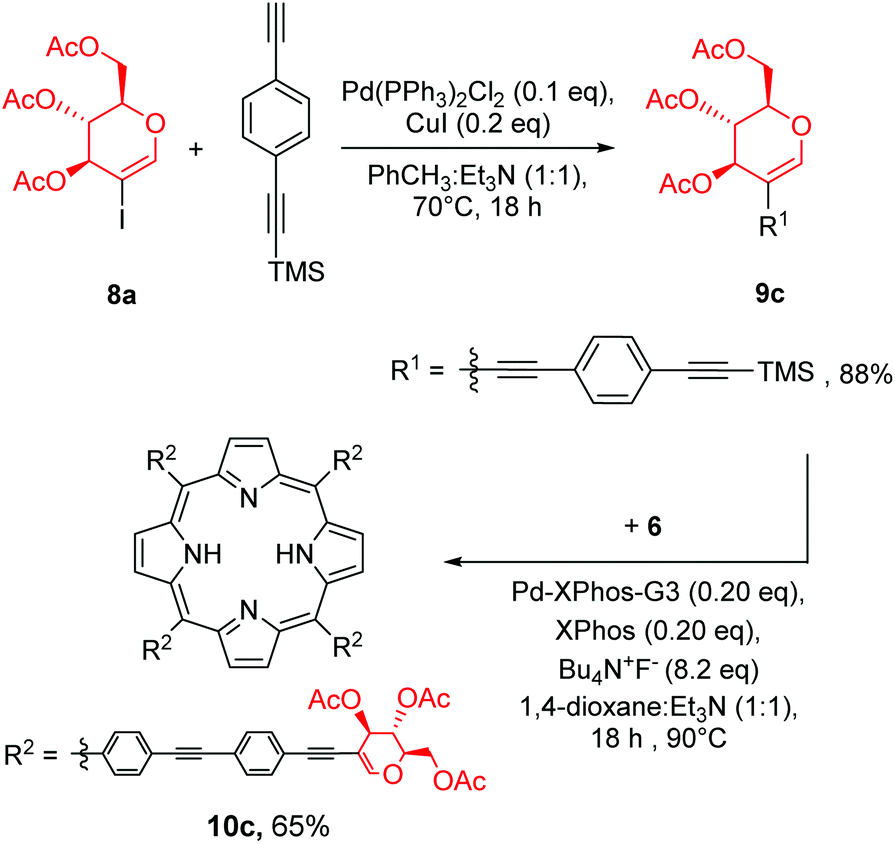 | ||
| Scheme 7 Developed methodology applied to a more complex linker. | ||
3. Conclusions
A new strategy to access glycoporphyrin hybrids has been developed using a methodology applying Sonogashira cross-coupling. The optimal conditions suitable for the cross-coupling of perbenzylated 2-iodoglycal with alkynes including 4-ethynylbenzaldehyde have been identified and after a slight modification, they were appropriate for coupling with halogenated porphyrins at the meso-aryl rings.The presented strategy is the first example of an organometallic cross-coupling between glycals and porphyrins leading to C–C conjugated products. In the context of the great importance of glycoporphyrins in the area of biomedicine, the reported strategy might be a starting point for diverse alternative strategies to be developed in the future. The biological evaluation of the deprotected hybrids is in progress.
A new, direct route towards glycoporphyrins has been established and will be expanded in future projects with respect to meeting the requirements of photodynamic therapy.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Warsaw University of Technology within the Excellence Initiative: Research University (IDUB) Program (NChem1). This publication is a part of the “Szkoła Orłów” project, co-financed by the European Social Fund under the Knowledge-Education-Development Operational Programme, Axis III, Higher Education for the Economy and Development, Measure 3.1, Competences In Higher Education.Notes and references
- (a) J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed; (b) J. Kou, D. Dou and L. Yang, Porphyrin photosensitizers in photodynamic therapy and its applications, Oncotarget, 2017, 8, 81591 CrossRef PubMed; (c) Handbook of Porphyrin Science, Vol. 1–25, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2010–2012 Search PubMed.
- (a) T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Recent Strategies to Develop Innovative Photosensitizers for Enhanced Photodynamic Therapy, Chem. Rev., 2021, 121, 13454 CrossRef CAS PubMed; (b) A. Oniszczuk, K. A. Wojtunik-Kulesza, T. Oniszczuk and K. Kasprzak, The potential of photodynamic therapy (PDT)—Experimental investigations and clinical use, Biomed. Pharmacother., 2016, 83, 912 CrossRef CAS PubMed.
- S. Pisarek, K. Maximova and D. Gryko, Strategies toward the synthesis of amphiphilic porphyrins, Tetrahedron, 2014, 70, 6685 CrossRef CAS.
- For reviews, see: (a) S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Glycosylated Porphyrins, Phthalocyanines, and Other Porphyrinoids for Diagnostics and Therapeutics, Chem. Rev., 2015, 115, 10261 CrossRef CAS PubMed; (b) H. Kataoka, H. Nishie, M. Tanaka, M. Sasaki, A. Nomoto, T. Osaki, Y. Okamoto and S. Yano, Potential of Photodynamic Therapy Based on Sugar-Conjugated Photosensitizers, J. Clin. Med., 2021, 10, 841 CrossRef CAS PubMed; (c) M. Lupu, Ph. Maillard, J. Mispelter, F. Poyer and C. D. Thomas, A glycoporphyrin story: from chemistry to PDT treatment of cancer mouse models, Photochem. Photobiol. Sci., 2018, 17, 1599 CrossRef CAS PubMed; (d) A. A. Aksenova, Y. L. Sebyakin and A. F. Mironov, Conjugates of Porphyrins with Carbohydrates, Russ. J. Bioorg. Chem., 2003, 29, 201 CrossRef CAS PubMed.
- Selected examples: (a) P. M. R. Pereira, W. Rizvi, N. V. S. D. K. Bhupathiraju, N. Berisha and C. M. Drain, Carbon-1 versus Carbon-3 Linkage of d-Galactose to Porphyrins: Synthesis, Uptake, and Photodynamic Efficiency, Bioconjugate Chem., 2018, 29, 306 CrossRef CAS PubMed; (b) H. Kataoka, H. Nishie, N. Hayashi, M. Tanaka, A. Nomoto, S. Yano and T. Joh, New photodynamic therapy with next-generation photosensitizers, Ann. Transl. Med., 2017, 5, 183 CrossRef PubMed; (c) P. M. R. Pereira, S. Silva, J. S. Ramalho, C. M. Gomes, H. Girão, J. A. S. Cavaleiro, C. A. F. Ribeiro, J. P. C. Tomé and R. Fernandes, The role of galectin-1 in in vitro and in vivo photodynamic therapy with a galactodendritic porphyrin, Eur. J. Cancer, 2016, 68, 60 CrossRef CAS PubMed.
- Selected examples: (a) M. Nakai, T. Maeda, T. Mashima, S. Yano, S. Sakuma, E. Otake, A. Morita and Y. Nakabayashi, Syntheses and photodynamic properties of glucopyranoside-conjugated indium(III) porphyrins as a bifunctional agent, J. Porphyrins Phthalocyanines, 2013, 17, 1173 CrossRef CAS; (b) J. P. C. Tomé, M. G. P. M. S. Neves, A. C. Tomé, J. A. S. Cavaleiro, A. F. Mendonça, I. N. Pegado, R. Duarte and M. L. Valdeira, Synthesis of glycoporphyrin derivatives and their antiviral activity against herpes simplex virus types 1 and 2, Bioorg. Med. Chem., 2005, 13, 3878 CrossRef PubMed; (c) D. Aicher, A. Wiehe and C. B. W. Stark, Synthesis of Glycoporphyrins Using Trichloroacetimidates as Glycosyl Donors, Synlett, 2010, 395 CAS; (d) V. Sol, V. Chaleix, Y. Champavier, R. Granet, Y.-M. Huang and P. Krausz, Glycosyl bis-porphyrin conjugates: Synthesis and potential application in PDT, Bioorg. Med. Chem., 2006, 14, 7745 CrossRef CAS PubMed; (e) G. Fülling, D. Schröder and B. Franck, Water-Soluble Porphyrin Diglycosides with Photosensitizing Properties, Angew. Chem., Int. Ed. Engl., 1989, 28, 1519 CrossRef; (f) D. Oulmi, P. Maillard, J.-L. Guerquin-Kern, C. Huel and M. Momenteau, Glycoconjugated Porphyrins. 3. Synthesis of Flat Amphiphilic Mixed meso-(Glycosylated aryl)arylporphyrins and Mixed meso-(Glycosylated aryl)alkylporphyrins Bearing Some Mono- and Disaccharide Groups, J. Org. Chem., 1995, 60, 1554 CrossRef CAS.
- Selected examples: (a) X. Chen, L. Hui, D. A. Foster and C. M. Drain, Efficient Synthesis and Photodynamic Activity of Porphyrin-Saccharide Conjugates: Targeting and Incapacitating Cancer Cells, Biochemistry, 2004, 43, 10918 CrossRef CAS PubMed; (b) V. Pandey, M. K. Raza, P. Joshi and I. Gupta, Synthesis of Water-Soluble Thioglycosylated trans-A2B2 Type Porphyrins: Cellular Uptake Studies and Photodynamic Efficiency, J. Org. Chem., 2020, 85, 6309 CrossRef CAS PubMed; (c) A. Fadlan, H. Tanimoto, T. Ito, Y. Aritomi, M. Ueno, M. Tokuda, S. Hirohara, M. Obata, T. Morimoto and K. Kakiuchi, Synthesis, photophysical properties, and photodynamic activity of positional isomers of TFPP-glucose conjugates, Bioorg. Med. Chem., 2018, 26, 1848 CrossRef CAS PubMed; (d) J.-F. Longevial, K. El Cheikh, D. Aggad, A. Lebrun, A. van der Lee, F. Tielens, S. Clément, A. Morère, M. Garcia, M. Gary-Bobo and S. Richeter, Porphyrins Conjugated with Peripheral Thiolato Gold(I) Complexes for Enhanced Photodynamic Therapy, Chem. – Eur. J., 2017, 23, 14017 CrossRef CAS PubMed; (e) I. Sylvain, R. Zerrouki, R. Granet, Y. M. Huang, J. F. Lagorce, M. Guilloton, J. C. Blais and P. Krausz, Synthesis and biological evaluation of thioglycosylated porphyrins for an application in photodynamic therapy, Bioorg. Med. Chem., 2002, 10, 57 CrossRef CAS PubMed; (f) S. Ahmed, E. Davoust, H. Savoie, A. N. Boa and R. W. Boyle, Thioglycosylated cationic porphyrins–convenient synthesis and photodynamic activity in vitro, Tetrahedron Lett., 2004, 45, 6045 CrossRef CAS.
- (a) P. Wyrębek, A. Osuch-Kwiatkowska, Z. Pakulski, S. Jarosz and S. Ostrowski, The synthesis of sugar-decorated hydrophilic porphyrins, J. Porphyrins Phthalocyanines, 2013, 17, 384 CrossRef; (b) H. Li and L. Czuchajowski, Ribofuranosides N-substituted with meso-porphyrin as nucleoside-like compounds, Tetrahedron Lett., 1994, 35, 1629 CrossRef CAS.
- Selected examples: (a) F. Figueira, L. M. O. Lourenço, M. G. P. M. S. Neves, J. A. S. Cavaleiro and J. P. C. Tomé, Synthesis and characterization of novel 5-monocarbohydrate-10,20-bis-aryl-porphyrins, J. Porphyrins Phthalocyanines, 2019, 23, 1 CrossRef; (b) S. Ballut, D. Naud-Martin, B. Loock and P. Maillard, A Strategy for the Targeting of Photosensitizers. Synthesis, Characterization, and Photobiological Property of Porphyrins Bearing Glycodendrimeric Moieties, J. Org. Chem., 2011, 76, 2010 CrossRef CAS PubMed; (c) S. Ballut, A. Makky, B. Loock, J.-P. Michel, P. Maillard and V. Rosilio, New strategy for targeting of photosensitizers. Synthesis of glycodendrimeric phenylporphyrins, incorporation into a liposome membrane and interaction with a specific lectin, Chem. Commun., 2009, 224 RSC; (d) Z.-J. Wang, B. Chauvin, P. Maillard, F. Hammerer, D. Carez, A. Croisy, C. Sandré, S. Chollet-Martin, P. Prognon, J.-L. Paul, J. Blais and A. Kasselouri, Glycodendrimeric phenylporphyrins as new candidates for retinoblastoma PDT: Blood carriers and photodynamic activity in cells, J. Photochem. Photobiol., B, 2012, 115, 16 CrossRef CAS PubMed.
- Selected examples: (a) C. F. Dixon, A. N. Nottingham, A. F. Lozano, J. A. Sizemore, L. A. Russell, C. Valiton, K. L. Newell, D. Babin, W. T. Bridges, M. R. Parris, D. V. Shchirov, N. L. Snyder and J. V. Ruppel, Synthesis and evaluation of porphyrin glycoconjugates varying in linker length: preliminary effects on the photodynamic inactivation of Mycobacterium smegmatis, RSC Adv., 2021, 11, 7037 RSC; (b) M. C. Bennion, M. A. Burch, D. G. Dennis, M. E. Lech, K. Neuhaus, N. L. Fendler, M. R. Parris, J. E. Cuadra, C. F. Dixon, G. T. Mukosera, D. N. Blauch, L. Hartmann, Ni. L. Snyder and J. V. Ruppel, Synthesis of Porphyrin and Bacteriochlorin Glycoconjugates through CuAAC Reaction Tuning, Eur. J. Org. Chem., 2019, 6496 CrossRef CAS PubMed; (c) R. Daly, G. Vaz, A. M. Davies, M. O. Senge and E. M. Scanlan, Synthesis and Biological Evaluation of a Library of Glycoporphyrin Compounds, Chem. – Eur. J., 2012, 18, 14671 CrossRef CAS PubMed; (d) G. T. Mukosera, T. P. Adams, R. F. Rothbarth, H. Langat, S. Akanda, R. G. Barkley, R. D. Dolewski, J. V. Ruppel and N. L. Snyder, Synthesis of glycosylated zinc(II) 5,15-diphenylporphyrin and zinc(II) 5,10,15,20-tetraphenylporphyrin analogs using Cu-catalyzed azide-alkyne 1,3-dipolar cycloaddition reactions, Tetrahedron Lett., 2015, 56, 73 CrossRef CAS.
- (a) M. Cornia, C. Valenty, S. Capacchi and P. Gozzini, Synthesis, characterisation and conformational studies of lipophilic, amphiphilic and water-soluble C-glyco-conjugated porphyrins, Tetrahedron, 1998, 54, 8091 CrossRef CAS; (b) N. Ono, M. Bougauchi and K. Maruyama, Water-Soluble Porphyrins with Four Sugar Molecules, Tetrahedron Lett., 1992, 33, 1629 CrossRef CAS; (c) P. Stepanek, M. Dukh, D. Saman, J. Moravcova, L. Kniezo, D. Monti, M. Venanzi, G. Mancini and P. Drasar, Synthesis and solvent driven self-aggregation studies of meso-“C-glycoside-porphyrin derivatives, Org. Biomol. Chem., 2007, 5, 960 RSC; (d) M. Malachowska, C. Sperduto, M. Darmostuk, D. Monti, M. Venanzi, G. Mancini, C. W. D'Acunto, J. Králová, T. Ruml, Z. Wimmer and P. B. Drašar, Porphyrins with directly meso-attached disaccharide moieties: Synthesis, self-assembly and cellular study, J. Porphyrins Phthalocyanines, 2016, 20, 773 CrossRef CAS; (e) G. Casiraghi, M. Cornia, F. Zanardi, G. Rassu, E. Ragg and R. Bortolini, Synthesis and Characterization of Porphyrin-Sugar Carbon Conjugates, J. Org. Chem., 1994, 59, 1801 CrossRef CAS.
- P. Pasetto, X. Chen, C. M. Drain and R. W. Franck, Synthesis of hydrolytically stable porphyrin C- and S-glycoconjugates in high yields, Chem. Commun., 2001, 81 RSC.
- For a review, see: N. Hussain, A. Ahmed and D. Mukherjee, 2-Halo Glycals as “Synthon” for 2-C-Branched Sugar: Recent Advances and Applications in Organic Synthesis, Asian J. Org. Chem., 2020, 9(6), 882 CrossRef CAS.
- S. Dharuman and Y. D. Vankar, N-Halosuccinimide/AgNO3-Efficient Reagent Systems for One-Step Synthesis of 2-Haloglycals from Glycals: Application in the Synthesis of 2C-Branched Sugars via, Heck Coupling Reactions, Org. Lett., 2014, 16, 1172 CrossRef CAS PubMed.
- (a) I. Cobo, M. L. Matheu, S. Castillón, O. Boutureira and B. G. Davis, Phosphine-Free Suzuki–Miyaura Cross-Coupling in Aqueous Media Enables Access to 2-C-Aryl-Glycosides, Org. Lett., 2012, 14, 1728 CrossRef CAS PubMed; (b) S. Jana and J. D. Rainier, The Synthesis of Indoline and Benzofuran Scaffolds Using a Suzuki–Miyaura Coupling/Oxidative Cyclization Strategy, Org. Lett., 2013, 15, 4426 CrossRef CAS PubMed.
- M. Malinowski, T. V. Tran, M. de Robichon, N. Lubin-Germain and A. Ferry, Mild Palladium-Catalyzed Cyanation of Unprotected 2-Iodoglycals in Aqueous Media as Versatile Tool to Access Diverse C2-Glycoanalogues, Adv. Synth. Catal., 2020, 362, 1184 CrossRef CAS.
- (a) J. Liu, P. Han, J.-X. Liao, Y.-H. Tu, H. Zhou and J.-S. Sun, Palladium-Catalyzed Cross-Coupling of 2-Iodoglycals with N-Tosylhydrazones: Access to 2-C-Branched Glycoconjugates and Oxadecalins, J. Org. Chem., 2019, 84(14), 9344 CrossRef CAS PubMed; (b) J. Mestre, A. Lishchynskyi, S. Castillon and O. Boutureira, Trifluoromethylation of Electron-Rich Alkenyl Iodides with Fluoroform-Derived “Ligandless” CuCF3, J. Org. Chem., 2018, 83, 8150 CrossRef CAS PubMed; (c) A. Ahmed, N. Hussain, M. Bhardwaj, A. K. Chhalodia, A. Kumar and D. Mukherjee, Palladium catalysed carbonylation of 2-iodoglycals for the synthesis of C-2 carboxylic acids and aldehydes taking formic acid as a carbonyl source, RSC Adv., 2019, 9, 22227 RSC.
- Selected examples: (a) N. Hussain, M. Bhardwaj, A. Ahmed and D. Mukherjee, Synthesis of Sugar-Based Enones and Their Transformation into 3,5-Disubstituted Furans and 2-Acyl-Substituted 1,2,3-Trideoxy Sugars in the Presence of Lewis Acids, Org. Lett., 2019, 21, 3034 CrossRef CAS PubMed; (b) A. A. Soares-Paulino and H. A. Stefani, Synthesis of Diverse C2-Glyco-Acyl Azides and -Ureas by Palladium-Catalyzed Carbonylation Coupling of 2-Iodoglycals, Eur. J. Org. Chem., 2020, 3847 CrossRef CAS; (c) A. Bordessa, A. Ferry and N. Lubin-Germain, Access to Complex C2-Branched Glycoconjugates via Palladium-Catalyzed Aminocarbonylation Reaction of 2-Iodoglycals, J. Org. Chem., 2016, 81, 12459 CrossRef CAS PubMed; (d) M. P. Darbem, K. S. Kanno, I. M. de Oliveira, C. H. A. Esteves, D. C. Pimenta and H. A. Stefani, Synthesis of amidoglucals and glucal esters via carbonylative coupling reactions of 2-iodoglucal using Mo(CO)6 as a CO source, New J. Chem., 2019, 43, 696 RSC; (e) M. Robichon, A. Bordessa, N. Lubin-Germain and A. Ferry, “CO” as a Carbon Bridge to Build Complex C2-Branched Glycosides Using a Palladium-Catalyzed Carbonylative Suzuki–Miyaura Reaction from 2-Iodoglycals, J. Org. Chem., 2019, 84, 3328 CrossRef PubMed; (f) M. P. Darbem, H. A. Esteves, I. M. de Oliveira, D. C. Pimenta and H. A. Stefani, Palladium-Catalyzed Thio- and Selenocarbonylation of 2-Iodoglycals, ChemCatChem, 2020, 12, 576 CrossRef CAS.
- (a) R. A. A. Al-Shuaeeb, D. Montoir, M. Alami and S. Messaoudi, Synthesis of (1→2)-S-Linked Saccharides and S-Linked Glycoconjugates via a Palladium-G3-XantPhos Precatalyst Catalysis, J. Org. Chem., 2017, 82, 6720 CrossRef CAS PubMed; (b) M. Malinowski, C. Banoun, M. de Robichon, N. Lubin-Germain and A. Ferry, Glycosamine derivatives through metal-catalyzed C-N bond formation on protected and unprotected 2-iodoglycals, Eur. J. Org. Chem., 2021, 1521 CrossRef CAS; (c) O. Monasson, M. Malinowski, N. Lubin-Germnain and A. Ferry, Hirao cross-coupling reaction as efficient tool to build non-natural C2-phosphonylated sugars, Synthesis, 2022 DOI:10.1055/a-1709-3305 , in press.
- K. Sonogashira, Y. Tohda and N. Hagihara, A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines, Tetrahedron Lett., 1975, 50, 4467 CrossRef.
- (a) R. Chinchilla and C. Nájera, Recent advances in Sonogashira reactions, Chem. Soc. Rev., 2011, 40, 5084 RSC; (b) R. Chinchilla and C. Nájera, The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- (a) A. Shamim, S. N. S. Vasconcelos, B. Ali, L. S. Madureira, J. Zukerman-Schpector and H. A. Stefani, Ligand and copper free Sonogashira coupling to achieve 2-alkynyl d-glucal derivatives: regioselective electrophile promoted nucleophilic 5-endo-dig cyclization, Tetrahedron Lett., 2015, 56, 5836 CrossRef CAS; (b) D. C. Koester and D. B. Werz, Sonogashira–Hagihara reactions of halogenated glycals, Beilstein J. Org. Chem., 2012, 8, 675 CrossRef CAS PubMed.
- (a) S. Hiroto, Y. Miyake and H. Shinokubo, Synthesis and Functionalization of Porphyrins through Organometallic Methodologies, Chem. Rev., 2017, 117, 2910 CrossRef CAS PubMed; (b) H. Shinokubo and A. Osuka, Marriage of porphyrin chemistry with metal-catalysed reactions, Chem. Commun., 2009, 1011 RSC.
- We have observed some differences in the reactivity depending on the source of 1. Usually, lower yields were obtained if 1 was synthesized from commercially available perbenzylated D-glucal, while best yields were achieved when perbenzylated D-glucal was synthesized according to the procedure described in the ESI.† The exact nature of this phenomenon is under investigation.
- G. Ji, Z. Yang, Y. Zhao, H. Zhang, B. Yu, J. Xu, H. Xua and Z. Liu, Synthesis of metalloporphyrin-based conjugated microporous polymer spheres directed by bipyridine-type ligands, Chem. Commun., 2015, 34, 7352 RSC.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions, J. Org. Chem., 1987, 5, 827 CrossRef.
- H. F. Chow, C. W. Wan, K. H. Low and Y. Y. Yeung, A Highly Selective Synthesis of Diarylethynes and Their Oligomers by a Palladium-Catalyzed Sonogashira Coupling Reaction under Phase Transfer Conditions, J. Org. Chem., 2001, 66, 1910 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's protective groups in organic synthesis, John Wiley & Sons, Inc., USA, 2007 Search PubMed.
- T. Ljungdahl, T. Bennur, A. Dallas, H. Emtenäs and J. Mårtensson, Two Competing Mechanisms for the Copper-Free Sonogashira Cross-Coupling Reaction, Organometallics, 2008, 27, 2490 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1qo01909k |
| ‡ Authors contributed equally. |
| This journal is © the Partner Organisations 2022 |