
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Room temperature synthesis of perylene diimides facilitated by high amic acid solubility†
Markus C.
Kwakernaak
 ab,
Marijn
Koel
a,
Peter J. L.
van den Berg
a,
Erik M.
Kelder
b and
Wolter F.
Jager
*a
ab,
Marijn
Koel
a,
Peter J. L.
van den Berg
a,
Erik M.
Kelder
b and
Wolter F.
Jager
*a
aDepartment of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: W.F.Jager@tudelft.nl
bDepartment of Radiation Science and Technology/Reactor Institute Delft, Delft University of Technology, 2629 JB Delft, The Netherlands
First published on 10th January 2022
Abstract
A novel protocol for the synthesis of perylene diimides (PDIs), by reacting perylene dianhydride (PDA) with aliphatic amines is reported. Full conversions were obtained at temperatures between 20 and 60 °C, using DBU as the base in DMF or DMSO. A “green” synthesis of PDIs, that runs at higher temperatures, was developed using K2CO3 in DMSO. The reaction sequence for the imidization process, via perylene amic acid intermediates (PAAs), has been confirmed experimentally aided by the synthesis and full characterization of stable model amic acid salts and amic esters. Kinetic studies, using absorption spectroscopy, have established that PDI formation proceeds via fast amic acid formation, followed by a slow conversion to imides. Solubility of the intermediate PAA salts is found to be low and rate-limiting. Based on this finding, quantitative PDI synthesis at room temperature was achieved by diluting the reaction mixture with water, the solvent in which PAA salts have better solubility. Thus, the otherwise harsh synthesis of PDIs has been transformed into an extremely convenient functional group tolerant and highly efficient reaction that runs at room temperature.
Introduction
Perylene-3,4,9,10-tetracarboxylic acid derivatives (PTCAs) form a class of organic dyes and pigments, from which perylene-3,4,9,10-tetracarboxylic diimides (PDIs) are the most abundantly used representatives.1 These compounds have very benign optical properties, which has resulted in large scale application of PDIs as dyes and pigments.2 Moreover, PDIs are well-suited for utilization as functional materials, mostly in the optoelectronic domain.3 Current research efforts are focussed on the use of PDIs as non-fullerene acceptors in photovoltaics,4 light harvesting antenna molecules,5 electrode materials in batteries,6 fluorescent probes7 and photocatalysts for the dehalogenation of aromatic compounds.8Despite this long history and intense interest by scientists from a wide range of disciplines, the synthesis of PDIs is generally performed under harsh conditions, with limited functional group tolerance.9 PDIs are routinely synthesised by the “Langhals method”, reacting perylene-3,4,9,10-tetracarcoxylic dianhydride (PDA) with primary amines in molten imidazole at 140–180 °C, using zinc acetate as a catalyst.10 This method has been the gold standard for decades and has proven itself to be very reliable and universally applicable. Nevertheless, a milder, greener and more user-friendly synthesis would be desirable, in order to make more PDIs accessible to a larger scientific audience.
The harsh conditions in the “Langhals method” appear to originate from either the low reactivity of the amic acid intermediates, the low solubility of starting compounds and intermediates, or both. The more soluble bay-halogenated PDAs generally undergo imidization reactions at milder conditions, typically using organic acids as catalyst and (co)solvent,11 while the imidization of 4-substituted naphthyl anhydrides proceeds at even milder conditions,12 typically in refluxing ethanol. These observations suggest that solubility is the major driver for applying harsh reaction conditions in the current PDI syntheses.
Greener methods for PDI synthesis have been reported recently, notably a solvent-less method using a twin screw extruder13 and a method in which water at high temperatures and elevated pressure is used.14 Both methods work particularly well for aliphatic amines and are definitely green(er). Still these reactions require high temperatures, which limits functional group tolerance. Interestingly, when using water at milder temperatures, around 60 °C, PDIs are not formed in significant quantities. Instead, perylene-3,4-dicarboxylic acid monoanhydride-9,10-dicarboxylic acid monoaimides (PMAMIs) are the major reaction products.15 Most likely, anhydride ring opening by hydroxide ions,16 resulting in the formation of adjacent dicarboxylates on the perylene scaffold is responsible for the observed PMAMI formation.17–19 It should be mentioned nevertheless, that partial hydrolysis of PDA to perylene-3,4-dicarboxylic monoanhydride-9,10-dicarboxyates, followed by a selective imidization at the anhydride position is a highly beneficial reaction that has been exploited for efficient syntheses of aliphatic PMAMIs and N-desymmetrized PDIs.20
In contrast to imidization, esterification of PDA has been achieved at mild reaction conditions,21 even at room temperature,22 see Scheme 1. In the first step of this reaction an alcohol opens the anhydride in the presence of the strong base DBU. The perylene-3,9-dicarboxylic acid-4,10-dicarboxylic dialkyl esters 2 resulting from the first reaction step are highly soluble and subsequently react with an alkyl halide in a slower rate-determining step to produce perylene-3,4,9,10-tetracarboxylic tetra-alkyl esters (PTEs 3) in almost quantitative yields. Interestingly, in a variation of this reaction, the synthesis of an imide at room temperature, postulated to proceed via an amic acid, has been reported. Although the reported synthetic procedure, exploiting a 3-day aminolysis reaction, may not be the most practical one, this finding suggests that PDI synthesis at mild reaction conditions is a realistic option.22
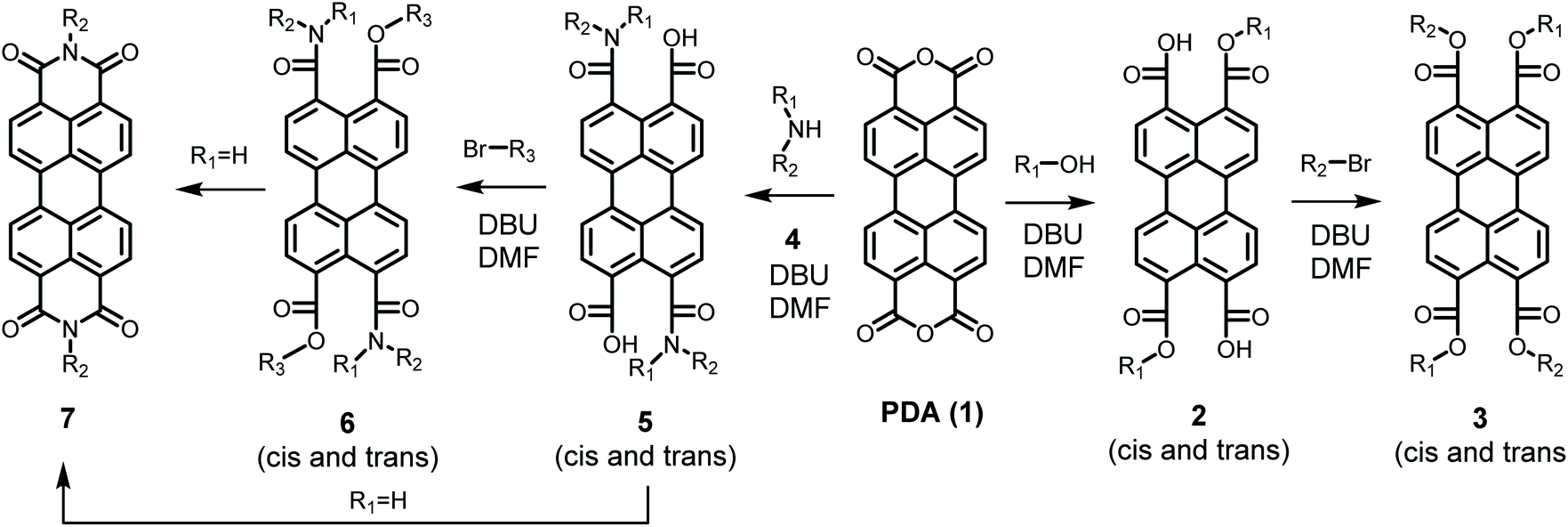 | ||
| Scheme 1 Room temperature synthesis of PTEs (3) PDAAs (5), PDAEs (6) and PDIs (7). | ||
Additional information suggesting that PDI synthesis at low temperatures is feasible, comes from mechanistic studies on naphthalene-1,8-dicarboxylic amic acid (NMAA)23 and naphthalene-1,3,4,8-tetracarboxylic diamic acid (NDAA).24 NMAA has been synthesised by a reaction of naphthalene-1,8-dicarboxylic anhydride (NMA) with butyl amine at low temperatures.25 At high pH values in water, NMAA forms the corresponding imide, naphthalene-1,8-dicarboxylic imide (NMI), while at low pH values the anhydride NMA is formed. These reactions are reversible and already proceeds at appreciable rates at temperatures as low as 30 °C. These studies suggest that formation of PDIs from PDA via corresponding amic acids may take place at low temperatures and high pH values, provided that a suitable polar solvent is identified. This may not be a trivial event in the light of the low solubility of most PTCA derivatives. Also it should be mentioned that the chemistry of PTCAs may differ from those of the corresponding naphthalene derivatives in other aspects than solubility. For example perylene-3,4,9,10-tetracarboxilic acid chloride has never been prepared10b whereas the analogous naphthyl-1,8-di26 and 1,3,4,8-tetracid chlorides27 are readily available by conventional means.
In this work we report the synthesis of PDIs by reacting PDA with primary aliphatic amines at low temperatures, in a manner analogous to the synthesis of PTEs depicted in Scheme 1. Reaction conditions for the imidization of PDA under “standard” conditions, with DBU as the base in the solvent DMF, were optimized. In addition an entirely Green synthesis of PDIs, using DMSO and K2CO3, was developed. To identify the reactive intermediates formed during the imidization reaction, stable model amic acids have been synthesised by the reaction of PDA with secondary amines. The (photo)physical properties of these compounds were determined and their spectra were compared with those of the reaction intermediates to support the proposed mechanism of imide formation. A kinetic model of the imidization reactions has been constructed and tested by monitoring imidization reactions in real time. Based on the mechanistic insights obtained from these experiments the reaction conditions were optimized further to achieve quantitative imide formation at room temperature.
Results and discussion
General reaction and proposed reaction mechanism
When secondary amines were reacted with PDA 1, using the synthetic protocol for the synthesis of PTEs, perylene-3,4,9,10-tetracarboxylic-diamic esters PDAE 6 were formed via perylene-3,4,9,10-tetracarboxylic-diamic acid PDAA 5, see Scheme 1. Both compounds have been isolated and fully characterized, vide infra. When primary amines were employed, using the same procedure, however, PDIs 7 were obtained in appreciable yields. The fact that PDIs are formed is not surprising as such, since amic acids and amic esters are known to react further to form imides. The observation that this reaction takes place at room temperature already was surprising to us and clearly indicates that PDI synthesis can be performed at very mild conditions.Next, it is worthwhile to establish whether alkylation of amic acids, i.e. amic ester formation, is a prerequisite for efficient imide formation. Reactions from amic esters to imides have been reported in the context of polyimide formation. This work, however, involved the synthesis of five-membered phthalimides and these imidization reaction have been performed by “brute force”. To the best of our knowledge, neither the temperature nor the solvent dependence of such reactions have been explored.28 In another work the alkylation of a perylene-3,4-monoamic acid has been reported. This reaction resulted in a double alkylation at the carboxylate and the amide nitrogen,29 but imide formation was not reported.
Whether the imide formation using our original procedure proceeds via the amic acid 5, the amic ester 6, or possibly both has been investigated by reacting 1 with 2-ethyl-hexyl amine 4b, in the presence and in the absence of butyl bromide. For both reactions PDI 7b was isolated, in yields of 60% and 25%, respectively, see Table 2 entries 3 and 4. This outcome demonstrated convincingly that addition of an alkylating reagent significantly increased the yield of PDI 7b, and thereby proofs that imidization via PDAE 6 is the faster route to form PDIs.30
Although significantly higher yields have been obtained by adding an alkylating reagent, we decided to further develop mild imidization reactions without the use of alkylating reagents. This choice has been made because alkylation requires an extra reaction step, uses a highly reactive alkylating reagent and thereby severely limits functional group tolerance. As for the reaction without an alkylating reagent, the imide formation itself already has a conversion around 50%,31 and that is a good starting point for obtaining satisfactory yields after optimizing the reaction conditions. It should be noted that the use of alkylating reagents for highly unreactive systems, such as electron deficient or sterically hindered amines, is still a viable option.
The detailed reaction schemes for the imidization reactions, starting from PDA 1 or model compound perylene-3,4-dicardoxylic anhydride-9,10-dicarboxylic dibutyl ester PMADE 11,32 are depicted in Schemes 2 and 3. The imidization reaction of PMADE 11 is included in this work, because this reaction proceeds via a single amic acid intermediate compound 12, and yields reaction products, perylene-3,4-dicarboxylic monoimide-9,10-dicarboxylic diesters (PMIDEs 13), that are soluble in common organic solvents. The more complex imidization of PDA (1) proceeds through four intermediates; compound 8, cis- and trans-5 and compound 9. In addition, the resulting PDIs 7 generally are not soluble in organic solvents, which prevents easy identification of the reaction product(s).
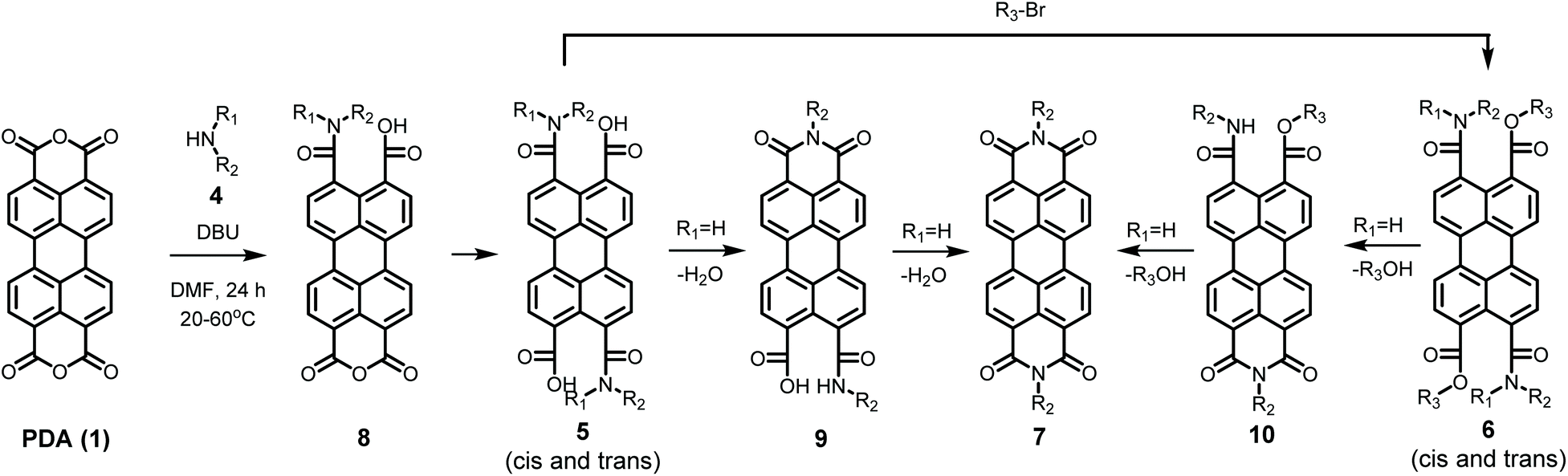 | ||
| Scheme 2 Imidization reaction starting from PDA (1) including all anticipated intermediates. | ||
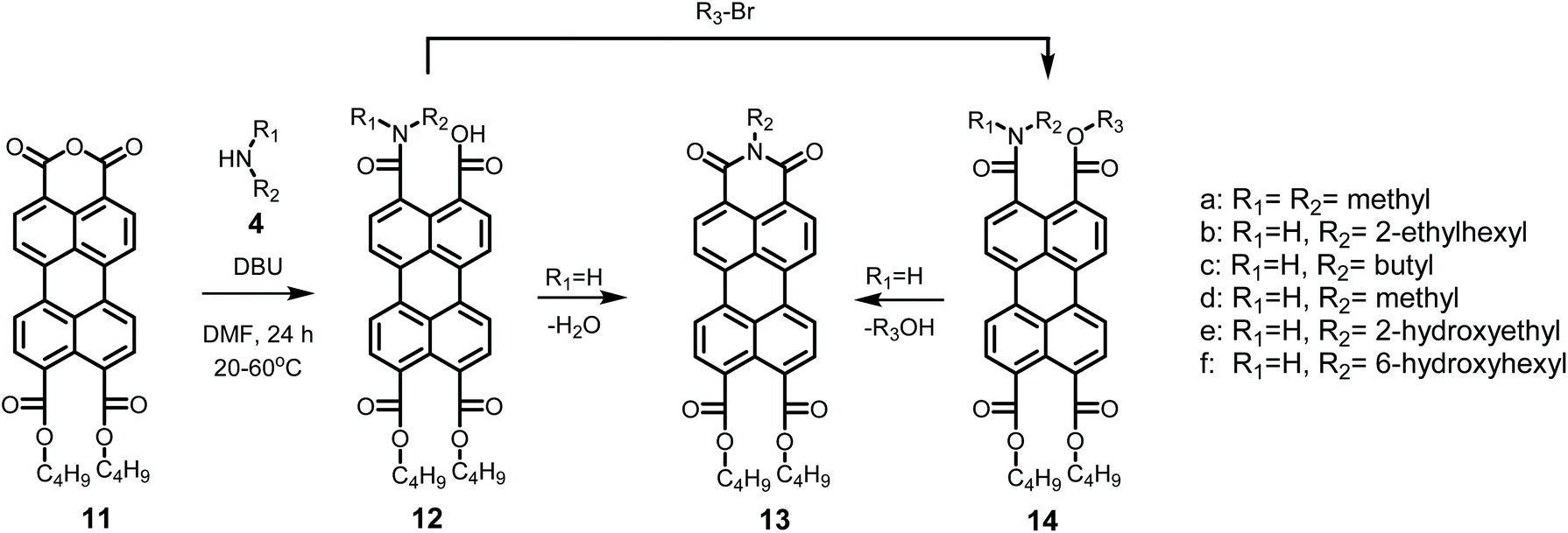 | ||
| Scheme 3 Imidization reaction starting from PMADE (11) including all anticipated intermediates. | ||
Synthesis and characterization of model amic acids
In order to validate the reaction mechanism depicted in Schemes 2 and 3, the potassium salts of the amic acids 12a, 5a and 9a were synthesised and characterised. These compounds are model compounds for the intermediate amic acid salts in the imidization reaction. Potassium salts were synthesised, because these compounds are easier to isolate and characterise than the corresponding DBU salts. It has been reported that hydrolysis of structurally similar amic acids yield anhydrides under acidic conditions.23,29 Under basic conditions amic acids derived from primary amines form imides, but this reaction is blocked for amic acids derived from secondary amines. Therefore amic acids salts K12a, K25a and K9a are expected to be stable under basic conditions.At room temperature the formation of the amic acid salt K12a from PMADE 11 proceeds within 60 minutes, as the pale reaction mixture, in which PMADE 11 is sparsely soluble, is converted to a bright orange solution. Isolation of compound K12a was achieved by precipitation, using water-free conditions, see entry 1, Table 3. Formation of the perylene monoamide triester 14a, a thermally and hydrolytically stable compound, was accomplished by adding butylbromide to the reaction mixture. This reaction is highly efficient and without any purification efforts compound 14a was isolated in 86% yield, see entry 2 in Table 3. Compounds K12a and 14a were characterised by NMR, absorption and emission spectroscopy and mass spectrometry. These techniques provided solid proof of the molecular structure of these compounds, see ESI.† With the successful synthesis and characterization of compounds K12a and 14a it has been proven that the amic acid 12 in an intermediate in the imidization of PMADE 11, which strongly supports the validity of Scheme 3. Also, the synthesis and spectral characterization of K12a facilitates the identification of other perylene monoamic acid diesters salts of 12.
When PDA 1 is reacted with dimethylamine 4a soluble amic acids are formed even faster. The initial reaction mixture, a red suspension, is transformed to a deep orange viscous liquid within 20 minutes. Amic acid salt K25a was isolated by precipitation in dry acetone, see entry 1, Table 2. The dibasic salt K25a is soluble in polar solvents, including water, and exhibits an absorption spectrum that is very similar to that of PTE 3 and perylene-3,4,9,10-tetracarboxylate.16 When butylbromide was added after formation of PDAA 5a, PDAE 6a was obtained in 76% yield, virtually free of contaminants, see Table 2, entry 2 and Fig. S1 and S2† in the ESI.† The cis and trans regioisomers of compound 6a were not distinguishable in the 1H NMR spectrum. Broadening of the resonances of the aromatic protons is the sole indication of the presence of two isomers, a result that is in line with the 1H NMR spectrum reported for a similar amic acid,33 see Fig. S1.† The 13C NMR spectrum, however, clearly revealed the formation of two isomers by the presence of two amide and two carboxylate carbons. Also the number of aromatic carbons atoms in the 13C NMR spectrum exceeds the 10 expected for either the cis or the trans isomer, see Fig. S2.† The successful isolation of compounds K25a and 6a confirms that diamic acid salt of 5 is an intermediate in the imidization of PDA and supports the reaction mechanism in Scheme 2.
Model compounds K9a and 10a were synthesized according to Scheme 4. This synthesis starts with the imidization of compound 11 using our mild imidization protocol and produced 13c in almost quantitative yield. Cleavage of the ester functionalities in concentrated sulphuric acid gave compound 15 in high yield.34 Ring opening, using dimethylamine 4a, yielded model compound K9a, that was isolated via an anhydrous precipitation. The hydrolytically stable and highly soluble compound 10a was obtained from K9ain situ, by addition of bromobutane. The identity of compounds 10a and K9a was unambiguously proven by NMR spectroscopy and mass spectrometry, see ESI.†
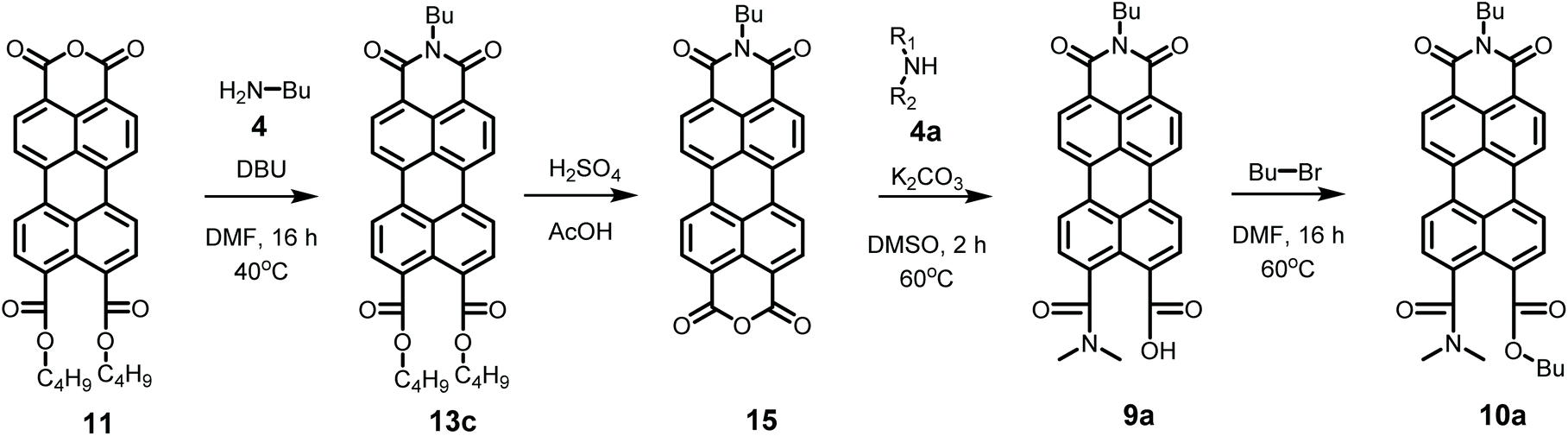 | ||
| Scheme 4 Synthesis of model compounds K9a and 10a. | ||
To provide proof of the intermittency of compound 9 in the imidization reaction we have reacted compound 15 with butylamine under standard conditions (DBU, DMF) to form compound 9cin situ, see Scheme S1.† The conversion of this compound to PDI 7c was followed by absorption spectroscopy, vide infra, and this experiment has confirmed the intermediary of compound 9c in Scheme 2.
The absorption and emission spectra of the amic acid salts K25a, K9a, K12a were recorded in water, DMF and ethanol, see Fig. S14 and S16,† while the spectra of the corresponding apolar model compounds 6a, 10a and 14a were recorded in ethanol and chloroform, see Fig. S15.† The spectra of all compounds in the common solvent EtOH are presented in Fig. 1, while photo physical data are presented in Table S3.† The absorption spectra of compounds 3, 13c and 7c in chloroform are depicted in Fig. S3.† The spectra in this Figure serve as a point of reference, for the spectra expected for molecularly dissolved perylene diamic acids, perylene monoamic acid monoimides and perylene diimides, respectively.
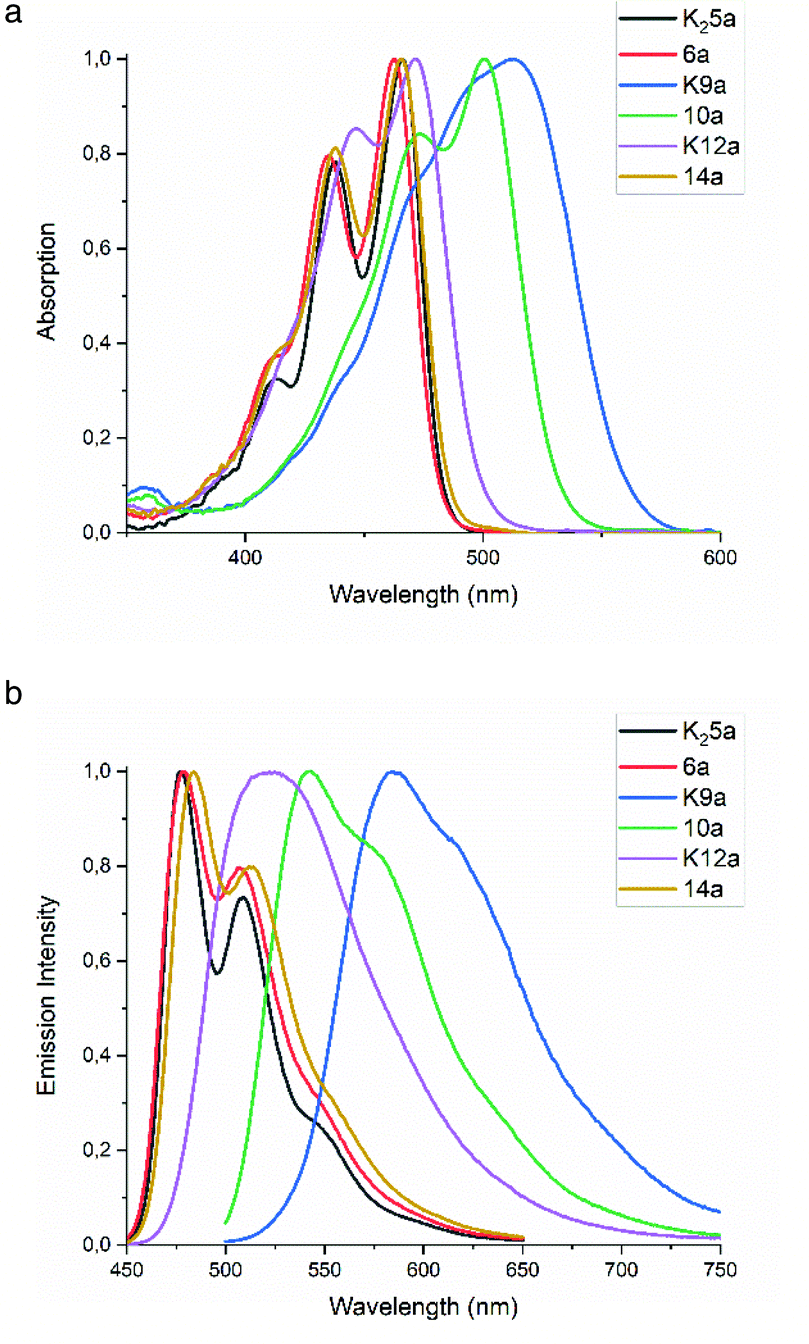 | ||
| Fig. 1 (a) Normalized UV-Vis absorption and (b) normalized fluorescence emission spectra of compounds 3, K25a, 6a, K9a, 10a, K12a and 14a in ethanol. | ||
The absorption spectra in ethanol of diamic acid salt K25a, diamic ester 6a and the diester monoamic ester 14a resemble that of tetraester 3, with a clear vibronic structure and absorption maxima around 470 and 440 nm. Substitution of a butyl ester by a dimethyl amide (3 to 14a, 6a) resulted in ∼2 nm blue shifts per substitution, whereas substitution of an amic ester by an amic acid salt (6a, 14a to 12a, 5a) resulted in ∼3 nm red shifts per substitution. The absorption of the monoamic ester diester 10a is 32 nm red shifted as compared to tetraester 3, see also Fig. S3.† The absorption spectra of the amic acid salts K12a and K9a are red shifted and show a diminished vibrational structure, as compared to their amic ester analogues. Amic acids, obtained after protonation of their potassium salts, exhibit blue shifted absorption spectra, but are too unstable for full characterisation, see Fig. 8, S12 and S13.†
The emission spectra of PTE 3 and compounds 6a, 9a and 14a are very similar. In all cases the emission spectra are mirror images of the absorption spectra and fluorescence quantum yields ΦF are around 0.85. The emission spectra of the amic acid salts K12a and K9a show diminished vibrational structure and quantum yields ΦF of 0.96 and 0.65, respectively.
Since the amic acid salts of 5 and 9 are intermediates in the PDI synthesis, it is worthwhile to investigate the (photo) physical data of compounds K25a and K9a in different solvents, see Table 1. Surprisingly, these amic acid salts have poor solubility in DMF, the solvent used for the imide formation, while solubility in water is much higher. Fluorescence quantum yields of compound K25a are very high in water and ethanol, which indicates that this dianionic compound is molecularly dissolved. In DMF, however, the fluorescence quantum yield is a meagre 0.16. For compound K9a the structure-less absorption and emission spectra, as compared to those of compound 10a and 13c, may be indicative for aggregate formation. Fluorescence quantum yields are high in ethanol, but in DMF and water quantum yields of 0.14 and 0.19 reflect substantial quenching. Solutions of K9a in water form a strong foam layer after shaking, which indicates that this compound acts as a surfactant and presumably forms micelles. The corresponding DBU salts of 5a and 9a have not been isolated, but spectra from diluted reaction mixtures containing these DBU salts have been recorded. Absorption and emission spectra as well as fluorescence quantum yields taken from these reaction mixtures closely resemble those of the corresponding potassium salts.
To determine the relative rates of the reaction steps in the imidization process, we have reacted PDA 1 with butyl amine 4c at room temperature and monitored the reaction by sampling the reaction mixture at regular intervals. Subsequently, these samples were diluted and analysed by absorption spectroscopy. Highly fluorescent yellow solutions were obtained after 20 minutes of reaction, both in water and chloroform, as seen in Fig. S4.† This result indicates that all PDA had reacted, and that all reaction products are soluble, both in water and chloroform. The absorption and emission spectra of the reaction product closely resembled those of diamic acid salt K25a, indicating that the reaction product is the DBU salt of 5c. Amic acid formation using K2CO3 in DMSO, proceeded at a similar rate.
Upon heating the reaction to 60 °C, the amount of diamic acid salt 5c slowly decreased. It is anticipated that the DBU salt of 9c is formed. However, this compound has not been identified in the absorption spectra. Poor solubility of compound 9c in chloroform, the solvent used for dilution, along with the presence of considerable quantities of compounds 5c and 7c, may be the reason for this. The formation of PDI 7c, however, is clearly visible when the imidization reaction is performed, see Fig. S4.†35
In summary, these preliminary studies of the reaction of PDA 1 with butylamine 4c, revealed a fast formation of diamic acid 5c in 15–20 minutes at room temperature. At prolonged reaction times and elevated temperatures, typically a few hours at 60 °C, the concentration of diamic acid 5 decreased and monoamic acid monoimide 9c was formed (but not detected). Subsequently the formation of PDI 7c has been observed. It was concluded that the fast ring opening reactions, forming amic acid 5c, and the subsequent slow imidization processes, forming intermediate 9c and eventually the PDI 7c, are decoupled processes that take place at markedly different time scales.
Reaction optimization by variation of reaction conditions
After validation of Scheme 3 and the establishment of the relative rates of the reaction steps, the imidization reaction was optimized by changing the reaction parameters. The reaction of PDA 1 with 2-ethyl-hexyl amine 4b was chosen for this purpose, because the product of this reaction, PDI 7b, is highly soluble in chloroform and conveniently characterised by NMR spectroscopy. Isolation of the reaction product was extremely convenient; pouring the reaction mixture in water, filtering the precipitate and washing the product with dilute base to remove water-soluble amic acids, yielded pure PDI.36Using the standard conditions (4 molar equivalents of amine and DBU and DMF as the solvent), increasing the reaction yields was achieved by increasing the temperature to 60 °C or the reaction time to 7 days, see entries 5 and S1† in Tables 2 and S1.† From a practical point of view the elevated temperature is preferred. In the original work by Langhals,10 Zinc Acetate Zn(OAc)2 was used to catalyse the reaction. In our experiments Zn(OAc)2 leads to higher yields at room temperature, but addition of this compound no longer has a pronounced effect at elevated temperatures, see entries 2 and 3 in Table S1.† Therefore, and also because we want to keep the reactions clean and simple, we refrain from the addition of Zn(OAc)2. In summary, the optimized imidization proceeds at 60 °C, achieves quantitative yields and takes approximately 24 hours.
| Entry | Perylene anhydride | Reagents | Base | Solvent | Reaction conditions | Yield, in % |
|---|---|---|---|---|---|---|
| a First hour kept at room temperature. b At 80 °C. c 2 ml of water added. d Water was added to the reaction after half an hour. | ||||||
| 1 | PDA (1) | 4a | K2CO3 | DMSO | 24 h | K25a |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | RT | 66% | |
| 2 | PDA (1) | 4a | DBU | DMF | 24 h | 6a |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | RT | 76% | |
| BuBr, 2 mmol | ||||||
| 3 | PDA (1) | 4b | DBU | DMF | 24 h | 7b |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | RT | 25% | |
| 4 | PDA (1) | 4b | DBU | DMF | 24 h | 7b |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | RT | 60% | |
| BuBr, 4 mmol | ||||||
| 5 | PDA (1) | 4b, 4c, 4da, 4f | DBU | DMF | 24 h | 7b, 7c, 7d, 7f |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | 60 °C | >97% | |
| 6 | PDA (1) | 4b, 4c | DBU | DMSO | 24 h | 4b, 4c |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | 60 °C | >97% | |
| 7 | PDA (1) | 4b, 4c, 4d,a4e,b4f | K2CO3 | DMSO | 24 h | 7b, 7c, 7d, 7e, 7f |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | 100 °C | >97% | |
| 8 | PDA (1) | 4b, 4c | K2CO3 | DMSO | 24 h | 7b 93% |
| 0.5 mmol | 1.1 mmol | 2 mmol | 4 ml | 100 °C | 7c >97% | |
| 9 | PDA (1) | 4e | DBU | DMF | 8 h | 7e |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | RT | >97% | |
| 10 | PDA (1) | 4b,c4c, 4d, 4f | DBU | DMSO | 24 h | 7b, 7c, 7d >97% |
| 0.5 mmol | 2 mmol | 2 mmol | 4 ml | RT | 4f 90% | |
| Waterd, 4 ml | ||||||
Our next objective is to convert the mild, convenient and efficient imidization reaction into a green(er) and more sustainable process.37 This has been accomplished by substitution of the solvent DMF and the base DBU by greener alternatives. Substitution of toxic DMF by DMSO, a non-toxic solvent with similar physical properties, did not affect the reaction yields (entry 6, Table 2). With regard to the substitution of DBU by organic bases, it was observed that DBU is distinctly the best base for this reaction, as was the case for the PTE formation as well.38 Reaction with TEA or DIPEA, resulted in significantly lower yields, typically around 20–25% after 24 hours at 60 °C, entry S4, Table S1.† Since most organic bases have toxicity issues similar to those of DBU, substitution of DBU by organic bases was not explored further. Finally, the use of the non-toxic inorganic base K2CO3 in combination with DMSO, was investigated. At 60 °C, significantly lower yield were observed than with DBU as a base, see entry S5, Table S1.† By raising the reaction temperature to 100 °C, however, the decreased reaction rate was fully compensated for and the reaction gave quantitative yields, see entry 7 in Table 2. This result demonstrates that an entirely green and sustainable process for the imidization of PDA has been developed. Apart from starting material PDA 1, the amine 4b and the reaction product PDI 7b, all inherent to this reaction, the solvent and other reactant are non-toxic and environmentally benign.
Next, imidization reactions of PDA 1 were undertaken using the amines 4c–4f. The PDIs obtained by these reactions bear small alkyl chains (7c–7d), or hydroxyalkyl chains (7e–7f) and only the PDIs 7c and 7f are sparsely soluble in chloroform.39 With butylamine 4c, the room temperature reaction was incomplete, but the yield of 7c, (48%) was considerably higher than that of 7b (25%). By increasing the reaction temperature to 60 °C, the reaction yielded 7c in quantitative yield, both in DMF and DMSO. Using the green conditions, DMSO as a solvent and K2CO3 as a base, quantitative yields were obtained at 100 °C, see entries 5–7 and S6 in Tables 2 and S1.† In order to make the DMSO/K2CO3 reaction entirely green, the excess of amine was limited to 10%. After facilitating the amic acid formation, by stirring one hour at room temperature, the reaction mixture was heated to 100 °C and PDI 7c was isolated in quantitative yield, see entry 8 Table 2. With this modification only minute amounts of unreacted amine are present in the final reaction mixture. This procedure of forming the amic acid at room temperature prior to imidization at higher temperatures, may also be extremely suited for working with volatile or expensive amines.
In the synthesis of 7d, methylamine 4d dissolved in methanol was used as a reactant. The room temperature reaction yielded 7d in 89% yield. When the reaction was stirred at room temperature for 1 hour, to ensure amic acid formation and prevent evaporation of the amine, quantitative yields were obtained for the DBU reactions at 60 °C as well as the K2CO3 reaction at 100 °C, see entries 5,7 and S7 in Tables 2 and S1.†19 For the reaction with 6-amino-1-hexanol 4f, the yield of the DMF/DBU reaction was 66% at room temperature. Quantitative yields are obtained by raising the temperature to 60 °C, while the K2CO3 reaction gave quantitative yields at 100 °C, see entries 5, 7 and S8 in Tables 2 and S1.†
The imidization reaction with ethanolamine 4e proceeded much faster and quantitative conversion at room temperature was achieved after 8 hours already. When this reaction was sampled and diluted in water, the resulting absorption spectra clearly showed that amic acid formation and imidization both take place at room temperature and that amic acid formation and imidization are no longer decoupled. The initial rise in the concentration of amic acid 5e is reversed within 20 minutes and a broad structure-less red-shifted absorption emerges, which closely resembles that of compound 9a, see Fig. 2 an S16.† Also, a purple precipitate originating from PDI 7e is formed upon diluting the reaction mixture in water early in the reaction. For the same reaction in DMSO, using K2CO3 as the base, amic acid formation took place within 20 minutes, but imidization was not observed at room temperature, see Fig. S6.† For this reaction quantitative yields were obtained at 80 °C, see entries 7 and 9, Table 2.
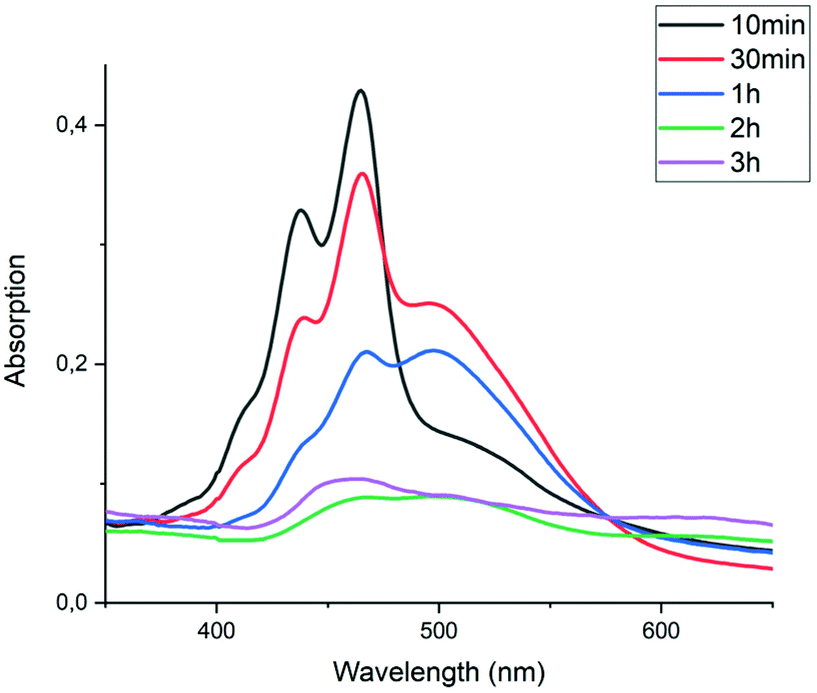 | ||
| Fig. 2 UV-Vis spectra of the reaction of PDA (1) with ethanolamine (4e) in DMF using DBU as the base. Spectra were taken from samples of the reaction mixture diluted in water. The baseline is lifted due to the presence of insoluble PDA 1 and/or PDI 7e. Absorptions do not accurately scale with concentrations in the reaction mixture due to sampling from a heterogeneous reaction mixture. | ||
The mild imidization reaction with model compound PMADE 11 and the aliphatic amines 4b–4f was also examined. Reactions were performed at room temperature and yields, after purification by column chromatography are displayed in Table 3. Purity and identity of compounds 13b–f were conveniently proven by NMR spectroscopy. From the reported yields in Table 3 it is concluded that in all cases the reaction yields are well over 50% at room temperature, and that ethanolamine 4e is the most reactive amine. These room temperature yields correlate well with those obtained for the bisimides and may be indicative for the reactivity of the amines in the imidization of perylene anhydrides. By increasing the reaction temperatures to 40 °C, quantitative yields can be obtained overnight, as demonstrated with the synthesis of compound 13c, vide supra.
| Entry | Perylene anhydride | Reagents | Base | Solvent | Reaction conditions | Yield, in % |
|---|---|---|---|---|---|---|
| 1 | PMADE (11) | 4a | K2CO3 | DMSO | 24 h | K12a |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 61% | |
| 2 | PMADE (11) | 4a | DBU | DMF | 24 h | 14a |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 86% | |
| BuBr, 0.76 mmol | ||||||
| 3 | PMADE (11) | 4b | DBU | DMF | 24 h | 13b |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 55% | |
| 4 | PMADE (11) | 4c | DBU | DMF | 24 h | 13c |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 65% | |
| 5 | PMADE (11) | 4d | DBU | DMF | 24 h | 13d |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 75% | |
| 6 | PMADE (11) | 4e | DBU | DMF | 24 h | 13e |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 80% | |
| 7 | PMADE (11) | 4f | DBU | DMF | 24 h | 13f |
| 0.19 mmol | 0.38 mmol | 0.38 mmol | 2 ml | RT | 75% |
In summary, the reactions of PDA 1 with all aliphatic amines are very convenient and quantitative yields are obtained with DBU in DMF or DMSO at 60 °C and with K2CO3 in DMSO at 100 °C. Quantitative PDI formation resulted in a convenient workup in which the filtrate is transparent, virtually colourless and free from PTCA derivatives. These results indicate that these reaction conditions are universally applicable for aliphatic amines. For all reactions the ring opening by the amines is fast while the imidization from the amic acid intermediates is rate-determining. Therefore, differences in amine reactivity must be due to differences in rates of imidization of the respective amic acids.
Reaction kinetics; modelling and measuring
Based on the reaction sequence for the imidization of PDA in Scheme 2, a kinetic model has been constructed. With this kinetic model the reaction mechanism can be validated and the rate constants for the different reaction steps determined. Furthermore, this model may also help to understand why our novel synthetic approach produces PDIs at room temperature already. In addition, factors that limit the rate of the imidization step, can be identified and potentially resolved to make the process even faster at mild and green conditions.The imidization of PDA consists of 4 reaction steps, two consecutive second-order amic acid forming ring opening reactions, followed by two intramolecular consecutive first-order imidization reactions, see eqn (1). These consecutive reaction steps are identical by mechanism and their rate constants should have near-identical values, i.e. k1 ≈ k′1 and k2 ≈ k′2. It should also be noted that the stable amic acids salts K25a and K9a, have been synthesised and characterized, which facilitated easy identification of analogous amic acid intermediates.
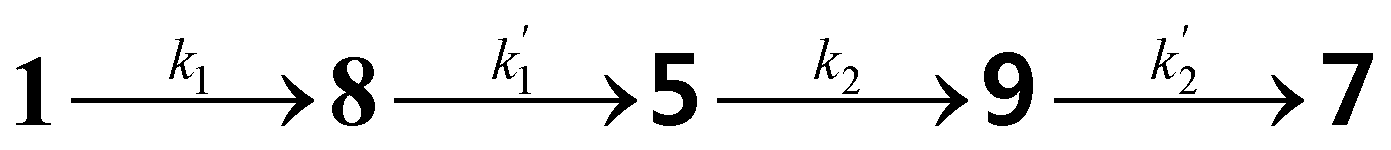 | (1) |
Bimolecular ring opening reactions to form amic acids are fast, vide supra, and we have observed that these ring opening reactions are generally decoupled from the slower unimolecular imidization reactions. Therefore, both reactions, the amic acid formation from 1 to 5 and the imidization from 5 to 7, will be described separately.
The first part of the reaction, from PDA to diamic acid 5, is treated in detail in the ESI.† In deriving the kinetic equations, we assume that compound 1 is insoluble, whereas compound 8 will be somewhat soluble. Therefore, the conversion from PDA 1 to compound 8 is the rate-limiting step in this sequence. Accumulation of compound 8, whose spectroscopic signature in molecular solutions is expected to be close to that of compound 9, apart from a ∼4 nm blue shift,20b is not anticipated. The rate of consumption of compound 1 and the rate of formation of compound 5 according to eqn (S3) and (S8),† are identical and depicted in Fig. S7.† Experimental data, shown in Fig. S3 and S6,† do not resemble those in Fig. S7,† most likely because the data used for constructing Fig. S3 and S6,† were obtained from sampling inhomogeneous reaction mixtures.
Compounds 5 and 9, participating in the second part of the reaction, are partly soluble in the reaction medium, as indicated by the deep red colour that is developed throughout the reaction. Eqn (2)–(4) describe the rates of the individual reaction steps.
 | (2) |
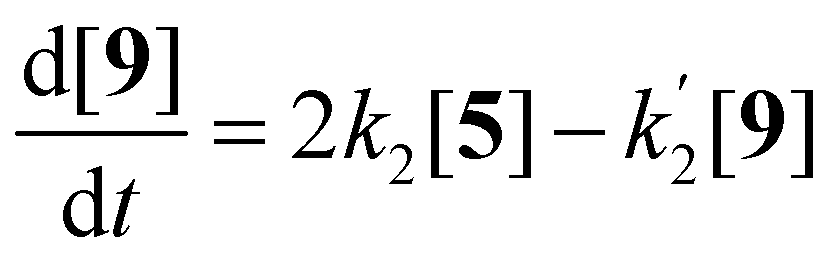 | (3) |
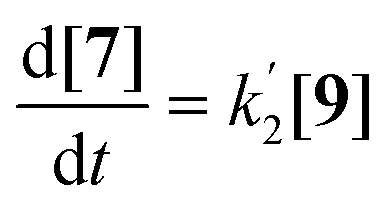 | (4) |
Solutions for eqn (2)–(4) are given by eqn (5)–(7).40
[5] = [5]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp−2k2t exp−2k2t | (5) |
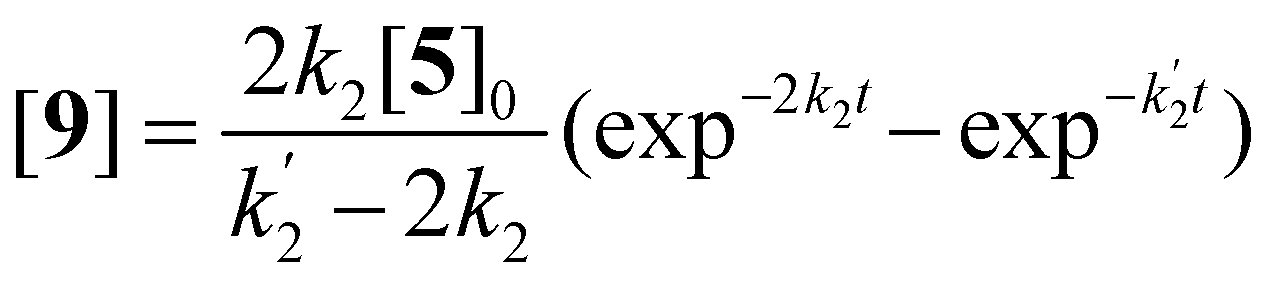 | (6) |
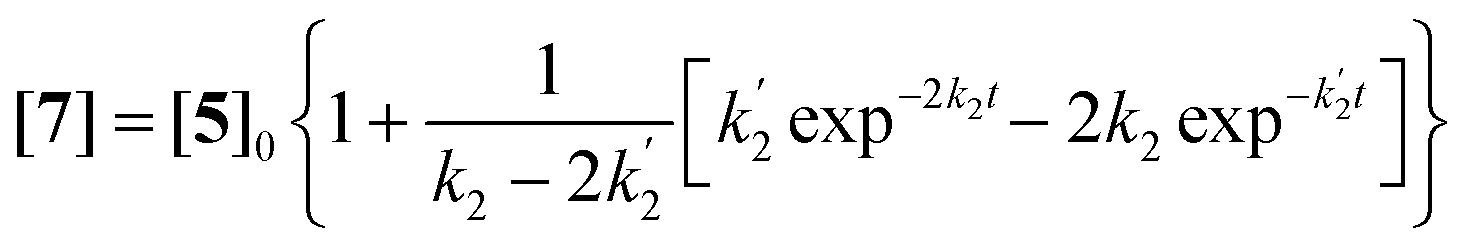 | (7) |
The kinetic model for the imidization of PMADE 11 or PMAMI 15, in which the same reaction rate constants k1 and k2 are employed, is described in the ESI (eqn (S9)–(S15)†).
A graphical representation of the composition of the reaction mixture, according to eqn (5)–(7), is given in Fig. 3. The decay of the diamic acid 5 follows first order kinetics, the amic acid monoimide 9 is an intermediate whose concentration builds up to nearly 50% and subsequently declines, while the formation of PDI 7 distinctly lags behind the disappearance of compound 5. Experimental data, to support our kinetic model have not been obtained so far. Qualitatively the consumption of compound 5c and formation of compound 7c have been demonstrated but detection, let alone quantification of the amount of compound 9c has not been achieved.
 | ||
| Fig. 3 Consumption of diamic acid 5, formation and disappearance of reaction intermediate 9 and formation of PDI 7 according to eqn (5)–(7). | ||
It should be noted that the amic acid salts of compounds 5 and especially 9 have limited solubility in DMF and DMSO and form aggregates and precipitates at the high concentrations used for the standard reactions. Thus solubility and possibly the type of aggregation of the amic acid salts of 5 and 9,41 will govern the rate of the imidization reaction. The solubility and aggregation behaviour of these salts depends on the reaction temperature, the solvent, the substituent R2 and the counter ions. Therefore, we anticipate that these are the major factors that determine rates of imide formation.
To test the hypothesis that solubility of amic acid salts limits the rate of imidization reaction, we decided to perform the second stage of this reaction in a highly diluted medium. The imidization reaction with butylamine 4c at room temperature was conducted under standard conditions (DMF, DBU) for 30 minutes, until the formation of the diamic acid 5c was completed. At this stage the reaction mixture was a thick orange slurry, a clear indication that the amic acid salts derived from compound 5c did not fully dissolve. By strongly diluting the reaction mixture with DMF, reaching concentrations in the 10−5 molar range,42 the remainder of the reaction at room temperature was followed in situ by absorption spectroscopy, see Fig. 4.43 While the normal reaction reaches 48% conversion in 24 hours, the diluted reaction reaches this conversion in approximately 6 hours and achieves a near-quantitative conversion in 24 hours. The diamic acid salt of 5c, absorbing at 438 and 466 nm, is consumed swiftly, and a broad absorption (extending up to 600 nm), which closely resembles the absorption of model compound K9a, emerges. In time this broad absorption, which originates from the salt of compound 9c, builds up and disappears, while eventually PDI 7c, with distinct absorptions at 488 and 524 nm, is formed. At the end of the reaction a clear, brightly fluorescent solution that contains PDI 7c is obtained. The good solubility of PDI 7c in DMF is really surprising, although it is obvious that these solutions are oversaturated.44
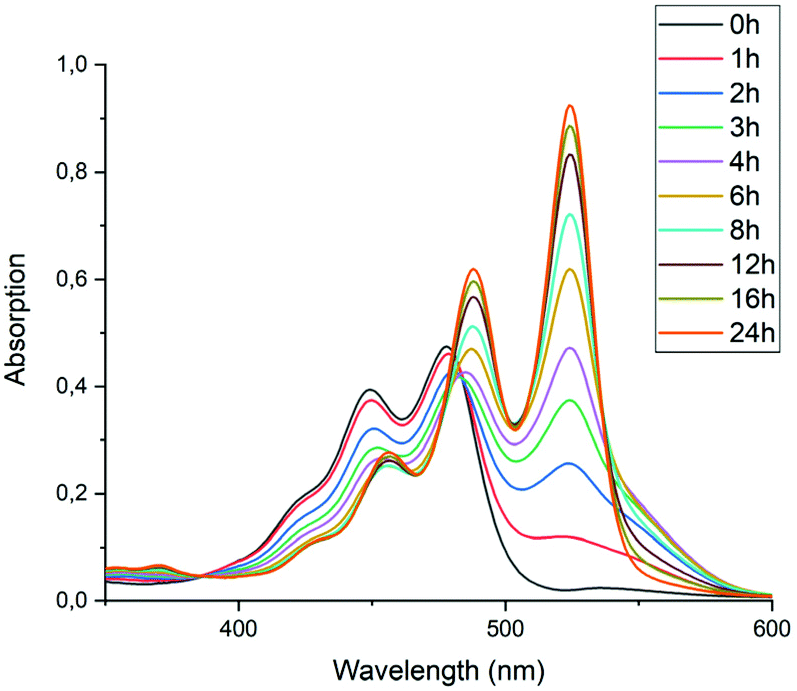 | ||
| Fig. 4 Absorption spectra as a function of time of the reaction of the DBU salt of diamic acid 5c in DMF at room temperature (20 °C) at concentrations around 10−5 M. | ||
In Fig. 5, the composition of the reaction mixture as a function of time is displayed. For determining the composition of the reaction mixture, it was assumed that the spectrum taken at t = 0 is that of the DBU salt of 5c and that the spectrum taken after 24 hours is from 7c. Relative concentrations of 5c and 7c were determined by manual fitting of the absorption spectra. The remaining absorption spectrum, obtained by subtracting the contributions from compounds 5c and 7c, is assigned to reaction intermediate 9c. The fitting was done such that the absorption spectra of compound 9c were consistent throughout the series, and closely resembled the spectra that we have previously obtained from compound K9a, see Fig. S14.† Using this procedure, determining the concentration of PDI 7c is fairly consistent and reliable. However, for determining [5c] and [9c] independently this procedure is not suited.45
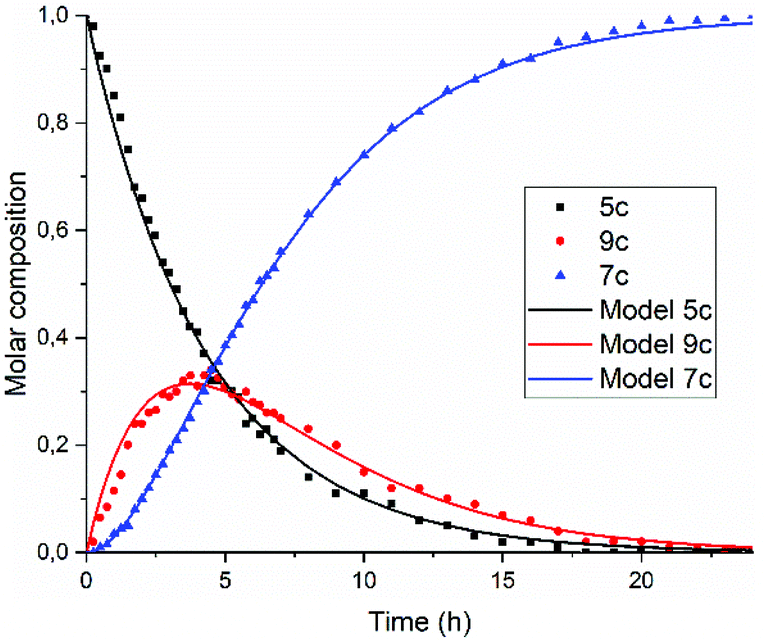 | ||
| Fig. 5 Composition of the reaction mixture obtained from the data displayed in Fig. 5. Solid lines represent the fitting curves obtained using eqn (5)–(7). The solid squires, circles and triangles are concentrations of compounds 5c, 9c and 7c, respectively obtained by analysing the absorption spectra. | ||
The experimental data in Fig. 5 resemble those predicted by our model depicted in Fig. 3, qualitatively; the monotonic decrease of compound 5c, the delayed formation of compound 7c and the intermediate formation of compound 9c are clearly visible. The curve fitting of the data in Fig. 5 was accomplished by using values for the rate constants k2 and k′2 of 3 × 10−5 s−1and 8 × 10−5 s−1, respectively. The blue curve for the formation of PDI 7 was constructed by taking identical values for k2 and k′2 of 5 × 10−5 s−1. We anticipate that these seemingly different values for the rate constants k2 and k′2 are artefacts that emerged because the concentrations of diamic acid 5c and the monoamic acid 9c could not be determined independently.
To find out if this assumption is correct, the rate constant k′2 was determined independently by monitoring the reaction from compound 9c to 7c. To this end, monoanhydride 15 was reacted with butylamine 4c under standard conditions for 30 minutes to form compound 9c quantitatively, see Scheme S1.† After dilution with DMF, the conversion from intermediate 9c to PDI 7c by a first order process was monitored, see Fig. 6 and 7. In Fig. 6 three isosbestic points at 499, 512 and 535 nm are observed, which proofs that 9c is converted to 7c directly. The rate constant k′2 was determined to be 5 × 10−5 s−1, as demonstrated in Fig. 7. In addition this reaction rate constant was also determined from the reaction of monoamic acid diester 12c to PMIDE 13c, see Fig. S9 and S10.† The imidization rate constant for this reaction had the same value, 5 × 10−5 s−1. Combining these results it is obvious that the rate constants k2/k′2 for the transformation of an amic acid to an imide on the perylene scaffold has a value of 5 × 10−5 s−1 and is not significantly affected by the substituent(s) at the opposing peri-position(s). It is also concluded that kinetic measurements, starting from compound 5c are not appropriate for determining k2 and k′2 independently. With the assumption that k2 = k′2, however, the reaction rate constant can be determined from such kinetic measurements. To investigate the stability of amic acids under acidic conditions, dilute solutions of the model amic acid salts K25a, K9a and K12a in DMF were acidified with HCl. In line with the reported behaviour of naphthyl-1,8-dicarboxylic amic acids, perylene amic acids were highly unstable.46 Protonation was visible by a distinct 9 nm blue shift in the absorption spectrum and formation of 5a was immediately followed by the appearance of an absorption around 505 nm, see Fig. 8. After 30 minutes the spectrum of PDA 1, the end product of the acid decomposition process, became visible and the reaction was finished within 5 hours. From this experiment, the spectrum of the so far undetected monoamic acid monoanhydride 8a was obtained, by subtracting the spectra of compounds 5a and 1 from those of the reaction mixture, in a procedure similar to the one used to analyse the imidization of compound 5c, see Fig. S11.† It appears that compound 8a is molecularly dissolved, as its absorption spectrum exhibits weak vibrational structure and is neither broadened nor red shifted.47 Similar experiments, monitoring the acid catalysed anhydride formation from amid acids 9a and 12a are depicted in Fig. S12 and S13.† For these reactions clear and unambiguous isosbestic points were observed, due to the absence of reaction intermediates. PDA, PMAMI and PMADE, formed by amic acid hydrolysis, were molecularly dissolved in DMF and formed highly fluorescent oversaturated solutions. Rate constants for these reactions were not determined, because pH values, on which these reaction rates depend, have not been determined.
 | ||
| Fig. 6 Absorption spectra as a function of time of the reaction of the DBU salt of amic acid monoimide 9c in DMF at concentrations around 5 × 10−6 M. The starting solution at t = 0 already contains traces of the product 7c. The sharp isosbestic points, at 499, 512 and 535 nm, clearly indicate the presence of only two compounds; 9c and 7c. | ||
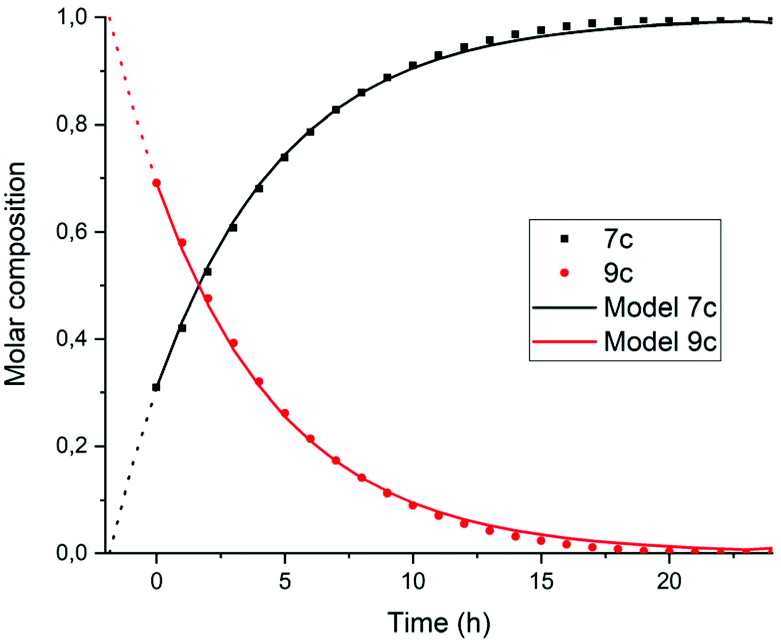 | ||
| Fig. 7 Composition of the reaction mixture obtained from the data displayed in Fig. 7. Solid lines represent the theoretical fitting curves obtained using a first-order decay obtained using eqn (S11) and (S12).† | ||
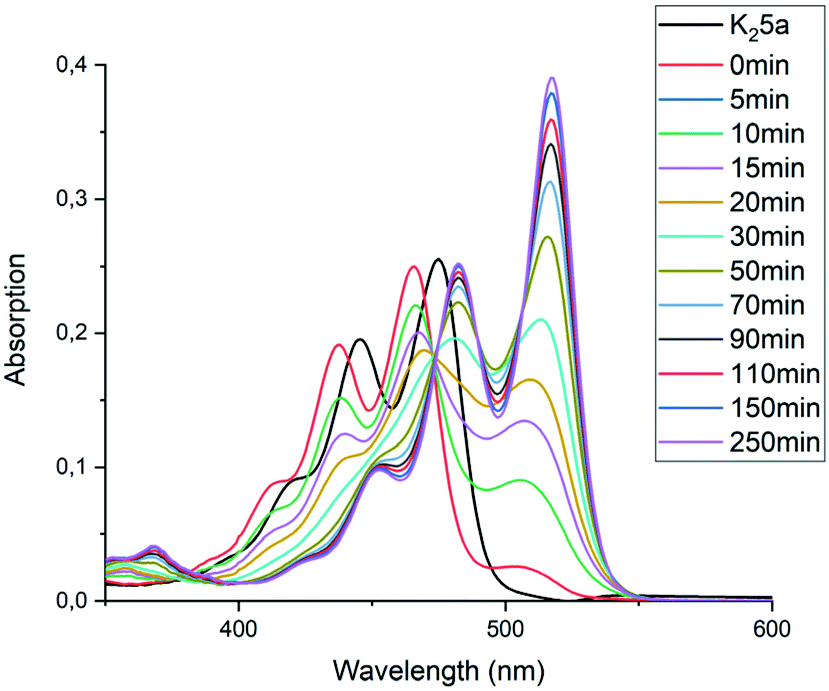 | ||
| Fig. 8 Absorption spectra of compound K25a (black) in DMF and 5a (red), formed after acidification with 1 drop 1 N HCl. In the time-dependent absorption spectra the reaction from 5a (λmax = 466 and 438 nm) via compound 8a to compound 1 (λmax = 517.5 and 482 nm) is visible. Please note that the first spectrum after acidification already contains traces of compound 8a. | ||
Finally it should be noted that fast anhydride formation from amic acids, implies that workup of reaction products in the PDI synthesis must be performed under basic conditions, until all amic acids are removed. Otherwise anhydrides are formed that cannot be removed.
Room temperature imidization
The dilution experiments depicted in Fig. 4–7 have demonstrated that imidizations of PDA proceed at room temperature and have low activation barriers for imide formation. The overall reaction rate under standard conditions is limited by the solubility of the intermediate amic acid salts of 5 and 9. At high dilution48 the imide formation for the DBU reaction in DMF is strongly accelerated, with a rate constants k2 around 5 × 10−5 s−1. This allows the reaction to proceed to a >97% conversion in 24 hours at room temperature. It appears, from visual inspection of the reaction mixtures, that the DBU salts of the amic acids 5 and 9 have better solubility, as compared to the potassium salts, and this increased solubility may be the cause of the apparent DBU catalysis of the imidization reaction.Our findings, that the rate of imidization is limited by the solubility of the amic acid salts 5 and 9, and that dilution has a highly beneficial effect on the overall reaction rate, can be readily exploited for improving the synthesis of PDIs. Taking into account that amic acids 5 and 9 have higher solubility in water (as compared to DMF and DMSO) and that for imidization of NMA and NDA water is an excellent solvent, dilution with water appears to be a viable option to increase the imidization rate even further.
When the synthesis of PDI 7c was started employing the normal reaction protocol (DBU in DMSO) quantitative formation of PDAA 5c was achieved within an hour at room temperature. Subsequently, one equivalent of water (4 ml) was added, resulting in instantaneous dissolution of the DBU salt of 5c to form a deep red solution.49 After 24 hours of reaction at room temperature, PDI 7c was obtained in quantitative yield, entry 10 Table 1. It should be noted that this reaction has a 50% yield without water addition. Water is the ideal solvent to add, because in our synthetic protocol water should be added anyway for separating the solid product from the reaction mixture. For amines 4b, 4d and 4f, the same procedure, adding water after amic acid formation was examined. For the formation of 7d, imide formation was quantitative as well, while for the formation of 7f, the yield was high, 90%, but not quantitative. For the formation of 7b, we only added 2 mL of water because the long hydrophobic 2 ethyl hexyl tails limit water solubility. The yield for this reaction was quantitative.50
These results imply that aliphatic PDIs can be synthesised in quantitative yields at room temperature, without (7e) or with the addition of water (7b–d). When stable aliphatic amines, such as 4b–f are used, synthesis at 60–100 °C is a reliable and practical option. As it has been reported that PDI synthesis with amino acids under Langhals conditions does not affect the stereochemistry of the appended amino acid,51 the 60–100 °C “high temperature” synthesis is appropriate for these compounds as well. However, when temperature sensitive amines are used, imidization at room temperature will be the preferred route.
Discussion
In this work, we have demonstrated that imidization of PDA, contrary to previous experience, is a process that is conveniently and efficiently performed at moderate temperatures, if need be at room temperature. The first step of the process, formation of diamic acid, is facile and fast in DMF or DMSO with all bases that we have explored. Conditions for PDAA synthesis found in the literature are generally harsh,33 but structurally similar amic acids, such as 1,8-naphthyl-dicarboxylic amic acids and 1,3,4,8-naphthyl-tetracarboxylic diamic acids, have been synthesised under much milder conditions.25 Also the formation of compound 2, the ester analogue of perylene diamic acid 5, is a process that runs at extremely mild conditions. Therefore the observation that amic acid formation is the easy step in the imide synthesis is not very surprising and in line with our expectations.Imide formation from amic acids, on the other hand, is anticipated to be much more difficult to achieve. It is reasonable to assume that limited solubility of PTCA derivatives is a major factor necessitating the high temperatures in the conventional PDI syntheses, since imidization from more soluble analogous (di)anhydride substrates is reported at lower temperatures.11,12 The activation barrier for the imidization step has been largely unexplored, since all PDI forming reactions have been performed far above room temperature. In contrast to the data obtained from synthetic sources, mechanistic research on the reactivity of 1,8-naphthyl-dicarboxylic amic acids in water indicates that the activation barriers for analogous imidization reactions are low. The conversion from napththyl amic acids to naphthyl imides is a process that takes place reversibly at temperatures as low as 30 °C. These data suggest that in water, a solvent in which PTCA derivatives tend to be poorly soluble, imidization is a process with a low activation barrier. So if polar solvents in which perylene amic acids are soluble can be found, imidization reactions of PDA may proceed at low temperatures.
Our experiments, in which diamic acid salts of 5 were fully dissolved in DMF at high dilution, gave full conversion to PDI 7 at room temperature. This proofs that amic acid salt solubility is the major rate limiting factor for the imization of PDA. Absorption spectra taken at high dilution have clearly demonstrated that all reaction products and intermediates are visible. The diamic acid salts of 5 and even PDI 7 are molecularly dissolved, but monoimide amic acid salts of 9 still forms aggregates at these high dilutions. This finding suggests that the solubility of compound 9 is the major factor limiting the imidization rate. Since we have observed that the solubility of compound 5 is also limited under the standard reaction conditions, the solubility of this compound may be rate limiting as well. For amic acid salts of 5 and 9 it has been observed that potassium salts are less soluble than DBU salts and this solubility difference correlates with a lower reactivity of the reactions when K2CO3 is used. Another case where high solubility and high reactivity are correlated is the unusual fast reaction of ethanolamine 4e with PDA under standard conditions. The more intense red colour of the reaction mixture observed in this synthesis obviously reflects a higher solubility of the intermediate amic acid salts. Once more, the rate for this reaction is higher when DBU is used as the base. Based on these observations, we conclude that the rate enhancing effect of DBU primarily originates from the higher solubility of the DBU amic acid salts in the reaction medium.
For optimal results, in terms of materials used and rate of the reaction, the bimolecular amic acid formation from PDA is best performed at high concentrations, using only a small excess of amine in as little solvent as possible. For this reaction DMF and DMSO are the only useful solvents identified so far. The second step, the unimolecular imide formation, is limited by the solubility of the amic acid salts and therefore dilution with an appropriate polar solvent is required to speed up this part of the reaction. Enhancing the yield has been proven by strongly diluting the reaction mixture with DMSO or DMF. By adding just one equivalent amount of water the same result, i.e. quantitative PDI formation at room temperature, was obtained as well. The use of water in this respect is extremely beneficial, as water is green, cheap and is already used for precipitating the reaction product PDI 7 out of the solution. Further reaction optimization along these lines will be the subject of coming research in our group.
Looking ahead further, attractive opportunity emerge in broadening the scope of the mild imidization reactions. For example in terms of the amines and anhydrides used as starting compounds. It is known from literature that aromatic amines are less nucleophilic and generally have lower yields in imidization reactions.10,13,14 Preliminary results of reactions on PMADE 11 have shown that at elevated temperatures aromatic imides can be formed.52 Reactions of NDI with butyl amine demonstrated the formation of dibutyl NDI at room temperature already. In the literature this reaction is reported in DMF or acetic acid at temperatures above 100 °C.53 Attaching delicate and sensitive (bio)molecules, such as amino terminated nucleic acids and proteins, to strongly fluorescent perylene anhydride moieties like compound 11 is another attractive prospect of our novel synthetic procedure. Very mild reaction conditions can be employed, which is advantageous in view of the high value and limited stability of NH2-terminated biomolecules. An additional advantage of the mild imidization protocol is that every step of the process can be monitored and examined in great detail using absorption spectroscopy. This facilitates optimization of the reaction by tuning the individual reaction steps.
Conclusions
The imidization of PDA 1 has been converted from a harsh “molten imidazole” process, performed at 140–180 °C, into a highly practical, efficient process that runs at strongly decreased temperatures. When using primary aliphatic amines, DBU and either DMF or DMSO as the solvent, full conversion within 24 hours is achieved in the temperature range between 20–60 °C. By using K2CO3 in DMSO, a truly green and user-friendly synthesis has been developed at reaction temperatures between 80–100 °C.The reaction sequence for the imidization process via various amic acid intermediates, as displayed in Scheme 2, has been confirmed experimentally, aided by the synthesis and full characterization of stable model amic acid salts and amic esters. The formation of diamic acid salts is a fast process that is completed within 30 minutes. Conversion from amic acids to imides is rate determining, due to the low solubility of the intermediate amic acid salts. Diluting the reaction mixture after amic acid formation, strongly increases the reaction rates. The reaction rate constant for the imidization process in DMF was determined to be 5 × 10−5 s−1 at 20 °C. An improved synthesis, that yields quantitative conversion at room temperature, was developed by adding water after amic acid is formed. An equivalent amount of water is enough to solubilise these amic acids and run the reaction to completion.
Finally, the convenience of the novel synthetic protocol needs to be emphasized. A protective atmosphere, special equipment and advanced synthetic skills are no longer required. When run at the proper temperature, full conversion is obtained and work-up comes down to product precipitation in water followed by a simple filtration. Current research efforts are focussed on broadening the scope of our synthetic protocol by exploring the synthesis of aromatic PDIs and the use of delicate, temperature sensitive amines.
Experimental
Perylene-3,4-dicarboxylic monoanhydride-9,10-dicarboxylic dibutyl ester (11) was synthesized according to literature procedures.32a All other reagents utilized in the syntheses were used as received from the manufacturers, unless otherwise stated. The solvents used were of reagent grade. PDA (1), 97% purity, was obtained from Sigma Aldrich.Instrumentation and characterization
The NMR spectra were recorded with 400 MHz pulsed Fourier transform NMR spectrometer in CDCl3, DMSO-d6 or MeOH-d4 at room temperature. The chemical shift values are given in ppm and J values in Hz. High-resolution mass spectra were collected on an AccuTOF GCv 4G, JMS-T100GCV, Mass spectrometer (JEOL, Japan). The FD/FI probe (FD/FI) was equipped with an FD Emitter, Carbotec (Germany), FD 10 μm. Typical measurement conditions were as follow: Current rate 51.2 mA min−1 over 1.2 min; Counter electrode −10 kV; Ion source 37 V. The samples were prepared in dichloromethane. Absorption measurements were performed using a PerkinElmer Lambda 365 UV−Vis spectrophotometer. Photoluminescence studies were done in Jobin Horiba SPEX Fluorolog 111 Spectrofluorometer. For quantum yield measurements, the formula for optically dilute solutions was used.54 Fluorescence quantum yields were determined by the comparative method using perylene-3,4,9,10-tetracarboxylic tetrabutylester (ΦF = 0.95 in CH2Cl2) and N,N′-di(1-hexylheptyl)perylene-3,4,9,10-tetracarboxy bisimide (ΦF = 0.99 in CHCl3) and perylene-3,4,9,10-tetracarboxylic tetrapotassium salt (ΦF = 1.0 in water) as reference compounds.16 Melting points (uncorrected) were recorded on a Gallenkamp melting point apparatus.Kinetic measurements were performed on the formation of imides from amic acids at low concentrations in 3 mL quartz cuvettes with a 10 mm pathway. Reactions from the anhydrides 1, 11 or 15 to the amic acid salts of 5c, 12c and 9c were performed at room temperature (20 °C) at the smaller scale (200 or 100 mg starting compound) in DMF using DBU as the base at the conditions specified in the synthetic section. For the synthesis of 9c, 100 mg of 15, 2.0 equivalents of DBU and amine in 2 mL DMF were used. After 1 hour of reaction a droplet of the reaction mixture was diluted with DMF and a few droplets of this solution were added to a cuvette placed in a dual beam spectrophotometer. Absorption spectra were taken at room temperature every 5 minutes over a period of 24 hours.
Synthesis
Yield: 188 mg, 0.34 mmol, 66%. Mp >350 °C. 1H NMR (MeOH-d4, 400 MHz): 8.40 (2H, J = 8 Hz, d), 8.38 (2H, J = 8 Hz, d), 7.79 (2H, J = 8 Hz, d), 7.51 (2H, J = 8 Hz, d), 3.10 (6H, s), 3.05 (6H, s). MS (ESI): [M]2− Calculated for C28H20N2O6, 240.0666; found: 240.0668 [KM]− Calculated for C28H20KN2O6: 519.0964; found: 519.0965.
Yield: 230 mg, 0.39 mmol, 76%. Mp = 190–198 °C. 1H NMR (CDCl3, 400 MHz): 8.24 (4H, m), 7.86 (4H, J = 8 Hz, d), 7.52 (2H, J = 8 Hz, d), 4.33 (4H, J = 8 Hz, t), 4.15 (6H, s), 3.12 (6H, s), 1.82 (4H, J = 8 Hz, quin), 1.52 (4H, m), 1.01 (6H, J = 8 Hz, t). 13C NMR (CDCl3, 101 MHz): 172.13, 172.09, 169.04, 169.02, 133.60, 133.29, 132.73, 131.12, 130.43, 129.44, 129.28, 128.82, 128.32, 127.81, 127.66, 120.97, 120.80, 120.75, 77.04, 76.72, 65.37, 39.80, 35.06, 30.68, 19.30, 13.83. MS (FD): [M]+ Calculated for C36H38N2O6, 594.2730; found: 594.2764, [M2]+ Calculated for C72H77N4O12 1188.5460; found: 1188.5600.
In a 50 mL round bottom flask equipped with a magnetic stirring bar, PDA 1 (200 mg, 0.51 mmol), base (DBU or K2CO3, 2.04 mmol) and amine 4 (2.04 mmol) were suspended in 4 mL solvent. Reactions with methylamine 4d used a 40% solution in MeOH and were stirred for one hour at RT prior to heating. The suspension was stirred for 24 hours at the desired temperature. The reaction mixture was cooled to room temperature. Water (40 mL) was added under strong stirring, which caused a precipitation of the crude product. The precipitate was left to settle for a maximum of 24 hours. The reaction product was filtered and the obtained solid was washed extensively with 0.01 M K2CO3 until almost no color was observed in the filtrate and subsequently with water. When the reaction involved the more hydrophobic amine 4b, the filtrate was washed two more times with dilute HCl (1 M, 10 mL) and two more times with demineralized water (25 mL). The solid was dried in a vacuum oven.
DBU/DMSO water addition. After addition of the reagents, the suspension was stirred for 1 hour at room temperature. Subsequently 4 mL of water was added (2mL for the synthesis of 7b) and the reaction mixture was stirred for another 23 hours at room temperature. Workup as described above.
DBU/DMF method, 60 °C, from 1.00 g (2.55 mmol) PDA: Purple solid, yield 1.534 g, 2.50 mmol, 98%.
K 2 CO 3 /DMSO method, 100 °C, from 1.00 g (2.55 mmol) PDA: Purple solid, yield 1.548 g, 2.52 mmol, 99%.
DBU/DMSO water addition, from 200 mg PDA: Purple solid, yield 307 mg, 0.50 mmol, 98%.
1H NMR (δH[ppm], CDCl3, 400 MHz): 8.61 (4H, J = 8 Hz, d), 8.52 (4H, J = 8 Hz, d), 4.14 (4H, m), 1.97 (2H, m), 1.42–1.32 (18H, m), 0.96 (6H, J = 8 Hz, t), 0.90 (6H, J = 8 Hz, t). 13C NMR (δC[ppm], CDCl3, 101 MHz): 163.70, 134.44, 131.35, 126.30, 123.25, 122.98, 44.34, 37.96, 30.77, 28.71, 24.08, 23.08, 14.10, 10.64.
DBU/DMF method, 60 °C, from 1.00 g (2.55 mmol) PDA: Purple solid, yield 1.249 g, 2.49 mmol, 98%.
K 2 CO 3 /DMSO method, 100 °C, from 1.00 g (2.55 mmol) PDA: Purple solid, yield 1.239 g, 2.47 mmol, 97%.
DBU/DMSO water addition, from 200 mg PDA: Purple solid, yield 249 mg, 0.50 mmol, 98%.
1H NMR (δH[ppm], CDCl3, 400 MHz): δ 8.74 (4H, J = 8 Hz, d), 8.67 (4H, J = 8 Hz, d), 4.23 (4H, J = 8 Hz, t), 1.74 (4H, J = 8 Hz, quin), 1.47 (4H, J = 8 Hz, sex), 0.99 (6H, J = 8 Hz, t).
DBU/DMF method, RT, from 200 mg PDA: Red solid, yield 210 mg, 0.50 mmol, 98%.
K 2 CO 3 /DMSO method, 100 °C, from 200 mg PDA: Red solid, yield 207 mg, 0.49 mmol, 92%.
DBU/DMSO water addition, from 200 mg PDA: Red solid, yield 214 mg, 0.51 mmol, 100%.
1H NMR (δH[ppm], D2SO4, 400 MHz): 9.15 (4H, J = 8 Hz, d), 9.10 (4H, J = 8 Hz, d), 3.93 (6H, s). FTIR: 742, 808, 850, 863, 1021, 1053, 1127, 1156, 1184, 1237, 1284, 1326, 1358, 1400, 1446, 1507, 1578, 1594, 1659, 1698.
K 2 CO 3 /DMSO method, 80 °C, 1.00 g (2.55 mmol) PDA: Dark purple solid, yield 1.177 g, 2.36 mmol, 97%.
1H NMR (δH[ppm], D2SO4, 400 MHz): 9.21–9.07 (8H, m), 5.64 (4H, J = 8 Hz, t), 4.85 (4H, J = 8 Hz, t).
DMF/DBU method, 60 °C, 200 mg PDA: Purple solid, yield 297 mg, 0.50 mmol, 97%.
K 2 CO 3 /DMSO method, 100 °C, 200 mg PDA: Purple solid, yield 296 mg, 0.50 mmol, 97%.
DBU/DMSO water addition, 200 mg PDA: Purple solid, yield 270 mg, 0.46 mmol, 90%.
1H NMR (CDCl3, 400 MHz): 8.77 (4H, J = 8 Hz, d), 8.72 (4H, J = 8 Hz, d), 4.37 (4H, J = 8 Hz, t), 4.25 (4H, J = 8 Hz, t), 8.69 (8H, m), 1.50 (8H, m).
:1 acetone/petroleum ether mixture was added. The precipitate is filtered off and washed with acetone/petroleum ether. The purple solid was dissolved in ethanol (5 mL) and filtered to remove the remaining K2CO3. The organic solvent was removed from the filtrate by rotary evaporation. This yielded a yellow solid that was dried in a vacuum oven.
Yield: 42 mg, 0.08 mmol, 73%. Mp >350 °C. 1H NMR (MeOD, 400 MHz): 8.59–8.45 (6H, m), 7.88 (1H, J = 8 Hz, d), 7.59 (1H, J = 4 Hz, d), 4.16 (2H, J = 8 Hz, t), 3.13 (3H, s). 3.10 (3H, s), 1.73 (2H, J = 8 Hz, quin), 1.47 (2H, J = 8 Hz, sex), 1.02 (3H, J = 8 Hz, t). MS (ESI): [M]− Calculated for C28H20N2O6, 240.0666; found: 240.0668 [KM]− Calculated for C30H23N2O5, 491.1612; found: 491.1598.
Yield: 53 mg, 0.097 mmol, 77%. Mp = 153–154 °C. 1H NMR (CDCl3, 400 MHz): 8.63 (2H, J = 8 Hz, d), 8.46 (4H, m), 7.95 (1H, J = 8 Hz, d), 7.63 (1H, J = 8 Hz, d), 4.35 (2H, J = 8 Hz, t), 4.22 (2H, J = 8 Hz, t), 3.18 (3H, s), 3.13 (3H, s), 1.84 (2H, m), 1.75 (2H, m), 1.47 (2H, m), 1.01 (6H, m). 13C NMR (CDCl3, 101 MHz): 168.56, 163.38, 163.34, 135.33, 135.14, 132.43, 131.51, 131.01, 130.90, 129.92, 129.22, 127.84, 125.48, 122.68, 122.44, 121.55, 121.31, 121.22, 120.94, 65.74, 40.24, 39.84, 35.12, 30.67, 30.20, 20.44, 19.31, 13.87, 13.85. MS (FD): [M]+ Calculated for C34H32N2O5, 548.2311; found: 548.2326, [M2]+ Calculated for C68H62N4O10, 1096.4622; found: 1096.4767.
:1 acetone/petroleum ether mixture was added. The resulting yellow precipitate was left to settle and filtered off. The yellow solid was dissolved in ethanol (5 mL) and filtered to remove the remaining K2CO3. The organic solvent was removed from the filtrate by rotary evaporation. This yielded a yellow solid that was dried in a vacuum oven.
Yield: 35 mg, 0.063 mmol, 61%. Mp >350 °C. 1H NMR (MeOD, 400 MHz): 8.40 (2H, J = 8 Hz, d), 8.36 (2H, J = 4 Hz, d), 7.76 (2H, J = 8 Hz, d), 7.81 (1H, J = 8 Hz, d), 7.51 (1H, J = 8 Hz, d), 4.30 (4H, J = 8 Hz, t), 3.12 (3H, s), 3.08 (3H, s), 1.78 (4H, J = 8 Hz, quin), 1.52 (4H, J = 8 Hz, sex), 1.01 (6H, J = 8 Hz, t). MS: (ESI): [M]− Calculated for C34H32NO7, 566.2148; found: 566.2165, [KM2]− Calculated for C68H62KN2O14, 1171.4000; found: 1171.3961.
Yield: 66 mg, 0.10 mmol 55%. Mp = 178 °C. 1H NMR (CDCl3, 400 MHz): 8.41 (2H, d, 8 Hz), 8.20 (2H, d, 4 Hz), 8.17 (2H, d, 4 Hz), 7.97 (2H, J = 8 Hz, d), 4.36 (4H, J = 8 Hz, t), 4.11 (2H, m), 1.96 (1H, J = 8 Hz, sep), 1.82 (4H, J = 8 Hz, quin), 1.53 (4H, J = 8 Hz, sex), 1.42–1.32 (8H, m), 1.02 (6H, J = 8 Hz, t), 0.96 (3H, J = 8 Hz, t), 0.90 (3H, J = 8 Hz, t). 13C NMR (CDCl3, 101 MHz): 168.20, 163.78, 135.06, 131.86, 131.78, 131.18, 130.15, 129.04, 128.93, 128.83, 125.67, 122.37, 121.95, 121.60, 77.31, 76.99, 76.67, 65.54, 44.18, 37.97, 30.78, 30.62, 28.71, 24.09, 23.08, 19.25, 14.09, 13.79, 10.64. MS (FD): [M]+ Calculated for C40H43NO6, 633.3090; found: 633.3073.
Synthesis for compound 15, performed at 40 °C overnight (16 h) starting from 200 mg (0.38 mmol) 11: Yield: 215 mg, 0.37 mmol, 97% red solid. Traces of PDI 7c are visible in the NMR spectrum of the crude reaction product, presumably originating from PDA contaminant in the starting material. Yield after column chromatography, 204 mg, 0.35 mmol, 92%.
1H NMR (CDCl3, 400 MHz): 8.17 (2H, J = 8 Hz, d), 7.91 (2H, J = 8 Hz, d), 7.84 (4H, m), 4.36 (4H, J = 8 Hz, t), 4.12 (2H, J = 8 Hz, t), 1.82 (4H, J = 8 Hz, quin), 1.74 (2H, J = 8 Hz, quin), 1.52 (6H, m), 1.03 (9H, m). 13C NMR (CDCl3, 101 MHz): 168.12, 163.15, 134.61, 131.67, 131.47, 130.73, 129.97, 128.81, 128.47, 128.44, 125.21, 122.14, 121.67, 121.27, 77.33, 77.01, 76.70, 65.56, 40.26, 30.64, 30.17, 20.43, 19.28, 13.86, 13.82. MS (FD): [M]+ Calculated for C38H39NO7, 577.2464; found: 577.2443.
Yield: 77 mg, 0.14 mmol, 75%. Mp = 263 °C. 1H NMR (CDCl3, 400 MHz): 8.32 (2H, J = 8 Hz, d), 8.11 (2H, J = 8 Hz, d), 8.07 (2H, J = 8 Hz, d), 7.93 (2H, J = 8 Hz, d), 4.35 (4H, J = 8 Hz, t), 3.50 (3H, s), 1.81 (4H, J = 8 Hz, quin), 1.51 (4H, J = 8 Hz, sex), 1.01 (6H, J = 8 Hz, t). 13C NMR (CDCl3, 101 MHz): 168.15, 163.53, 135.09, 131.83, 131.69, 131.02, 130.13, 128.96, 128.66, 128.63, 125.52, 122.37, 121.63, 121.48, 77.30, 76.99, 76.67, 65.57, 30.61, 26.98, 19.25, 13.79. MS (FD): [M]+ Calculated for C33H29NO6, 535.1995; found: 535.1983.
Yield: 82 mg, 0.14 mmol, 80%. Mp = 305 °C. 1H NMR (CDCl3, 400 MHz): 7.92 (2H, J = 8 Hz, d), 7.69 (2H, J = 8 Hz, d), 7.59 (2H, J = 8 Hz, d), 4.36–4.31 (6H, m), 4.03 (2H, J = 4 Hz, t), 2.89 (br, 1H), 1.84 (4H, J = 8 Hz, quin), 1.54 (4H, J = 8 Hz, sex), 1.04 (6H, J = 8 Hz, t). 13C NMR (CDCl3, 101 MHz): 168.00, 163.73, 134.40, 131.68, 130.84, 130.63, 129.84, 128.46, 127.95, 127.95, 124.61, 122.12, 120.99, 120.98, 77.33, 77.01, 76.69, 65.62, 61.27, 42.72, 30.63, 19.28, 13.84. MS (FD): [M]+ Calculated for C34H31NO7, 565.2101; found: 565.2100.
Yield: 88 mg, 0.14 mmol, 75%. Mp = 195 °C. 1H NMR (CDCl3, 400 MHz): 8.09 (2H, J = 8 Hz, d), 7.84 (2H, J = 8 Hz, d), 7.80 (2H, J = 8 Hz, d), 7.75 (2H, J = 8 Hz, d), 4.36 (4H, J = 8 Hz, t), 4.08 (2H, J = 8 Hz, t), 3.86 (2H, J = 8 Hz, t), 1.83 (6H, m), 1.76 (2H, m), 1.64 (2H, m), 1.52 (6H, m), 1.03 (6H, J = 8 Hz, t). 13C NMR (CDCl3, 101 MHz): 168.11, 163.11, 134.57, 103.66, 129.96, 128.74, 128.37, 128.30, 125.06, 122.17, 121.47, 121.19, 65.58, 62.70, 40.22, 32.56, 30.64, 27.88, 26.64, 25.22, 19.28, 13.83. MS (FD): [M]+ Calculated for C38H39NO7, 621.2727; found: 621.2703.
Yield: 103 mg, 0.17 mmol, 86%. Mp = 123–127 °C. 1H NMR (CDCl3, 400 MHz): 8.06 (4H, m), 7.92 (2H, m), 7.90 (1H, J = 8 Hz, d), 7.75 (1H, J = 8 Hz, d), 7.42 (1H, d), 4.34 (6H, J = 8 Hz, t), 3.15 (3H, s), 3.13 (3H, s), 1.87–1.76 (6H, m), 1.57–1.48 (6H, m), 1.05–0.99 (9H, m). 13C NMR (CDCl3, 101 MHz): 171.92, 168.90, 168.65, 134.11, 133.22, 133.07, 132.63, 130.92, 130.41, 130.31, 130.00, 129.68, 129.31, 128.89, 128.83, 128.69, 128.43, 127.75, 121.42, 121.33, 120.95, 120.68, 65.49, 65.23, 39.80, 35.08, 30.66, 19.29, 13.80. MS (FD): [M]+ Calculated for C38H41NO7, 623.2883; found: 623.2886.
Yield: 215 mg, 93%. 1H NMR (CDCl3, 400 MHz): 8.81 (8H, m), 4.26 (2H, J = 8 Hz, t), 1.76 (2H, J = 8 Hz, quin), 1.49 (2H, J = 8 Hz, sex), 1.00 (3H, J = 8 Hz, t).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was in part funded by The Netherlands Organization for Scientific Research (NWO) by the Materials for Sustainability project SPEAR under Grant No. 739.017.012. The authors thank Dr Georgy Filonenko and Ed Zuidinga for the mass spectrometry measurements.Notes and references
- M. Kardos, Über einige Aceanthrenchinon- und 1.9-Anthracen-Derivate, Ber. Dtsch. Chem. Ges., 1913, 46, 2086–2091 CrossRef CAS.
- W. Herbst and K. Hunger, Industrial Organic Pigments, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) F. Würthner, Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures, Chem. Commun., 2004, 1564–1579 RSC; (b) H. Langhals, Control of the interactions in multichromophores: Novel concepts. Perylene bis-imides as components for larger functional units, Helv. Chim. Acta, 2005, 88, 1309–1343 CrossRef CAS; (c) C. Huang, S. Barlow and S. R. Marder, Perylene-3,4,9,10-tetracarboxylic Acid Diimides: Synthesis, Physical Properties, and Use in Organic Electronics, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- (a) C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Non-Fullerene Electron Acceptors for Use in Organic Solar Cells, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed; (b) Z. Liu, Y. Wu, Q. Zhang and G. Xiang, Non-fullerene small molecule acceptors based on perylene diimides, J. Mater. Chem., 2016, 4, 17604–17622 RSC; (c) N. Zink-Lorre, E. Font-Sanchis, A. Sastre-Santos and F. Fernandez-Lazaro, Perylenediimides as more than just non-fullerene acceptors: versatile components in organic, hybrid and perovskite solar cells, Chem. Commun., 2020, 56, 3824–3838 RSC.
- (a) R. F. Kelly, W. S. Shin, B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, Photoinitiated charge transport in supramolecular assemblies of a 1,7,N,N ‘-tetrakis(zinc porphyrin)-perylene-3,4,9,10-bis(dicarboximide), J. Am. Chem. Soc., 2007, 129, 3173–3181 CrossRef PubMed; (b) J. H. Hurenkamp, W. R. Browne, R. Augulis, A. Pugzlys, P. M. H. van Loosdrecht, J. H. van Esch and B. L. Feringa, Intramolecular energy transfer in a tetra-coumarin perylene system: influence of solvent and bridging unit on electronic properties, Org. Biomol. Chem., 2007, 5, 3354–3362 RSC; (c) M. Fulitsuka, K. Harada, A. Sugimoto and T. Majima, Excitation Energy Dependence of Photoinduced Processes in Pentathiophene-Perylene Bisimide Dyads with a Flexible Linker, J. Phys. Chem. A, 2008, 112, 10193–10199 CrossRef PubMed; (d) M. R. Wasielewski, Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed; (e) V. M. Blas-Ferrando, J. Ortiz, L. Bouissane, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and A. Sastre-Santos, Rational design of a phthalocyanine-perylenediimide dyad with a long-lived charge-separated state, Chem. Commun., 2012, 48, 6241–6243 RSC; (f) P. D. Frischmann, K. Mahata and F. Wurthner, Powering the future of molecular artificial photosynthesis with light-harvesting metallo-supramolecular dye assemblies, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC; (g) K. Dubey, D. Inan, S. Sengupta, E. J. R. Sudhölter, F. C. Grozema and W. F. Jager, Tunable and Highly Efficient Light-Harvesting Antenna Systems Based on 1,7-Perylene-3,4,9,10-Tetracarboxylic Acid Derivatives, Chem. Sci., 2016, 7, 3517–3532 RSC.
- (a) P. D. Frischmann, L. C. H. Gerber, S. E. Doris, E. Y. Tsai, F. Y. Fan, X. Qu, A. Jain, K. A. Persson, Y.-M. Chiang and B. A. Helms, Supramolecular Perylene Bisimide-Polysulfide Gel Networks as Nanostructured Redox Mediators in Dissolved Polysulfide Lithium-Sulfur Batteries, Chem. Mater., 2015, 27, 6765–6770 CrossRef CAS; (b) A. Iordache, V. Delhorbe, M. Bardet, L. Dubois, T. Gutel and L. Picard, Perylene-Based All-Organic Redox Battery with Excellent Cycling Stability, ACS Appl. Mater. Interfaces, 2016, 8, 22762–22767 CrossRef CAS PubMed; (c) P. Ranque, C. George, R. K. Dubey, R. van der Jagt, D. Flahaut, R. Dedryvère, M. Fehse, P. Kassanos, W. F. Jager, E. J. R. Sudhölter and E. M. Kelder, Scalable route to Electroactive and Light active Perylene diimide dye polymer Binder for Li ion Batteries, ACS Appl. Energy Mater., 2020, 3, 2271–2277 CrossRef CAS PubMed.
- (a) T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, The Rylene Colorant Family—Tailored Nanoemitters for Photonics Research and Applications, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed; (b) D. Aigner, S. A. Freunberger, M. Wilkening, R. Saf, S. M. Borisov and I. Klimant, Enhancing Photoinduced Electron Transfer Efficiency of Fluorescent pH-Probes with Halogenated Phenols, Anal. Chem., 2014, 86, 9293 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Reduction of aryl halides by consecutive visible light-induced electron transfer processes, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- For a recent review on the synthesis of PDIs and other PTCA derivatives, see: A. Nowak-Król and F. Würthner, Progress in the synthesis of perylene bisimide dyes, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- (a) H. Langhals, Synthese von hochreinen Perylen-Fluoreszenzfarbstoffen in großen Mengen – gezielte Darstellung von Atrop-Isomeren, Chem. Ber., 1985, 118, 4641 CrossRef CAS; (b) H. Langhals, Cyclic Carboxylic Imide Structures as Structure elements of High Stability. Novel Developments in Perylene Dye Chemistry, Heterocycles, 1995, 40, 477–500 CrossRef CAS.
- F. Würthner, J. Sautter and J. Schilling, Synthesis of diazadibenzoperylenes and characterization of their structural, optical, redox, and coordination properties, J. Org. Chem., 2002, 67, 3037–3044 CrossRef PubMed.
- D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and D. Sacchi, A two-channel chemosensor for the optical detection of carboxylic acids, including cholic acid, J. Mater. Chem., 2005, 15, 2670–2675 RSC.
- Q. Cao, D. E. Crawford, C. Shi and S. L. James, Greener Dye Synthesis: Continuous, Solvent-Free Synthesis of Commodity Perylene Diimides by Twin-Screw Extrusion, Angew. Chem., Int. Ed., 2020, 59, 4478–4483 CrossRef CAS PubMed.
- B. Baumgartner, A. Svirkova, J. Bintinger, C. Hametner, M. Marchetti-Deschmann and M. M. Unterlass, Green and highly efficient synthesis of perylene and naphthalene bisimides in nothing but water, Chem. Commun., 2017, 53, 1229–1232 RSC.
- Y. Nagao and T. J. Misono, Synthesis and Reactions of Perylenecarboxylic Acid-derivatives 8. Synthesis of N-alkyl-3,4–9,10-Perylenetetracarboxylic monoanhydride monoimide, Bull. Chem. Soc. Jpn., 1981, 54, 1191–1194 CrossRef CAS.
- H. Langhals, J. Karolin and L. B-Å. Johansson, Spectroscopic properties of new and convenient standards for measuring fluorescence quantum yields, J. Chem. Soc., Faraday Trans., 1998, 94, 2919–2922 RSC.
- PMAMI was obtained after acid work-up, which converts a dicarboxylic acid into an anhydride.
- At low temperatures imide formation is prevented, because amic acid formation from dicarboxylates requires elevated temperatures, as in ref. 14.
- Using a 40% methylamine solution in refluxing water, a quantitative PDI synthesis was reported, which makes sense in view of the extremely high concentration and the huge excess of the amine, see: P. J. Stang, D. H. Cao, S. Saito and A. M. Arif, Self-assembly of Cationic Tetranuclear, Pt(II) and Pd(II) Macrocylic Squares, J. Am. Chem. Soc., 1995, 117, 6273–6283 CrossRef CAS.
- Formation of monoimides from perylene dicarboxylate monoanhydride has been reported, see: (a) H. Tröster, Untersuchungen zur Protonierung von Perylen-3,4,9,10-tetracarbonsäurealkalisalzen, Dyes Pigm., 1983, 4, 171–177 CrossRef; (b) H. Kaiser, J. Linder and H. Langhals, Synthese von nichtsymmetrisch substituierten Perylen-Fluoreszenzfarbstoffen, Chem. Ber., 1991, 124, 529–535 CrossRef CAS.
- (a) T. Hassheider, S. A. Benning, H. S. Kitzerow, M. F. Achard and H. Bock, Color-tuned electroluminescence from columnar liquid crystalline alkyl arenecarboxylates, Angew. Chem., Int. Ed., 2001, 40, 2060–2063 CrossRef CAS; (b) R. K. Dubey, N. Westerveld, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, Facile Synthesis of Pure 1,6,7,12-Tetrachloroperylene-3,4,9,10-tetracarboxy Bisanhydride and Bisimide, Org. Lett., 2015, 17, 1882–1885 CrossRef CAS PubMed.
- J. Kelber, H. Bock, O. Thiebaut, E. Grelet and H. Langhals, Room-Temperature Columnar Liquid-Crystalline Perylene Imido-Diesters by a Homogeneous One-Pot Imidification–Esterification of Perylene-3,4,9,10-tetracarboxylic Dianhydride, Eur. J. Org. Chem., 2011, 707–712 CrossRef CAS.
- T. C. de Barros, P. B. Filho, M. Loos, M. J. Politi, H. Chaimovich and I. M. Cuccovia, Formation and decomposition of N-alkylnaphthalimides: experimental evidences and ab initio description of the reaction pathways, J. Phys. Org. Chem., 2011, 24, 385–397 CrossRef CAS.
- (a) M. Kim and D. W. Dixon, Hydrolysis of aliphatic naphthalene diimides: effect of charge placement in the side chains, J. Phys. Org. Chem., 2008, 21, 731–737 CrossRef CAS; (b) D. Shukla, D. M. Meyer and W. G. Ahearn, Aromatic Amic Acid Salts and Composition, US2011/295010, 2011, A1.
- T. C. Barros, G. R. Molinari, P. Berci Filho, V. G. Toscano and M. J. Politi, Photophysical properties of N-alkylnaphthalimides and analogs, J. Photochem. Photobiol., A, 1993, 76, 55–60 CrossRef CAS.
- F. A. Mason, Derivatives of 1 8-naphthalic acid Part I The preparation and properties of 1 8-naphthalyl chloride, J. Chem. Soc., 1924, 125, 2116–2119 RSC.
- K. Strey and J. Voes, EPR Studies on Carboxylic Esters. Part 15. Spin Density Distribution in the Radical Anions of Naphthalenecarboxylic Esters, J. Chem. Res., Miniprint, 1998, 3, 648 Search PubMed.
- Y. Kim, M. Ree, T. Chang, C. S. Ha, T. L. Nunes and J. S. Lin, Rodlike Flexible Polyimide Composite Films Prepared from Soluble Poly(amic diethyls ester) precursors – Miscibility, Structure and Properties, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 2075–2082 CrossRef CAS.
- L. Feiler, H. Langhals and K. Polborn, Synthesis of Perylene-3,4-dicarboximides- Novel Highly Photostable Fluorescent Dyes, Liebigs Ann., 1995, 1229–1244 CrossRef CAS.
- This higher reactivity of amic esters is attributed by us to the higher solubility of amic esters as compared to amic acids, vide infra.
- With 25% of PDI isolated, the conversion of the imidization reaction is expected to be at least 50%, as the remainder of the reaction mixture consists mainly of perylene monoimide monoamic acid 9b (∼5(0%) and lesser amounts of perylene diamic acid 5b (∼25%). See also the “Reaction kinetics” section in this manuscript.
- (a) C. Xue, R. Sun, R. Annab, D. Abadi and S. Jin, Perylene monoanhydride diester: a versatile intermediate for the synthesis of unsymmetrically substituted perylene tetracarboxylic derivatives, Tetrahedron Lett., 2009, 50, 853–856 CrossRef CAS; (b) R. Wang, Z. Shi, C. Zhang, A. Zhang, J. Chen, W. Guo and Z. Sun, Facile synthesis and controllable bromination of asymmetrical intermediates of perylene monoanhydride/monoimide diester, Dyes Pigm., 2013, 98, 450–458 CrossRef CAS.
- The 1H NMR spectrum of compound 6a is similar to the one reported for a similar perylene diamic acid. For these compounds the cis and trans isomers could not be distinguished either, see: A. C. Boccia, V. Lukes, A. Eckstein-Andicsova and E. Kozma, Solvent- and concentration-induced self-assembly of an amphiphilic perylene dye, New J. Chem., 2020, 44, 892–899 RSC.
- Generally chlorosulfonic acid or sulphuric acid are used for this transformation. Mixtures of acetic acid and sulfuric acid have been used at elevated temperatures: V. Kunz, V. Stepanenko and F. Würthner, Embedding of a ruthenium(II) water oxidation catalyst into nanofibers via self-assembly, Chem. Commun., 2015, 51, 290–293 RSC.
- When sampled in water the formation of 7c was deduced from an elevated baseline in the absorption spectra and a purple solid precipitate in the reaction mixture.
- For PDI 7b we initially used an alternative workup procedure, involving extraction with chloroform, followed by filtration. However, this procedure was tedious less efficient and only suitable for soluble PDIs.
- P. Anastas and N. Eghbali, Green Chemistry: Principles and Practice, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- For the synthesis of PTEs in acetonitrile, we have attempted to replace DBU by other tertiary amines, but none of these bases gave satisfactory yields. In this specific case, apart from the solubility of the intermediate salts, base strength may also have been critical.
- For the solubility of aliphatic PDIs, see: S. Demming and H. Langhals, Leichtlösliche, lichtechte Perylen-Fluoreszenz-farbstoffe, Chem. Ber., 1988, 121, 225–230 CrossRef.
- Practical Chemical Kinetics in Solution: O. A. El Seoud, W. J. Baader and E. L. Bastos, in Encyclopedia of Physical Organic Chemistry, ed. Z. Wang, John Wiley & Sons, Inc., 2017 Search PubMed.
- (a) M. Hecht and F. Würthner, Supramolecularly Engineered J-Aggregates Based on Perylene Bisimide Dyes, Acc. Chem. Res., 2021, 54, 642–653 CrossRef CAS PubMed; (b) Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Self-assembled pi-stacks of functional dyes in solution: structural and thermodynamic features, Chem. Soc. Rev., 2009, 38, 564–584 RSC; (c) H. Wang, T. E. Kaiser, S. Uemura and F. Würthner, Perylene bisimide J-aggregates with absorption maxima in the NIR, Chem. Commun., 2008, 1181–1183 RSC.
- Extinction coefficient of diamic acids like K25a are 33000 M−1 cm−1.
- In Fig. 5 it is clearly visible that the first spectrum primarily contains compound K25c. Absorption above 500 nm, indicates that compound K9c is already present in the reaction mixture. The last spectrum consists mostly of compound 7c, but the tail in the absorption above 500 nm may indicate that not all K9c has been consumed.
- PDA, PDIs and PMAMIs are virtually insoluble in DMF. When these compounds are formed in situ by reaction in DMF, oversaturated solutions are formed. These compound are molecularly dissolved and highly fluorescent, but precipitate in due time.
- Extracting the relative concentrations of 5c, 9c and 7c from the absorption spectra has been performed manually in a few independent runs. In all cases the relative concentration of 7c was consistent. The relative concentrations of 5c and 9c, on the other hand, showed a larger variation between the different runs.
- Reactions of naphthyl amic acids as a function of the pH in water has been investigated by absorption spectroscopy, see ref. 23: B. S. Souza, J. R. Mora, E. H. Wanderlind, R. M. Clementin, J. C. Gesser, H. D. Fiedler, F. Nome and F. M. Menger, Transforming a Stable Amide into a Highly Reactive One: Capturing the Essence of Enzymatic Catalysis, Angew. Chem., Int. Ed., 2017, 56, 5345–5348 CrossRef CAS PubMed.
- The apparent isosbestic point at 473 nm is confusing. Close inspection of the spectra shows that an isosbestic point for compounds 5a and 1 is found at this wavelength, but that the extinction coefficient of the reaction intermediate 8a at 473 nm is slightly higher.
- The high dilution used here comes down to 1 gram of PDA in 750 mL DMF.
- Water addition was also accompanied by a spontaneous ∼23 °C rise in temperature, see: N. Friedman, Hydrogen Bonding and Heat of Solution, J. Chem. Educ., 1977, 54, 248 CrossRef CAS . This temperature increase was short-lived and has not contributed to the increased yield.
- For the “green” reaction, with K2CO3 and DMSO, adding 4 ml of water after 1 hour increased solubility, but the orange potassium salt of compound 5c did not fully dissolve. Continuation of the reaction at room temperature did result in a higher PDI yield, ∼75%, but the reaction yield was not quantitative.
- (a) Y. Xu, S. Leng, C. Xue, R. Sun, J. Pan, J. Ford and S. Jin, A room-temperature liquid-crystalline phase with crystalline pi stacks, Angew. Chem., Int. Ed., 2007, 46, 3896–3899 CrossRef CAS PubMed; (b) J. Gershberg, M. R. Stojković, M. Škugor, S. Tomić, T. H. Rehm, S. Rehm, C. R. Saha-Möller, I. Piantanida and F. Würthner, Sensing of Double-Stranded DNA/RNA Secondary Structures by Water Soluble Homochiral Perylene Bisimide Dyes, Chem. – Eur. J., 2015, 21, 7886–7895 CrossRef CAS PubMed.
- For the reaction of 11 with 4-dimethylamino aniline using DBU in DMF under argon, a 60% yield was obtained after reacting overnight at 60 °C.
- (a) Y. Matsunaga, K. Goto, K. Kubono, K. Sako and T. Shinmyozu, Photoinduced Color Change and Photomechanical Effect of Naphthalene Diimides Bearing Alkylamine Moieties in the Solid State, Chem. – Eur. J., 2014, 20, 7309–7316 CrossRef CAS PubMed; (b) S. Saha and P. Sahoo, Luminescence turn-on response of naphthalene diimide based chemosensor with Formaldehyde: A novel stratagem for estimation of formaldehyde in storage fish samples, Bioorg. Med. Chem. Lett., 2021, 49, 128287 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic and Plenum Publishers, New York, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extra figures, schemes and tables to support the main text. Procedure for kinetic measurements and modelling. Absorption and emission spectra and fluorescence quantum yields of all new compounds. 1H and 13C NMR spectra of all synthesized compounds Mass spectra of all the new compounds. See DOI: 10.1039/d1qo01723c |
| This journal is © the Partner Organisations 2022 |