
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antidiabetic profiling of veramycins, polyketides accessible by biosynthesis, chemical synthesis and precursor-directed modification†
Denis
Dardić
a,
Nils
Böhringer
be,
Alberto
Plaza
a,
Florian
Zubeil
a,
Juliane
Pohl
ab,
Svenja
Sommer
ab,
Leo
Padva
ab,
Jonathan
Becker
b,
Maria A.
Patras
a,
Mona-Katharina
Bill
a,
Michael
Kurz
c,
Luigi
Toti
d,
Sven W.
Görgens
c,
Sören M. M.
Schuler
a,
André
Billion
a,
Oliver
Schwengers
 b,
Paulus
Wohlfart
d,
Alexander
Goesmann
b,
Norbert
Tennagels
d,
Andreas
Vilcinskas
ab,
Peter E.
Hammann
d,
Till F.
Schäberle
*abe and
Armin
Bauer
*c
b,
Paulus
Wohlfart
d,
Alexander
Goesmann
b,
Norbert
Tennagels
d,
Andreas
Vilcinskas
ab,
Peter E.
Hammann
d,
Till F.
Schäberle
*abe and
Armin
Bauer
*c
aFraunhofer Institute for Molecular Biology and Applied Ecology (IME), Branch for Bioresources, 35392 Gießen, Germany. E-mail: Till.F.Schaeberle@agrar.uni-giessen.de
bJustus-Liebig-University Gießen, 35392 Gießen, Germany
cSanofi-Aventis Deutschland GmbH, R&D Integrated Drug Discovery, 65926 Frankfurt am Main, Germany. E-mail: Armin.Bauer@sanofi.com
dSanofi-Aventis Deutschland GmbH, R&D German Hub, 65926 Frankfurt am Main, Germany
eGerman Center of Infection Research (DZIF), Partner Site Gießen-Marburg-Langen, 35392 Gießen, Germany
First published on 9th February 2022
Abstract
Seven new polyketides, termed veramycins, were isolated from a Streptomyces sp. from the Sanofi microbial strain collection along with their known congeners NFAT-133 and TM-123. Veramycin A, an α-pyrone congener of TM-123 and NFAT-133 showed an increased baseline deoxy-glucose uptake in the absence of insulin in a modified L6 rat skeletal muscle cell line (L6 GLUT4 AS160-like cells). In addition, both compounds slightly increased the sensitivity to insulin in this cell line. Total syntheses of NFAT-133, TM-123 and veramycin A were accomplished starting from a central building block, which bears the three contiguous stereogenic centers of this polyketide family. Our approach enables an efficient, selective and flexible access to all possible isomers of the stereotriad for further exploration of this series as a potential anti-diabetic lead structure as exemplified by the synthesis of an NFAT-133 epimer. Finally, the corresponding biosynthetic gene cluster (BGC) was identified by genome sequencing and gene inactivation. Based on feeding experiments, a biosynthetic pathway was proposed, which enabled access to new veramycin A analogs by precursor-directed biosynthesis.
Introduction
5′-Adenosine monophosphate-activated protein kinase (AMPK) is a highly conserved and ubiquitously expressed protein. It plays a central role in the regulation of cellular energy homeostasis by interacting with numerous metabolic pathways and is denoted the “fuel gauge” of the cell. Cellular energy levels are precisely monitored by AMPK through sensing AMP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ATP and ADP:ATP ratios. Hence, the global effect of AMPK activation leads to an increase of catabolic processes with concomitant down-regulation of non-essential anabolic processes in order to maintain cellular energy homeostasis. Furthermore, AMPK plays a decisive role in the coordination of metabolism at the level of the whole body.1
:ATP and ADP:ATP ratios. Hence, the global effect of AMPK activation leads to an increase of catabolic processes with concomitant down-regulation of non-essential anabolic processes in order to maintain cellular energy homeostasis. Furthermore, AMPK plays a decisive role in the coordination of metabolism at the level of the whole body.1
AMPK activation modulates multiple pathways involved in metabolic processes (e.g., skeletal muscle glucose uptake by phosphorylation of downstream targets). Beyond its metabolic role, involvement of AMPK has been identified in onco-suppression, regulation of appetite and circadian rhythm, anti-inflammatory response and longevity.2–5
Due to its central role in energy regulation, modulation of AMPK signalling became an attractive target for the treatment of metabolic disorders. Skeletal muscle contraction leads to AMPK activation and, consequently, to e.g. increased glucose uptake and insulin sensitivity.6 Since skeletal muscle AMPK signalling is likely not impaired in type 2 diabetes patients, AMPK activation has gained particular interest as a potential therapeutic intervention for the treatment of this disease.7 Furthermore, AMPK is gaining more and more interest as a target in other indications (e.g. non-alcoholic fatty liver disease (NAFLD) and oncology).8,9
Numerous natural products (NPs) and NP-derived drugs act as indirect activators of AMPK, mainly by inhibiting mitochondrial respiration, leading to shutdown of ATP synthesis and increase of AMP levels.10
One such NP from microbial origin is the small aromatic polyketide NFAT-133. It was isolated in 1995 from the culture broth of Streptomyces sp. AB 2184C-502 during a bioactivity-guided screening campaign in search of immunosuppressant agents. The compound was initially discovered as an inhibitor of NFAT transcription factor activation, an early step in T cell activation.11 NFAT-133 was later re-isolated from Streptomyces sp. PM0324667, again by a bioactivity-guided approach, due to its ability to induce glucose uptake in insulin-stimulated differentiated L6 rat skeletal myotubes in the presence of insulin. In db/db mice, the compound showed a significant reduction of plasma glucose and insulin levels when administered intraperitoneally as well as a significant body weight reduction.12 Detailed mechanistic investigations revealed that NFAT-133 decreases the mitochondrial membrane potential in a dose-dependent manner, which leads to a reduced ATP production and, consequently, an increase of the AMP:ATP ratio.13 NFAT-133 was also isolated from Streptomyces sp. K07-0010 along with the two structurally related isochromans panowamyin A and B, and displayed moderate antitrypanosomal activity.14 Very recently, NFAT-133 and several related small polyketides, the benwamycins, were isolated from another soil-derived Streptomyces species.15 While the preparation of our manuscript about investigations on the isolation, characterization, biosynthesis and total synthesis of NFAT-133 and its congeners, the BGC corresponding to NFAT-133 production in Streptomyces pactum was published along with the disclosure of further congeners (Chart 1).16
 | ||
| Chart 1 Structures of NFAT-133 and known related polyketides. | ||
The 2D structure of NFAT-133 was determined in 199511 but only in 2016, a first proposal of the absolute configuration of NFAT-133 was published based on data obtained from NOESY experiments, J-based configuration analysis (JBCA) and advanced Mosher's analysis.17 For the contiguous three stereogenic centers the absolute configurations were proposed as 10R, 11R and 12S, respectively, representing a syn,anti (threo,erythro) stereotriad (or stereotriad “B” according to the Hoffmann nomenclature).18 The first total synthesis of NFAT-133, however, led to the revised configuration of 10S, 11R and 12S, representing a syn,syn (threo,threo) stereotriad.19
During our ongoing screening program for antibacterial compounds, we have isolated and characterized NFAT-133 from the crude extract of Streptomyces sp. ST157608 from Sanofi's microbial strain collection. After bioassay and UPLC-HRMS guided fractionation, the antibacterial activity of the extract against E. coli was assigned to the presence of pactamycin. After re-fermentation of the strain, the recently disclosed16 TM-123 (13) as well as 7 new polyketide natural products 17–23 were isolated from larger batches of the extract (Chart 2).
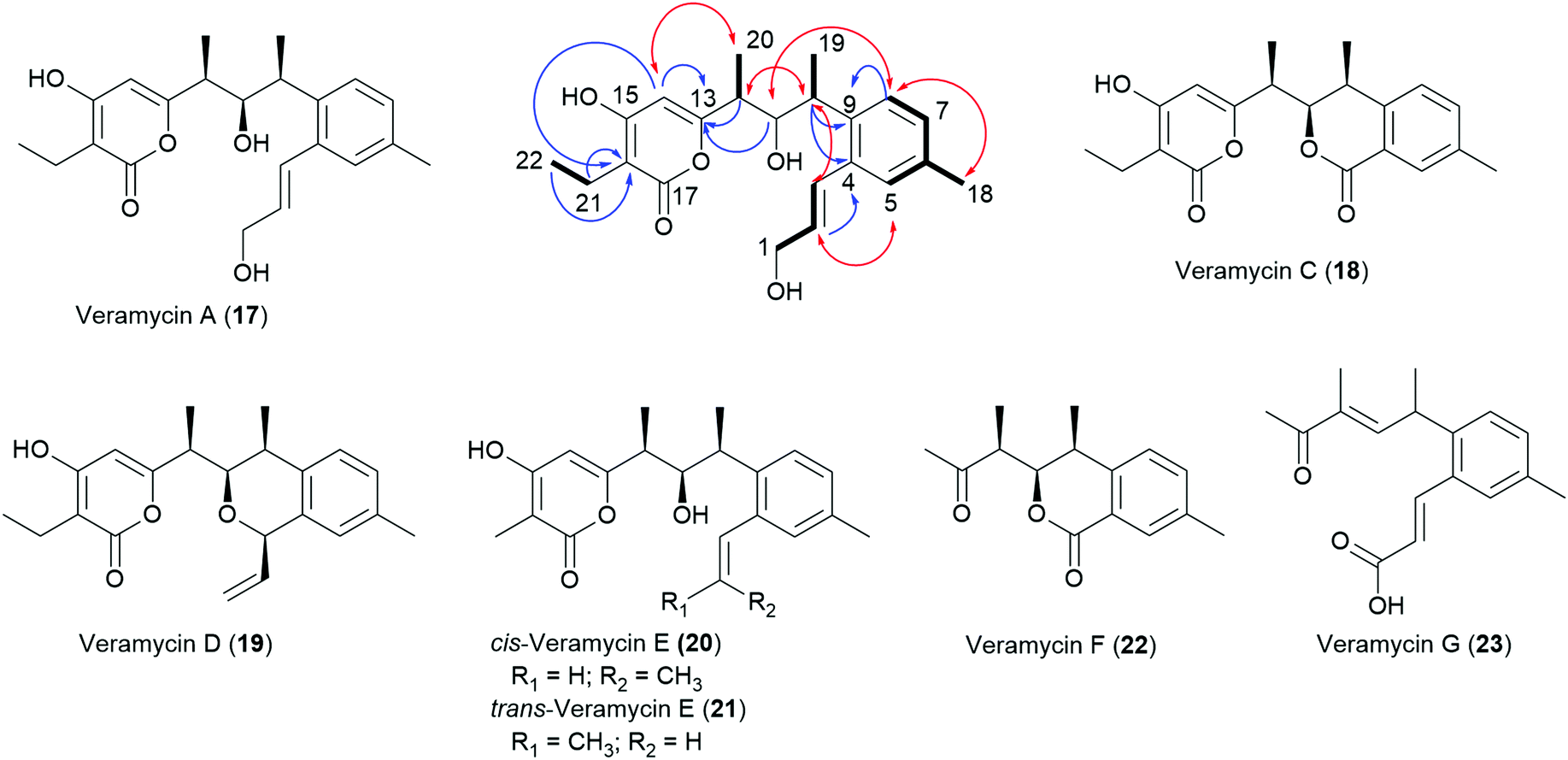 | ||
| Chart 2 New polyketides isolated from Streptomyces sp. ST157608 and key HMBC (blue arrows), COSY (bold lines), and ROE (red double headed arrows) correlations of compound 17. | ||
A closely related congener termed veramycin A (17) with an α-pyrone motif particularly gained our attraction, since it showed a comparable effect to NFAT-133 on glucose uptake in L6 rat skeletal myotubes. Further investigations culminated in total syntheses of NFAT-133, TM-123 and veramycin A, detailed biosynthetic studies and the identification of further congeners by precursor-directed biosynthesis.
Results and discussion
Isolation and structure elucidation
Compounds 1, 13 and 17–23 were isolated from culture broths of large-scale fermentations of Streptomyces sp. ST157608 by SPE with Amberlite™ XAD followed by semi-preparative RP-HPLC.Veramycin A (17) was obtained as a white powder and its molecular formula was determined as C22H28O5 based on HR-ESI-MS, indicating nine degrees of unsaturation. The examination of the 1H and 13C NMR data together with the HSQC spectrum showed resonances ascribable to four methyl groups (δH 2.26 s, δC 21.1; δH 1.27 d, δC 18.5; δH 1.12 d, δC 13.0; δH 1.02 t, δC 12.8), one methylene (δH 2.36 q, δC 17.3), and four aromatic methines (δH 7.02, δC 129.6; δH 7.13, δC 128.3; δH 7.20, δC 128.5; δH 5.88, δC 101.3). Besides, the 13C NMR spectrum showed resonances ascribable to one carbonyl at δC 168.6, and twelve olefinic carbons at δC 101.2–167.2. Moreover, the 1D and 2D NMR data revealed the presence of a 1,2,4-trisubstituted benzene ring with H-5 (δH 7.20), H-7 (δH 7.02) and H-8 (δH 7.13) together with a hydroxylated allylic residue at H-1 (δH 4.23, δC 63.8), H-2 (δH 6.13, δC 132.8) and H-3 (δH 6.86, δC 129.5) connected to position 2 of the trisubstituted benzene ring. The planar structure of 17 was determined from 2D NMR experiments and the connectivity of the residues was confirmed by key COSY and long-range HMBC correlations. In comparison to TM-123 (13), an additional methyl group is present in 17, which explains the 14.0156 amu difference. Furthermore, COSY correlations of 17 revealed a spin system pointing to an ethyl moiety. The comparison of 3JH–H coupling constants and chemical shifts of 13 and 17 revealed an identical relative stereochemistry – and hence the configurations of the stereogenic centers were assigned as 10S, 11R and 12S for 17 as well.
TM-123 (13), originally termed veramycin B during our isolation campaign, was the most abundant compound in the extract and thereby assumed to represent the final product of the biosynthesis. It was obtained as a white powder. Its molecular formula was determined as C21H26O5 based on HR-ESI-MS and its 1H and 13C NMR data showed complete identity with those reported for TM-123.16
Veramycin C (18), obtained as a white powder was determined to have the molecular formula C20H22O5 based on HR-ESI-MS, indicating ten degrees of unsaturation. A closer examination of the NMR data revealed the absence of a hydroxylated allylic residue at the aromatic ring in contrast to 13 and 17. Instead, the presence of a second lactone moiety at δC 167.5 ppm for C-3 was assigned. The two consecutive stereogenic centers of 18 were determined to be 10S, 11R, 12S as key ROE correlations between H-10/H-11, H-11/H-14, and H-12/H-19 have been observed. Furthermore, a vicinal coupling constant of 3J = 10.4 Hz between H-11 and H-12 was observed, corroborating a cis configuration. These findings were in line with the configurations observed for the structurally related isochromans panowamycin A and B.14
Veramycin D (19) was isolated as a white powder. Its molecular formula was determined as C22H26O4 by HR-ESI-MS. The 2D NMR data of 19 revealed the absence of a lactone moiety at position C-3 but the presence of a cyclic ether. Furthermore, a vinyl moiety was observed. The relative stereochemistry of 19 was also characterized by the observation of the key ROE correlations between H-10/H-11, H-11/H-14, and 10-CH3/H-12 as well as in the case of 18 and assigned as 10S,11R,12S.
cis-Veramycin E (20), a white powder with a molecular formula of C21H26O4 determined by HR-ESI-MS displayed a structure very close to 13 based on the NMR data obtained for this compound, suggesting the lack of a terminal oxidation (at C-1) of the allylic side chain. The 3JH–H coupling constant of 11.2 Hz between H-2 and H-3 is pointing to the cis configuration of the allylic residue differing from the one observed in 1, 13 and 17.
After large-scale fermentations, 20 was isolated as a 7:4 mixture with its trans isomer 21. trans-Veramycin E (21) displays a 3JH–H coupling constant of 15.4 Hz between H-2 and H-3 in line with those observed for 1, 13 and 17. Additionally, two new compounds related to the Panowamycins14 and NFAT-133 (1) were isolated and characterized. The new panowamycin derivative, a white powder, was termed veramycin F (22). Its molecular formula was determined as C15H18O3 based on HR-ESI-MS, indicating seven degrees of unsaturation. A closer examination of the NMR data revealed the absence of a hydroxylated allylic residue but the presence of an isochroman-1-one moiety (lactone δC 167.0 ppm at C-3) as in veramycin C (18).
The molecular formula of the NFAT-133 congener, which was termed veramycin G (23), was determined as C17H20O3 based on HR-ESI-MS, indicating eight degrees of unsaturation. The examination of the 1D and 2D NMR data revealed the presence of a carboxylic acid (δC 170.1) at position C-1 instead of the hydroxylated allylic residue. Furthermore, the secondary alcohol at position C-11 was missing as the 13C NMR spectrum showed resonances ascribable to an additional olefinic carbon (δH 6.77, δC 149.4.).
Biological profiling
Cellular glucose uptake measurements were performed in a modified L6 rat skeletal muscle cell line (L6 GLUT4 AS160-like cells).20 This modified cell line is characterized by an improved insulin sensitivity compared to the L6 cell line initially employed by Kulkarni-Almeida et al. in their studies on NFAT-133 (5 nM vs. 200 nM).12 After differentiation into myotubes, the L6 GLUT4 AS160-like cells were pre-incubated for 24 hours with either control solvent dilution or NPs, followed by a 30 min incubation with insulin dilutions and finally 20 min with 14C 2-deoxy-glucose (Fig. 1 and Table 1). In this modified L6 cell line, insulin incubation resulted in a concentration dependent deoxy-glucose uptake with an EC50 of approximately 9.85 × 10−9 mol L−1. Pre-incubation with TM-123 (veramycin B) had no effect on insulin-dependent deoxy-glucose uptake. In contrast, veramycin A and NFAT-133 resulted in a concentration-dependent enhancement of deoxy-glucose-uptake (curve up-shift). Baseline deoxy-glucose uptake in the absence of insulin was already increased by both NPs. Also both veramycin A and NFAT-133 slightly increased the sensitivity to insulin as depicted by the EC50 values in Table 1. | ||
| Fig. 1 Effect of 24 min incubation of veramycin A, TM-123 (veramycin B), and NFAT-13 on insulin-inducted uptake of 14C-2-deoxyglucose (14C-DOG) in L6 GLUT4 AS160 myotubes. | ||
| Curve | EC50 [M] | EC50 95% CI [M] | Hill slope | Hill 95% CI | Bottom | Bottom 95% CI | Top | Top 95% CI |
|---|---|---|---|---|---|---|---|---|
| Control 1 | 8.34 × 10−9 | 6.55–10.53 × 10−9 | 0.89 | 0.73–1.09 | 622 | 518–706 | 2289 | 226–2360 |
| Veramycin A | 8.82 × 10−9 | 5.51–8.43 × 10−9 | 1.22 | 0.96–1.54 | 1296 | 1175–1401 | 3381 | 3303–3465 |
| Control 2 | 1.08 × 10−8 | 8.06 × 10−9–1.41 × 10−8 | 0.91 | 0.70–1.17 | 656 | 537–748 | 2228 | 2154–2318 |
| TM-123 (veramycin B) | 9.62 × 10−9 | 7.66 × 10−9–1.20 × 10−8 | 0.82 | 0.70–0.99 | 651 | 545–736 | 2435 | 2370–2511 |
| Control 3 | 1.04 × 10−8 | 8.26 × 10−9–1.31 × 10−8 | 1.08 | 0.85–1.38 | 621 | 530–699 | 2172 | 2106–2246 |
| NFAT-133 | 5.42 × 10−9 | 4.06 × 10−9–7.08 × 10−9 | 1.01 | 0.77–1.31 | 1343 | 1193–1457 | 3099 | 3025–3182 |
Total synthesis
In order to provide sufficient amounts of material of NFAT-133 and the veramycins for in-depth biological profiling, we set out for a total synthesis of this compound class that would in addition provide an access to non-natural analogs and derivatives for further mechanistic and structure–activity relationship (SAR) studies.Based on the presence of NFAT-133 (1) in the same strain, a common biosynthetic pathway with TM-123 and the veramycins was assumed. Therefore, it seemed obvious that the absolute configurations of the centers in the stereotriad of all polyketide metabolites of ST157608 should be the same. However, when the present studies on the structure elucidation of compounds 13 and 17–23 were initiated in our laboratories, only the first, tentative proposal of 10R, 11R and 12S (syn,anti or threo,erythro) configuration for the three consecutive stereogenic centers of NFAT-133 was published (Scheme 1, epi-1).17 The synthesis and stereochemical revision of 1 was disclosed while our own studies on the total synthesis of NFAT-133 and the Veraymcins were already in progress.19
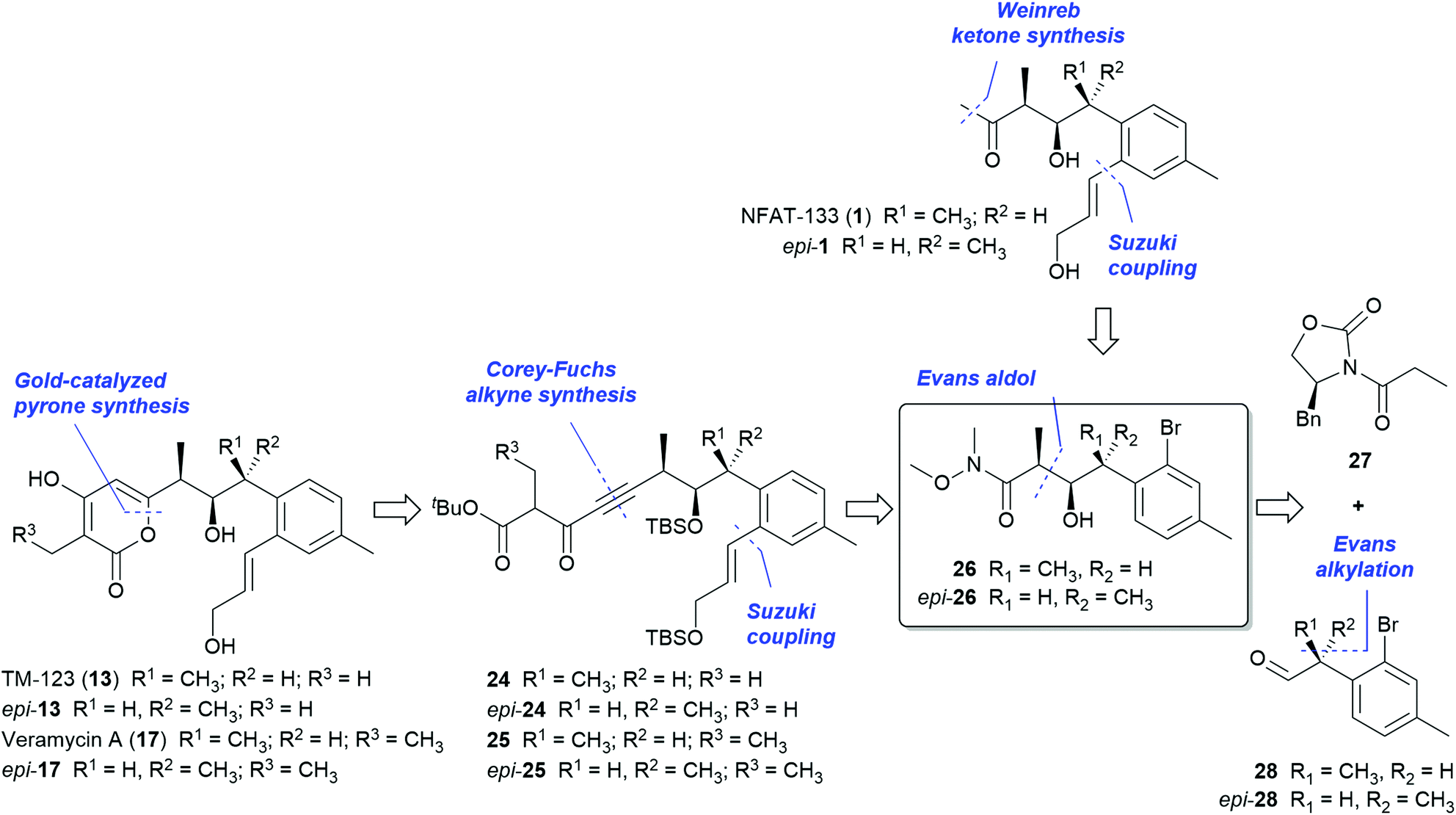 | ||
| Scheme 1 Retrosynthetic analysis. | ||
In line with the initial proposal for 117 relying upon on JBCA for 1,2-hydroxymethyl dimethine systems21,22 and own initial analyses of 3JH–H and 2,3JC–H coupling constants between H-10/H11, H-11/C-10 and H-10/C-12, the configurations of the three stereogenic centers of 13 and 17 were thus at first tentatively assigned as 10R, 11R and 12S, too (Scheme 1, epi-13 and epi-17). We realized, however, that the configurational analyses based on standard JBCA parameters led to controversial results with regard to the open-chain congeners 1, 13, 17, 20 and 21 on the one hand and the cyclic derivatives 18, 19 and 22 on the other hand.
The conformationally rigid isochroman rings render the determination of the relative configurations of these cyclized systems based on ROE and 3JH–H data very predictive. We therefore reasoned that the apparent discrepancy compared to the open-chain congeners may be attributed to an incorrect configurational assignment resulting from the limitations of JBCA. In contrast to more common polyketides, there are exceptional conformational preferences and substituent effects of the compounds 1, 13, 17, 20 and 21 that make the accurate prediction of the configuration of these particular vicinal alkyl aryl systems by experimental NMR parameters alone very difficult.‡
In the light of the limited predictivity of the JBCA results, we devised a flexible, convergent approach that would also enable the controlled synthesis of all isomers of the central stereotriad. We focused first on NFAT-133 (1), TM-123 (13) and veramycin A (17) as target molecules for the synthetic approach but taking into account that the strategy employed herein would also be applicable to the synthesis of their congeners 20, 21 and 23 as well as the isochroman derivatives 18, 19 and 22. Key to our approach is the central building block 26, which bears the three contiguous stereogenic centers (Scheme 1).
Both syn,syn and syn,anti configurations of the stereotriad (26 and epi-26) should be accessible by an Evans aldol reaction of acyl oxazolidinone 27 with either the R- or the S-configured α-methyl phenylacetaldehyde 28 (or epi-28, respectively) by an Evans alkylation route. The o-bromo function of the aryl moiety of 26 represents a handle for the installation of the allylic side chains by C–C cross coupling reactions under mild conditions while the N-methoxy amide should be both amenable to the ketone function of 1 and, via a sequence of reduction to the corresponding aldehyde, Corey–Fuchs alkine synthesis and acylation to the ynedione intermediates 24 and 25, required for the envisaged gold-catalyzed cyclization23–25 to the pyrone systems of 13 and 17, respectively.
For the synthesis of epi-1, the presumed structure of NFAT-133 based on the proposed17 absolute configuration at the time when our efforts started, (R)-aldehyde epi-28 was employed. Its synthesis started from commercially available acid 29, which was reacted with (R)-4-benzyloxazolidin-2-one via the acid chloride to provide 30 in good yields. Alkylation of 30 with MeI and NaHMDS furnished a mixture of diastereomers in favor of 31 (de = 9:1) which were separated by flash chromatography. The auxiliary was cleaved off with excellent recovery under reductive conditions with NaBH4 to furnish alcohol 32, which was then oxidized to aldehyde epi-28 with Dess–Martin periodinane (Scheme 2). Aldehyde epi-28 was reacted with the (Z)-boron enolate generated from oxazolidinone 27 under standard Evans aldol conditions: enolate formation with n-Bu2BOTf and DIPEA as the base at 0 °C followed by reaction with the aldehyde at −78 °C to 0 °C, yielding the 10R,11R,12S configured Evans-syn product 33 as the major diastereomer as expected for this matching case of double stereodifferentiation (intrinsically favored diastereofacial anti selectivity of the α-chiral aldehyde epi-28).26
 | ||
| Scheme 2 Synthesis of epi-1. | ||
Other diastereomers were only observed in traces in the 1H NMR spectrum of the crude product. Pure 33 was obtained in 78% yield as a crystalline solid after flash chromatography. Cleavage of the auxiliary proceeded smoothly with MeONHMe·HCl and Me3Al to furnish the central Weinreb amide intermediate epi-26 in 88% yield with excellent auxiliary recovery (97%). The absolute configurations of 33 and epi-26 were unambiguously confirmed by single-crystal X-ray analysis (see ESI†). The allylic side chain was introduced by reacting epi-26 and boronic ester 34 under mild Suzuki conditions using 6 mol% XPhos Pd G4 pre-catalyst. The resulting product 35 was obtained in 71% yield. Reacting 35 with MeMgBr followed by desilylation of the resulting ketone 36 furnished epi-1 as a viscous oil.
However, the optical rotation of this material ([α]23589 = −15.4, c = 0.85, MeOH) did not match the reported value for NFAT-133 ([α]22D = +24, c = 0.45, MeOH). Furthermore, the 1H and 13C NMR spectra of epi-1 did not match with those reported for NFAT-133,11 in particular regarding the 13C shifts of the stereogenic centers and their adjacent methyl carbons. These findings were confirmed by the report of the total synthesis and structural revision of NFAT-133 published during the course of our work.19
The corresponding building block for the syn,syn stereotriad series with the absolute 10S,11R,12S configurations required (S)-aldehyde 28 for the subsequent key aldol addition. Aldehyde 28 was prepared essentially the same way as its (R) enantiomer epi-28. Applying the same conditions as for the synthesis of 33 (reaction of 28 with the boron enolate of 27 at −78 °C to 0 °C in the presence of DIPEA), aldol product 37 was only obtained in 53% yield and the non-Evans-anti diastereomer was isolated as major side product (31%). This is likely a consequence of stereochemical mismatch.‡ Under optimized conditions (performing the reaction at 0 °C in the presence of NEt3), 37 was obtained in 81% yield. The central Weinreb amide building block 26, resulting from oxazolidinone cleavage of 37, was reacted with freshly prepared27 pinacol boronate 34 under further improved Suzuki conditions (with 3 mol% of XPhos Pd G4 as catalyst) to give intermediate 38 in 96% yield. Conversion of 38 to the corresponding methyl ketone 39 followed by removal of the TBS group finally furnished NFAT-133 (1) in 9 steps and 25% overall total yield (from commercially available starting materials) with all spectroscopic data11,12,15,17 and optical rotation ([α]22D = +40, c = 0.45, MeOH; lit.17 [α]22D = +24, c = 0.45, MeOH) in accordance with those reported for the isolated NP.
For the synthesis of TM-123 (13) and veramycin A (17), the transformation of intermediate 38 to the corresponding cyclization precursors required protection of the secondary alcohol of 38. The resulting TBS protected Weinreb amide 40 was smoothly reduced to aldehyde 41 with DIBAL-H in THF at −78 °C. Compound 41 was further elaborated into the propiolic acid methyl ester 43 obtained directly by quenching the lithiated Corey–Fuchs alkine intermediate (generated from dibromoalkene 42) with methyl chloroformate.
Reaction of 43 with either tert-butyl ester 44 or TMSE ester 45 under directed Claisen conditions28 furnished the cyclization precursors 24 and 46 in 84% and 94% yield, respectively. The cyclization to the corresponding pyrones was performed under Fürstner conditions23,25 in MeNO2/AcOH (10:1) in the presence of 5 mol% of the gold(I) catalyst SPhosAuNTf2. Final deprotection delivered TM-123 (13) and veramycin A (17) in 50% and 69% yield over 2 steps, respectively; corresponding to total yields of 16% (over 15 steps) for veramycin A (17) and 12% (over 14 steps) for TM-123 (13) from commercially available starting materials (Scheme 3).
 | ||
| Scheme 3 Synthesis of NFAT-133 (1), TM-123 (13) and veramycin A (17). | ||
All spectroscopic data of synthetic 13 and 17 were in accordance with those reported or determined for the isolated natural products. Optical rotations were determined as [α]22D = +114 (c = 1.02, MeOH), lit.16 [α]25D = +101.6 (c = 0.23, MeOH) for 13 and [α]22D = +128 (c = 0.6; MeOH) for 17, respectively.
Biosynthesis
To obtain insights into the biosynthetic origin of the compounds isolated from Streptomyces sp. ST157608 (1, 13, 17–23, cf. fig. 1), feeding studies were performed. Experiments with deuterium-labeled [2,2,3,3,3-2H5]propionate, [3,3,4,4,4-2H5]butyrate, and L-[methyl-2H3]-methionine were performed and target compounds 1, 13 and 17 were analysed by HR-UPLC/MS. The incorporation of 2H-labeled propionate and butyrate was confirmed. However, 2H-labeled methionine was not incorporated. Next, the stable isotopes [1-13C] acetate, [2-13C] acetate, and [1-13C] propionate were supplemented to fermentations. This revealed 13 to be biosynthesized by assembly of five methylmalonyl- and three malonyl-CoA units (Fig. 2). The observed 13C labeling of C-14 indicates this carbon to be acetate-derived, whereby in 1 a decarboxylation of this end-standing building block took place.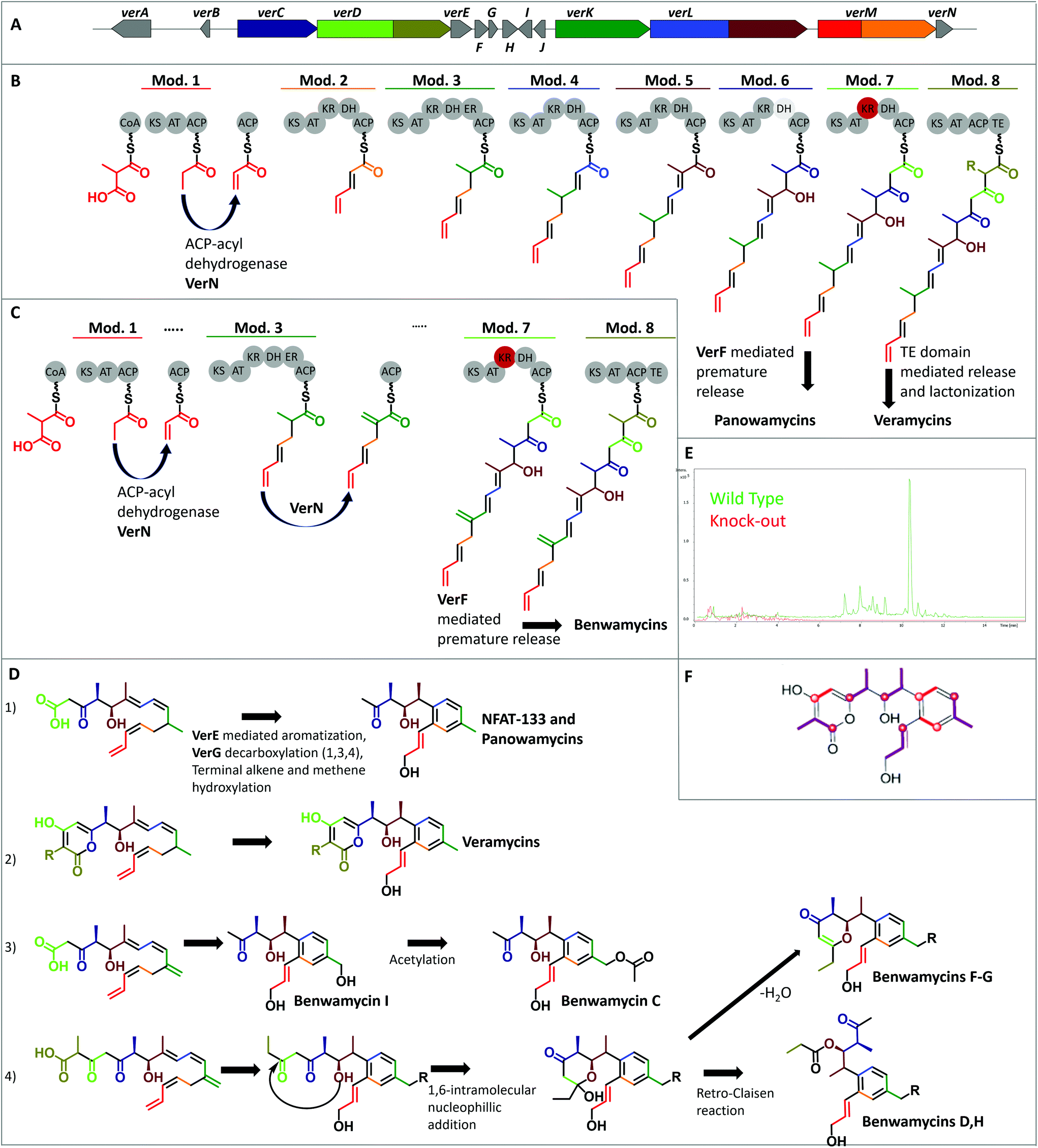 | ||
| Fig. 2 Biosynthetic gene cluster and putative biosynthesis. (A) Biosynthetic genes. The genes are colored according to the building blocks incorporated. (B) Mod. = Modules and corresponding domains including the nascent molecule chain. ACP: acyl carrier protein, AT: acyl transferase, CoA: Coenzyme A, DH: dehydratase, ER: enoylreductase, KR: ketoreductase, TE: thioesterase, grey shaded DH6: can be skipped, red colored KR7: inactive. (C) VerN-catalyzed double bond formation and VerF-catalyzed chain release that forms the Benwamycin precursor. (D) Biosynthetic steps towards the final molecules. (E) Extracted Ion Chromatogram (EIC) of molecular formula C22H28O5 (17), considering [M + H]+, [M–H2O + H]+ and [M + Na]+ adducts of wild type and verD deletion mutant extracts. (F) Identification of 13C-labelled carbon atoms from stable isotope feeding experiment for 13. Overlay of [1-13C] acetate (red dots) and [1-13C] propionate (violet dots). | ||
The same labeling pattern of the polyketides that possess structural similarity with subtle but intriguing differences indicate a common biosynthetic pathway with an array of chemical reactions generating this compound family. To determine the corresponding BGC, strain Streptomyces sp. ST157608 was genome sequenced. In the assembled genome, we in silico identified a putative corresponding BGC (Tables S13 and S14†), which was subsequently verified by a gene knockout experiment. It was experimentally linked to the production of the veramycins by disruption of the PKS gene verD, encoding for module seven and eight. Introduction of a resistance cassette led to production abolition of all derivatives described so far, observed by LC/MS analysis (Fig. 2 and Fig. S2–16†). During preparation of this manuscript, the group of Taifo Mahmud independently published the sequencing results as well as gene inactivation and genetic complementation experiments of the NFAT-133 BGC of Streptomyces pactum ATCC 27456. Here, we propose a biosynthetic pathway of these polyketides, considering all the available results. Thereby, the biosynthesis of veramycins, panowamycins and benwamycins can be unified (Fig. 2). Based on that, the veramycins can be recognised as the final full size molecules of the biosynthesis. Therefore, the genes are named verA-verN, even though the nomenclature of the various known molecules of this family is manifold.
The BGC was identified as a type I PKS BGC, consisting of five genes coding for eight PKS modules and five genes encoding other biosynthetic functions. Peculiarly, out of these five additional genes only one can be considered a tailoring enzyme, while the other four directly participate in the biosynthesis of the core structures of vera-, panowa- and benwamycins, respectively. The BGC does not follow the co-linearity rule, i.e. the order of the genes encoding individual modules does not represent the order of the building blocks observed in the final molecules. Compared to the previously reported S. pactum BGC, the BGC from Streptomyces sp. ST157608 has the same organisation and an overall high identity on nucleotide level (>95%). However the genes nftO and nftP, situated downstream of nftN (homologue of verN), are not present in the genome of strain ST157608.
The starter module 1 consists of a ketosynthase (KS), an acyl transferase (AT) and an acyl carrier protein (ACP) domain, respectively. The starter unit loaded is methylmalonyl-CoA that is decarboxylated by KS1 and subsequently modified by the dedicated acyl-ACP dehydrogenase VerN, yielding the terminal alkene group (Fig. 2). In the previously proposed biosynthetic pathway, the same module was identified as starter module. However, the 13C labelling experiments are not in line with the proposed orientation of the polyketide chain in the final molecule. It was reasoned that lack of cysteine in the CHH catalytic triad leads to inactivity of this KS domain; hence, to incorporation of a terminal carboxy group that becomes part of the western lactone ring. For the ver BGC, we also observe lack of the catalytic triad (QHH instead of CHH). However, the active site Cys is presenting the growing PKS chain to the decarboxylated extender unit. Decarboxylation itself is mainly mediated by the two amino acids His307 and Asn345, as well as a Phe that acts as a gatekeeper.29
Since module 1 represents the starter, no PKS chain can be presented and therefore, no conserved Cys is necessary for decarboxylation activity. This is consistent with other reported polyketide biosynthetic pathways such as the starter generation in tetronomycin biosynthesis, where acetate can be found in the final antibiotic molecule, while the AT-domain is specific for malonyl-CoA.30 In accordance with that, the respective tetronomycin KS1 also carries the QHH instead of CHH motif. Hence, we propose an unusual but consistent starter generation to yield finally hydroxylated PKS products in majority; whereby, few show the terminal alkene moiety. Module 2, consisting of a KS, an AT, a ketoreductase (KR), a dehydratase (DH) and an ACP domain, extends this unusual starter by an acetate unit and reduces the starter carbonyl group to a double bond. Module 3 (KS-AT-KR-DH-ER-ACP) incorporates methylmalonyl-CoA into the growing carbon chain and reduces the carbonyl group of the second building block to full saturation. Module 4, 5 and 6 (all KS-AT-KR-DH-ACP) elongate the molecule by an acetate (malonyl-CoA-derived) and two propionate (methylmalonyl-CoA-derived) units, respectively. The thereby introduced carbonyl groups are reduced to double bonds by modules 4 and 5. However, the reduction state catalysed by VerC (module 6) seems to be variable to a certain degree: nearly all derivatives show a reduction to a hydroxyl group at C-11, while only 23 exhibits an enol moiety. The latter case would be expected based on the in silico prediction, since a DH domain is present in module 6. Amino acid sequence analysis of this DH6 domain indicated it to be intact; however, dehydration can only be observed in one out of ten compounds isolated. Module 7 (KS-AT-KR-DH-ACP) of VerD elongates the polyketide chain by a further acetate unit and despite the presence of KR and DH domains, no reduction takes place. Here, sequence analysis predicted the KR7 domain to be inactive; thereby, blocking subsequent dehydration. The final module 8 (KS-AT-ACP; also part of VerD) incorporates the last building unit into the nascent polyketide chain. This module seems to show a certain degree of promiscuity towards the CoA-extender incorporated, resulting in either a C-21, or C-22 chain. It also carries the thioesterase (TE) domain, responsible for chain release and keto–enol tautomerism-dependent lactonization of the western part of the veramycins. It is proposed that the eastern aromatic ring is formed by a VerE-dependent mechanism. This was experimentally proven by verE knockout experiments that resulted in molecules in which only the western lactone ring was present and the eastern one absent.16 VerE was annotated as a cytochrome P450-dependent aromatase and shows similarity to Orf1–257, which is proposed to catalyse the ring closure in lorneic acid biosynthesis by a C−C coupling within the polyketide chain via epoxid formation. For veramycins, the ring is closed between C-4 and C-9 with subsequent aromatisation. In addition, the conjugated C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 and C3C4 double bond system is dissipated, yielding a C2C3 double bond. Hydroxylation of the terminal C atom seems to be favoured once the ring is closed, even though hydroxylated variants of the linear chain were observed.16 Veramycin D can be generated by non-enzymatic intramolecular ether formation. In this case, isomerization of the terminal double bond and hydroxylation are not induced. Veramycin E is another non-hydroxylated variant. If the intramolecular ring closure is a concerted action with hydoxylation, has to be determined in future studies.
C2 and C3C4 double bond system is dissipated, yielding a C2C3 double bond. Hydroxylation of the terminal C atom seems to be favoured once the ring is closed, even though hydroxylated variants of the linear chain were observed.16 Veramycin D can be generated by non-enzymatic intramolecular ether formation. In this case, isomerization of the terminal double bond and hydroxylation are not induced. Veramycin E is another non-hydroxylated variant. If the intramolecular ring closure is a concerted action with hydoxylation, has to be determined in future studies.
The biosynthesis of 1–12 and 22, which possess a smaller polyketide chain, is essentially identical to the veramycins. However, the premature polyketide is cleaved off from the assembly line from ACP7 by the MBL fold metallo-hydrolase VerF. The released premature polyketide is subsequently decarboxylated by the putative decarboxylase VerG to yield NFAT-133 (1), or other compounds without the western pyrone ring. For the latter, dependent on the reduction state of C-13, either 2 and 22, or 3 are the final compounds. The hydroxylation of the benwamycins at position C-18 (as in 5, 6, 8, 9, 12) is likely analogous to the hydroxylation of the terminal alkene group in the veramycins. The acyl-ACP dehydrogenase VerN introduces a C6C18 double bond, which gets subsequently hydroxylated, possibly during formation of the aromatic system. This hydroxy group is then accessible for further augmentation by acylation of the molecule as observed in 6. Benwamycins F and G (11, 12) represent molecules with a different heterogenic western ring system. Formation of these molecules starts by release of the full length nascent polyketide chain as in veramycin B. However, keto–enol tautomerism-dependent lactonization does not take place, yielding a terminal carboxy group. Then, this carboxy function is cleaved off, similar to the generation of NFAT133, and the C-10 hydroxy group enables the energetically favoured 1,6-intramolecular nucleophilic addition to the remaining keto function at position C-15. The intermediate hemiacetal product can then either undergo water elimination generating the 4-pyrone unit present in the structures of benwamycins F-G (11 and 12), or undergo retro-Claisen reaction, which transfers the C-15–C-17 propionyl unit onto the C-10 hydroxy unit, generating the benwamycins D and H (7 and 8). Such a retro-Claisen reaction can happen in only slightly acidic conditions. This is a hint that some of the compounds observed are not biosynthesized by the producer organism, rather they are non-enzymatically formed during purification. Anyway, this reaction pathway explains therefore the precise and selective propionylation at this position. The formation of the aromatic system, and if present of the 4-pyrone ring, is analogous to the veramycins. In the biosynthesis of 18 and 22, the starter module is skipped and therefore malonyl-CoA represents the starter unit. The resulting terminal carboxyl group subsequently undergoes lactonization. Benwamycin E (10), carrying a terminal carboxy group and thus blurring the sight on the starter generation can be obtained by incorporation of malonyl-CoA as starter unit. Due to the different sizes of malonyl-CoA and the canonical starter methylmalonyl-CoA, decarboxylation and subsequent dehydration might be blocked, resulting in the observed terminal carboxyl group. Since only one compound with this terminal moiety was observed until now, it can be assumed that the starter AT possesses detectible, but limited promiscuity.
Precursor-directed biosynthesis
Due to the postulated low specificity of module 8, it seemed feasible to produce non-natural veramycins by precursor-directed biosynthesis amenable to further derivatizations, e.g. by click-chemistry. Allyl-malonic acid, propargyl-, (3-chloropropyl)- and isobutyl-malonic acid were utilized as building blocks after saponification of their commercially available diesters (see ESI† for further information). After fermentation, feeding and extraction the samples were analyzed via HPLC-HR/MS for identification of a successful incorporation of the used malonic acid derivatives. Corresponding signals of allyl- (49) and propargyl-veramycin (50) were present (Chart 3), whereas neither incorporation of (3-chloropropyl)-nor isobutyl-malonic acid could be detected. The successful incorporation of allyl- and propargyl-malonic acid prove the low substrate-specificity of the last biosynthetic module. | ||
| Chart 3 Allyl- and propargyl derivatives from precursor-directed biosynthesis. | ||
After refermentation, 2.4 mg of 49 were isolated as a colorless powder with a molecular formula of C23H28O5, indicating ten degrees of unsaturation. Indeed, the analysis of the 1D and 2D NMR data revealed the presence of an additional second allylic chain located at the lactone system, (Table S10†).
Conclusions
7 new polyketides, termed veramycins, were isolated from a Streptomyces sp. from the Sanofi microbial strain collection. These polyketides are structurally closely related to NFAT-133, TM-123, the panowamycins and the benwamycins. In particular, veramycin A, a closely related α-pyrone congener of TM-123 may represent a novel lead structure for the development of antidiabetic indirect AMPK activators, since this compound and NFAT-133 showed an increased baseline deoxy-glucose uptake in the absence of insulin in a modified L6 rat skeletal muscle cell line and furthermore increased slightly the sensitivity to insulin in this cell line.We established a robust and scalable total synthesis of NFAT-133, TM-123 and veramycin A, which ensures compound supply for further lead profiling and mechanistic investigations. Our approach represents a flexible route towards other naturally occurring related polyketides as well as synthetic congeners for extended structure–activity relationship investigations in the context of hit and lead exploration programs in this series of compounds with a rich activity profile comprising antidiabetic, immunosuppressive and antitrypanosomal activities. Investigation of the corresponding BGC revealed that the biosynthesis of plenty compounds is encoded by this gene locus. Building on that, precursor-directed biosynthesis experiments culminated in the identification of new veramycin congeners. One of this congeners bears a terminal alkine motif, suitable for further exploration in 1,3-dipolar cycloaddition reactions (“click chemistry”).
Conflicts of interest
C. Pöverlein, M. Kurz, P. Hammann, and A. Bauer are or have been employed by Sanofi S.A. or one of its affiliates and are shareholders of Sanofi S.A.Author contributions
Conceptualization: FZ, SWG, SMMS, AB, PW, AG, NT, AV, TFS, AB; Funding acquisition: AV, PEH; Investigation: DD, NB, JP, SS, LP, JB, MAP, MKB, MK, LT, SWG, AB, OS, PW, AB; Resources: AV, PEH; Supervision: AP, FZ, AG, NT, AV, PEH, TFS, AB; Writing – original draft: DD, NB, JP, SS, PW, TFS, AB; Writing – review & editing: TFS, AB.Acknowledgements
This study was financially supported by the Hessen State Ministry of Higher Education, Research, and the Arts (HMWK) via generous grant for the LOEWE Research Center Insect Biotechnology and Bioresources. Sanofi contributed in the framework of Sanofi Fraunhofer Natural Product Center of Excellence. We thank the NMR department of the Justus-Liebig-University Giessen and Chiara Presenti, Ute Messinger and Christoph Hartwig for technical assistance.Notes and references
- S. Olivier, M. Foretz and B. Viollet, Promise and challenges for direct small molecule AMPK activators, Biochem. Pharmacol., 2018, 53, 147–158 CrossRef PubMed.
- K. Burkewitz and Y. Zhang, AMPK at the Nexus of Energetics and Aging, Cell Metab., 2014, 20, 10–25 CrossRef CAS PubMed.
- S. Umezawa, T. Higurashi and A. Nakajima, AMPK: Therapeutic Target for Diabetes and Cancer Prevention, Curr. Pharm. Des., 2017, 23, 3629–3644 CrossRef CAS PubMed.
- D. G. Hardie, Molecular Pathways: Is AMPK a Friend or a Foe in Cancer?, Clin. Cancer Res., 2015, 21, 3836–3840 CrossRef CAS PubMed.
- M. Webb, D. P. Sideris and M. Biddle, Modulation of mitochondrial dysfunction for treatment of disease, Bioorg. Med. Chem. Lett., 2019, 29, 1270–1277 CrossRef CAS PubMed.
- M. Friedrichsen, B. Mortensen, C. Pehmøller, J. B. Birk and J. F. P. Wojtaszewski, Exercise-induced AMPK activity in skeletal muscle: role in glucose uptake and insulin sensitivity, Mol. Cell. Endocrinol., 2013, 366, 204–214 CrossRef CAS PubMed.
- R. Kjøbsted, J. R. Hingst, J. Fentz, M. Foretz, M.-N. Sanz, C. Pehmøller, M. Shum, A. Marette, R. Mounier, J. Treebak, F. P. Wojtaszewski, B. Viollet and L. Lantier, AMPK in skeletal muscle function and metabolism, FASEB J., 2018, 32, 1741–1777 CrossRef PubMed.
- A. L. De Souza Almeida Matos, J. S. Oakhill, J. Moreira, K. Loh, S. Galic and J. W. Scott, Allosteric regulation of AMP-activated protein kinase by adenylate nucleotides and small-molecule drugs, Biochem. Soc. Trans., 2019, 47, 733–741 CrossRef PubMed.
- D. Garcia and R. J. Shaw, AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance, Mol. Cell, 2017, 66, 789–800 CrossRef CAS PubMed.
- T. Joshi, A. K. Singh, P. Haratipour, A. N. Sah, A. K. Pandey, R. Naseri, V. Juyal and M. H. Farzaei, Targeting AMPK signaling pathway by natural products for treatment of diabetes mellitus and its complications, J. Cell Physiol., 2019, 234, 17212–17231 CrossRef CAS PubMed.
- N. S. Burres, U. Premachandran, S. Hoselton, D. Cwik, J. E. Hochlowski, Q. Ye, G. N. Sunga, J. P. Karwowski, M. Jackson, D. N. Whittern and J. B. McAlpine, Simple Aromatics Identified with a NFAT-lacZ Transcription Assay for the Detection of Immunosuppressants, J. Antibiot., 1995, 48, 380–386 CrossRef CAS PubMed.
- A. A. Kulkarni-Almeida, M. K. Brahma, P. Padmanabhan, P. D. Mishra, R. R. Parab, N. V. Gaikwad, C. S. Thakkar, P. Tokdar, P. V. Ranadive, A. S. Nair, A. A. Damre, U. A. Bahirat, N. J. Deshmukh, L. S. Doshi, A. V. Dixit, S. D. George, R. A. Vishwakarma, K. V. S. Nemmani and G. B. Fermentation, Isolation, Structure, and antidiabetic activity of NFAT-133 produced by Streptomyces strain PM0324667, AMB Express, 2011, 1, 42 CrossRef PubMed.
- C. S. Thakkar, A. S. Kate, D. C. Desai, A. R. Ghosh and A. A. Kulkarni-Almeida, NFAT-133 increases glucose uptake in L6 myotubes by activating AMPK pathway, Eur. J. Pharmacol., 2015, 769, 117–126 CrossRef CAS PubMed.
- J. Hashida, M. Niitsuma, M. Iwatsuki, M. Mori, A. Ishiyama, M. Namatame, A. Nishihara-Tsukashima, A. Matsumoto, I. Ara, Y. Takahashi, H. Yamada, K. Otoguro, K. Shiomi and S. Ōmura, Panowamycins A and B, new antitrypanosomal isochromans produced by Streptomyces sp. K07-0010, J. Antibiot., 2012, 65, 197–202 CrossRef CAS PubMed.
- F.-X. Yang, J.-P. Huang, Z. Liu, Z. Wang, J. Yang, J. Tang, Z. Yu, Y. Yan, G. Kai and S.-X. Huang, Benwamycins A–G, Trialkyl-Substituted Benzene Derivatives from a Soil-Derived Streptomyces, J. Nat. Prod., 2020, 83, 111–117 CrossRef CAS PubMed.
- W. Zhou, P. Posri, M. E. Abugrain, A. J. Weisberg, J. H. Chang and T. Mahmud, Biosynthesis of the Nuclear Factor of Activated T Cells Inhibitor NFAT-133 in Streptomyces pactum, ACS Chem. Biol., 2020, 15, 3217–3226 CrossRef CAS PubMed.
- Y. Yang, L. Yu, H. Komaki, N. Oku and Y. Igarashi, Absolute configuration of NFAT-133, an aromatic polyketide with immunosuppressive and antidiabetic activity from actinomycetes, J. Antibiot., 2016, 69, 69–71 CrossRef CAS PubMed.
- R. W. Hoffmann, Stereoselective Syntheses of Building Blocks with Three Consecutive Stereogenic Centers: Important Precursors of Polyketide Natural Products, Angew. Chem., Int. Ed. Engl., 1987, 26, 489–594 CrossRef.
- H. Sato, E. Kwon, Y. Taguchi, S. Yoshida, S. Kuwahara and Y. Ogura, Synthesis and Stereochemical Revision of the Aromatic Polyketide NFAT-133, J. Nat. Prod., 2019, 82, 1791–1796 CrossRef CAS PubMed.
- M. Korn, P. Wohlfart, T. Gossas, M. Kullman-Magnusson, B. Niederhaus, J. Dedio and N. Tennagels, Comparison of metabolic and mitogenic response in vitro of the rapid-acting insulin lispro product SAR342434, and US- and EU-approved Humalog®, Regul. Toxicol. Pharmacol., 2019, 109, 104497 CrossRef CAS PubMed.
- N. Matsumori, D. Kaneno, M. Murata, H. Nakamura and K. Tachibana, Stereochemical Determination of Acyclic Structures Based on Carbon−Proton Spin-Coupling Constants. A Method of Configuration Analysis for Natural Products, J. Org. Chem., 1999, 64, 866–876 CrossRef CAS PubMed.
- G. Bifulco, P. Dambruoso, L. Gomez-Paloma and R. Riccio, Determination of Relative Configuration in Organic Compounds by NMR Spectroscopy and Computational Methods, Chem. Rev., 2007, 107, 3744 CrossRef CAS PubMed.
- W. Chaładaj, M. Corbet and A. Fürstner, Total Synthesis of Neurymenolide A Based on a Gold-Catalyzed Synthesis of 4-Hydroxy-2-pyrones, Angew. Chem., Int. Ed., 2012, 51, 6929 CrossRef.
- J. S. Lee, J. Shin, H. J. Shin, H.-S. Lee, Y.-J. Lee, H.-S. Lee and H. Won, Total Synthesis and Configurational Validation of (+)-Violapyrone C, Eur. J. Org. Chem., 2014, 4472 CrossRef CAS.
- A. Fürstner, Gold Catalysis for Heterocyclic Chemistry: A Representative Case Study on Pyrone Natural Products, Angew. Chem., Int. Ed., 2018, 57, 4215 CrossRef PubMed.
- W. R. Roush, Concerning the diastereofacial selectivity of the aldol reactions of .alpha.-methyl chiral aldehydes and lithium and boron propionate enolates, Org. Chem., 1991, 56, 4151 CrossRef CAS.
- Y. D. Wang, G. Kimball, A. S. Prashad and Y. Wang, Zr-Mediated hydroboration: stereoselective synthesis of vinyl boronic esters, Tetrahedron Lett., 2005, 46, 8777 CrossRef CAS.
- Z. Thang, Y. Kitamura and A. G. Myers, An Efficient Directed Claisen Reaction Allows for Rapid Construction of 5,6-Disubstituted 1,3-Dioxin-4-ones, Synthesis, 2015, 47, 2709 CrossRef.
- M. P. Austin and J. P. Noel, The chalcone synthase superfamily of type III polyketide synthases, Nat. Prod. Rep., 2003, 20, 79 RSC.
- Y. Demydchuk, Y. Sun, H. Hong, J. Staunton, J. B. Spencer and P. F. Leadlay, Analysis of the tetronomycin gene cluster: insights into the biosynthesis of a polyether tetronate antibiotic, ChemBioChem, 2008, 9(7), 1136–1145 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2093273–2093275. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01652k |
| ‡ See ESI† for a detailed discussion. |
| This journal is © the Partner Organisations 2022 |