DOI:
10.1039/D1QO01580J
(Research Article)
Org. Chem. Front., 2022,
9, 163-172
A mechanistic study on the regioselective Ni-catalyzed methylation–alkenylation of alkyne with AlMe3 and allylic alcohol†
Received
21st October 2021
, Accepted 16th November 2021
First published on 17th November 2021
Abstract
The recently reported Ni-catalyzed methylation–allylation of alkynes with allylic alcohols and AlMe3 reagents delivers valuable tetrasubstituted alkene units in a highly regioselective fashion. Motivated by the experimental significance and the mechanistic ambiguity (e.g. the details of C–O bond activation, the function of the Lewis acid AlMe3, etc.), we conducted a detailed mechanistic study using density functional theory (DFT) calculations. The reaction was found to occur via the C–O bond activation of allylic alcohol, allylation of alkyne, transmetalation, reductive elimination and catalyst regeneration steps. AlMe3 could easily react with the allylic alcohol to form a cyclodialuminoxane species bearing two groups of Lewis acid–base interactions. In this regard, the C(allyl)–O bond is remarkably activated, resulting in a relatively facile C–O activation (compared to the typical Lewis-acid mediated reactions). In addition, the Al–Me interaction is a double-edged sword to the methylation step: the cyclodialuminoxane group functions as the methyl resource, while the full dissociation of the Al–Me interaction (via a cis-to-trans isomerization) is a requisite to fully release the methyl group. In this context, the “Me-relay” pathway involving the [Ni]–Me intermediate is found to be more plausible than all other mechanistic possibilities (such as the typical concerted Me-transfer mechanism in previous studies).
1. Introduction
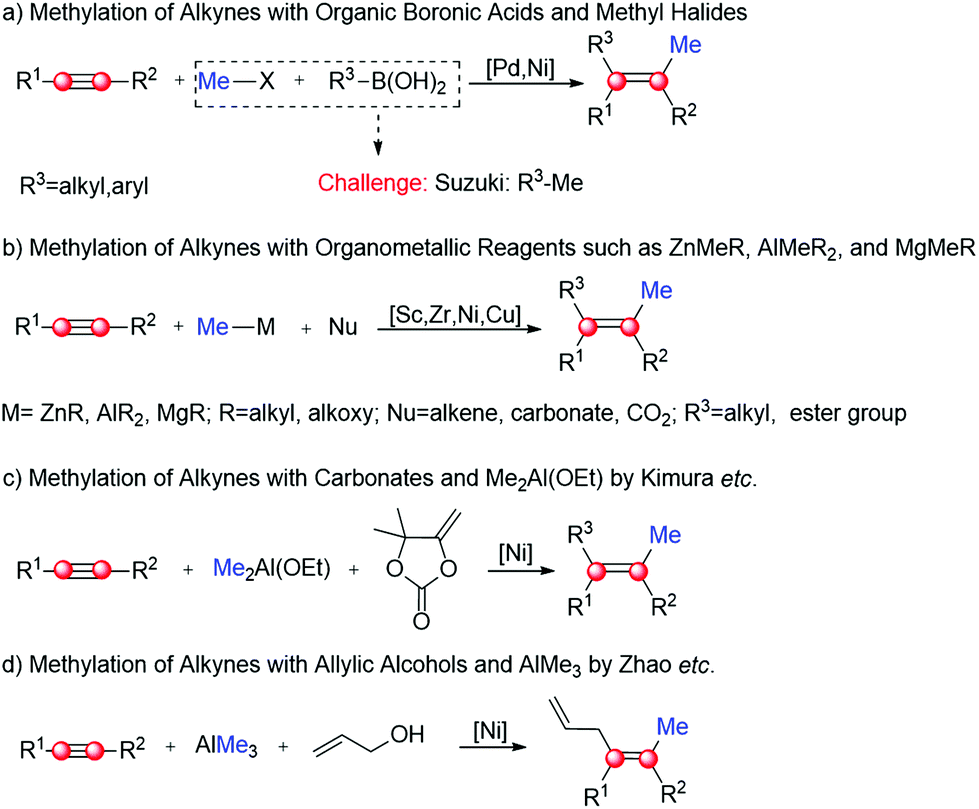
Dicarbofunctionalization of alkynes represents a practical strategy to prepare tetrasubstituted alkenes.1–7 In this context, the introduction of a Me group into the alkyne substrate (i.e. methylation) has been successfully utilized to synthesize a series of biologically active natural compounds, such as Nexium, Edurant and Ranexa.6,8–12 The typical methylation using organic boronic acids and methyl halides has advantages of functional group tolerance and high reaction rates (Scheme 1a),6,7,13–15 but is frequently limited by side reactions such as Suzuki coupling. This issue could be addressed by adding organometallic reagents into the reaction system (Scheme 1b).16–23 As one of the most attractive reagents, AlR3 has been successfully used to mediate a large number of methylation, with high regio- and stereo-selectivity in many cases.16,19,24–28 For example, in the presence of Me2Al(OEt) and carbonates, Kimura's group accomplished a series of Ni-catalyzed methylation–alkenylation of alkynes with mild to high regioselectivity (Scheme 1c).19 Using a similar strategy, Zhao's group recently developed a Ni-catalyzed methylation–allylation of alkynes using AlMe3 and allylic alcohol as substrates (Scheme 1d).29 In their study, the syn-methylation–allylation product was obtained exclusively. Of note, this is the first report on the highly regioselective methylation–alkenylation of alkynes directly with allylic alcohols.
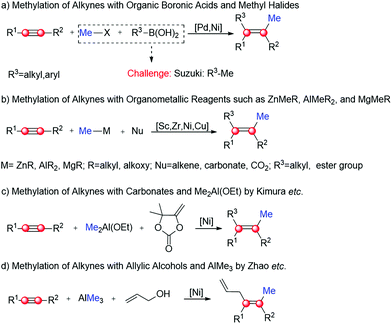 |
| | Scheme 1 Methylation of alkynes with (a) methyl halides, (b) organometallic reagents, (c) Me2Al(OEt) and (d) AlMe3 as the methylation reagent. | |
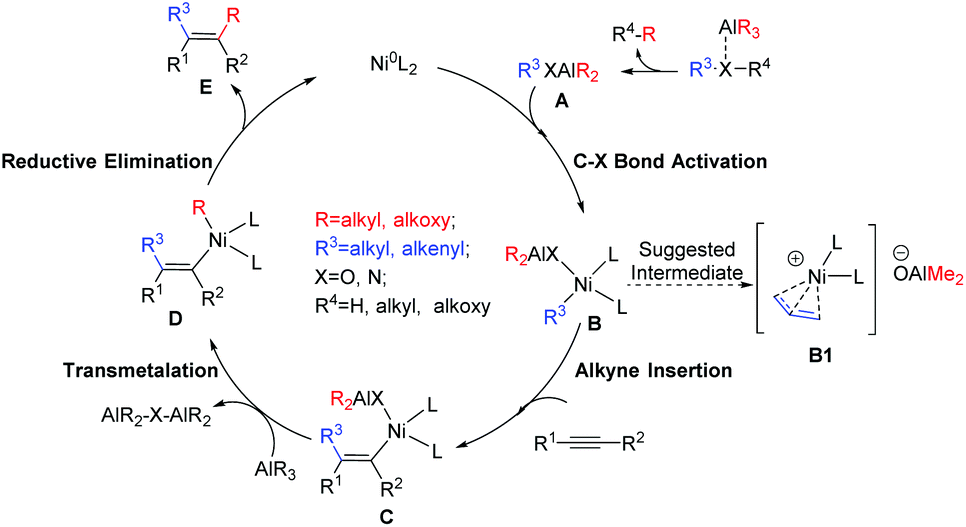
For the alkyl aluminum mediated dicarbofunctionalization of alkynes, it is generally accepted that AlR3 first reacts with the substrate to form the Al-incorporated intermediate A (Scheme 2). After that, C–X bond activation30–32 occurs to form the Ni(II) species B. Alkyne insertion into the newly formed Ni–C bond then generates the alkenyl-Ni(II) species C.7,10,33,34 Finally, the second functionalization of alkyne occurs via successive transmetalation–reductive elimination steps24–26,35–38 (C → D → E + NiL2), with simultaneous regeneration of the Ni(0) catalyst. In accordance with this mechanism, the reaction in Scheme 1d was suggested to proceed via a key allylic Ni(II) intermediate (B1), and the overall catalytic cycle occurs through an allylation–methylation mechanism.
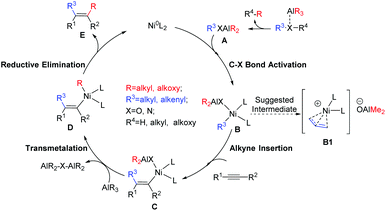 |
| | Scheme 2 The general mechanism of the alkyl aluminum mediated dicarbofunctionalization of alkynes, and the suggested key intermediate (B1) in the reaction system of Scheme 1d. | |
Under the premise of the proposed mechanism, the main mechanistic issue lies in the sequence of allylation and methylation steps, and the inherent reason for the reaction sequence. Meanwhile, according to recent studies, the reaction of AlMe3 with alcohol could possibly occur in different modes39–44 (e.g. replacing one or two Me groups with the alkyloxy group), and the structure responsible for the allylation/methylation step remains unclear. The uncertainty of the structure of the Lewis acid further results in the different C–O bond activity. Meanwhile, it remains unknown whether the inner-sphere or outer-sphere C–O activation is more favorable. To obtain answers to these questions, we carried out density functional theory (DFT) calculations on the reaction system in Scheme 1d. Comparing the relative facility of different mechanistic possibilities, the overall catalytic cycle was proposed. The cyclodialuminoxane species (formed by the reaction of AlMe3 with allylic alcohol with subsequent dimerization) and the “Me-relay” step were found to be pivotal for the overall transformation. This study provides an in-depth understanding of the methylation reaction of alkynes, and will hopefully benefit the expansion of more dicarbofunctionalization reaction systems.
2. Computational methods and the model reaction
2.1 Computational methods
All calculations were performed using Gaussian 16 software, Rev. C01. The DFT functional B3LYP,45,46 including the Grimme empirical dispersion correction (GD3BJ),45 was used for geometry optimization. The SDD47 basis set (including the pseudopotential) was employed on Ni, including an extra f polarization function with the exponent of 3.130.48 The 6-31G(d)49,50 basis set was employed on other elements (the combination of SDD and 6-31G(d) was designated as BS1). Frequency analysis was performed using the same B3LYP-D3BJ/BS1 method to confirm that the optimized structures are local minima or transition states. The intrinsic reaction coordinate (IRC)47 calculations were performed to ensure that the obtained transition states connect the correct reactants and products. Solution-phase single-point energy calculations were conducted on the basis of the gas-phase-optimized structures by using the M0651 functional including the Grimme empirical dispersion correction (GD3)45 method and the SMD52 solvation model (solvent = toluene). The SDD basis set was used for Ni, and 6-311+G(2d,p)53 was employed on other elements (referred to as BS2). The combination of B3LYP for geometry optimization and M06 for single-point energy calculations has also been used in recent studies.30,54–57 The chosen method is verified by the consistency of the calculated enthalpy changes for the dimerization of AlMe3 with the experimental data58,59 (−21.4 vs. −20.4 kcal mol−1). WBI bond order analysis60–62 was performed using the M06-GD3/BS2 method. The noncovalent interaction (NCI) analysis63–66 was carried out using the Multiwfn program67 and visualized using VMD software.68 The 3D structure of the optimized geometries was plotted using CYLview software.69
2.2 Computational models
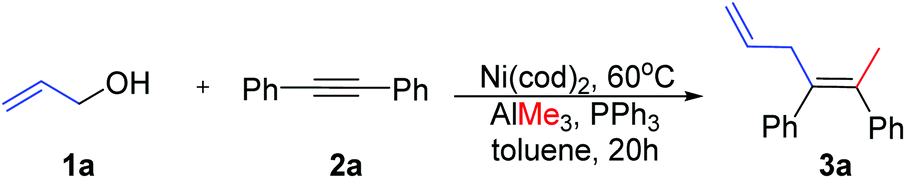
In accordance with Zhao's experiments,29 Ni-catalyzed methylation–allylation of 2a with 1a and AlMe3 in the presence of the PPh3 ligand (toluene solvent) was used as the modelling reaction in theoretical calculations (Scheme 3). Upon heating at 60 °C, this reaction produced 3a with 92% yield after 20 hours.
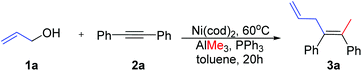 |
| | Scheme 3 Model reaction in theoretical calculations. | |
3. Results and discussion
3.1 Formation of alkoxyaluminum species
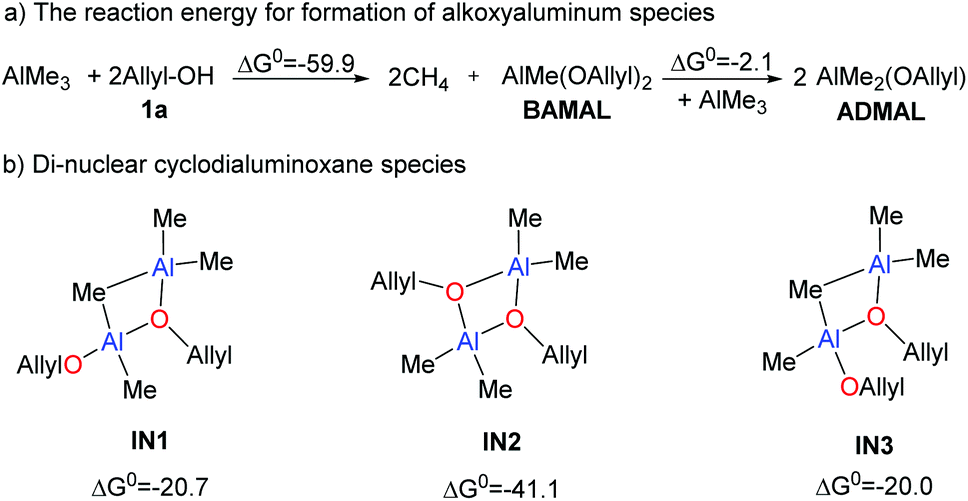
In view of the recent studies in using Lewis acids as catalysts,39,40 the aggregation of AlMe3 with alcoholic substrate 1a could occur via different modes (Scheme 4). When AlMe3 was slowly added into the reaction mixture, AlMe3 might react with 2 fold 1a to form bis(allyloxy)methylaluminum (BAMAL), releasing 2 fold CH4 simultaneously.41–44 The reaction energy of this step is −59.9 kcal mol−1 (Scheme 4). BAMAL could further react with another AlMe3 to form two ADMAL. This process is exergonic by 2.1 kcal mol−1, indicating the reversibility of the transformation and the co-existence of BAMAL and ADMAL.
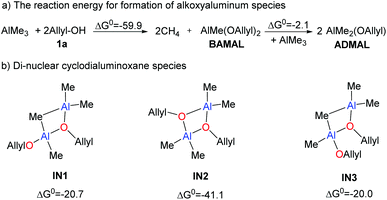 |
| | Scheme 4 Formation of alkoxyaluminum species. The reaction Gibbs free energies are given in kcal mol−1. | |
Due to the presence of both Lewis acidic and Lewis basic sites, the mono-nuclear aluminoxane species (ADMAL and BAMAL) is likely to undergo self-assembly.39,40 For clarity, the dimeric assembly of ADMAL, BAMAL and AlMe3 was taken into account. Considering equivalent amounts of 1a and AlMe3 in Zhao's experiment,29 we mainly focus on the dimeric aggregation of BAMAL with AlMe3 and the self-aggregation of ADMAL. In this context, three structures, i.e.IN1, IN2, and IN3 (Scheme 4), could be possibly formed. The results indicate that the formation of IN2 is more appealing (exergonic by 41.1 kcal mol−1 relative to 2 fold BAMAL), because it allows for the formation of two groups of dative bonding between the donor-O and Al center.39,40 Therefore, cyclodialuminoxane IN2 was used as the main alkoxyaluminum species in the following calculations.
3.2 C–O bond activation
In accordance with the recent studies,70–73 Ni(PPh3)2 is used as the starting state of the Ni(0) catalyst. To this end, the coordination of the allylic group of IN2 to the Ni(0) center could generate IN4 (energy reference point) or IN5 (Fig. 1), depending on whether the oxygen atom is coordinated to the Ni center or not. IN4 and IN5 could then lead to the outer-sphere and inner-sphere C–O activation pathway, respectively (outer-sphere and inner-sphere refer to the orientation of the cyclodialuminoxane species and Ni(0)L2 moieties). The energy barriers are 14.8 kcal mol−1 (IN4 → TS1) and 24.6 kcal mol−1 (IN5 → TS2). As shown in Fig. 1, the C1–O bond length of TS1 (2.518 Å) is much longer than that of TS2 (2.100 Å). Therefore, the feasibility of the outer-sphere pathway over the inner-sphere one is predominantly caused by the reduced steric hindrance between the cyclodialuminoxane group and the PPh3 ligand. The outer- and inner-sphere pathways converge after the exergonic coordination of the cyclodialuminoxane group to the Ni center (IN6). From IN6, the dissociation of the anionic cyclodialuminoxane group is endergonic by 8.3 kcal mol−1 (IN7), predominantly due to the use of a non-polar solvent of toluene in experiments. In contrast, dissociation of one PPh3 ligand from IN6 is thermodynamically more feasible, generating a more stable ally-Ni intermediate IN8. Compared to the aforementioned pathways, the direct C–O bond activation of 1a, ADMAL and BAMAL is remarkably much more difficult (with energy barriers >35 kcal mol−1, Scheme S1†). The reason mainly relates to the weaker C–O bond in the allylic group coordinated structure of IN4 (compared to all other precursors, evidenced by the bond length and WBI bond order analysis in Fig. S1†). Meanwhile, the typical Lewis acid-mediated C–O activation,30,74–76i.e. AlMe3/ADMAL/BAMAL mediated C–O bond cleavage of 1a, was also examined but was excluded due to high energy barriers (for more details, please see Scheme S2†). The activation of the allylic alcohol substrate via forming the dimeric aluminum complex has not been reported in earlier studies.
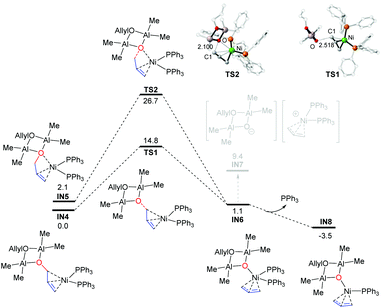 |
| | Fig. 1 Gibbs free energy profiles for the C–O bond activation starting from IN4 and IN5. Optimized geometries with selected structural parameters for the C–O bond activation transition states. The relative Gibbs free energies are given in kcal mol−1. The bond distances are given in Å. | |
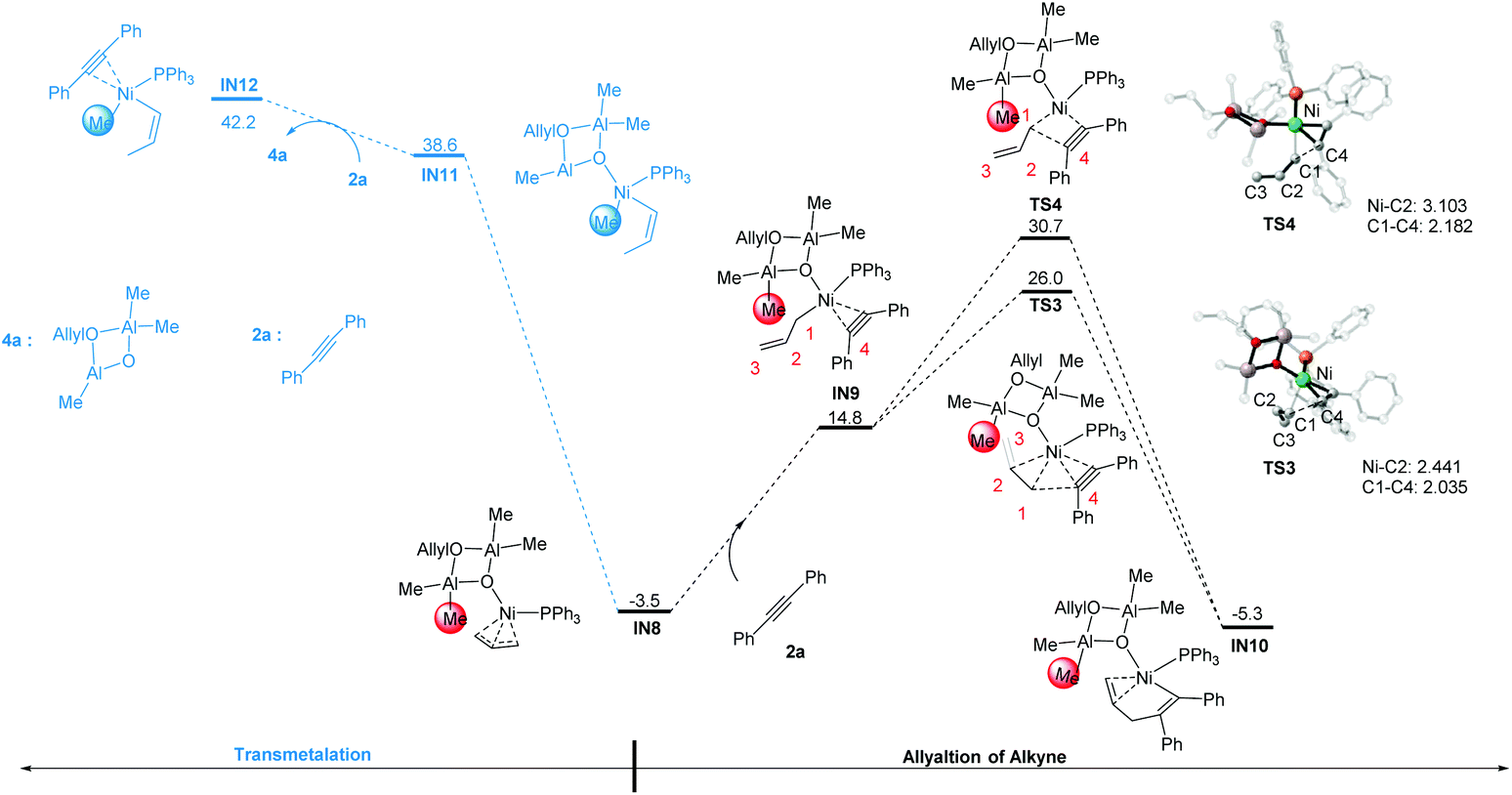
3.3 Allylation of alkyne and competitive pathways
According to Scheme 2, the C–X bond activation is followed by the alkyne insertion step. In this regard, IN8 could first undergo the coordination of the alkyne substrate 2a to the Ni center to give intermediate IN9. After that, there are two allylation pathways to produce the alkenyl–Ni species IN10. In one pathway, the C2 and C3 atoms are proximal to the Ni-alkyne group in the transition state TS3, forming a distorted C3-coordination. In the other pathway, the C2 and C3 atoms are away from the Ni center in the transition state TS4. TS3 is 4.7 kcal mol−1 lower in free energy than TS4. This may be related to the different interaction between the Ni center and the allyl group. In TS3, the short C2–Ni distance of 2.441 Å implies the bonding interactions therein. Due to the extra stability of the C2–C3 bond coordination, the C1–C4 bond distance in TS3 is slightly shorter than that in TS4 (2.035 vs. 2.182 Å, Fig. 2).
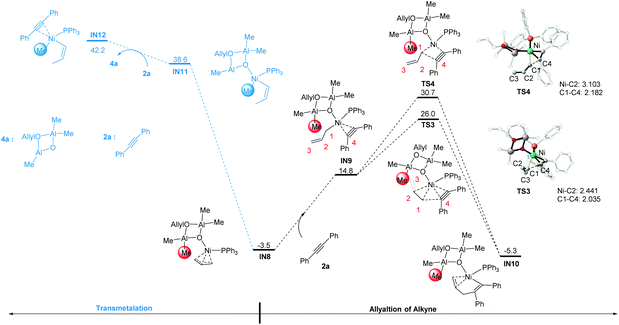 |
| | Fig. 2 Gibbs free energy profiles of allylation and methylation pathways starting from IN8. The optimized geometries with selected structural parameters for the transition states involved in the allylation of alkyne. The relative Gibbs free energies are given in kcal mol−1. The bond distances are given in Å. | |
In addition to the alkyne insertion step, we also considered the possibility for direct methylation on IN8, with the cyclodialuminoxane group acting as the methyl source (possibly occurs via a first transmetalation step). Nevertheless, the free energy of the intermediates involved in this pathway is too high (IN11: 38.6 kcal mol−1, IN12: 42.2 kcal mol−1). The results indicate that from IN8, the allylation is more feasible than the methylation step.
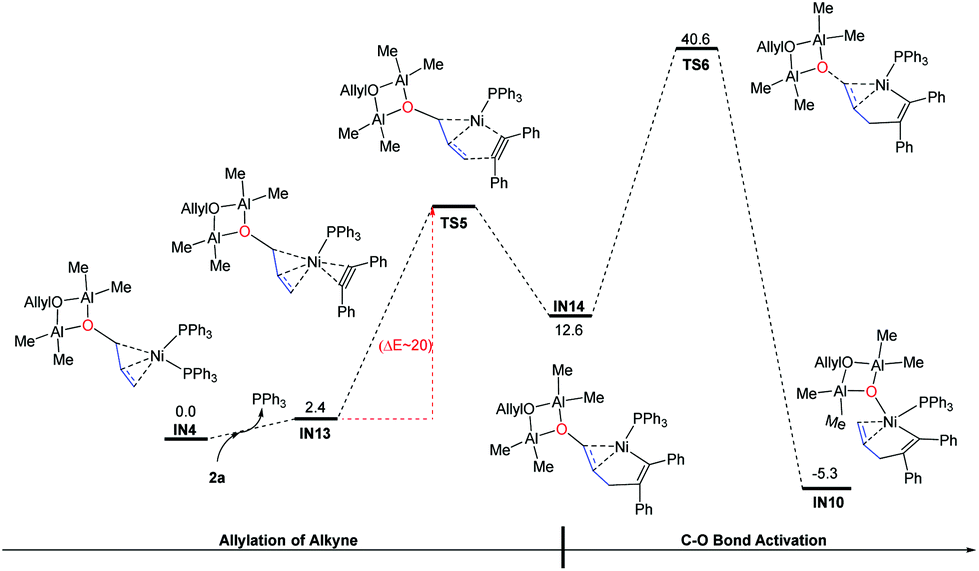
In addition to the C–O bond activation–allylation/methylation mechanism, we also examined another possible pathway in which allylation occurs before C–O bond activation.38 In this mechanism, the alkyne will react directly with IN4 (Fig. 3). Taking into account the influence of steric hindrance, only the outer-sphere pathway was probed. Starting from IN4, ligand exchange of 2a with one PPh3 gives intermediate IN13. After that, C(allyl)–C(alkyne) gradually approaches each other to form the intermediate IN14via transition state TS5 (Note, all efforts in locating the transition state TS5 were failed, partial optimization is conducted to give relative electronic energies in Fig. 3 (ΔE ∼ 20 kcal mol−1). For more details, see Fig. S2†). From IN14, the C–O bond is activated to produce the intermediate IN10 after transition state TS6. However the high activation energy barrier of this pathway (IN4 → TS6: 40.6 kcal mol−1) excludes the possibility of this pathway.
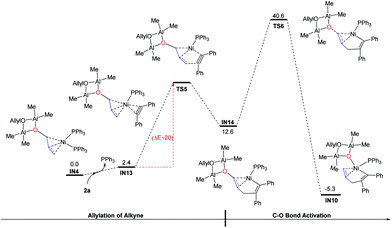 |
| | Fig. 3 Gibbs free energy profiles for the allylation and C–O bond activation starting from IN4. The relative Gibbs free energies and electronic energies are given in kcal mol−1. | |
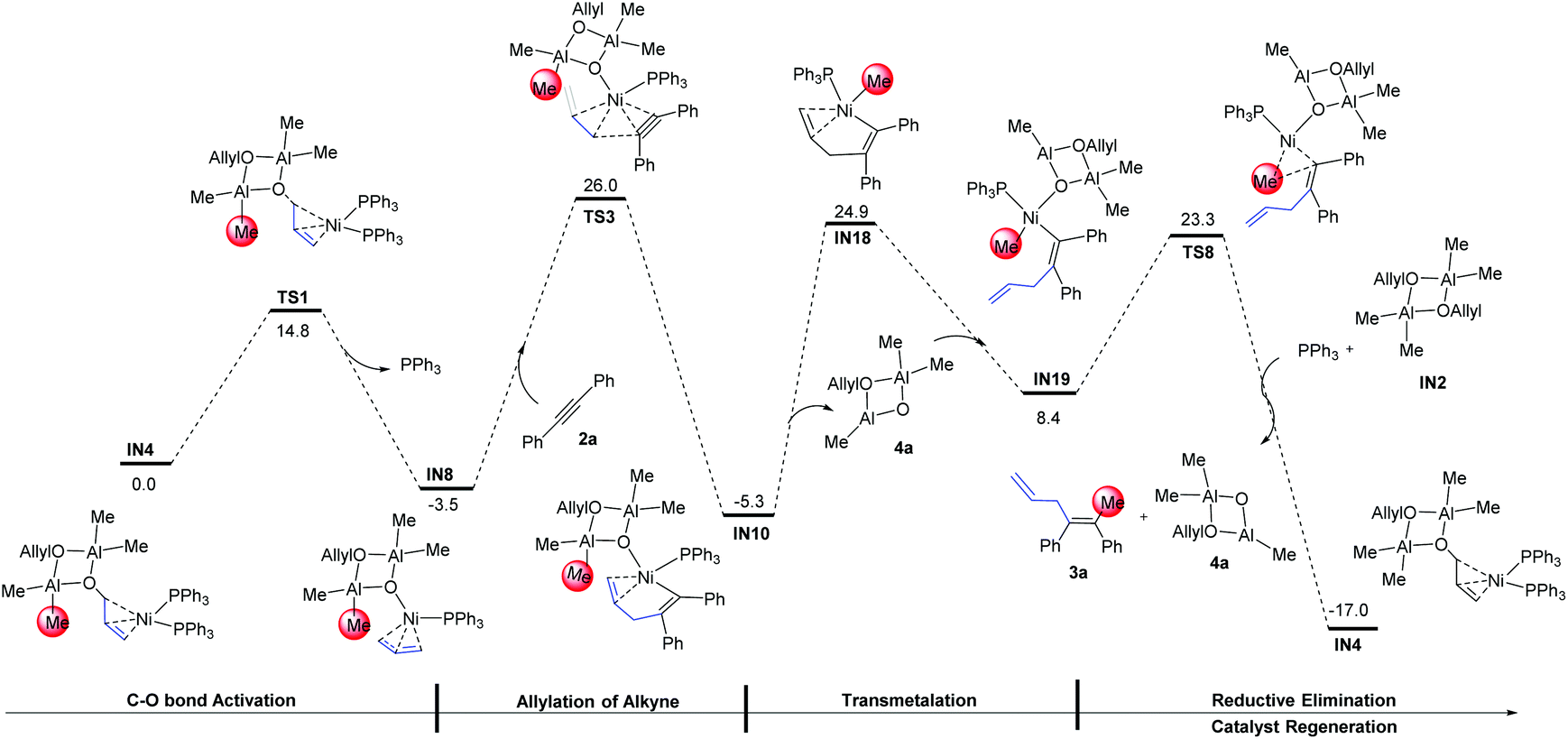
3.4 Methylation: transmetalation and reductive elimination
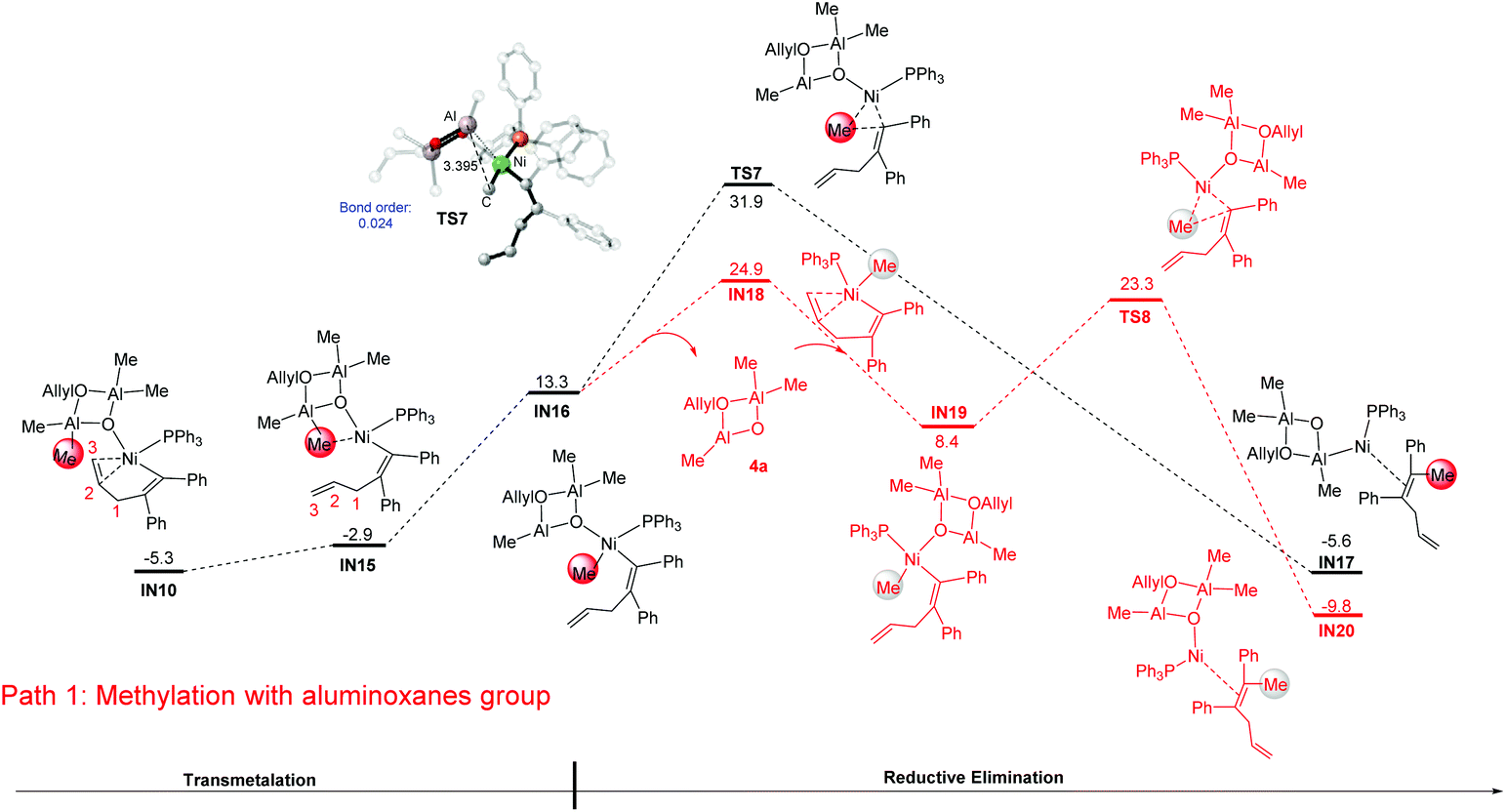
From the alkenyl-Ni species IN10, the methylation was proposed to occur via a successive transmetalation–reduction elimination step in previous studies.24,25,36,37 According to the calculation results, the methylation occurs with a spontaneous C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond dissociation of the C2–C3 bond, forming IN16 as a result (a detailed energy profile for the methylation is given in Fig. S3†). Then reductive elimination from IN16 occurs and produces the intermediate IN17 (IN16 → TS7: 18.6 kcal mol−1, black line). Of note, the relative energy of the reductive elimination transition state TS7 is already 37.2 kcal mol−1 higher than that of IN10, excluding the possibility of this pathway. Examining the optimized geometry of TS7, we noticed that the difficulty in reductive elimination might be caused by the strong Lewis acid–base interaction between the cyclodialuminoxane group and the migrating Me group (bond length of Al–C: 3.395 Å; bond order of Al–C: 0.024). Such interaction could be eliminated by changing the location of the cyclodialuminoxane group with the PPh3 ligand in IN16. The calculation indicates that the isomerization occurs via a dissociative pathway. That is, the dissociation of the cyclodialuminoxane group first occurs with a simultaneous re-coordination of the alkene group, and the formed intermediate IN18 then undergoes tautomerization and re-coordination of cyclodialuminoxane to generate IN19. From IN19, reductive elimination might be more likely to occur to produce the intermediate IN20, as the related transition state TS8 is 8.6 kcal mol−1 lower in energy than TS7. According to this mechanism, the overall barrier of the methylation step is 30.2 kcal mol−1 (IN10 → IN18).
C bond dissociation of the C2–C3 bond, forming IN16 as a result (a detailed energy profile for the methylation is given in Fig. S3†). Then reductive elimination from IN16 occurs and produces the intermediate IN17 (IN16 → TS7: 18.6 kcal mol−1, black line). Of note, the relative energy of the reductive elimination transition state TS7 is already 37.2 kcal mol−1 higher than that of IN10, excluding the possibility of this pathway. Examining the optimized geometry of TS7, we noticed that the difficulty in reductive elimination might be caused by the strong Lewis acid–base interaction between the cyclodialuminoxane group and the migrating Me group (bond length of Al–C: 3.395 Å; bond order of Al–C: 0.024). Such interaction could be eliminated by changing the location of the cyclodialuminoxane group with the PPh3 ligand in IN16. The calculation indicates that the isomerization occurs via a dissociative pathway. That is, the dissociation of the cyclodialuminoxane group first occurs with a simultaneous re-coordination of the alkene group, and the formed intermediate IN18 then undergoes tautomerization and re-coordination of cyclodialuminoxane to generate IN19. From IN19, reductive elimination might be more likely to occur to produce the intermediate IN20, as the related transition state TS8 is 8.6 kcal mol−1 lower in energy than TS7. According to this mechanism, the overall barrier of the methylation step is 30.2 kcal mol−1 (IN10 → IN18).
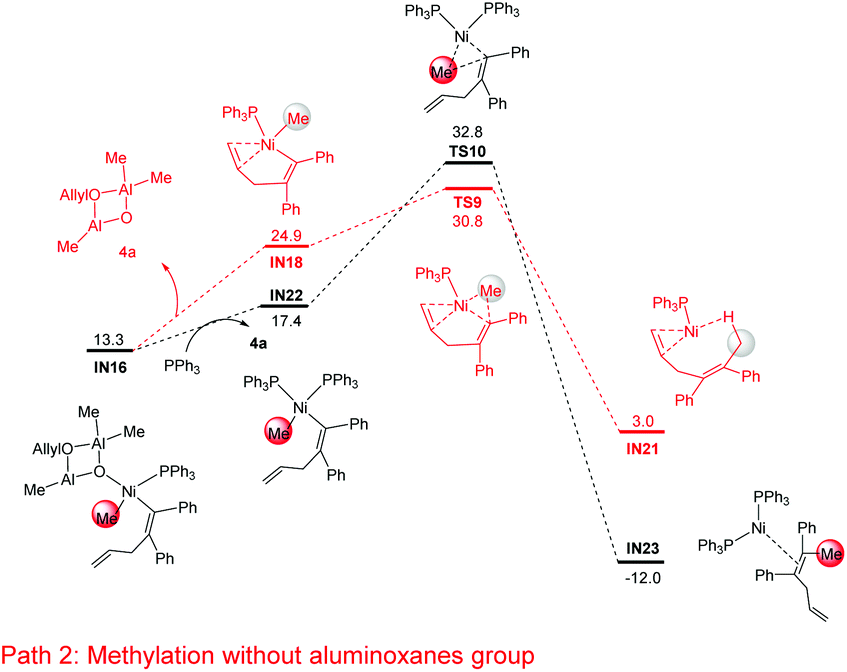
In addition to the isomerization mechanism in Fig. 4, the possibility of methylation directly on IN16 (i.e. releasing the cyclodialuminoxane group from IN16, IN16 → IN18, Fig. 5) was examined. Meanwhile, the mechanistic possibility of replacing the cyclodialuminoxane group with another PPh3 ligand was also examined (IN16 → IN22, Fig. 5). However, the free energies of transition states in these pathways (TS9: 30.8 kcal mol−1; TS10: 32.8 kcal mol−1) are both higher than that of TS8 (Fig. 4: 23.3 kcal mol−1). Therefore, these pathways were excluded.
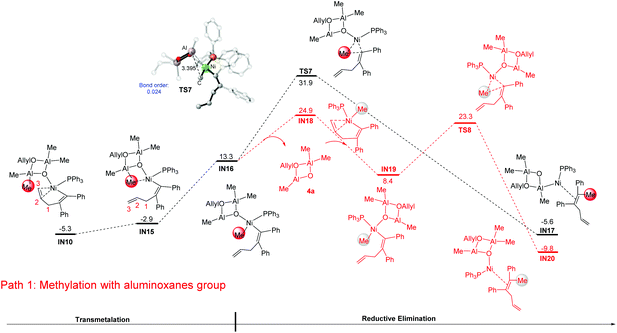 |
| | Fig. 4 Gibbs free energy profiles for transmetalation and reductive elimination starting from the alkenyl-Ni species IN10. Optimized geometries with selected structural parameters of TS7. The relative Gibbs free energies are given in kcal mol−1. The bond distances are given in Å. | |
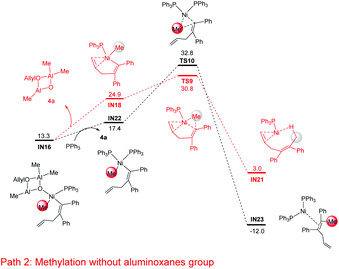 |
| | Fig. 5 Gibbs free energy profiles for methylation without the aluminoxane group starting from the Ni–Me species IN16. The relative Gibbs free energies are given in kcal mol−1. | |
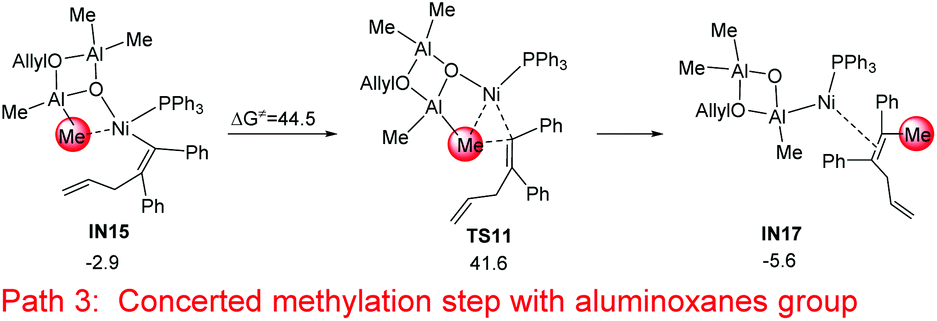
In addition to the stepwise methylation (transmetalation–reductive elimination) mechanism, a concerted methylation step, which occurs via a direct transfer of the methyl group to the alkyne group, was examined (without the formation of a Ni–Me intermediate).38 However the concerted step is excluded because of the high energy barrier of 44.5 kcal mol−1 (IN15 → TS11, Fig. 6).
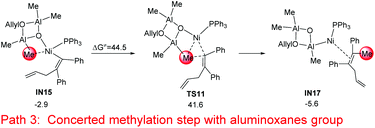 |
| | Fig. 6 Relative Gibbs free energies for the concerted methylation step with the aluminoxane group starting from IN15. The relative Gibbs free energies are given in kcal mol−1. | |
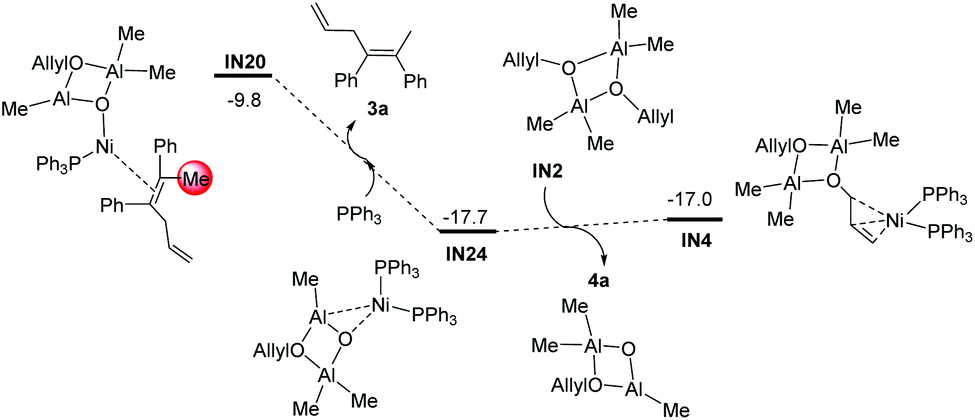
After the methylation step, the intermediate IN20 could easily release the dicarbofunctionalization product 3a (IN20 + PPh3→IN24 + 3a, Fig. 7), and regenerate the intermediate IN4via ligand exchange procedures (Fig. 7).
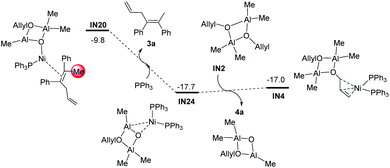 |
| | Fig. 7 Gibbs free energy profiles of the catalyst regeneration step starting from IN20. The relative Gibbs free energies are given in kcal mol−1. | |
3.5 Summary of the most feasible mechanism
According to the aforementioned results and discussion, an illustrative diagram for the most feasible pathway of the Ni(0)-catalyzed allylmethylation of an alkyne is shown in Fig. 8 (some intermediates were omitted for clarity). The C–O bond activation (IN4 → TS1 → IN8) occurs first. Then the intermediate IN8 reacts with the alkyne to give alkenyl-Ni species IN10 through allylation of alkyne. After that, transmetalation of intermediate IN10 might give a Ni–Me species IN19. The reductive elimination occurs subsequently to release intermediate 3a and the final ligand exchange occurs to regenerate the intermediate IN4. Throughout the catalytic cycle, IN10 is the most stable intermediate, the rate-determining step is the transmetalation step of methylation, and the overall activation barrier is 30.2 kcal mol−1 (IN10 → IN18). Due to the relatively high energy barrier of the allylation step compared to that of the subsequent steps, the allylation is expected as the regioselectivity determining step for unsymmetric alkynes.
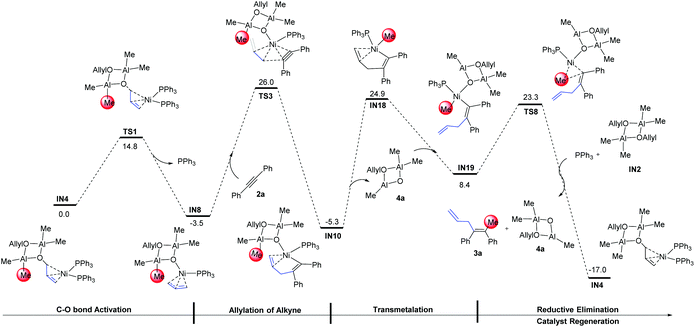 |
| | Fig. 8 Gibbs free energy profiles for the overall transformation in the most feasible mechanism. The relative Gibbs free energies are given in kcal mol−1. | |
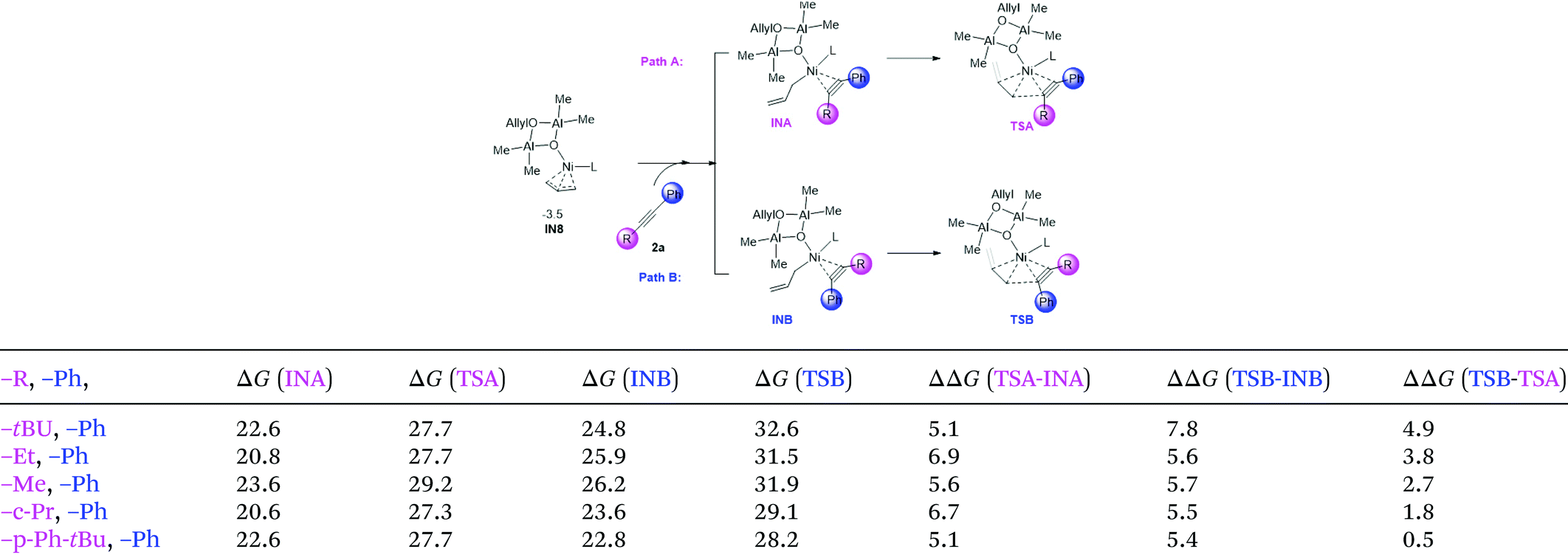
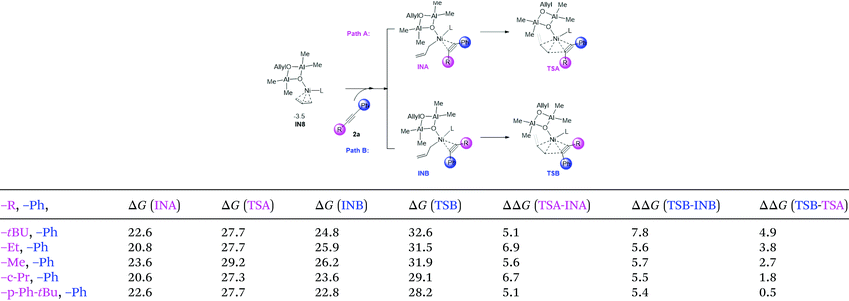
3.6 Origin of regioselectivity
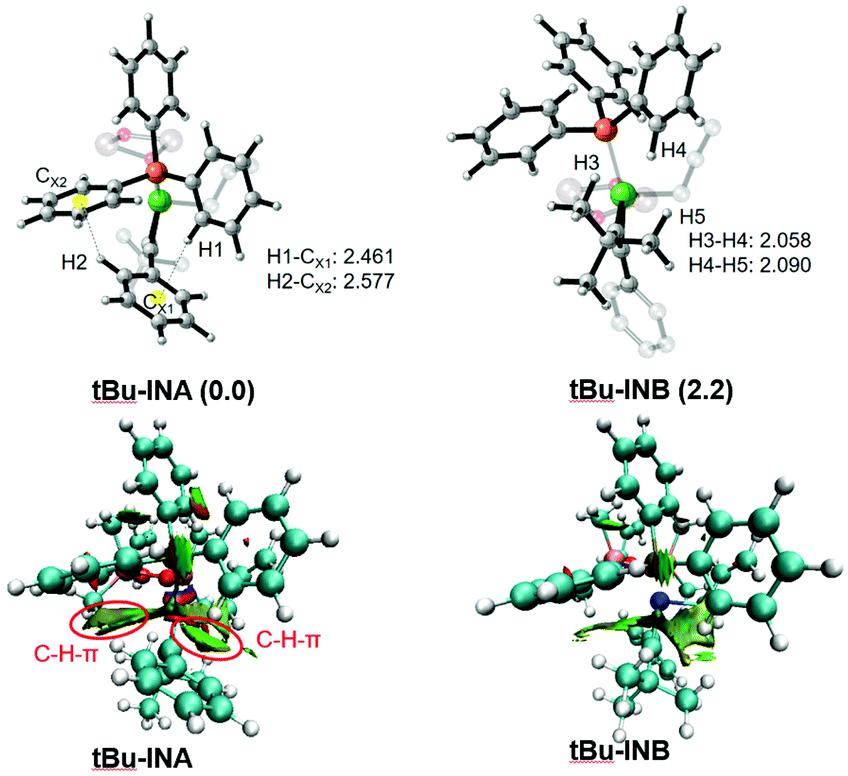
In Zhao's experiments, for unsymmetric alkynes bearing one alkyl and one aromatic substituent, the allylation and methylation occur on the alkyl and aryl substituted sites, respectively.29 To elucidate the origin of the regioselectivity, we carried out calculations on a series of di-substituted alkynes (with –tBu, –Et, –Me, –c-Pr, –p-Ph-tBu, and –Ph substituents, Table 1). The allylation on both sites of the substrate (Path A: allylation of the alkyl substituted carbon and Path B: allylation of the Ph substituted carbon) is mainly considered. The related intermediate and transition state were denoted as INA/B and TSA/B, respectively. From the calculation results (Table 1), the free energies of INB and TSB are higher than those of the related INA and TSA in all systems. The results correlate with Zhao's experimental outcome and indicate that the observed regioselectivity could be regulated by the coordination step (IN8 → INA/B). To verify this assumption, we selected tBu-, Ph-disubstituted alkyne for a detailed structural analysis.
Table 1 Illustrative diagram of the allylation on di-substituted asymmetric alkynes, and the relative energy of the related intermediates and transition states
| The relative Gibbs free energies are given in kcal mol−1. |
|
According to the optimized geometry in Fig. 9, the C–H⋯π interaction in tBu-INA could be easily identified (H1-CX1: 2.461; H2-CX2: 2.577), and could be evidenced by a topological NCI (noncovalent interaction) analysis.63–66 Such C–H⋯π interaction is absent in tBu-INB. Meanwhile, there are two pairs of H–H distances within the van der Waals radius (H3–H4: 2.058; H4–H5: 2.090), indicating the steric repulsion therein. According to these results, both the lower steric hindrance and the presence of the non-covalent C–H⋯π interactions facilitate a prior allylation on the alkyl-substituted carbon atom.
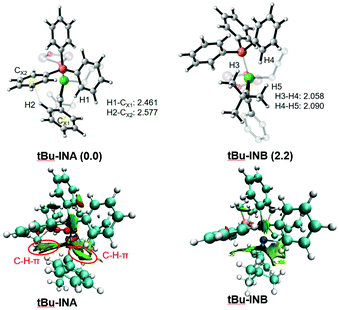 |
| | Fig. 9 The optimized structures and visualization of the main noncovalent interaction (NCI, density cutoff of 0.6 au) of tBu-INA and tBu-INB. Only selected atoms are shown for clarity. Green indicates weak interaction. The bond distances are given in Å. The relative free energies are given in kcal mol−1 (relative to tBu-INA). | |
4. Conclusion
The present study focuses on the detailed mechanism of methylation of alkyne and the origin of regioselectivities of the Ni-catalyzed dicarbofunctionalization reported by Zhao et al. earlier. This proposed mechanism consists of several steps: C–O bond activation, allylation of alkyne, transmetalation, reductive elimination and catalyst regeneration. Cyclodialuminoxane, formed by the reaction of AlMe3 with allylic alcohol with a subsequent dimerization step, is a pivotal intermediate. Compared to the typical Lewis acid mediated C–O activation, the C(allyl)–O bond is remarkably activated, resulting in a relatively facile C–O activation. Meanwhile, a “Me-relay” (the Me group is first transferred to the Ni center, and then to the adjacent C (alkyne) atom) is found to be responsible for the methylation step, while all other methylations (such as a concerted methylation step in which the Me group is directly transferred to the alkyne) were excluded. The mechanistic proposal demonstrates the great potential of AlR3 in mediating the C–O bond activation and alkylation reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study is supported by the National Natural Science Foundation of China (51961135104, 21732006, 51821006), the National Key R&D Program of China (2017YFA0303502) and Natural Science Foundation of Anhui Province (2108085J08). The calculations have been done on the supercomputing system in the Supercomputing Center of University of Science and Technology of China and Anhui University. We thank Prof. Mårten Ahlquist from KTH Royal Institute of Technology for the professional helps on calculation methods.
References
- J.-J. Wu, J. Xu and X. Zhao, Selenide-Catalyzed Stereoselective Construction of Tetrasubstituted Trifluoromethylthiolated Alkenes with Alkynes, Chem. – Eur. J., 2016, 22, 15265–15269 CrossRef.
- J. Chen, S. Chen, X. Xu, Z. Tang, C.-T. Au and R. Qiu, Nickel-Catalyzed Regioselective Cleavage of Csp2-S Bonds: Method for the Synthesis of Tri- and Tetrasubstituted Alkenes, J. Org. Chem., 2016, 81, 3246–3255 CrossRef CAS.
- T. Ichitsuka, T. Fujita and J. Ichikawa, Nickel-Catalyzed Allylic C(sp3)-F Bond Activation of Trifluoromethyl Groups via β–Fluorine Elimination: Synthesis of Difluoro-1,4-dienes, ACS Catal., 2015, 5, 5947–5950 CrossRef CAS.
- Y. Nakao, T. Yukawa, Y. Hirata, S. Oda, J. Satoh and T. Hiyama, Allylcyanation of Alkynes: Regio- and Stereoselective Access to Functionalized Di- or Trisubstituted Acrylonitriles, J. Am. Chem. Soc., 2006, 128, 7116–7117 CrossRef CAS PubMed.
- A. B. Flynn and W. W. Ogilvie, Stereocontrolled Synthesis of Tetrasubstituted Olefins, Chem. Rev., 2007, 107, 4698–4745 CrossRef CAS.
- N. F. McKinley and D. F. O'Shea, Carbolithiation of Diphenylacetylene as a Stereoselective Route to (Z)-Tamoxifen and Related Tetrasubstituted Olefins, J. Org. Chem., 2006, 71, 9552–9555 CrossRef CAS.
- Y. Hirata, T. Yukawa, N. Kashihara, Y. Nakao and T. Hiyama, Nickel-Catalyzed Carbocyanation of Alkynes with Allyl Cyanides, J. Am. Chem. Soc., 2009, 131, 10964–10973 CrossRef CAS.
- R. B. Williams, A. Norris, C. Slebodnick, J. Merola, J. S. Miller, R. Andriantsiferana, V. E. Rasamison and D. G. I. Kingston, Cytotoxic Sesquiterpene Lactones from Vernonia pachyclada, from the Madagascar Rainforest, J. Nat. Prod., 2005, 68, 1371–1374 CrossRef CAS.
- S. Kraft, K. Ryan and R. B. Kargbo, Recent Advances in Asymmetric Hydrogenation of Tetrasubstituted Olefins, J. Am. Chem. Soc., 2017, 139, 11630–11641 CrossRef CAS.
- D.-W. Ji, Y.-C. Hu, H. Zheng, C.-Y. Zhao, Q.-A. Chen and V. M. Dong, A regioselectivity switch in Pd-catalyzed hydroallylation of alkynes, Chem. Sci., 2019, 10, 6311–6315 RSC.
- Y. Yamamoto, E. Ohkubo and M. Shibuya, Selective synthesis of trisubstituted (trifluoromethyl)alkenes via ligand-free Cu-catalyzed syn hydroarylation, hydroalkenylation and hydroallylation of (trifluoromethyl)alkynes, Green Chem., 2016, 18, 4628–4632 RSC.
- P. Prasit, Z. Wang, C. Brideau, C.-C. Chan, S. Charleson, W. Cromlish, D. Ethier, J. F. Evans, A. W. Ford-Hutchinson, J. Y. Gauthier, R. Gordon, J. Guay, M. Gresser, S. Kargman, B. Kennedy, Y. Leblanc, S. Léger, J. Mancini, G. P. O'Neill, M. Ouellet, M. D. Percival, H. Perrier, D. Riendeau, I. Rodger, P. Tagari, M. Thérien, P. Vickers, E. Wong, L.-J. Xu, R. N. Young, R. Zamboni, S. Boyce, N. Rupniak, M. Forrest, D. Visco and D. Patrick, The discovery of rofecoxib, [MK 966, VIOXX, 4-(4′-METHYLSULFONYLPHENYL)-3-PHENYL-2(5H)-FURANONE], an orally active cyclooxygenase-2 inhibitor, Bioorg. Med. Chem. Lett., 1999, 9, 1773–1778 CrossRef CAS.
- F. Xue, J. Zhao, T. S. A. Hor and T. Hayashi, Nickel-Catalyzed Three-Component Domino Reactions of Aryl Grignard Reagents, Alkynes, and Aryl Halides Producing Tetrasubstituted Alkenes, J. Am. Chem. Soc., 2015, 137, 3189–3192 CrossRef CAS PubMed.
- E. E. Lin, J.-Q. Wu, F. Schäfers, X.-X. Su, K.-F. Wang, J.-L. Li, Y. Chen, X. Zhao, H. Ti, Q. Li, T.-M. Ou, F. Glorius and H. Wang, Regio- and stereoselective synthesis of tetra- and triarylethenes by N-methylimidodiacetyl borondirected palladium-catalysed three-component coupling, Commun. Chem., 2019, 2, 34 CrossRef.
- Z. Li, A. García-Domínguez and C. Nevado, Nickel-Catalyzed Stereoselective Dicarbofunctionalization of Alkynes, Angew. Chem., Int. Ed., 2016, 55, 6938–6941 CrossRef CAS PubMed.
- T. Mori, Y. Akioka, H. Kawahara, R. Ninokata, G. Onodera and M. Kimura, Efficient and Selective Formation of Unsaturated Carboxylic and Phenylacetic Acids from Diketene, Angew. Chem., Int. Ed., 2014, 53, 10434–10438 CrossRef CAS.
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, Efficient and Selective Formation of Unsaturated Carboxylic and Phenylacetic Acids from Diketene, J. Am. Chem. Soc., 2009, 131, 5070–5071 CrossRef CAS PubMed.
- Y. Mori, T. Mori, G. Onodera and M. Kimura, Efficient and Selective Formation of Unsaturated Carboxylic and Phenylacetic Acids from Diketene, Synthesis, 2014, 46, 2287–2292 CrossRef CAS.
- R. Ninokata, T. Yamahira, G. Onodera and M. Kimura, Nickel-Catalyzed CO2 Rearrangement of Enol Metal Carbonates for the Efficient Synthesis of β-Ketocarboxylic Acids, Angew. Chem., Int. Ed., 2017, 56, 208–211 CrossRef CAS.
- M. Kimura, K. Kojima, Y. Tatsuyama and Y. Tamaru, Nickel-Catalyzed Multicomponent Connection of Dimethylzinc, Alkynes, 1,3-Butadiene, Aldehydes, and Amines, J. Am. Chem. Soc., 2006, 128, 6332–6333 CrossRef CAS.
- M. Kimura, M. Togawa, Y. Tatsuyama and K. Matsufuji, Nickel-catalyzed multi-component coupling reaction of aldimine, alkyne, and dimethylzinc via dimerization of butadiene, Tetrahedron Lett., 2009, 50, 3982–3984 CrossRef CAS.
- J. Wu and N. Yoshikai, Cobalt-Catalyzed Alkenylzincation of Unfunctionalized Alkynes, Angew. Chem., Int. Ed., 2016, 55, 336–340 CrossRef CAS PubMed.
- Q.-W. Song, Q.-N. Zhao, J.-Y. Li, K. Zhang and P. Liu, Selective Conversion of CO2 and Switchable Alcohols into Linear or Cyclic Carbonates via Versatile Zinc Catalysis, Synthesis, 2019, 51, 739–746 CrossRef CAS.
- S. Ogoshi, M. Ueta, T. Arai and H. Kurosawa, AlMe3-Promoted Oxidative Cyclization of η2-Alkene and η2-Ketone on Nickel(0). Observation of Intermediate in Methyl Transfer Process, J. Am. Chem. Soc., 2005, 127, 12810–12811 CrossRef CAS.
- M. Ohashi, O. Kishizaki, H. Ikeda and S. Ogoshi, Ni(0)-Catalyzed Formation of Azaaluminacyclopentenes via Azanickelacyclopentenes: A Unique Nickel/Aluminum Double Transmetalation Reaction, J. Am. Chem. Soc., 2009, 131, 9160–9161 CrossRef CAS PubMed.
- M. Ohashi, H. Saijo, T. Arai and S. Ogoshi, Nickel(0)-Catalyzed Formation of Oxaaluminacyclopentenes via an Oxanickelacyclopentene Key Intermediate: Me2AlOTf-Assisted Oxidative Cyclization of an Aldehyde and an Alkyne with Nickel(0), Organometallics, 2010, 29, 6534–6540 CrossRef CAS.
- M. Takimoto and Z. Hou, Cu-Catalyzed Formal Methylative and Hydrogenative Carboxylation of Alkynes with Carbon Dioxide: Efficient Synthesis of α,β-Unsaturated Carboxylic Acids, Chem. – Eur. J., 2013, 19, 11439–11445 CrossRef CAS.
- M. Takimoto, S. Usami and Z. Hou, Scandium-Catalyzed Regio-and Stereospecific Methylalumination of Silyloxy/Alkoxy-Substituted Alkynes and Alkenes, J. Am. Chem. Soc., 2009, 131, 18266–18268 CrossRef CAS.
- W. Li, S. Yu, J. Li and Y. Zhao, Nickel-Catalyzed Allylmethylation of Alkynes with Allylic Alcohols and AlMe3: Facile Access to Skipped Dienes and Trienes, Angew. Chem., Int. Ed., 2020, 59, 14404–14408 CrossRef CAS PubMed.
- D. Liu, Z. Xu, H. Yu and Y. Fu, Mechanistic Insights into the Nickel-Catalyzed Regioselective Carboxylation of Allylic Alcohols, Organometallics, 2021, 40, 869–879 CrossRef CAS.
- Y. Naganawa, H. Guo, K. Sakamoto and Y. Nakajima, Nickel-Catalyzed Selective Cross-Coupling of Chlorosilanes with Organoaluminum Reagents, ChemCatChem, 2019, 11, 3756–3759 CrossRef CAS.
- X. Zhang, Y. Liu, G. Chen, G. Pei and S. Bi, Theoretical Insight into C(sp3)-F Bond Activations and Origins of Chemo- and Regioselectivities of “Tunable” Nickel-Mediated/-Catalyzed Couplings of 2–Trifluoromethyl-1-alkenes with Alkynes, Organometallics, 2017, 36, 3739–3749 CrossRef CAS.
- M. Mailig, A. Hazra, M. K. Armstrong and G. Lalic, Catalytic Anti-Markovnikov Hydroallylation of Terminal and Functionalized Internal Alkynes: Synthesis of Skipped Dienes and Trisubstituted Alkenes, J. Am. Chem. Soc., 2017, 139, 6969–6977 CrossRef CAS.
- P. Xie, Z. Sun, S. Li, L. Zhang, X. Cai, W. Fu, X. Yang, Y. Liu, X. Wo and T.-P. Loh, Catalytic Anti-Markovnikov Hydroallylation of Terminal and Functionalized Internal Alkynes: Synthesis of Skipped Dienes and Trisubstituted Alkenes, Org. Lett., 2020, 22, 1599–1604 CrossRef CAS.
- X. Kang, Y. Luo, G. Zhou, X. Wang, X. Yu, Z. Hou and J. Qu, Theoretical Mechanistic Studies on the trans-1,4-Specific Polymerization of Isoprene Catalyzed by a Cationic La-Al Binuclear Complex, Macromolecules, 2014, 47, 4596–4606 CrossRef CAS.
- M. E. Rudkin, P. M. Joensuu, W. S. MacLachlan and H. W. Lam, Diastereoselective Cobalt-Catalyzed Alkylative Aldol Cyclizations Using Trialkylaluminum Reagents, Org. Lett., 2008, 10, 2939–2942 CrossRef CAS.
- X. Liu, C.-C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Lewis Acid Assisted Nickel-Catalyzed Cross-Coupling of Aryl Methyl Ethers by C-O Bond-Cleaving Alkylation: Prevention of Undesired β-Hydride Elimination, Angew. Chem., Int. Ed., 2016, 55, 6093–6098 CrossRef CAS.
- X. Zhang, Y. Liu, G. Chen, G. Pei and S. Bi, Theoretical Insight into C(sp3)-F Bond Activations and Origins of Chemo- and Regioselectivities of “Tunable” Nickel-Mediated/-Catalyzed Couplings of 2–Trifluoromethyl-1-alkenes with Alkynes, Organometallics, 2017, 36, 3739–3749 CrossRef CAS.
- M. Linnolahti, A. Laine and T. A. Pakkanen, Screening the Thermodynamics of Trimethylaluminum-Hydrolysis Products and Their Co-Catalytic Performance in Olefin-Polymerization Catalysis, Chem. – Eur. J., 2013, 19, 7133–7142 CrossRef CAS PubMed.
- R. Glaser and X. Sun, Thermochemistry of the Initial Steps of Methylaluminoxane Formation. Aluminoxanes and Cycloaluminoxanes by Methane Elimination from Dimethylaluminum Hydroxide and Its Dimeric Aggregates, J. Am. Chem. Soc., 2011, 133, 13323–13336 CrossRef CAS PubMed.
- A. S. Lisovenko, K. Morokuma and A. Y. Timoshkin, Initial Gas Phase Reactions between Al(CH3)3/AlH3 and Ammonia: Theoretical Study, J. Phys. Chem. A, 2015, 119, 744–751 CrossRef CAS PubMed.
- H. Hagihara, K. Tsuchihara, J. Sugiyama, K. Takeuchi and T. Shiono, Copolymerization of Propylene and Polar Allyl Monomer with Zirconocene/Methylaluminoxane Catalyst: Catalytic Synthesis of Amino-Terminated Isotactic Polypropylene, Macromolecules, 2004, 37, 5145–5148 CrossRef CAS.
- P. K. Vardhanapu, V. Bheemireddy, M. Bhunia, G. Vijaykumar and S. K. Mandal, Cyclic (Alkyl)amino Carbene Complex of Aluminum(III) in Catalytic Guanylation Reaction of Carbodiimides, Organometallics, 2018, 37, 2602–2608 CrossRef CAS.
- R. Fernández-Galán, A. Ramos, E. Huergo, A. Antiñolo, F. Carrillo-Hermosilla, A. Rodríguez-Diéguez and D. García-Vivó, Cyclic (Alkyl)amino Carbene Complex of Aluminum(III) in Catalytic Guanylation Reaction of Carbodiimides, Chem. Commun., 2019, 55, 2809–2812 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initioparametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104–154123 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, Reaction Path
Following In Mass-Weighted Internal Coordinates, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, A set of f-polarization functions for pseudo-potential basis sets of the transition metals, Sc-Cu, Y-Ag and La-Au, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. HGllwarth, M. Biihme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Kijhler, R. Stegmann, A. Veldkamp and G. Frenking, A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al-B1 and f-type polarization functions for Zn, Cd, Hg, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- Q. Zhang, Z.-Q. Zhang, Y. Fu and H.-Z. Yu, Mechanism of the Visible Light-Mediated Gold-Catalyzed Oxyarylation Reaction of Alkenes, ACS Catal., 2016, 6, 798–808 CrossRef CAS.
- Q. Lu, B. Wang, H. Yu and Y. Fu, Mechanism of the Visible Light-Mediated Gold-Catalyzed Oxyarylation Reaction of Alkenes, ACS Catal., 2015, 5, 4881–4889 CrossRef CAS.
- J. Liu, W. Nie, H. Yu and J. Shi, A mechanistic study on Cu(I) catalysed carboxylation of the C–F bond with CO2: a DFT study, Org. Biomol. Chem., 2020, 18, 9065–9071 RSC.
- J.-L. Yu, S.-Q. Zhang and X. Hong, Mechanisms and Origins of Chemo- and Regioselectivities of Ru(II)-Catalyzed Decarboxylative C-H Alkenylation of Aryl Carboxylic Acids with Alkynes: A Computational Study, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef CAS PubMed.
- C. H. Henrickson and D. P. Eyman, Lewis acidity of alanes. Interactions of trimethylalane with sulphides, Inorg. Chem., 1967, 6, 1461–1465 CrossRef CAS.
- M. B. Smith, The monomer−dimer equilibria of liquid aliminum alkyls: III. Trimethylaluminum: the monomer−dimer equilibria of liquid and gaseous trimethylaluminum and triethylaluminum, J. Organomet. Chem., 1972, 46, 31–49 CrossRef CAS.
- A. E. Reed and F. Weinhold, Mechanisms and Origins of Chemo- and Regioselectivities of Ru(II)-Catalyzed Decarboxylative C-H Alkenylation of Aryl Carboxylic Acidswith Alkynes: A Computational Study, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A.-E. Reed, L.-A. Curtiss and F. Weinhold, Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- J. A. Mendes, P. Merino, T. Soler, E. J. Salustiano, P. R. R. Costa, M. Yus, F. Foubelo and C. D. Buarque, Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint, J. Org. Chem., 2019, 84, 2219–2233 CrossRef CAS PubMed.
- Y. Wang and Y. Lan, Mechanism and origin of diastereoselectivity of N-heterocyclic carbene-catalyzed cross-benzoin reaction: A DFT study, Chin. Chem. Lett., 2020, 31, 736–738 CrossRef CAS.
- S. Jerhaoui, J.-P. Djukic, J. Wencel-Delord and F. Colobert, AsymmetricNearly Barrierless C(sp3)-H Activation Promoted by Easily-Accessible N-Protected Aminosulfoxides as New Chiral Ligands, ACS Catal., 2019, 9, 2532–2542 CrossRef CAS.
- J. Wang, Z. Liu, J. Li, Z. Song, C. Hu and Z. Su, exo/endo Selectivity Control in Diels-Alder Reactions of Geminal Bis(silyl) Dienes: Theoretical and Experimental Studies, J. Org. Chem., 2019, 84, 3940–3952 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
-
C. Y. Legault, CYLview20, Université de Sherbrooke, 2020 (http://www.cylview.org) Search PubMed.
- S.-J. He, J.-W. Wang, Y. Li, Z.-Y. Xu, X.-X. Wang, X. Lu and Y. Fu, Nickel-Catalyzed Enantioconvergent Reductive Hydroalkylation of Olefins with α–Heteroatom Phosphorus or Sulfur Alkyl Electrophiles, J. Am. Chem. Soc., 2020, 142, 214–221 CrossRef CAS.
- W. Shu, A. García-Domínguez, M. T. Quirós, R. Mondal, D. J. Cárdenas and C. Nevado, Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights, J. Am. Chem. Soc., 2019, 141, 13812–13821 CrossRef CAS PubMed.
- M. Liu, Z. Zhang, H. Liu, T. Wu and B. Han, Dehydroxyalkylative halogenation of C(aryl)–C bonds of aryl alcohols, Chem. Commun., 2020, 56, 7120–7123 RSC.
- A. Tortajada, R. Ninokata and R. Martin, Ni-Catalyzed Site-Selective Dicarboxylation of 1,3-Dienes with CO2, J. Am. Chem. Soc., 2018, 140, 2050–2053 CrossRef CAS PubMed.
- S. Li, J. Qiu, B. Li, Z. Sun, P. Xie and T.-P. Loh, Practical allylation with unactivated allylic alcohols under mild conditions, Org. Chem. Front., 2021, 8, 3354–3359 RSC.
- H. Zhang, X.-Y. Zhan, Y. Dong, J. Yang, S. He, Z.-C. Shi, X.-M. Zhang and J.-Y. Wang, Dehydration in water: frustrated Lewis pairs directly catalyzed allylization of electron-rich arenes and allyl alcohols, RSC Adv., 2020, 10, 16942–16948 RSC.
- X. Huo, G. Yang, D. Liu, Y. Liu, I. D. Gridnev and W. Zhang, Palladium-Catalyzed Allylic Alkylation of Simple Ketones with Allylic Alcohols and Its Mechanistic Study, Angew. Chem., Int. Ed., 2014, 53, 6776–6780 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete Gaussian 16 reference, Cartesian coordinates and computed energies of calculated structures. See DOI: 10.1039/d1qo01580j |
|
| This journal is © the Partner Organisations 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Wan
Nie
a,
Haizhu
Yu
a,
Wan
Nie
a,
Haizhu
Yu