
Key role of a π–π complex in diaryl cross-coupling between aryldiazonium salts and arylboronic acids using photosensitizer-free gold/photoredox catalysis†
Yanhong
Liu
 ,
Rongxiu
Zhu
,
Chengbu
Liu
and
Dongju
Zhang
*
,
Rongxiu
Zhu
,
Chengbu
Liu
and
Dongju
Zhang
*
Key Laboratory of Colloid and Interface Chemistry, Ministry of Education, Institute of Theoretical Chemistry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: zhangdj@sdu.edu.cn; Tel: +86-531-88365833
First published on 17th November 2021
Abstract
DFT and TD-DFT calculations were performed to better understand the photosensitizer-free visible-light-mediated Au-catalyzed cross-coupling between aryldiazonium salts and arylboronic acids. The π–π type complex between the aryldiazonium salt and the arylboronic acid, rather than either the generally accepted gold(I)–aryldiazonium salt complex or the aryldiazonium salt itself, was shown to play the role of a virtual photoinitiator. The oxidation of Au(I) to Au(III), the key aspect of the dual gold/photoredox catalytic aryl–aryl cross-coupling, occurs via the radical addition of an aryl radical rather than an aryldiazo radical on Au(II) species. The transmetalation of the arylboronic acid to Au(III) species was identified as the rate-determining step, and the presence of a tetrafluoroborate anion can assist this process remarkably. The experimentally observed effect of substituents on the aryldiazonium salt and boronic acid on the reactivity was also rationalized.
1. Introduction
Biaryl compounds are important structural motifs in agrochemicals, pharmaceuticals, materials and organic products.1,2 Over the last few years, synthetic chemists have made great efforts to develop efficient methods for the construction of biaryl scaffolds via C–C cross-coupling reactions catalyzed by transition metals,3–6 in particular, Pd-based complexes, the most successful species for the coupling between aryl electrophiles and aryl nucleophiles due to the readily accessible Pd(0)/Pd(II) cycle.7,8 In contrast, the use of gold catalysts for such reactions has remained highly challenging for a long time because of the high redox potential of the AuI/AuIII couple (E0 = +1.41 V),9 which means that strong external oxidants,10–12 suitable ligands13 or reactive electrophiles (such as aryldiazonium chlorides)14–16 are required to access AuI/AuIII catalytic cycles. In recent years, to avoid the use of strong oxidants in gold catalysis, a new strategy for C–C cross-coupling has been developed by Glorius,17,18 Toste19,20 and Fensterbank21 that involves merging gold catalysis with photoredox catalysis under mild conditions.22The groups of Lee23 and Fouquet24 reported examples of dual gold/photoredox catalytic aryl–aryl cross-coupling in the presence of a photosensitizer. Furthermore, it was demonstrated by Hashmi25–27 and Bandini28 that gold-catalyzed C(sp2)–C(sp2) cross-coupling can be even achieved by only using an aryl radical source (aryldiazonium salts) combined with visible light irradiation without an exogenous photosensitizer, and several elegant works have been reported recently by Patil,29,30 Wong31 and Echavarren.32 In particular, the cross-coupling of aryldiazonium salts with electron-rich arylboronic acids, ideal coupling partners for dual catalytic systems,33–36 has been shown to be a highly efficient approach for the synthesis of biaryl compounds. Scheme 1 shows a prototype of the pioneering works of Hashmi et al.26,27,37 on the synthesis of diaryl compounds via photosensitizer-free dual gold/photoredox catalysis.
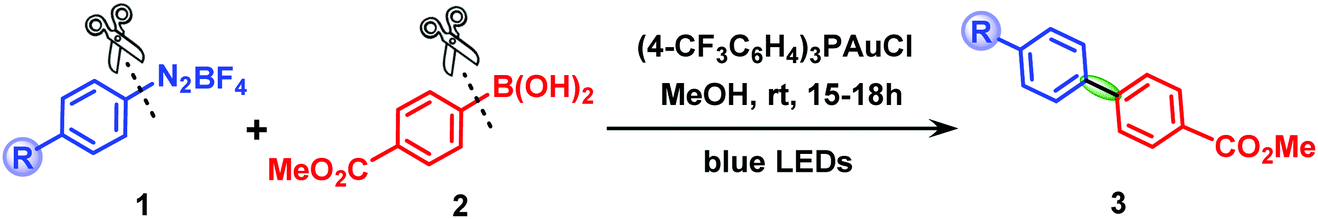 | ||
| Scheme 1 Photosensitizer-free visible-light-mediated gold-catalyzed diaryl cross-coupling between aryldiazonium salts and arylboronic acids. | ||
It is particularly interesting how the gold/photoredox reaction works without the presence of a photosensitizer. Hashmi et al.26,27,37 have proposed two potential mechanisms, as shown by catalytic cycles I and II in Scheme 2. In cycle I, the reaction starts from the single electron transfer (SET) between AuI species A and aryldiazonium salt Ar1N2+, resulting in AuII species B and an aryldiazo radical Ar1N2˙. Subsequently, B captures Ar1N2˙ to form AuIII species C, which then evolves into key aryl AuIII species D by extrusion of N2 under light irradiation. An alternative pathway (cycle II) for the generation of D could be via direct addition of the aryldiazonium salt to A to form N2–AuIII–Ar1 species E, which then carries out elimination of N2 under light irradiation to afford D. Next, the transmetalation of arylboronic acid 2 to D gives diaryl Ar2–AuIII–Ar1 species F, from which the aryl–aryl reductive elimination delivers biaryl product Ar1–Ar23 and regenerates A. In these two mechanisms, the role of light irradiation is believed to be assisting extrusion of N2 from C or E (in the absence of light, the formation of a C–N bond to diazonium salts was observed under the same conditions38).
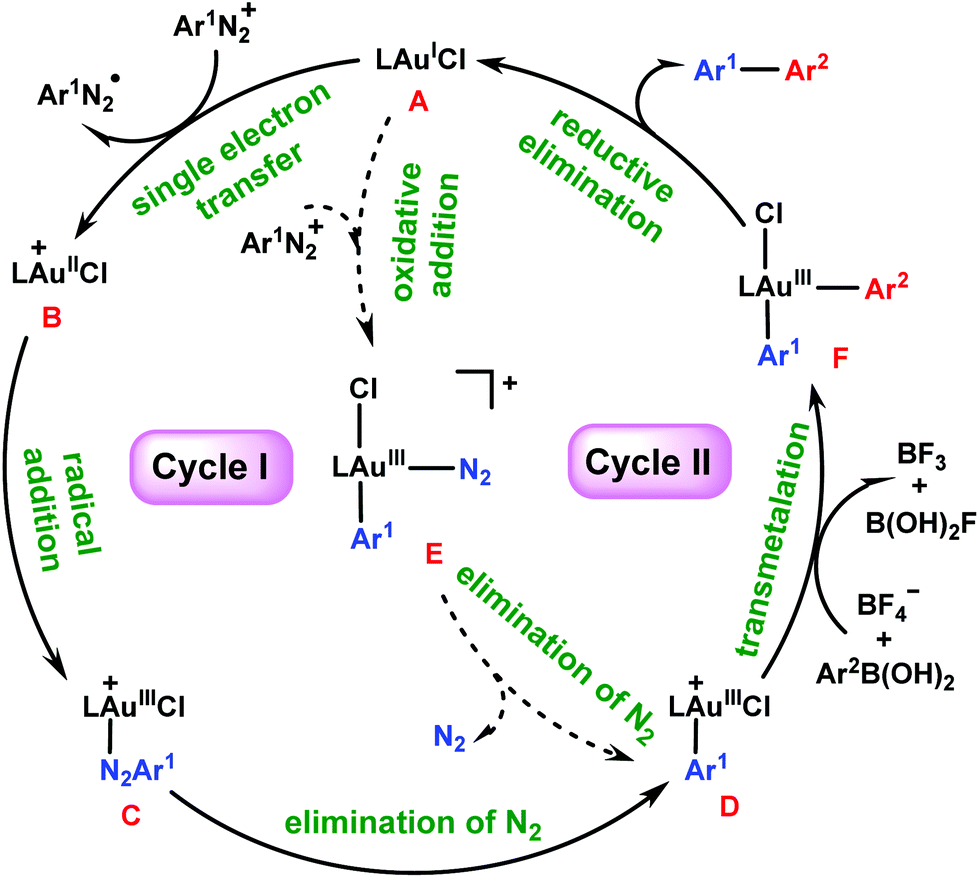 | ||
| Scheme 2 Possible mechanisms for photosensitizer-free visible-light-mediated gold-catalyzed diaryl coupling between aryldiazonium salts and arylboronic acids. | ||
Intriguingly, here, by performing density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations, we show that both of these cycles are not accessible. Alternatively, we propose a new mechanism in which the reaction requires the assistance of a virtual photoinitiator that is identified as the π–π complex between the aryldiazonium salt and the arylboronic acid, rather than either the generally accepted complex of AuI with the aryldiazonium salt or the aryldiazonium salt itself. In the newly proposed mechanism, the reaction starts from denitrification of the excited π–π complex to form a triplet aryl cation, which then performs SET with the AuI complex and the oxidation of AuI to AuIII, and the reaction proceeds through the radical addition of an aryl radical rather than an aryldiazo radical on AuII species. We hope that the theoretical results will provide some insights into future reaction protocols.
2. Results and discussion
Our attention first focuses on the catalytic cycles shown in Scheme 2. Calculations were carried out for the reaction shown in Scheme 1 with R = F. Fig. 1 shows the calculated energy profiles for the first steps of cycles I and II. The SET from (4-CF3C6H4)3PAuICl to aryldiazonium salt Ar1N2+ to give AuII and aryldiazonium radical Ar1N2˙ needs to overcome a barrier as high as 53.7 kcal mol−1 (the blue pathway in Fig. 1). Although such a high energy requirement is within the photon energy that blue LEDs can provide,39 our TD-DFT calculations show that neither (4-CF3C6H4)3PAuICl nor Ar1N2BF4 falls in the blue LED region (Table 1). Moreover, the AuIII–N2Ar1 complex C involved in this pathway cannot be successfully located as a minima due to the weak interaction in the metal–azo bond.40–42 Thus cycle I in Scheme 2 seems not to be viable.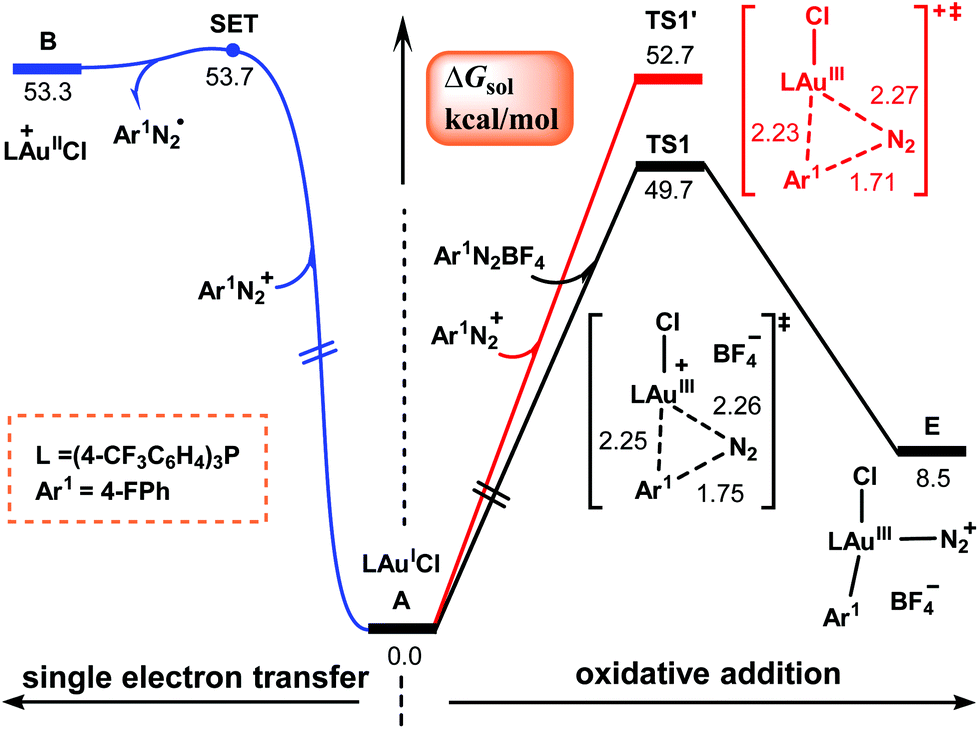 | ||
| Fig. 1 Calculated Gibbs energy profiles for the first steps of cycles I and II shown in Scheme 2. Bond distances are in Å. | ||
| Entry | Species | E ex | f | λ max | Characters |
|---|---|---|---|---|---|
| 1 | Aryldiazonium salt (1) | 4.25 | 0.0341 | 292 | H−1 → L (96.2%) |
| 2 | Arylboronic acid (2) | 4.74 | 0.0316 | 261 | H → L (84.1%) |
| 3 | (4-CH3C6H4)3PAuICl (AuI) | 4.86 | 0.1819 | 255 | H → L (90.5%) |
| 4 | MeOH | 8.21 | 0.0008 | 151 | H → L (98.9%) |
| 5 | Complex of 1 with AuI | 2.87 | 0.0147 | 432 | H → L (98.2%) |
| 6 | Complex of 1 with MeOH | 3.05 | 0.0074 | 406 | H → L (99.7%) |
| 7 | Complex of 1 with 2 | 3.05 | 0.0158 | 406 | H → L (89.0%) |
| 8 | Complex of 2 with AuI | 4.47 | 0.0128 | 277 | H → L (58.3%), H → L+1 (20.1%) |
| 9 | Complex of 2 with MeOH | 4.72 | 0.0333 | 263 | H → L (84.0%) |
| 10 | Complex of MeOH with AuI | 4.76 | 0.1521 | 261 | H → L (94.0%) |
Alternatively, according to cycle II, the reaction is initiated from the direct oxidative addition of Ar1N2+ to the AuI center to form AuIII species E. Two potential pathways (the black and red lines in Fig. 1) have been located, which proceed viaTS1 and TS1′ with and without the assistance of a tetrafluoroborate anion, respectively. The relative Gibbs energies of these two transition states were calculated to be 49.7 and 52.7 kcal mol−1, which are consistent with previous reports in the literature.42–44 However, these values are too high for a reaction that proceeds at room temperature. Therefore, the mechanism described in cycle II also seems to be problematic.
Our present calculations propose a new energetically viable mechanism, which emphasizes that the reaction proceeds via a photoinitiation mechanism where the π–π complex between the aryldiazonium salt and the arylboronic acid is identified as a virtual photoinitiator. The relevant details are discussed as follows.
2.1 Potential photoinitiators
To verify which species plays a substantial role in the light absorption as a virtual photoinitiator, we carried out TD-DFT calculations for various possible species in the present catalytic system, including four isolated components, aryldiazonium salt 1, arylboronic acid 2, AuI catalyst and MeOH solvent, as well as the various complexes between them. As shown in Table 1, the maximum absorptions of all four isolated species are beyond the range of visible light (390–780 nm);45,46 however, the maximum absorptions of the complexes of aryldiazonium salt with 2, the AuI catalyst and MeOH fall in the visible region. Therefore, only these three complexes are considered as potential photoinitiators under blue light irradiation.31,43,47 The calculated excitation–decay energy profiles of three complexes are shown in Fig. 2, S1 and S2 in the ESI,† where the complexes can be excited vertically under irradiation with blue LEDs to their respective lowest singlet excited states, which then decay to the corresponding lowest triplet states via intersystem crossing (ISC).48 Examination of orbital contributions indicates that the excitation of all three complexes corresponds to a HOMO–LUMO electron transition to form the excited triplet charge transfer complexes (CTCs). Which of these three complexes is the most likely photoinitiator? To answer this question, we studied the subsequent reactions of the excited triplet CTCs, which can be quenched either oxidatively or reductively via SET with the redox-active components in the system,45,49 resulting in the generation of the aryl radical followed by the oxidation of AuI species.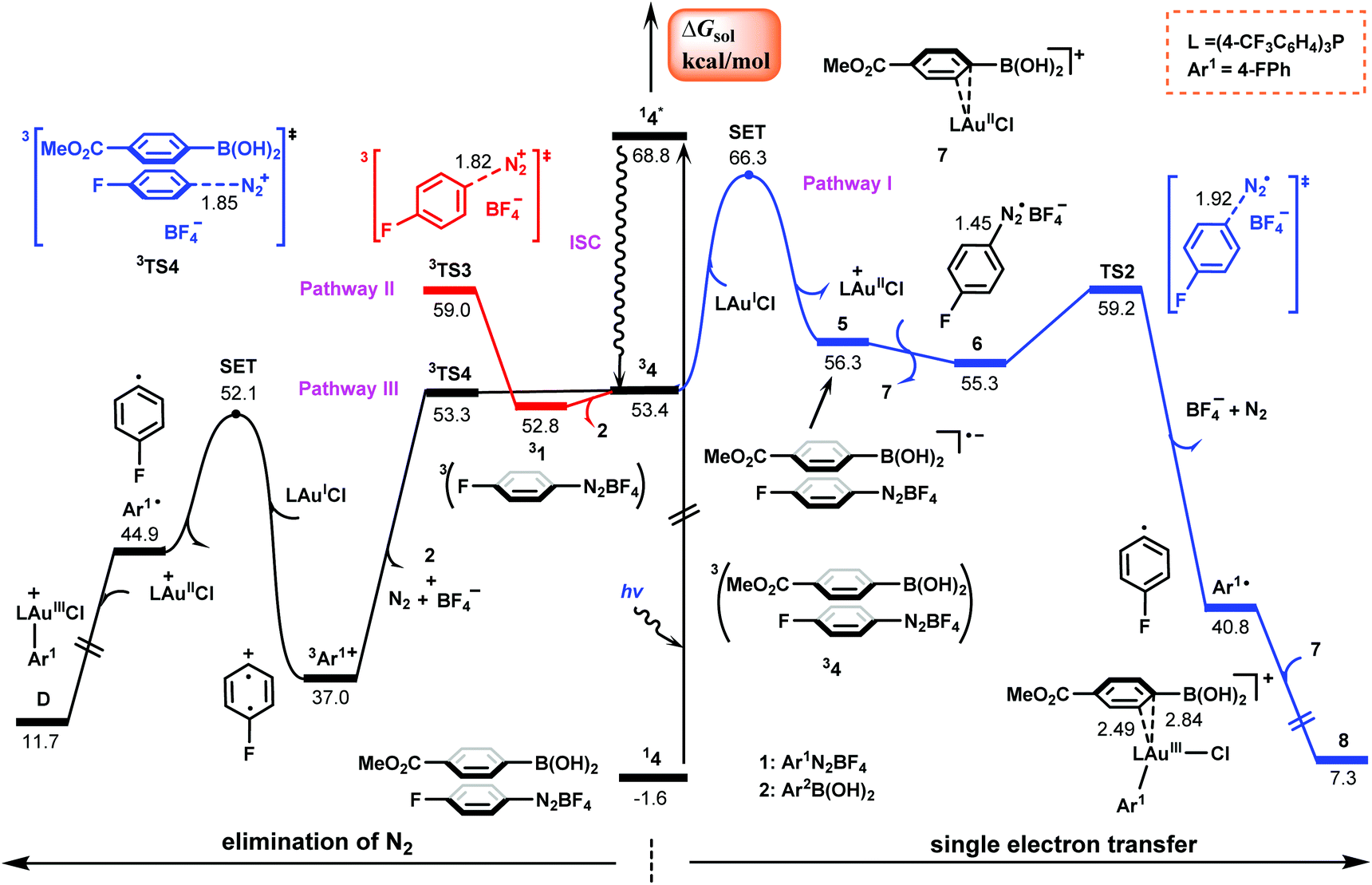 | ||
| Fig. 2 Calculated Gibbs energy profiles of the generation of aryl radical Ar1˙ (Ar1 = 4-FPh) and the oxidation of gold(I) using the complex of the aryldiazonium salt with the arylboronic acid as a photoinitiator. Bond distances are in Å. | ||
2.2 Generation of the aryl radical followed by oxidation of AuI species
In the newly proposed mechanism, the generation of the aryl radical is a key process in the present gold/photoredox catalysis. Based on the three excited triplet complexes of the aryldiazonium salt (Table 1), we studied various possible pathways for the generation of the aryl radical, and Fig. 2 summarizes the energetically most favorable one which originates from the complex of the aryldiazonium salt with the arylboronic acid. Those originating from the other two complexes are given in Fig. S1 and S2† for simplification.As shown in Fig. 2, from the triplet complex between the aryldiazonium salt and arylboronic acid, 34, there are three possible pathways that may yield aryl radicals.43,50 Pathway I starts from the SET of the AuI catalyst to 34, giving the reduced state complex 5 and AuII species with an energy barrier of 12.9 kcal mol−1 calculated according to the Marcus–Hush theory.51,52 This process is determined to be endothermic by 2.9 kcal mol−1. Subsequently, 5 undergoes ligand exchange with the AuII species, leading to the anionic aryldiazonium salt radical 6 and the complex of AuII with the arylboronic acid complex, 7. By carrying out elimination of N2, 6 affords the aryl radical Ar1˙viaTS2 with 5.8 kcal mol−1. Finally, the aryl radical preferentially associates the AuII species 7 with an unpaired electron to form AuIII species 8, and this process is determined to be exothermic by 33.5 kcal mol−1.
Alternatively, pathway II occurs via decomposition of 34 into singlet state arylboronic acid and triplet aryldiazonium salt, which then performs the elimination of N2via3TS3 with a barrier of 6.2 kcal mol−1. A third pathway initiates from the direct elimination of N2 from 34via3TS4 and this process is identified as a barrierless process with an exothermicity of 16.4 kcal mol−1, generating a triplet aryl cation 3Ar1+, which then performs SET with the AuI catalyst to obtain Ar1˙ and AuII species. This barrier is estimated to be 15.1 kcal mol−1. In comparison with pathways I and II, pathway III is the most energetically favorable for the generation of Ar1˙.
The generation of Ar1˙ from the excited complex of the aryldiazonium salt with the AuI catalyst (Fig. S1†) cannot compete with that from the excited complex of the aryldiazonium salt with the arylboronic acid (Fig. 2) due to the relative higher energy requirement (12.1 kcal mol−1 for N2 extrusion and 15.1 kcal mol−1 for SET, respectively, Fig. S1†). It should be noted that the calculated barrier for N2 extrusion, 12.1 kcal mol−1, is comparable with that calculated for the Au-catalyzed 1,2-difunctionalization of alkynes.42 In addition, it was found that the complex between the aryldiazonium salt and MeOH solvent could also act as a photoinitiator, as indicated by the calculated low barrier (6.5 kcal mol−1) for the elimination of N2 from its excited triplet complex (Fig. S2†). From an energy point of view, the π–π complex between the two reactants most likely acts as a photoinitiator, which carries out the π → π* (HOMO–LUMO) transition upon vertical excitation under blue LED irradiation (Fig. S3†) to induce the generation of Ar1˙ and AuII. Once formed, Ar1˙ can immediately perform oxidative addition on the in situ formed AuII complex, resulting in AuIII complex D.
2.3 Transmetalation and reductive elimination
Fig. 3 shows the calculated results for the subsequent reaction of the AuIII–Ar1 species with arylboronic acid, which delivers the final diaryl cross-coupling product via two elementary steps: transmetalation and reductive elimination.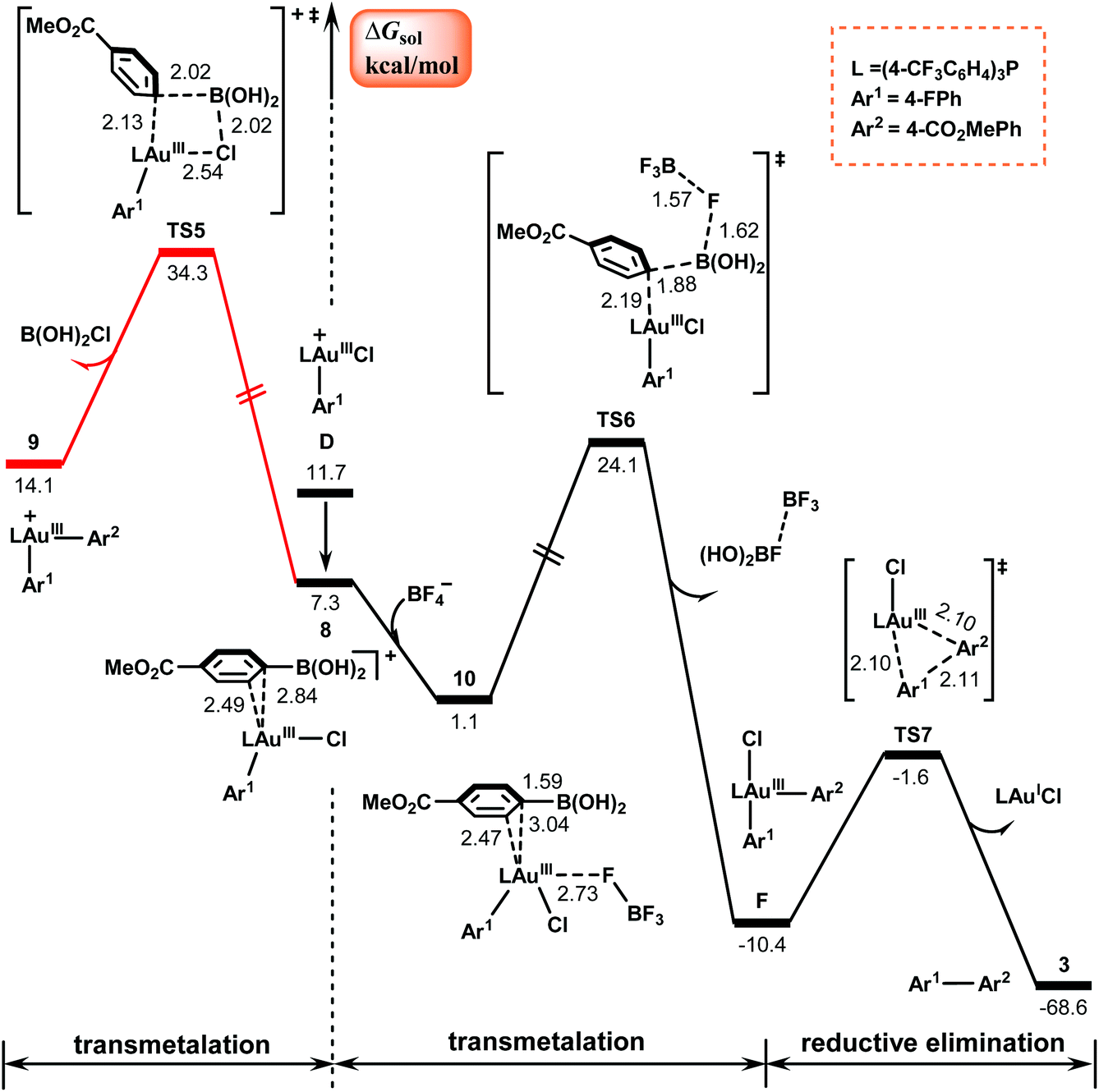 | ||
| Fig. 3 Calculated Gibbs energy profiles for the transmetalation and the subsequent reductive elimination to form the final product. Bond distances are in Å. | ||
The left Gibbs energy profile in Fig. 3 describes a direct transmetalation pathway of the arylboronic acid to the cationic π-coordinated AuIII complex 8. This pathway involves the rupture of the Au–Cl bond viaTS5, a four-centered transition state, and involves a barrier of 34.3 kcal mol−1, resulting in cationic diaryl AuIII species 9 with the release of B(OH)2Cl. The barrier is too high for a reaction at room temperature. The high energy requirement is attributed to the high energy requirement for breaking the strong Au–Cl bond.53
Following the experimental observation that the presence of the BF4− counterion remarkably improves the yield, we calculated the BF4−-assisted transmetalation process, as shown by the right black energy profile in Fig. 3. The transition state involved is denoted as TS6, where as indicated by the calculated bond distances, a three-center–two-electron B–F–B bond is formed via the interaction between the negatively charged fluorine atom in BF4− and the positively charged boron atom in the arylboronic acid. In the presence of the BF4− counterion, the transmetalation process was determined to be exothermic by 11.5 kcal mol−1 and the barrier of the reaction was reduced to 24.1 kcal mol−1 in comparison with the reaction without the assistance of the BF4− counterion. These calculated results are consistent with the previous studies by Toste et al.,54,55 who substantiated the fluorine-assisted transfer of aryl groups from boron to gold.
The BF4−-assisted transmetalation results in the neutral diaryl AuIII species F, which then undergoes reductive elimination viaTS7 to obtain the desired C(sp2)–C(sp2) cross-coupling products 3 and regenerate the AuI catalyst. The barrier of this process is estimated to be only 8.8 kcal mol−1, indicating that the reductive elimination is facile.
Based on the above results, we summarize the mechanism in Scheme 3. In this mechanism, the π–π complex 1G between aryldiazonium salt 1 and arylboronic acid 2 plays the role of photoinitiator. Under blue LED irradiation, the complex 1G is excited to its triplet excited state 3G, which then undergoes elimination of N2 and SET with gold(I) catalyst A to give the cationic gold(II) species B and aryl radical Ar1˙. Subsequently, the addition of Ar1˙ to B results in the key AuIII–Ar1 intermediate D, from which the biaryl Ar1–AuIII–Ar2 complex F can be obtained via transmelalation with the assistance of a BF4− anion. Finally, F undergoes reductive elimination to release the diaryl cross-coupling product 3 and regenerate the AuI species A. From the present calculations, it is clear that the transmetalation process with an energy barrier of 24.1 kcal mol−1 can be identified as the rate-determining step of the reaction and the overall reaction is estimated to be exothermic by 68.6 kcal mol−1.
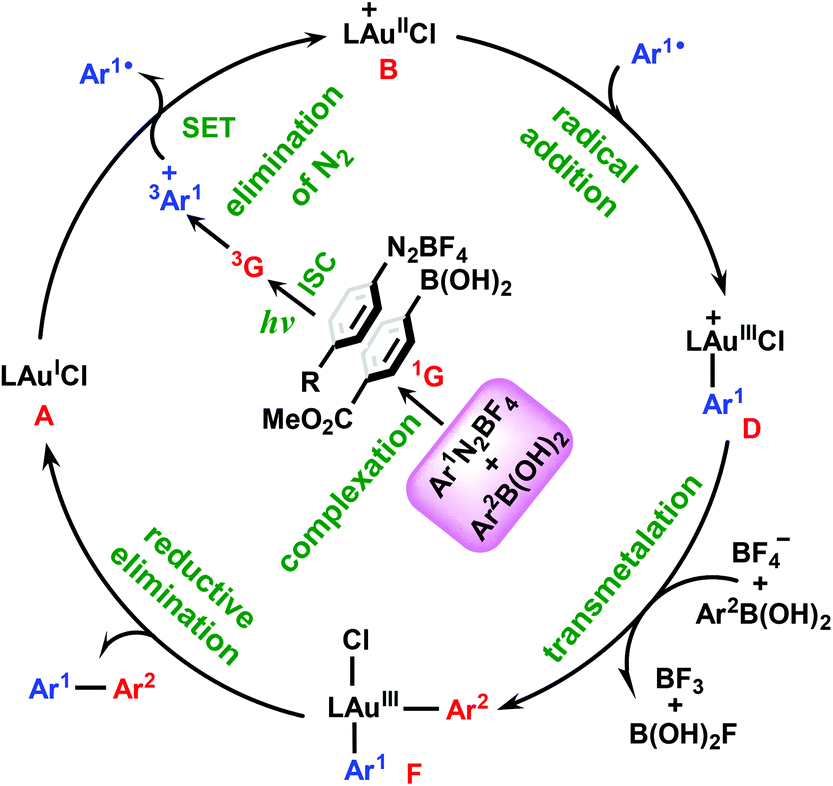 | ||
| Scheme 3 Newly proposed mechanism. | ||
2.4 Substituent effect
According to Hashmi et al.,26,27 the substituent on the aryldiazonium salt significantly affects the reactivity: electron-poor aryldiazonium salts afford the biaryl products in high yields (77–84%), while electron-rich aryldiazonium salts result in the product in low yields (∼30%). To elucidate the effect of the different substituent groups on the reactivity, we carried out calculations for the reactions of two other representative aryldiazonium salts that bear the electron-withdrawing CF3 group and the electron-donating OMe group, respectively. The calculated results of two key steps, generation of an aryl radical and transmetalation, are shown in Fig. 4, and the results of the reaction with the F substituent discussed above are also given for comparison. The calculated barriers of these two steps are 8.5 and 19.7 kcal mol−1 for the CF3-substituted aryldiazonium salt (1CF3), 15.1 and 24.1 kcal mol−1 for the F-substituted aryldiazonium salt (1F), and 26.2 and 27.4 kcal mol−1 for the OMe-substituted aryldiazonium salt (1OMe). Clearly, the electron-poor aryldiazonium salt is significantly more favorable for both the elementary steps than the electron-rich aryldiazonium salt, which is in good agreement with experimental observation. The much lower energy requirement of the SET process in the CF3-substitutent-involving reaction is due to the strong electron-withdrawing property of the CF3 group, which makes the C atom in the para-position of CF3 substituent more positively charged in comparison with F and OMe substitutents and hence more favorable for the oxidation of the AuI species (Fig. 4a). On the other hand, the lower barrier of the transmetalation in the CF3-substitutent-involving reaction (Fig. 4b) can be attributed to the stronger electrostatic interaction between the Au center and the C2 atom, making the corresponding transition state (TS8) more stable than those in the reactions involving F and OMe substituents (TS6 and TS9). Additionally, we also performed calculations for the rate-determining step (transmetalation) of the systems involving boronic acids with electron-donating substituents (OMe and NMe2) and the results are shown in Fig. S6.† For these two systems, the calculated barriers of the transmetalation are 32.0 and 31.8 kcal mol−1, which are clearly much higher than those for the systems involving boronic acids with electron-withdrawing substituents, such as COOMe in Fig. 4. This can be attributed to the weaker electrostatic interaction between the Au center and the C2 atom induced by the electron-donating substituents, making the transmetalation process less favorable. These calculated results are consistent with the experimental observation that boronic acids bearing electron-withdrawing substituents appear to react more efficiently than those bearing electron-donating substituents.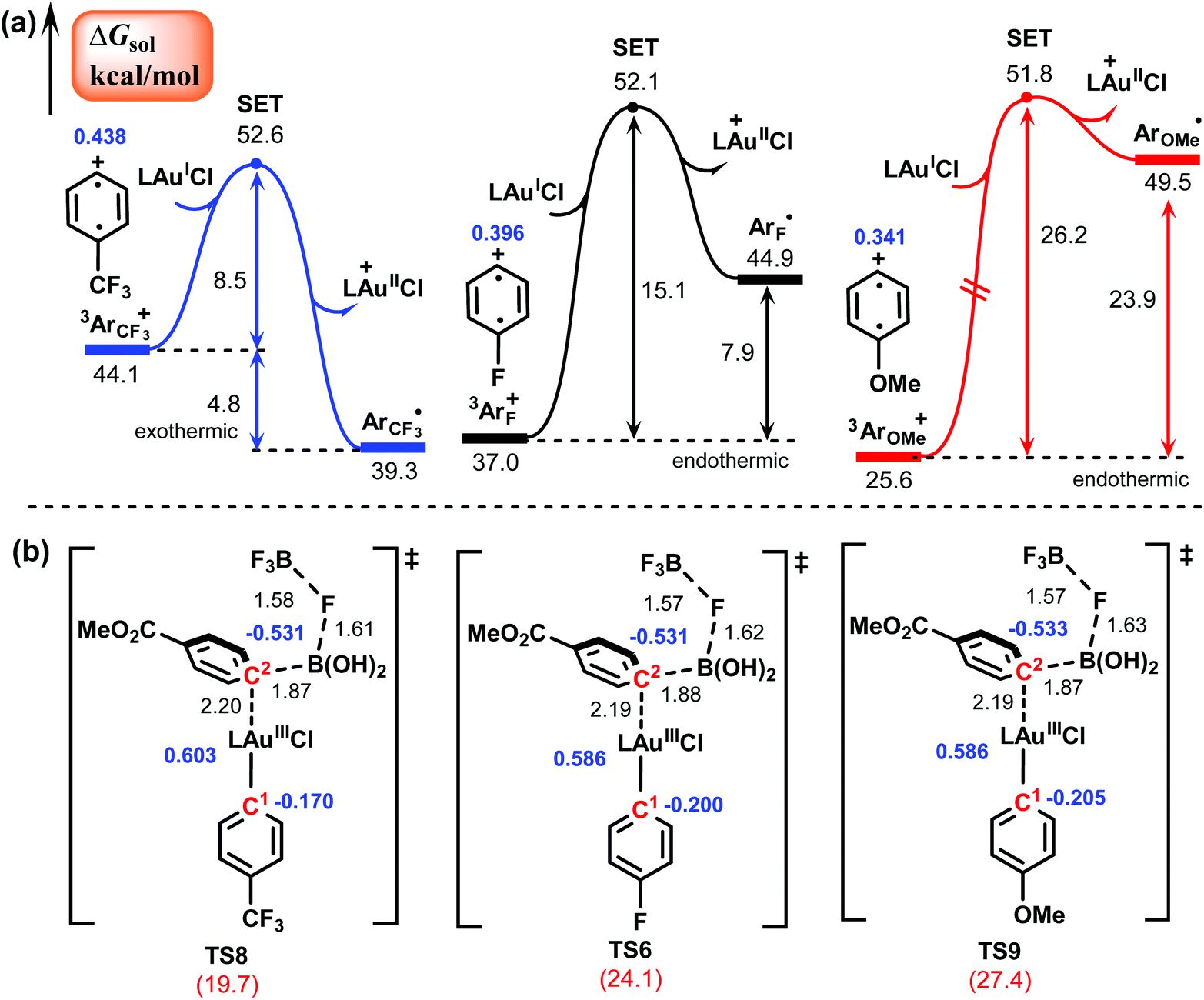 | ||
| Fig. 4 Generation of an aryl radical (a) and transmetalation transition states (b) involved in the reactions of aryldiazonium salts bearing –CF3, –F, and –OMe groups, respectively. Gibbs free energy barriers (red), bond distances (black) and NPA charges (blue) are in kcal mol−1, Å and e, respectively. | ||
3. Conclusions
In summary, by performing systematic DFT and TD-DFT calculations we proposed a new mechanism for the photosensitizer-free visible-light-mediated gold-catalyzed cross-coupling between aryldiazonium salts and arylboronic acids. In this new mechanism, the π–π complex between the aryldiazonium salt and the arylboronic acid is indicated to act as a photoinitiator to generate the key aryl radical and the AuIII intermediate. The rate-determining step of the reaction is the transmetalation of the arylboronic acid with the assistance of a tetrafluoroborate anion to the AuIII center. The calculated substituent effect of aryldiazonium salts and arylboronic acids on the reactivity is consistent with the experimental observation. The theoretical results provide new understanding for the reaction under study.4. Computational details
The reaction shown in Scheme 1 was chosen as the prototype reaction of the photosensitizer-free gold/photoredox-catalyzed diaryl cross-coupling between arylboronic acids and aryldiazonium salts reported by Hashmi et al.26,27,37 All DFT and TD-DFT calculations were carried out using the Gaussian 16 program.56 Geometry optimizations of stationary points were performed using the M06 suite57 of meta-GGA functionals with the spin-restricted method for closed-shell species and the spin-unrestricted method for open-shell species. The basis set used was the LANL2DZ effective core potential (ECP) basis set58 augmented by an f-type polarization function (ζf = 1.050)59 for gold atoms and 6-31G(d,p)60 for other atoms. The solvent effect of methanol was taken into account using the polarizable continuum model (PCM)61 in the geometry optimization calculations. Frequency analyses were performed at the same level of theory to confirm the optimized structures to be local minima (with zero imaginary frequency) or first-order saddle points (with one imaginary frequency). The intrinsic reaction coordinate (IRC)62 calculations were also performed to ensure that the transition states connected the reactant or product properly. All reported energies of intermediates and transition states were solvated Gibbs energies. The excitation energies and absorption wavelengths of several species were obtained by performing TD-DFT calculations63,64 at the M06/LANL2DZ(f)/6-31G(d,p) level with the PCM solvation effect of methanol solvent. Natural population analyses (NPA)65 were carried out using the NBO 3.1 program as implemented in the Gaussian 16 package.Author contributions
Yanhong Liu performed the calculations and wrote the original draft; Rongxiu Zhu analyzed the results, Chengbu Liu supervised this work and Dongju Zhang wrote and reviewed this paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21773139 and 21833004).References
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, C−H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed.
- J. Wencel-Delord, C. Nimphius, F. W. Patureau and F. Glorius, [RhIIICp*]-Catalyzed Dehydrogenative Aryl–Aryl Bond Formation, Angew. Chem., Int. Ed., 2012, 51, 2247–2251 CrossRef CAS PubMed.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Gold-Catalyzed Direct Arylation, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed.
- L. Zhang, L. Zhu, Y. Zhang, Y. Yang, Y. Wu, W. Ma, Y. Lan and J. You, Experimental and Theoretical Studies on Ru(II)-Catalyzed Oxidative C−H/C−H Coupling of Phenols with Aromatic Amides Using Air as Oxidant: Scope, Synthetic Applications, and Mechanistic Insights, ACS Catal., 2018, 8, 8324–8335 CrossRef CAS.
- A. H. Cherney, N. T. Kadunce and S. E. Reisman, Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C−C Bonds, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- P. Hu, Y. Shang and W. Su, A General Pd-Catalyzed Decarboxylative Cross-Coupling Reaction between Aryl Carboxylic Acids: Synthesis of Biaryl Compounds, Angew. Chem., Int. Ed., 2012, 51, 5945–5949 CrossRef CAS PubMed.
- J. Yu, J. Liu, G. Shi, C. Shao and Y. Zhang, Ligand-Promoted Oxidative Cross-Coupling of Aryl Boronic Acids and Aryl Silanes by Palladium Catalysis, Angew. Chem., Int. Ed., 2015, 54, 4079–4082 CrossRef CAS.
- S. G. Bratsch, Standard Electrode Potentials and Temperature Coefficients in Water at 298.15 K, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- M. N. Hopkinson, A. D. Gee and V. Gouverneur, AuI/AuIII Catalysis: An Alternative Approach for C–C Oxidative Coupling, Chem. – Eur. J., 2011, 17, 8248–8262 CrossRef CAS.
- G. Zhang, L. Cui, Y. Wang and L. Zhang, Homogeneous Gold-Catalyzed Oxidative Carboheterofunctionalization of Alkenes, J. Am. Chem. Soc., 2010, 132, 1474–1475 CrossRef CAS.
- S. Witzel, A. S. K. Hashmi and J. Xie, Light in Gold Catalysis, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS.
- M. Joost, A. Zeineddine, L. Estévez, S. Mallet−Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Facile Oxidative Addition of Aryl Iodides to Gold(I) by Ligand Design: Bending Turns on Reactivity, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS.
- E. O. Asomoza-Solís, J. Rojas-Ocampo, R. A. Toscano and S. Porcel, Arenediazonium Salts as Electrophiles for the Oxidative Addition of Gold(I), Chem. Commun., 2016, 52, 7295–7298 RSC.
- I. Medina-Mercado, E. O. Asomoza-Solís, E. Martínez-González, V. M. Ugalde-Saldívar, L. G. Ledesma-Olvera, J. E. Barquera-Lozada, V. Gómez-Vidales, J. Barroso-Flores, B. A. Frontana-Uribe and S. Porcel, Ascorbic Acid as an Aryl Radical Inducer in the Gold-Mediated Arylation of Indoles with Aryldiazonium Chlorides, Chem. – Eur. J., 2020, 26, 634–642 CrossRef CAS.
- U. A. Carrillo-Arcos and S. Porcel, Gold Promoted Arylative Cyclization of Alkynoic Acids with Arenediazonium Salts, Org. Biomol. Chem., 2018, 16, 1837–1842 RSC.
- B. Sahoo, M. N. Hopkinson and F. Glorius, Combining Gold and Photoredox Catalysis: Visible Light-Mediated Oxy- and Aminoarylation of Alkenes, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed.
- A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo and F. Glorius, Dual Gold/Photoredox-Catalyzed C(sp)–H Arylation of Terminal Alkynes with Diazonium Salts, Chem. Sci., 2016, 7, 89–93 RSC.
- X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, Dual Visible Light Photoredox and Gold-Catalyzed Arylative Ring Expansion, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS PubMed.
- S. Kim, J. Rojas-Martin and F. D. Toste, Visible Light-Mediated Gold-Catalysed Carbon(sp2)–Carbon(sp) Cross-Coupling, Chem. Sci., 2016, 7, 85–88 RSC.
- Z. Xia, V. Corcé, F. Zhao, C. Przybylski, A. Espagne, L. Jullien, T. Le Saux, Y. Gimbert, H. Dossmann, V. Mouriès-Mansuy, C. Ollivier and L. Fensterbank, Photosensitized Oxidative Addition to Gold(I) Enables Alkynylative Cyclization of o-Alkylnylphenols with Iodoalkynes, Nat. Chem., 2019, 11, 797–805 CrossRef CAS PubMed.
- I. Medina-Mercado and S. Porcel, Insights into the Mechanism of Gold(I) Oxidation with Aryldiazonium Salts, Chem. – Eur. J., 2020, 26, 16206–16221 CrossRef CAS.
- V. Gauchot and A.-L. Lee, Dual Gold Photoredox C(sp2)–C(sp2) Cross Couplings – Development and Mechanistic Studies, Chem. Commun., 2016, 52, 10163–10166 RSC.
- T. Cornilleau, P. Hermange and E. Fouquet, Gold-Catalysed Cross-Coupling between Aryldiazonium Salts and Arylboronic Acids: Probing the Usefulness of Photoredox Conditions, Chem. Commun., 2016, 52, 10040–10043 RSC.
- S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, New Transmetalation Reagents for the Gold-Catalyzed Visible Light Enabled C(sp or sp2)−C(sp2) Cross-Coupling with Aryldiazonium Salts in the Absence of a Photosensitizer, Chem. Commun., 2018, 54, 13802–13804 RSC.
- J. Xie, K. Sekine, S. Witzel, P. Krämer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Light-Induced Gold-Catalyzed Hiyama Arylation: A Coupling Access to Biarylboronates, Angew. Chem., Int. Ed., 2018, 57, 16648–16653 CrossRef CAS.
- S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Photosensitizer-Free, Gold-Catalyzed C–C Cross-Coupling of Boronic Acids and Diazonium Salts Enabled by Visible Light, Adv. Synth. Catal., 2017, 359, 1522–1528 CrossRef CAS.
- C. Sauer, Y. Liu, A. De Nisi, S. Protti, M. Fagnoni and M. Bandini, Photocatalyst-Free, Visible Light Driven, Gold Promoted Suzuki Synthesis of (Hetero)biaryls, ChemCatChem, 2017, 9, 4456–4459 CrossRef CAS.
- M. O. Akram, P. S. Mali and N. T. Patil, Cross-Coupling Reactions of Aryldiazonium Salts with Allylsilanes under Merged Gold/Visible-Light Photoredox Catalysis, Org. Lett., 2017, 19, 3075–3078 CrossRef CAS.
- I. Chakrabarty, M. O. Akram, S. Biswas and N. T. Patil, Visible Light Mediated Desilylative C(sp2)–C(sp2) Cross-Coupling Reactions of Arylsilanes with Aryldiazonium Salts under Au(I)/Au(III) Catalysis, Chem. Commun., 2018, 54, 7223–7226 RSC.
- J.-R. Deng, W.-C. Chan, N. C.-H. Lai, B. Yang, C.-S. Tsang, B. C.-B. Ko, S. L.-F. Chan and M.-K. Wong, Photosensitizer-Free Visible Light-Mediated Gold-Catalysed cis-Difunctionalization of Silyl-Substituted Alkynes, Chem. Sci., 2017, 8, 7537–7544 RSC.
- J. G. Mayans, J.-S. Suppo and A. M. Echavarren, Photoredox-Assisted Gold-Catalyzed Arylative Alkoxycyclization of 1,6-Enynes, Org. Lett., 2020, 22, 3045–3049 CrossRef CAS PubMed.
- A. Tabey, M. Berlande, P. Hermange and E. Fouquet, Mechanistic and Asymmetric Investigations of the Au-Catalysed Cross-Coupling between Aryldiazonium Salts and Arylboronic Acids Using (P,N) Gold Complexes, Chem. Commun., 2018, 54, 12867–12870 RSC.
- F. Mo, D. Qiu, L. Zhang and J. Wang, Recent Development of Aryl Diazonium Chemistry for the Derivatization of Aromatic Compounds, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed.
- F.-X. Felpin and S. Sengupta, Biaryl Synthesis with Arenediazonium Salts: Cross-coupling, CH-Arylation and Annulation Reactions, Chem. Soc. Rev., 2019, 48, 1150–1193 RSC.
- S. R. Dubbaka and P. Vogel, Organosulfur Compounds: Electrophilic Reagents in Transition-Metal-Catalyzed Carbon–Carbon Bond-Forming Reactions, Angew. Chem., Int. Ed., 2005, 44, 7674–7684 CrossRef CAS PubMed.
- L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Photosensitizer-Free Visible-Light-Mediated Gold-Catalyzed 1,2-Difunctionalization of Alkynes, Angew. Chem., Int. Ed., 2016, 55, 4808–4813 CrossRef CAS.
- S. Taschinski, R. Döpp, M. Ackermann, F. Rominger, F. de Vries, M. F. S. J. Menger, M. Rudolph, A. S. K. Hashmi and J. E. M. N. Klein, Light-Induced Mechanistic Divergence in Gold(I) Catalysis: Revisiting the Reactivity of Diazonium Salts, Angew. Chem., Int. Ed., 2019, 58, 16988–16993 CrossRef CAS.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- J. G. Taylor, A. V. Moro and C. R. D. Correia, Evolution and Synthetic Applications of the Heck–Matsuda Reaction: The Return of Arenediazonium Salts to Prominence, Eur. J. Org. Chem., 2011, 1403–1428 CrossRef CAS.
- B. Schmidt, N. Elizarov, N. Riemer and F. Hölter, Acetamidoarenediazonium Salts: Opportunities for Multiple Arene Functionalization, Eur. J. Org. Chem., 2015, 5826–5841 CrossRef CAS.
- Y. Liu, Y. Yang, R. Zhu, C. Liu and D. Zhang, The Dual Role of Gold(I) Complexes in Photosensitizer-Free Visible-Light-Mediated Gold-Catalyzed 1,2-Difunctionalization of Alkynes: A DFT Study, Chem. – Eur. J., 2018, 24, 14119–14126 CrossRef CAS.
- Y. Liu, Y. Yang, R. Zhu, C. Liu and D. Zhang, DFT Study on Photosensitizer-Free Visible-Light-Mediated Au-Catalyzed cis-Difunctionalization of Alkynes: Mechanism and Selectivities as Compared to Rh Catalysis, J. Org. Chem., 2019, 84, 16171–16182 CrossRef CAS PubMed.
- I. A. Sanhueza, F. J. R. Klauck, E. Senol, S. T. Keaveney, T. Sperger and F. Schoenebeck, Base-Free Cross-Couplings of Aryl Diazonium Salts in Methanol: PdII–Alkoxy as Reactivity-Controlling Intermediate, Angew. Chem., Int. Ed., 2021, 60, 7007–7012 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- H. Ren, G.-F. Li, B. Zhu, X.-D. Lv, L.-S. Yao, X.-L. Wang, Z.-M. Su and W. Guan, How Does Iridium(III) Photocatalyst Regulate Nickel(II) Catalyst in Metallaphotoredox-Catalyzed C−S Cross-Coupling? Theoretical and Experimental Insights, ACS Catal., 2019, 9, 3858–3865 CrossRef CAS.
- S. Witzel, M. Hoffmann, M. Rudolph, M. Kerscher, P. Comba, A. Dreuw and A. S. K. Hashmi, Excitation of Aryl Cations as the Key to Catalyst-Free Radical Arylations, Cell Rep. Phys. Sci., 2021, 2, 100325 CrossRef CAS.
- P. Xiao, C.-X. Li, W.-H. Fang, G. Cui and W. Thiel, Mechanism of the Visible-Light-Mediated Copper-Catalyzed Coupling Reaction of Phenols and Alkynes, J. Am. Chem. Soc., 2018, 140, 15099–15113 CrossRef CAS.
- K. Kalyanasundaram, Photophysics, Photochemistry and Solar Energy Conversion with Tris(bipyridyl)ruthenium(II) and its Analogues, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- G. J. Sherborne, A. G. Gevondian, I. Funes-Ardoiz, A. Dahiya, C. Fricke and F. Schoenebeck, Modular and Selective Arylation of Aryl Germanes (C−GeEt3) over C−Bpin, C−SiR3 and Halogens Enabled by Light-Activated Gold Catalysis, Angew. Chem., Int. Ed., 2020, 59, 15543–15548 CrossRef CAS.
- R. A. Marcus, On the Theory of Electrochemical and Chemical Electron Transfer Processed, Can. J. Chem., 1959, 37, 155–163 CrossRef CAS.
- R. A. Marcus and N. Sutin, Electron Transfers in Chemistry and Biology, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- A. Kempter, C. Gemel and R. A. Fischer, Insertion of Ga(DDP) into the Au−Cl Bond of (PPh3)AuCl: A First Structurally Characterized Au−Ga Bond, Inorg. Chem., 2005, 44, 163–165 CrossRef CAS PubMed.
- N. P. Mankad and F. D. Toste, C−C Coupling Reactivity of an Alkylgold(III) Fluoride Complex with Arylboronic Acids, J. Am. Chem. Soc., 2010, 132, 12859–12861 CrossRef CAS PubMed.
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard III and F. D. Toste, Two Metals Are Better Than One in the Gold Catalyzed Oxidative Heteroarylation of Alkenes, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- P. J. Hay and W. R. Wadt, Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, A Set of f-Polarization Functions for Pseudo-Potential Basis Sets of the Transition Metals Sc-Cu, Y-Ag and La-Au, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- S. Huzinaga, Basis Sets for Molecular Calculations, Comput. Phys. Rep., 1985, 2, 281–339 CrossRef.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- K. Fukui, The Path of Chemical Reactions−The IRC Approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, An Efficient Implementation of Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational results and Cartesian coordinates of all optimized structures located in this work (PDF). See DOI: 10.1039/d1qo01464a |
| This journal is © the Partner Organisations 2022 |