
Transition-metal-free hydroamination/defluorination/cyclization of perfluoroalkyl alkynes with amidines†
Li-Wen
Sun
a,
Zi-Lun
Yu
a,
Xin-Long
Luo
a,
Mengtao
Ma
 b,
Zhi-Liang
Shen
*a and
Xue-Qiang
Chu
*a
b,
Zhi-Liang
Shen
*a and
Xue-Qiang
Chu
*a
aInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: ias_zlshen@njtech.edu.cn; xueqiangchu@njtech.edu.cn
bDepartment of Chemistry and Materials Science, College of Science, Nanjing Forestry University, Nanjing 210037, China
First published on 16th November 2021
Abstract
An efficient defluorinative net-[3 + 3]-cyclization strategy for the construction of perfluoroalkyl-substituted pyrimidine derivatives by using a series of perfluoroalkyl alkynes and amidines as starting materials was developed. The present reaction proceeded successfully under transition-metal-free conditions to form two new C–N bonds and a new heterocyclic ring through a sequence of hydroamination, defluorination, and annulation. The desired pyrimidines could be obtained with good functional group tolerance and moderate to good yields. Moreover, the distinctive fluorine effects of perfluoroalkyl substituents are vital for tuning the reactivity of alkynes for the anticipated defluorinative annulation. The pendant π system would lower associated bond dissociation energy significantly compared to that of a nonactivated C(sp3)–F bond.
Introduction
Organofluorine compounds are of special importance in fields ranging from modern agrochemistry, drug discovery, clinical medicines, and materials science to organic synthesis due to the unique properties of fluorine.1 It has been well recognized that the introduction of fluorine atoms or higher homologue fluoroalkyl groups (Rf) into organic molecules can lead to dramatic changes in physical and chemical properties.2 During the past decades, considerable progress has been accomplished in trifluoromethylation using various CF3 reagents. However, its bulkier analogue, the perfluoroalkyl (Rf) group, has attracted vastly less attention, although many biologically and pharmaceutically active molecules, such as anticancer drug (Faslodex), fungicide (ZJ5337), and fungicide (Nicofluprole), contain this privileged subunit (Fig. 1).3 Additionally, highly substituted pyrimidines represent one of the most varied and largest families of N-heterocycles, which are associated with interesting activities such as PI3K inhibitor (NVP-BKM 120), antidiabetic activity (Gemigliptin), and fungicide (Fluoxastrobin).4 In this context, the development of practical and reliable approaches for the facile incorporation of both perfluoroalkyl group and pyrimidine structural motif in one molecule is urgently demanded, especially starting from readily available starting materials under mild reaction conditions.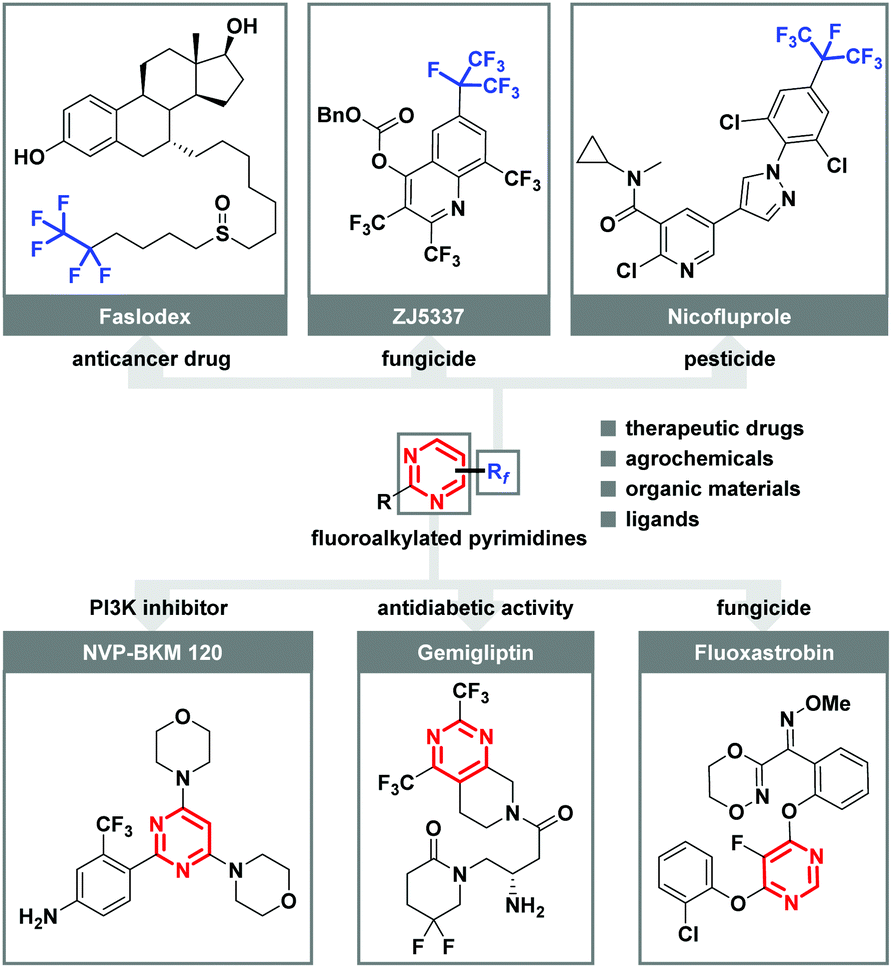 | ||
| Fig. 1 Representative biologically active perfluoroalkylated molecules and pyrimidines. | ||
Amidine, as a dual nucleophile bearing two nitrogen atoms, has been extensively used as a versatile building block in the preparation of diversified N-heterocyclic compounds.5 Considerable advancements pertaining to transition metal-catalyzed or metal-free condensation/oxidation cascade of amidines with 1,3-dicarbonyl derivatives,6 α,β-unsaturated ketones/aldehyde,7 propiophenones,8 allylbenzene,9 propargylic alcohols,10 or propargylic amines,11 have been made (Scheme 1, 1). Alternatively, lots of efforts have been spent on the late-stage modification of pre-existing pyrimidine counterparts through direct C–H bond functionalization by employing extra Rf-source (Scheme 1, 2a).12 However, these transformations generally suffered from the requirement of heterocyclic precursors, expensive metal catalysts, and harsh reaction conditions. In particular, the site-selectivity of perfluoroalkylation is predominantly dictated by the innate reactivity of the substrate. Recently, we found that multi-component cyclization through sequential fluoroalkylation, defluorination, and heteroannulation could provide chemists an efficient strategy to prepare perfluoroalkyl substituted pyrimidines (Scheme 1, 2b).13 In these cases, the in situ formed α-perfluoroalkylated ketones served as the key intermediates. However, transition metal-catalyst or photocatalyst were essential for the success.
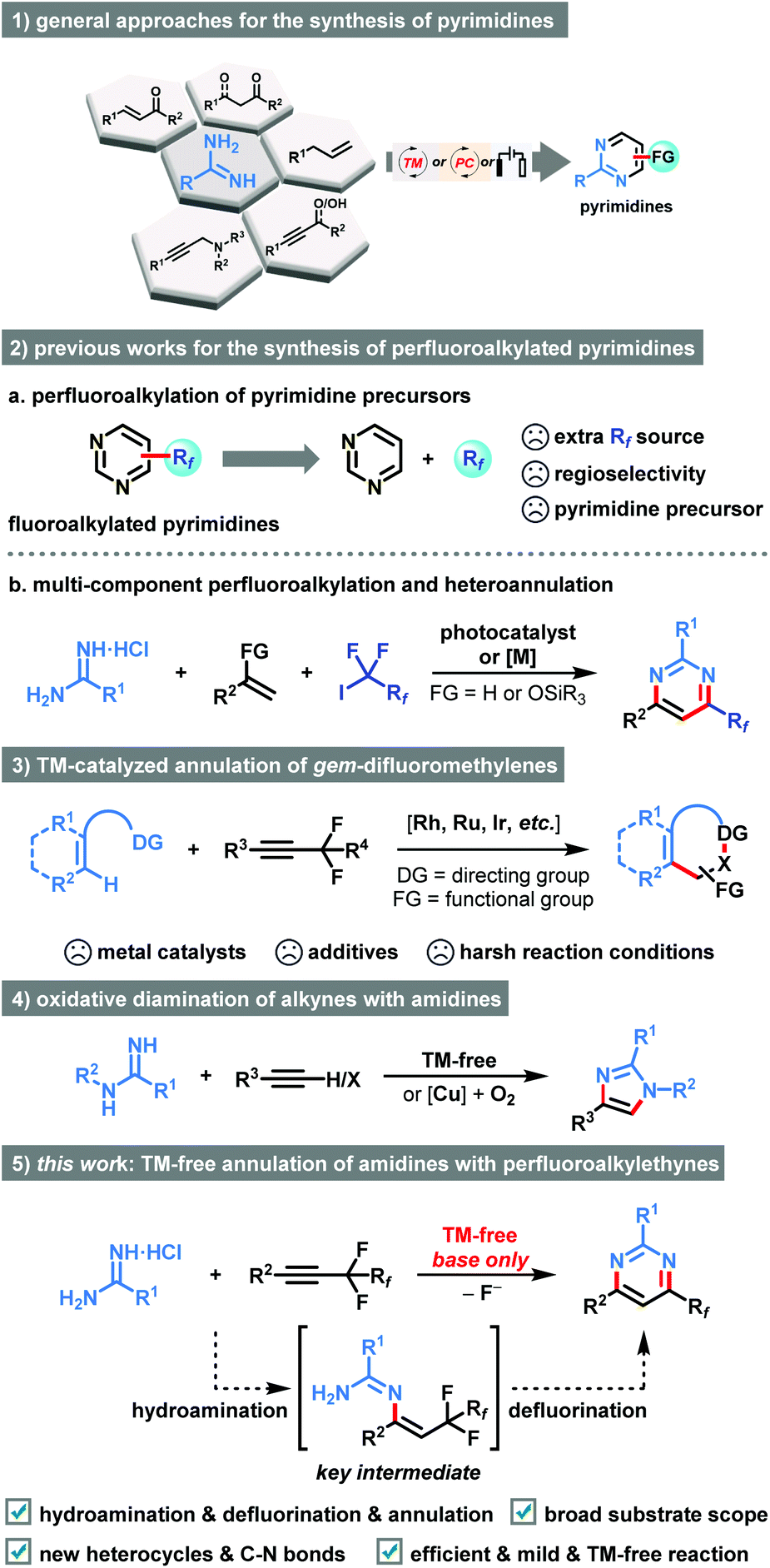 | ||
| Scheme 1 Amidine as a versatile building block in the preparation of diversified N-heterocyclic compounds and related approaches for the synthesis of functionalized pyrimidines. | ||
The introduction of fluorine atoms adjacent to the alkyne moiety can tune the reactivity of the triple C–C bonds.14 Diverse reaction modes involving one or two folds C–F bond cleavage and transformation of alkynyl functionality have been realized by the research groups of Loh,14a Feng,14a,b Wang,14c Yi,14d,e Fan,14f and Rovis14g (Scheme 1, 3). Nevertheless, the utilities of costly metal catalysts, extra additives, and harsh reaction conditions were necessary. While research interest had also been directed towards the exploration of oxidative cyclization of simple alkynes with amidines for the synthesis of imidazoles (Scheme 1, 4),15 as far as we know, the direct exploitation of perfluorinated internal alkynes via two-fold C–F bond functionalization for rapidly accessing six-membered heterocycles with spatially defined perfluoroalkyl substituent has never been reported till now. The transition-metal-free net-[3 + 3]-condensation of perfluoroalkyl alkynes and amidines has the potential to advance green and sustainable chemistry.
Continuing with our recent interests in heterocyclic chemistry,16 herein we describe a transition metal-free method for the synthesis of perfluoroalkylated pyrimidines commencing from perfluoroalkyl alkynes and amidines through a convenient sequence of hydroamination, defluorination, and annulation cascade (Scheme 1, 5). In this newly developed protocol, the perfluoroalkyl alkyne acted as an active unsaturated substrate because it could easily undergo the hydroamination under basic conditions. The success of the intramolecular ring-closure step could be attributed to the activation of C(sp3)–F bonds under the effect of the alkali–metal base.17 Moreover, the perfluoroalkyl substituents are vital for the net-[3 + 3]-heteroannulation due to their distinctive fluorine effects in tuning the reactivity of alkynes. As a result, the perfluoroalkyl group exerts both activating effect and directing effect on the annulation. This method involved new C–N bonds formation and a new six-membered ring construction, which featured broad substrate scope, good functional group tolerance, and synthetic simplicity in an eco-friendly manner.
Results and discussion
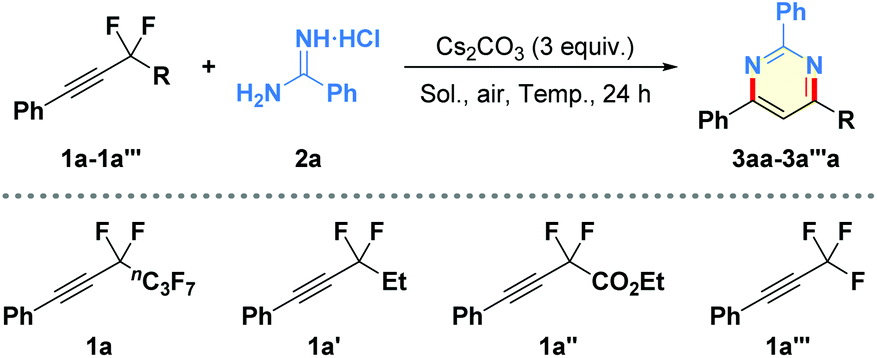
There are few examples of the intermolecular hydroamination of fluorine-substituted internal alkynes.18 Initially, we examined the reaction of (perfluorohex-1-yn-1-yl)benzene (1a)19 with benzamidine hydrochloride (2a)20 in the presence of copper catalyst, additive, and different bases in THF at 60 °C for 24 h under air atmosphere (see Table S1 in the ESI† for more details). To our delight, the proposed cyclization could be successfully realized by the sole use of Cs2CO3 as the base, leading to the desired functionalized pyrimidine 3aa in 63% NMR yield (Table 1, entry 1). Evaluation of various solvents, including MeCN, DMF, DMSO, tBuOH, DCE, H2O, and toluene, led to the discovery of DMF as the optimal solvent for the aminated reaction (entries 2–8). Of different reaction temperatures examined (entries 9–12), performing the model reaction at 70 °C gave the product 3aa in the highest NMR yield (73% isolated yield; entry 11). Furthermore, DMA was found to be superior to DMF or NMP as the reaction solvent (entries 13 and 14). In addition, shortening reaction time from 24 h to 6 h was unfavorable for the complete conversion of starting material 1a, affording the corresponding product 3aa in slightly decreased yield (entry 15). Finally, the importance of perfluoroalkyl moiety and the unique fluorine effects for the net-[3 + 3]-annulation was clarified by using 1,1-difluoropropyl-, 2-ethoxy-1,1-difluoro-2-oxoethyl-, or trifluoromethyl-substituted alkynes (1a′–1a′′′) in the control experiments, which delivered low yields of the corresponding defluorinative products (entries 16–18).|
|
||||
|---|---|---|---|---|
| Entry | R | Solvent | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: 1a–1a′′′ (0.3 mmol), 2a (0.36 mmol), and Cs2CO3 (0.9 mmol) in solvent (1.5 mL) under air for 24 h. b Yields were determined by NMR analysis with 1,4-dimethoxybenzene (0.25 mmol) as an internal standard. c Isolated yield. d 6 h. e At 100 °C. | ||||
| 1 | n C3F7 (1a) | THF | 60 | 63 |
| 2 | n C3F7 (1a) | MeCN | 60 | 37 |
| 3 | n C3F7 (1a) | DMF | 60 | 67 |
| 4 | n C3F7 (1a) | DMSO | 60 | 50 |
| 5 | n C3F7 (1a) | t BuOH | 60 | 37 |
| 6 | n C3F7 (1a) | DCE | 60 | 33 |
| 7 | n C3F7 (1a) | H2O | 100 | 0 |
| 8 | n C3F7 (1a) | Toluene | 60 | 58 |
| 9 | n C3F7 (1a) | DMF | 25 | 37 |
| 10 | n C3F7 (1a) | DMF | 40 | 54 |
| 11 | n C3F7 (1a) | DMF | 70 | 74 (73)c |
| 12 | n C3F7 (1a) | DMF | 80 | 70 |
| 13 | n C3F7 (1a) | NMP | 70 | 76 |
| 14 | n C3F7 (1a) | DMA | 70 | 79 (77) |
| 15 | n C3F7 (1a) | DMA | 70 | 74d (72)c |
| 16 | C2H5 (1a′) | DMA | 70 | Trace (<5)e |
| 17 | CO2Et (1a′′) | DMA | 70 | <5 |
| 18 | F (1a′′′) | DMA | 70 | <5 (<5)e |
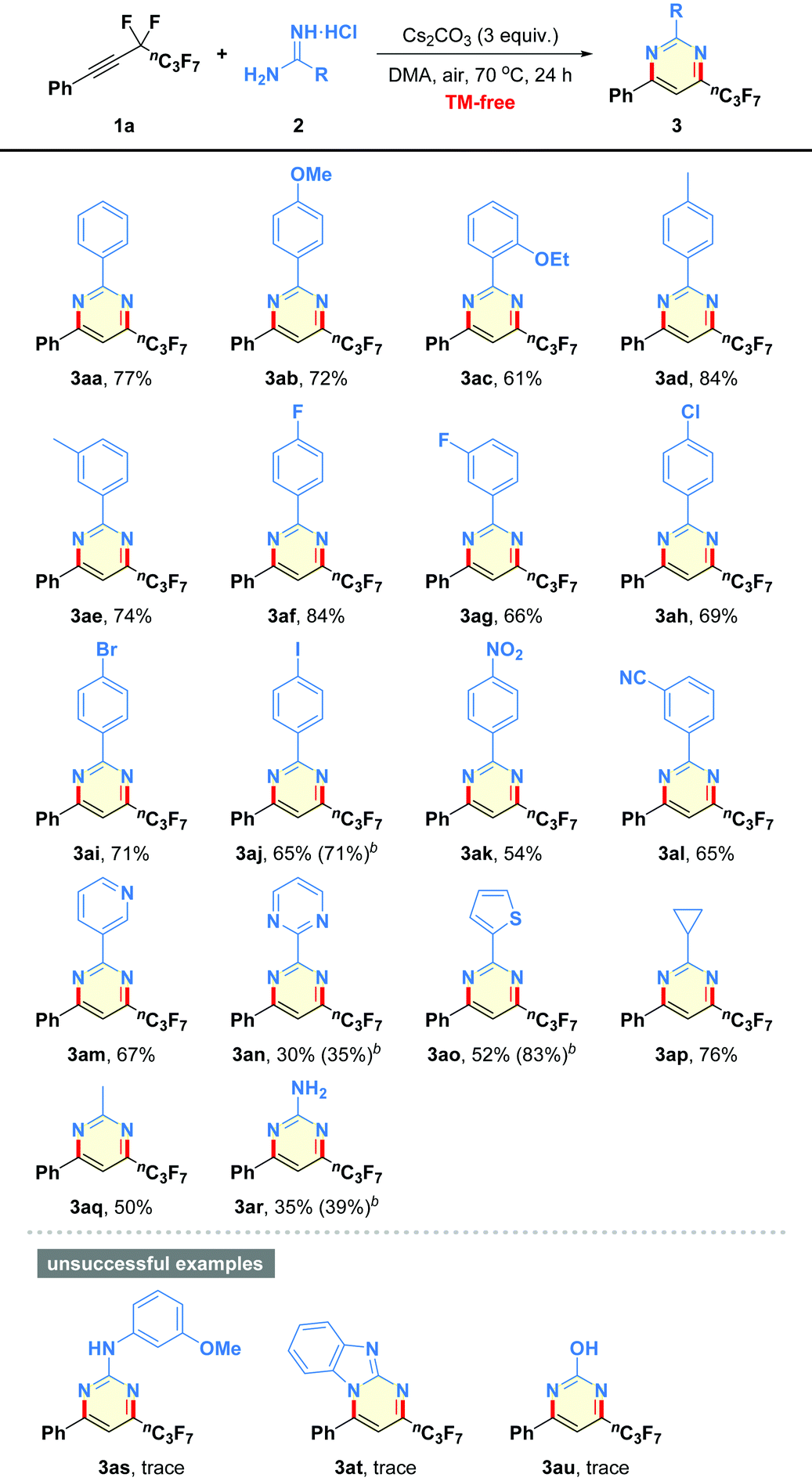
After obtaining the optimized reaction conditions, we began to evaluate the generality of this defluorinative cyclization with regard to diverse amidines, and the results are summarized in Table 2. First of all, a variety of substituents of varying electronic character and steric hindrance on the phenyl moieties in amidines were investigated (3ab–3al). It was found that substrates containing either electron-donating (e.g., OMe, OEt, Me) or electron-withdrawing (e.g., halogen, NO2, CN) groups could provide the desired pyrimidines 3ab–3al in moderate to good yields (54–84% yields). Notably, the ortho steric hindrance of substrate 2c did not obviously interfere with the reaction outcome (3ac). Functional groups, such as F, Cl, Br, I, NO2, and CN were well tolerated under the mild reaction conditions, which offered good opportunities for the late-stage functionalization (3af–3al). Other heteroaromatic motifs, such as pyridine-3-yl (2m), pyrimidine-2-yl (2n), and thiophen-2-yl (2o), were smoothly incorporated into the pyrimidine-based products 3am–3ao in 35–83% yields. Even alkyl-containing amidines participated well in the present transformation, leading to the corresponding products 3ap and 3aq in 76% and 50% yields, respectively. Additionally, guanidine hydrochloride was also proven to be a good candidate to give the aminated product 3ar in 39% yield. However, dual-nucleophilic 1-(3-methoxyphenyl)guanidine (2s), 1H-benzo[d]imidazol-2-amine (2t), and urea (2u) failed to produce any fluorinated products 3as–3au.
Encouraged by the above results, the general applicability of the present method for the synthesis of highly functionalized pyrimidines by employing a wide array of perfluorobutyl alkynes 1 as substrates was studied. As outlined in Table 3, not only perfluorobutyl alkynes bearing electron-donating groups but also substrates containing electron-withdrawing substituents in the aryl rings could react well with amidines 2i or 2n in a remarkably regioselective manner to produce trisubstituted products 3bi–3ii in 28–80% yields. Although 2-amidinopyrimidine hydrochloride (2n) was found to be less reactive than 2i, this resulting polycyclic aza-aromatic 3bn could not be easily accessed through traditional synthetic routes.12 Moreover, the mild reaction showed good tolerance to several functional groups, such as methoxy (1b), methyl (1c–1e), tert-butyl (1f), and halogens (1g–1i). However, the presence of a methyl group at the C2-position of phenyl ring in the substrate 1e seemed to have a negative effect on the reaction performance (3ei, 29% yield). Furthermore, alkyne 1j derived from naphthalene was tested, furnishing product 3ji in 33% yield, which is mainly due to the steric effect. Similarly, the reactions worked equally well with heteroaryl perfluorobutylethynes 1k–1l under the standard reaction conditions to give the corresponding heterocyclic variants 3ki–3li in reasonable yields.13 In addition, when (E)-(5,5,6,6,7,7,8,8,8-nonafluorooct-1-en-3-yn-1-yl)benzene (1m) was prepared and subjected to the net-[3 + 3] heteroannulation, the desired product 3mi was obtained in 60% yield. As for alkyl substrate, 5,5,6,6,7,7,8,8,8-nonafluoro-2-methyloct-3-yn-2-ol (1n) could participate into the reaction to produce the desired alkyl-substituted pyrimidine 3ni in 41% yield.
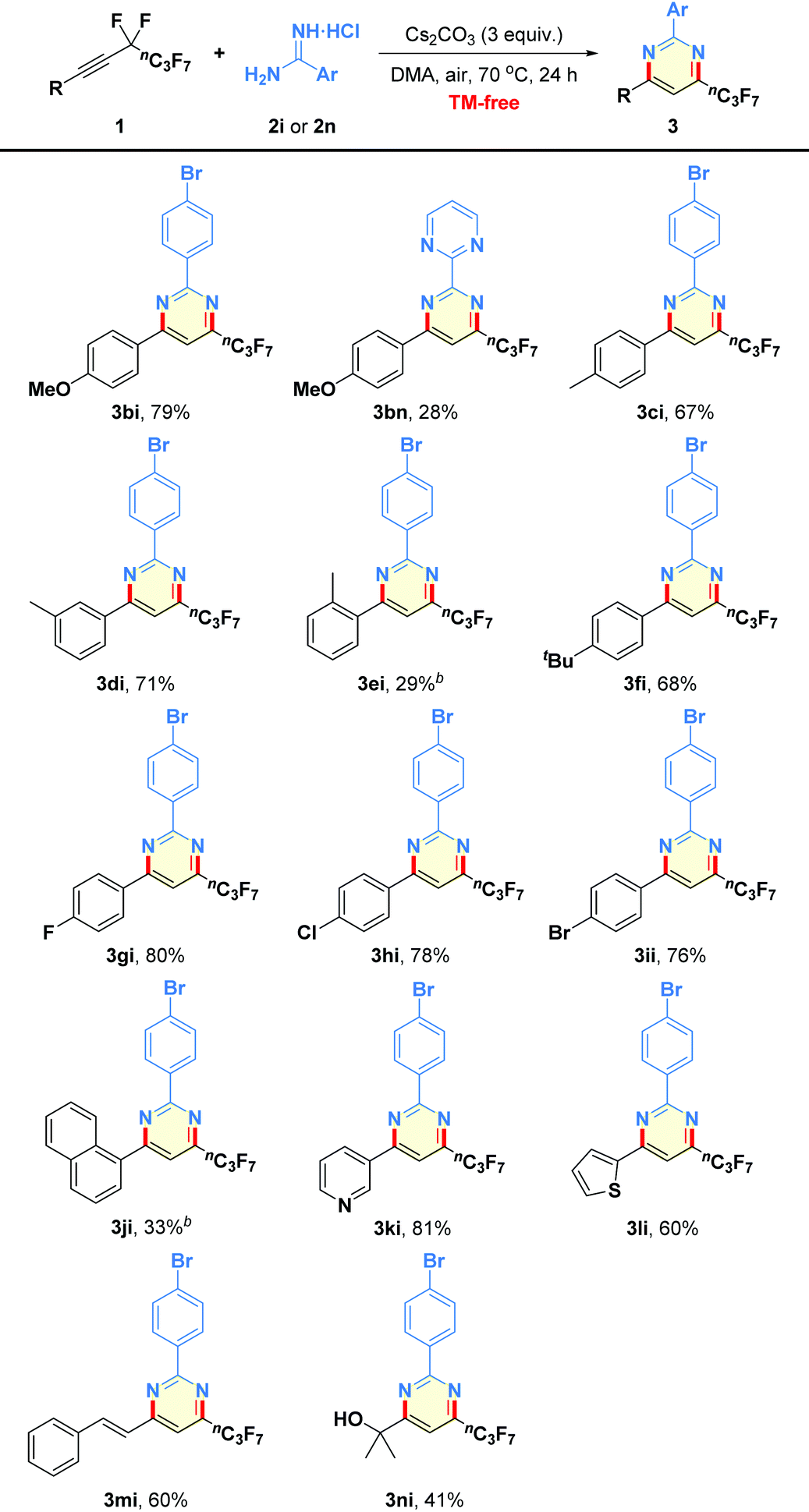
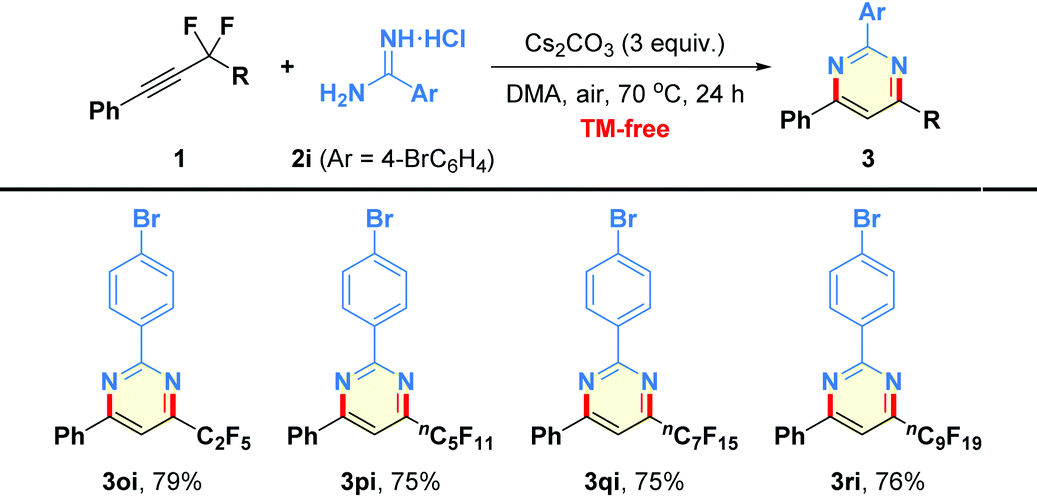
Next, we turned our attention to expand the substrate scope of the reaction to encompass a spectrum of fluoroalkyl alkynes 1o–1r (Table 4). We were delighted to find that the substrates possessing different perfluoroalkyl chains (3–10 carbon) worked well to produce the products 3oi–3ri in 75%–79% yields.
| a Reaction conditions: 1 (0.3 mmol), 2i (0.36 mmol), Cs2CO3 (0.9 mmol), and DMA (1.5 mL) at 70 °C for 24 h under air; isolated yields. |
|---|
|
To further demonstrate the procedural simplicity and synthetic practicability of the present method, an efficient procedure was established for the scale-up synthesis of product 3aa (Scheme 2A, 80% yield). In addition, the present method could be applied to the late-stage modification of complex molecules. As shown in Scheme 2B, perfluorobutyl derivatives of Estrone and diacetone-D-glucose could be readily accommodated with the well-established conditions to generate the target heterocycles 3sa and 3ta in 38% and 72% yields, respectively. Meanwhile, palladium-catalyzed coupling reaction of the resulting 3ai with thiophen-2-ylboronic acid (4) provided the heterocycle 5 in 96% yield (Scheme 2C), thereby having a chance for potential applications in materials science.
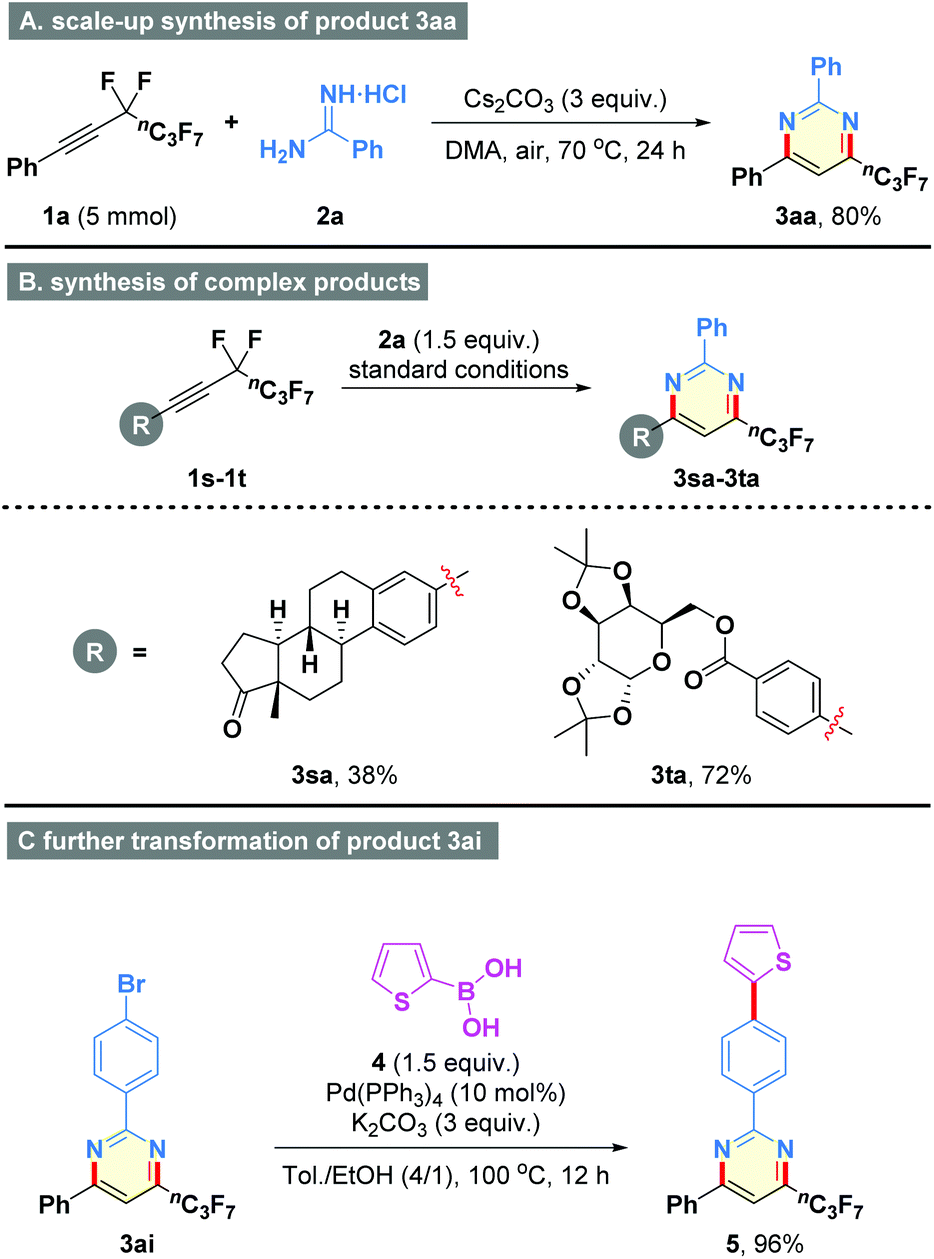 | ||
| Scheme 2 Scale-up synthesis and further transformations. | ||
In order to clarify the possible mechanism of this transformation, some control experiments were carried out (Scheme 3). First, free-radical intermediate was not involved in the reaction because the addition of radical inhibitor TEMPO (2.5 equiv.) did not affect the reaction performance (Scheme 3A). Second, the deuterium-labeling experiment resulted in the generation of compound 3aa-D, suggesting that the proton atom of product might originate from water in the hydroamination process (Scheme 3B). Third, (perfluorohex-1-yn-1-yl)benzene (1a) could not be converted to fluoroalkylated ynone 6 under the standard conditions, revealing that defluorination step occurred after the intermolecular interaction of 1a and 2a (Scheme 3C). Fourth, the reaction of pre-synthesized 6 with amidine 2a generated the corresponding product 3aa in 77% yield, which further supported the above conjecture (Scheme 3D).
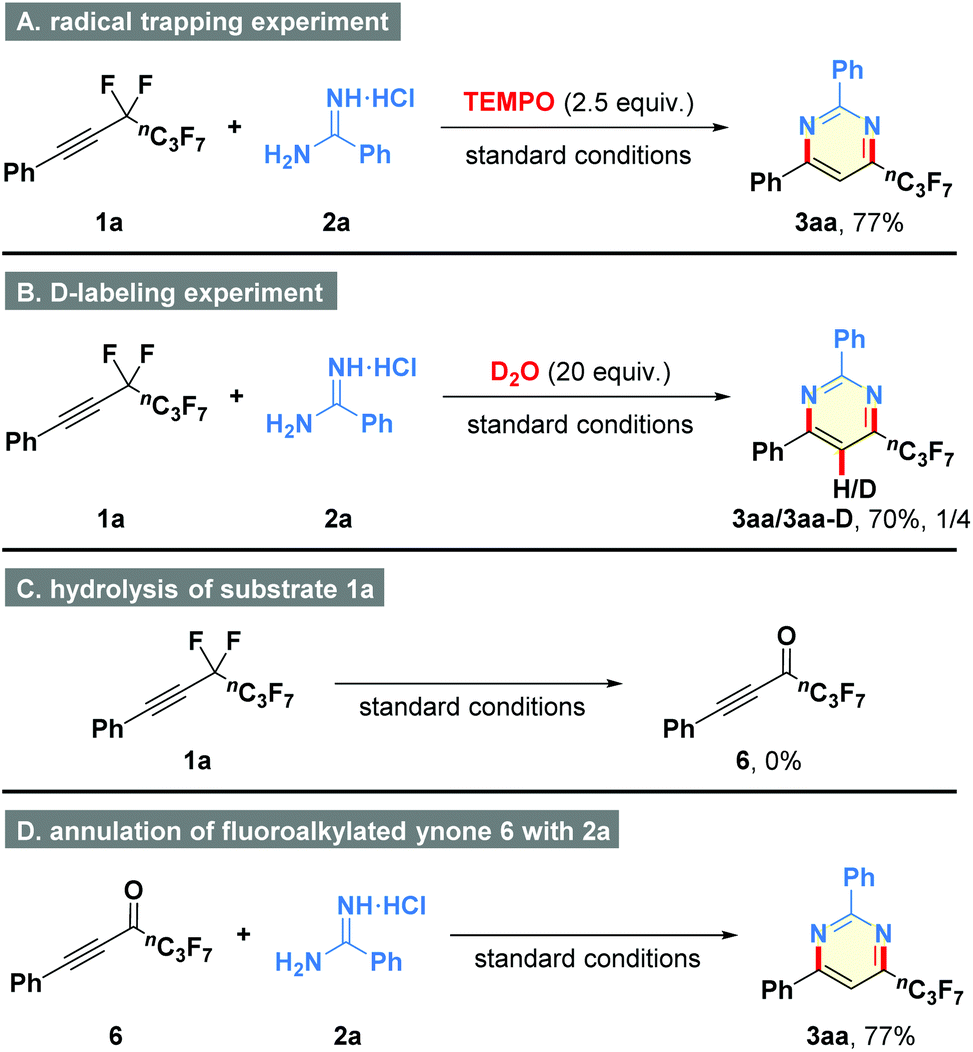 | ||
| Scheme 3 Mechanistic studies. | ||
On the basis of these mechanistic insights, a plausible mechanism was proposed in Scheme 4.13–18 Initially, selective addition of amidine 2 to the triple bond of perfluoroalkyl alkyne 1 under basic conditions provides the vinyl anion A, which then abstracts a proton from water to give the hydroaminated intermediate B. The related intermediate could be detected by 1H NMR and HRMS analysis of the model reaction mixture.21 Next, an intramolecular nucleophilic substitution via the cleavage of the C(sp3)–F bond affords the ring-closure species C, which further undergoes aromatization through removing a molecule of HF with the assistance of alkali–metal base to produce the final product 3.17 Notably, the existence of the Rf group is essential for the desired net-[3 + 3]-heteroannulation of perfluoroalkyl alkynes with amidines.
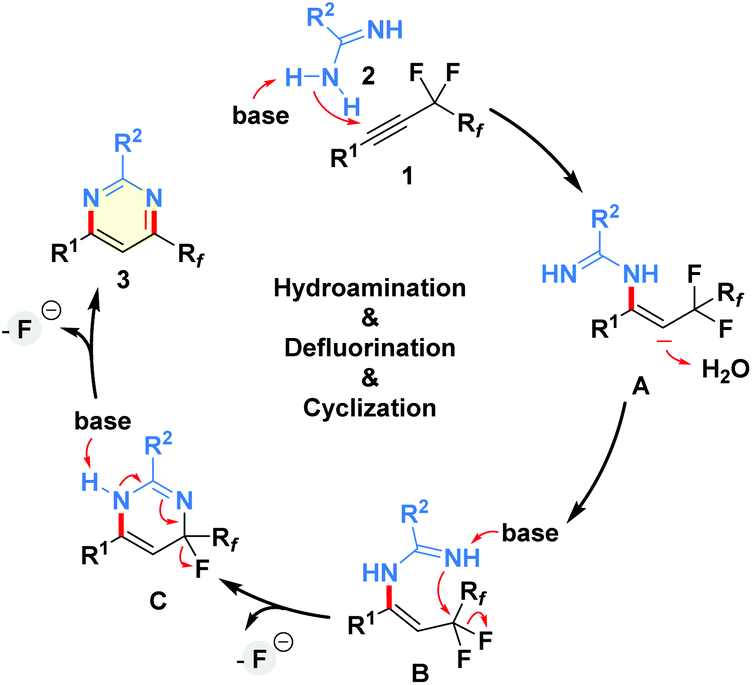 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusion
In summary, we have developed an efficient defluorinative reaction of perfluoroalkyl alkynes with amidines for the facile synthesis of potentially biologically and pharmaceutically active perfluoroalkylated pyrimidines through a sequence of hydroamination, defluorination, and annulation. This modular and regioselective C–F bond functionalization, which worked efficiently under transition metal-free conditions, featured broad scope of both coupling partners, excellent functional group tolerance, synthetic simplicity, and scale-up synthesis. In the present reaction, two new C–N bonds and a new six-membered ring were simultaneously constructed in a one-pot manner. The perfluoroalkyl group exerts both activating effect and directing effect on the annulation.Experimental
General procedures for the synthesis of perfluoroalkylated pyrimidines 3
A solution of perfluoroalkylethyne 1 (0.3 mmol), amidine hydrochloride 2 (0.36–0.45 mmol), and Cs2CO3 (293.2 mg, 0.9 mmol) in DMA (1.5 mL) was stirred at 70 °C under air for 24 h. The reaction was then quenched by saturated NH4Cl solution (20 mL) and extracted with EtOAc (20 mL × 3). The organic layer was washed with saturated brine twice, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (300–400 mesh) using petroleum ether/ethyl acetate as eluent to afford the pure products 3.General procedure for the scale-up synthesis of product 3aa
A solution of (perfluorohex-1-yn-1-yl)benzene (1.60 g, 5 mmol, 1a), benzamidine hydrochloride (0.94 g, 6 mmol, 2a), and Cs2CO3 (4.89 g, 15 mmol) in DMA (30 mL) was stirred at 70 °C under air for 24 h. The reaction was then quenched by saturated NH4Cl solution (100 mL) and extracted with EtOAc (100 mL × 3). The organic layer was washed with saturated brine twice, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (300–400 mesh) using petroleum ether/ethyl acetate as eluent to afford the pure product 3aa (1.60 g, 80% yield).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from National Natural Science Foundation of China (No. 22001121 and 21772093), Natural Science Foundation of Jiangsu Province (BK20180690), National Students’ Platform for Innovation and Entrepreneurship Training Program (202110291019Z), and Nanjing Tech University (Start-up Grant No. 39837118 and 39837146).References
- (a) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, U.K., 2009 CrossRef; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications, Wiley-VCH, Weinheim, Germany, 2nd edn, 2013 CrossRef; (c) X.-Q. Chu, D. Ge, Y.-Y. Cui, Z.-L. Shen and C.-J. Li, Desulfonylation via Radical Process: Recent Developments in Synthetic Applications, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS.
- (a) M. Hird, Fluorinated Liquid Crystals-Properties and Applications, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC; (b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- (a) E. Prchalova, O. Stepanek, S. Smrcek and M. Kotora, Medicinal Applications of Perfluoroalkylated Chain-Containing Compounds, Future Med. Chem., 2014, 6, 1201–1229 CrossRef CAS; (b) J. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O'Hagan, Next Generation Organofluorine Containing Blockbuster Drugs, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS.
- (a) B. Chai, S. Wang, W. Yu, H. Li, C. Song, Y. Xu, C. Liu and J. Chang, Synthesis of Novel Strobilurin-Pyrimidine Derivatives and Their Antiproliferative Activity Against Human Cancer Cell Lines, Bioorg. Med. Chem. Lett., 2013, 23, 3505–3510 CrossRef CAS; (b) S. Lee, D. Lim, E. Lee, N. Lee, H. Lee, J. Cechetto, M. Liuzzi, L. H. Freitas-Junior, J. S. Song, M. A. Bae, S. Oh, L. Ayong and S. B. Park, Discovery of Carbohybrid-Based 2-Aminopyrimidine Analogues as a New Class of Rapid-Acting Antimalarial Agents Using Image-Based Cytological Profiling Assay, J. Med. Chem., 2014, 57, 7425–7434 CrossRef CAS; (c) J.-Q. Zhang, Y.-J. Luo, Y.-S. Xiong, Y. Yu, Z.-C. Tu, Z.-J. Long, X.-J. Lai, H.-X. Chen, Y. Luo, J. Weng and G. Lu, Design, Synthesis, and Biological Evaluation of Substituted Pyrimidines as Potential Phosphatidylinositol 3-Kinase (PI3K) Inhibitors, J. Med. Chem., 2016, 59, 7268–7274 CrossRef CAS.
- (a) W. Guo, M. Zhao, W. Tan, L. Zheng, K. Tao and X. Fan, Developments Towards Synthesis of N-Heterocycles from Amidines via C–N/C–C Bond Formation, Org. Chem. Front., 2019, 6, 2120–2141 RSC; (b) W.-B. Cao, X.-Q. Chu, Y. Zhou, L. Yin, X.-P. Xu and S.-J. Ji, Copper-Catalyzed Construction of Eight-membered Rings via Oxidative Ring Expansion and Intermolecular Cyclization Sequencing of Indoles with Amidines: Efficient Synthesis of Benzo[1,3,5]triazocin-6(5H)-ones, Chem. Commun., 2017, 53, 6601–6604 RSC.
- For selected reviews, see: (a) M. Radi, S. Schenone and M. Botta, Recent Highlights in the Synthesis of Highly Functionalized Pyrimidines, Org. Biomol. Chem., 2009, 7, 2841–2847 RSC; (b) M. Mahfoudh, R. Abderrahim, E. Leclerc and J.-M. Campagne, Recent Approaches to the Synthesis of Pyrimidine Derivatives, Eur. J. Org. Chem., 2017, 2856–2865 CrossRef CAS.
- (a) M. C. Bagley, D. D. Hughes and P. H. Taylor, Highly Efficient Synthesis of Pyrimidines under Microwave-assisted Conditions, Synlett, 2003, 259–261 CrossRef CAS; (b) K. S. Vadagaonkar, H. P. Kalmode, S. Prakash and A. C. Chaskar, Greener [3 + 3] Tandem Annulation–Oxidation Approach Towards the Synthesis of Substituted Pyrimidines, New J. Chem., 2015, 39, 3639–3645 RSC; (c) D. L. Cousins, P. Fricero, K. P. M. Kopf, E. J. McColl, W. Czechtizky, Y. H. Lim and J. P. A. Harrity, Pyrimidin-6-yl Trifluoroborate Salts as Versatile Templates for Heterocycle Synthesis, Angew. Chem., Int. Ed., 2021, 60, 9412–9415 CrossRef CAS.
- X.-Q. Chu, W.-B. Cao, X.-P. Xu and S.-J. Ji, Iron Catalysis for Modular Pyrimidine Synthesis through β–Ammoniation/Cyclization of Saturated Carbonyl Compounds with Amidines, J. Org. Chem., 2017, 82, 1145–1154 CrossRef CAS PubMed.
- W. Guo, C. Li, J. Liao, F. Ji, D. Liu, W. Wu and H. Jiang, Transition Metal Free Intermolecular Direct Oxidative C–N Bond Formation to Polysubstituted Pyrimidines Using Molecular Oxygen as the Sole Oxidant, J. Org. Chem., 2016, 81, 5538–5546 CrossRef CAS.
- M. Lin, Q. Chen, Y. Zhu, X. Chen, J. Cai, Y. Pan and Z. Zhan, Copper(II)-Catalyzed Synthesis of Pyrimidines from Propargylic Alcohols and Amidine: A Propargylation-Cyclization-Oxidation Tandem Reaction, Synlett, 2011, 1179–1183 CAS.
- J. Chen, R. Properzi, D. P. Uccello, J. A. Young, R. G. Dushin and J. T. Starr, One-Pot Oxidation and Rearrangement of Propargylamines and in Situ Pyrazole Synthesis, Org. Lett., 2014, 16, 4146–4149 CrossRef CAS PubMed.
- E. V. Verbitskiy, G. L. Rusinova, O. N. Chupakhin and V. N. Charushin, Recent Advances in Direct C-H Functionalization of Pyrimidine, Synthesis, 2018, 50, 193–210 CrossRef CAS.
- (a) X.-Q. Chu, T. Xie, L. Li, D. Ge, Z.-L. Shen and T.-P. Loh, Combining Fluoroalkylation and Defluorination to Enable Formal [3 + 2 + 1] Heteroannulation by Using Visible-Light Photoredox Organocatalysis, Org. Lett., 2018, 20, 2749–2752 CrossRef CAS; (b) X.-Q. Chu, B.-Q. Cheng, Y.-W. Zhang, D. Ge, Z.-L. Shen and T.-P. Loh, Copper-Catalyzed Three-Component Cyclization of Amidines, Styrenes, and Fluoroalkyl Halides for the Synthesis of Modular Fluoroalkylated Pyrimidines, Chem. Commun., 2018, 54, 2615–2618 RSC; (c) B.-Q. Cheng, D. Ge, X. Wang and X.-Q. Chu, Perfluoroalkyl Halides as Fluorine-Containing Building Blocks for the Synthesis of Fluoroalkylated Heterocycles, Chin. J. Org. Chem., 2021, 41, 1925–1938 CrossRef.
- (a) C.-Q. Wang, L. Ye, C. Feng and T.-P. Loh, C-F Bond Cleavage Enabled Redox-Neutral [4 + 1] Annulation via C-H Bond Activation, J. Am. Chem. Soc., 2017, 139, 1762–1765 CrossRef CAS PubMed; (b) C.-Q. Wang, Y. Zhang and C. Feng, Fluorine Effects on Group Migration via a Rhodium(V) Nitrenoid Intermediate, Angew. Chem., Int. Ed., 2017, 56, 14918–14922 CrossRef CAS; (c) T. Li, C. Zhou, X. Yan and J. Wang, Solvent-Dependent Asymmetric Synthesis of Alkynyl and Monofluoroalkenyl Isoindolinones by CpRhIII-Catalyzed C−H Activation, Angew. Chem., Int. Ed., 2018, 57, 4048–4052 CrossRef CAS; (d) H. Gao, S. Lin, S. Zhang, W. Chen, X. Liu, G. Yang, R. A. Lerner, H. Xu, Z. Zhou and W. Yi, gem-Difluoromethylene Alkyne-Enabled Diverse C–H Functionalization and Application to the on-DNA Synthesis of Difluorinated Isocoumarins, Angew. Chem., Int. Ed., 2021, 60, 1959–1966 CrossRef CAS PubMed; (e) J. Yang, W. Shi, W. Chen, H. Gao, Z. Zhou and W. Yi, Rh(III)-Catalyzed Chemoselective C–H Alkenylation and [5 + 1] Annulation with Gem-Difluoromethylene Enabled by the Distinctive Fluorine Effect, J. Org. Chem., 2021, 86, 9711–9722 CrossRef CAS PubMed; (f) Y. Yang, N. Li, J. Zhao, Y. Jiang, X. Zhang and X. Fan, Selective Synthesis of 3-(α-Fluorovinyl)indoles and 3-Acylindoles via the Cascade Reactions of 1-Phenylpyrazolidinones with α,α-Difluoromethylene Alkynes, Adv. Synth. Catal., 2021, 363, 3600–3606 CrossRef CAS; (g) F. Romanov-Michailidis, B. D. Ravetz, D. W. Paley and T. Rovis, Ir(III)-Catalyzed Carbocarbation of Alkynes through Undirected Double C–H Bond Activation of Anisoles, J. Am. Chem. Soc., 2018, 140, 5370–5374 CrossRef CAS PubMed.
- (a) J. Li and L. Neuville, Copper-Catalyzed Oxidative Diamination of Terminal Alkynes by Amidines: Synthesis of 1,2,4-Trisubstituted Imidazoles, Org. Lett., 2013, 15, 1752–1755 CrossRef CAS PubMed; (b) X. Y. Chen, U. Englert and C. Bolm, Base-Mediated Syntheses of Di- and Trisubstituted Imidazoles from Amidine Hydrochlorides and Bromoacetylenes, Chem. – Eur. J., 2015, 21, 13221–13224 CrossRef CAS PubMed.
- (a) T. Xie, G.-Q. Wang, Y.-W. Wang, W. Rao, H. Xu, S. Li, Z.-L. Shen and X.-Q. Chu, Selective Quadruple C(sp3)-F Functionalization of Polyfluoroalkyl Ketones, iScience, 2020, 23, 101259 CrossRef CAS PubMed; (b) S.-Z. Cai, D. Ge, L.-W. Sun, W. Rao, X. Wang, Z.-L. Shen and X.-Q. Chu, Three-Component Heteroannulation for Tetrasubstituted Furan Construction Enabled by Successive Defluorination and Dual Sulfonylation Relay, Green Chem., 2021, 23, 935–941 RSC; (c) Q.-D. Wang, Y.-W. Wang, T. Xie, Y.-Y. Cui, M. Ma, Z.-L. Shen and X.-Q. Chu, Three-Component Bisannulation for the Synthesis of Trifluoromethylated Tetracyclic Aza-Aromatics through Six C(sp3)-F Bond Cleavage and Four C-N Bond Formation, J. Org. Chem., 2021, 86, 8236–8247 CrossRef CAS PubMed; (d) Q.-D. Wang, C. Zhang, L.-W. Sun, X.-L. Luo, W. Rao, Z.-L. Shen and X.-Q. Chu, Defluorinative Phosphorylation of Perfluoroalkyl Ketones: Synthesis of Fluoroalkylated and Phosphorylated Furan Derivatives, Org. Chem. Front., 2021, 8, 1503–1509 RSC; (e) Y.-C. Guo, X.-D. Song, W. Deng, W. Rao, H. Xu and Z.-L. Shen, An Efficient Synthesis of 4,5-Diaryl-3,4-dihydropyrimidin-2(1H)-one via a Cesium Carbonate-promoted Direct Condensation of 1-Aryl-2-propanone with 1,1′-(Arylmethylene)diurea, RSC Adv., 2020, 10, 30062–30068 RSC; (f) X.-D. Song, X.-R. Li, Y.-W. Wang, X.-Q. Chu, W. Rao, H. Xu, G.-Z. Han and Z.-L. Shen, Indium-mediated Difunctionalization of Iodoalkyl-tethered Unactivated Alkenes via an Intramolecular Cyclization and an Ensuing Palladium-catalyzed Cross-coupling Reaction with Aryl Halides, Org. Chem. Front., 2020, 7, 2703–2709 RSC; (g) Y.-Y. Cui, W.-X. Li, N.-N. Ma, C. Shen, X. Zhou, X.-Q. Chu, W. Rao and Z.-L. Shen, Nickel-catalyzed Direct Cross-coupling of Heterocyclic Phosphonium Salts with Aryl Bromides, Org. Chem. Front., 2021 10.1039/d1qo01474a.
- (a) J. Mao, Z. Wang, X. Xu, G. Liu, R. Jiang, H. Guan, Z. Zheng and P. J. Walsh, Synthesis of Indoles through Domino Reactions of 2-Fluorotoluenes and Nitriles, Angew. Chem., Int. Ed., 2019, 58, 11033–11038 CrossRef CAS; (b) S. Chen, L. Yang, Y. Shang, J. Mao and P. J. Walsh, Base-Promoted Tandem Synthesis of 2-Azaaryl Tetrahydroquinolines, Org. Lett., 2021, 23, 1594–1599 CrossRef CAS; (c) Z.-P. Ye, Y.-Z. Hu, J.-P. Guan, K. Chen, F. Liu, J. Gao, J.-A. Xiao, H.-Y. Xiang, X.-Q. Chen and H. Yang, Photocatalytic Cyclization/Defluorination Domino Sequence to Access 3–Fluoro-1,5-dihydro–2H–pyrrol-2-one Scaffold, Org. Lett., 2021, 23, 4754–4758 CrossRef CAS.
- T. Ishikawa, T. Sonehara, M. Minakawa and M. Kawatsura, Copper-Catalyzed Intermolecular Hydroamination of Internal Alkynes with Anilines and Amines, Org. Lett., 2016, 18, 1422–1425 CrossRef CAS PubMed.
- (a) T. Konno, J. Chae, M. Kanda, G. Nagai, K. Tamura, T. Ishihara and H. Yamanaka, Facile Syntheses of Various Per- or Polyfluoroalkylated Internal Acetylene Derivatives, Tetrahedron, 2003, 59, 7571–7580 CrossRef CAS; (b) L. Chu and F.-L. Qing, Copper-Mediated Aerobic Oxidative Trifluoromethylation of Terminal Alkynes with Me3SiCF3, J. Am. Chem. Soc., 2010, 132, 7262–7263 CrossRef CAS PubMed; (c) J. Li, L. Liu, K. Zheng, C. Zheng, H. Xiao and S. Fan, Silver-Mediated Perfluoroalkylation of Terminal Alkynes with Perfluoroalkyl Iodides, J. Org. Chem., 2020, 85, 8723–8731 CrossRef CAS.
- F. C. Schaefer and A. P. Krapcho, Preparation of Amidine Salts by Reaction of Nitriles with Ammonium Salts in the Presence of Ammonia, J. Org. Chem., 1962, 27, 1255–1258 CrossRef CAS.
- See the ESI† for more details of in situ HRMS and 1H NMR analysis of possible intermediates.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental details, mechanistic studies, characterization data for products, 1H, 13C, and 19F spectra of products. See DOI: 10.1039/d1qo01439k |
| This journal is © the Partner Organisations 2022 |