
Recent advances in catalytic enantioselective direct C–H bond functionalization of electron-deficient N-containing heteroarenes
Xiao-Lan
Liu†
c,
Luo-Bin
Jiang†
a,
Mu-Peng
Luo
 a,
Zhi
Ren
*b and
Shou-Guo
Wang
*a
a,
Zhi
Ren
*b and
Shou-Guo
Wang
*a
aShenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, 1068 Xueyuan Avenue, Shenzhen, China. E-mail: shouguo.wang@siat.ac.cn
bCollege of Pharmacy, Shenzhen Technology University, 3002 Lantian Road, Shenzhen, China. E-mail: renzhi@sztu.edu.cn
cSchool of Medical Imaging, Xuzhou Medical University, Xuzhou, China
First published on 22nd September 2021
Abstract
Catalytic enantioselective direct C–H bond functionalization of electron-deficient N-containing heteroarenes represents one of the most straightforward and powerful protocols to construct diverse enantioenriched highly functionalized N-heteroarene products. Efficient catalytic systems and a variety of strategies for highly chemo- and stereoselective C–H functionalization of electron-deficient N-containing heteroarenes have been developed over the past few decades. This review summarized the rapid progress in the catalytic asymmetric C–H functionalization of N-containing heteroarenes and discussed the challenges and potential of the field in the current stage.
 Xiao-Lan Liu | Xiao-Lan Liu received her PhD degree in 2020 from Fudan University. Then she joined Xuzhou Medical University in March 2020 as a lecturer. Her research interests mainly focus on the design and synthesis of photochemical materials and bioactive compounds. |
 Luo-Bin Jiang | Luo-Bin Jiang obtained his M.S. degree from the China State Institute of Pharmaceutical Industry in 2020. Then he joined Prof. Shou-Guo Wang's group as a research assistant at CADD center, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research interest focuses on photocatalytic asymmetric radical reactions and drug synthesis. |
 Mu-Peng Luo | Mu-Peng Luo obtained his bachelor's degree from Shanxi Medical University in 2014 and PhD degree from Shanghai Advanced Research Institute, Chinese Academy of Sciences, under the supervision of Prof. Wanguo Wei in 2019. After that, he joined Prof. Shuguang Yuan's group for postdoctoral study at Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research interest mainly focuses on the photocatalytic asymmetric radical reactions. |
 Zhi Ren | Zhi Ren obtained his M.S. degree from the Illinois Institute of Technology in 2011 and PhD degree from the University of Texas at Austin in United States under the supervision of Prof. Guangbin Dong in 2016. Starting from 2016, he went to Emory University for postdoctoral research under the guidance of Prof. Huw Davies and Prof. Djamaladdin (Jamal) Musaev. In 2020, he joined Shenzhen Technology University, and he is now an associate professor in the College of Pharmacy. His research interests include the transition metal-catalyzed C–H bond functionalization and related computational studies and the application of new methodologies for drug discovery and development. |
 Shou-Guo Wang | Shou-Guo Wang obtained his PhD from the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences in 2015 under the supervision of Prof. Shu-Li You before doing postdoctoral research with Prof. Nicolai Cramer at École Polytechnique Fédérale de Lausanne (EPFL). In 2020, he joined the Shenzhen Institutes of Advanced Technology (SIAT), Chinese Academy of Sciences, as a principal investigator. His main research interest encompasses the discovery of new catalytic systems in organic chemistry and beyond to enable their implementation in the synthesis of biologically active molecules. |
1. Introduction
Functionalized electron-deficient N-containing heteroarenes are common motifs in natural products (Fig. 1) and more importantly in numerous bioactive compounds and approved drugs (Fig. 2). The development of asymmetric synthetic strategies for constructing such frameworks has thus attracted significant attentions, and a broad range of synthetic methods have been established. In the field of catalytic enantioselective C–H bond functionalization, numerous achievements have been witnessed over the past few years,1 and these methodologies have emerged as increasingly robust tools and highly atom- and step-economical approaches toward various functionalized chiral electron-deficient N-containing heteroarenes. Recently, various research groups with different perspectives have made tremendous efforts to develop many elegant methods and reactions, and outstanding progress in new types of N-containing heteroarene substrates, new reactivities, catalytic systems, and the corresponding applications in the synthesis of bioactive compounds has been demonstrated (Fig. 3). Although impressive progress has been achieved in this field, compared to the well-developed and summarized asymmetric C–H bond functionalization of electron-rich heteroarenes,1c,2 the investigations on direct asymmetric C–H bond functionalization of electron-deficient heteroarenes remain underdeveloped and further advances are highly sought after.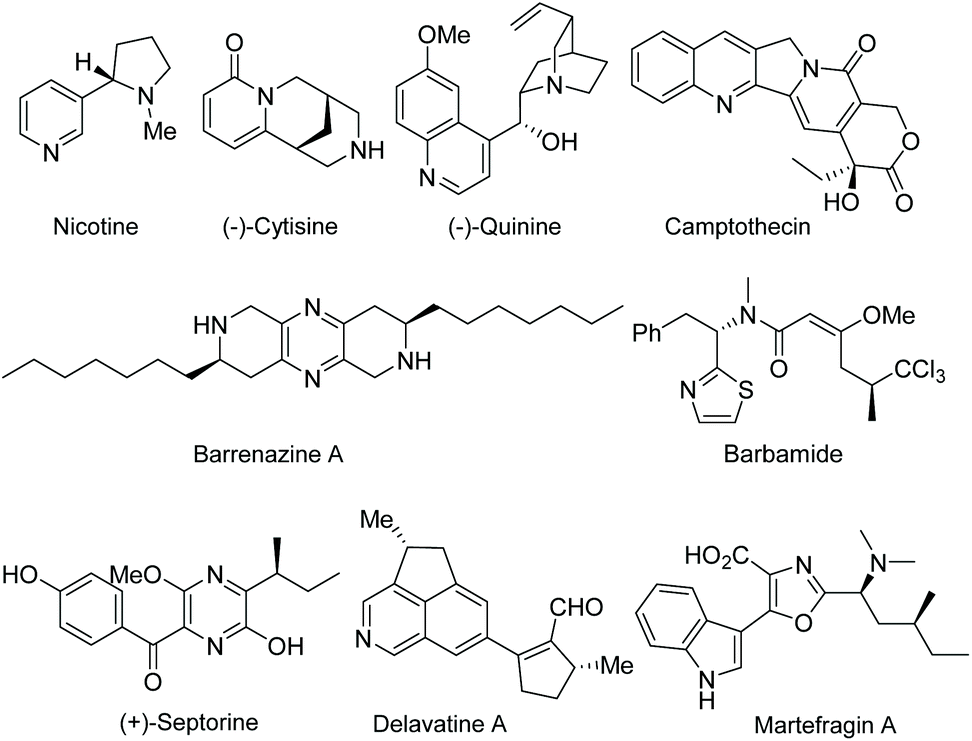 | ||
| Fig. 1 Representative natural products containing electron-deficient N-heteroarenes. | ||
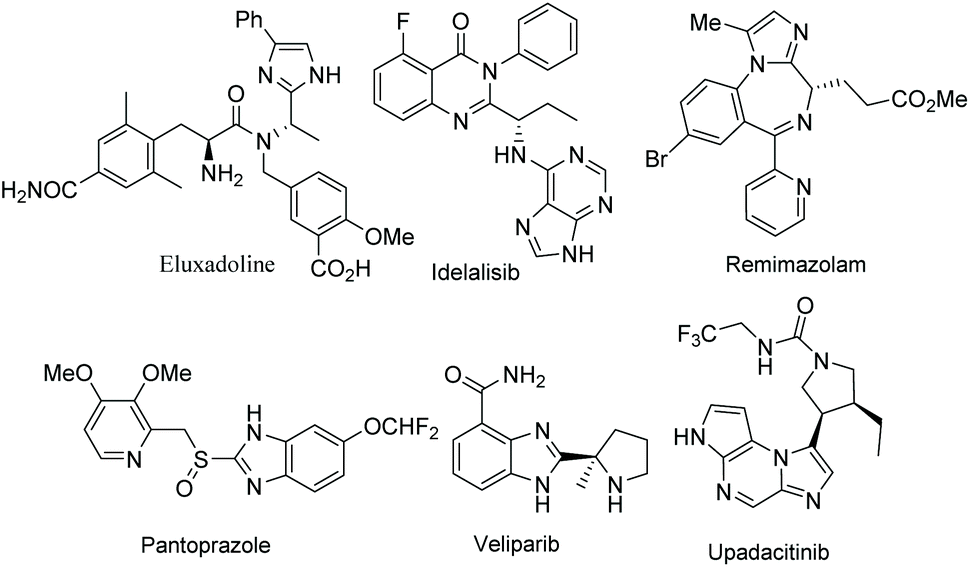 | ||
| Fig. 2 Representative approved drugs containing electron-deficient N-heteroarenes. | ||
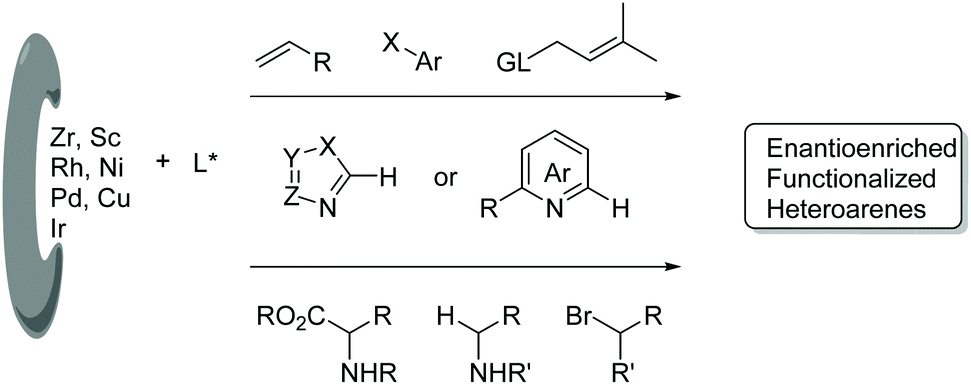 | ||
| Fig. 3 Direct enantioselective C–H bond functionalization on electron-deficient N-heteroarenes. | ||
Owing to the importance of highly functionalized N-containing heteroarenes, recent advancement in constructing functionalized N-containing heteroarenes through a direct C–H functionalization protocol has been summarized in several reviews. In 2013, Blackmond and Baran reported an overview of direct C–H bond functionalization of electron-poor aromatic heterocycles through radical addition processes.3 In 2017, Murakami, Itami and coworkers summarized the recent advances in the C–H functionalization of azines.4 In 2018, Kapur reviewed the progress in transition metal-catalyzed C–H functionalization of electron-deficient heteroarenes.5 However, these reviews mainly focused on non-asymmetric transformations and relevant catalytic asymmetric transformations have rarely been discussed. To the best of our knowledge, there is no comprehensive review published on catalytic asymmetric direct C–H functionalization of electron-deficient N-containing heteroarenes. Therefore, there is no doubt that a review on enantioselective direct C–H functionalization of electron-deficient N-containing heteroarenes would lead to a deeper understanding and ultimately promote further development in this field.
In this review, we mainly focus on the progress in the direct enantioselective C–H bond functionalization of electron-deficient N-heteroarenes, whereas the asymmetric C–H functionalization involving an electron-deficient N-heteroarene as a directing group6 is not included. Herein, diverse developments in enantioselective C–H functionalization of electron-deficient N-heteroarenes are summarized and classified according to the catalytic system and reaction type. Based on the mechanistic difference among these methods, this review is divided into two sections. The first section mainly describes transition metal-catalyzed reactions, and these reactions are organized in the order of the metal employed, such as rare-earth metals, Rh, Ni, Pd, and Cu. In this section, many reaction types and diverse heteroarenes are given. The second section focuses on light-promoted catalytic enantioselective Minisci-type reactions of diverse N-heteroarenes.
2. Transition metal catalysis
2.1 Rare-earth metal catalysis
The first example of rare-earth metal-catalyzed enantioselective C–H functionalization of electron-deficient heterocycles was developed in 1994 by the group of Jordan (Scheme 1).7 The extensive stoichiometric studies of cationic zirconium catalysis indicated that the Zr–C bond preferred 2,1-migratory insertion to give a branched insertion product due to the steric influence of the bulky Zr complex. The C–H cleavage was achieved by ortho-C–H metalation with the help of the pyridine nitrogen. The chiral cationic Cat-1 was prepared using a Brintzinger's C2-symmetric chiral biscyclopentadienyl ligand, and when Cat-1 was combined with hydrogen gas and 1-hexene, 2-picoline was converted to the alkylated product with low turnovers in 58% ee. The hydrogen gas was used to enable the turnover of the catalyst by hydrogenolysis of the newly formed Zr–C bond.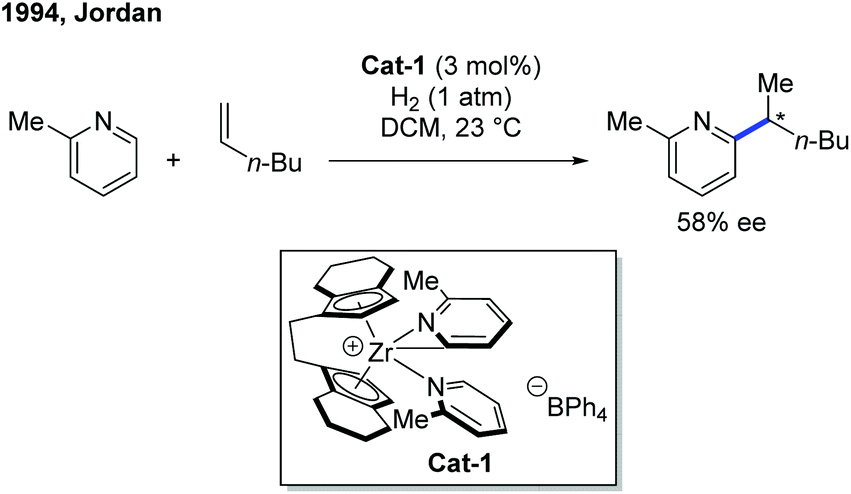 | ||
| Scheme 1 Zr-Catalyzed asymmetric C–H alkylation of 2-methylpyridine. | ||
Enlightened by Jordan's elegant work and their continuing interest in this field, in 2014, Hou and co-workers realized an enantioselective alkylation of pyridines with terminal olefins by the use of a cationic scandium catalyst (Scheme 2).8 A handful of chiral half-sandwich rare-earth metal catalysts with different rare-earth metals and binaphthyl-substituted cyclopentadiene ligands were prepared and evaluated. Particularly, by taking advantage of chiral Sc-catalyst Cat-2, a diversity of pyridine derivatives can be transformed into chiral alkylated products through the C–H functionalization process in high yields and good to excellent enantioselectivities. Unlike Zr catalysis, the Sc–C bond formed after the migratory insertion can be protonated by the incoming substrate to give the alkylation product (Int-3 to Int-1, Scheme 2). This non-metal hydride mechanism ensured that 2-iodo- and 2-bromopyridines remained intact during the reaction.
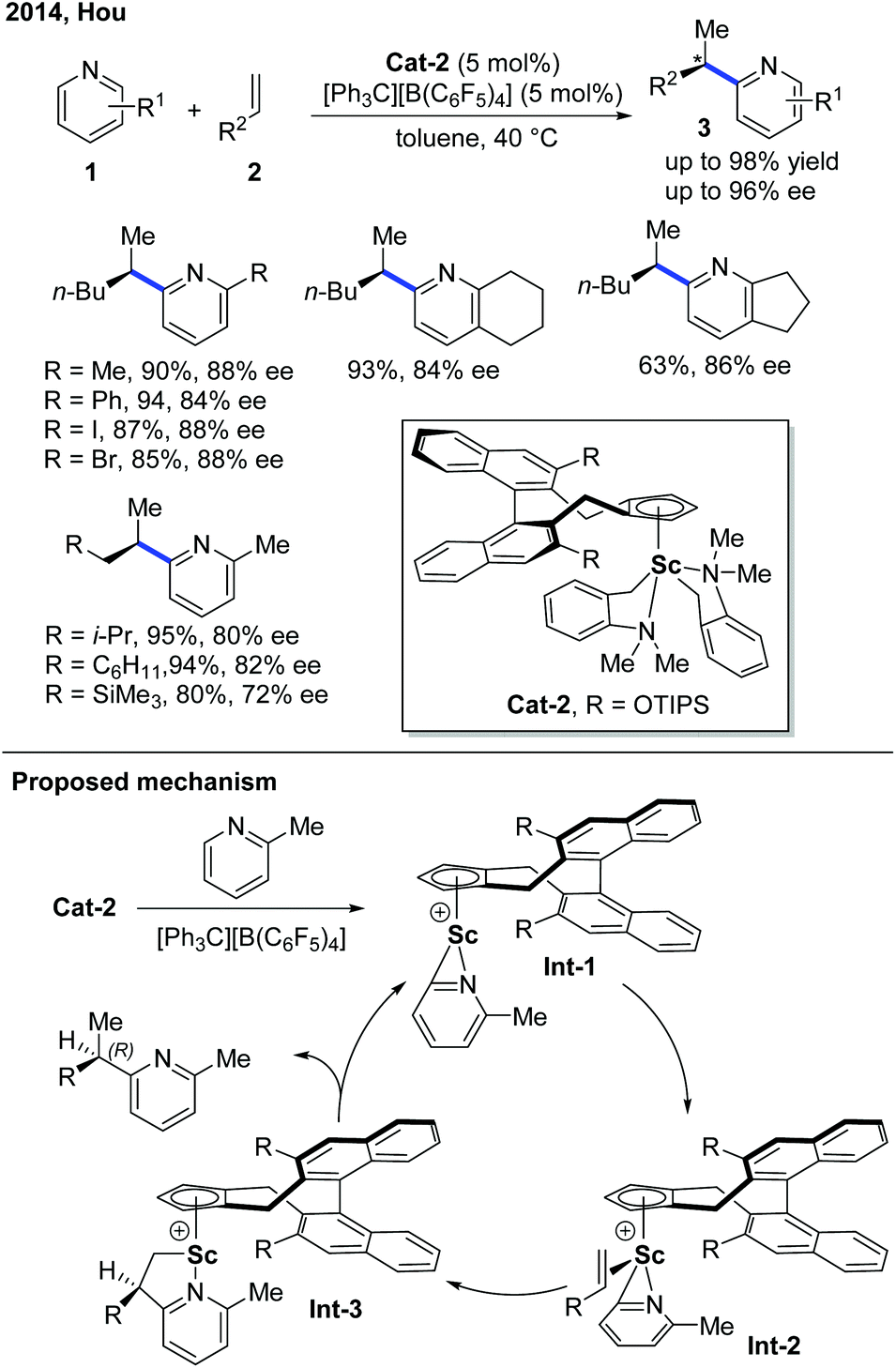 | ||
| Scheme 2 Sc-Catalyzed enantioselective C–H alkylation of pyridine derivatives with various alkenes. | ||
Six years later, Hou and co-workers further extended the Sc-catalyzed alkylation reaction to imidazoles for an asymmetric intramolecular cyclization (Scheme 3).9 The kinetic isotope effect (KIE) studies indicated a non-metal hydride mechanism with the C–H cleavage step being part of the rate-determining step. Different from most of the Rh- and Ni-catalyzed intramolecular endo-alkylation reactions, the exo-selectivity feature made Sc-catalyzed alkylation a complimentary asymmetric synthesis method of polycyclic imidazoles 5. In comparison with Cat-2, Cat-3 was furnished with a more sterically bulky TBDPS group for better enantiocontrol of the migratory insertion. The application of the product has been demonstrated by the Pd-catalyzed C–H oxidation and arylation of the proximal methyl group using the imidazole group as a directing group.
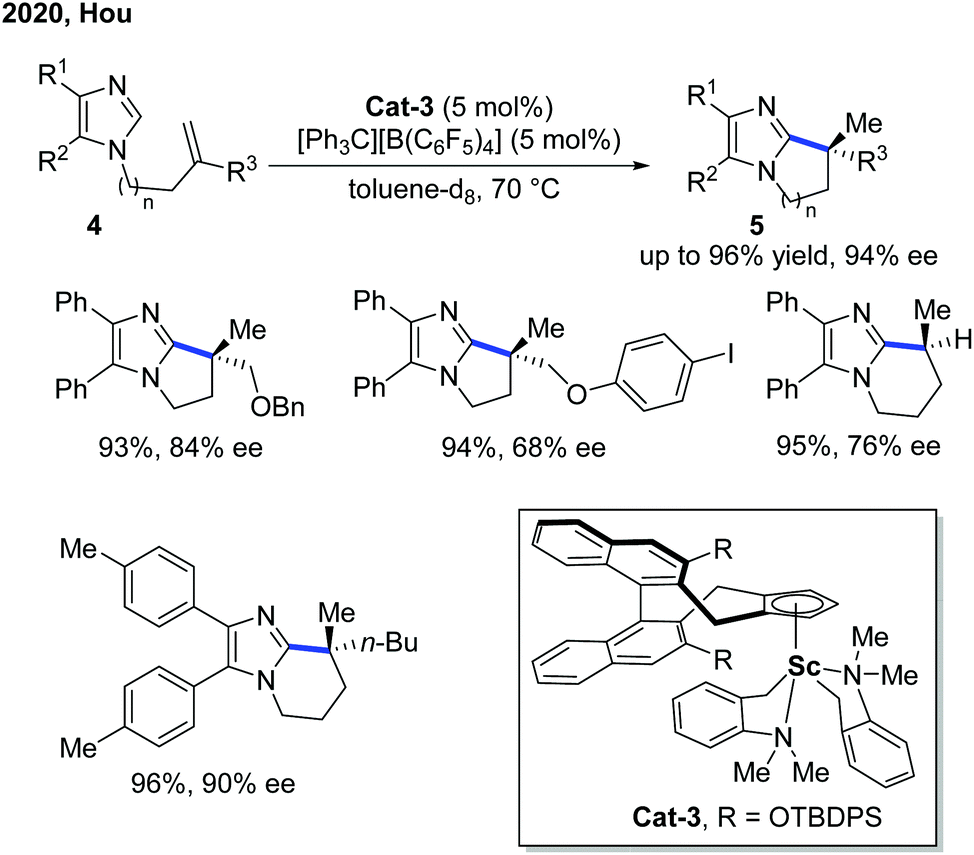 | ||
| Scheme 3 Sc-Catalyzed intramolecular C–H cyclization of imidazoles with tethered alkenes. | ||
In summary, rare-earth metal catalysis plays an important role in the asymmetric C–H alkylation of electron-deficient N-heterocycles, and the alkylation of pyridines and imidazoles has been successfully developed. As a consequence, it is still of great demand for establishing more diverse reaction types and N-heterocycle scopes, as well as a higher enantioselectivity. Nonetheless, one major disadvantage of rare-earth metal catalysis resides in the requirement for premade Cp catalysts, thus imposing restrictions on the widespread usage of these catalysts compared to others in situ generated by achiral pre-catalysts and chiral ligands.
2.2 Rh catalysis
Although the Rh-catalyzed C–H alkylation has been extensively studied, asymmetric transformations with electron-deficient heterocycles are still very limited. The first example based on Rh was reported by Ellman, Bergman and co-workers in 2009, in which they have disclosed an enantioselective endo-selective intramolecular alkylation for imidazoles using a Rh catalyst (Scheme 4).10 Bidentate ligands were found to be inhibitory in the previous works on non-enantioselective reactions. Indeed, most of the bidentate ligands tested gave no desired product during the reaction optimization. However, Tangphos, a bidentate phosphine ligand, is an interesting exception. In combination with a Rh pre-catalyst, the asymmetric cyclization of imidazoles via C–H alkylation was successfully performed in high yield and moderate to excellent ee (up to 90% yield and 98% ee) at an elevated temperature. Its reactivity was explained by the fact that Tangphos underwent a partial dissociation to form a monodentate ligand, releasing a coordination site.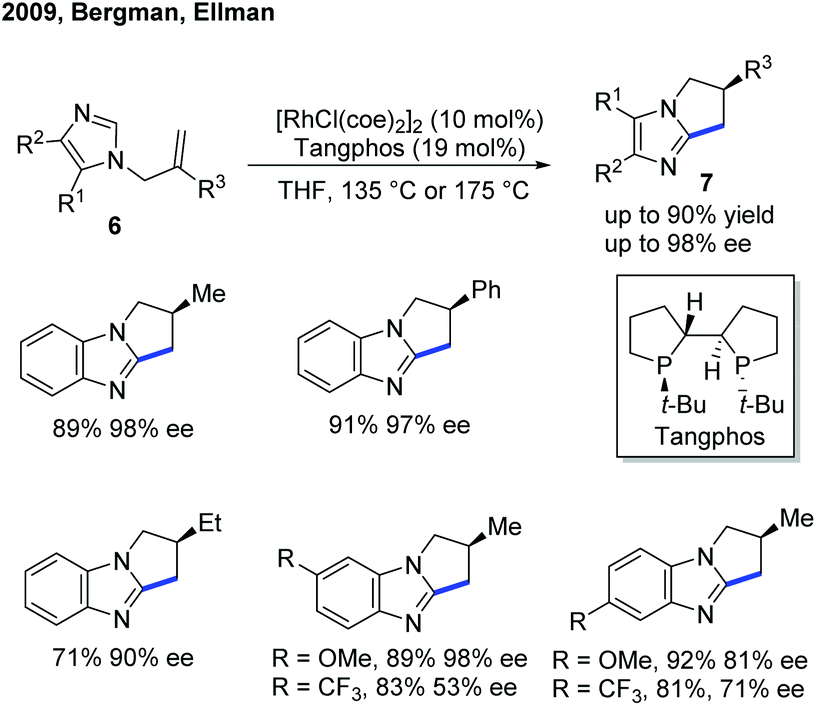 | ||
| Scheme 4 Rh(I)-Catalyzed intramolecular C–H cyclization of imidazoles with tethered alkenes. | ||
Another work on the Rh-catalyzed enantioselective intermolecular C–H alkylation of benzoxazoles with α-substituted acrylates was presented by the Rovis group in 2015 (Scheme 5).11 In the presence of a Rh pre-catalyst and a bulky chiral bisphosphine ligand (CTH-Xylyl-P-Phos, L1), the α-stereocenter of the ester group was controlled. Through well-designed deuterium labelling experiments, the classical enantioselective protonation mechanism was excluded. Based on these insightful mechanistic investigations, a significantly different mechanism for the asymmetric induction and epimerization process was proposed for this transformation.
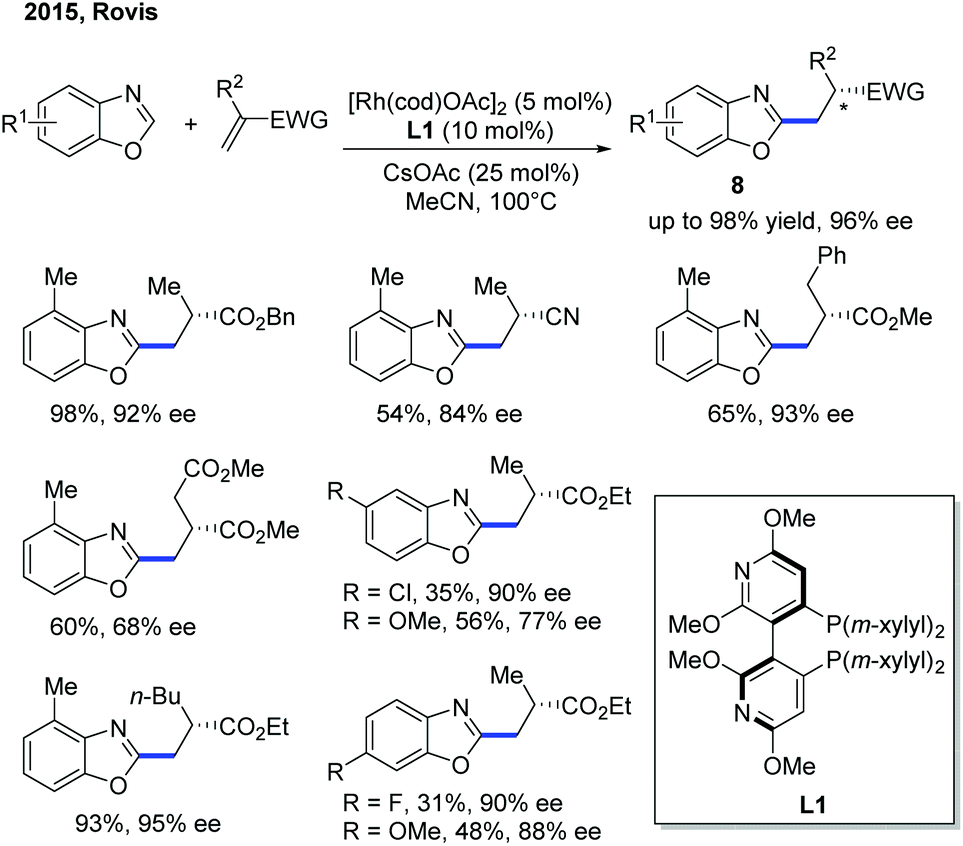 | ||
| Scheme 5 Rh(I)-Catalyzed intermolecular C–H alkylation of benzoxazoles with acrylates. | ||
In summary, despite the numerous studies on the Rh-catalyzed C–H alkylation of arenes, examples with electron-deficient heterocycles are unexpectedly rare. The scope of the heterocycles is limited to imidazoles and benoxazoles, and only the alkylation reactions have been developed. Therefore, further exploration is needed to expand the methodology to more reaction types and to incorporate a wider variety of heteroarene substrates.
2.3 Ni catalysis
The related example of Ni catalysis emerged relatively late than other metal catalysts. In 2015, the first example of Ni-catalyzed asymmetric C–H cyclization of pyridones by the Cramer group set a milestone in this aspect (Scheme 6).12 AlMe3 was employed as a Lewis acid, presumably to facilitate the C–H cleavage step. The switching of exo selectivity to endo by adding N-heterocyclic carbenes (NHCs) was found to be critical for the concise and elegant synthesis of lupin alkaloid (±)-cytisine, and this transformation could also be developed into an enantioselective version with asymmetric nickel catalysis. Using Hong's chiral NHC ligand, L2,13 two endo-selective asymmetric C–H cyclizations were accomplished in good yield with 57% ee.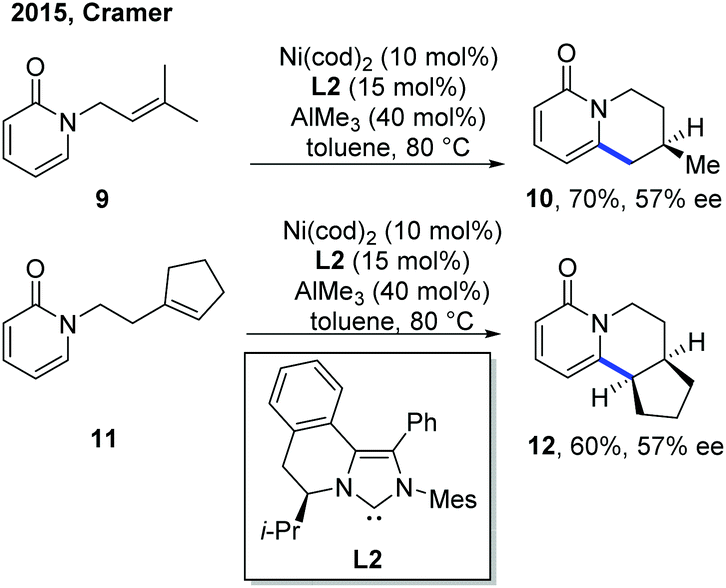 | ||
| Scheme 6 Two enantioselective examples of Ni-catalyzed intramolecular C–H cyclization of pyridones with tethered alkenes. | ||
To improve the enantioselectivity in the previous Ni(0)-catalyzed endo-selective cyclization, the Cramer group developed a Ni-catalytic system ligated with a bulky and electron-rich NHC ligand L314a–f and the reaction worked well in the presence of MAD, methylaluminum bis(2,6-di-t-butyl-4-methylphenoxide), as a Lewis acid (Scheme 7).15 In general, the products 14 can be obtained in good yield with good to excellent ee. Interestingly, the internal alkene substrate with the Ph substituent at the end gave the product as a racemic mixture, while the one with the cyclopentenyl substituent produced the exo product. The alkene conformation also contributed to the enantioselectivity of the product, and (E)-alkenes gave a higher ee.
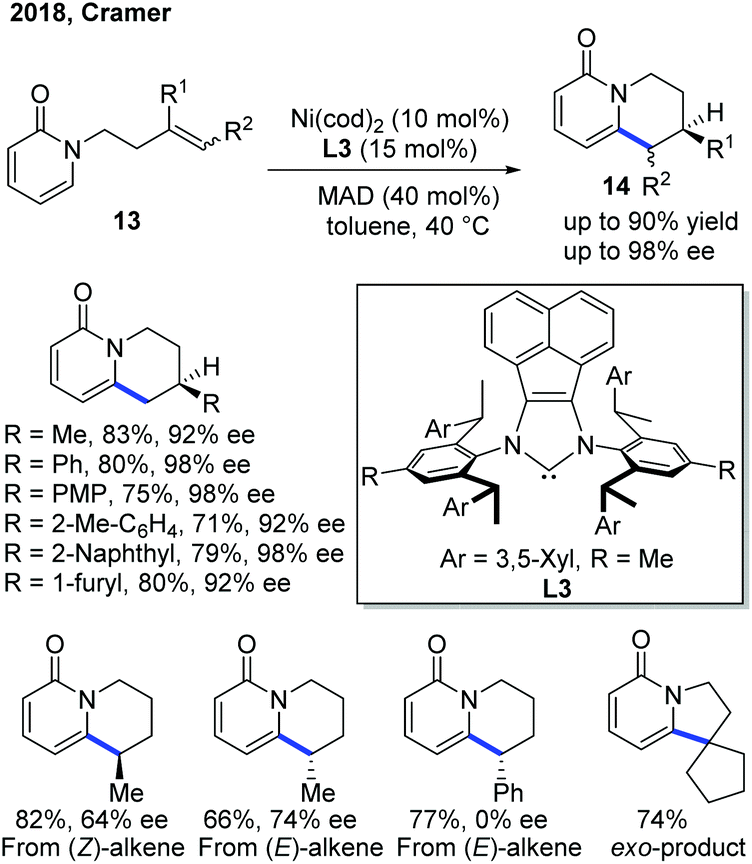 | ||
| Scheme 7 Improvement of enantioselectivity in Ni-catalyzed C–H cyclization using a combination of a bulky NHC ligand and a Lewis acid. MAD: (2,6-tBu2-4-Me-C6H2O)2AlMe. | ||
In 2018, the Ye group demonstrated the first enantioselective Ni-catalyzed exo-selective alkylation of imidazoles (Scheme 8).16 Although the achiral exo-selective cyclization of imidazoles with alkenes has been developed by Bergman and Ellman,17 the enantioselective reaction is still quite challenging. The Ye group's interest in the secondary phosphine oxide (SPO) ligand enabled them to discover the solution for this problem. The combination of a nickel pre-catalyst, AlMe3 as a Lewis acid, and SPO ligand L4 was essential for the stereocontrol of this transformation, giving good to excellent yields with a high ee. The SPO ligand was proposed to link the nickel catalyst and the aluminium Lewis acid for the asymmetric induction. The deuterium labelling studies showed that the reaction proceeded through a metal hydride intermediate, which was the first example that involved such a mechanism.
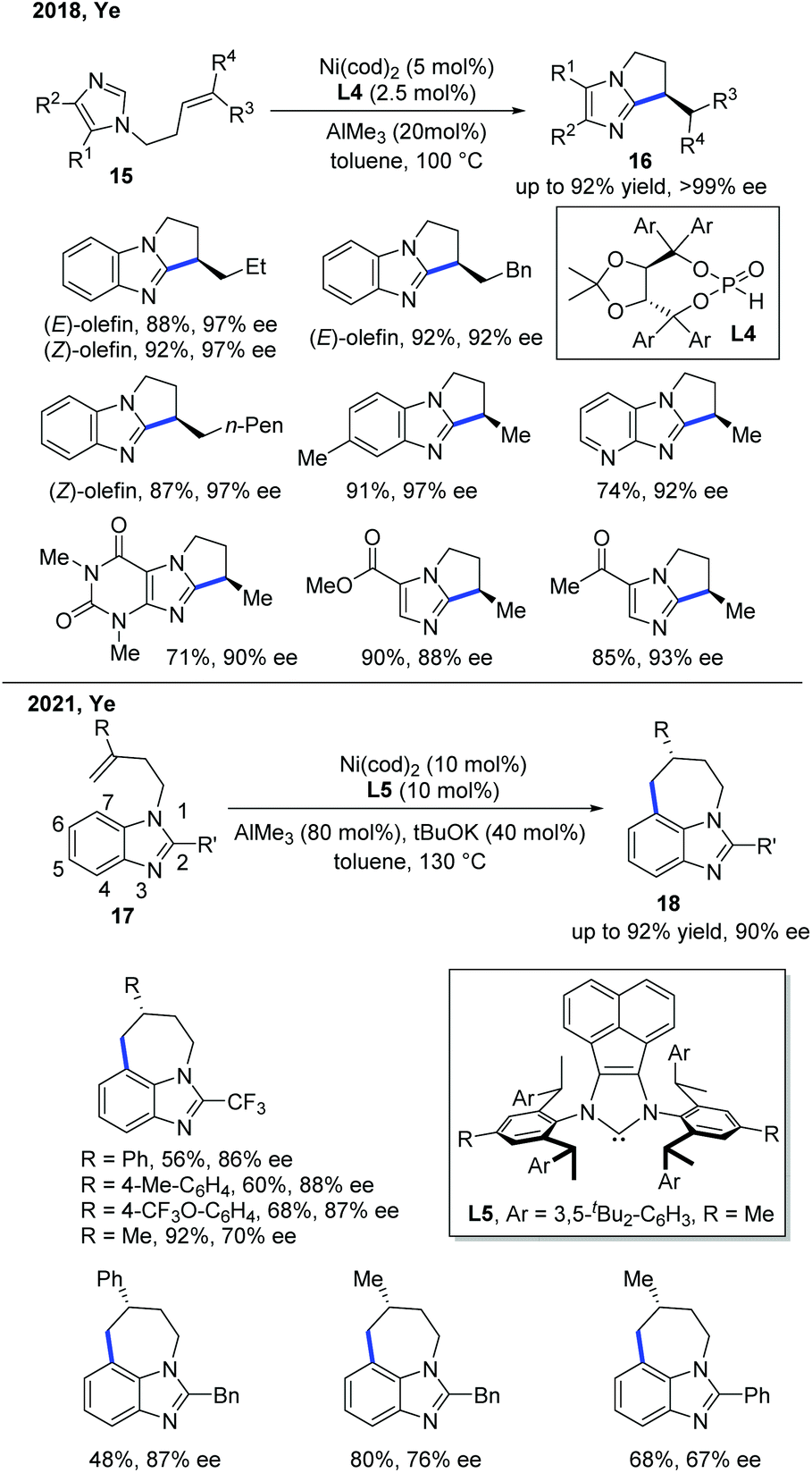 | ||
| Scheme 8 Ni-Catalyzed enantioselective C–H cyclization of imidazoles. | ||
Very recently, the Ye group reported another relevant Ni-catalyzed enantioselective C–H alkylation of benzoimidazoles to construct 7-membered tricyclic imidazole products (Scheme 8).18 To achieve the desired site-selectivity, the more reactive C2 position was occupied with other functional groups, and the cleavage of the C–H bond at the C7 position was accessed by employing a Ni catalyst with the help of AlMe3. The role of AlMe3 was believed to facilitate the reaction in two aspects: (1) promoting C–H bond activation by decreasing the electron density of the aromatic ring, and (2) increasing the steric hindrance to reduce the cost of entropy for 7-membered ring formation. Interestingly, a catalytic amount of base was needed to suppress the isomerization of the alkene, preventing the decomposition of the substrate. The chiral NHC ligand L5 was found to be optimal, providing high levels of enantiocontrol in this transformation. The intramolecular C–H cyclization was proposed to proceed via a metal-hydride mechanism.
The following year, the Ackermann group discovered an endo-selective C–H cyclization of imidazoles proceeding in high enantioselectivity under an aluminum-free condition (Scheme 9).19 Under the previous reaction conditions, aluminum served as the Lewis acid during the reaction, but it complicated the work-up process. By utilizing a better ligand, JoSPOphos (L6) with a classic Ni pre-catalyst, the intramolecular cyclization occurred in high yield and high enantioselectivity. Deuterium-labelling studies showed quite different results compared to Ye's work with the SPO ligand. In addition, a Ni–JoSPOphos–cod complex was prepared, characterized, and tested for reactivity to gain more insights into the reaction. Based on these studies, the authors proposed a non-metal hydride mechanism, and the C–H bond cleavage occurred by a ligand-to-ligand hydrogen transfer in support of the kinetic results.
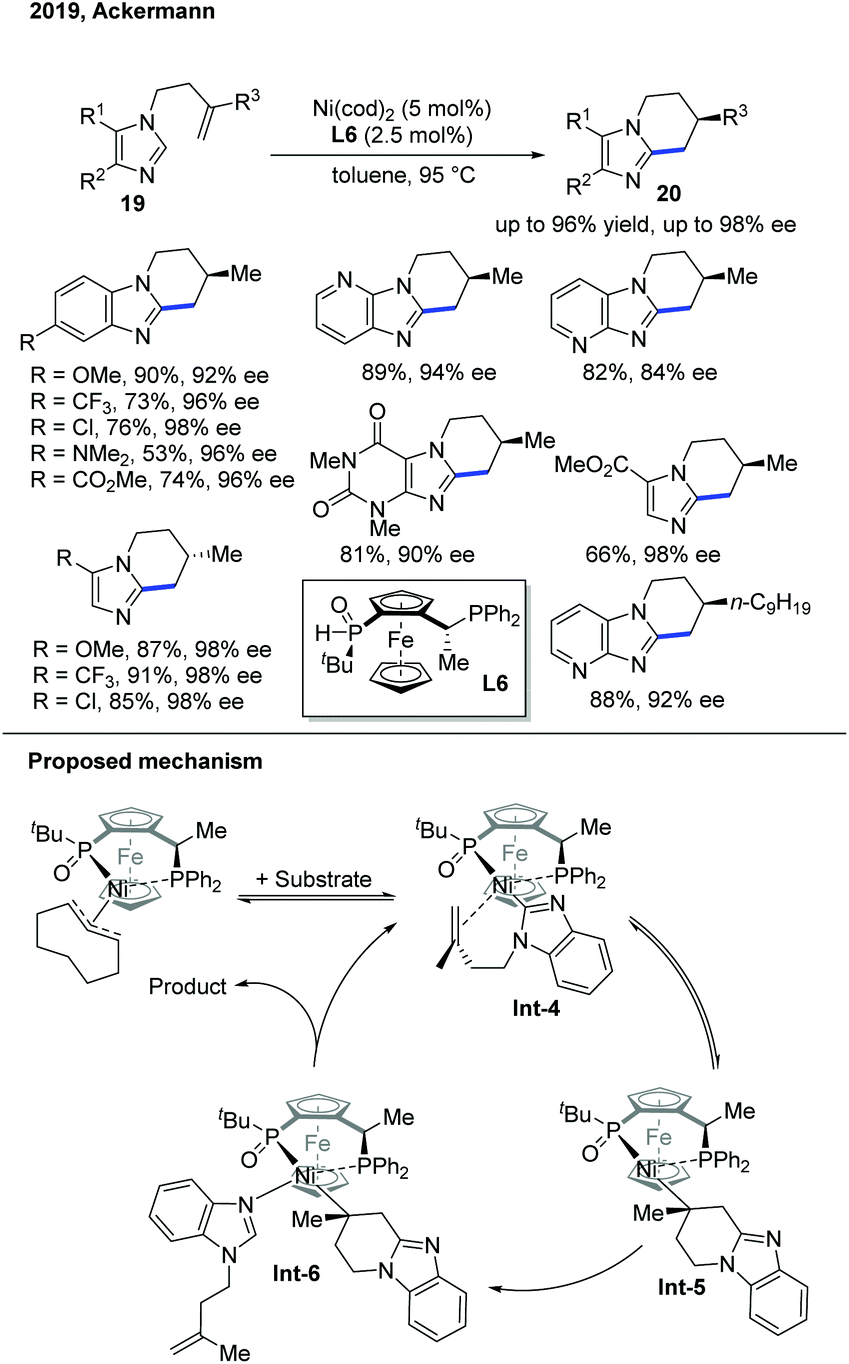 | ||
| Scheme 9 Ni-Catalyzed enantioselective endo C–H cyclization of imidazoles with a chiral SPO ligand under aluminium-free conditions. | ||
In 2019, the Shi group presented a nickel-catalyzed regio- and enantioselective endo C–H cyclization of pyridines with alkenes (Scheme 10).20 The optimal conditions were a Ni pre-catalyst, chiral NHC/hydrochloride acid salt, a catalytic base, and MAD to synthesize polycyclic pyridines. A Gawley-type NHC ligand with a saturated backbone (L7) was found to be the optimal ligand to achieve high yield and good stereocontrol. The alkenes tethered to three different positions of pyridines were all successfully cyclized. Pyridine derivatives with a C3-tethered alkene group were the most interesting substrates as the cyclization could occur at either the C2 or C4 position of the pyridine core, yet only the C4 cyclized product 22 was observed with high ee. The deuterium-labelling experiment implied that the reaction involved a metal-hydride mechanism.
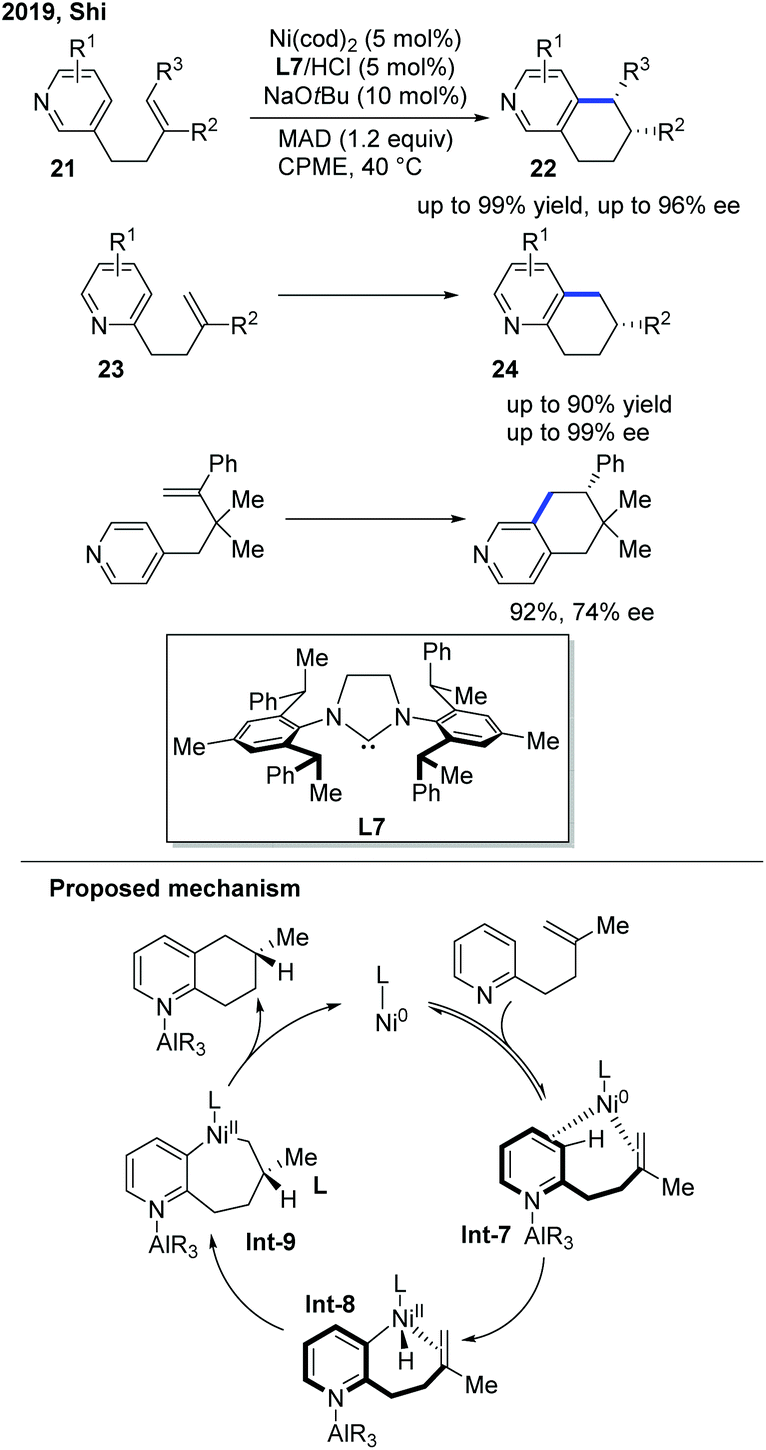 | ||
| Scheme 10 Ni-Catalyzed enantioselective endo regio- and enantioselective C–H cyclization of pyridines with alkenes. MAD: (2,6-tBu2-4-Me-C6H2O)2AlMe. | ||
Recently, Shi and Xu provided an alternative and more practical method for enantioselective intramolecular C–H alkylation of pyridones by the use of easily prepared NHC ligand L7, with commercially available AlEt3 (Scheme 11).21 Moreover, the authors further extended the scope of heterocycles to 4-pyrimidone derivatives 27. A metal hydride mechanism was also proposed, which was supported by deuterium-labelling experiments.
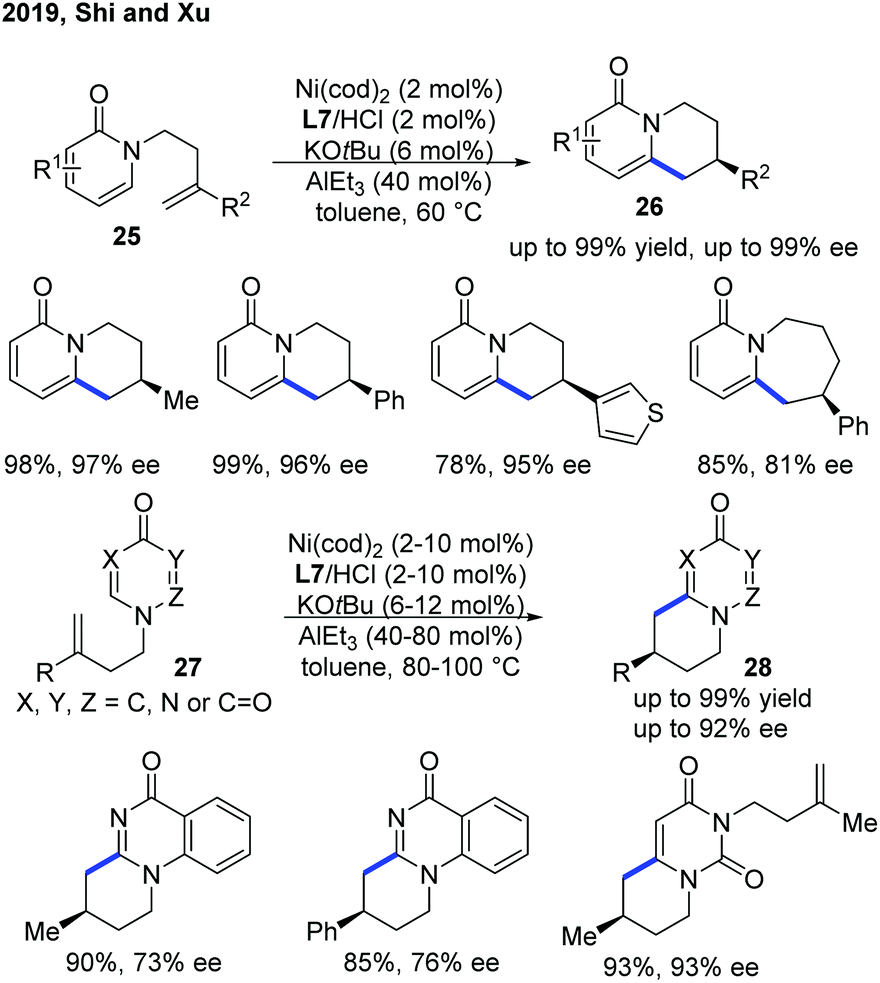 | ||
| Scheme 11 Practical Ni-catalyzed enantioselective endo C–H cyclization of pyridones and pryimidones with tethered alkenes. | ||
In summary, as the abundant first-row metal, nickel is widely used in the asymmetric C–H functionalization of electron-deficient N-heteroarenes. The scope of heteroarenes covers pyridones, imidazoles, pyridines, and pyrimidones. Many groups have made great contributions in this aspect to enable both endo and exo cyclization of N-heterocycles. However, the reaction type of Ni catalysis is rather limited to intramolecular alkylation reactions, and thus intermolecular reaction mode as well as more diverse functionalization types are still in high demand.
2.4 Pd catalysis
In 2015, the Zhu group disclosed the first Pd-catalyzed enantioselective Heck/intermolecular C–H functionalization of electron-deficient heterocycles (Scheme 12).22 This domino process starts with enantioselective intramolecular carbopalladation of alkenes followed by nucleophilic trapping of the σ-C(sp3)–Pd complex with heteroarenes via a C–H activation. The stereocontrolling step was the migratory insertion step in the Heck reaction, and the C–H activation step was not the rate-limiting step. The scope of heteroarenes used in the reaction involves oxadiazoles, benoxazole and benzothiazole. The application of this methodology was showcased by further transformations of the oxadiazole products into two natural products, (+)-esermethole and (+)-physostigmine. | ||
| Scheme 12 Pd-Catalyzed enantioselective domino Heck/intermolecular C–H cyclization with oxadiazoles, benoxazole and benzothiazole. TMG: N,N,N,N-tetramethylguanidine. | ||
As a continuation of their previous work, the Zhu group developed a double carbopalladation/intermolecular C–H alkylation sequence of N-(2-iodophenyl)-N-methyl methacrylamide derivatives 32 with oxadiazole (Scheme 13).23 While the previously used monophosphine ligand L8 showed no activity towards this transformation, the chiral biphosphine ligand L9 provided the dimer products 34 in uniformly excellent enantioselectivities. Furthermore, the addition of Ag3PO4 could enhance the diastereoselectivity of the reaction. Notably, the enantioselectivity of the products could be easily predicted by measuring the dr of the reaction by 1H NMR, which greatly helped in hastening the screening process. Further transformation of the products into key intermediates for the synthesis of natural products was also demonstrated to emphasize the significance of this new method.
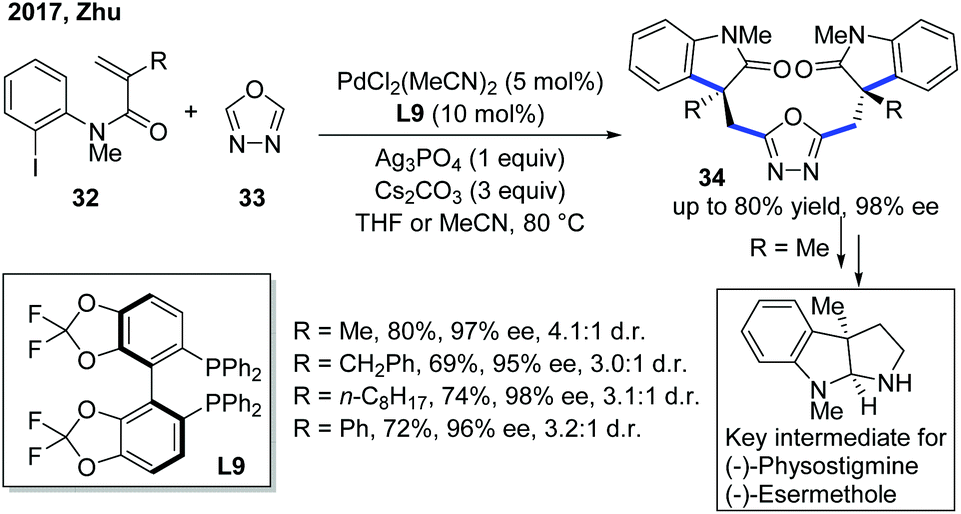 | ||
| Scheme 13 Pd-Catalyzed double Heck/intermolecular C–H cyclization with oxadiazoles to enhance the enantiopurity of key intermediates for natural product synthesis. | ||
In 2017, the Zhu group demonstrated the enantioselective domino Narasaka–Heck/C–H alkylation of electron-deficient heterocycles (Scheme 14).24 The first step of this reaction involves the oxidative addition of Pd(0) into the labile N–O bond to form an iminopalladium(II) complex Int-10. The Pd(II) complex goes through an enantioselective Heck reaction with the alkene to give dihydropyrrole and forms a new Pd–C(sp3) bond, which is the stereodetermining step governed by the chiral biaryl bisphosphine ligand, Synphos, L10. The Pd–C(sp3) bond was constructed through deprotonation of the oxadiazole and then transmetallation to give Pd complex Int-12. The subsequent reductive elimination delivered the desired product. The mechanistic investigation indicated that the radical background reaction was concurrently occurring and explained the difficulty of developing a highly enantioselective reaction.
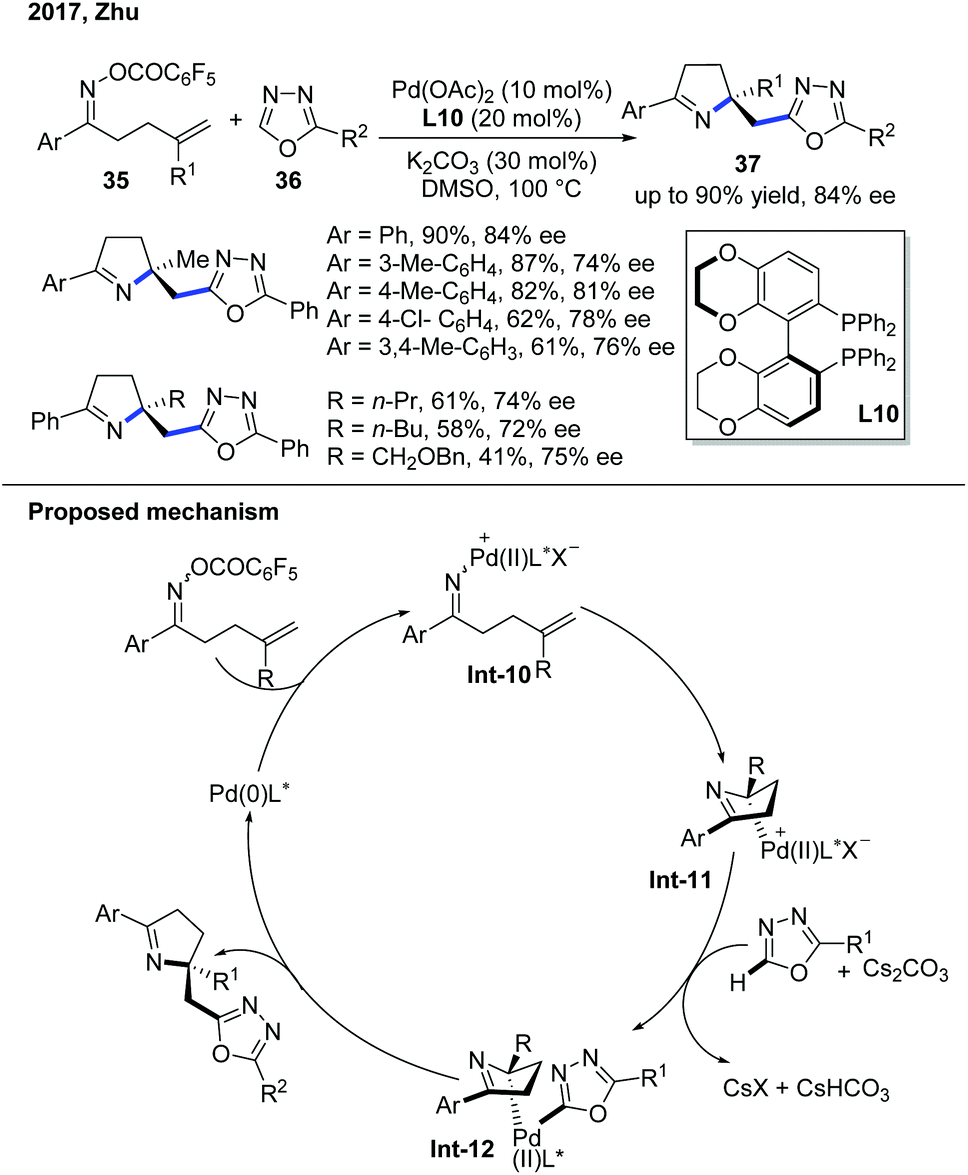 | ||
| Scheme 14 Pd-Catalyzed enantioselective Narasaka–Heck/C–H alkylation with oxadiazoles. | ||
Apart from centrally chiral compounds, Pd catalysis could also enable an access to axially chiral products via C–H functionalization of electron-deficient N-heterocycles. In 2020, Cramer, Baudoin and co-workers developed the first intermolecular Pd-catalyzed atropo-enantioselective C–H arylation of N-heteroarenes with aryl bromides (Scheme 15).25 Using a Pd(0) precursor with H8-BINAPO as a chiral ligand (L11), the scope of N-heteroarenes encompassed triazoles, pyrazoles and an imidazo[1,2-a]pyrimidine. While triazoles and pyrazoles generally delivered the atropisomeric heterobiaryls 40 in excellent enantioselectivities (up to 94% ee), the use of imidazo[1,2-a]pyrimidine as a substrate resulted in a low ee value along with two regioisomers. A deuterium kinetic isotope experiment suggested a concerted metalation–deprotonation mechanism for C–H activation, which was also the rate-determining step. Furthermore, the stereocontrolling step likely pointed at the reductive elimination as indicated by the effect of the biaryl dihedral angle of the bisphosphine monoxide ligand on the enantioselectivity.
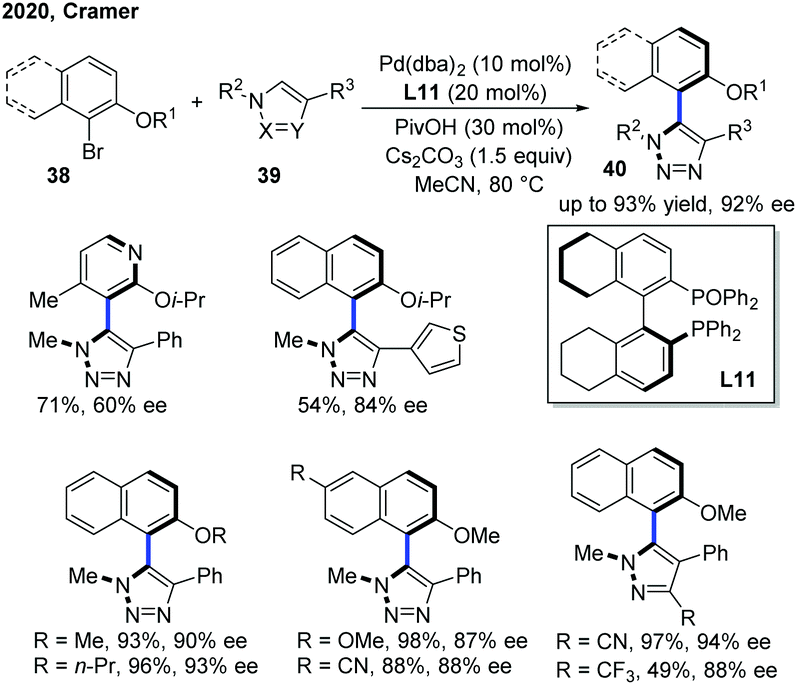 | ||
| Scheme 15 Pd-Catalyzed intermolecular enantio- and atroposelective C–H arylation of heteroarenes. | ||
In summary, there are two main types of Pd-catalyzed enantioselective C–H functionalizations of electron-deficient heterocycles. One is the domino alkylation reaction developed by the Zhu group, and another one is the atroposelective C–H arylation developed by Cramer and Baudoin. The heterocycle scope of this work involves oxadiazoles, triazoles, pyrazoles, and imidazo[1,2-a]pyrimidine. Unlike other metal catalysts, the C–H cleavage step here is not the first step in these Pd-catalyzed reactions.
Different transition metals, including Zr, Sc, Rh, Ni, and Pd, have been employed for asymmetric C–H functionalization of electron-deficient heteroarenes. After decades of development, the scope of heteroarenes has been expanded from heteroarenes with mono nitrogen, e.g. pyridines, to heteroarenes with multiple heteroatoms, e.g. imidazoles. Most of these reactions are intra- or intermolecular alkylation of heteroarenes, with one exception of arylation from the Cramer group. Therefore, more reaction types are highly desirable, and another significant direction for future development is to include different heteroarenes, especially electron-deficient ones.
2.5 Cu catalysis
In 2016, Ohmiya, Sawamura and co-workers reported a copper-catalyzed enantioselective C(sp2)–H allylation of various azoles with γ,γ-disubstituted primary allylic phosphates, constructing an all-carbon quaternary stereogenic center at the α-position of the electron-deficient azole heteroaromatic ring (Scheme 16).26 The combination of a copper(I) salt with a newly developed chiral NHC ligand L12 bearing a naphtholic hydroxy group was demonstrated to play an important role in achieving favorable reactivity, high regioselectivity for branched products 43 (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to >99:1 b:l) and good to excellent enantioselective introduction (74–93% ee), providing a convenient and efficient access to a range of enantioenriched branched α-chiral alkylated benzothiazoles and benzoxazoles. However, the C–H functionalization of 4-phenyloxazole proceeded with a slightly decreased enantioselectivity (66–78% ee) and low levels of branch regioselectivity (54:46 to 96:4 b:l). Consistent with the previous studies,27 a stoichiometric amount of tBuOLi as a base was essential for the success of this transformation, and the catalytic cycle was initiated with a tBuOLi-assisted C–H cupration of azoles to generate the key heterocuprate species.
:4 to >99:1 b:l) and good to excellent enantioselective introduction (74–93% ee), providing a convenient and efficient access to a range of enantioenriched branched α-chiral alkylated benzothiazoles and benzoxazoles. However, the C–H functionalization of 4-phenyloxazole proceeded with a slightly decreased enantioselectivity (66–78% ee) and low levels of branch regioselectivity (54:46 to 96:4 b:l). Consistent with the previous studies,27 a stoichiometric amount of tBuOLi as a base was essential for the success of this transformation, and the catalytic cycle was initiated with a tBuOLi-assisted C–H cupration of azoles to generate the key heterocuprate species.
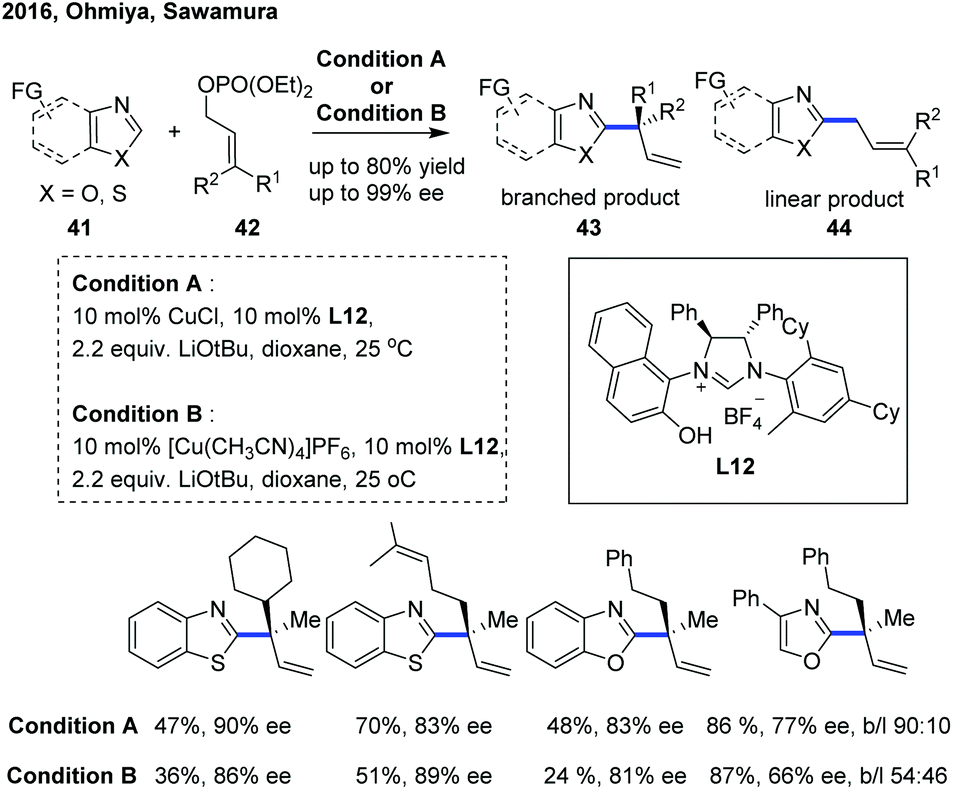 | ||
| Scheme 16 Copper-catalyzed enantioselective allylic alkylation of azoles. | ||
Recently, Liu and co-workers disclosed an enantioselective C(sp2)–H alkylation of azoles with racemic alkyl bromides (Scheme 17).28 With their newly developed copper(I)/cinchona alkaloid-derived N,N,P-ligand catalytic system, a range of α-chiral alkylated azoles 47 were obtained in good to high yields (up to 85% yield) and high enantioselectivities (up to 93% ee). Various alkyl bromides and different types of electron-deficient azoles, including 1,3,4-oxadiazoles, oxazoles, and benzo[d]oxazoles, were well compatible with this transformation. In addition, benzo[d]thiazole was also found to be a competent substrate, thus delivering the chiral product in good enantiocontrol (87% ee), albeit in a low yield (12%). The authors proposed that the in situ formed Cu(I)/N,N,P-ligand complex first reacted with azole via base-assisted C(sp2)–H metallation to give a Cu-azole species Int-13 (Scheme 19).27 The obtained Cu-azole species was able to reduce the alkyl bromide to alkyl radical Int-14 with concomitant generation of a Cu(II) species, which then combined to give a Cu(III) intermediate. Finally, reductive elimination from the Cu(III) intermediate proceeded to deliver the chirally alkylated azoles. In this transformation, the chiral Cu(I)/N,N,P-ligand catalyst exhibited an improved reducing capability to convert the alkyl halides to alkyl radicals under facile reaction conditions, thus suppressing the racemization of the obtained product.
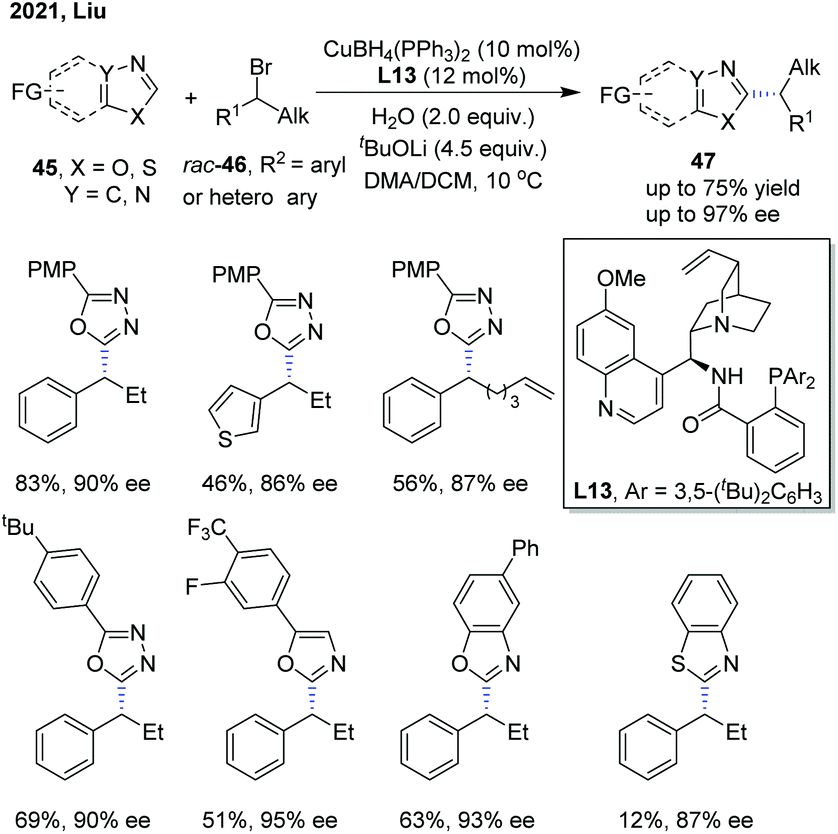 | ||
| Scheme 17 Copper-catalyzed asymmetric alkylation of azoles. | ||
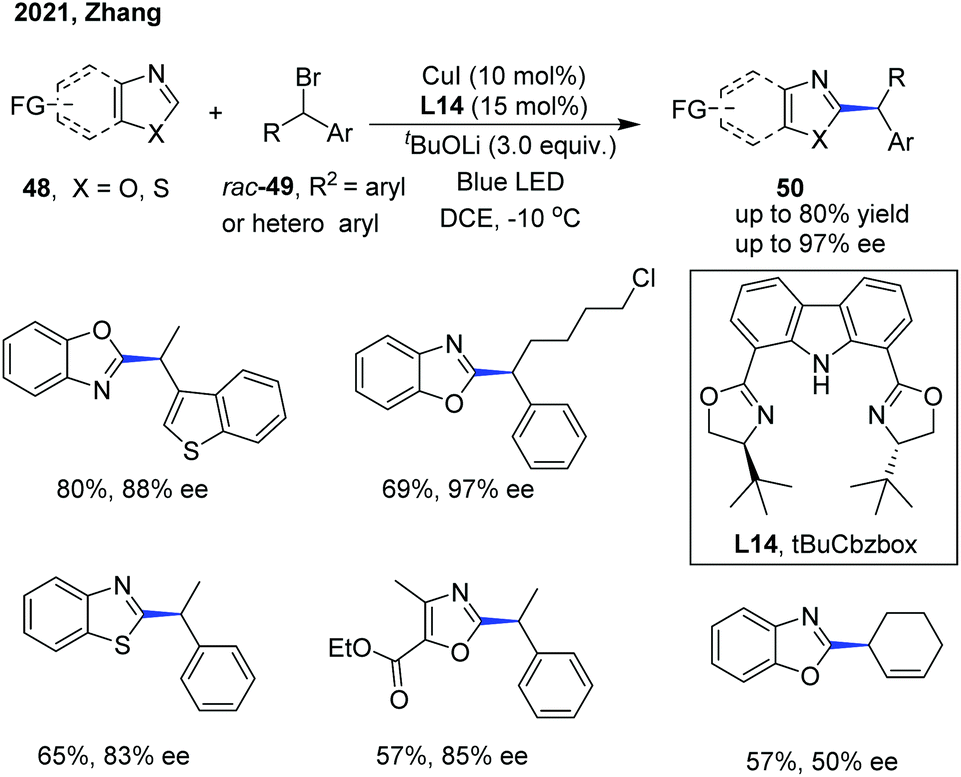 | ||
| Scheme 18 Visible light-promoted copper-catalyzed asymmetric alkylation of azoles. | ||
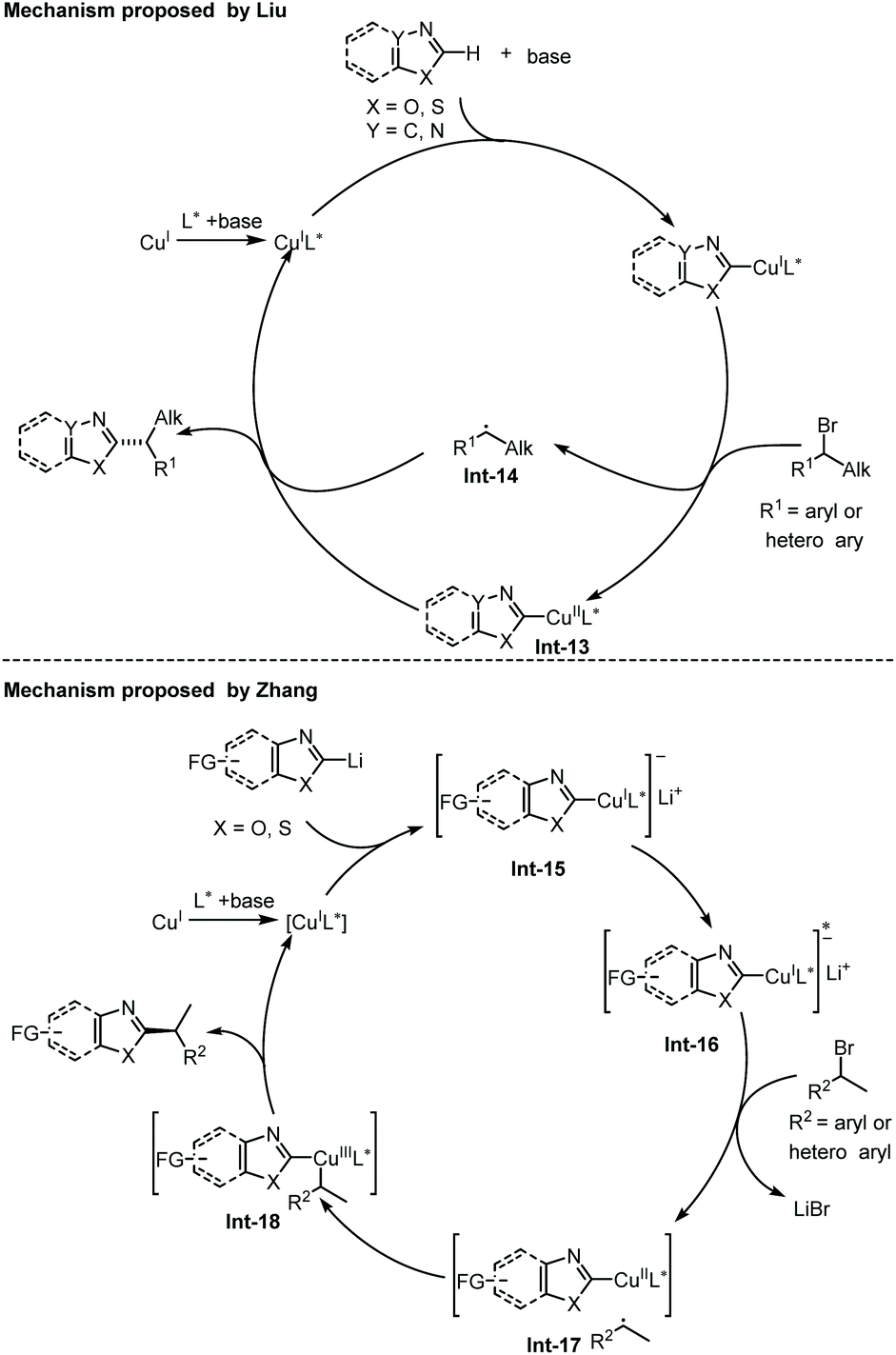 | ||
| Scheme 19 Proposed mechanism of copper-catalyzed asymmetric alkylation of azoles. | ||
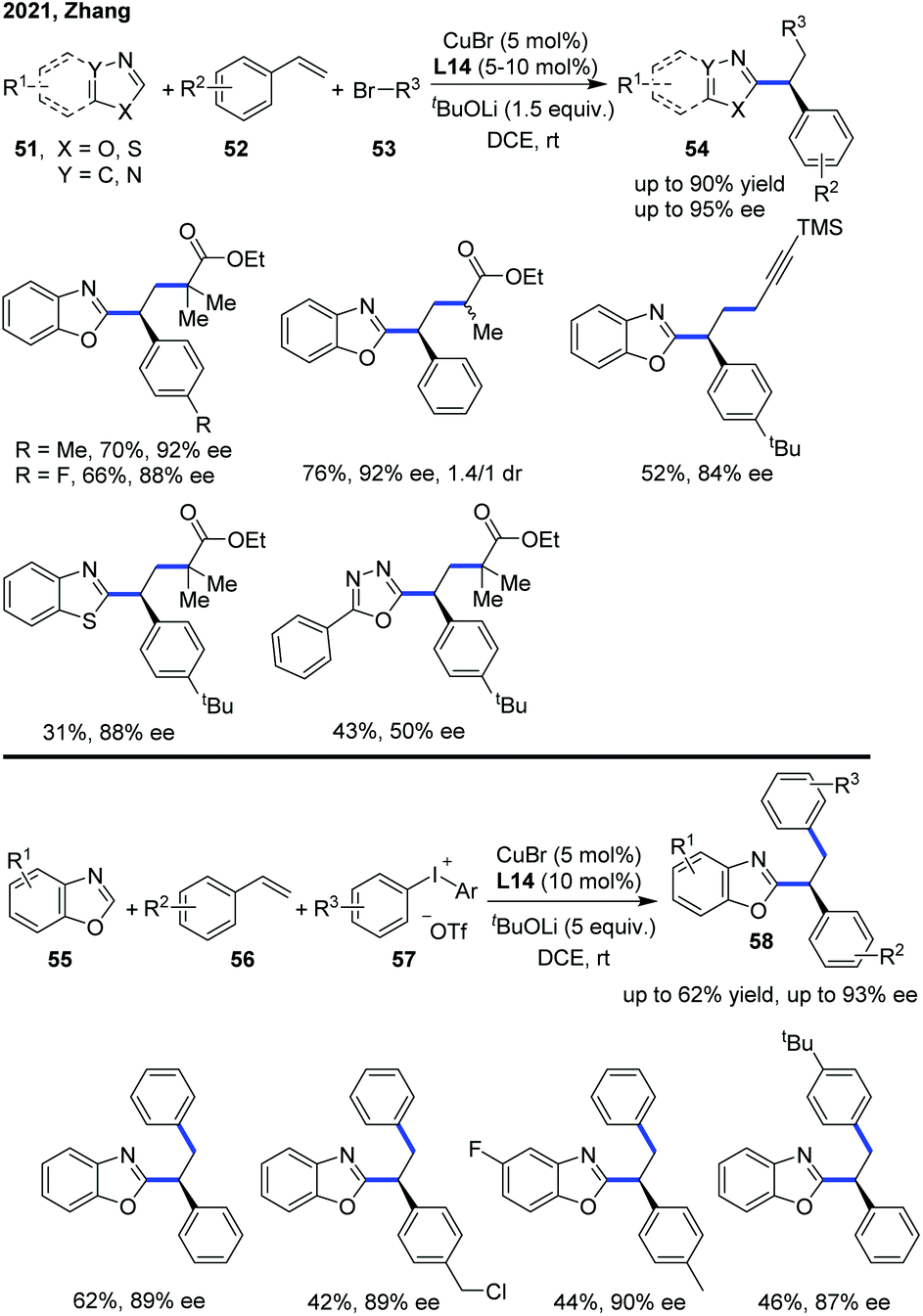
The Zhang group has also disclosed a blue-light-promoted copper-catalyzed asymmetric C–H functionalization of azoles with racemic secondary 1-arylalkyl bromides (Scheme 18).29 The enantioselective reaction was enabled by employing a copper(I)/chiral carbazole-based bisoxazoline (CbzBox) catalytic system, giving access to the C(sp2)–H alkylated azole products 50 in up to 80% yield and 99% ee. The blue-light-promoted photochemical protocol exhibited a significant enhancement in reaction efficiency and reactivity compared with the reactions conducted without light or under green LED irradiation. Interestingly, the copper–CbzBox complex was demonstrated to play a dual role as both a chiral catalyst and to form the photoactive complexes. The reaction showed broad functional group tolerance, with respect to both secondary 1-arylalkyl bromides and heteroaromatic azoles. Additionally, 3-bromocyclohex-1-ene was also demonstrated as a competent coupling partner, affording the allylic alkylation product in 57% yield and 50% ee. The catalytic cycle was initiated with a tBuOLi-promoted C–H cupration of azoles, generating the photoactive heterocycle-cuprate species Int-15 through the transmetallation process with the in situ generated Cu–CbzBox complex. Subsequent photoexcitation of Int-15 delivered intermediate Int-16, which further promoted the generation of alkyl radicals with the concomitant formation of species Int-17via a single-electron reduction of alkyl bromides. Then, an enantioselective radical trapping of intermediate Int-17 proceeded to form the corresponding chiral Cu(III) species Int-18. Finally, the reductive elimination of Int-18 or a direct single-electron transfer process of Int-17 occurred to provide the coupled chiral alkyl azole product and regenerated the active catalyst (Scheme 19).29
Lately, the Zhang group expanded the enantioselective copper-catalyzed asymmetric azole C–H functionalization strategy to a three-component radical cascade of azoles with alkenes and alkyl bromides (Scheme 20).30 The copper(I)/carbazole-based bisoxazoline (CbzBox) catalytic system proved to be essential to reduce the alkyl bromide to a corresponding alkyl radical, which undergoes a radical addition with styrene to generate a stabilized benzylic radical. This radical could subsequently be converted into a Cu(III) intermediate and the asymmetric radical cross-coupling of racemic alkyl bromides with azole C(sp2)–H bonds was performed. A wide range of valuable C–H functionalized azole products 54 were constructed with a high level of chemo-, regio-, and enantioselectivities from readily available coupling partners, under mild reaction conditions. In addition, diaryliodonium salt also served as an aryl radical precursor in the enantioselective copper catalytic system to bring about the catalytic enantioselective aryl/azolation of alkenes with moderate yields and high enantioselectivity.
 | ||
| Scheme 20 Proposed reaction mechanism of copper-catalyzed asymmetric alkylation of azoles. | ||
2.6 Minisci reaction
The Minisci-type reaction is well-known as a powerful tool for the direct C–H functionalization of electron-deficient N-heteroarenes by adding carbon-centered radicals to electrophilic heteroarenes.31 With the rapid development of photochemistry, the Minisci-type reaction has made a significant progress in constructing various substituted N-heteroarenes. Recently, the catalytic asymmetric Minisci reaction of N-heteroarenes under photoredox catalysis conditions has been successfully developed.In 2016, Fu and co-workers disclosed an interesting Minisci-type α-aminoalkylation of N-heteroarenes using an iridium photoredox catalyst (Ir[dF(CF3)ppy]2(dtbbpy)PF6) and phosphoric acid as the co-catalyst (Scheme 21).32 A variety of α-aminoalkylated N-heteroarenes 61 were obtained with moderate to good yields using α-amino acid-derived redox-active esters (N-(acyloxy)phthalimide) 60 as the α-aminoalkyl radical precursors. The proposed mechanism reveals that the α-aminoalkyl radical was obtained from the decarboxylation of N-(acyloxy)phthalimides in the iridium photoredox catalyzed single-electron reduction. The α-aminoalkyl radical that is generated can attack an N-heteroarene activated by a phosphoric acid co-catalyst to form a radical cation Int-20, which can be further oxidized by an excited photoredox catalyst and then be deprotonated by a counteranion to afford the α-aminoalkylation product.
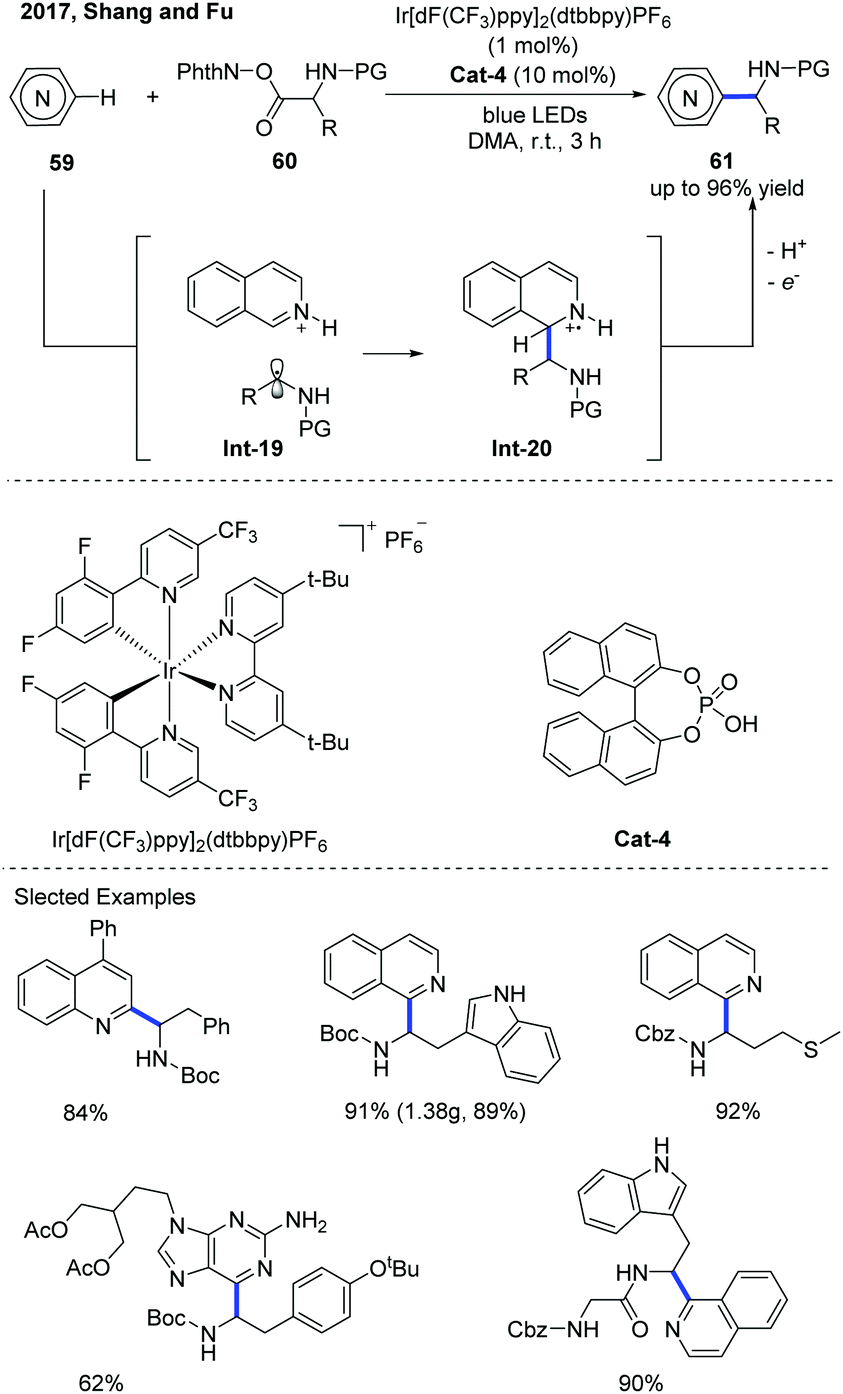 | ||
| Scheme 21 Light-promoted catalytic Minisci reaction of N-containing heteroarenes. | ||
The first example of catalytic enantioselective Minisci-type addition to N-heteroarenes was reported by Phipps and co-workers in 2018 (Scheme 22).33 Prochiral α-aminoalkyl radicals, generated from α-amino acid-derived redox-active esters, added to pyridines and quinolines giving a variety of α-aminoalkylated N-heteroarenes 64 with excellent enantioselectivity and regioselectivity. The chiral phosphate acid catalyst serves to both activate the substrate and induce asymmetry, while an iridium photocatalyst mediates the required electron transfer processes. Mechanistic studies33 reveal that both computational and experimental evidence supports the Curtin–Hammett control in this reaction, with the initial radical addition being quick and reversible (Int-22 to Int-23) and the enantioselectivity being achieved in the subsequent slower, irreversible deprotonation step (Int-23 to Int-24) (Scheme 23).34 A number of different possibilities for selectivity-determining deprotonation of the radical cation intermediate were assessed by the detailed DFT calculations. The computations pointed out that the internal deprotonation enacted by the amide group was a crucial structural feature of the radical precursor, with the assistance of the associated chiral phosphate (Int-24).
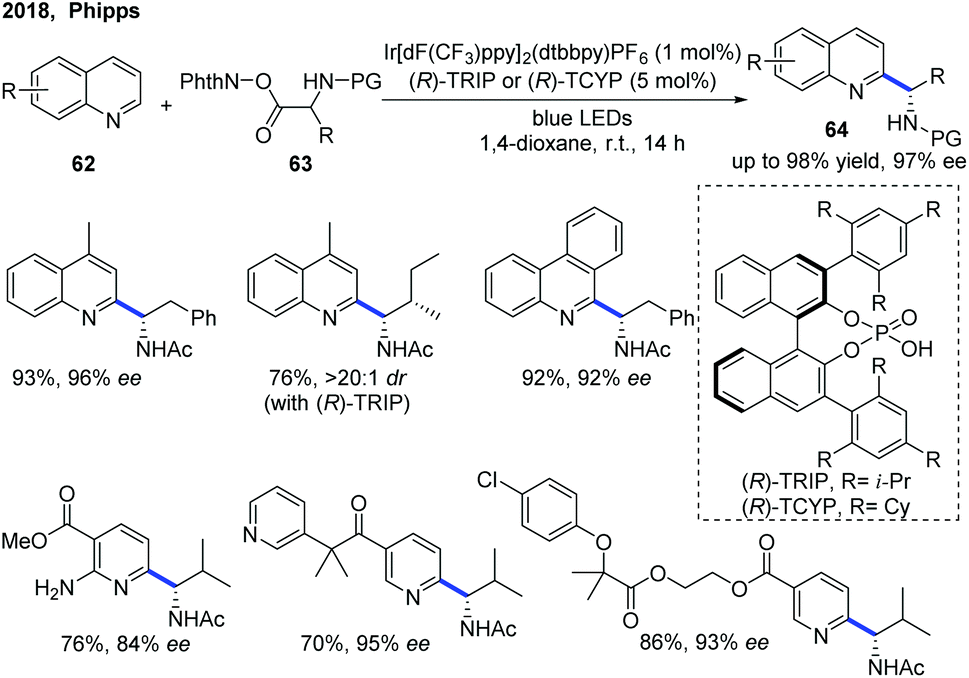 | ||
| Scheme 22 Light-promoted catalytic enantioselective Minisci reaction of N-containing heteroarenes. | ||
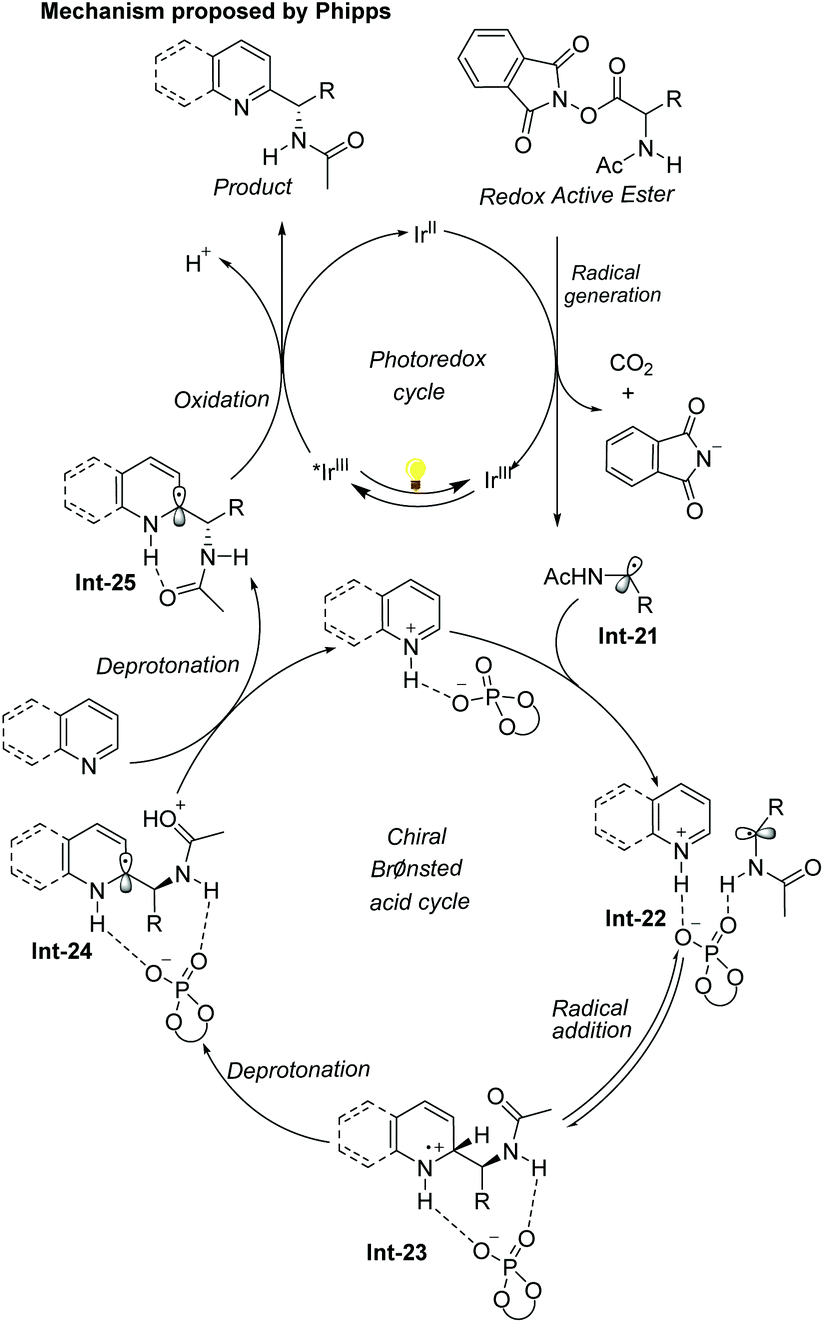 | ||
| Scheme 23 Proposed mechanism of the catalytic enantioselective Minisci reaction of N-heteroarenes. | ||
Shortly after, Jiang and co-workers independently developed a similar Minisci-type addition to N-heteroarenes using DPZ as a metal-free photoredox catalyst and a SPINOL-derived CPA as the chiral catalyst (Scheme 24).35 A wide scope of α-aminoalkylated isoquinolines 67 were obtained in high yields with good enantioselectivities. It is worth mentioning that the protocol can be well applied to quinoline substrates. In contrast, the method developed by Phipps and co-workers was not applicable to isoquinoline substrates.
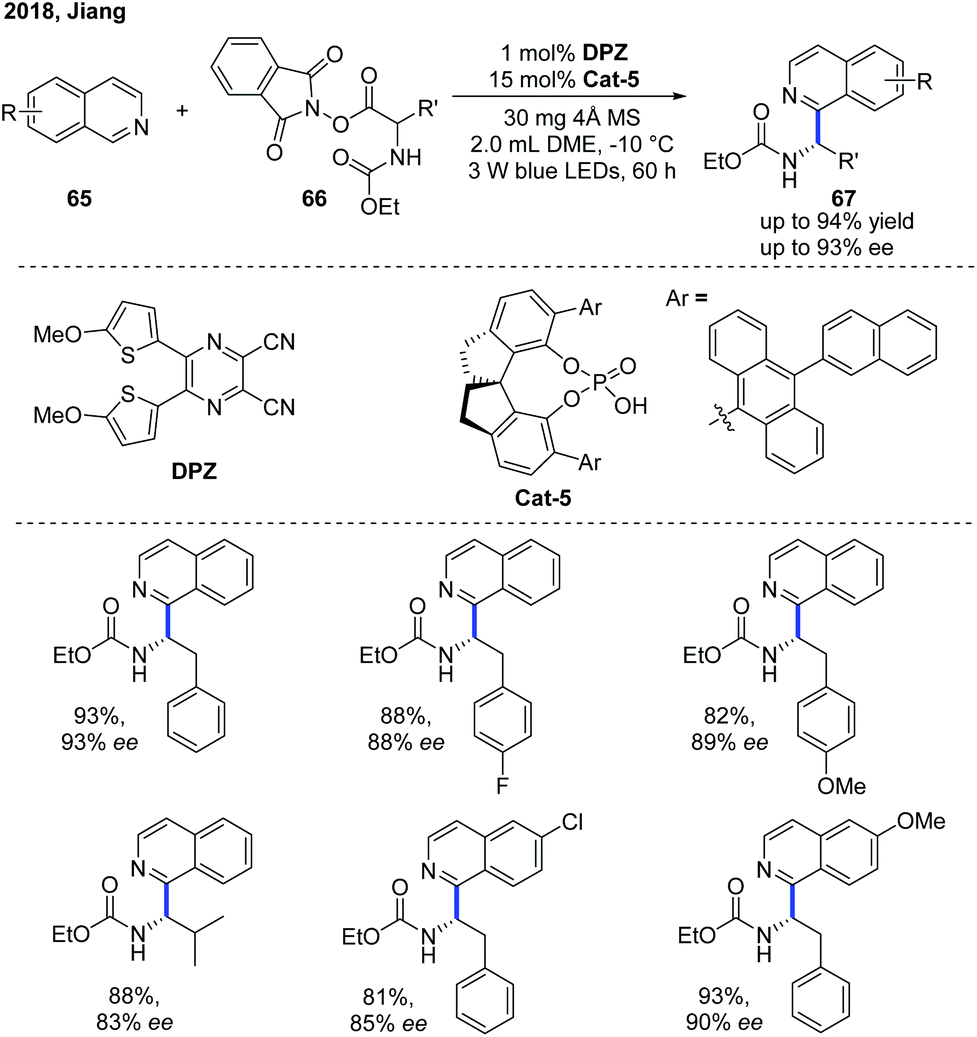 | ||
| Scheme 24 Catalytic enantioselective Minisci reaction between α-amino acid-derived redox-active esters and isoquinolines. | ||
In 2019, Fu and co-workers developed an asymmetric Minisci-type alkylation of N-heterocycles catalyzed by a phosphine/iodide-based photoredox system, which no longer needed other metal complex or organic dye-based photoredox catalysts (Scheme 25).36 The proposed mechanism revealed that initially a charge transfer complex (CTC) was formed by the assembly of NaI and PPh3 with RAE, and then an alkyl radical was generated after the fragmentation of CTC under blue LED irradiation, which attacked N-heteroarene activated by the chiral phosphoric acid to form a radical cation intermediate. The radical cation intermediate was oxidized by the PPh3-I radical intermediate and further deprotonated by the counteranion to afford the α-aminoalkylation product, while simultaneously regenerating NaI and PPh3. It is worth noting that this photoredox system can also be applied to Katritzky's N-alkylpyridinium salts and Togni's reagent-based deaminative alkylation.
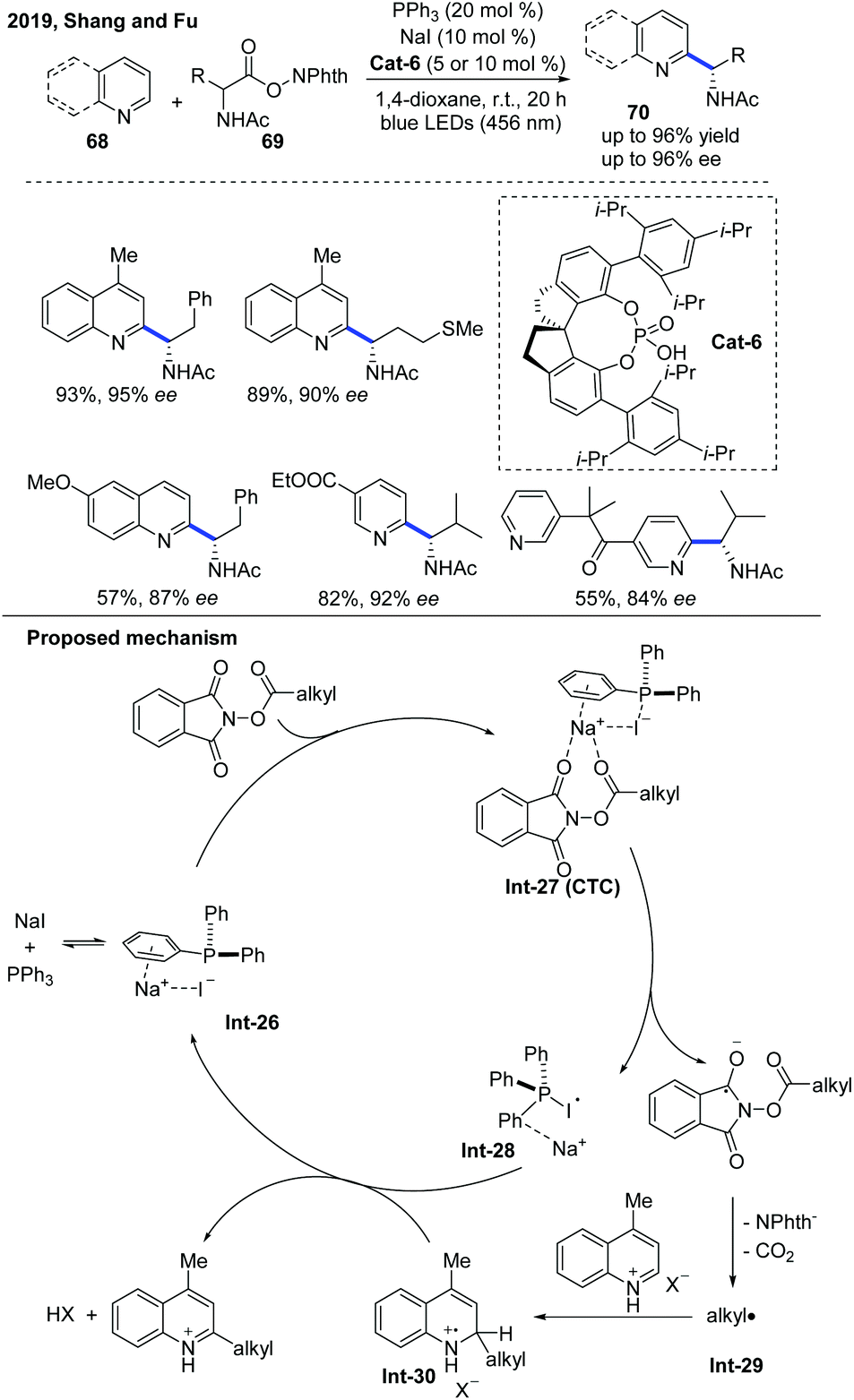 | ||
| Scheme 25 Photocatalytic enantioselective Minisci reaction of N-heteroarenes mediated by triphenylphosphine and sodium iodide. | ||
In 2019, the Phipps group developed a predictive, mathematical model to guide a successful catalytic enantioselective Minisci reaction of diazines using multivariate linear regression (MLR) analysis (Scheme 26).37 The model was established by the careful evaluation of catalyst/substrate training sets and parameter acquisition platforms upon the enantioselective Minisci addition of N-acyl, α-amino radicals to pyridines and quinolines. The MLR analysis results suggested that pyrimidines, with larger NBO values than pyridines, may be particularly amenable for the same reaction conditions. These hypotheses are well confirmed from the experimental results, and a series of α-amino-substituted pyrimidines were obtained in moderate to good yields with excellent enantioselectivity. However, when pyrazines and 2-aminopyridine were used as substrates, the yield and stereoselectivity of the reaction were relatively poor.
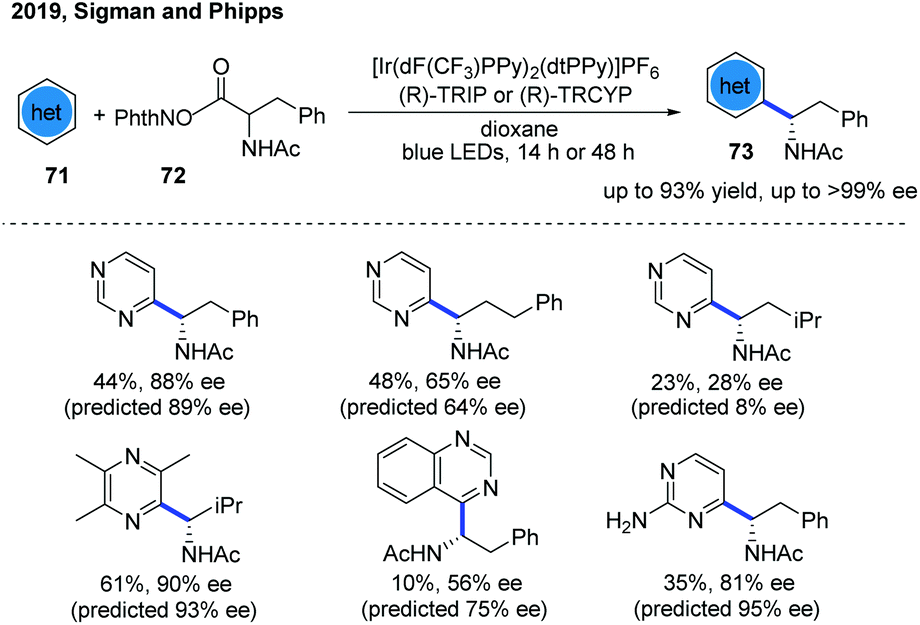 | ||
| Scheme 26 Catalytic enantioselective Minisci reactions of diazines. | ||
Very recently, the Phipps group disclosed an enantioselective Minisci reaction using a chiral phosphoric acid catalyst in conjunction with a hydrogen atom transfer reagent (Scheme 27).38 In this protocol, diacetyl was identified as a mild, chemoselective reagent for the generation of α-aminoalkyl radicals, which precluded the need for an added photocatalyst and was crucial to the success of this transformation. With the help of the catalyst, a variety of valuable chiral α-aminoalkyl-substituted pyridine and quinoline derivatives 76 were obtained with excellent regioselectivity and enantioselectivity, albeit in moderate yields.
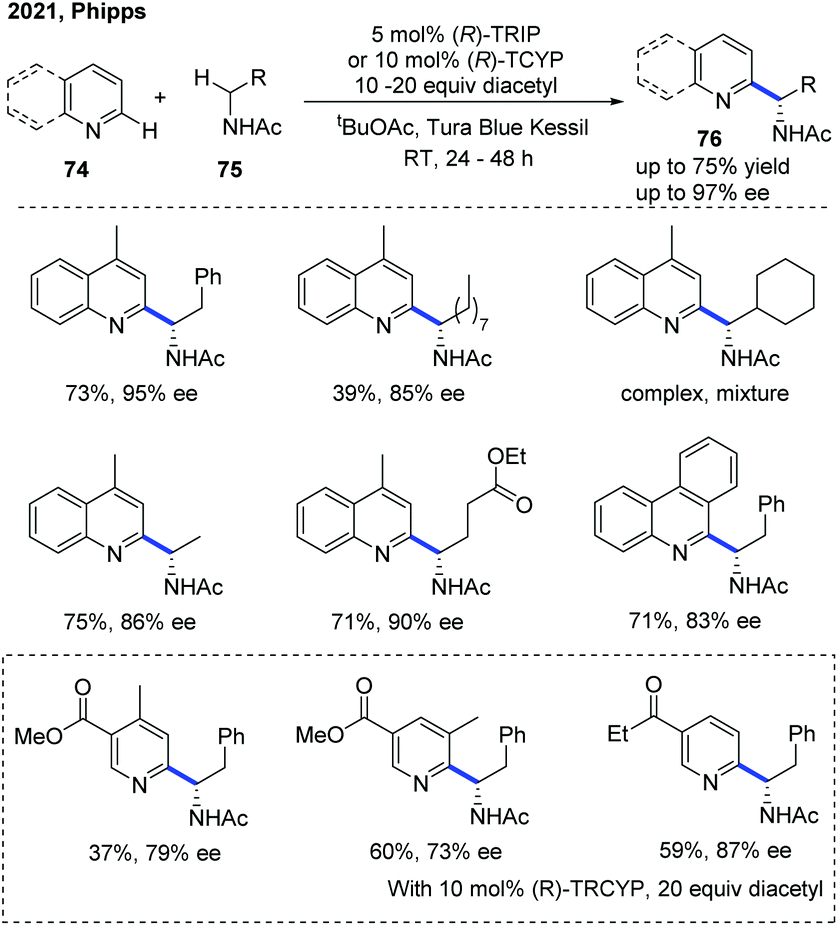 | ||
| Scheme 27 Photocatalytic enantioselective Minisci reaction between N-heteroarenes and amides. | ||
In 2019, the Studer group developed an enantioselective three-component Minisci reaction for the catalytic asymmetric synthesis of chiral heterocyclic γ-amino acid 79 and 1,2-diamine derivatives using a dual photoredox and chiral Brønsted acid as the co-catalyst (Scheme 28).39 The kinetic isotope effect (KIE) experiment suggested that the deprotonation might be the rate-determining step and the stereoselectivity was more likely controlled in the deprotonation step of the radical cation intermediate rather than in the initial C–C bond formation step, which is in accordance with the proposed mechanism in the two-component asymmetric Minisci reaction.34,37 This protocol allows straightforward synthesis of a wide range of valuable chiral heterocyclic γ-amino acids and 1,2-diamine derivatives with good to excellent enantioselectivities using simple, readily available starting materials.
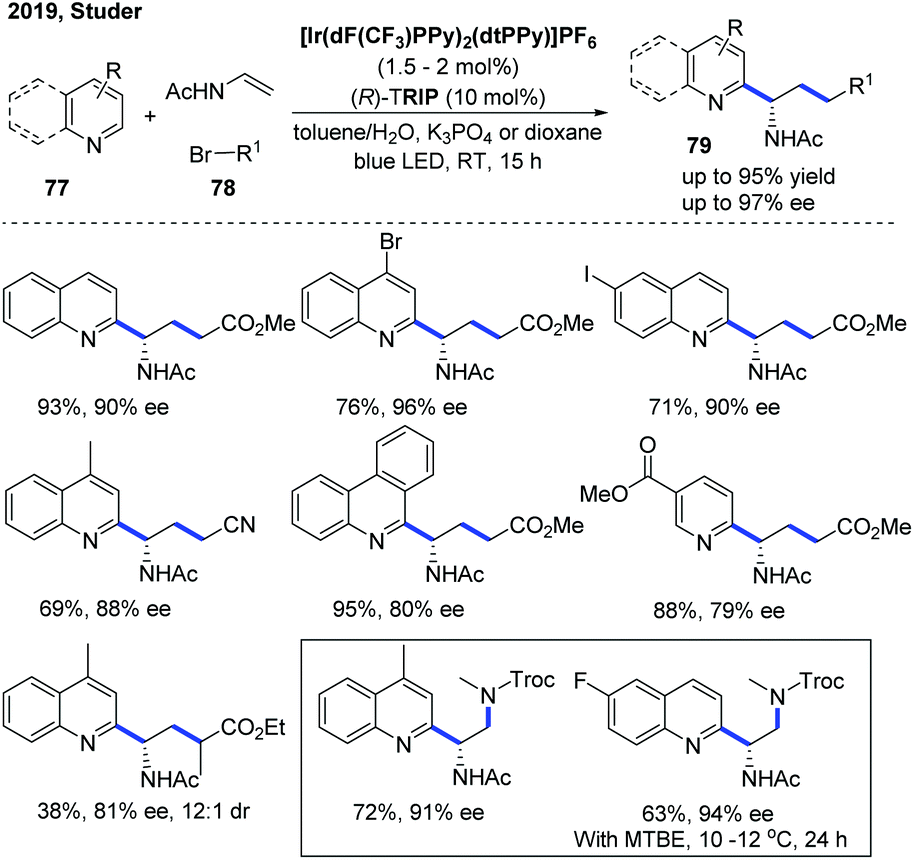 | ||
| Scheme 28 Photocatalytic enantioselective three-component Minisci reaction. | ||
3. Conclusion and outlook
Catalytic enantioselective direct C–H bond functionalization of electron-deficient N-containing heteroarenes has achieved enormous development over the past decade. A number of efficient catalytic systems with different strategies in highly chemo- and stereoselective C–H functionalization of electron-deficient N-containing heteroarenes provided a wide range of highly functionalized N-heteroarenes as attractive chiral building blocks. Despite the recent impressive progress, the catalytic enantioselective direct C–H bond functionalization of electron-deficient N-heteroarenes still faces significant challenges and further explorations in this field are needed. Effective and powerful catalytic systems with high catalytic activity and selectivity are still greatly required to explore the challenging and unprecedented heteroarenes and transformations. Furthermore, enantioselective catalytic systems based on Earth-abundant metals, such as Fe, Co, and Ni, are expected to be investigated since such catalytic systems might help in enhancing the practicality and versatility of the current asymmetric C–H functionalization processes. More efforts are needed for further mechanistic studies in combination with computational investigations into the reaction processes and details to provide guidance for developing novel catalysts and new transformations. In addition, the rapid development of photochemistry and electrochemistry provides new possibilities for expanding the applicability of asymmetric direct C–H functionalization of electron-deficient N-containing heteroarenes. Despite these challenges, there is no doubt that catalytic asymmetric direct C–H functionalization of electron-deficient N-containing heteroarenes will have extensive progress and satisfy the demands of valuable applications in the process of bioactive compound synthesis and drug discovery in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 22001260), the Natural Science Foundation of Guangdong Province (No. 2021A1515010016) and the Outstanding Talents Research Start-up Foundation of Xuzhou Medical University (No. D2020016).Notes and references
- (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Transition metal-catalyzed C-H activation reactions: diastereoselectivity and enantioselectivity, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (b) C. Zheng and S.-L. You, Recent development of direct asymmetric functionalization of inert C-H bonds, RSC Adv., 2014, 4, 6173–6214 RSC; (c) Asymmetric Functionalization of C–H Bonds, ed. S. L. You, Royal Society of Chemistry, Cambridge, UK, 2015 Search PubMed; (d) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Catalytic Enantioselective Transformations Involving C-H BondCleavage by Transition-Metal Complexes, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS.
- (a) T. B. Poulsen and K. A. Jørgensen, Catalytic Asymmetric Friedel-Crafts Alkylation Reactions-Copper Showed the Way, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS; (b) Catalytic Asymmetric Friedel-Crafts Reactions, ed. M. Bandini and A. Umani-Ronchi, Wiley-VCH, Weinheim, 2009 Search PubMed; (c) Y. Zhang, X.-H. Liu, L.-L. Lin and X.-M. Feng, Recent Advance in Catalytic Asymmetric Friedel-Crafts Reactions, Prog. Chem., 2018, 30, 491–504 Search PubMed.
- F. O'Hara, D. G. Blackmond and P. S. Baran, Radical-Based Regioselective C–H Functionalization of Electron Deficient Heteroarenes: Scope, Tunability, and Predictability, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef.
- K. Murakami, S. Yamada, T. Kaneda and K. Itami, C–H Functionalization of Azines, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS PubMed.
- R. Das and M. Kapur, Transition-Metal-Catalyzed C-H Functionalization Reactions of π-Deficient Heterocycles, Asian J. Org. Chem., 2018, 7, 1217–1235 CrossRef CAS.
- (a) Z.-K. Chen, B.-J. Wang, J.-T. Zhang, W.-L. Yu, Z. X. Liu and Y.-H. Zhang, Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (b) M. Zhang, Y.-F. Zhang, X.-M. Jie, H.-Q. Zhao, G. Li and W.-P. Su, Recent advances in directed C–H functionalizations using monodentate nitrogen-based directing groups, Org. Chem. Front., 2014, 1, 843–895 RSC.
- S. Rodewald and R. F. Jordan, Stereoselective Olefin Insertion Reactions of Chiral (EBI)Zr(ETA(2)-Pyrid-2-yl)(+) and (EBTHI)Zr(ETA(2)-Pyrid-2-yl)(+) Complexes, J. Am. Chem. Soc., 1994, 116, 4491–4492 CrossRef CAS.
- G.-Y. Song, W. N. O. Wylie and Z.-M. Hou, Enantioselective C-H Bond Addition of Pyridines to Alkenes Catalyzed by Chiral Half-Sandwich Rare-Earth Complexes, J. Am. Chem. Soc., 2014, 136, 12209–12212 CrossRef CAS.
- S.-J. Lou, Z.-B. Mo, M. Nishiura and Z.-M. Hou, Construction of All-Carbon Quaternary Stereocenters by Scandium Catalyzed Intramolecular C-H Alkylation of Imidazoles with 1,1-Disubstituted Alkenes, J. Am. Chem. Soc., 2020, 142, 1200–1205 CrossRef CAS PubMed.
- A. S. Tsai, R. M. Wilson, H. Harada, R. G. Bergman and J. A. Ellman, Rhodium Catalyzed Enantioselective Cyclization of Substituted Imidazoles via C-H Bond Activation, Chem. Commun., 2009, 3910–3912 RSC.
- C. M. Filloux and T. Rovis, Rh(I)-Bisphosphine-Catalyzed Asymmetric, Intermolecular Hydroheteroarylation of α-Substituted Acrylate Derivatives, J. Am. Chem. Soc., 2015, 137, 508–517 CrossRef CAS.
- P. A. Donets and N. Cramer, Ligand-Controlled Regiodivergent Nickel-Catalyzed Annulation of Pyridones, Angew. Chem., Int. Ed., 2015, 54, 633–637 CAS.
- D. Hirsch-Weil, K. A. Abboud and S. Hong, Isoquinoline-based chiral monodentate N-heterocyclic carbenes, Chem. Commun., 2010, 46, 7525–7527 RSC.
- (a) A. Albright and R. E. Gawley, Application of a C2-Symmetric Copper Carbenoid in the Enantioselective Hydrosilylation of Dialkyl and Aryl–Alkyl Ketones, J. Am. Chem. Soc., 2011, 133, 19680–19683 CrossRef CAS; (b) A. Albright, D. Eddings, R. Black, C. J. Welch, N. N. Gerasimchuk and R. E. Gawley, Design and Synthesis of C2-Symmetric N-Heterocyclic Carbene Precursors and Metal Carbenoids, J. Org. Chem., 2011, 76, 7341–7351 CrossRef CAS; (c) Y. Cai, X.-T. Yang, S.-Q. Zhang, F. Li, Y.-Q. Li, L.-X. Ruan, X. Hong and S.-L. Shi, Copper-Catalyzed Enantioselective Markovnikov Protoboration of α-Olefins Enabled by a Buttressed N-Heterocyclic Carbene Ligand, Angew. Chem., Int. Ed., 2018, 57, 1376–1380 CrossRef CAS; (d) Y. Cai, J.-W. Zhang, F. Li, J.-M. Liu and S.-L. Shi, Nickel/N-Heterocyclic Carbene Complex-Catalyzed Enantioselective Redox-Neutral Coupling of Benzyl Alcohols and Alkynes to Allylic Alcohols, ACS Catal., 2019, 9(1), 1–6 CrossRef; (e) D. Shen, Y.-J. Xu and S.-L. Shi, A Bulky Chiral N-Heterocyclic Carbene Palladium Catalyst Enables Highly Enantioselective Suzuki–Miyaura Cross-Coupling Reactions for the Synthesis of Biaryl Atropisomers, J. Am. Chem. Soc., 2019, 141, 14938–14945 CrossRef CAS PubMed; (f) J. Diesel, D. Grosheva, S. Kodama and N. Cramer, A Bulky Chiral N-Heterocyclic Carbene Nickel Catalyst Enables Enantioselective C–H Functionalizations of Indoles and Pyrroles, Angew. Chem., Int. Ed., 2019, 58, 11044–11048 CrossRef CAS.
- J. Diesel, A. M. Finogenova and N. Cramer, Nickel-Catalyzed Enantioselective Pyridone C-H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand, J. Am. Chem. Soc., 2018, 140, 4489–4493 CrossRef CAS PubMed.
- Y.-X. Wang, S.-L. Qi, Y.-X. Luan, X.-W. Han, S. Wang, H. Chen and M.-C. Ye, Enantioselective Ni-Al Bimetallic Catalyzed exo-Selective C-H Cyclization of Imidazoles with Alkenes, J. Am. Chem. Soc., 2018, 140, 5360–5364 CrossRef CAS.
- (a) K. L. Tan, R. G. Bergman and J. A. Ellman, Annulation of Alkenyl-Substituted Heterocycles via Rhodium-Catalyzed Intramolecular C–H Activated Coupling Reactions, J. Am. Chem. Soc., 2001, 123, 2685–2686 CrossRef CAS PubMed; (b) K. L. Tan, R. G. Bergman and J. A. Ellman, Intermediacy of an N-Heterocyclic Carbene Complex in the Catalytic C–H Activation of a Substituted Benzimidazole, J. Am. Chem. Soc., 2002, 124, 3202–3203 CrossRef CAS.
- J.-F. Li, W.-W. Xu, R.-H. Wang, Y. Li, G. Yin and M.-C. Ye, Construction 7-membered ring via Ni–Al bimetal-enabled C–H cyclization for synthesis of tricyclic imidazoles, Nat. Commun., 2021, 12, 3070–3078 CrossRef CAS PubMed.
- J. Loup, V. Muller, D. Ghorai and L. Ackermann, Enantioselective Aluminum-Free Alkene Hydroarylations through C-H Activation by a Chiral Nickel/JoSPOphos Manifold, Angew. Chem., Int. Ed., 2019, 58, 1749–1753 CrossRef CAS.
- W.-B. Zhang, X.-T. Yang, J.-B. Ma, Z.-M. Su and S.-L. Shi, Regio- and Enantioselective C-H Cyclization of Pyridines with Alkenes Enabled by a Nickel/N-Heterocyclic Carbene Catalysis, J. Am. Chem. Soc., 2019, 141, 5628–5634 CrossRef CAS PubMed.
- D. Shen, W. B. Zhang, Z.-Y. Li, S.-L. Shi and Y.-J. Xu, Nickel/NHC-Catalyzed Enantioselective Cyclization of Pyridones and Pyrimidones with Tethered Alkenes, Adv. Synth. Catal., 2020, 362, 1125–1130 CrossRef CAS.
- W.-Q. Kong, Q. Wang and J.-P. Zhu, Palladium-Catalyzed Enantioselective Domino Heck/Intermolecular C-H Bond Functionalization: Development and Application to the Synthesis of (+)-Esermethole, J. Am. Chem. Soc., 2015, 137, 16028–16031 CrossRef CAS PubMed.
- S. Tong, A. Limouni, Q. Wang, M.-X. Wang and J.-P. Zhu, Catalytic Enantioselective Double Carbopalladation/C-H Functionalization with Statistical Amplification of Product Enantiopurity: A Convertible Linker Approach, Angew. Chem., Int. Ed., 2017, 56, 14192–14196 CrossRef CAS PubMed.
- X. Bao, Q. Wang and J.-P. Zhu, Palladium-Catalyzed Enantioselective Narasaka-Heck Reaction/Direct C-H Alkylation of Arenes: Iminoarylation of Alkenes, Angew. Chem., Int. Ed., 2017, 56, 9577–9581 CrossRef CAS PubMed.
- Q.-H. Nguyen, S.-M. Guo, T. Royal, O. Baudoin and N. Cramer, Intermolecular Palladium(0)-Catalyzed Atropo-enantioselective C-H Arylation of Heteroarenes, J. Am. Chem. Soc., 2020, 142, 2161–2167 CrossRef CAS.
- H. Ohmiya, H. Zhang, S. Shibata, A. Harada and M. Sawamura, Construction of Quaternary Stereogenic Carbon Centers through Copper-Catalyzed Enantioselective Allylic Alkylation of Azoles, Angew. Chem., Int. Ed., 2016, 55, 4777–4780 CrossRef CAS.
- Y. Makida, H. Ohmiya and M. Sawamura, Regio- and Stereocontrolled Introduction of Secondary Alkyl Groups to Electron-Deficient Arenes through Copper-Catalyzed Allylic Alkylation, Angew. Chem., Int. Ed., 2012, 51, 4122–4127 CrossRef CAS.
- X.-L. Su, L. Ye, J.-J. Chen, X.-D. Liu, S.-P. Jiang, F.-L. Wang, L. Liu, C.-J. Yang, X.-Y. Chang, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Copper-Catalyzed Enantioconvergent Cross-Coupling of Racemic Alkyl Bromides with Azole C(sp2)-H Bonds, Angew. Chem., Int. Ed., 2021, 60, 380–384 CrossRef CAS PubMed.
- C. Li, B. Chen, X.-D. Ma, X.-L. Mo and G.-Z. Zhang, Light-Promoted Copper-Catalyzed Enantioselective Alkylation of Azoles, Angew. Chem., Int. Ed., 2021, 60, 2130–2134 CrossRef CAS PubMed.
- X. Ma and G. Zhang, Asymmetric Alkyl and Aryl/Azolation of Alkenes via a Single Cu(I) Complex, ACS Catal., 2021, 11, 5108–5118 CrossRef CAS.
- R. S. J. Proctor and R. J. Phipps, Recent Advances in Minisci-Type Reactions, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- W.-M. Cheng, R. Shang and Y. Fu, Photoredox/Brønsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes, ACS Catal., 2016, 7, 907–911 CrossRef.
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Catalytic enantioselective Minisci-type addition to heteroarenes, Science, 2018, 360, 419–422 CrossRef CAS PubMed.
- K. Ermanis, A. C. Colgan, R. S. J. Proctor, B. W. Hadrys, R. J. Phipps and J. M. Goodman, A Computational and Experimental Investigation of the Origin of Selectivity in the Chiral Phosphoric Acid Catalyzed Enantioselective Minisci Reaction, J. Am. Chem. Soc., 2020, 142, 21091–21101 CrossRef CAS PubMed.
- X.-Y. Liu, Y. Liu, G. Chai, B.-K. Qiao, X.-W. Zhao and Z.-Y. Jiang, Organocatalytic Enantioselective Addition of α-Aminoalkyl Radicals to Isoquinolines, Org. Lett., 2018, 20, 6298–6301 CrossRef CAS PubMed.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide, Science, 2019, 363, 1429–1434 CrossRef CAS.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, Predictive Multivariate Linear Regression Analysis Guides Successful Catalytic Enantioselective Minisci Reactions of Diazines, J. Am. Chem. Soc., 2019, 141, 19178–19185 CrossRef CAS PubMed.
- R. S. J. Proctor, P. Chuentragool, A. C. Colgan and R. J. Phipps, Hydrogen Atom Transfer-Driven Enantioselective Minisci Reaction of Amides, J. Am. Chem. Soc., 2021, 143, 4928–4934 CrossRef CAS PubMed.
- D.-Q. Zheng and A. Studer, Asymmetric Synthesis of Heterocyclic gamma-Amino-Acid and Diamine Derivatives by Three-Component Radical Cascade Reactions, Angew. Chem., Int. Ed., 2019, 58, 15803–15807 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2022 |