
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the boundaries of ferrocenesulfonyl fluoride chemistry†
William
Erb
 *a,
Jean-Pierre
Hurvois
*a,
Yury S.
Halauko
*b,
Vadim E.
Matulis
b and
Thierry
Roisnel
a
*a,
Jean-Pierre
Hurvois
*a,
Yury S.
Halauko
*b,
Vadim E.
Matulis
b and
Thierry
Roisnel
a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: william.erb@univ-rennes1.fr; jean-pierre.hurvois@univ-rennes1.fr
bBelarusian State University, 4, Nezavisimosti Avenue, 220030 Minsk, Belarus. E-mail: hys@tut.by
First published on 28th September 2022
Abstract
Ferrocenesulfonyl fluorides represent a new subfamily of metallocenes barely explored. Here, we describe how the use of deprotolithiation-electrophilic trapping sequences and ‘halogen dance’ reactions can afford up to 1,2,3,4,5-hetero pentasubstituted derivatives. While the orthogonal reactivity of ferrocenesulfonyl fluorides was demonstrated in a variety of transformations, SuFEx chemistry afforded various sulfur-based ferrocene derivatives. The electrochemical behaviour of many polysubstituted derivatives was studied and discussed as well as weak interactions identified in the solid state.
Introduction
Aromatic sulfonyl fluorides are a subfamily of aryl-SVI fluorides characterized by the lowest degree of fluorination with only one fluorine directly attached to sulfur.1 The first studies related to their chemistry were reported by Steinkopf in the late 1920s,2,3 and their ability to react with enzymes was described by Myers and Kemp in 1954.4 This ultimately led to the discovery of phenylmethanesulfonyl fluoride (PMSF) and 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF),5 two non-specific serine protease inhibitors, widely used in biochemistry.6 In 2014, Sharpless introduced the concept of sulfur(VI) fluoride exchange (SuFEx)7 as a novel opportunity for click chemistry and thus renewed the interest of chemists for these compounds.8 This results from the unique properties of this functional group such as resistance to reduction, high thermodynamic stability and exclusive substitution at sulfur. The sulfonyl fluorides are therefore stable under an amazing range of reaction conditions but they can also react with a nucleophile in smooth conditions, if properly activated via hydrogen-bonding or by a Lewis acid. As a consequence, sulfonyl fluorides have found applications in chemical biology,9–15 the functional group acting as a warhead to react with enzymes, and in material science to reach original materials.16,17 This was accompanied with main developments in the synthesis of sulfonyl fluorides,18,19 the study of their orthogonal reactivity,20 and the optimization of various SuFEx reaction conditions.21–24Since its discovery in 1951,25,26 ferrocene and its derivatives have found a pivotal place in chemistry with numerous applications in sensing,27–29 catalysis,30–34 medicinal chemistry35–41 and materials science.42–44 Looking at the number and the variety of ferrocene derivatives prepared over the years, it is surprising that only two ferrocenesulfonyl fluoride derivatives have been reported up to 2021. Indeed, in 1959, Nesmeyanov obtained 1′-methoxycarbonylferrocenesulfonyl and 1′-carboxyferrocenesulfonyl fluorides from their respective sulfonyl chlorides by reaction with potassium bifluoride (KHF2) in acetic acid (Fig. 1, top left).45 In 2021, Butenschön reported the reaction of ferrocenesulfonyl chloride with potassium fluoride in acetonitrile in the presence of 18-crown-6 ether, leading to the fluoride derivative in 79% yield (Fig. 1, top right).46 Further functionalization was demonstrated by a deprotolithiation-electrophilic trapping sequence using lithium 2,2,6,6-tetramethylpiperidide (LiTMP) as the base and iodomethane as the electrophile. At the same time, we independently reported our own efforts towards ferrocenesulfonyl fluoride and a few derivatives in a preliminary communication (Fig. 1, bottom).47 Here, we report a comprehensive study of the chemistry and properties of polysubstituted ferrocenesulfonyl fluorides.
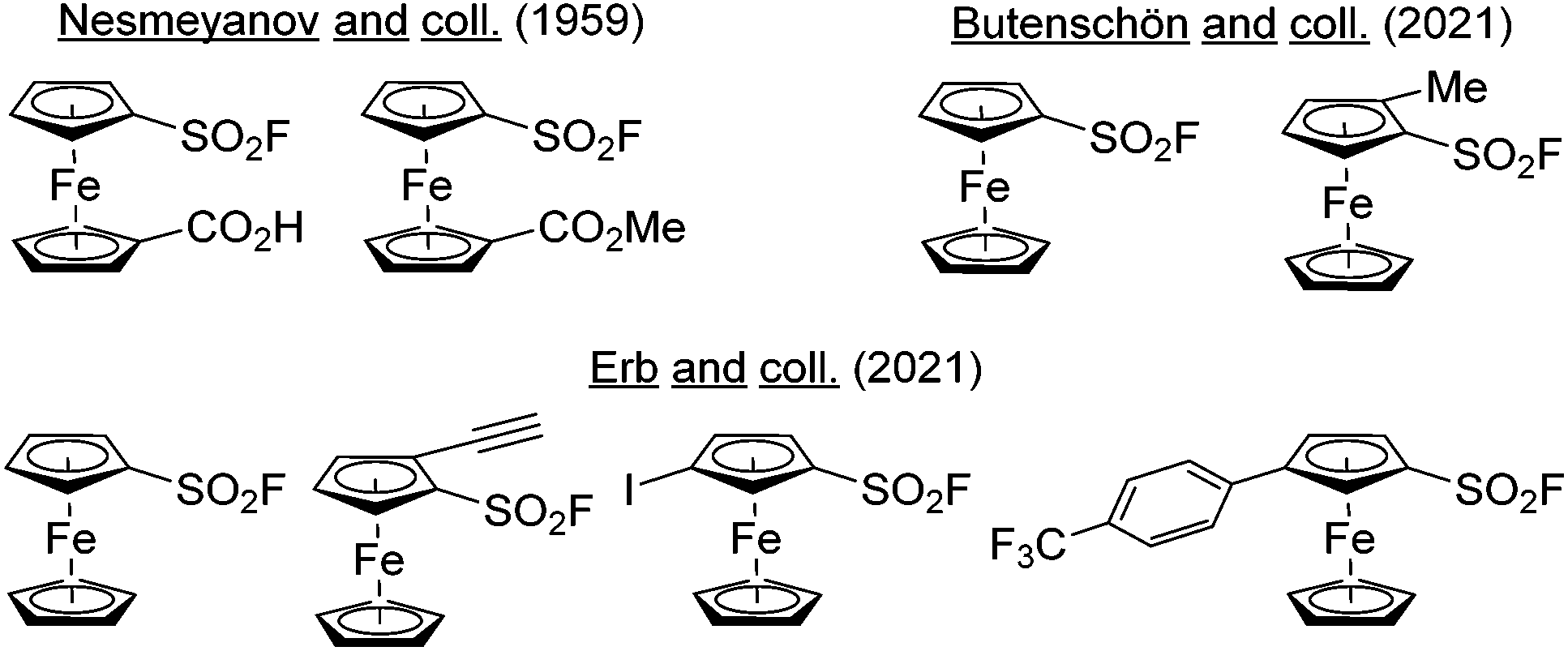 | ||
| Fig. 1 Ferrocenesulfonyl fluorides already reported. | ||
Results and discussion
Exploring the chemistry of ferrocenesulfonyl fluorides
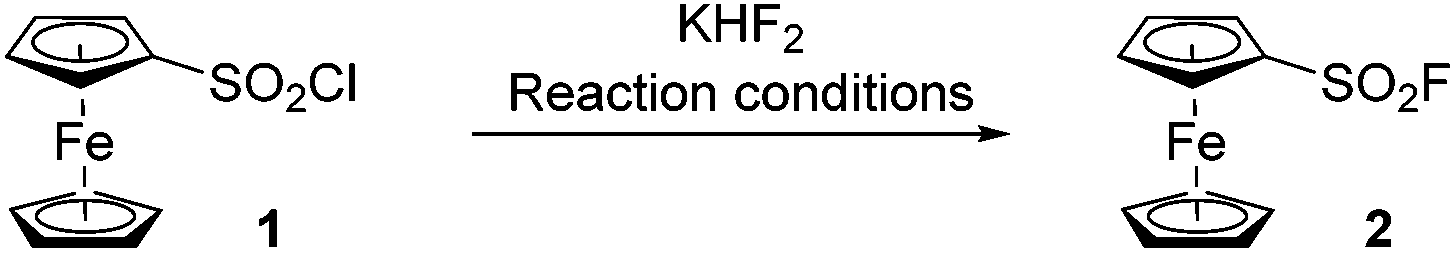
Inspired by the work of Willis and Ball, who prepared arenesulfonyl fluorides from the sulfinates by using N-fluorobenzenesulfonimide (NFSI) and Selectfluor respectively,48,49 we initially reacted lithium ferrocenesulfinate (prepared in situ from lithioferrocene50 and 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct) with NFSI. However, no title product was detected, and degradation was mainly observed.Therefore, we turned to a more classical approach with the reaction between ferrocenesulfonyl chloride (prepared by sulfonation-chlorination of ferrocene)51,52 and a fluoride. We initially explored reaction conditions inspired by Barbasiewicz, and reacted ferrocenesulfonyl chloride (1) with KHF2 in a water-acetonitrile mixture in the presence of tetrabutylammonium bromide (Table 1, entry 1).53 However, a moderate 32% yield of the desired compound 2 was reached. Adding more catalyst or performing the reaction at higher temperature did not improve the reaction outcome (entries 2 and 3). Concerned with the hydrolysis of ferrocenesulfonyl chloride, we evaluated an anhydrous mixture of acetonitrile and acetone and isolated 2 in 63% yield after 48 h at 60 °C (entry 4). We then decided to reassess the reaction conditions described by Nesmeyanov in 1959 and reacted 1 with KHF2 in acetic acid at 25 °C (entry 5). After 14 h, we pleasingly isolated the desired derivative 2 in 52% yield. While the addition of acetic anhydride as a dehydrating reagent was without effect (entry 6), we found that reaction time could be reduced to only 30 min when performing the substitution at 60 °C (63% yield, entry 7). Pleasingly, we were able to scale the reaction up to 0.3 mol, leading to 52 g of ferrocenesulfonyl fluoride (2) isolated in a single batch (64% yield).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Additive | T (°C) | Time (h) | Yield (%) |
a All reactions performed on a 10 mmol scale with 3 equivalents of KHF2.
b Reaction performed using 2 equiv. of KHF2.
c 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio.
d 1:1 ratio.
e Reaction performed on a 0.1 mmol scale. :4 ratio.
d 1:1 ratio.
e Reaction performed on a 0.1 mmol scale.
|
|||||
| 1b | H2O–MeCNc | nBu4NBr (1 mol%) | 25 | 14 | 32 |
| 2 | H2O–MeCNc | nBu4NBr (2 mol%) | 25 | 14 | 22 |
| 3 | H2O–MeCNc | nBu4NBr (2 mol%) | 60 | 14 | 24 |
| 4 | MeCN–acetoned | nBu4NBr (5 mol%) | 60 | 48 | 63 |
| 5 | AcOH | — | 25 | 14 | 52 |
| 6 | AcOH | Ac2O (0.5 equiv.) | 25 | 14 | 55 |
| 7e | AcOH | — | 60 | 0.5 | 63 |
Able to reach large quantities of ferrocenesulfonyl fluoride in a fast and inexpensive way, we next explored its functionalization by deprotolithiation-trapping. Indeed, the pKa values calculated within the DFT framework (see ESI†) for compound 2 show that sulfonyl fluoride group behaves as a powerful acidifying group, much stronger than N,N-dimethylsulfonamide,54O-isopropylsulfonate55 and fluorine,56 all three already being strong directing groups (Fig. 2).
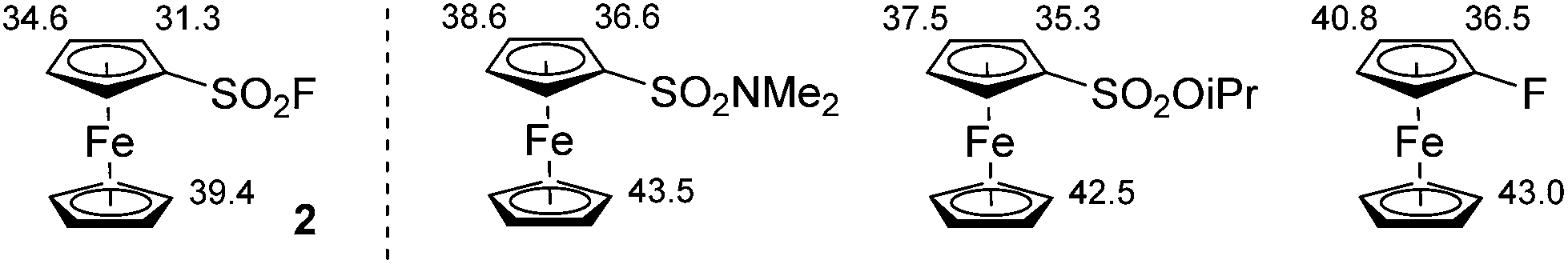 | ||
| Fig. 2 Calculated pKa values of compound 2 and selected ferrocene derivatives. | ||
In 2019, Barbasiewicz reported that alkyllithiums were not suitable bases to perform the ortho-deprotolithiation of arenesulfonyl fluorides, and that the use of lithium diisopropylamide in the presence of an in situ trap to intercept the lithiated intermediate was required.57 In 2021, Butenschön succeded in accumulating lithiated derivatives of 2 in tetrahydrofuran (THF) at −78 °C by using LiTMP (3 equiv.) as a hindered non-nucleophilic base.46 Indeed, subsequent addition of iodomethane led to the isolation of 2-methyl and 2,5-dimethylferrocenesulfonyl fluorides in 72 and 9% yields, respectively. Inspired by these results, we initially used LiTMP (1.1 equiv.) in the presence of trimethylsilyl chloride (1.1 equiv.) as an in situ trap in THF at −80 °C and isolated the 2-silylated derivative 3a (see Scheme 1) in a moderate 45% yield (42% recovery of 2). In 2017, Mukherjee and Grimster showed that the electrophilicity of the sulfur atom of arenesulfonyl fluorides can be reduced by the presence of electron-donating substituents on the phenyl ring.58 We similarly reasoned that the ferrocene core might be electron-rich enough to make ferrocenesulfonyl fluoride compatible with highly basic and nucleophilic alkyllithiums. Therefore, we performed the deprotolithiation of 2 with n-butyllithium in THF at −90 °C and isolated the derivative 3a in 80% yield after addition of trimethylsilyl chloride (Scheme 1). The use of the more reactive nBuLi·N,N,N′,N′-tetramethylethylenediamine (TMEDA) chelate was detrimental to the yield (68%) as the sulfone 4a was also isolated in 6% yield. We finally evaluated sBuLi for the deprotolithiation of 2 and isolated 3a in a good 82% yield despite the concomitant formation of 10% of the 2,5-bis(trimethylsilyl) derivative 5a.
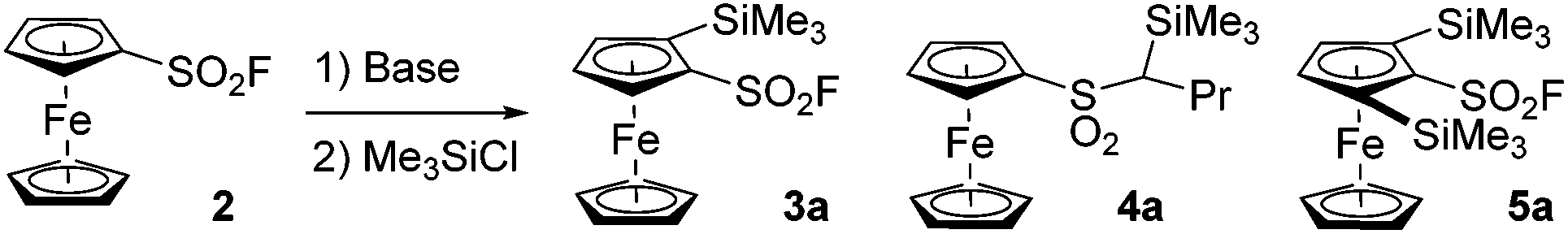 | ||
| Scheme 1 Deprotolithiation of 2. Reaction conditions: (i) nBuLi (1.2 equiv.) or either nBuLi·TMEDA (1.2 equiv.), sBuLi (1.2 equiv.), THF, −90 °C, 15 min; (b) Me3SiCl (1.2 equiv.), −90 to 0 °C. | ||
While functionalization of compound 2 by deprotolithiation was easily achieved, being able to control the stereochemical outcome of the reaction represents another challenge. In the past, the nBuLi·(−)-sparteine59 and nBuLi·(+)-sparteine surrogate60,61 chelates have been employed to perform the enantioselective deprotolithiation of a few ferrocenecarboxamides with up to 99% ee. Under close reaction conditions, the ee dropped to 58% starting from O-isopropylferrocenesulfonate.55 Other chelates made of alkyllithiums and derivatives of chiral cyclohexane-1,2-diamine were also used to functionalize (dialkylaminomethyl)ferrocenes62,63 and boron trifluoride-activated aminoferrocenes64,65 with ee ranging from 80 to 98%. Albeit less efficient, chiral lithium amides can also be used to reach enantioenriched 1,2-disubstituted ferrocenes. This was demonstrated by Price and Simpkins who reported the asymmetric deprotolithiation of P,P-diphenylferrocenephosphine oxide in 54% ee by using lithium bis[(S)-1-phenylethyl]amide [(S)-PEALi] in the presence of trimethylsilyl chloride as an in situ trap.66 We recently demonstrated that the use of more efficient chiral zinc-based in situ traps could be beneficial for some substrates and managed to reach enantioenriched N,N-dialkylferrocenecarboxamides,67O-alkylferrocenecarboxylates68 and O-isopropylferrocenesulfonate55 with ee ranging from 64 to 80%.
Here, we initially evaluated the use of alkyllithium·chiral diamine chelates to perform the enantioselective deprotolithiation of the substrate 2 before adding a suitable electrophile. However, the use of nBuLi in combination with either (+)-sparteine or its surrogate only afforded traces of the desired silylated product 3* in modest enantioselectivities (Table 2, entries 1 and 2). Using the more reactive sBuLi resulted in better yields but at the expense of the enantioselectivities (entries 3 and 4). We next evaluated chelates of either nBuLi or sBuLi with N,N,N′,N′-tetramethylcyclohexane-1,2-diamine ((R,R)-TMCDA). Again, better yields were encountered with the more reactive sBuLi but enantioselectivities remained low in both cases (entries 5 and 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | E | Yield (%) | er |
| a One equivalent of base and in situ trap were used. b Two equivalents of base and in situ trap were used. | |||||
| 1 | nBuLi·(+)-sparteine | Et2O | Me3Si | 2 | 33:67 |
| 2 | nBuLi·(+)-sparteine surrogate | Et2O | Me3Si | 3 | 36:64 |
| 3 | sBuLi·(+)-sparteine | Et2O | Me3Si | 61 | 54:46 |
| 4 | sBuLi·(+)-sparteine surrogate | Et2O | Me3Si | 48 | 54:46 |
| 5 | nBuLi·(R,R)-TMCDA | Et2O | Me3Si | 37 | 59:41 |
| 6 | sBuLi·(R,R)-TMCDA | Et2O | Me3Si | 64 | 56:44 |
| 7a | (R)-PEALi with TMSCl | THF | Me3Si | 38 | 58:42 |
| 8a | (S)-PEALi with TMSCl | THF | Me3Si | 33 | 42:58 |
| 9b | (R)-PEALi with TMSCl | THF | Me3Si | 42 | 57:43 |
| 10a | (R)-PEALi with [{(R)-PEA}2Zn] | THF | I | Traces | — |
| 11b | (R)-PEALi with [{(R)-PEA}2Zn] | THF | I | 33 | 29:71 |
|
|||||
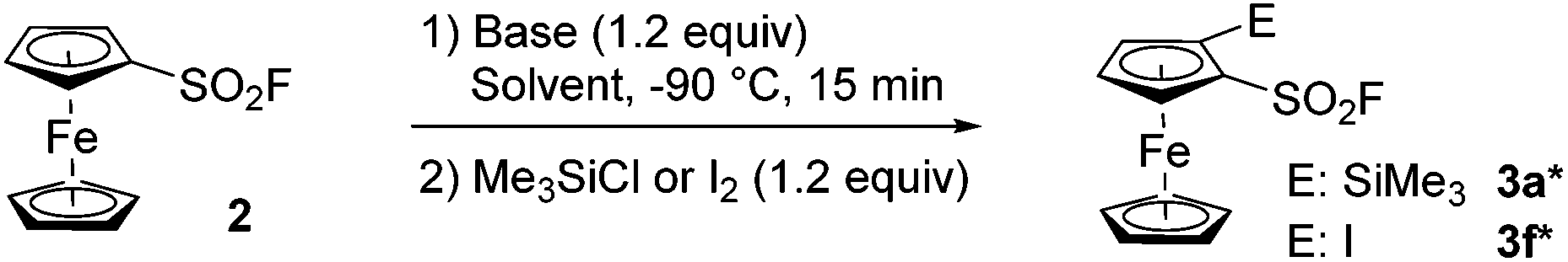
We finally evaluated the use of chiral lithium amides in combination with an in situ trap. Employing either (R)- or (S)-PEALi (1.2 equiv.) in the presence of trimethylsilyl chloride afforded the product 3a* in moderate yield and selectivity (entries 7 and 8), not improved when an excess of base was used (entry 9). Surprisingly, employing stoichiometric amounts of (R)-PEALi and [{(R)-PEA}2Zn] only afforded traces of the 2-iodoferrocenesulfonyl fluoride 3f* after iodolysis (entry 10). Repeating the reaction with 2 equivalents of (R)-PEALi led to 3f* in a moderate 33% yield but with the best enantioselectivities so far (entry 11). The relatively high acidity of the positions adjacent to the directing group and a weak interaction between the chiral base and the directing group, usually required to reach decent level of enantiocontrol,69 might account for these disappointing results. These results tend to indicate that the asymmetric deprotometallation of ferrocenesulfonyl fluoride (2) would require the development of original chiral bases. As this fall out of the scope of the present study, we decided to focus our efforts on the synthesis of racemic 2-substituted derivatives. The deprotolithiation was performed in our optimized conditions before the addition of a range of electrophiles. The use of trimethylsilyl chloride, diphenyldisulfide, chlorodiphenylphosphine respectively afforded the silane 3a (80% yield, Scheme 2), the sulfide 3b (67% yield) and the phosphine 3c (63% yield) while the use of diethyl chlorophosphate led to the phosphate 3d in 53% yield. The use of NFSI or iodine as the electrophile delivered the fluorinated 3e (48% yield) and the iodinated 3f (76% yield) derivatives. Using methyl chloroformate, N,N-diethylcarbamoyl chloride or Eschenmoser's salt70 as the electrophiles led to the methyl ester 3g (60% yield), the N,N-diethylcarboxamide 3h (44% yield) and the dimethylaminomethyl derivative 3i (75% yield).
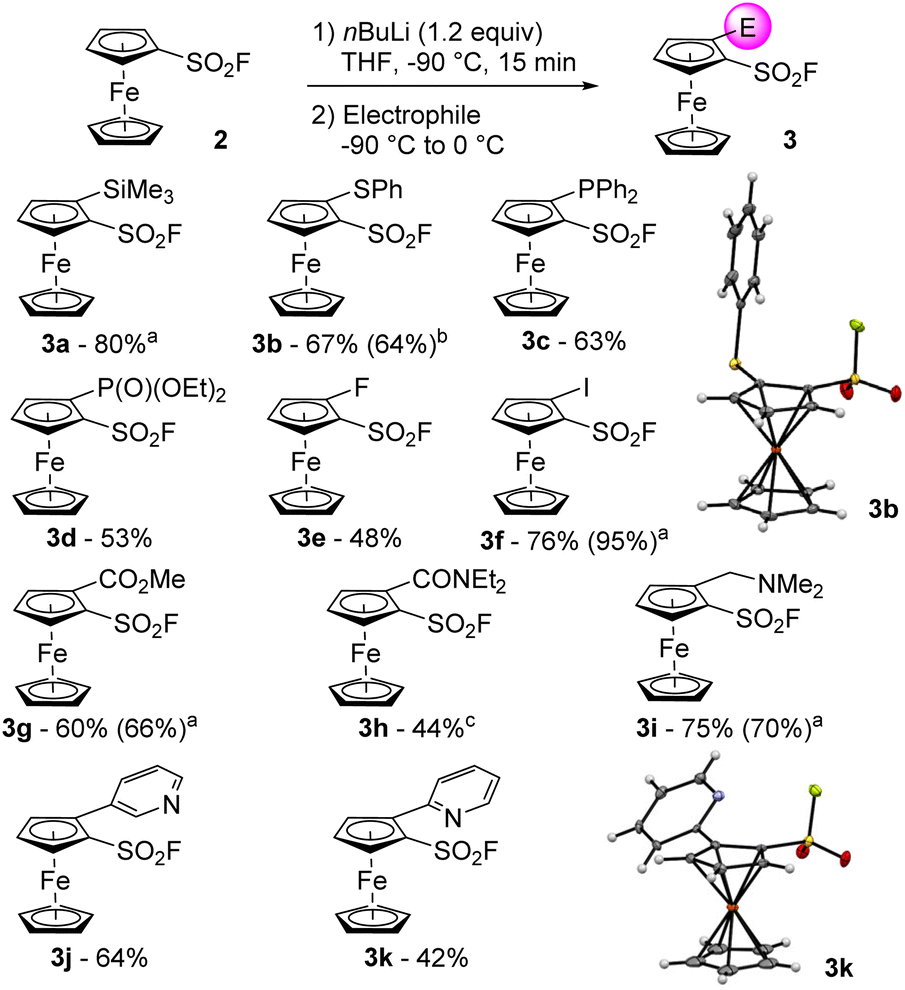 | ||
| Scheme 2 Deprotolithiation-electrophilic trapping sequences from the substrate 2 (for electrophiles used, see text above). All reactions performed on a 2 mmol scale. aReaction performed on a 10 mmol scale. bReaction performed on a 12 mmol scale. cReaction performed on a 25 mmol scale. | ||
It was also possible to perform a transmetallation from the lithiated intermediate to a zinc species before performing a Negishi cross-coupling with either 3-iodo- or 2-chloropyridine (compounds 3j and 3k; 64 and 42% yields, respectively).71,72
In some cases, we also observed the nucleophilic substitution of fluorine by nBuLi followed by the deprotolithiation of the C-sp3 next to the in situ formed sulfone. This led to the compounds 4a, b and c, all three isolated in 6% yield (Fig. 3). We also attempted to intercept 2-lithioferrocenesulfonyl fluoride with benzophenone, carbon dioxide and N,N-dimethylformamide (DMF). However, the desired products were not obtained from these attempts. It is possible that a transient alkoxide or carboxylate reacted with the fluorosulfonyl function to give sultone derivatives, prone to further reactions in the presence of a base. Similar cyclizations have already been reported by Davies and Dick in the benzene series.73
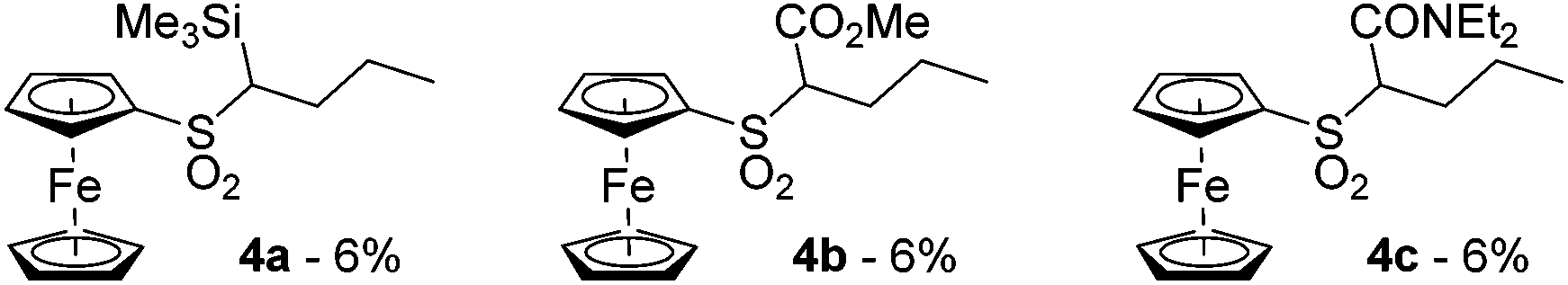 | ||
| Fig. 3 n-Butylsulfonylferrocene derivatives obtained during ferrocenesulfonyl fluoride deprotometallation-electrophilic trapping. | ||
To progress towards 1,2,3-trisubstituted ferrocenes, we engaged 2-trimethylsilylferrocenesulfonyl fluoride (3a) in another deprotolithiation-electrophilic trapping sequence (Scheme 3). By using nBuLi (1.5 equiv.) in THF at −90 °C for 15 min before the addition of iodine, the trisubstituted compound 5b was isolated in 85% yield. We also observed the formation of 2-(trimethylsilyl)-1-(1-iodobutylsulfonyl)ferrocene 4d (6%, see ESI†), coming from the attack of nBuLi on the sulfonyl fluoride group and the subsequent deprotometallation-iodolysis in α-position to the sulfone. Therefore, we tested the use of sBuLi under the same reaction conditions and obtained 5b in a similar 90% yield without traces of a sulfone derivative. Pleasingly, it was possible to scale the reaction up to 20 mmol with similar results (95% yield). The scope of this deprotolithiation-electrophilic trapping sequence was then studied by using other electrophiles such as NFSI, methyl chloroformate and N,N-diethylcarbamoyl chloride. The corresponding fluoride 5c, methyl ester 5d and N,N-diethylcarboxamide 5e were obtained in moderate to good yields. As phenylthio substituent is compatible with deprotolithiation,74 the substrate 3b was engaged into the same deprotolithiation-electrophilic trapping sequence. The use of iodine and hexachloroethane afforded the halogenated derivatives 5f and 5g in good yields while using NFSI furnished 5h in a moderate 45% yield, together with the inseparable remaining substrate 3b (9.5% estimated yield by 1H and 19F NMR). Trapping the lithiated intermediate with Eschenmoser's salt led to the (dimethylaminomethyl)ferrocene derivative 5i in 65% yield. We were also eager to assess the behaviour of a ferrocene substituted by two directing groups in deprotolithiation and therefore selected the substrate 3i. Furthermore, to evaluate the effect of the coordinating dimethylamino moiety onto the nucleophilic addition onto the fluorosulfonyl group, we came back to the use of nBuLi. The deprotolithiation was thus carried out with 1.5 equivalents of nBuLi at −90 °C before the addition of trimethylsilyl chloride. Apart from the recovery of 6% of starting material 3i, we isolated the two isomers 5j and 5k (20 and 9% yield, respectively), as well as the sulfone 4e in 17% yield. The increased yield of the butylsulfone 4e when compared to 4a–d might results from a favored attack of the nucleophilic base guided by the amine, as suspected.
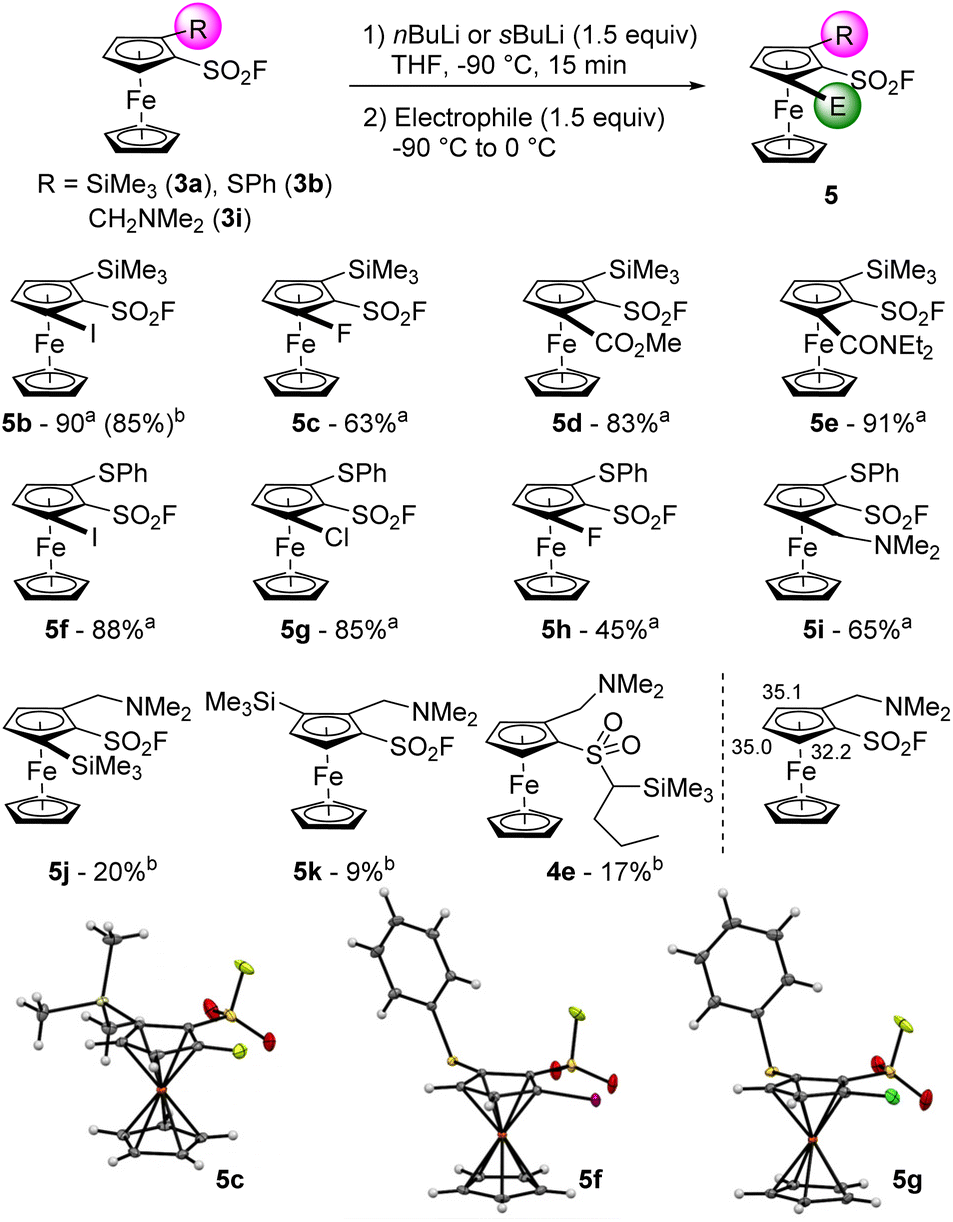 | ||
| Scheme 3 Deprotolithiation-electrophilic trapping sequence from the 1,2-disubstituted ferrocenes 3. aReaction performed with sBuLi. bReaction performed with nBuLi. | ||
The ‘halogen dance’ reaction is a stability-driven isomerization able to convert a 2-halogeno substituted compound into its 3-halogeno isomer.75–80 It usually requires the use of bulky lithium amides such as LiTMP or lithium diisopropylamide (LiDA) to perform an initial deprotolithiation which is followed by iterative lithium-halogen exchanges. Evidenced in the ferrocene series in 2010,81 the ‘halogen dance’ reaction has been successfully extended to various ferrocene derivatives and currently represents a valuable synthetic tool to reach original derivatives.47,54–56,82–87 Typical substrates combine a powerful directing group (DG) and a protecting group (PG) to prevent a competitive deprotolithiation of the otherwise activated free position (Scheme 4, top). A smart way to compare the ability of different directing groups to trigger the ‘halogen dance’ reaction is to compare the calculated pKa values of a series of isomerized product (Scheme 4, bottom). It appeared that moving from fluorine56 to sulfonamide54 and sulfonate55 reduces the pKa of the position between iodine and the directing group. The acidifying properties of the sulfonyl fluoride group were even more striking as a value of 27.7 was calculated for 4-iodo-2-trimethylsilylferrocenesulfonyl fluoride (6a). The isomerization from the substrate 5b should therefore be highly favoured.
 | ||
| Scheme 4 Top: The ‘halogen dance’ reaction. Bottom: calculated pKa values of selected ferrocene derivatives obtained by the ‘halogen dance’ reaction. | ||
Starting from the substrate 5b, the ‘halogen dance’ reaction was carried out by using LiTMP (1.1 equiv.) in THF at −50 °C for 1 h (Scheme 5). After methanolysis, the desired product 6a was isolated in 62% yield together with 22% of the deiodinated product 3a, resulting from a competitive lithium/iodine exchange, as already reported in the literature.55,56,82,83,87 Pleasingly, it was possible to perform the reaction on a 16 mmol scale with slightly better results (6a, 79% yield). Using Eschenmoser's salt to trap the lithiated intermediate afforded the 1,2,3,4-tetrasubstituted derivative 6b in 68% yield while the use of methyl chloroformate only afforded the ester 6c in a surprising low 2% yield. We also found possible to perform a lithium to zinc transmetallation using ZnCl2·TMEDA before performing a Negishi cross-coupling. The desired product 6d was isolated in 27%, together with 9% of the 1,2,4-trisubstitued ferrocene 6a. Although this is the first time that a one-pot ‘halogen dance’ reaction-Negishi cross-coupling has been reported in the ferrocene series, related examples from 2,5- or 2,3-dibromothiophenes have been recently documented by Okano.88,89 We next attempted to apply the same isomerization conditions to the substrate 5f and isolated the isomers 6e and 6f in 38 and 13% yield, respectively, after methanolysis (16% of deiodinated product 3b was also obtained). The combination of the long-range acidifying properties of the sulfonyl fluoride group, combined with the moderate adjacent protecting group ability of the phenylthio substituent, could explain why both the 3 and 4 positions of the substrate 5f can be deprotolithiated. The ‘halogen dance’ reaction of the resulting lithiated intermediates leads to the isomers 6e–f. When the methanolysis was replaced by an electrophilic trapping with Eschenmoser's salt, we only obtained 12% of the 1,2,3,4-tetrasubstituted ferrocene 6g with the recovery of 21% of the starting material.
 | ||
| Scheme 5 Deprotolithiation-electrophilic trapping sequence from the 1,2-disubstituted ferrocenes 5b and 5f. aReaction performed on a 16 mmol scale. | ||
Another approach towards polysubstituted ferrocenes relies on iterative deprotolithiation-electrophilic trapping sequences, provided that the substituents already present are compatible with strong bases and that a new directing group is introduced at each step.
We initially selected the N,N-diethylferrocenecarboxamide derivative 5e as a substrate in this approach. The deprotolithiation was carried out with LiTMP in the presence of ZnCl2·TMEDA as an in situ trap before performing the iodolysis of the resulting zinc species (Scheme 6, top). The expected derivative 7a was obtained in a moderate 56% yield, together with its isomer 7b (7% yield) with the iodine atom on the other cyclopentadienyl ring. Although similar remote functionalization has previously been reported from N,N-diisopropylferrocenecarboxamide59,90 or (4-isopropyl-2-oxazolyl)-ferrocene,91 both bearing an adjacent trimethylsilyl group, it was not expected starting from such a substrate. Therefore, to favour a deprotolithiation next to a directing group, we moved from the carboxamide to a fluorine atom (Scheme 6, bottom). The substrate 5c was deprotolithiated with sBuLi before the addition of hexachloroethane as the electrophile towards the tetrasubstituted ferrocene 8a (75% yield). The trimethylsilyl group preventing any adjacent deprotolithiation, it was first removed using a slight excess of tetrabutylammonium fluoride (TBAF) towards the 1,2,3-trisubstituted ferrocene 9 (70% yield). With two positions adjacent to a directing group, it was not clear which would be deprotolithiated first. To get an insight of the regioselectivity of the deprotolithiation, 9 was treated with sBuLi before the addition trimethylsilyl chloride. An inseparable 1:1 mixture of the two isomers 8a and 8b was obtained in 50% yield, indicating the similar directing group properties of the chlorine atom and the fluorosulfonyl group, at least on this substrate.
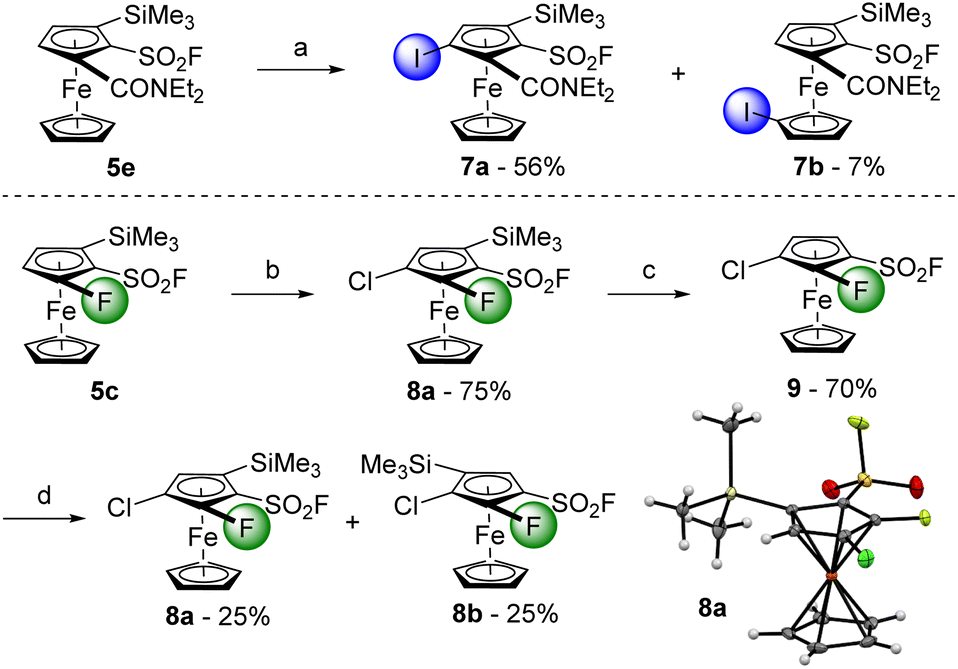 | ||
| Scheme 6 Deprotolithiation-electrophilic trapping sequence towards polysubstituted ferrocenes. Reaction conditions: (a) (i) ZnCl2·TMEDA (1.0 equiv.), THF, −30 °C; (ii) LiTMP (2.0 equiv.), 30 min; (iii) I2 (2.0 equiv.), −30 °C to rt; (b) (i) sBuLi (1.2 equiv.), THF, −80 °C, 1 h; (ii) C2Cl6 (1.2 equiv.), −80 °C to rt; (c) TBAF (1.5 equiv.), THF, 0 °C, 5 min; (d) (i) sBuLi (1.5 equiv.), THF, −90 °C, 1 h; (ii) Me3SiCl (1.2 equiv.), −80 °C to rt. | ||
Thus, to avoid any regioselectivity issue, we replaced the chlorine atom of 8a by a methyl group with the substrate 10, obtained in 85% yield from 5c (Scheme 7). A desilylation was performed with TBAF towards the compound 11 (89% yield), which was treated with sBuLi before an iodolysis delivered the 1,2,3,4-tetrasubstituted derivative 12 in 87% yield. The latter was finally engaged into the ‘halogen dance’ reaction in the presence of LiTMP before the addition of Eschenmoser's salt. The first 1,2,3,4,5-pentasubstituted ferrocenesulfonyl fluoride 13 was therefore obtained in 31% yield while the 1,2,3,4-tetrasubstituted ferrocene 14, resulting from a lithium-iodine exchange, was also isolated in 21% yield.
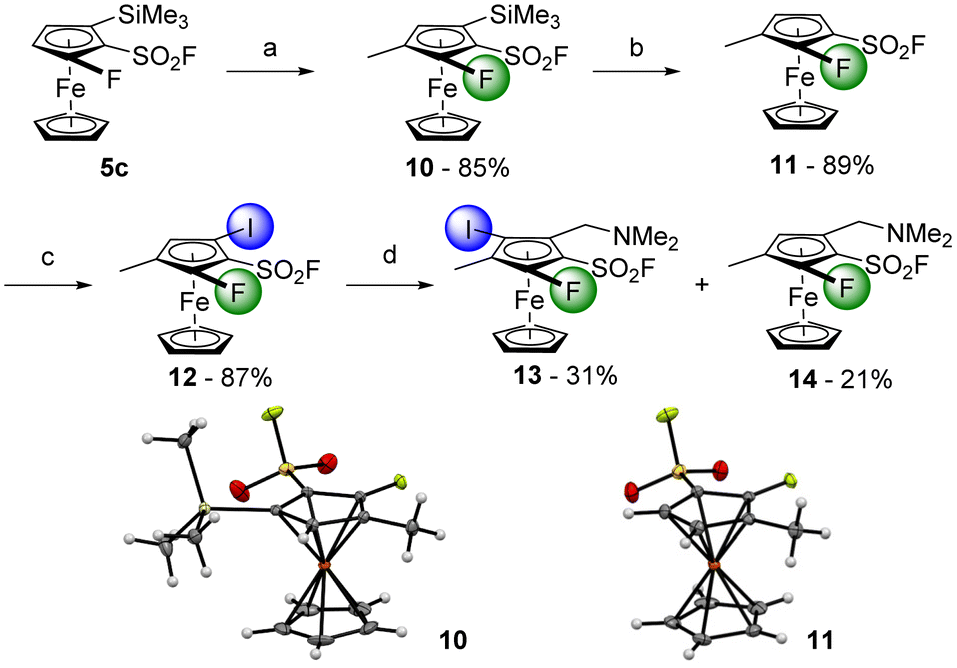 | ||
| Scheme 7 Deprotolithiation-electrophilic trapping sequence towards the 1,2,3,4,5-pentasubstituted ferrocene 13. Reaction conditions: (a) (i) sBuLi (1.2 equiv.), THF, −80 °C, 1 h; (ii) CH3I (1.2 equiv.), −80 °C to rt; (b) TBAF (1.5 equiv.), THF, 0 °C, 5 min; (c) (i) sBuLi (1.5 equiv.), THF, −90 °C, 1 h; (ii) I2 (1.2 equiv.), −80 °C to rt; (d) (i) LiTMP (1.1 equiv.), THF, −50 °C, 1 h: (ii) H2C=NMe2I (1.1 equiv.), −50 °C to rt. | ||
To reduce the step count towards 1,2,3,4,5-pentasubstituted ferrocenes, we tried to replace the trimethylsilyl group by a phenylthio substituent. The 1,2,3-trisubstituted ferrocene 5g was thus deprotolithiated with sBuLi (1.2 equiv.) before methyl chloroformate was added towards the 1,2,3,4-tetrasubstituted ferrocene 15, isolated in 68% yield (Scheme 8, top). We planned to introduce iodine as the fifth substituent and therefore reacted the substrate 15 with an excess of LiTMP in the presence of ZnCl2·TMEDA as an in situ trap before performing an iodolysis. However, degradation mainly occurred under these reaction conditions, and we only isolated 5% of the desired 1,2,3,4,5-pentasubstituted ferrocene 16 (3% of the starting material 15 were also recovered).
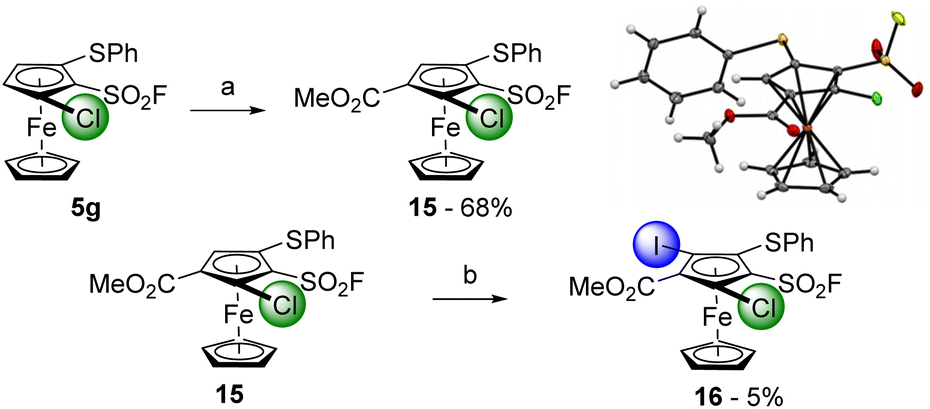 | ||
| Scheme 8 Deprotolithiation-electrophilic trapping sequence towards the 1,2,3,4,5-pentasubstituted ferrocene 16. Reaction conditions: (a) (i) sBuLi (1.2 equiv.), THF, −80 °C, 1 h; (ii) ClCO2Me (4.0 equiv.), −80 °C to 0 °C; (b) (i) ZnCl2·TMEDA (1.0 equiv.), THF, −50 °C; (ii) LiTMP (2.0 equiv.), 20 min; (iii) I2 (2.0 equiv.), −30 °C to 0 °C. | ||
We finally evaluated the possibility to replace the methyl ester by a dimethylaminomethyl group and therefore prepared the derivative 17 in 64% from the substrate 5g (Scheme 9). However, a competitive chlorine/lithium exchange occurred, leading to 10% of the inseparable 1,2,3-trisubstituted ferrocene 5i, identified by comparison with the NMR spectrum of the pure compound (Scheme 3). A final deprotolithiation-electrophilic trapping sequence was performed on the mixture of 17 and 5i with sBuLi and iodine, successively. The former afforded the expected 1,2,3,4,5-pentasubstituted ferrocene 18 in 44% yield while the latter led to the ferrocene 6g, previously obtained by the ‘halogen dance’ reaction (Scheme 5), and to its isomer 6h (32 and 54% yield, respectively). The structures of the ferrocenes 17 and 18 were unambiguously assigned by X-ray diffraction analysis (Fig. 4).
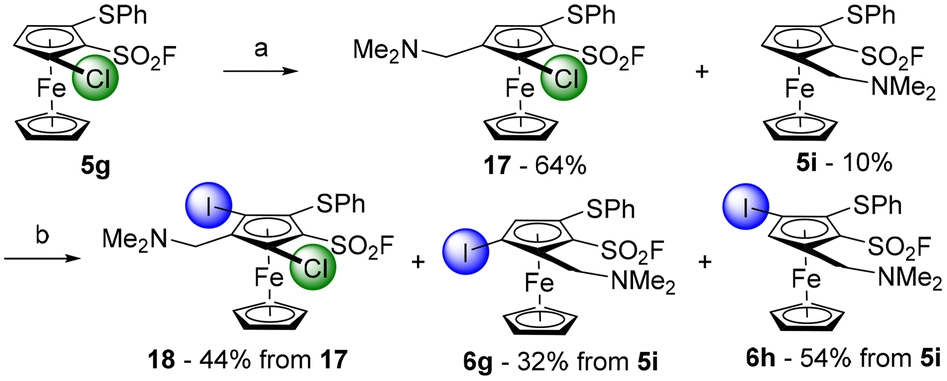 | ||
| Scheme 9 Deprotolithiation-electrophilic trapping sequence towards the 1,2,3,4,5-pentasubstituted ferrocene 18. Reaction conditions: (a) (i) sBuLi (1.2 equiv.), THF, −80 °C, 1 h; (ii) H2C=NMe2I (1.2 equiv.), −50 °C to 0 °C; (b) (i) sBuLi (1.2 equiv.), THF, −80 °C, 1 h; (ii) I2 (1.2 equiv.), −80 °C to 0 °C. | ||
 | ||
| Fig. 4 Solid-state structures of the polysubstituted ferrocenes 17 and 18. | ||
With the variety of ferrocenesulfonyl fluoride derivatives available, we were eager to evaluate their orthogonal reactivity in reactions other than deprotolithiation. To increase the chemical space available around the ferrocenesulfonyl fluoride scaffold, we first performed some desilylation reactions. The compounds 6a, 6b and 7a were thus treated with TBAF (1.5 equiv.) in THF at 0 °C (Scheme 10). Full conversion was observed after 5 min and the compounds 19a and 19b were isolated in good yields. Pleasingly, it was possible to scale up the desilylation of 6a and 6b from 1 to 10 mmol with similar results (84 and 79% yields, respectively). However, only 65% of 19c were obtained due to the degradation tendency of the N,N-diethylcarboxamide group.
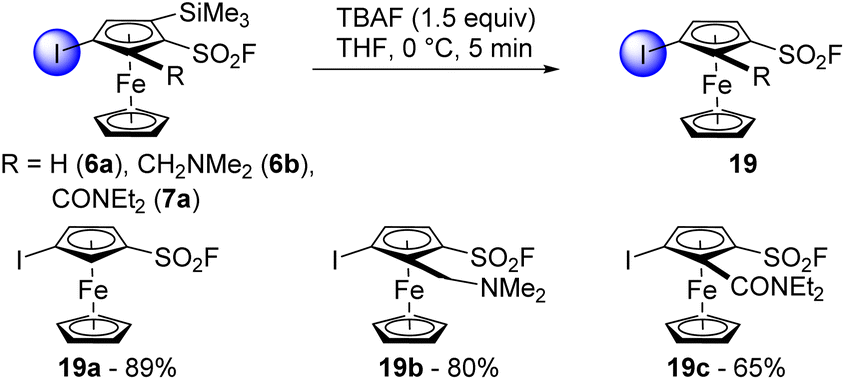 | ||
| Scheme 10 Desilylation of substituted ferrocenesulfonyl fluoride derivatives. | ||
The halogen/lithium exchange is a useful reaction to convert aromatic bromides and iodides into a range of derivatives depending on the electrophile used. Therefore, to reach original 1,2,4-trisubstituted ferrocenes, we first treated the substrate 6a with of tBuLi (2 equiv.) in THF at −90 °C for 5 min (Scheme 11).92,93 The addition of DMF led to the aldehyde 20a (83% yield) while the use of triisopropylborate afforded the derivative 20b (70% yield), easily purified as a pinacol ester. The same reaction conditions were applied to the 1,3-disubstituted ferrocene 19a, and the use of Eschenmoser's salt or methyl chloroformate as electrophiles afforded the derivatives 20c and 20d in 72 and 86% yield, respectively. No sign of competitive deprotolithiation or nucleophilic addition of the lithiated product was noticed during these transformations. Only of few percent of the compounds 3a and 2, resulting from the in situ hydrolysis of the lithiated intermediate were noticed. Furthermore, while the use of DMF to reach 2-formylferrocenesulfonyl fluoride previously failed (Scheme 2), the good yield obtained for the ferrocene 20a clearly indicates that the position on which the functional group is introduced is of main importance in this series.
 | ||
| Scheme 11 Iodine/lithium exchange from the ferrocenes 6a and 19a. | ||
The quaternarization of the dimethylaminomethylated derivative 3i was performed with an excess of iodomethane in acetonitrile and afforded the ammonium salt 21 in quantitative yield (Scheme 12, top). However, performing the substitution of dimethylamine to an acetate proved more challenging. Indeed, when the same 1,2-disubstituted ferrocene 3i was reacted with an excess of acetic anhydride at 120 °C for 1 h, the corresponding acetate 22a was isolated in a moderate 52% yield while degradation was observed (Scheme 12, bottom). Performing the substitution at lower temperature required longer reaction time and resulted in lower yields. As the 1,3-disusbtitued isomer 22b was obtained in 60% yield after 1 h at 120 °C from 20c, it can be proposed that the position of the substituent has a limited influence on the reaction outcome and that the main issue is probably the limited stability of the compounds in these conditions.
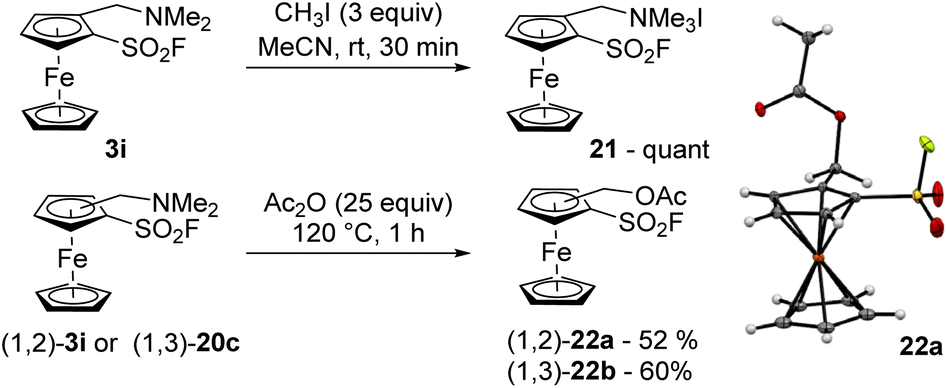 | ||
| Scheme 12 Quaternarization and substitution of amines from ferrocenes 3i and 20c. | ||
Arenesulfonyl fluorides are known to be compatible with the use of reductants such as sodium cyanoborohydride,94 sodium triacetoxyborohydride,95 zinc in acetic acid,96 hydrogen and palladium,97 or DIBALH at −40 °C.95 In the ferrocene series, we were able to carry out the reduction of the ester 3g with an excess of DIBALH in THF at 0 °C towards the alcohol 23 (49% yield; Scheme 13, top). The poor stability of the latter was nevertheless a limiting factor which could be overcome by using the crude alcohol in a further step as demonstrated by the synthesis of the silyl ether 24a in 68% yield. The 1,3-disubstiuted isomer 24b was similarly obtained (70% yield) starting from the ester 20d. Moses also demonstrated that arenesulfonyl fluorides could be stable in the presence of borane at room temperature.98 In our hands, we were able to perform the reduction of the N,N-diethylcarboxamide derivative 3h using an excess of borane at 80 °C (compound 25). The sulfonyl fluoride remained mostly unaffected by either the reductant used, or the strong basic aqueous conditions used during the work-up procedure.
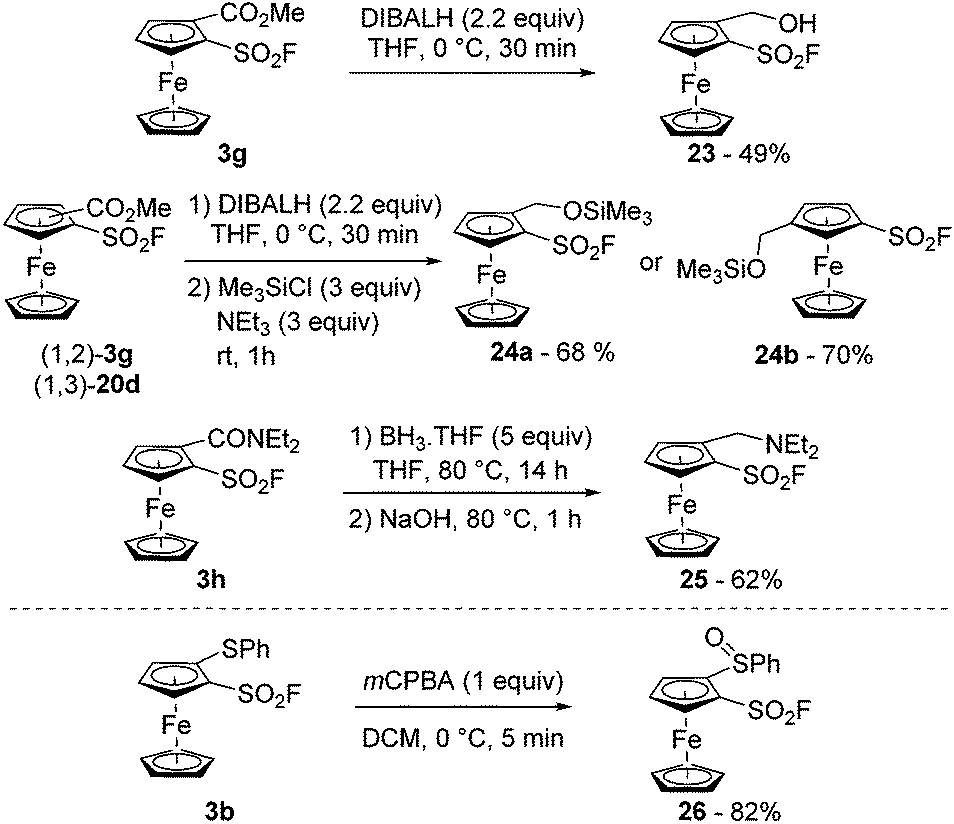 | ||
| Scheme 13 Ferrocenesulfonyl fluoride derivatives in reductive (top) and oxidative (bottom) reaction conditions. | ||
Up to now, aromatic sulfonyl fluorides have been demonstrated to be stable in the presence of various oxidative agents such as chromium trioxide,99N-methylmorpholine oxide,100 the Dess–Martin periodinane,95 nitric acid,101tert-butyl hypochlorite,102 osmium tetroxide103 and sodium periodate.104 However, to our knowledge, the compatibility of sulfonyl fluoride with peracids has never been studied. Thus, we reacted 2-(phenylthio)ferrocenesulfonyl fluoride (3b) with meta-chloroperbenzoic acid (mCPBA) in dichloromethane at 0 °C towards the corresponding sulfoxide 26 (Scheme 13, bottom). With only 1 equivalent of oxidant, full conversion was reached within minutes and the desired product was isolated in 82% yield. However, when the reaction was repeated with 5 equivalents of mCPBA in view to get the corresponding sulfone, degradation mainly occurred and the sulfoxide 26 was the only isolable product (8% yield). The origin of this degradation currently remains unclear. However, the increased electrophilicity of the fluorosulfonyl group upon oxidation of the sulfide to the sulfoxide or sulfone as well as the light sensitivity of the sulfoxide 26 in solution might be involved.
Sulfonyl fluorides are usually inert in cross-coupling reactions in the benzene series.105 Thus we decided to study the behaviour of some of our ferrocenesulfonyl fluoride derivatives in a few representative reactions, starting with the Suzuki–Miyaura cross-coupling.106,107 Indeed, arenesulfonyl fluorides have been successfully used in this coupling using ligand-free or palladium-ligand combinations in water,108,109 water-dioxane mixtures104 or anhydrous DMF110 and cyclic ethers.111 We initially attempted the coupling of 2-iodoferrocenesulfonyl fluoride (3f) with various phenylboronic acids in the presence of Pd(dba)2 (dba: dibenzylideneacetone), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos)112 and cesium fluoride in toluene at 110 °C (Scheme 14). Pleasingly, both electron-rich and -poor phenylboronic acids were compatible as the coupling products 27a–c were obtained in good yields. The 3-iodo isomer 19a behaved in very similar way, even when starting from sterically hindered 2,6-dimethoxyphenylboronic acid (94 and 79% yields for the compounds 28a and 28b, respectively). When the 1,2,4-trisubstituted ferrocene 6a was evaluated under similar reaction conditions, the products 29a and 29b were obtained in moderate yields. The reduced yield of 29b when compared to 28b suggests some negative effect of the trimethylsilyl substituent on the reaction outcome. Although additional studies would be required, a steric effect might not be predominant here regarding the remote position of the iodine atom. The same reaction conditions were also applied to the 1,2,3,4-tetra- and 1,2,3-trisubstituted substrates 6b and 19b but the starting materials were recovered unchanged (not shown). The position of the amino substituent next to the iodine atom, able to trap a catalytic species, might explain this lack of reactivity. Moran and Ding recently reported that the usual inertness of sulfonyl fluoride in cross-coupling can be overcome, and proposed an original desulfonative Suzuki–Miyaura coupling.113,114 However, when we treated ferrocenesulfonyl fluoride (2) with 4-methoxyphenylboronic acid under Moran's reaction conditions (Pd(acac)2, SPhos, dioxane, 130 °C), we did not observed any reaction and the starting material was fully recovered (not shown).
 | ||
| Scheme 14 Ferrocenesulfonyl fluorides in Suzuki–Miyaura cross-coupling. | ||
We previously demonstrated the compatibility of ferrocenesulfonyl fluorides with organozinc derivatives in the Negishi cross-coupling towards 1,2-disubstituted (Scheme 2) and 1,2,3,4-tetrasubstituted derivatives (Scheme 5). However, we also wanted to involve other substrates bearing reactive groups in such coupling. We first started with the ester 3g which was deprotolithiated with LiTMP in the presence of ZnCl2·TMEDA as an in situ trap in THF at −50 °C (Scheme 15, top). The resulting zinc species was then engaged in the cross-coupling with 3-iodopyridine in the presence of PdCl2 and dppf towards the 1,2,3-trisubstituted ferrocene 30a. Although isolated in a moderate 36% yield, there was no evidence of functionalization next to the ester. We also engaged the iodinated derivative 3f in a similar sequence and obtained the desired ferrocene 30b in 45% yield. Although moderate, to our knowledge, this constitutes the first example of a palladium-catalysed Negishi coupling from an iodinated zinc species. We finally engaged 30b in a Suzuki–Miyaura cross-coupling under the same reaction conditions as before and obtained the 2,5-bis-arylated ferrocenesulfonyl fluoride 31 in 75% yield (Scheme 15, bottom).
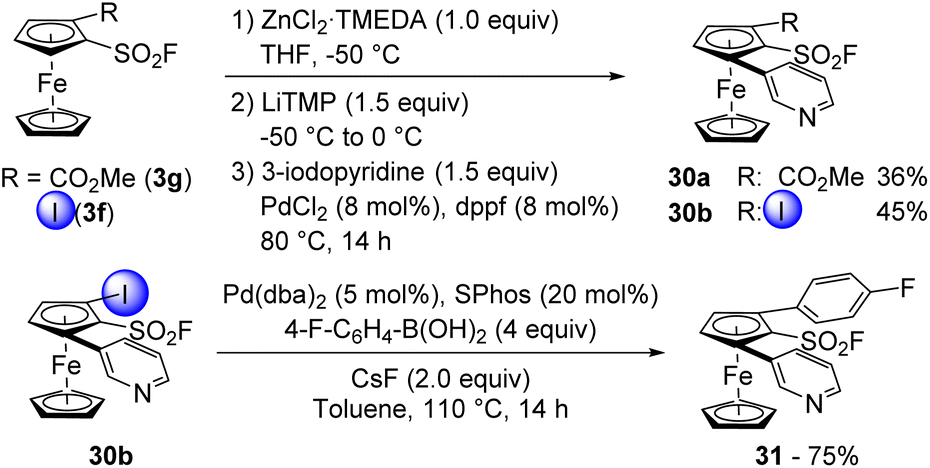 | ||
| Scheme 15 Sensitive ferrocenesulfonyl fluorides in Negishi cross-coupling and subsequent Suzuki–Miyaura cross-coupling. | ||
There are only two reports on the involvement of aromatic sulfonyl fluorides in a Sonogashira cross-coupling.115,116 One from Robinson using copper(I) iodide and bis(triphenylphosphine)palladium(II) dichloride and triethylamine,95 the other from Melnyk using the same catalytic system but with potassium carbonate as the base.117 In the ferrocene series, Albrecht and Long reported very efficient Sonogashira couplings using a combination of PdCl2(MeCN)2 and P(tBu)3.118 We were thus eager to evaluate the ability of this system in the Sonogashira coupling of some iodinated ferrocenesulfonyl fluorides with trimethylsilylacetylene. The 2-iodinated derivative 3f was first reacted in the presence of commercially available bis(tri-tert-butylphosphine)palladium(0) and copper(I) iodide in a THF-diisopropylamine mixture at 25 °C (Scheme 16, top). After 14 h, the desired coupling product 32a was isolated in a very good 90% yield. The use of the 3-iodinated isomer 19a as substrate similarly afforded the compound 32b in 84% yield. The Sonogashira coupling was finally extended to more complex structures with the phenylthiol-substituted ferrocenesulfonyl fluorides 5f and 6e. Under similar reaction conditions, the 1,2,3-trisubstituted ferrocene 32c was isolated in 77% yield while its 1,2,4-trisubstituted isomer 32d was obtained in 73% yield. Although moderate yields were recorded for these two last products, to our knowledge, it is the first time that sulfide-substituted ferrocenes are successfully used in a Sonogashira cross-coupling. Desilylation was finally performed in mild conditions using TBAF to deliver the terminal alkynes 33a–c in moderate to good yields (Scheme 16, bottom).
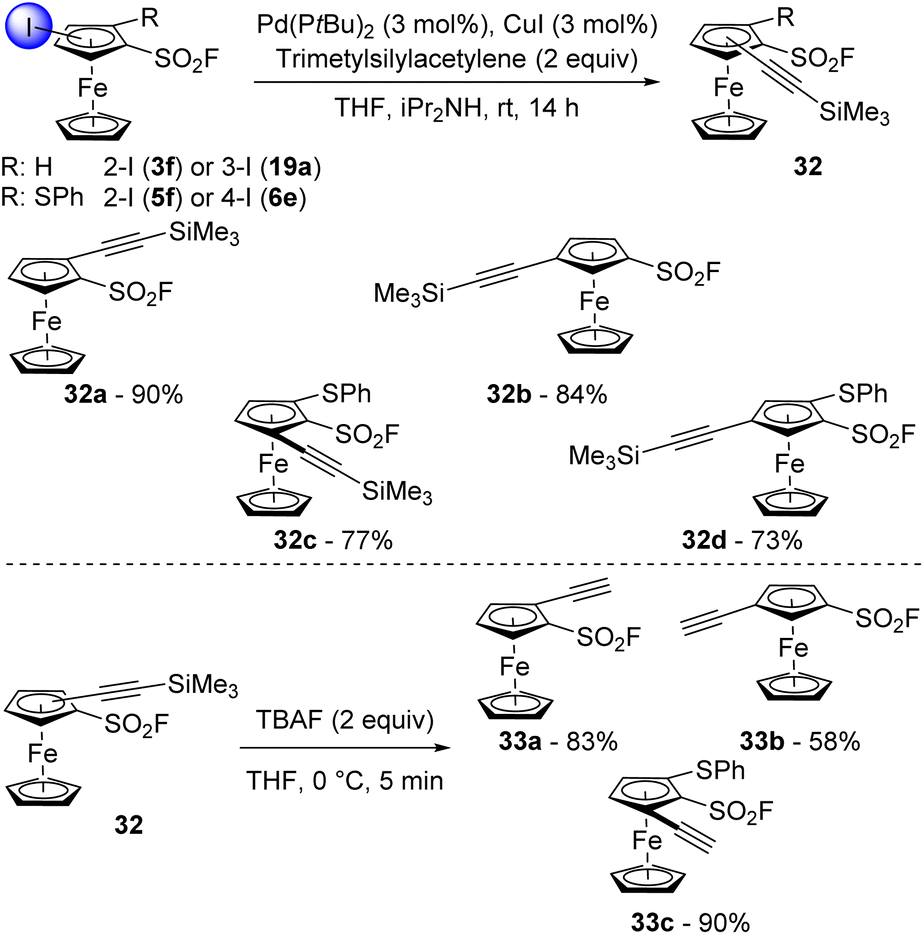 | ||
| Scheme 16 Ferrocenesulfonyl fluorides in Sonogashira cross-coupling and subsequent desilylation. | ||
To our knowledge, halogenated arenesulfonyl fluorides have never been engaged into a metal-catalysed amidation. We were thus eager to evaluate the compatibility of some ferrocenesulfonyl fluorides in this coupling. We first selected 4-iodo-2-trimethylsilylferrocenesulfonyl fluoride (6a) and attempted a copper(I) oxide promoted coupling with phthalimide in acetonitrile at 80 °C, but the substrate 6a was fully recovered.119 Unfortunately, using an acetonitrile/pyridine mixture as solvent120 did not favour any reaction. We recently demonstrated that iodoferrocenes could react with a range of amides using a copper(I) iodide-N,N′-dimethylethylenediamine (DMEDA) catalytic system in the presence of tribasic potassium phosphate in dioxane.121,122 Using these reaction conditions, the coupling between the 1,2,4-trisubstitued ferrocene 6a and pyrrolidinone was first attempted and afforded the desired product 34a in a moderate 44% yield (Scheme 17). We also isolated 14% of the starting material 6a and 28% of the deiodinated product 3a. When the 1,3-disusbstituted ferrocene 19a was engaged in the same coupling conditions, the yield of the expected product 34b dropped to 19% while 32% of the starting material was recovered (27% of deiodinated product 2 were also obtained). As already observed in the Suzuki–Miyaura coupling of iodoferrocenesulfonyl fluorides (Scheme 14), the trimethylsilyl group has also an impact on the amidation outcome. However, while a negative effect was observed in the palladium-catalysed Suzuki coupling, a positive effect was noticed in this copper-promoted amidation.
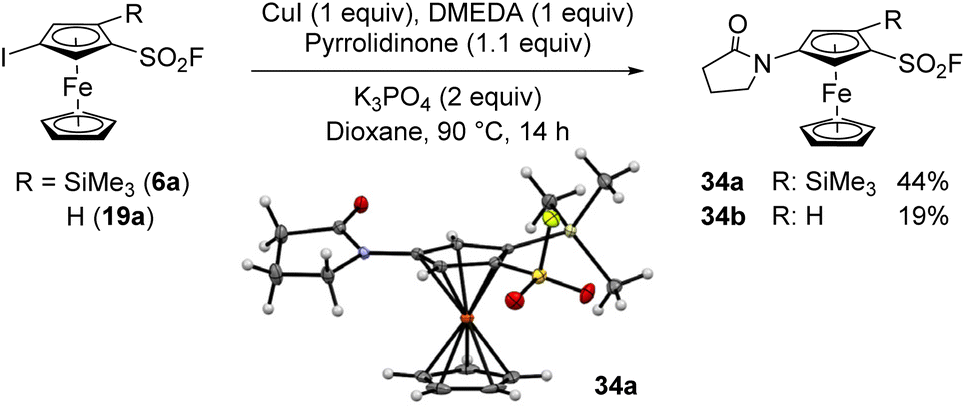 | ||
| Scheme 17 Ferrocenesulfonyl fluorides in Goldberg-type cross-coupling. | ||
The coupling of 3-iodoferrocenesulfonyl fluoride (19a) with carboxylic acids in the presence of copper(I) oxide,61,86 as well as a copper-catalysed Hirao coupling with diethylphosphite were also attempted but failed to deliver any expected product (not shown).123
Apart from their robustness in various reaction conditions, sulfonyl fluorides can react specifically at the sulfur atom under specific conditions. We were thus eager to evaluate the ability of ferrocenesulfonyl fluorides to act as a general precursor of various sulfur-based ferrocene derivatives. Indeed, while the electron-rich ferrocene core reduces the electrophilicity of the sulfonyl fluorine allowing the use of alkyllithiums in deprotolithiation, it might also impair substitution by nucleophiles. Therefore, we first evaluated the reaction between 2 and morpholine in acetonitrile in the presence of cesium carbonate or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
However, no desired product could be detected after 14 h at 25 or 80 °C (Table 3, entries 1 and 2). Similar results were reported in the benzene series by Ende and Balls who further showed that the reactivity of sulfonyl fluorides can be improved in the presence of calcium triflimide.124,125 However, concerned by the price of this reagent, we took inspiration from the 1-hydroxybenzothiazole (HOBt)-mediated amidation of sulfonyl fluorides described by Li.126 The reaction between the substrate 2, HOBt and morpholine in dimethylsulfoxide (DMSO) afforded an encouraging 21% yield of sulfonamide 35a after 14 h at 50 °C (entry 3). While the addition of 1,1,3,3-tetramethyldisiloxane (TMDS), as described by Li,126 was without effect (entry 4), we found that increasing the amount of HOBt and using triethylsilane as an additive slightly increased the yield of 35a (entry 5). While increasing the reaction time to 72 h was beneficial (entry 6), performing the reaction at 110 °C led to complete decomposition. When the substitution was attempted from 3-iodoferrocenesulfonyl fluoride (19a), the corresponding sulfonamide 35b was isolated in 76% yield (entry 7), suggesting that the electron-withdrawing properties of iodine might improve the reaction outcome.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent (n equiv.) | Temp (°C) | Time (h) | 35 (%) | 2 (%) |
| a Reaction performed in MeCN instead of DMSO. b Compound 19a. | |||||
| 1a | Cs2CO3 (3) | 25–80 | 14 | nd (35a) | 76–38 |
| 2a | DBU (3) | 25–80 | 14 | nd (35a) | 89–91 |
| 3 | HOBt (1) | 50 | 14 | 21 (35a) | 50 |
| 4 | HOBt (1)–TMDS (2) | 50 | 14 | 20 (35a) | 48 |
| 5 | HOBt (1.5)–Et3SiH (2) | 50 | 14 | 34 (35a) | 50 |
| 6 | HOBt (1.5)–Et3SiH (2) | 50 | 72 | 61 (35a) | 11 |
| 7 | HOBt (1.5)–Et3SiH (2) | 50 | 16 | 76 (35b) | ndb |

Azoles are known to be more reactive than aliphatic amines in the SuFEx reaction in the benzene series.49 A similar trend was also observed in the ferrocene series as the reaction between ferrocenesulfonyl fluoride (2) and imidazole in the presence of cesium carbonate at room temperature led to the compound 36a in 91% yield (Scheme 18). While the reaction between 3-methoxycarbonylferrocenesulfonyl fluoride (20d) and pyrazole was as efficient (compound 36b, 87% yield), the less reactive 1,2,4-triazole only afforded 36c in 25% yield despite the presence of iodine on the substrate 19a, recovered in 65% yield. The SuFEx reaction was also found to be sensitive to steric hindrance as the reaction between 4-iodo-2-trimethylsilylferrocenesulfonyl fluoride (6a) and 1,2,4-triazole led to only 3% of the product 36c lacking the silyl group (not shown). As we also isolated 33% of the desilylated ferrocene 19a and 39% of starting material, it is likely that the desilylation occurred before the substitution.
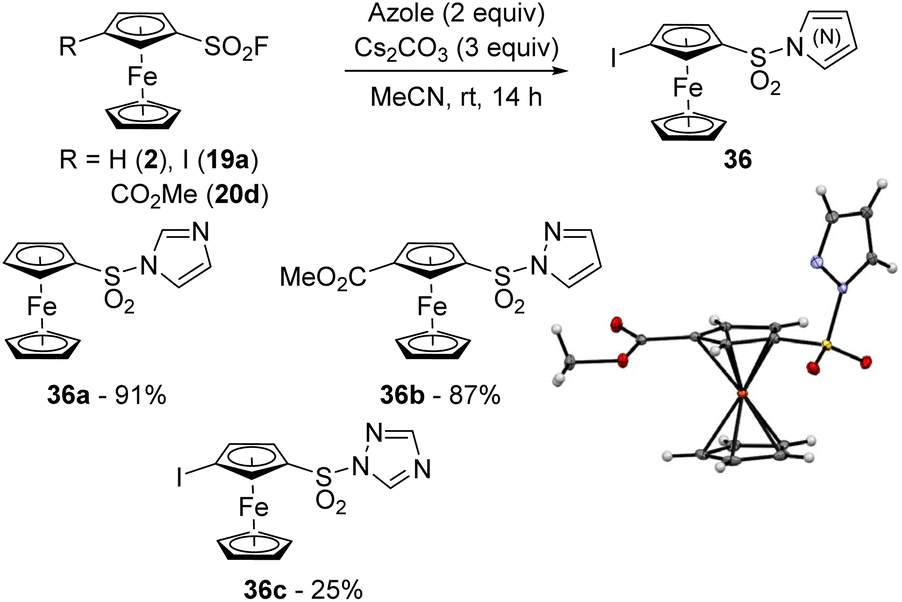 | ||
| Scheme 18 SuFEx reaction of ferrocenesulfonyl fluorides with azoles. | ||
In 2019, Ma, Xu and Yang reported the similar reactivity of aromatic fluorosulfonates and sulfonyl fluorides with trimethylsilyloxyl imidates towards N-acylsulfamates and N-acylsulfonamides.127 In a related work, De Borgraeve prepared N-acylsulfamate derivatives from aryl fluorosulfonates and carboxamides in the presence of sodium hydride.128 Inspired by this work, we reacted ferrocenesulfonyl fluoride (2) with acetamide in the presence of 2.5 equivalent of sodium hydride (Scheme 19). After 14 h at room temperature, the desired product 37a was isolated in a moderate 45% yield. Pleasingly, increasing both the amount of sodium hydride to 4 equivalents and the temperature to 50 °C afforded the N-acylsulfonamide 37a in a better 63% yield. Although the reaction was successfully extended to 3-iodoferrocenesufonyl fluoride 19a (product 37b, 70% yield), all our attempts to react benzamide derivatives into this SuFEx reaction failed.
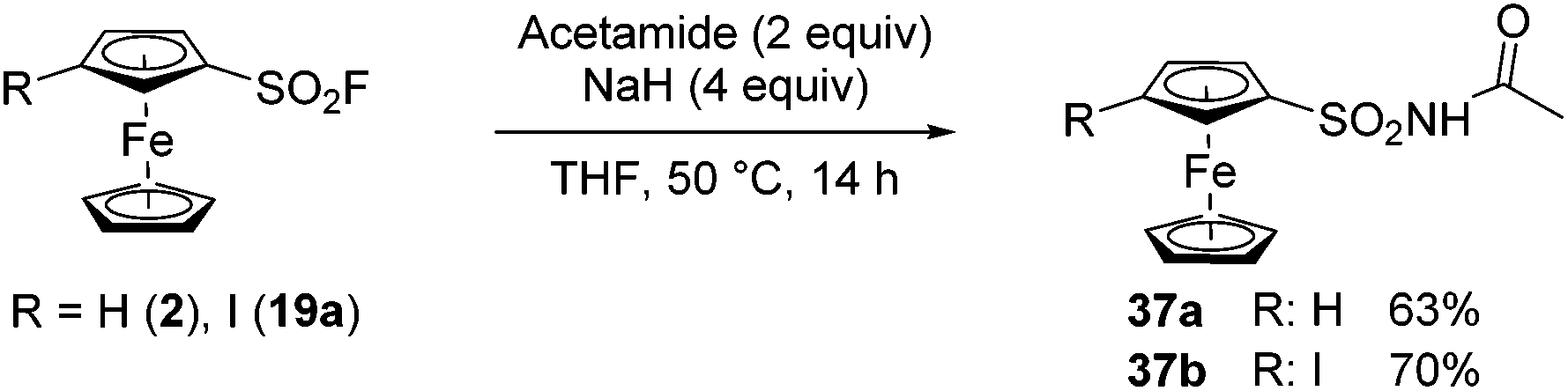 | ||
| Scheme 19 SuFEx reaction of ferrocenesulfonyl fluorides with acetamide. | ||
Aromatic sulfonates can be obtained from the corresponding sulfonyl fluorides by reaction with either silyl ethers, in the presence of an activator such as an ammonium fluoride,129 or DBU,130 or alcohols in the presence of a 2-tert-butyl-1,1,3,3-tetramethylguanidine-hexamethyldisilazane mixture,131 or cesium carbonate.49 In our hands, while the reaction between ferrocenesulfonyl fluoride (2) and 4-methoxyphenol in the presence of DBU at room temperature in acetonitrile only afforded 20% of the sulfonate 38a, performing the reaction at 80 °C led to a much better 97% yield. However, we found that the use of cesium carbonate at room temperature was more efficient as the product 38a was obtained in 97% yield (Scheme 20). Under similar reaction conditions, the less acidic isopropanol was unreactive and required prior deprotonation with sodium hydride in THF to deliver the sulfonate 38b in a quantitative yield after only 30 min at room temperature. Under similar reaction conditions, the 3-iodosulfonate 38c was isolated in 89% yield. In 2017, Ball reported the reaction between 2,2,2-trifluoroethanol and 1-naphthalenesulfonyl fluoride in the presence of cesium carbonate.49 As we recently shown that 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) can sometimes behave as a weak nucleophile,132 we attempted the reaction between 2 and this fluorinated alcohol. As one could have expected, reactions performed with cesium carbonate in acetonitrile at room temperature or sodium hydride in THF were sluggish (5% of desired product isolated 38d under the latter conditions). Performing the reaction at 80 °C in acetonitrile was slightly more efficient, leading to the sulfonate 38d in 11% yield. We finally attempted the reaction from 3-chloro-2-fluoroferrocenesulfonyl fluoride (9), cyclopentanol and sodium hydride in THF. While degradation was mainly observed, we were nevertheless able to isolate 31% of the desired product 38e and 8% of the original ferrocene 38f which results from both SuFEx and ipso-substitution of the fluorine atom.133
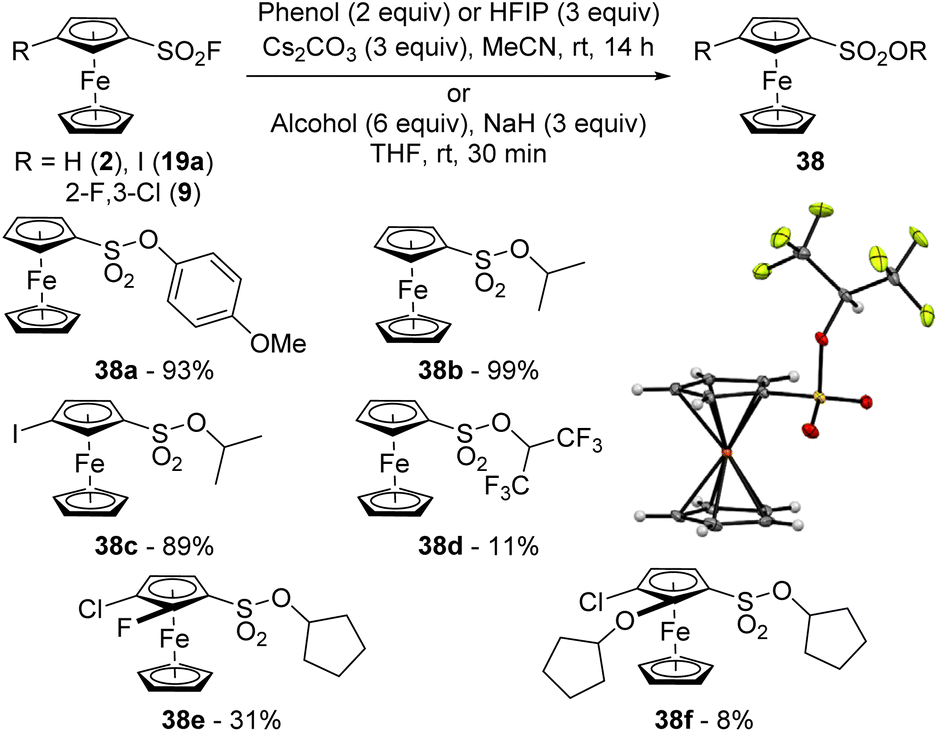 | ||
| Scheme 20 SuFEx reaction of ferrocenesulfonyl fluorides with alcohols. | ||
Due to the exclusive reaction at sulfur, the addition of organometallics onto arenesulfonyl fluorides represents a straightforward approach toward arenesulfones. Furthermore, depending on the nature of the organometallic reagent and the reaction conditions, it is possible to reach mono- or disulfone derivatives.134–136 During the deprotolithiation of ferrocenesulfonyl fluorides with nBuLi in THF at −90 °C, we sometimes observed the formation of the corresponding butylsulfone. Therefore, to favour the substitution, we reacted the substrate 2 with 1.5 equivalent of either methyl- or phenyl-lithium at 0 °C and obtained the methyl- and phenyl-sulfones 39a–b in 84 and 72% yield, respectively (Scheme 21, top). We were also eager to evaluate the effect of steric hindrance on this substitution and therefore selected the bulky 2,5-bis(trimethylsilyl)ferrocenesulfonyl fluoride (5a) as a substrate. The addition of methyllithium occurred smoothly and the methylsulfone 39c was isolated in 66% yield. Therefore, although the steric shielding by the two adjacent trimethylsilyl groups has an impact on the substitution with alkyllithium, it cannot prevent it. Another route towards sulfones relies on the SuFEx reaction between a sulfonyl fluoride derivative and aromatics, as studied by Hyatt,137 and used sporadically in post-functionalization reactions.109,138,139 Therefore, we finally reacted the ferrocenesulfonyl fluorides 2, 3f and 19a with 1,4-dimethoxybenzene in the presence of aluminium trichloride (Scheme 21, bottom). While the sulfones 40a and 40c were isolated in very good yields, the 1,2-disubstituted ferrocene 40b was obtained in 74% due to a competitive deiodination (compound 40a, 15%).
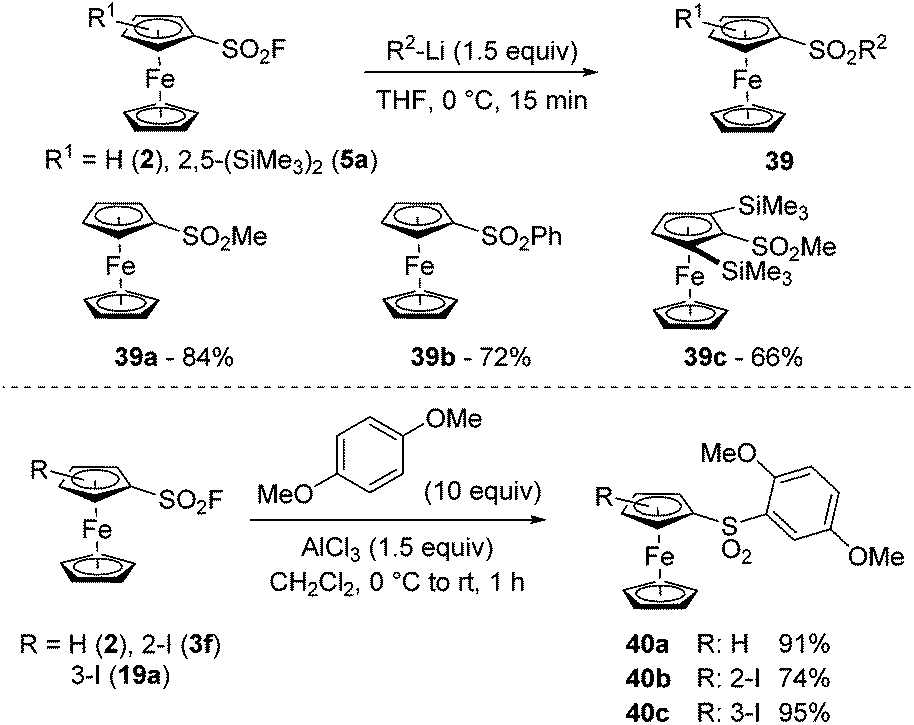 | ||
| Scheme 21 SuFEx reaction of ferrocenesulfonyl fluorides with nucleophilic organometallics and aromatics. | ||
Electrochemical characterization
As ferrocenesulfonyl fluorides represent a new family of ferrocene derivatives, their electrochemical behaviour has never been studied. In 1997, Kagan determined the redox potential of a series of ferrocene sulfides, sulfoxides and sulfones.140 All compounds studied exhibited a one-electron reversible oxidation with a E1/2 value up to 695 mV (vs. SCE) for phenylferrocenesulfone in acetonitrile. With the fluorine directly attached to the sulfur atom, higher redox potential values were expected for ferrocenesulfonyl fluorides, which also raised the question of the ferricenium species stability. The redox behaviour of 22 representative ferrocenesulfonyl fluorides was thus studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in dry, oxygen-free, dichloromethane, using nBu4NPF6 (0.1 M) as the supporting electrolyte (Tables 4–6). Measurements were performed with a glassy carbon disk working electrode, an Ag/AgCl reference electrode and a glassy carbon rod counter electrode.| Compound | E pa | E pc | i pa/ipcb | E 1/2 |
|---|---|---|---|---|
| a Potentials values given relative to FcH/FcH+, scan rate = 100 mV s−1. b From CV experiments. c From DPV experiments. | ||||
| 2 | 0.58 | 0.48 | 1.00 | 0.56 |
| 3b | 0.57 | 0.47 | 0.91 | 0.57 |
| 3e | 0.70 | 0.58 | 1.40 | 0.66 |
| 3f | 0.68 | 0.58 | 0.95 | 0.67 |
| 3g | 0.73 | 0.63 | 0.90 | 0.72 |
| 3k | 0.65 | 0.56 | 1.00 | 0.64 |
| 33a | 0.71 | 0.60 | 0.92 | 0.68 |
| 19a | 0.72 | 0.62 | 0.92 | 0.69 |
| 20d | 0.80 | 0.70 | 0.92 | 0.79 |
| 33b | 0.72 | 0.62 | 0.95 | 0.70 |
| 5f | 0.67 | 0.56 | 1.03 | 0.65 |
| 6e | 0.72 | 0.62 | 0.95 | 0.71 |

|
||||
| Compound | E pa | E pc | i pa/ipcb | E 1/2 |
|---|---|---|---|---|
| a Potentials values given relative to FcH/FcH+, scan rate = 100 mV s−1. b From CV experiments. c From DPV experiments. | ||||
| 5c | 0.60 | 0.50 | 1.00 | 0.58 |
| 10 | 0.59 | 0.49 | 0.90 | 0.58 |
| 8a | 0.80 | 0.70 | 0.88 | 0.79 |
| 11 | 0.63 | 0.53 | 1.10 | 0.61 |
| 12 | 0.74 | 0.64 | 1.10 | 0.73 |
| 13 | 0.67/0.94 | —/0.85 | 1.03 | 0.64/0.94 |

|
||||
a
Pleasingly, apart for the 2-fluorinated derivative 3e (Fig. 5), all 1,2- and 1,3-disubstituted ferrocenesulfonyl fluorides exhibited a reversible one-electron oxidation (Table 4). As a strong electron-withdrawing group, the fluorosulfonyl substituent raised the E1/2 value of compound 2 up to 0.56 V vs. FcH/FcH+, which therefore appears to be one of the monosubstituted ferrocene with the highest redox potential.141,142 All the substituents introduced next to the fluorosulfonyl group increased the redox potential, except for the phenylthio derivative 3b. The origin of this lack of effect remains unclear as this substituent usually raises the E1/2 value by 0.13 V.140,143 In the ferrocene series, the E1/2 value of substituted ferrocenes usually correlates with the sum of the Hammett values σp.141,144–147 However, when we evaluated a related relationship for the 2-substituted ferrocenesulfonyl fluorides, we found that the fluorinated derivative 3e falls out the expected trend. This probably results from the strong resonance effect of fluorine which counterbalances its inductive effect. As the development of a multi parameters analysis was out of the scope of the present study, we temporarily excluded this fluorinated compound. As a result, a modest linear correlation between the E1/2 values and the sum of σp constants was noticed (E1/2 = 2.2624Σσp − 0.3413, R2 = 0.8397, plot SI1). However, no better correlation between the redox potential and the sum of the σm or resonance factor (R) could be found. As already observed in the ferrocenesulfonamide and sulfonate series,54,55 the effect of the substituent is more pronounced on the 1,3-disubstituted ferrocenes than on the 1,2-disubstituted isomers. A similar trend was also noticed with the 1,2,3-trisubstituted ferrocene 5f and its 1,2,4-trisusbstituted isomer 6e. To account for the effect of an ester at the 3-position of a homo disubstituted ferrocene, Klett, Förster and Heinze successfully used the sum σp + σm.148 To get a general view of the position of a substituent on the redox potential, we similarly tried to correlate the E1/2 value with the Σσp for the 1,2-substituted ferrocenes and with the sum σp (SO2F) + σm (substituent) for the 1,3-disusbtituted isomers. However, no general linear relationship was observed. Focusing only on ferrocenesulfonyl fluoride (2) and the 1,3-disusbtituted ferrocenes 2, 19a and 33b, we found a better correlation between the E1/2 value and the Σσp (E1/2 = 1.9182Σσp − 0.189, R2 = 0.9619, plot SI2) than for the Σσm (E1/2 = 1.6264Σσm − 0.0816, R2 = 0.8153, plot SI3). Taking together, these results suggest that although the sum σp + σm can account for the effect of a substituent at the 3 position of a disubstituted ferrocene in some cases,148 it is not possible to extend this analysis to all 1,3-disubstituted ferrocenes.
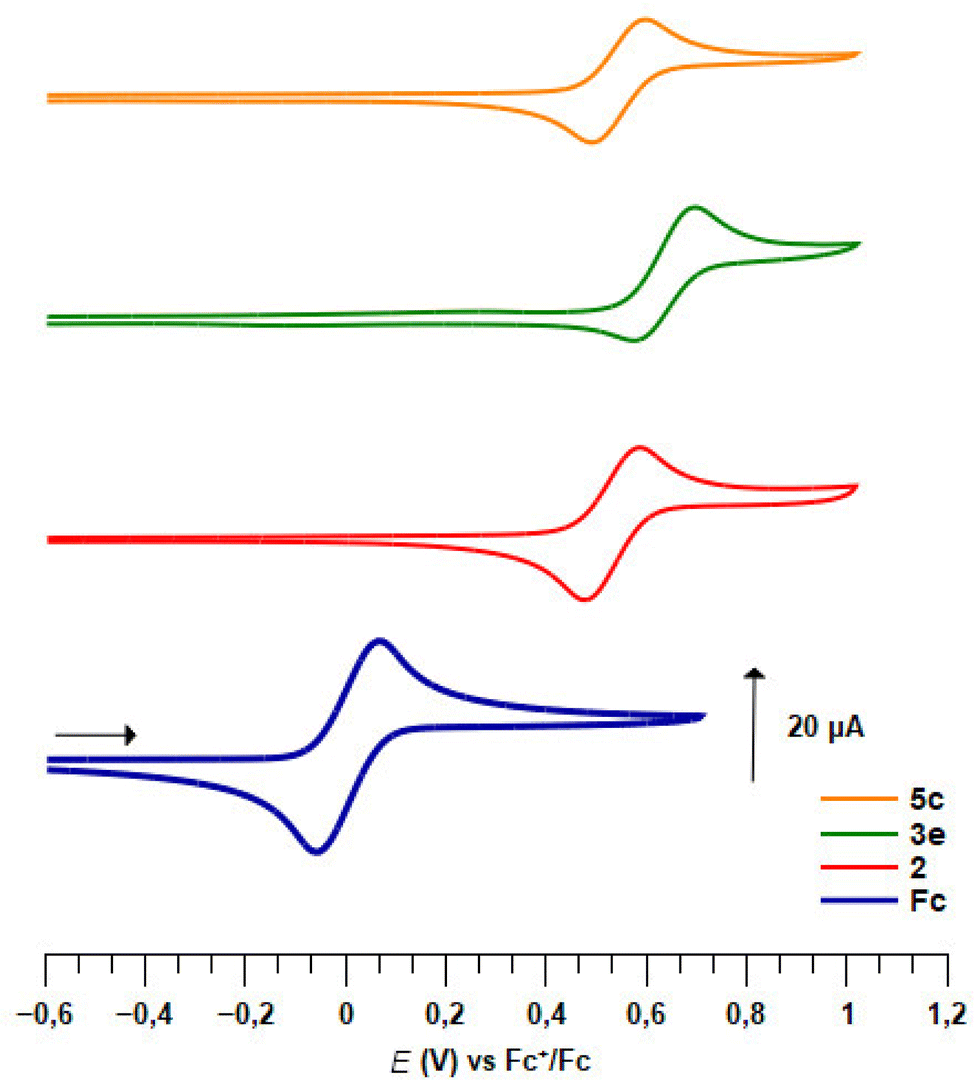 | ||
| Fig. 5 Plots of cyclic voltammograms of ferrocene (Fc) and the derivatives 2, 3e and 5c. | ||
To our knowledge, the redox potential evolution of a ferrocene derivative upon the introduction of five different substituents has never been studied. Therefore, we first focused our efforts on the polysubstituted ferrocenesulfonyl fluorides described on Scheme 7 (Table 5). From the 2-fluorinated ferrocene 3e, the introduction of a trimethylsilyl group has a higher effect than expected on the redox potential which was lowered by 0.08 V.149,150 Furthermore, while ferrocene 3e underwent an irreversible oxidation, the silyl group on compound 5c appeared to stabilize the ferricenium species, as the oxidation of the latter was totally reversible (Fig. 5). From the work of Sauvage,145 the addition of a methyl substituent was expected to lower the redox potential by 0.05 V. However, in our hands, no change in the E1/2 value was noticed between the compounds 5c and 10. The introduction of a chlorine instead of a methyl group increased the redox potential by 0.21 V (compound 8a), slightly more than the +0.16 V anticipated from the work of Albrecht and Long.151 Probably due to the effect of the trimethylsilyl substituent, all three ferrocenes 5c, 8a and 10 underwent a one-electron totally reversible oxidation.
While we recorded no evolution of the E1/2 value between ferrocenes 5c and 10, the expected effect of a methyl substituent (−0.05 V)145 was observed between the ferrocenes 3e and 11. The introduction of iodine with the compound 12 resulted in a smaller increase of the redox potential than expected (+0.11 V vs. +0.15 V).151 Upon the introduction of the dimethylaminomethyl substituent (compound 13), an irreversible oxidation process was observed at +0.64 V, which was attributed to the oxidation of the tertiary amine into an iminium (Fig. 6).152 The latter also raised the redox potential of the ferrocene core by +0.21 V.152 In this series of fluorinated ferrocenesulfonyl fluorides, it appeared that the evolution of the redox potential does not follow a purely additive effect of each substituent. Indeed, we further tried to plot the E1/2 values of the ferrocene 2, 3e, 11, 12 and 13 against the Σσp or Σσm but failed to obtain any correlation (plots SI4–5). However, a linear correlation was observed between the redox potential and the sum σp (SO2F) + σp (substituents adjacent to SO2F) + σm (substituents remote from SO2F) with the equation E1/2 = 0.9977Σσ + 0.3256, R2 = 0.9698 (plot SI6).
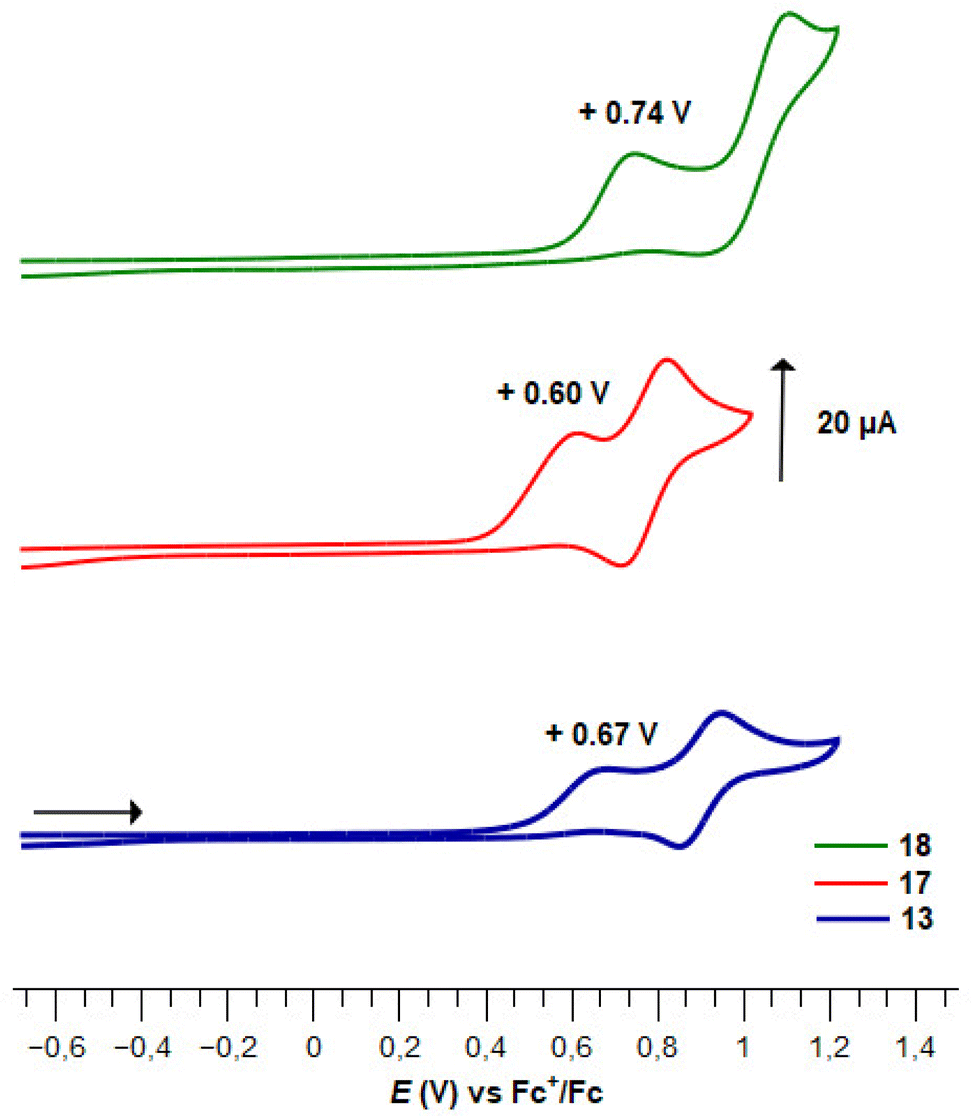 | ||
| Fig. 6 Plots of cyclic voltammograms of ferrocene derivatives 13, 17 and 18. | ||
Finally, we evaluated the electrochemical behaviour of the polysubstituted ferrocenes described on Scheme 8 (Table 6). Surprisingly, the introduction of the different substituents modified the redox potentials in an unexpected way. Indeed, from the 2-phenylthioferrocenesulfonyl fluoride 3b, the addition of a chlorine atom resulted in the irreversible oxidation of the compound 5g and in a large increase of the redox potential (+0.27 V observed vs. +0.16 V expected, Fig. 5). Surprisingly, the introduction of an ester (compound 15) was without effect on the redox potential. As already observed with the compound 13, upon the introduction of the dimethylaminomethyl substituent (compound 17) an irreversible oxidation process of the tertiary amine occurred at +0.60 V (Fig. 6). A final increase of the redox potential was noticed upon the introduction of iodine towards the ferrocene 18 (+0.25 V vs. +0.15 expected). The irreversible oxidation of the tertiary amine into an iminum was observed at a higher potential (+0.74 V), and an irreversible bi-electronic oxidation of the ferrocene core was also recorded (Fig. 6). The pentasubstituted ferrocene 18 has one of the highest redox potentials recorded to date (+1.05 V vs. FcH/FcH+) for a pentasubstituted ferrocene, as even the E1/2 value of pentachloro (+0.77 V)153 and pentafluoroferrocene (+0.82 V)154 were below 1 V. To our knowledge, only three ferrocene derivatives have a higher redox potential: the decabromo-147 and decachloroferrocenes153 (+1.09 and +1.24 V, respectively) and the 1,1′,2,2′-tetra(formyl)ferrocene (+1.14 V).155 Although the modification of redox potential upon the introduction of the five substituents were often unexpected, we tried to plot the E1/2 value against different Σσ. Contrary to the previous series, while no correlation was observed between the redox potential and the sum σp (SO2F) + σp (substituents adjacent to SO2F) + σm (substituents remote from SO2F) (plot SI7), we obtained good correlations between the E1/2 values and either the Σσp (E1/2 = 0.9588Σσp + 0.4115, R2 = 0.9789, plot SI8) or Σσm (E1/2 = 1.7567Σσm – 0.0661, R2 = 0.9529, plot SI9).
Although some relationships between the redox potentials and the Hammett constants emerged from this work on hetero polysubstituted ferrocenes, we need to emphasize that a general trend cannot be deduced. Indeed, as relationships were sometimes better with the Σσp, the Σσm or even the Σσp/σm, great care should be taken when trying to fit redox potentials with a simple sum of Hammett parameters on such highly functionalized substrates. Indeed, many other factors such as orbitals ordering, changes in hybridization and frontier orbital composition, through-space interaction between the metal and the substituents as well as electrostatic repulsion between the metal and the Cp ring orbitals need to be considered.147,151,154 Therefore, a better understanding of the substituent effect on the redox potential of a hetero polysubstituted ferrocene would require a dedicated study, which falls out the scope of this work.
Specific solid-state structures and weak interactions of some ferrocenesulfonyl fluorides
Some of the many ferrocenesulfonyl fluorides afforded crystals suitable for X-ray diffraction analysis and a few of them deserve an additional description regarding weak interactions. Indeed, chalcogen–chalcogen interactions were identified for the compounds 5g and 15.156,157 For the former, a sulfur-sulfur interaction between two molecules is likely to occur with a S2⋯S2 bond length (3.1452(12) Å) below the sum of the van der Waals radii (3.60 Å) and a C8–S2⋯S2 angle of 154.19(7) ° (Fig. 7, top).158 For the latter, an interaction between one lone-pair of the oxygen and the σ-hole of the sulfur atom159 is observed (O16⋯S2 2.9812(18) Å, C18–S2⋯O16 156.40(9) °, Fig. 7, bottom). This might be at the origin of the peculiar orientation of the phenylthio substituent on the compound 15. Indeed, while the phenyl ring of this substituent is usually observed pointing up in the ferrocene series,56,84,160–163 it is here tilted, probably due to this oxygen-sulfur interaction.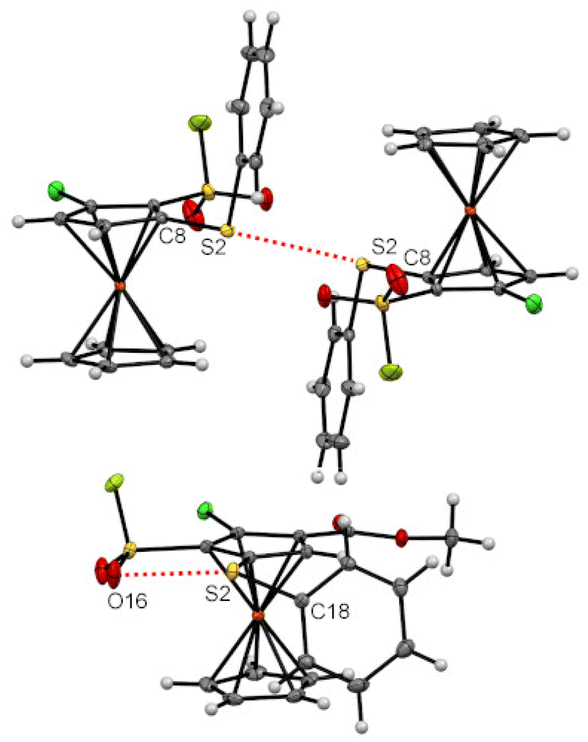 | ||
| Fig. 7 Chalcogen bonds observed at the solid state for the compounds 5g (top) and 15 (bottom). | ||
Various halogen bonds (XB) were also identified at the solid state,164 mainly for iodinated ferrocenesulfonyl fluorides. Indeed, although iodoferrocene derivatives usually do not behave as halogen bond donors, the presence of strong electron-withdrawing groups on the organometallic can increase the positive potential of the σ-hole of the halogen atom and thus favour XB, as observed in the benzene series.165,166 A similar trend has also been observed for ferrocene iodoalkyne derivatives.167 As a result, XB have recently been observed for iodoferrocenes substituted with sulfonamides,54 sulfonates55 and sulfoxides.87 For the 1,2,3,4-tetrasubstituted ferrocene 6d, halogen bonds were observed between the iodine atom and the nitrogen lone-pair of the pyridine ring (I1⋯N15 3.219(4) Å, C6–I1⋯N15 158.27(11) °). As a result of the iodine and nitrogen atom pointing in opposite directions, linear strings of XB, one for each enantiomer, are observed in the crystal structure (Fig. 8, top). For the pentasubstituted ferrocene 18, an interaction between the lone pair of the amine and the σ-hole of the iodine atom is also observed (I1⋯N12 3.092(3) Å, C6–I1⋯N12 157.53(11)°). The proximity between the two atoms involved in the interaction here led to a zig-zag chain of molecules (Fig. 8, bottom). For both compounds 6d and 18, the values of bond lengths and angles fall within the range of expected values.168
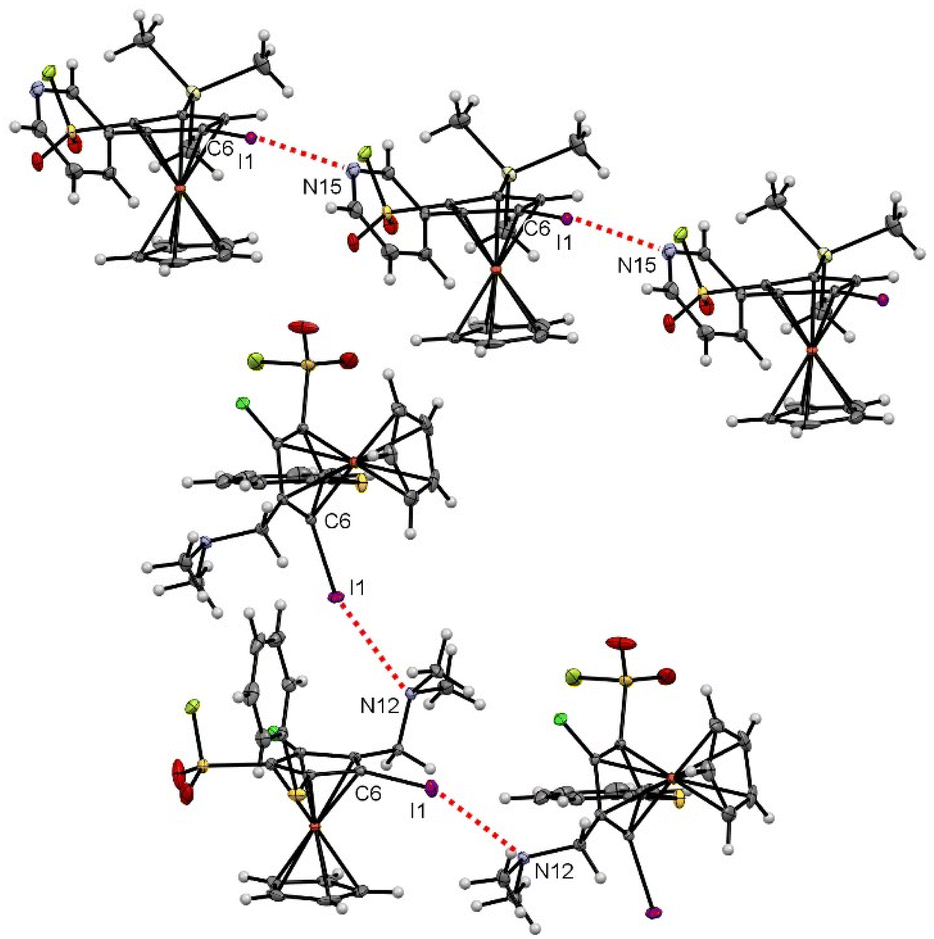 | ||
| Fig. 8 Halogen bonds observed at the solid state for the compounds 6d (top) and 18 (bottom). | ||
For compound 6e, the interaction between the iodine at the 4 position and one oxygen of the fluorosulfonyl group led to a string of XB for each enantiomer (I1⋯O11 3.219(4) Å, C6–I1⋯O11 175.90(6)°, Fig. 9, top). While longer XB were noticed for the 1,2,3-trisubstituted ferrocene 30b (I1⋯O11 3.342(3) Å, C6–I1⋯O11 143.58(11) °, the proximity between the halogen and the fluorosulfonyl group led to a zig–zag network of halogen bonds, each enantiomer engaged in a single chain (Fig. 9, middle). In addition of the chalcogen–chalcogen interaction discussed above, the 1,2,3,4-tetrasubstituted ferrocene 15 also features halogen bonds, this time between the chlorine atom and the ester group, with both bond length (CI1⋯O12 3.0184(2) Å) and angle (C6–Cl1⋯O12 175.04(9)°) in the range of typical values.169 A zig-zag chain of XB, involving both enantiomers, is observed at the solid-state and probably results from the proximal position of the chlorine and ester substituents (Fig. 9, bottom). As expected, the oxygen-iodine and oxygen-chlorine bonds lengths were below the sum of the corresponding van der Waals radii (3.50 Å for OI and 3.27 Å for OCl).158
 | ||
| Fig. 9 Halogen bonds observed at the solid state for the compounds 6e (top), 30b (middle) and 15 (bottom). | ||
We finally observed an iodine–sulfur halogen bond for the 1,2,3-trisubstitued ferrocene 5f. The I2⋯S2 bond length of 3.4903(6) Å and a C6–I1⋯S2 angle of 171.31(5) ° is characteristic of the interaction between a lone pair of the sulfur atom and the σ-hole of the iodine atom.170 The remote position of the two atoms involved in XB led to the formation of a string of interactions involving both enantiomers (Fig. 10).
 | ||
| Fig. 10 Halogen bonds observed at the solid state for the compound 5f. | ||
Conclusions
First reported in 1959, the chemistry of ferrocenesulfonyl fluoride derivatives has remained a dormant field for six decades. With the development of a multigram scale synthesis of ferrocenesulfonyl fluoride, the chemistry of these original compounds could be fully explored. Their functionalization was easily achieved by using deprotometallation-electrophilic trapping sequences and the “halogen dance” reaction to introduce up to five different substituents on the same Cp ring. DFT calculations were used to explain some of the reaction outcome. The orthogonal reactivity of the sulfonyl fluoride was demonstrated in various transformations including iodine/lithium exchanges, pseudo-benzylic nucleophilic substitutions, both oxidations and reductions and various cross-couplings. The possibility to carry out SuFEx reactions was also explored on several substrates towards various sulfur-based functional groups. The electrochemical evaluation of twenty-two ferrocenesulfonyl fluorides was also performed. While we easily established the strong electron-withdrawing properties of this functional group, finding simple relationships between the redox potentials of hetero polysubstituted ferrocenes and the Hammett parameters of the substituents proved highly challenging. We finally highlighted various weak interactions observed at the solid state for these original ferrocene derivatives. Considering the still growing applications of sulfonyl fluorides in chemical biology and material science, we hope that this first general study dedicated to ferrocenesulfonyl fluorides will favour the widespread development of this original scaffold.Author contributions
Investigation, W. E., J.-P. H., T. R., Y. S. H. and V. E. M.; funding acquisition, project administration and supervision, W. E.; writing–original draft preparation, W. E.; writing–review and editing, W. E., J. P. H., Y. S. H., T. R. and V. E. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded, in part, by the Agence Nationale de la Recherche (Ferrodance project). A CC-BY public copyright licence has been applied by the authors to present document and will be applied to all subsequent versions up to the Authors Accepted Manuscript arising from this submission, in accordance with the grant's open access conditions. This work was also supported by the Université de Rennes 1 and CNRS. We gratefully acknowledge the Fonds Européen de Développement Régional (FEDER; D8 VENTURE Bruker AXS diffractometer), Thermofisher (generous gift of 2,2,6,6-tetramethylpiperidine) and BASF (generous gift of (R,R)- and (S,S)-bis(α-phenylethyl)amine). W.E. would like to thank Prof. F. Mongin for support, critically reviewing this document and making valuable suggestions.References
- M. Magre, S. Ni and J. Cornella, (Hetero)aryl-SVI Fluorides: Synthetic Development and Opportunities, Angew. Chem., Int. Ed., 2022, 61, e202200904 CrossRef CAS PubMed
.
- W. Steinkopf, Über Aromatische Sulfofluoride, J. Prakt. Chem., 1927, 117, 1 CrossRef CAS
- W. Steinkopf and P. Jaeger, Über Aromatische Sulfofluoride. II. Mitteilung, J. Prakt. Chem., 1930, 128, 63 CrossRef CAS
- D. K. Myers and A. Kemp, Inhibition of Esterases by the Fluorides of Organic Acids, Nature, 1954, 173, 33 CrossRef CAS PubMed
- A. M. Gold and D. Fahrney, Sulfonyl Fluorides as Inhibitors of Esterases. II. Formation and Reactions of Phenylmethanesulfonyl α-Chymotrypsin, Biochemistry, 1964, 3, 783 CrossRef CAS PubMed
- J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases, Chem. Rev., 2002, 102, 4639 CrossRef CAS
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry, Angew. Chem., Int. Ed., 2014, 53, 9430 CrossRef CAS PubMed
- T. S.-B. Lou and M. C. Willis, Sulfonyl fluorides as targets and substrates in the development of new synthetic methods, Nat. Rev. Chem., 2022, 6, 146 CrossRef CAS
- A. Narayanan and L. H. Jones, Sulfonyl fluorides as privileged warheads in chemical biology, Chem. Sci., 2015, 6, 2650 RSC
- M. Gehringer and S. A. Laufer, Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology, J. Med. Chem., 2019, 62, 5673 CrossRef CAS PubMed
- L. H. Jones and J. W. Kelly, Structure-based design and analysis of SuFEx chemical probes, RSC Med. Chem., 2020, 11, 10 RSC
- O. O. Grygorenko, D. M. Volochnyuk and B. V. Vashchenko, Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances, Eur. J. Org. Chem., 2021, 6478 CrossRef CAS
- N. Shindo and A. Ojida, Recent progress in covalent warheads for in vivo targeting of endogenous proteins, Biorg. Med. Chem., 2021, 47, 116386 CrossRef CAS
- R. J. Grams and K.-L. Hsu, Reactive chemistry for covalent probe and therapeutic development, Trends Pharmacol. Sci., 2022, 43, 249 CrossRef CAS PubMed
- G. M. Kline, K. Nugroho and J. W. Kelly, Inverse Drug Discovery identifies weak electrophiles affording protein conjugates, Curr. Opin. Chem. Biol., 2022, 67, 102113 CrossRef CAS PubMed
- J. Yatvin, K. Brooks and J. Locklin, SuFEx Click: New Materials from SOxF and Silyl Ethers, Chem. – Eur. J., 2016, 22, 16348 CrossRef CAS PubMed
-
L. Xu, P. Wu and J. Dong, in Synthetic Polymer Chemistry: Innovations and Outlook, ed. Z. Zhao, R. Hu, A. Qin and B. Z. Tang, The Royal Society of Chemistry, 2019, pp. 1 Search PubMed
- A. Chelagha, D. Louvel, A. Taponard, R. Berthelon and A. Tlili, Synthetic Routes to Arylsulfonyl Fluorides, Catalysts, 2021, 11, 830 CrossRef CAS
- T. Zhong, Z. Chen, J. Yi, G. Lu and J. Weng, Recent progress in the synthesis of sulfonyl fluorides for SuFEx click chemistry, Chin. Chem. Lett., 2021, 32, 2736 CrossRef CAS
- P. K. Chinthakindi and P. I. Arvidsson, Sulfonyl Fluorides (SFs): More Than Click Reagents?, Eur. J. Org. Chem., 2018, 3648 CrossRef CAS
- A. Dondoni and A. Marra, SuFEx: a metal-free click ligation for multivalent biomolecules, Org. Biomol. Chem., 2017, 15, 1549 RSC
- T. Abdul Fattah, A. Saeed and F. Albericio, Recent advances towards sulfur(VI) fluoride exchange (SuFEx) click chemistry, J. Fluorine Chem., 2018, 213, 87 CrossRef CAS
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, The growing applications of SuFEx click chemistry, Chem. Soc. Rev., 2019, 48, 4731 RSC
- C. Lee, A. J. Cook, J. E. Elisabeth, N. C. Friede, G. M. Sammis and N. D. Ball, The Emerging Applications of Sulfur(VI) Fluorides in Catalysis, ACS Catal., 2021, 11, 6578 CrossRef CAS
- T. J. Kealy and P. L. Pauson, A New Type of Organo-Iron Compound, Nature, 1951, 168, 1039 CrossRef CAS
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, 114. Dicyclopentadienyliron, J. Chem. Soc., 1952, 632 RSC
- A. J. McConnell and P. D. Beer, Heteroditopic Receptors for Ion-Pair Recognition, Angew. Chem., Int. Ed., 2012, 51, 5052 CrossRef CAS PubMed
- P. Molina, A. Tárraga and M. Alfonso, Ferrocene-based multichannel ion-pair recognition receptors, Dalton Trans., 2014, 43, 18 RSC
- R. Sun, L. Wang, H. Yu, Z.-u. Abdin, Y. Chen, J. Huang and R. Tong, Molecular Recognition and Sensing Based on Ferrocene Derivatives and Ferrocene-Based Polymers, Organometallics, 2014, 33, 4560 CrossRef CAS
- T. J. Colacot, A Concise Update on the Applications of Chiral Ferrocenyl Phosphines in Homogeneous Catalysis Leading to Organic Synthesis, Chem. Rev., 2003, 103, 3101 CrossRef CAS
- R. Gómez Arrayás, J. Adrio and J. C. Carretero, Recent Applications of Chiral Ferrocene Ligands in Asymmetric Catalysis, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed
- N. A. Butt, D. Liu and W. Zhang, The Design and Synthesis of Planar Chiral Ligands and Their Application to Asymmetric Catalysis, Synlett, 2014, 615 CAS
- J. C. Zhu, D. X. Cui, Y. D. Li, R. Jiang, W. P. Chen and P. A. Wang, Ferrocene as a Privileged Framework for Chiral Organocatalysts, ChemCatChem, 2018, 10, 907 CrossRef CAS
- L. Cunningham, A. Benson and P. J. Guiry, Recent developments in the synthesis and applications of chiral ferrocene ligands and organocatalysts in asymmetric catalysis, Org. Biomol. Chem., 2020, 18, 9329 RSC
- M. Navarro, W. Castro and C. Biot, Bioorganometallic Compounds with Antimalarial Targets: Inhibiting Hemozoin Formation, Organometallics, 2012, 31, 5715 CrossRef CAS
- M. Patra, G. Gasser and N. Metzler-Nolte, Small organometallic compounds as antibacterial agents, Dalton Trans., 2012, 41, 6350 RSC
- G. Jaouen, A. Vessieres and S. Top, Ferrocifen type anti cancer drugs, Chem. Soc. Rev., 2015, 44, 8802 RSC
- M. Patra and G. Gasser, The medicinal chemistry of ferrocene and its derivatives, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS
- B. S. Ludwig, J. D. G. Correia and F. E. Kühn, Ferrocene derivatives as anti-infective agents, Coord. Chem. Rev., 2019, 396, 22 CrossRef CAS
- A. Singh, I. Lumb, V. Mehra and V. Kumar, Ferrocene-appended pharmacophores: an exciting approach for modulating the biological potential of organic scaffolds, Dalton Trans., 2019, 48, 2840 RSC
- B. Sharma and V. Kumar, Has Ferrocene Really Delivered Its Role in Accentuating the Bioactivity of Organic Scaffolds?, J. Med. Chem., 2021, 64, 16865 CrossRef CAS PubMed
- R. Pietschnig, Polymers with pendant ferrocenes, Chem. Soc. Rev., 2016, 45, 5216 RSC
- S. Ø. Scottwell and J. D. Crowley, Ferrocene-containing non-interlocked molecular machines, Chem. Commun., 2016, 52, 2451 RSC
- M.-L. Hu, M. Abbasi-Azad, B. Habibi, F. Rouhani, H. Moghanni-Bavil-Olyaei, K.-G. Liu and A. Morsali, Electrochemical Applications of Ferrocene-Based Coordination Polymers, ChemPlusChem, 2020, 85, 2397 CrossRef CAS PubMed
- A. N. Nesmeyanov and O. A. Reutov, Derivatives of ferrocene-1-carboxy-1′-sulfonic acid, Izv. Akad. Nauk SSSR, Ser. Khim., 1959, 926 CAS
- G. M. R. Boston, H. M. Philipp and H. Butenschön, Fluorosulfonylferrocene, (Trifluoromethylsulfonyl)ferrocene and New Ferrocenyl Sulfonates: Directed ortho Lithiation and New Anionic Thia-Fries Rearrangements at Ferrocene, Eur. J. Inorg. Chem., 2021, 4903 CrossRef CAS
- W. Erb and T. Roisnel, The chemistry of ferrocenesulfonyl fluoride revealed, Dalton Trans., 2021, 50, 16483 RSC
- A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, One-pot palladium-catalyzed synthesis of sulfonyl fluorides from aryl bromides, Chem. Sci., 2017, 8, 1233 RSC
- A. L. Tribby, I. Rodríguez, S. Shariffudin and N. D. Ball, Pd-Catalyzed Conversion of Aryl Iodides to Sulfonyl Fluorides Using SO2 Surrogate DABSO and Selectfluor, J. Org. Chem., 2017, 82, 2294 CrossRef CAS PubMed
- F. Rebiere, O. Samuel and H. B. Kagan, A convenient method for the preparation of monolithioferrocene, Tetrahedron Lett., 1990, 31, 3121 CrossRef CAS
- D. W. Slocum and W. Achermann, The Preparation of Several N-Substituted and N,N-Disubstituted Ferrocenesulfonamides, Synth. React. Inorg. Met.-Org. Chem., 1982, 12, 397 CrossRef CAS
- W. Erb, M. Wen, T. Roisnel and F. Mongin, Synthesis of Ferrocenesulfonyl Chloride: Key Intermediate toward Ferrocenesulfonamides, Synthesis, 2021, 2612 CrossRef CAS
- A. Talko and M. Barbasiewicz, Nucleophilic Fluorination with Aqueous Bifluoride Solution: Effect of the Phase-Transfer Catalyst, ACS Sustainable Chem. Eng., 2018, 6, 6693 CrossRef CAS
- M. Wen, W. Erb, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel and V. Dorcet, Functionalization of N,N-Dialkylferrocenesulfonamides toward Substituted Derivatives, Organometallics, 2021, 40, 1129 CrossRef CAS
- W. Erb, M. Wen, J.-P. Hurvois, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and T. Roisnel,
O-Isopropylferrocenesulfonate: Synthesis of Polysubstituted Derivatives and Electrochemical Study, Eur. J. Inorg. Chem., 2021, 2021, 3165 CrossRef CAS
- M. Tazi, M. Hedidi, W. Erb, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet, G. Bentabed-Ababsa and F. Mongin, Fluoro- and Chloroferrocene: From 2- to 3-Substituted Derivatives, Organometallics, 2018, 37, 2207 CrossRef CAS
- A. Talko, D. Antoniak and M. Barbasiewicz, Directed ortho-Metalation of Arenesulfonyl Fluorides and Aryl Fluorosulfates, Synthesis, 2019, 2278 CAS
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, A study of the reactivity of S(VI)–F containing warheads with nucleophilic amino-acid side chains under physiological conditions, Org. Biomol. Chem., 2017, 15, 9685 RSC
- M. Tsukazaki, M. Tinkl, A. Roglans, B. J. Chapell, N. J. Taylor and V. Snieckus, Direct and Highly Enantioselective Synthesis of Ferrocenes with Planar Chirality by (−)-Sparteine-Mediated Lithiation, J. Am. Chem. Soc., 1996, 118, 685 CrossRef CAS
- C. Genet, S. J. Canipa, P. O'Brien and S. Taylor, Catalytic Asymmetric Synthesis of Ferrocenes and P-Stereogenic Bisphosphines, J. Am. Chem. Soc., 2006, 128, 9336 CrossRef CAS PubMed
- W. Erb, T. Roisnel and V. Dorcet, Practical Chromatography-Free Synthesis of 2-Iodo-N,N-diisopropylferrocenecarboxamide and Further Transformations, Synthesis, 2019, 3205 CAS
- Y. Nishibayashi, Y. Arikawa, K. Ohe and S. Uemura, Enantioselective ortho-Lithiation of Substituted Ferrocenes, J. Org. Chem., 1996, 61, 1172 CrossRef CAS
- P. Steffen, C. Unkelbach, M. Christmann, W. Hiller and C. Strohmann, Catalytic and Stereoselective ortho-Lithiation of a Ferrocene Derivative, Angew. Chem., Int. Ed., 2013, 52, 9836 CrossRef CAS PubMed
- C. Metallinos, J. Zaifman and L. Dodge, Aminoferrocene Lithiation by Boron Trifluoride Activation, Org. Lett., 2008, 10, 3527 CrossRef CAS PubMed
- M. Costa, Z. Josh, D. Travis, V. B. Lori and T. Keivan, Asymmetric Lithiation of Boron Trifluoride–Activated Aminoferrocenes: An Experimental and Computational Investigation, Adv. Synth. Catal., 2010, 352, 1967 CrossRef
- D. Price and N. S. Simpkins, Concerning the asymmetric metallation of ferrocenes by chiral lithium amide bases, Tetrahedron Lett., 1995, 36, 6135 CrossRef CAS
- M. Hedidi, G. Dayaker, Y. Kitazawa, Y. Tatsuya, M. Kimura, W. Erb, G. Bentabed-Ababsa, F. Chevallier, M. Uchiyama, P. C. Gros and F. Mongin, Enantioselective deprotometalation of N,N-dialkyl ferrocenecarboxamides using metal amides, New J. Chem., 2019, 43, 14898 RSC
- G. Dayaker, W. Erb, M. Hedidi, F. Chevallier, M. Blot, P. C. Gros, G. Hilmersson, T. Roisnel, V. Dorcet, G. Bentabed-Ababsa and F. Mongin, Enantioselective deprotometalation of alkyl ferrocenecarboxylates using bimetallic bases, New J. Chem., 2021, 45, 22579 RSC
- J. A. S. Howell, P. C. Yates, N. Fey, P. McArdle, D. Cunningham, S. Parsons and D. W. H. Rankin, Molecular Mechanics Analysis of Structure and Diastereoselectivity toward Lithiation in Amido- and α-Aminoferrocene Complexes, Organometallics, 2002, 21, 5272 CrossRef CAS
- J. Schreiber, H. Maag, N. Hashimoto and A. Eschenmoser, Dimethyl(methylene)ammonium Iodide, Angew. Chem., Int. Ed. Engl., 1971, 10, 330 CrossRef CAS
- E. Negishi, A. O. King and N. Okukado, Selective carbon-carbon bond formation via transition metal catalysis. 3. A highly selective synthesis of unsymmetrical biaryls and diarylmethanes by the nickel- or palladium-catalyzed reaction of aryl- and benzylzinc derivatives with aryl halides, J. Org. Chem., 1977, 42, 1821 CrossRef CAS
- E. Negishi, Palladium- or nickel-catalyzed cross coupling. A new selective method for carbon-carbon bond formation, Acc. Chem. Res., 1982, 15, 340 CrossRef CAS
- W. Davies and J. H. Dick, 285. Benzenesulphonyl fluoride derivatives, J. Chem. Soc., 1932, 2042 RSC
- C. Pichon, B. Odell and J. M. Brown, A direct meta-lithiation route to 1,3-disubstituted ferrocenes, Chem. Commun., 2004, 598 RSC
-
J. Fröhlich, in Prog. Heterocycl. Chem, ed. H. Suschitzky and E. F. V. Scriven, Elsevier, 1994, vol. 6, pp. 1 Search PubMed
- X.-F. Duan and Z.-B. Zhang, Recent Progress of Halogen-Dance Reactions in Heterocycles, Heterocycles, 2005, 65, 2005 CrossRef CAS
- M. Schlosser, The 2×3 Toolbox of Organometallic Methods for Regiochemically Exhaustive Functionalization, Angew. Chem., Int. Ed., 2005, 44, 376 CrossRef CAS PubMed
- M. Schnurch, M. Spina, A. F. Khan, M. D. Mihovilovic and P. Stanetty, Halogen dance reactions-A review, Chem. Soc. Rev., 2007, 36, 1046 RSC
-
M. Schnürch, in Halogenated Heterocycles: Synthesis, Application and Environment, ed. J. Iskra, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 185 Search PubMed
- W. Erb and F. Mongin, Halogen ‘dance’: a way to extend the boundaries of arene deprotolithiation, Tetrahedron, 2016, 72, 4973 CrossRef CAS
- G. Dayaker, A. Sreeshailam, F. Chevallier, T. Roisnel, P. Radha Krishna and F. Mongin, Deprotonative metallation of ferrocenes using mixed lithium–zinc and lithium–cadmium combinations, Chem. Commun., 2010, 46, 2862 RSC
- M. Tazi, W. Erb, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, V. Dorcet and F. Mongin, From 2- to 3-Substituted Ferrocene Carboxamides or How to Apply Halogen “Dance” to the Ferrocene Series, Organometallics, 2017, 36, 4770 CrossRef CAS
- W. Erb and T. Roisnel, Asymmetric synthesis of hetero-1,2,3,4,5-pentasubstituted ferrocenes, Chem. Commun., 2019, 55, 9132 RSC
- M. Tazi, W. Erb, T. Roisnel, V. Dorcet, F. Mongin and P. J. Low, From ferrocene to fluorine-containing penta-substituted derivatives and all points in-between; or, how to increase the available chemical space, Org. Biomol. Chem., 2019, 27, 9352 RSC
- T. Blockhaus, S. Bernhartzeder, W. Kempinger, C. Klein-Heßling, S. Weigand and K. Sünkel, Evidence for “Halogen-Dance” and Ring-Exchange Reactions in Chloro-methylthio-ferrocenes, Eur. J. Org. Chem., 2020, 6576 CrossRef CAS
- W. Erb, L. Kadari, K. Al-Mekhlafi, T. Roisnel, V. Dorcet, P. Radha Krishna and F. Mongin, Functionalization of 3-Iodo-N,N-Diisopropylferrocene-Carboxamide, a Pivotal Substrate to Open the Chemical Space to 1,3-Disubstituted Ferrocenes, Adv. Synth. Catal., 2020, 362, 832 CrossRef CAS
- M. Wen, W. Erb, F. Mongin, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and T. Roisnel, Synthesis of Polysubstituted Ferrocenesulfoxides, Molecules, 2022, 27, 1798 CrossRef CAS PubMed
- K. Okano, K. Sunahara, Y. Yamane, Y. Hayashi and A. Mori, One-Pot Halogen Dance/Negishi Coupling of Dibromothiophenes for Regiocontrolled Synthesis of Multiply Arylated Thiophenes, Chem. – Eur. J., 2016, 22, 16450 CrossRef CAS
- K. Morii, Y. Yasuda, D. Morikawa, A. Mori and K. Okano, Total Synthesis of Lamellarins G, J, L, and Z Using One-Pot Halogen Dance/Negishi Coupling, J. Org. Chem., 2021, 86, 13388 CrossRef CAS
- L. Kadari, T. Roisnel, W. Erb, P. R. Krishna and F. Mongin, Remote Deprotometalation-Iodolysis of N,N-Diisopropyl-2-trimethylsilylferrocenecarboxamide: A New Route Toward 1,1′-Disubstituted Ferrocenes, Synthesis, 2020, 3153 CAS
- C. J. Richards, T. Damalidis, D. E. Hibbs and M. B. Hursthouse, Synthesis of 2-[2-(Diphenylphosphino)ferrocenyl]oxazoline Ligands, Synlett, 1995, 74 CrossRef CAS
- E. J. Corey and D. J. Beames, Mixed cuprate reagents of type R1R2CuLi which allow selective group transfer, J. Am. Chem. Soc., 1972, 94, 7210 CrossRef CAS
- D. Seebach and H. Neumann, Brom-Lithium-Austausch an Vinyl- und Aryl-bromiden mit tert-Butyllithium Zur Ringerweiterung über Dibromcarbenaddukte, Chem. Ber., 1974, 107, 847 CrossRef CAS
- D. A. Shannon, C. Gu, C. J. McLaughlin, M. Kaiser, R. A. L. van der Hoorn and E. Weerapana, Sulfonyl Fluoride Analogues as Activity-Based Probes for Serine Proteases, ChemBioChem, 2012, 13, 2327 CrossRef CAS PubMed
- O. Fadeyi, M. D. Parikh, M. Z. Chen, R. E. Kyne Jr., A. P. Taylor, I. O'Doherty, S. E. Kaiser, S. Barbas, S. Niessen, M. Shi, S. L. Weinrich, J. C. Kath, L. H. Jones and R. P. Robinson, Chemoselective Preparation of Clickable Aryl Sulfonyl Fluoride Monomers: A Toolbox of Highly Functionalized Intermediates for Chemical Biology Probe Synthesis, ChemBioChem, 2016, 17, 1925 CrossRef CAS PubMed
- A. A. Filatov, V. N. Boiko and Y. L. Yagupolskii, Interaction of 2,4,6-tris(fluorosulfonyl)chlorobenzene with O-, N-, S-, C-nucleophiles and F-anion, J. Fluorine Chem., 2012, 143, 123 CrossRef CAS
- K. A. Tolmachova, Y. S. Moroz, A. Konovets, M. O. Platonov, O. V. Vasylchenko, P. Borysko, S. Zozulya, A. Gryniukova, A. V. Bogolubsky, S. Pipko, P. K. Mykhailiuk, V. S. Brovarets and O. O. Grygorenko, (Chlorosulfonyl)benzenesulfonyl Fluorides—Versatile Building Blocks for Combinatorial Chemistry: Design, Synthesis and Evaluation of a Covalent Inhibitor Library, ACS Comb. Sci., 2018, 20, 672 CrossRef CAS
- L.-C. Han, P. A. Stanley, P. J. Wood, P. Sharma, A. I. Kuruppu, T. D. Bradshaw and J. E. Moses, Horner–Wadsworth–Emmons approach to piperlongumine analogues with potent anti-cancer activity, Org. Biomol. Chem., 2016, 14, 7585 RSC
- S. Park, H. Song, N. Ko, C. Kim, K. Kim and E. Lee, SuFEx in Metal–Organic Frameworks: Versatile Postsynthetic Modification Tool, ACS Appl. Mater. Interfaces, 2018, 10, 33785 CrossRef CAS PubMed
- A. J. van der Zouwen, J. Lohse, L. H. E. Wieske, K. F. Hohmann, R. van der Vlag and M. D. Witte, An in situ combinatorial methodology to synthesize and screen chemical probes, Chem. Commun., 2019, 55, 2050 RSC
- J. Koh, A. J. Greaves and J. P. Kim, Synthesis and spectral properties of alkali-clearable azo disperse dyes containing a fluorosulfonyl group, Dyes Pigm., 2003, 56, 69 CrossRef CAS
- R. Cui, J. Ye, J. Li, W. Mo, Y. Gao and H. Chen, Construction of Bisindolines via Oxidative Coupling Cyclization, Org. Lett., 2020, 22, 116 CrossRef CAS PubMed
- P. Dupau, R. Epple, A. A. Thomas, V. V. Fokin and K. B. Sharpless, Osmium-Catalyzed Dihydroxylation of Olefins in Acidic Media: Old Process, New Tricks, Adv. Synth. Catal., 2002, 344, 421 CrossRef CAS
- T. S.-B. Lou and M. C. Willis, Arylsulfonyl fluoride boronic acids: Preparation and coupling reactivity, Tetrahedron, 2020, 76, 130782 CrossRef CAS
-
N. D. Ball, in Emerging Fluorinated Motifs, ed. J.-A. Ma and D. Cahard, Wiley–VCH, 2020, vol. 2, pp. 621 Search PubMed
- N. Miyaura, K. Yamada and A. Suzuki, A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides, Tetrahedron Lett., 1979, 20, 3437 CrossRef
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457 CrossRef CAS
- P. K. Chinthakindi, H. G. Kruger, T. Govender, T. Naicker and P. I. Arvidsson, On-Water Synthesis of Biaryl Sulfonyl Fluorides, J. Org. Chem., 2016, 81, 2618 CrossRef CAS
- Y. Liu, D. Yu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Arenesulfonyl Fluoride Synthesis via Copper-Catalyzed Fluorosulfonylation of Arenediazonium Salts, Org. Lett., 2020, 22, 2281 CrossRef CAS PubMed
- H. Mukherjee, N. Su, M. A. Belmonte, D. Hargreaves, J. Patel, S. Tentarelli, B. Aquila and N. P. Grimster, Discovery and optimization of covalent Bcl-xL antagonists, Bioorg. Med. Chem. Lett., 2019, 29, 126682 CrossRef CAS PubMed
- A. Y. Cherepakha, K. O. Stepannikova, B. V. Vashchenko, M. V. Gorichko, A. A. Tolmachev and O. O. Grygorenko, Hetaryl Bromides Bearing the SO2F Group – Versatile Substrates for Palladium-Catalyzed C–C Coupling Reactions, Eur. J. Org. Chem., 2018, 6682 CrossRef CAS
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, Catalysts for Suzuki−Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed
- P. Chatelain, C. Muller, A. Sau, D. Brykczyńska, M. Bahadori, C. N. Rowley and J. Moran, Desulfonative Suzuki–Miyaura Coupling of Sulfonyl Fluorides, Angew. Chem., Int. Ed., 2021, 60, 25307 CrossRef CAS
- G. Zhang, C. Guan, Y. Zhao, H. Miao and C. Ding, ‘Awaken’ aryl sulfonyl fluoride: a new partner in the Suzuki–Miyaura coupling reaction, New J. Chem., 2022, 46, 3560 RSC
- K. Sonogashira, Y. Tohda and N. Hagihara, A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines, Tetrahedron Lett., 1975, 16, 4467 CrossRef
- R. Chinchilla and C. Nájera, The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed
- J. Tam, M. Henault, L. Li, Z. Wang, A. W. Partridge and R. A. Melnyk, An activity-based probe for high-throughput measurements of triacylglycerol lipases, Anal. Biochem., 2011, 414, 254 CrossRef CAS PubMed
- M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Rapid Sonogashira cross-coupling of iodoferrocenes and the unexpected cyclo-oligomerization of 4-ethynylphenylthioacetate, Chem. Commun., 2013, 49, 5663 RSC
- P. Srinivas, S. Prabhakar, F. Chevallier, E. Nassar, W. Erb, V. Dorcet, V. Jouikov, P. Radha Krishna and F. Mongin, Synthesis of ferrocene amides and esters from aminoferrocene and 2-substituted ferrocenecarboxylic acid and properties thereof, New J. Chem., 2016, 40, 9441 RSC
- B. Bildstein, M. Malaun, H. Kopacka, K. Wurst, M. Mitterböck, K.-H. Ongania, G. Opromolla and P. Zanello,
N,N′-Diferrocenyl-N-heterocyclic Carbenes and Their Derivatives, Organometallics, 1999, 18, 4325 CrossRef CAS
- L. Kadari, W. Erb, T. Roisnel, P. Radha Krishna and F. Mongin, Iodoferrocene as a partner in N-arylation of amides, New J. Chem., 2020, 44, 15928 RSC
- L. Kadari, W. Erb, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, D. Lyakhov, T. Roisnel, P. Radha Krishna and F. Mongin, On the N-Arylation of Acetamide Using 2-, 3- and 1′-Substituted Iodoferrocenes, Eur. J. Inorg. Chem., 2021, 2021, 377 CrossRef CAS
- K. Abaid, W. Erb, M. Blot, T. Roisnel, F. Mongin and S. Touil, Ferrocenephosphonates: Copper-Promoted Synthesis and Further Functionalization, Synthesis, 2022, 2799 CAS
- P. Mukherjee, C. P. Woroch, L. Cleary, M. Rusznak, R. W. Franzese, M. R. Reese, J. W. Tucker, J. M. Humphrey, S. M. Etuk, S. C. Kwan, C. W. am Ende and N. D. Ball, Sulfonamide Synthesis via Calcium Triflimide Activation of Sulfonyl Fluorides, Org. Lett., 2018, 20, 3943 CrossRef CAS PubMed
- S. Mahapatra, C. P. Woroch, T. W. Butler, S. N. Carneiro, S. C. Kwan, S. R. Khasnavis, J. Gu, J. K. Dutra, B. C. Vetelino, J. Bellenger, C. W. am Ende and N. D. Ball, SuFEx Activation with Ca(NTf2)2: A Unified Strategy to Access Sulfamides, Sulfamates, and Sulfonamides from S(VI) Fluorides, Org. Lett., 2020, 22, 4389 CrossRef CAS PubMed
- M. Wei, D. Liang, X. Cao, W. Luo, G. Ma, Z. Liu and L. Li, A Broad-Spectrum Catalytic Amidation of Sulfonyl Fluorides and Fluorosulfates, Angew. Chem., Int. Ed., 2021, 60, 7397 CrossRef CAS PubMed
- S. Zhang, H. Xiong, F. Lu, F. Ma, Y. Gu, P. Ma, H. Xu and G. Yang, Synthesis of N-Acyl Sulfamates from Fluorosulfonates and Potassium Trimethylsilyloxyl Imidates, J. Org. Chem., 2019, 84, 15380 CrossRef CAS
- P. Gilles, C. Veryser, S. Vangrunderbeeck, S. Ceusters, L. Van Meervelt and W. M. De Borggraeve, Synthesis of N-Acyl Sulfamates from Fluorosulfates and Amides, J. Org. Chem., 2019, 84, 1070 CrossRef CAS PubMed
- E. Hirsch, S. Hünig and H.-U. Reißig, Darstellung von Enolsulfonsäureestern aus Trimethylsilylenolethern—Synthetische Konsequenz eines bemerkenswerten Kationeneffekts, Chem. Ber., 1982, 115, 3687 CrossRef CAS
- V. Gembus, F. Marsais and V. Levacher, An Efficient Organocatalyzed Interconversion of Silyl Ethers to Tosylates Using DBU and p-Toluenesulfonyl Fluoride, Synlett, 2008, 1463 CrossRef CAS
- C. J. Smedley, J. A. Homer, T. L. Gialelis, A. S. Barrow, R. A. Koelln and J. E. Moses, Accelerated SuFEx Click Chemistry For Modular Synthesis, Angew. Chem., 2022, 61, e202112375 CAS
- W. Erb, V. Carré and T. Roisnel, HFIP-Promoted Substitution in the Ferrocene Series: Smooth Approach towards Original Catalysts, Eur. J. Org. Chem., 2021, 5702 CrossRef CAS
- Z. You, K. Higashida, T. Iwai and M. Sawamura, Phosphinylation of Non-activated Aryl Fluorides through Nucleophilic Aromatic Substitution at the Boundary of Concerted and Stepwise Mechanisms, Angew. Chem., Int. Ed., 2021, 60, 5778 CrossRef CAS PubMed
- H. Fukuda, F. J. Frank and W. E. Truce, Synthesis of β-Disulfones from Sulfonyl Fluorides and Organometallic Compounds, J. Org. Chem., 1963, 28, 1420 CrossRef CAS
- A. I. Khodair, A. A. Abdel-Wahab and A. M. El-Khawaga, Synthesis of Sulphones from Sulphonyl Fluorides and Organometallic Compounds, Z. Naturforsch., B, 1978, 33, 403 CrossRef
- L. L. Frye, E. L. Sullivan, K. P. Cusack and J. M. Funaro, Sulfonylation of organometallic reagents with arenesulfonyl fluorides: a simple one-step synthesis of sulfones, J. Org. Chem., 1992, 57, 697 CrossRef CAS
- J. A. Hyatt and A. W. White, Synthesis of Aryl Alkyl and Aryl Vinyl Sulfones via Friedel-Crafts Reactions of Sulfonyl Fluorides, Synthesis, 1984, 214 CrossRef CAS
- H.-L. Qin, Q. Zheng, G. A. L. Bare, P. Wu and K. B. Sharpless, A Heck–Matsuda Process for the Synthesis of β-Arylethenesulfonyl Fluorides: Selectively Addressable Bis-electrophiles for SuFEx Click Chemistry, Angew. Chem., Int. Ed., 2016, 55, 14155 CrossRef CAS PubMed
- X.-Y. Chen, Y. Wu, J. Zhou, P. Wang and J.-Q. Yu, Synthesis of β-Arylethenesulfonyl Fluoride via Pd-Catalyzed Nondirected C–H Alkenylation, Org. Lett., 2019, 21, 1426 CrossRef CAS PubMed
- A. Gref, P. Diter, D. Guillaneux and H. B. Kagan, Electrochemical behavior of various sulfur and phophorus derivatives of ferrocene, New J. Chem., 1997, 21, 1353 CAS
- W. E. Britton, R. Kashyap, M. El-Hashash, M. El-Kady and M. Herberhold, The anomalous electrochemistry of the ferrocenylamines, Organometallics, 1986, 5, 1029 CrossRef CAS
- M. Toma, T. Kuvek and V. Vrček, Ionization Energy and Reduction Potential in Ferrocene Derivatives: Comparison of Hybrid and Pure DFT Functionals, J. Phys. Chem. A, 2020, 124, 8029 CrossRef CAS
- D. A. Khobragade, S. G. Mahamulkar, L. Pospíšil, I. Císařová, L. Rulíšek and U. Jahn, Acceptor-Substituted Ferrocenium Salts as Strong, Single-Electron Oxidants: Synthesis, Electrochemistry, Theoretical Investigations, and Initial Synthetic Application, Chem. – Eur. J., 2012, 18, 12267 CrossRef CAS PubMed
- W. F. Little, C. N. Reilley, J. D. Johnson and A. P. Sanders, Chronopotentiometric Studies of Ferrocene Derivatives. II. Directly Substituted Ferrocenes, J. Am. Chem. Soc., 1964, 86, 1382 CrossRef CAS
- A. Paul, R. Borrelli, H. Bouyanfif, S. Gottis and F. Sauvage, Tunable Redox Potential, Optical Properties, and Enhanced Stability of Modified Ferrocene-Based Complexes, ACS Omega, 2019, 4, 14780 CrossRef CAS
- D. M. Evans, D. D. Hughes, P. J. Murphy, P. N. Horton, S. J. Coles, F. F. de Biani, M. Corsini and I. R. Butler, Synthetic Route to 1,1′,2,2′-Tetraiodoferrocene That Avoids Isomerization and the Electrochemistry of Some Tetrahaloferrocenes, Organometallics, 2021, 40, 2496 CrossRef CAS
- S. M. Rupf, I. S. Dimitrova, G. Schröder and M. Malischewski, Preparation and One-Electron Oxidation of Decabromoferrocene, Organometallics, 2022, 41, 1261 CrossRef CAS
- S. D. Waniek, J. Klett, C. Förster and K. Heinze, Polysubstituted ferrocenes as tunable redox mediators, Beilstein J. Org. Chem., 2018, 14, 1004 CrossRef CAS
- G. L. K. Hoh, W. E. McEwen and J. Kleinberg, Substituent Effects in the Chronopotentiometric Oxidation of Ferrocenes, J. Am. Chem. Soc., 1961, 83, 3949 CrossRef CAS
- O. J. Curnow, G. M. Fern, S. Klaib, U. Böhme, H. Lang and R. Holze, Structure–molecular orbital energy correlations in Me3Si-substituted bis(indenyl)iron complexes evidenced with UV–vis spectroscopy and cyclic voltammetry, J. Electroanal. Chem., 2005, 585, 167 CrossRef CAS
- M. S. Inkpen, S. Du, M. Hildebrand, A. J. P. White, N. M. Harrison, T. Albrecht and N. J. Long, The Unusual Redox Properties of Fluoroferrocenes Revealed through a Comprehensive Study of the Haloferrocenes, Organometallics, 2015, 34, 5461 CrossRef CAS
- Ž. Petrovski, M. R. P. Norton de Matos, S. S. Braga, C. C. L. Pereira, M. L. Matos, I. S. Gonçalves, M. Pillinger, P. M. Alves and C. C. Romão, Synthesis, characterization and antitumor activity of 1,2-disubstituted ferrocenes and cyclodextrin inclusion complexes, J. Organomet. Chem., 2008, 693, 675 CrossRef
- K. N. Brown, P. T. Gulyas, P. A. Lay, N. S. McAlpine, A. F. Masters and L. Phillips, Electrochemistry of chlorinated ferrocenes: stability of chlorinated ferrocenium ions, J. Chem. Soc., Dalton Trans., 1993, 835 RSC
- W. Erb, N. Richy, J.-P. Hurvois, P. J. Low and F. Mongin, From ferrocene to 1,2,3,4,5-pentafluoroferrocene: halogen effect on the properties of metallocene, Dalton Trans., 2021, 50, 16933 RSC
- A. Hildebrandt, K. Al Khalyfeh, D. Schaarschmidt and M. Korb, Multi-functionalized ferrocenes: –Synthesis and characterization –, J. Organomet. Chem., 2016, 804, 87 CrossRef CAS
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, From Noncovalent Chalcogen–Chalcogen Interactions to Supramolecular Aggregates: Experiments and Calculations, Chem. Rev., 2018, 118, 2010 CrossRef CAS PubMed
- X. Zhang, Z. Gong, J. Li and T. Lu, Intermolecular Sulfur⋯Oxygen Interactions: Theoretical and Statistical Investigations, J. Chem. Inf. Model., 2015, 55, 2138 CrossRef CAS PubMed
- A. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441 CrossRef CAS
- M. R. Koebel, A. Cooper, G. Schmadeke, S. Jeon, M. Narayan and S. Sirimulla, S⋯O and S⋯N Sulfur Bonding Interactions in Protein–Ligand Complexes: Empirical Considerations and Scoring Function, J. Chem. Inf. Model., 2016, 56, 2298 CrossRef CAS PubMed
- S. Kumar, H. B. Singh and G. Wolmershäuser, Protection against Peroxynitrite-Mediated Nitration Reaction by Intramolecularly Coordinated Diorganoselenides, Organometallics, 2006, 25, 382 CrossRef CAS
- L. Bernardi, B. F. Bonini, E. Capitò, G. Dessole, C. Femoni, M. Fochi, M. Comes-Franchini, A. Mincio and A. Ricci, Diastereoselective additions of organometallic reagents to (SFc)-2-p-tolylsulfanylferrocene carboxyaldehyde and to (SFc)-2-p-tolylsulfanyl ferrocenyl imines. Synthesis of new central and planar chiral ferrocenyl alcohols and amines, ARKIVOC, 2003, 2004, 72 Search PubMed
- E. P. Sánchez-Rodríguez, F. Hochberger-Roa, R. Corona-Sánchez, J. E. Barquera-Lozada, R. A. Toscano, M. Urrutigoïty, M. Gouygou, M. C. Ortega-Alfaro and J. G. López-Cortés, Chiral bidentate [N,S]-ferrocene ligands based on a thiazoline framework. Synthesis and use in palladium-catalyzed asymmetric allylic alkylation, Dalton Trans., 2017, 46, 1510 RSC
- T. Blockhaus, C. Klein-Heßling, P. M. Zehetmaier, F. L. Zott, H. Jangra, K. Karaghiosoff and K. Sünkel, Ferrocenes with a Persulfurated Cyclopentadienyl Ring: Synthesis, Structural Studies, and Optoelectronic Properties, Chem. – Eur. J., 2019, 25, 12684 CrossRef CAS PubMed
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed
- C. Präsang, A. C. Whitwood and D. W. Bruce, Halogen-Bonded Cocrystals of 4-(N,N-Dimethylamino)pyridine with Fluorinated Iodobenzenes, Cryst. Growth Des., 2009, 9, 5319 CrossRef
- K. Raatikainen, M. Cametti and K. Rissanen, The subtle balance of weak supramolecular interactions: The hierarchy of halogen and hydrogen bonds in haloanilinium and halopyridinium salts, Beilstein J. Org. Chem., 2010, 6, 4 Search PubMed
- V. Mamane, P. Peluso, E. Aubert, R. Weiss, E. Wenger, S. Cossu and P. Pale, Disubstituted Ferrocenyl Iodo- and Chalcogenoalkynes as Chiral Halogen and Chalcogen Bond Donors, Organometallics, 2020, 39, 3936 CrossRef CAS
- H. R. Khavasi, M. Hosseini, A. A. Tehrani and S. Naderi, Strengthening N⋯X halogen bonding via nitrogen substitution in the aromatic framework of halogen-substituted arylpyrazinamides, CrystEngComm, 2014, 16, 4546 RSC
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions, Angew. Chem., Int. Ed., 2011, 50, 314 CrossRef CAS PubMed
- E. Bartashevich, S. Mukhitdinova, I. Yushina and V. Tsirelson, Electronic criterion for categorizing the chalcogen and halogen bonds: sulfur-iodine interactions in crystals, Acta Crystallogr., Sect. B: Struct. Sci., 2019, 75, 117 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: General considerations, safety consideration, crystallography, electrochemical measurements, experimental section (procedures, compounds analyses and X-ray crystallographic details), NMR spectra, selected NOESY correlations, DFT calculations, cartesian coordinates of DFT optimized structures, HPLC data, voltammograms, additional plots. CCDC 2189751–2189770. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi01854c |
| This journal is © the Partner Organisations 2022 |