
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An amorphous sodium aluminate hydrate phase mediates aluminum coordination changes in highly alkaline sodium hydroxide solutions†
Trent R.
Graham
 *a,
Jian Zhi
Hu
ab,
Nicholas R.
Jaegers
a,
Xin
Zhang
a,
Carolyn I.
Pearce
ac and
Kevin M.
Rosso
*a
*a,
Jian Zhi
Hu
ab,
Nicholas R.
Jaegers
a,
Xin
Zhang
a,
Carolyn I.
Pearce
ac and
Kevin M.
Rosso
*a
aPacific Northwest National Laboratory, Richland, WA 99354, USA. E-mail: trenton.graham@pnnl.gov; Kevin.rosso@pnnl.gov
bThe Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA 99164, USA
cDepartment of Crop & Soil Sciences, Washington State University, Pullman, WA 99164, USA
First published on 5th September 2022
Abstract
Aluminum (Al) transformations between solid and liquid states in the Na2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3:H2O system often involve changes in coordination and polymerization, with the intermediate molecular states challenging to resolve. To detect Al transformations in situ, a solid-state mixture of sodium hydroxide monohydrate (NaOH·H2O) and boehmite (AlOOH) was heated above the melting point of NaOH·H2O to dissolve AlOOH and prepare nonasodium bis(hexahydroxyaluminate) trihydroxide hexahydrate (NSA). In situ27Al magic angle spinning, nuclear magnetic resonance (MAS NMR) spectroscopy was used to monitor Al speciation and coordination during AlOOH dissolution into a homogenous melt, and the crystallization of NSA during cooling to room temperature, supported with ex situ X-ray diffraction, Raman spectroscopy, and 27Al multiple-quantum, 3QMAS NMR spectroscopy. Novel metastable aluminate species were identified during the transformation. Dissolution of AlOOH in molten NaOH·H2O entails a transition from octahedral Al in AlOOH to tetrahedral Al in the aluminate anion [Al(OH)4]− and mu-oxo aluminate dimer [Al2O(OH)6]2− present in solution. These tetrahedral solution-state species then precipitate to form an intermediate, amorphous, tetrahedrally coordinated, sodium aluminate hydrate phase which is stable at 70 °C, and subsequentially crystallizes during cooling to form monomeric octahedral Al in the NSA structure. These transformations and associated intermediates provide insight into the molecular scale mechanisms of Al coordination changes, which in this case appear to be mediated by an amorphous precursor containing oligomerized, tetrahedral Al.
:Al2O3:H2O system often involve changes in coordination and polymerization, with the intermediate molecular states challenging to resolve. To detect Al transformations in situ, a solid-state mixture of sodium hydroxide monohydrate (NaOH·H2O) and boehmite (AlOOH) was heated above the melting point of NaOH·H2O to dissolve AlOOH and prepare nonasodium bis(hexahydroxyaluminate) trihydroxide hexahydrate (NSA). In situ27Al magic angle spinning, nuclear magnetic resonance (MAS NMR) spectroscopy was used to monitor Al speciation and coordination during AlOOH dissolution into a homogenous melt, and the crystallization of NSA during cooling to room temperature, supported with ex situ X-ray diffraction, Raman spectroscopy, and 27Al multiple-quantum, 3QMAS NMR spectroscopy. Novel metastable aluminate species were identified during the transformation. Dissolution of AlOOH in molten NaOH·H2O entails a transition from octahedral Al in AlOOH to tetrahedral Al in the aluminate anion [Al(OH)4]− and mu-oxo aluminate dimer [Al2O(OH)6]2− present in solution. These tetrahedral solution-state species then precipitate to form an intermediate, amorphous, tetrahedrally coordinated, sodium aluminate hydrate phase which is stable at 70 °C, and subsequentially crystallizes during cooling to form monomeric octahedral Al in the NSA structure. These transformations and associated intermediates provide insight into the molecular scale mechanisms of Al coordination changes, which in this case appear to be mediated by an amorphous precursor containing oligomerized, tetrahedral Al.
1. Introduction
Despite the importance of aluminum (oxy)hydroxide reactivity in highly alkaline systems for industrial processes, including high-level radioactive waste processing and aluminum (Al) refining, molecular-scale details of the Al transformation pathways during dissolution and crystallization remain challenging to resolve experimentally due to rapid coordination changes and the potential presence of metastable intermediates.During crystallization of Al (oxy)hydroxides from aqueous solutions, the speciation of Al is often substantially different in the solution versus in the solid phase.1 Under acidic conditions (pH < 4), Al3+ ions form octahedrally coordinated hexa-aquo complexes [Al(OH2)6]3+ and aquohydroxo complexes, e.g., [Al(OH)1(OH2)5]2+. These aquohydroxo complexes condense by olation (loss of water and formation of hydroxo bridges) to form dimers, trimers, tetramers, etc. At near-neutral and higher pH, four hydrolyzed trimers [Al3(OH)7(OH2)6]2+ assemble around a central [Al(OH2)6]3+ ion, and subsequent olation leads to the formation of a Keggin-like multimeric polycation [(Al13O4)(OH)24(OH2)12]7+, consisting of a central tetrahedrally coordinated Al surrounded by twelve edge-sharing octahedrally coordinated AlO6 units. In concentrated alkaline solutions Al coordination is tetrahedral, and the dominant species is the aluminate [Al(OH)4]− hydroxy anion. There is no water in the inner coordination sphere of this hydroxo complex and water must be formed as a leaving group prior to formation of an oxo bridge, leading to additional oxolated, polynuclear, solution-state species (e.g., Al2O(OH)62−).2 Because the partial charge on the Al is low, this condensation reaction is limited in concentrated alkaline solutions and the tetrahedral monomer remains the dominant species, even at high concentrations.3
On the other hand, solid Al(OH)3 phases tend to be based on extended networks of octahedrally coordinated Al3+. These phases are typically layered structures based on hexagonal rings of edge-sharing octahedra, e.g., gibbsite, which can precipitate at temperatures under 80 °C such as by addition of sodium hydroxide (NaOH) to acidic aluminum nitrate (Al(NO3)3) solutions or by acidification of basic sodium aluminate NaAl(OH)4 solutions.4–9 The neutralization of acidic aluminate nitrate solutions initially produces an amorphous gel in which octahedral [Al(OH2)6]3+ complexes can oligomerize to form hexameric rings isostructural with gibbsite sheets.10 Transformation into gibbsite then proceeds via a multi-step process involving metastable intermediates and several competing oligomeric Al-hydroxide species, not all of which have been identified. Examination of solids extracted during gibbsite crystallization revealed the presence of low-coordinated (tetrahedral and pentahedral) transitional Al species at the interface, whose concentration gradually diminished over time as they interacted with oxygen in water to become octahedrally coordinated and were incorporated into gibbsite.11 However, these pentahedral Al species have not been observed in the absence of an interface, e.g., during acidification of NaAl(OH)4 solutions, when a coordination change is required for the tetrahedral [Al(OH)4]− anion, although it is hypothesized that water is also involved in facilitating addition reactions that lead to octahedral oligomers.12
Higher temperatures (>80 °C) favor the formation of oxyhydroxides, e.g., boehmite AlOOH, with dissolution of the gibbsite releasing sufficient concentrations of [Al(OH)4]− ions into NaOH solution to reach a saturation state that overcomes the nucleation barrier to precipitate AlOOH.13 This dissolution pathway also proceeds via an Al octahedral to tetrahedral coordination change, and vice-a-versa for reprecipitation. Similar dissolution and reprecipitation pathways have been shown in lithium hydroxide (LiOH) solutions. In caustic LiOH solutions at room temperature, where the solubility of Al is low (ca. 10 mM), gibbsite dissolves and produces solution-state, metastable, tetrahedral aluminates that subsequently precipitate octahedral lithium dialuminate hydroxide. This dissolution-reprecipitation pathway occurs despite the structural similarities between the quasi-2D octahedral Al frameworks of these two materials, which should allow for solid-state intercalation of Li+ without the need for a coordination change.14
The alkali metals at later periods than sodium in Group 1 show different behavior, with tetrahedral μ-oxo aluminate dimer M2Al2O(OH)6 salts (M = K+ or Rb+) directly crystallizing from concentrated potassium and rubidium hydroxide, or the cesium salt of the tetrahedral aluminate monomer, CsAl(OH)4·2H2O, crystallizing from cesium hydroxide.15 A comparison of concentrated Na2O:Al2O3:H2O and K2O:Al2O3:H2O systems reveals that counterions influence solvent-solute clustering dynamics to either promote or frustrate crystallization.16,17 In potassium aluminate solutions, cation–anion prenucleation clusters promote liquid–liquid separation events, resulting in fast diffusion dynamics, nucleation, and rapid precipitation of K2[Al2O(OH)6]. With supersaturated sodium aluminate solutions, prenucleation clusters manifest as densely packed arrangements that locally minimize the hydration enthalpy but frustrate the precipitation of crystalline phases. As a result, sodium aluminate solutions can remain supersaturated for months at room temperature, over which time they form a semitransparent gel with a mixture of coprecipitated crystalline phases, including nonasodium bis(hexahydroxyaluminate) trihydroxide hexahydrate (NSA; Na9[Al(OH)6]2(OH)3·6H2O) and monosodium aluminate hydrate (MSA; Na2[Al2O3(OH)2]·1.5H2O).18–20
Given their relatively low melting temperature, these crystalline sodium aluminate hydrates (NSA and MSA) comprise a useful model system to probe: (i) solid-state recrystallization pathways and intermediate phases independent of solution-state aluminates; and (ii) the significance of water in the coordination change, given that only structural hydration waters are present.21 NSA is triclinic (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and is composed of a three-dimensional matrix of edge-sharing sodium octahedra coordinated with water and hydroxyls which encapsulate monomeric, Al3+ octahedra ligated by hydroxides (Fig. 1).4,5 Thus, crystallization of NSA allows the precipitation mechanism of tetrahedral, solution state aluminate species to be scrutinized without additional complexity attributed to condensation of aluminate into the octahedral layers of gibbsite and boehmite. Recent investigations of NSA, coprecipitated with sodium hydroxide monohydrate (NaOH·H2O), revealed that loss of water within the ion–ion networks in the crystallizing mother liquor led to condensation of [Al(OH)4]− and precipitation in trace amounts of approximately 0.5 ± 0.1% of the total Al content of the sodium salt of the tetrahedral μ-oxo aluminate dimer Na2Al2O(OH)6.22 Na2Al2O(OH)6 had not previously been observed in the NaOH system as a solid phase but could be measured as a trace defect in NaOH·H2O, or as an interface- or surface-associated moiety in NSA.22
) and is composed of a three-dimensional matrix of edge-sharing sodium octahedra coordinated with water and hydroxyls which encapsulate monomeric, Al3+ octahedra ligated by hydroxides (Fig. 1).4,5 Thus, crystallization of NSA allows the precipitation mechanism of tetrahedral, solution state aluminate species to be scrutinized without additional complexity attributed to condensation of aluminate into the octahedral layers of gibbsite and boehmite. Recent investigations of NSA, coprecipitated with sodium hydroxide monohydrate (NaOH·H2O), revealed that loss of water within the ion–ion networks in the crystallizing mother liquor led to condensation of [Al(OH)4]− and precipitation in trace amounts of approximately 0.5 ± 0.1% of the total Al content of the sodium salt of the tetrahedral μ-oxo aluminate dimer Na2Al2O(OH)6.22 Na2Al2O(OH)6 had not previously been observed in the NaOH system as a solid phase but could be measured as a trace defect in NaOH·H2O, or as an interface- or surface-associated moiety in NSA.22
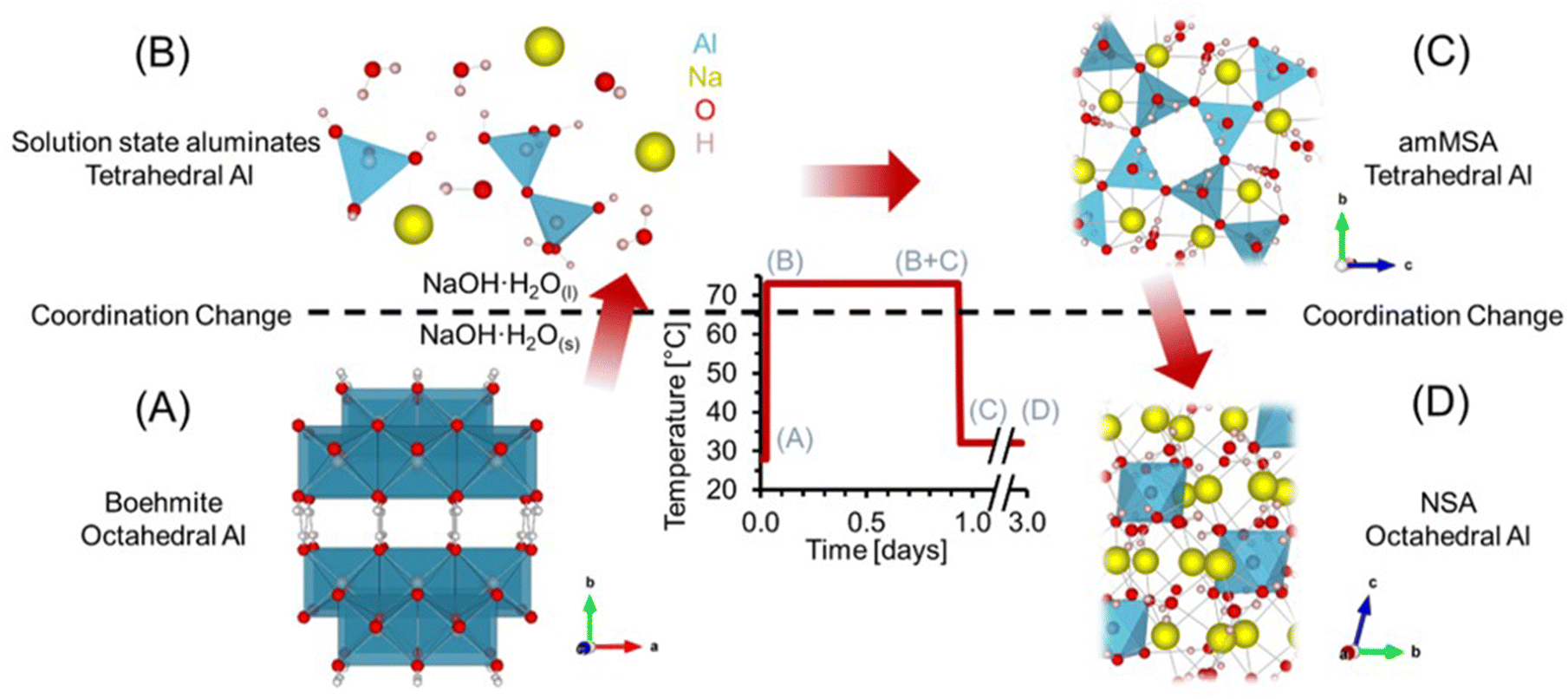 | ||
| Fig. 1 Experimental scheme and corresponding Al3+ crystal structures and solution species. (A) Boehmite (B) solution-state aluminates comprising both monomeric Al(OH)4− and Al2O(OH)62− structures, (C) Amorphous monosodium aluminate hydrate (amMSA), and (D) Nonasodium bis(hexahydroxyaluminate) trihydroxide hexahydrate (NSA) are shown. The temperature of the phase transition of NaOH·H2O is marked with a dashed line. | ||
Here, we examine the dissolution of AlOOH in molten NaOH·H2O, which involves transformation of edge sharing octahedra in AlOOH (Fig. 1a) to solution species of tetrahedral coordination (Fig. 1b). Following dissolution of AlOOH at 70 °C, we observed a metastable, amorphous, tetrahedral Al3+ species that formed as a result of the precipitation of solution-state aluminates. We subsequently identified this species as amorphous monosodium aluminate hydrate (amMSA). Crystalline MSA is tetragonal (space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m) and contains extended networks of tetrahedrally coordinated Al fulfilled by three bridging oxo bonds linking to adjacent Al and a single hydroxy termination (Fig. 2c and d).20,23 While Al in both the solution and the amMSA phases is tetrahedrally coordinated, in the latter amMSA phase, the Al polymerization while less extensive than in crystalline MSA is nonetheless greater than in the aluminate dimer. Cooling of this mixture from 70 °C to 30 °C results in crystallization of the amMSA phase into NSA, requiring depolymerization of tetrahedral Al into octahedrally coordinated Al monomers in the NSA structure.4,5
21m) and contains extended networks of tetrahedrally coordinated Al fulfilled by three bridging oxo bonds linking to adjacent Al and a single hydroxy termination (Fig. 2c and d).20,23 While Al in both the solution and the amMSA phases is tetrahedrally coordinated, in the latter amMSA phase, the Al polymerization while less extensive than in crystalline MSA is nonetheless greater than in the aluminate dimer. Cooling of this mixture from 70 °C to 30 °C results in crystallization of the amMSA phase into NSA, requiring depolymerization of tetrahedral Al into octahedrally coordinated Al monomers in the NSA structure.4,5
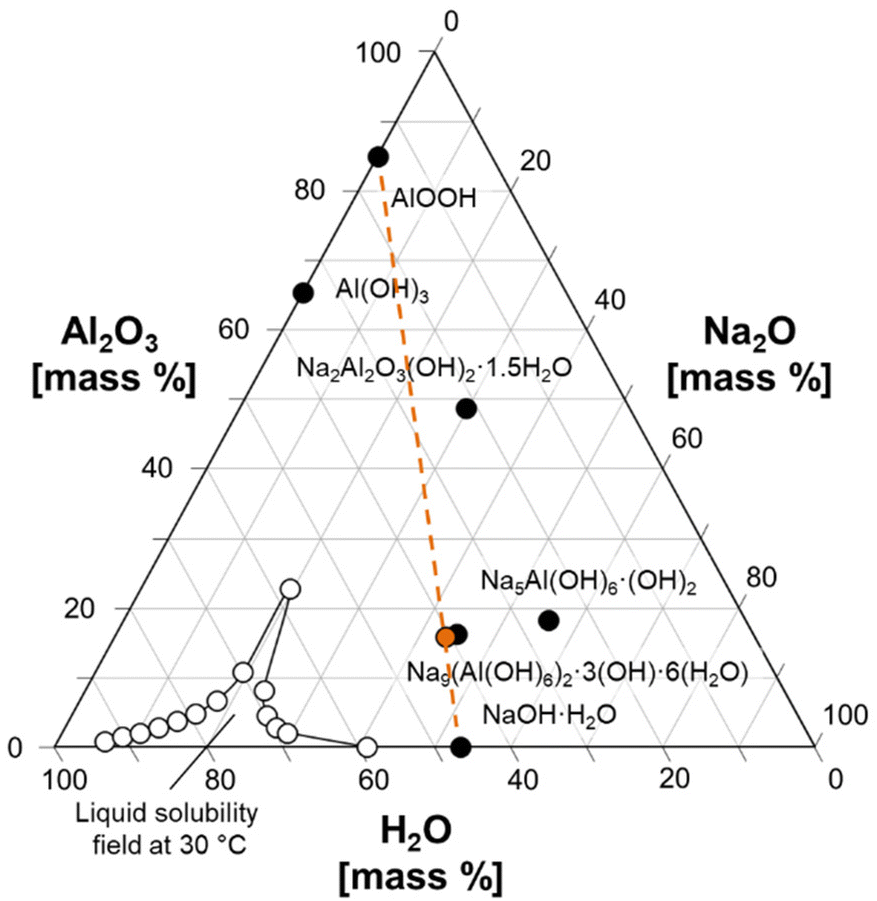 | ||
| Fig. 2 Ternary diagram of the Na2O:Al2O3:H2O system, showing how a physical mixture of AlOOH and NaOH·H2O can approximate the composition of NSA. Several known phases in the ternary system are marked with filled symbols and annotated. The composition of the mixture of NaOH·H2O and AlOOH is marked with an orange symbol, and an orange line is drawn to guide the eyes. The compositions defining the solubility field of solutions at a temperature of 30 °C are drawn with unfilled symbols.8,30 | ||
This work establishes that amMSA is an intermediate aluminate species that mediates the exchange of octahedral, polymerized Al in AlOOH with the monomeric, octahedral Al of NSA. In situ magic angle spinning (MAS) 27Al nuclear magnetic resonance (NMR) spectroscopy was used to follow the Al coordination changes during the heating and cooling process using high-pressure NMR rotors24 capable of preventing loss of water during this process. These studies were complemented by X-ray diffraction (XRD), Raman spectroscopy and 27Al MAS NMR spectroscopy, and 27Al multiple-quantum (3Q) MAS NMR experiments at high field to inspect the spectroscopic qualities of amMSA. Characterization of these sodium aluminate hydrates is of industrial importance both for Al ore processing and refining, and for the processing of Al-rich highly alkaline radioactive waste produced during cold war activities currently stored in underground tanks at the Department of Energy's Hanford site, Washington State. In the later example, studies of the reactivity of MSA are of interest because MSA forms through caustic leaching of these Al-bearing waste sludges and has recently been shown to recrystallize into NSA under these conditions.21,25,26
2. Experimental methods’
All chemicals were used as received. The purity and vendor of the used chemicals are listed following their first description.2.1. Synthesis of aluminum oxyhydroxide (boehmite, AlOOH)
AlOOH was crystallized by hydrothermal treatment of an amorphous aluminum hydroxide gel prepared from addition of NaOH solution to aluminum nitrate solution.27 A 0.25 M aluminum nitrate solution was prepared from dissolution of aluminum nitrate nonahydrate (Al(NO3)3·9H2O, ≥98.5%, Sigma Aldrich) in deionized water (18 MΩ cm, Barnstead™ Genpure™ pro water purification system, Thermofisher). The pH of the aluminum nitrate solution was increased from 2 to 10 via dropwise addition of NaOH (≥98%, Sigma-Aldrich) dissolved in deionized (DI) water to form a 1 M solution. The aqueous suspension of amorphous aluminum hydroxide was stirred for 1 h. The Al(OH)3 gel-like precipitates were collected by centrifugation at 8600 rpm and washed three times with DI water. The precipitates were dispersed in DI water, the pH was titrated to 10 with 1 M NaOH, then additional water was added to form 16 mL of a 0.5 m Al(OH)3 suspension. The suspension was added to a Teflon lined Parr vessel and placed in a rotating oven (10 rpm) at 200 °C for 48 h. The resulting boehmite nanoparticles were collected by centrifugation at 8600 rpm and washed with DI water three times. The boehmite solids were dried overnight at 80 °C in an electric oven. Characterization of boehmite nanoparticles with 27Al MAS NMR spectroscopy indicated that only 6-coordinate Al is present, which is in agreement with the crystal structure (see Fig. S1 in the ESI†). Additional characterization of the boehmite is available in literature, including techniques such as X-ray diffraction, Raman spectroscopy, Brunauer–Emmett–Teller (BET) measurements of surface area, and also synchrotron X-ray and Neutron scattering.27,28 Note that 27Al MAS NMR data of the initial boehmite solids in the absence of NaOH·H2O were acquired at a field of 14.1 T, and subsequent fitting of the data with quadrupolar line shape parameters demonstrated that the parameters (isotropic chemical shift, quadrupolar coupling parameter, and asymmetry parameter) were in good agreement with the work of others.292.2. Sample preparation for NMR spectroscopy and complementary characterization
A physical mixture of NaOH·H2O (99.99%, Sigma-Aldrich) and AlOOH was mixed in an N2-filled glovebox to give a 4.5:1:14.5 mole ratio of Na2O:Al2O3:H2O, which approximates the stoichiometry of NSA (4.5:1:13.5, Na2O:Al2O3:H2O) as shown in Fig. 2. This physical mixture was loaded into a 7.5 mm zirconia NMR rotor for variable temperature MAS NMR spectroscopy at 7.05 T. Additional samples of the same composition were prepared, and ex situ Raman spectroscopy, X-ray diffraction, and 27Al MAS NMR spectroscopy at 14.1 T were used to characterize the ambient temperature crystallization process, following the dissolution of AlOOH in molten NaOH·H2O. For the ex situ studies, a mixture of NaOH·H2O and AlOOH was prepared such that the mole ratio of the mixture was (4.5:1:14.5 mole ratio of Na2O:Al2O3:H2O) and placed in a Teflon-lined Parr vessel, then heated in a rotary oven operating at 70 °C and 10 rpm for 24 hours. The Parr vessel was removed from the oven and the solids were removed from the vessel after 1 hour. The use of pressurize-able Parr vessels insured that there was no loss of water during the heating of these materials. Therefore, the composition of the solids is defined by the initial mixture of AlOOH and NaOH·H2O.
2.3. NMR spectroscopy
Variable temperature, in situ27Al MAS NMR spectroscopy at 7.05 T was conducted to investigate both the octahedral to tetrahedral Al coordination during dissolution of AlOOH, the precipitation of an unidentified amorphous phase, and the tetrahedral to octahedral Al coordination change during crystallization of the amorphous phase into NSA. In situ27Al MAS NMR spectra were acquired on a Varian-Inova NMR spectrometer operating at temperatures between 20 and 70 °C using a 7.5 mm MAS probe. The temperature was calibrated with the 1H chemical shifts of ethylene glycol under equivalent spinning rates.31In situ NMR spectroscopy samples were prepared by loading a physical mixture of AlOOH and NaOH·H2O into a 7.5 mm custom designed zirconia rotor.24 Note that prior studies tracking the mass of these sealed rotors filled with 70% by volume water, indicated that the rotors provide a gas-tight seal to temperatures of 245 °C which generates a vapor pressure of H2O of 35 atm.24 The use of pressurize-able NMR spectroscopy rotors insured that there was no loss of water during the heating of AlOOH and NaOH·H2O. Therefore, the composition of the solids is defined by the initial mixture of AlOOH and NaOH·H2O. The sample spinning rate was approximately 3.4 kHz. 27Al MAS NMR spectra were collected using a single pulse with a π/10 pulse length to balance the uniformity of the excitation of liquid and solid-state Al resonances with sensitivity, a 1 s recycle delay and 100 ms acquisition time. The 27Al chemical shift was externally referenced to 1 M Al(NO3)3·9H2O in D2O (δ = 0 ppm).Additional 27Al MAS NMR studies of the tetrahedral to octahedral transformation following the crystallization of the amorphous phase into NSA at ambient temperature were conducted at 14.1 T and 20 kHz spinning rate to provide spectra with increased resolution. A sample, prepared in a Parr vessel as described above, was loaded into commercial 2.5 mm Bruker rotors with Vespel drive and bottom caps. In situ27Al MAS NMR spectroscopy was then conducted using a 14.1 T Bruker NMR spectrometer. The corresponding 27Al Larmor frequency is 156.375 MHz. A MASDVT600W2 BL2.5 X/Y/H probe was inserted into the spectrometer at a temperature of approximately 25 °C with the temperature regulated by flowing N2 gas. A series of single pulse, direct excitation, 27Al MAS NMR spectra were acquired and a 27Al multiple quantum (3Q)MAS NMR spectrum was collected.
Single pulse, direct excitation 27Al NMR spectra were obtained at 14.1 T through the collection of 1024 transients with an 18.5184 ms acquisition time, a 1 s delay between transients, a 20 kHz MAS spin rate, and a π/20 liquid-state pulse corresponding to a duration of 0.45 μs. An excitation length nutation experiment on 1 M Al(H2O)63+ solution prepared via aluminum chloride hexahydrate (AlCl3·6H2O, ≥99%, Sigma-Aldrich) dissolution in H2O was used to measure the liquid-state π/20 pulse width. In addition, chemical shifts are referenced to the 1 M Al(H2O)63+ solution (δ = 0 ppm).
27Al 3QMAS spectra were also acquired at a field of 14.1 T using the Bruker, 2.5 mm MAS probe. For the amMSA sample, the spectrum was acquired with a z-filter, 27Al 3QMAS pulse sequence (three pulse z-filter sequence, mp3qzqf) at a spinning rate of 20.0 kHz. The excitation pulse power level and conversion pulse were −20 dB, the selective pulse power level was 5.23 dB, the delay between the second and third pulse (D4) was 20 μs, the acquisition time was about 14.88 ms and the spectral width for the evolution dimension (F1) was 20.0 kHz. Optimized P1, P2, and P3 pulse durations were 3.8, 1.6, and 17.0 μs. The recycle delay between scans was 20 seconds, a collection of 96 transients were acquired with 72 increments. Acquisition utilized States-Time Proportional Phase Incrementation (TPPI) processing.
2.4. Post acquisition processing of 27Al MAS NMR spectra and 27Al 3QMAS NMR spectra
In situ 27Al MAS NMR spectra obtained at a field of 7.05 T were first processed in Mestrenova (version 14.01-23559, released 2019-06-07, Mestrelab Research S.L.) where the spectra were zero-filled once and 50 Hz of exponential line broadening was applied. 27Al MAS NMR spectra acquired at a field of 14.1 T were also first processed in Mestrenova wherein the free induction decay was zero-filled to 52.5 ms, after which 20 Hz of exponential line broadening was applied. The 2D 3QMAS spectrum was sheared in Topspin (v3.5.7), 20 Hz of exponential line broadening was applied, and slices along the F2-dimension were then extracted.2.5. Simulation of 27Al MAS NMR spectra and 27Al 3QMAS NMR spectra
Quadrupolar line shape parameters including the spinning side band components for AlOOH, amMSA, and NSA were regressed by deconvoluting experimental 27Al MAS NMR spectra at 14.1 and 7.05 T. For amMSA, the regression of the 27Al MAS NMR spectra at 14.1 T and 7.05 T were guided by the analysis of 27Al 3QMAS NMR spectra. The 27Al MAS NMR spectra were fit to obtain the quadrupolar line shape parameters in ssNAKE (v 1.3)32 using the finite MAS quadrupole model through which the quadrupolar line shape parameters were fit through the minimization of the residual between the data and the summation of the components using the Powell minimization. The 3QMAS extracted slices were then simulated in ssNAKE using the 3QMAS fitting routine for I = 5/2 nuclei, MQ = 3, and with a finite MAS of 20 kHz. These quadrupolar line shapes of AlOOH, amMSA, and NSA were assumed to be temperature independent and were used to fit the in situ27Al MAS NMR data acquired at a field of 7.05 T by algebraic combination with a Lorentzian line attributed to a solution aluminate component. The peak position, fwhm and signal intensity of the Lorentzian line could vary.2.6. X-ray diffraction (XRD)
The transformation of the initial MSA and NaOH·H2O phase mixture to NSA was analyzed by XRD, where the solids were loaded into an analytical cell, equipped with Kapton windows, in an N2-filled glovebox. The cell was transferred to a Philips X'pret multi-purpose diffractometer (PANAlytical Almelo, The Netherlands), utilizing a fixed copper anode, operating at 40 mA and 50 kV. Phase identification and quantification were performed in TOPAS (v7, (Bruker AXS)). The Rietveld fits of the powder XRD patterns were carried out using TOPAS using literature NSA4 and sodium hydroxide monohydrate33 crystal structures with the atomic coordinates not refined. Note that the shapes of the reflections were calculated using the fundamental parameters approach, and the instrumental factors were established with patterns obtained from NIST silicon (SRM 640D) and LaB6 (SRM 660A) powders. Line broadening from finite crystallite size was modeled using the double-Voigt approach.342.7. Raman spectroscopy
Raman spectra were acquired on samples prepared ex situ. The Raman spectra were collected on a Horiba LabRam HR spectrometer, installed on a Nikon Ti-E inverted microscope with a 40× objective through the application of a continuous 632.81 nm laser at a temperature of approximately 25 °C. Spectra were acquired between 100–3800 cm−1. Diagnostic Raman peaks for NSA and MSA occur between 390–550 cm−1, and this window was deconvoluted using a single linear spline to remove the background and then followed by regression of purely Lorentzian line shape parameters. The regression utilized the fewest number of Lorentzian peaks required to fit the data.3. Results and discussion
The results and discussion are organized as follows. First, the dissolution of AlOOH in molten NaOH·H2O after heating to 70 °C, and the subsequent precipitation of a solid phase after cooling to near ambient temperature, are presented in terms of in situ27Al MAS NMR spectroscopy at a low field of 7.05 T. Then, the ex situ characterizations of the unidentified phases (amMSA and NSA) formed during the reaction using X-ray diffraction, Raman spectroscopy, and high field (14.1 T) 27Al MAS NMR spectroscopy are described. The order of these results shows the in situ results first, and then unravels the complexity of the in situ NMR spectroscopy results with the ex situ characterizations. Lastly, using a combination of single pulse direct excitation NMR spectra at 14.1 T and 7.05 T and 3QMAS NMR spectra at 14.1 T, the quadrupolar line shape parameters of the amorphous phase are identified.3.1. In situ 27Al MAS NMR spectroscopy of AlOOH dissolution in molten NaOH·H2O
Dissolution of AlOOH in molten NaOH·H2O was followed with in situ27Al MAS NMR spectroscopy. The chemical shifts of 27Al MAS NMR resonances are sensitive to the coordination of the Al nucleus, and were used to monitor the dissolution of edge sharing Al octahedra in AlOOH to form Al tetrahedra in solution. Tetrahedrally coordinated Al exhibits an apparent chemical shift near 80 ppm, whereas octahedrally coordinated Al exhibits a chemical shift of approximately 0 ppm.35,36 In addition, the 27Al nucleus is also quadrupolar (spin 5/2) and 100% abundant.At a relatively low field of 7.0 T, NMR spectroscopy was used to track Al speciation in a mixture of AlOOH and NaOH·H2O as it was heated to 70 °C (Fig. 3). The initial spectrum acquired at 30 °C shows the presence of trace (<1%) tetrahedral Al and dominant octahedral Al. Given that the melting point of NaOH·H2O is 64 °C,37 and that tetrahedral Al is not present in as synthesized AlOOH (based on 27Al MAS NMR spectroscopy at 14.1 T and 20.0 kHz, shown in Fig. S1 in the ESI†), the presence of trace tetrahedral species at 30 °C indicates some limited transformation of AlOOH upon physical mixing with NaOH·H2O upon increasing the temperature to only 30 °C.
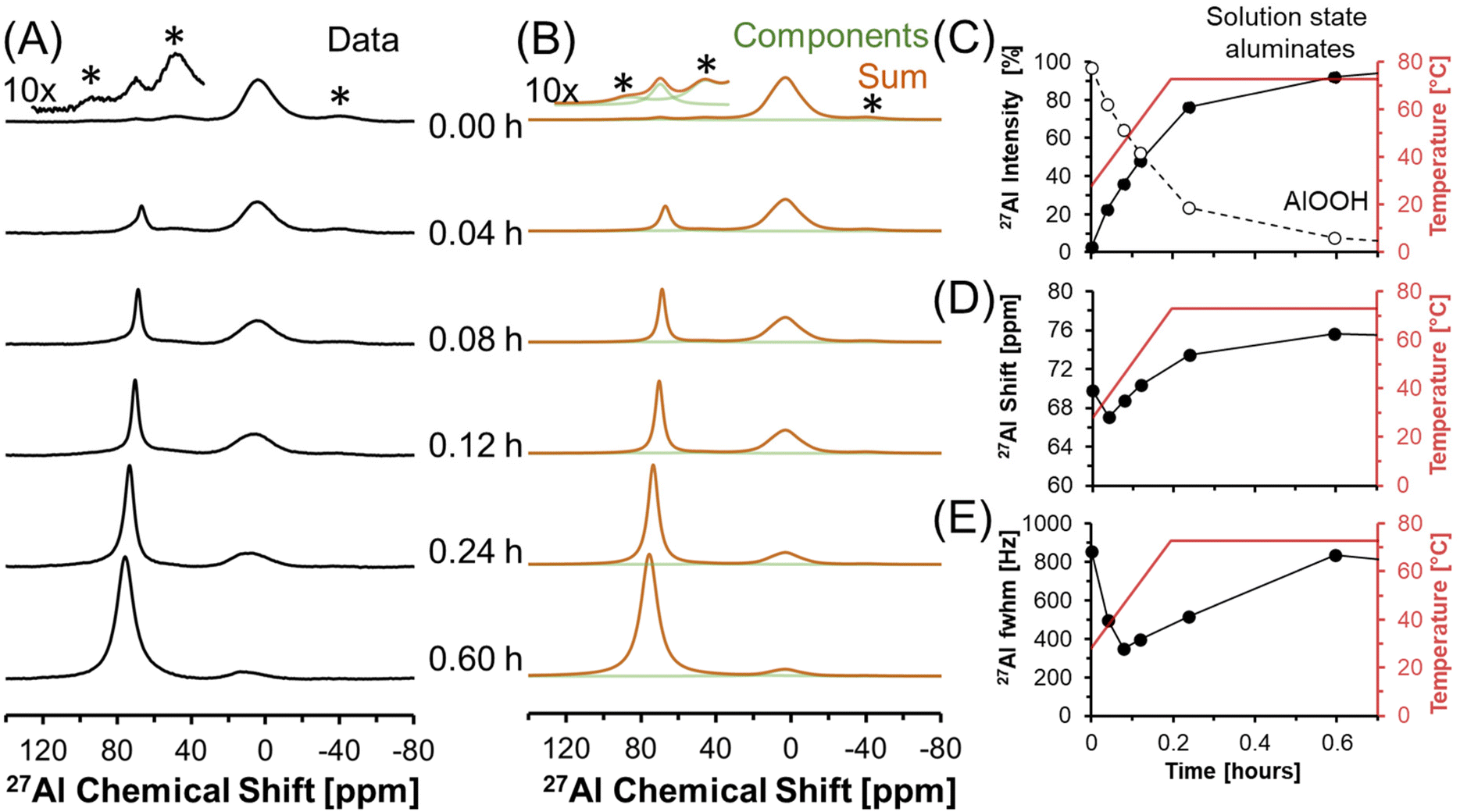 | ||
| Fig. 3 (A) Temporal progression of single pulse, direct excitation, in situ27Al MAS NMR spectra acquired at 7.05 T of the dissolution of AlOOH in NaOH·H2O at 3.4 kHz and temperatures increasing from 30 to 70 °C. (B) Corresponding simulation of the data with fixed quadrupolar line shapes for the solids, and a flexible Lorentzian line for the liquid component. Note that a 10x magnified spectrum is offset from the initial spectrum acquired at t = 0 hours. (C) Relative intensity of the 27Al NMR signals and Lorentzian line shape parameters for the solution component such as the (D) chemical shift and (E) full-width at half maximum (fwhm). Note that lines connect the data points in the charts and are only drawn to guide the eyes. Spinning side bands are annotated with an asterisk (*). | ||
Raising the temperature to 70 °C at a rate of approximately 3 °C min−1 resulted in a dramatic increase in the concentration of tetrahedral species relative to octahedral AlOOH. The increase in the magnitude of the tetrahedral Al resonance occurred along with an downfield chemical shift, from approximately 70 to 73 ppm, which corresponds to deshielding of the tetrahedral species (Fig. 3D). In addition, there is an increase in the chemical shift of AlOOH as the temperature increases likely due to chemical exchange with the incipient tetrahedral phase. An increase in the chemical shift of the tetrahedral 27Al nucleus was also observed when the Al3+ concentration in solution was increased in LiOH, KOH, and NaOH solutions, and was attributed to changes in Al speciation, from predominantly Al(OH)4− monomers, to mixtures with Al2O(OH)62− dimers.38 The final spectrum after 0.6 hours (Fig. 3) shows that the tetrahedral resonance is predominantly Lorentzian in shape. However, over the next 20 hours, systematic changes in the tetrahedral line shape occur, consistent with the emergence of a second tetrahedral Al resonance, which is assigned to the precipitation of an unknown tetrahedral Al3+ species (see section 3.4† for characterization including details regarding the deconvolution of the quadrupolar line shape).
3.2. Precipitation of an amorphous tetrahedral phase at 70 °C
Transformation of solution state aluminates to an unknown phase where Al is tetrahedrally-coordinated was followed in a series of direct excitation, single pulse in situ27Al MAS NMR spectra collected at 7.05 T and 70 °C (Fig. 4).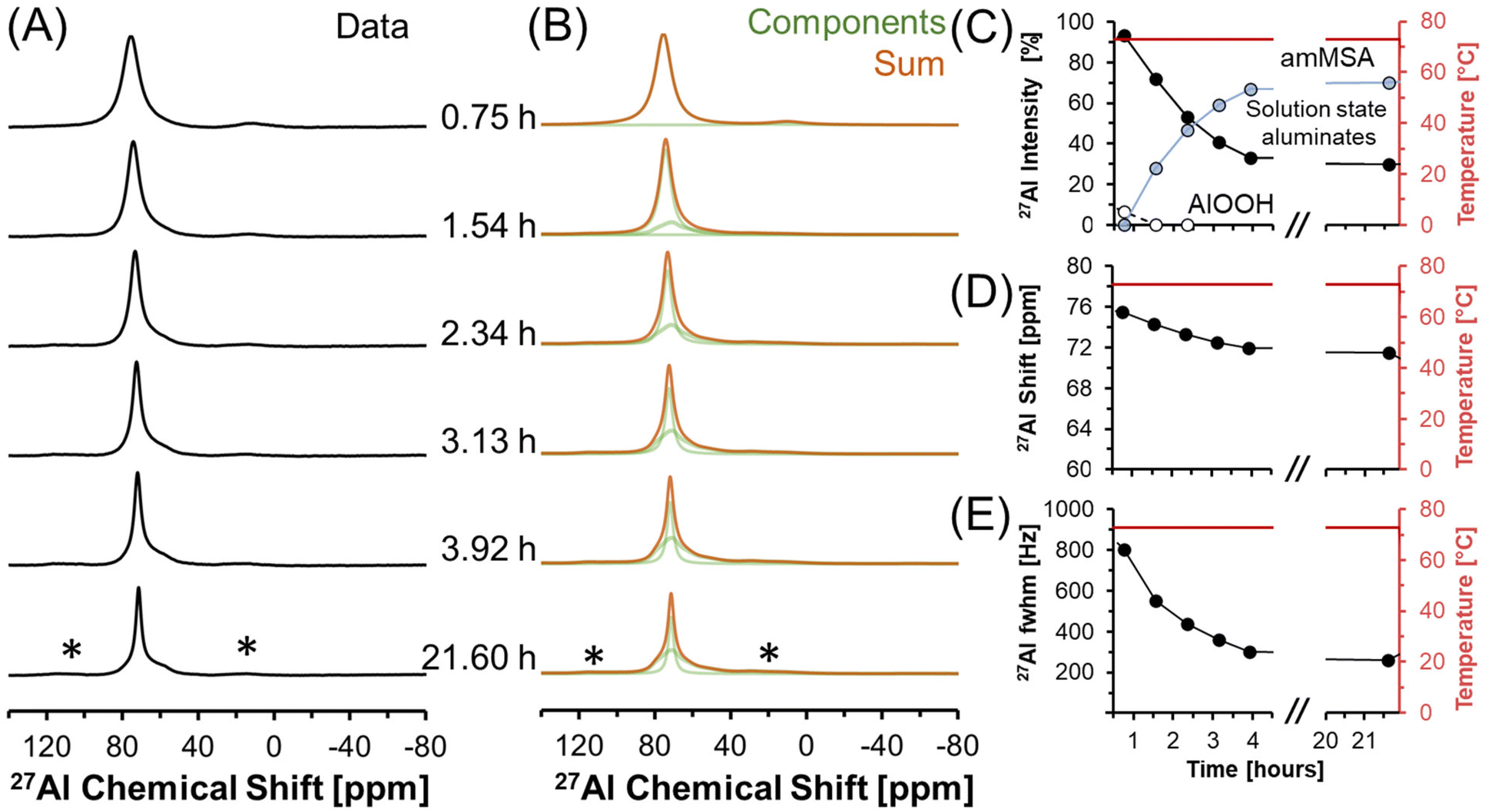 | ||
| Fig. 4 (A) Single pulse, direct excitation, in situ27Al MAS NMR spectra at a field of 7.05 T with 3.4 kHz spinning rate and at 70 °C tracking solution state aluminates precipitating to form an unknown phase with Al in tetrahedral coordination. (B) Corresponding deconvolution of the spectra using the quadrupolar line shape of a boehmite spectrum, a Lorentzian line attributed to solution-state aluminates, and the quadrupolar line shape attributed to the unknown phase (amMSA). (C) Relative intensity of the 27Al NMR signals and Lorentzian line shape parameters for the solution component such as the (D) chemical shift and (E) full-width at half maximum (fwhm). Note that lines connect the data points in the charts and are only drawn to guide the eyes. Spinning side bands of very weak intensity are annotated with an asterisk (*). | ||
In Fig. 4, the tetrahedral component progressively resonates at lower chemical shift (decreasing from 76 to 72 ppm) and, over the course of 4 hours, the full width at half maximum progressively narrows from 800 Hz to 200 Hz. The relative integral of the Lorentzian line shape of the 27Al MAS NMR spectral component assigned to the solution-phase sodium aluminates decreases from ∼100% at 0.7 hours, to ∼30% at 4 hours, and remains approximately constant over the course of one day. The spectroscopic qualities of the Lorentzian component are consistent with a reduction in the concentration of solution-state tetrahedral species. The reduction in the Lorentzian signal intensity is also inversely correlated with the emergence of a prominent shoulder which creates an asymmetry in the resonance. The assignment of the Lorentzian component to a solution phase and the prominent shoulder to a precipitated phase can be rationalized with 27Al MAS NMR experiments where the excitation pulse length is nutated, as shown in Fig. S2 in the ESI.†
Analysis of the nutation experiments indicates that the Lorentzian component exhibits sinusoidal dependence on the excitation pulse length, which is commonly seen in solution state species, and that the asymmetric component has a drastically reduced 90° pulse length, which is commonly seen in solid phases. While it is possible that 27Al sites in solid phases with weak quadrupolar interactions, such as having a quadrupolar coupling coefficient between 0–2 MHz, can also exhibit quasi-sinusoidal nutation behavior, this is unlikely because of the low field that the NMR data was collected at. For example, the octahedral Al site in lithium aluminate layered double hydroxides at the same field strength has a quadrupolar coupling coefficient of 1.4 MHz, and was observed to have a reduced 90° pulse length compared to solution state aluminates at the same magnetic field.14
The spectra were deconvolved with three components: (i) solid phase octahedral boehmite; (ii) a solution phase tetrahedral Lorentzian line shape; and (iii) an unknown tetrahedral solid phase based on the tetrahedral region of the final spectrum in Fig. 4, which is demonstrated later to be amMSA. This deconvolution allowed the relative abundance of the unknown phase to be estimated, and shows that it increased to approximately 70% of the Al signal over 4 hours. After 4 hours, the proportion of the signal intensity of the solution-state aluminates attributed to the Lorentzian component and the unknown phase (amMSA) remains constant over the course of one day. Note that the amMSA component is a combination of Al site 1 and Al site 2, as shown in Fig. 10.
3.3. In situ 27Al MAS NMR spectroscopy of transformations occurring during the temperature quench
27Al MAS NMR spectra acquired at 7.05 T tracking the changes in Al speciation upon reducing the temperature of the sample from approximately 70 to 30 °C are shown in Fig. 5. In the spectra, the Lorentzian component assigned to solution-state species decreases in intensity from approximately 30 to 10% of the total Al signal, whereas the relative integral corresponding to the unknown tetrahedral phase increases. During the process, the chemical shift of the Lorentzian component also reduces from 72 to 60 ppm and the fwhm significantly broadens from 300 to 1700 MHz. The increase in the fwhm upon lowering temperature is likely due to an increase in the viscosity, and thus a decrease in 27Al mobility, as the mixture cools. Also, while the reduction in the chemical shift of the Lorentzian component follows the same trend observed by others,38 which is that a reduction in the chemical shift occurs when the concentration of Al is reduced, but the magnitude of the change in chemical shift exceeds what was observed previously. Specifically, prior literature has observed a decrease in the chemical shift from near 80 ppm to approximately 70 ppm when comparing 5 mM Al in dilute NaOH and 12 M NaOH,38 while in these spectra, a tetrahedral Al chemical shift of 60 ppm is observed.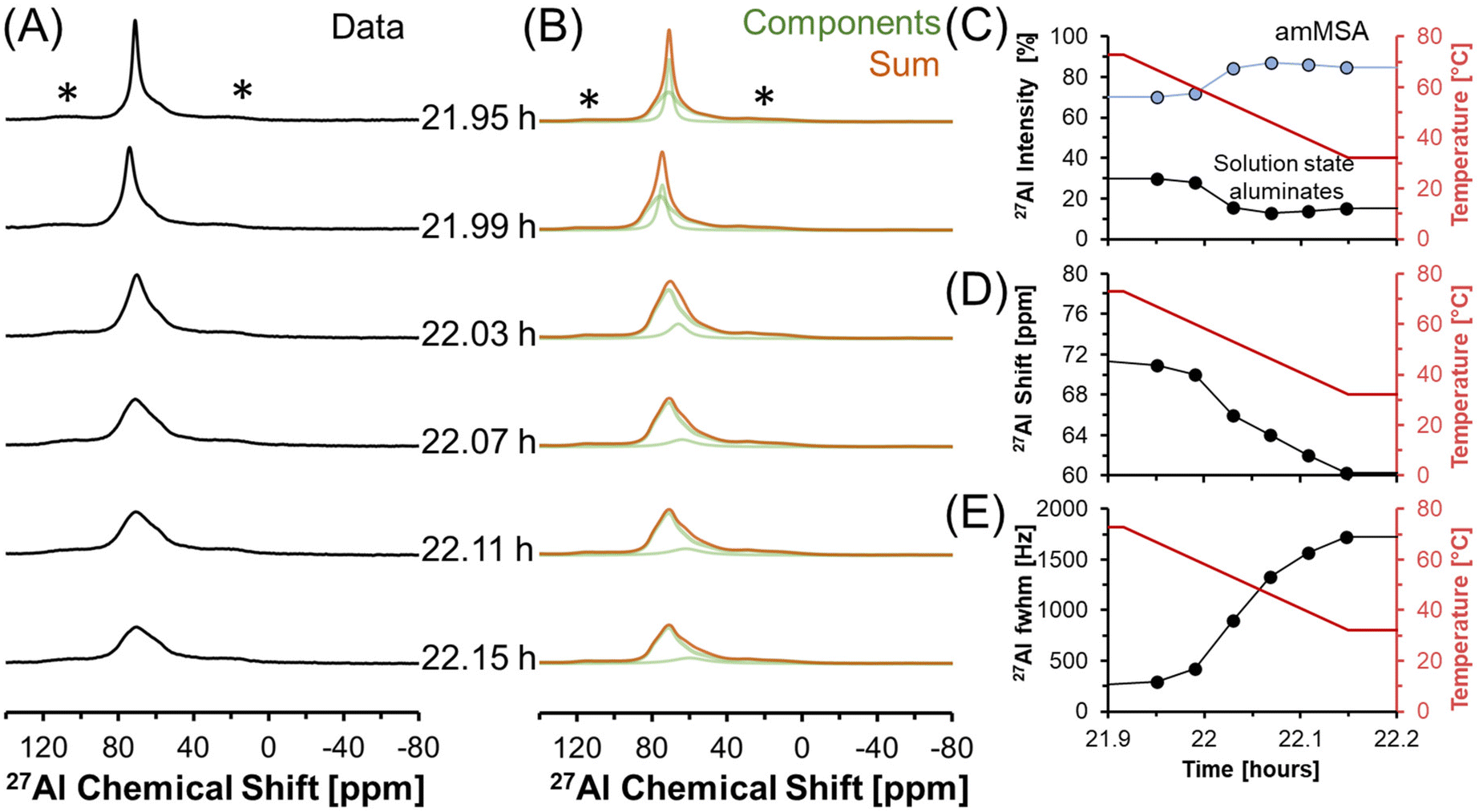 | ||
| Fig. 5 (A) Single pulse, direct excitation, in situ27Al MAS NMR spectra at a field of 7.05 T with 3.4 kHz spinning rate tracking the decrease in temperature from 70 to 30 °C to monitor the precipitation of solution state aluminates precipitating to form an unknown phase (amMSA) with Al in tetrahedral coordination. (B) Spectra were deconvoluted with a Lorentzian line attributed to solution-state aluminates and the quadrupolar lineshape attributed to the unknown phase with Al in tetrahedral coordination. (C) Relative intensity of the 27Al NMR signals and Lorentzian line shape parameters for the solution component such as the (D) chemical shift and (E) full-width at half maximum (fwhm). Note that lines connect the data points in the charts and are only drawn to guide the eyes. Spinning side bands of very weak intensity are annotated with an asterisk (*). Note that the amMSA component is a combination of Al site 1 and Al site 2, as shown in Fig. 10. | ||
Fig. 6 shows in situ27Al MAS NMR spectra following the tetrahedral to octahedral Al coordination change at a field of 7.05 T. Over the course of 2 days, the tetrahedral component decreased in intensity while the octahedral component increased. In addition, the Lorentzian-shaped tetrahedral component assigned to solution state species reduced in prevalence from approximately 10% to trace amounts below 1%, and the peak position and full width at half maxima of the Lorentzian line is approximately constant over the course of the reaction. Spectra acquired at a field of 14.1 T and a spinning rate of 20 kHz indicated that there are no pentahedral species observable with 27Al MAS NMR spectroscopy during the coordination change, and that the line shape of tetrahedral and octahedral species are approximately constant during the temperature quench, as shown in Fig. S3 in the ESI.† Additional characterization was carried out to determine the structures of: (i) the unknown intermediate tetrahedral phase (amMSA) that persisted for several days; and (ii) the octahedral phase that formed during the temperature quench (shown subsequentially to be NSA).
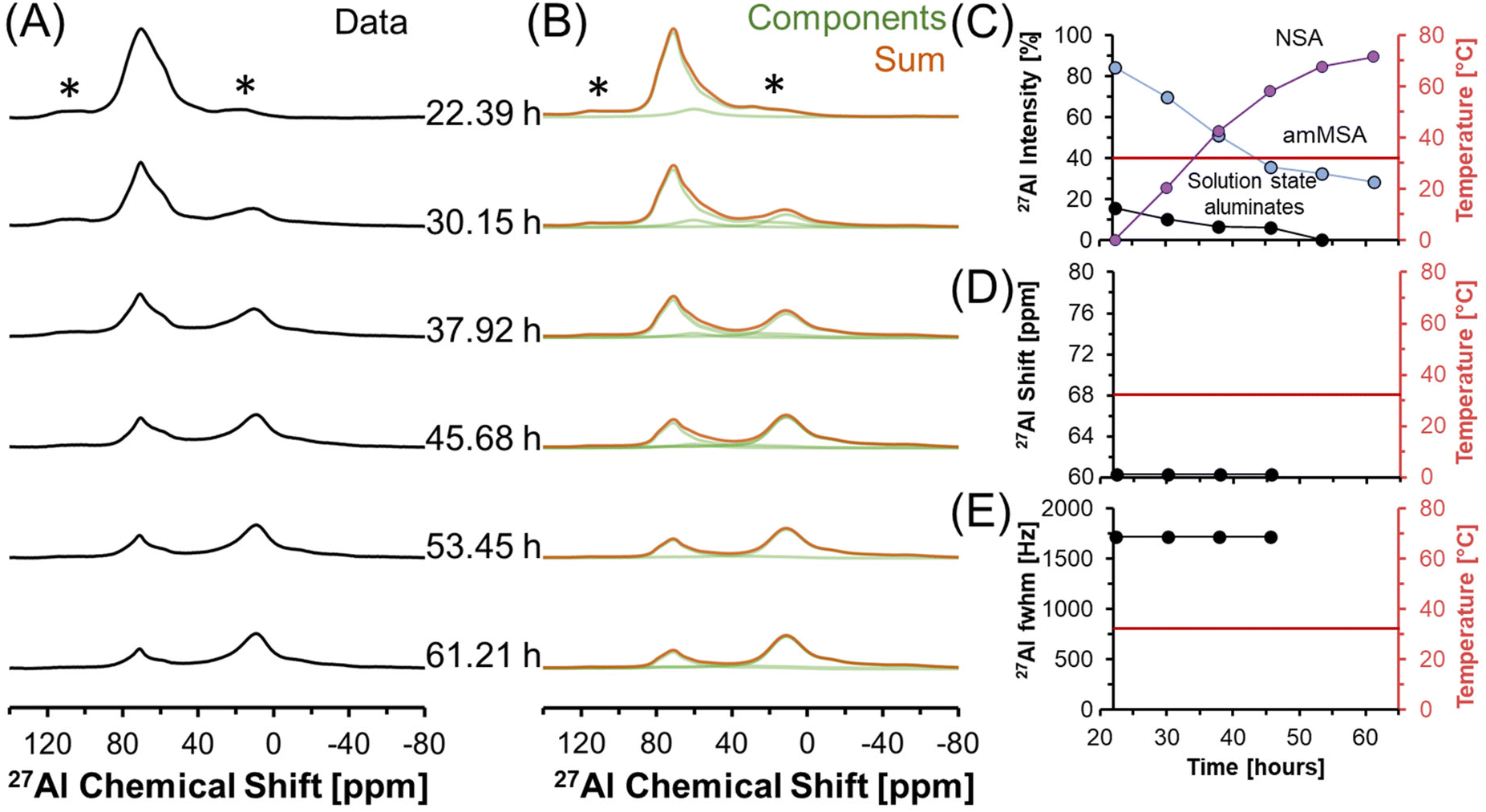 | ||
| Fig. 6 (A) Single pulse, direct excitation, in situ27Al MAS NMR spectra at a field of 7.05 T with 3.4 kHz spinning rate tracking the decrease in temperature from 70 to about 30 °C to track the coordination change between tetrahedral and octahedral Al. (B) Spectra were deconvoluted with a Lorentzian line attributed to solution-state aluminates, the quadrupolar lineshape attributed to the unknown phase with Al in tetrahedral coordination (amMSA), and the quadrupolar lineshape attributed to the octahedral phase (NSA). (C) Relative intensity of the 27Al NMR signals and Lorentzian line shape parameters for the solution component such as the (D) chemical shift and (E) full-width at half maximum (fwhm). Note that lines connect the data points in the charts and are only drawn to guide the eyes. Spinning side bands are annotated with an asterisk (*). Note that the amMSA component is a combination of Al site 1 and Al site 2, as shown in Fig. 10. | ||
3.4. Additional characterization of the intermediate tetrahedral-Al phase and final octahedral-Al phase
To better understand the amorphous tetrahedral phase and final octahedral phase formed in the in situ study, additional characterization was performed using X-ray diffraction, Raman spectroscopy and 27Al MAS NMR spectroscopy at a field of 14.1 T. The XRD diffractograms for samples prepared ex situ in a Parr vessel and promptly analyzed upon cooling is shown in Fig. 7. The X-ray diffractogram could be fit with a broad background corresponding to an amorphous component, and two crystalline phases: (i) NSA, with Al in octahedral coordination; and (ii) sodium hydroxide monohydrate. Thus, the crystalline octahedral Al phase formed during the temperature quench is identified as NSA. Based on the width of the diffraction peaks, the crystallite size of the NSA and sodium hydroxide monohydrate components are 49 and 52 nm, respectively, and the relative crystalline phase abundance is 83% NSA and 17% sodium hydroxide monohydrate. Note that the XRD was collected promptly after cooling the mixture of AlOOH and NaOH·H2O, based on NMR spectroscopy, the majority of the aluminum is present in the form of the amorphous monosodium aluminate phase. Consequentially, this leads to acquisition of a fairly noisy XRD diffractogram. Analysis of scanning electron microscopy, shown in Fig. S4 of the ESI,† showed that the samples were composed of approximately 10-micron size columnar aggregates decorated with a rough surface composed of smaller crystallites. There are no peaks in the diffraction pattern corresponding to crystalline monosodium aluminate hydrate (MSA), or any other crystalline phase with Al in tetrahedral coordination. Given this lack of crystalline phases with Al in tetrahedral coordination, and the broad amorphous background, we propose that the intermediate tetrahedral component is amorphous.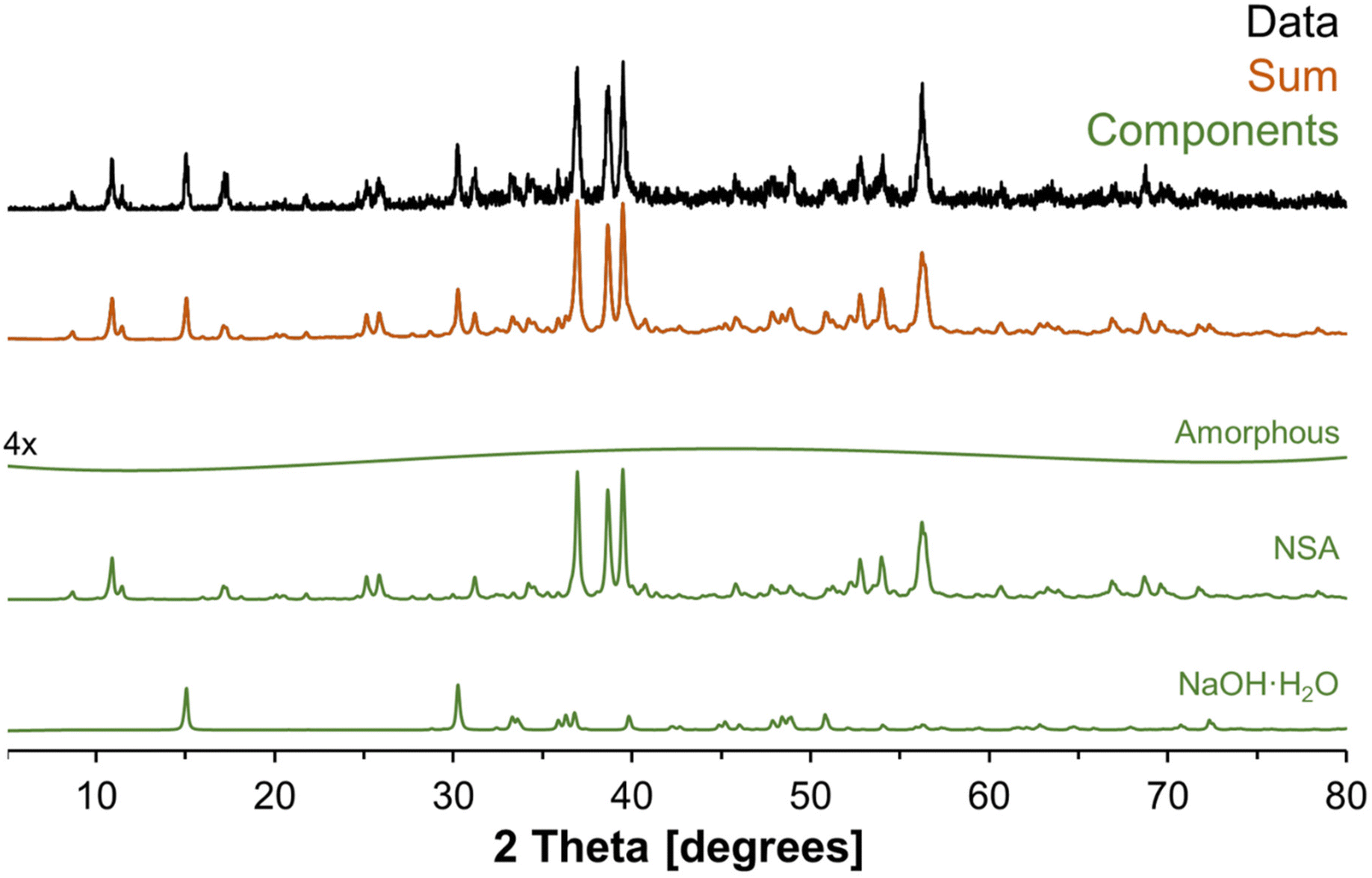 | ||
| Fig. 7 Further characterization of the products in an ex situ sample using X-ray diffraction. The XRD-apparent phases are vertically offset, and the components are NSA, sodium hydroxide monohydrate, and a weak background component. The relative phase abundance was determined in TOPAS (v6) from Rietveld refinements using the literature crystal structures of NSA4 and sodium hydroxide monohydrate33 with the atomic coordinates not refined. The relative phase abundance of the crystalline components are 83% NSA and 17% sodium hydroxide monohydrate. Note that the weak amorphous component was vertically magnified by a factor of 4. | ||
Unlike XRD, the Raman spectra shown in Fig. 8 can provide insight into the local chemical environment of amorphous species. Raman spectroscopy is particularly useful for identifying between aluminate in the form of crystalline monosodium aluminate (MSA) and aluminate in the form of NSA because in the region between 390 and 550 cm−1, the sodium aluminates can be discriminated by the AlO6 vibration at 500 cm−1 in NSA and the Al(OAl)3 vibration at 450 cm−1 in MSA.21 The vibrational band (v2) for the phases produced from the dissolution of AlOOH in molten NaOH·H2O has a Lorentzian line shape with a fwhm of 35 cm−1, which is much greater than the narrow band in the Raman spectrum of crystalline MSA hydrate, with a fwhm of 10 cm−1. Similarly, a component of the mixture has a Raman band (v1) at 500 cm−1 which is consistent with the position of the prominent band assigned to AlO6 in the NSA spectra. The v1 band also is broader than the comparable band in the NSA reference compound. The presence of the v1 band in the Raman spectrum is assigned to NSA which forms as the final product following dissolution of AlOOH in NaOH·H2O, which is also evident in the XRD diffractograms (Fig. 7), and is consistent with the emergence of the octahedral-coordinated Al phase in the in situ27Al MAS NMR spectra (Fig. 6). The difference in the fwhm of the v1 band compared to the reference spectrum in Fig. 8 is likely due to the small crystallite size of NSA indicated by XRD as the reference compound spectra was acquired on crystals of approximately 100 micron in length.22 Comparison of a time-series of Raman spectra of the reaction mixture acquired after 24.5 and 41.4 hours shown in Fig. S5 in the ESI,† indicates that the v1 becomes more prominent upon aging at room temperature, whereas the v2 band decreases in intensity. The changes in intensity of the v1 and v2 Raman bands are in agreement with the NMR results which indicate that the octahedral phase assigned to NSA and visible with XRD crystallizes via consumption of the tetrahedral Al phase with the later tetrahedral Al phase not observable via XRD.
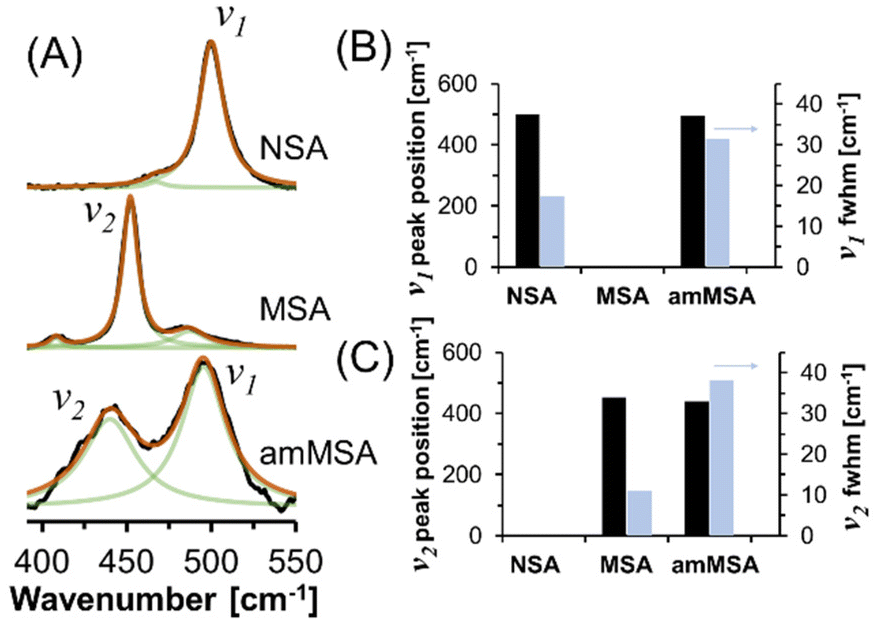 | ||
| Fig. 8 (A) Further characterization of the products in ex situ samples via Raman spectroscopy showing the increase in FWHM of the amMSA peak in the unknown tetrahedral phase relative to the crystalline MSA and NSA reference spectra. The reference spectra were previously published.21 The fit components are shown in green, the summation of the components is shown in orange, and the data is shown in black. (B) and (C) Line shape parameters of prominent vibrational modes. The fwhm is shown on a second y-axis. The Raman spectra of crystalline MSA and NSA in part (A) and corresponding line fits in part (B) were adapted with permission from T.R. Graham et al., Inorg. Chem., 2020, 59, 6857–6865 (ref. 21). Copywrite 2020 American Chemical Society. Based on an analysis of the variation in the line shape to changes in the parameters, the uncertainty in peak position and fwhm are approximately 2 and 3 cm−1, respectively. Note that the amMSA sample is crystallizing into NSA, which explains the presence of the v1 band in that sample. | ||
A 27Al 3QMAS NMR spectrum was also acquired at 14.1 T to probe the unique spectroscopic signatures of the tetrahedral phase (Fig. 9). Acquisition of the 27Al 3QMAS NMR spectrum of the amorphous phase provided additional resolution. 3QMAS acquisitions utilize the correlation between the excitation of multiple quantum coherences and their subsequent conversion into single quantum coherences in NMR-active nuclei that are not spin ½ to produce spectra that are of greater resolution than single pulse direct excitation spectra.39,40 The 27Al 3QMAS NMR spectrum of the tetrahedral region of the amorphous tetrahedral phase is shown in Fig. 8. The 27Al 3QMAS NMR spectrum shows that the amorphous tetrahedral phase has at least two Al sites that have different peak shapes and locations in the F1 and F2 dimension. Based on the asymmetry parameter for the two sites, which are both near 0.7 and 0.8 for site 1 and site 2 respectively, and the quadrupolar coupling coefficient in excess of 2.9 and 4.7 MHz, respectively, these resonances are assigned to tetrahedral aluminum in a distorted environment, such as in a partially condensed Al(OH)nO4−n network, where n = 0, 1, 2 or 3.
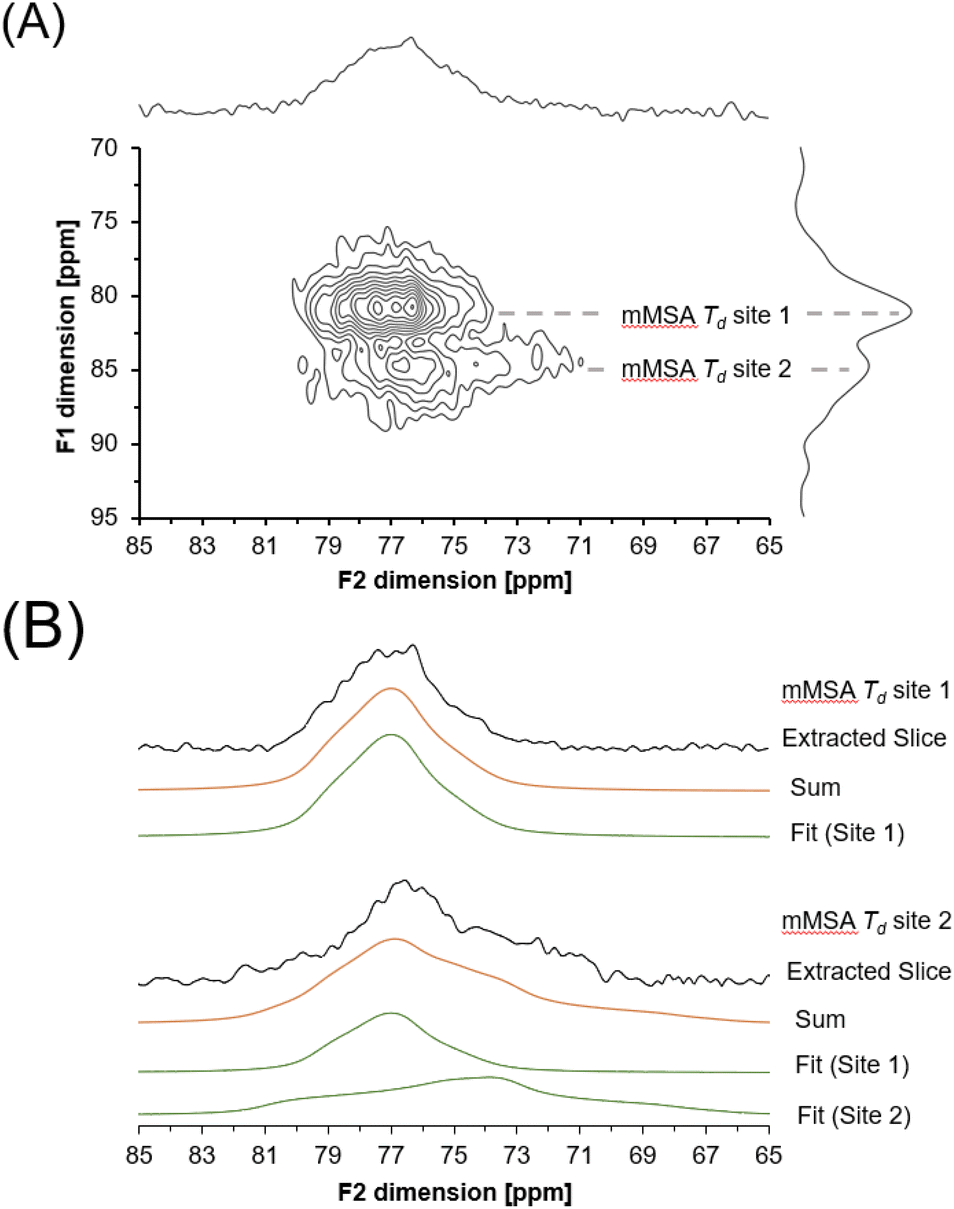 | ||
| Fig. 9 (A) 27Al 3QMAS NMR spectrum of amMSA showing the tetrahedral region. (B) Extracted slices and 3QMAS NMR line shape fits. Note that the slices were chosen at the maxima of each peak in the F1 dimension, and this leads to some contribution from site 1 in the extracted slice for site 2. For site 1, δiso (isotropic chemical shift) is 79 ppm, CQ (quadrupolar coupling coefficient) is 2.9 MHz, η (asymmetry parameter) is 0.7, LB (Lorentzian broadening) is 200 Hz and GB (Gaussian broadening) is 200 Hz. For site 2, δiso is 81 ppm, CQ is 4.7 MHz, η is 0.8, LB is 300 Hz and GB is 100 Hz. Based on an analysis of the variation in the line shape to changes in the parameters, the uncertainty in these parameters are estimated to be approximately 1 ppm for δiso, 0.3 MHz for CQ, 0.2 for η, 100 Hz for LB, and 100 Hz for GB. | ||
3.5. Determination of the quadrupolar line shape parameters for aMSA
Description of the quadrupolar line shape parameters of the two tetrahedral sites in amMSA provided insight into the local structure of Al in the reactive intermediate. The strategy to deconvolute the amMSA component in the 27Al NMR data required a multistep analysis. First, the two identifiable sites of amMSA viewable in the 27Al 3QMAS NMR data acquired at 14.T with 20 kHz of spinning were analyzed graphically to obtain the isotropic chemical shift and quadrupolar product (PQ) based on the apparent center of mass of the two peaks in ssNAKE. Then, deconvolutions of the 27Al MAS NMR spectrum of amMSA acquired at 14.1 T with 20 kHz of spinning rate were performed, the isotropic chemical shift was held constant, and PQ was used to constrain the possible values of CQ (quadrupolar coupling coefficient) and η (asymmetry parameter). The δiso (isotropic chemical shift), CQ and η parameters for amMSA, as well as NSA and AlOOH are reported in Table 1. Using the values of the δiso, CQ and η obtained from fitting the 27Al MAS NMR spectra of amMSA at 14.1 T, the 27Al MAS NMR spectra acquired at 7.05 T with 3.4 kHz of spinning during the in situ NMR study were simulated and inspected for agreement, as shown with similar comparisons for AlOOH and NSA in Fig. 10. The simulated and experimental spectra at 7.05 T were in good agreement with each other. Then to further validate the quadrupolar line shape parameters of the two Al sites in amMSA, extracted slices of the 3QMAS NMR spectra were also simulated using the same quadrupolar line shape parameters, and the resonances simulated with these line shape parameters are shown in Fig. 9. The extracted 3QMAS slices simulated with the regressed line shape parameters for the two Al sites in amMSA were also in good agreement with the experimental data.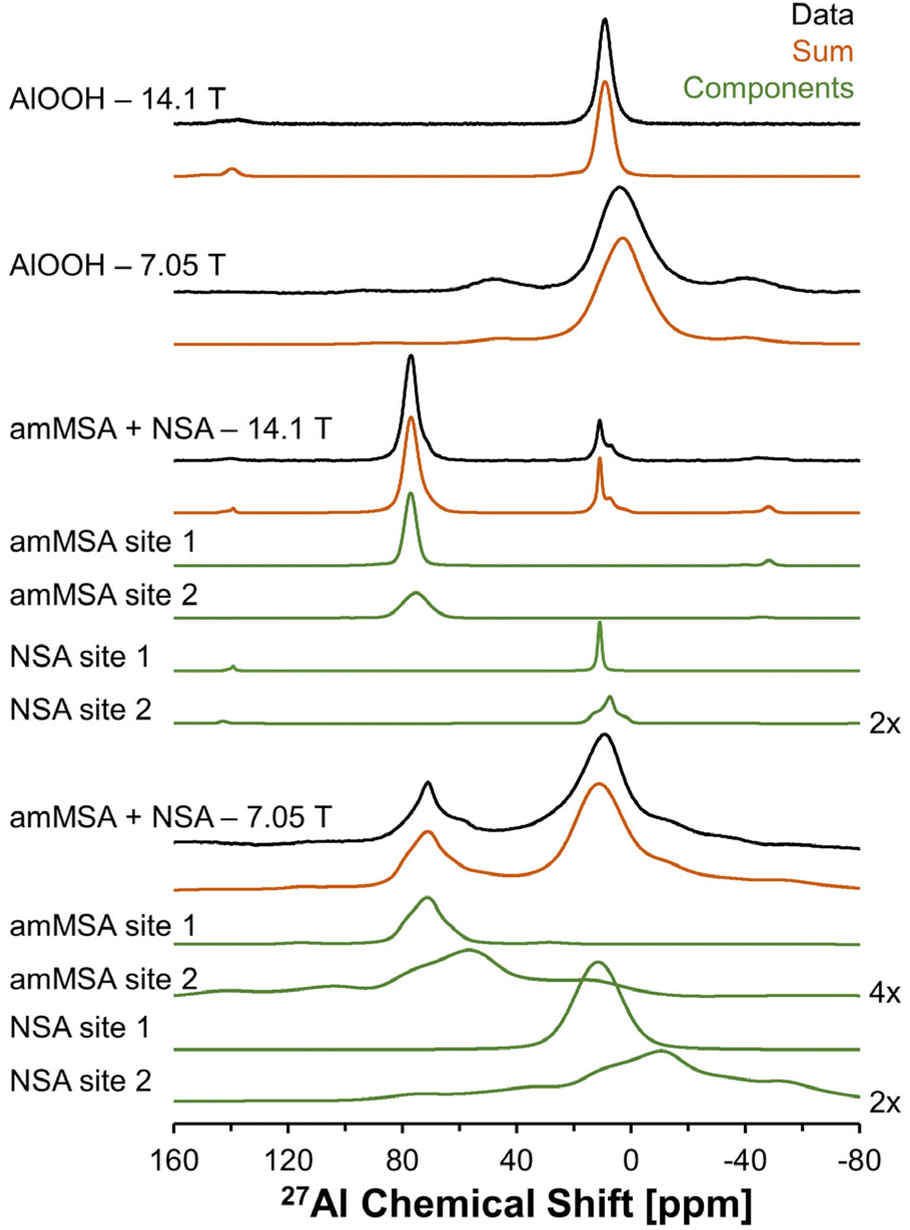 | ||
| Fig. 10 Summary of 27Al MAS NMR data acquired at a field of 14.1 or 7.05 T. The resonances were fit to acquire quadrupolar line shape parameters, shown in Table 1. The fit resonances were generated in ssNAKE using the finite spinning model, which allows for simulation of the spinning side band manifold which overlapped with the central transitions for the spectra at 7.05 T. Note that a select number of components are magnified by a factor annotated on the figure to allow for inspection of the broad components. The spectrum of amMSA + NSA at 14.1 T corresponds to the spectrum acquired immediately upon inserting the sample into the magnet, with spectra acquired at later reaction times shown in Fig. S3, in the ESI.† The spectrum of amMSA + NSA at 7.05 T corresponds to the spectrum acquired at 61.1 h as shown in Fig. 6. | ||
| 14.1 T and 20 kHz spinning rate | 7.05 T and 3.4 kHz spinning rate | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Site # | δ iso [ppm] | C Q [MHz] | η | LB [Hz] | GB [Hz] | Relative intensitya [%] | δ iso [ppm] | C Q [MHz] | η | LB [Hz] | GB [Hz] | Relative intensitya [%] |
| Note: δiso is the isotropic chemical shift, CQ is the quadrupolar coupling coefficient, η is the asymmetry parameter, LB is Lorentzian broadening and GB is Gaussian broadening. Based on an analysis of the variation in the line shape to changes in the parameters, the uncertainty in these parameters are estimated to be approximately 1 ppm for δiso, 0.3 MHz for CQ, 0.2 for η, 100 Hz for LB, and 100 Hz for GB.a The relative intensity of AlOOH, NSA, and amMSA sites are relative to the total octahedral and tetrahedral signal intensity in the respective phases.b The isotropic chemical shift of the spectra acquired at 7.05 T is offset by approximately a constant of 1 ppm, likely due to the use of a perdeuterated reference solution prepared from aluminum nitrate nonahydrate. | |||||||||||||
| AlOOH | 1 | 12 | 2.9 | 0.9 | 300 | 600 | 100 | 13 | 2.9 | 0.9 | 300 | 800 | 100 |
| NSA | 1 | 11 | 1.3 | 0.0 | 200 | 100 | 50 | 13 | 1.3 | 0.0 | 400 | 1200 | 50 |
| NSA | 2 | 14 | 4.5 | 0.9 | 200 | 200 | 50 | 16 | 4.5 | 0.9 | 1200 | 0 | 50 |
| amMSA | 1 | 80 | 2.9 | 0.7 | 200 | 400 | 57 | 81 | 2.9 | 0.7 | 400 | 0 | 55 |
| amMSA | 2 | 82 | 4.7 | 0.8 | 400 | 800 | 43 | 83 | 4.7 | 0.8 | 0 | 1200 | 45 |
A combination of XRD, Raman spectroscopy and high-field NMR spectroscopy indicates that the Al species in the amorphous tetrahedral phase exhibit Al coordination environments similar to Al(OAl)3(OH) in crystalline MSA, but the phase lacks the long range order necessary for diffraction peaks to appear in XRD. While prior research has shown that the crystalline tetrahedral MSA phase has an apparent chemical shift near 71 ppm and a well-defined quadrupolar line shape,21 in contrast, the XRD-amorphous tetrahedral phase has an less-defined quadrupolar line shape with a peak maxima near 80 ppm and a prominent shoulder at 74 ppm as shown in Fig. 10. The octahedral region of the XRD-amorphous phase was also inspected, but only a pair of resonances matching the line shape of NSA was found. The contrast in the location of the Al site assigned to Al(OAl)3 in the amorphous phase in the 3QMAS spectra indicates that the chemical environment around this unit is distinct from Al in MSA.21 As shown in Fig. S6 and S7 in the ESI,†in situ23Na MAS NMR spectroscopy and 1H MAS NMR spectroscopy at 7.05 T indicated that the sodium, water and hydroxide in the structure are undergoing chemical exchange at elevated temperatures between amMSA and the solution component because only a single resonance is observable. At temperatures of 30 °C, while there is evidence for a broad 23Na MAS NMR component attributed to a solid phase, the water and hydroxide in the structures are still undergoing chemical exchange and only one, ensemble 1H MAS NMR resonance is observable. We hypothesize that the difference in the chemical environment between MSA and amorphous tetrahedral phase is due to Al and Na vacancies, or other defect structures that result in poor long-range order. Based on the analysis, the amorphous tetrahedral phase is called amorphous monosodium aluminate hydrate (amMSA).
This observation of the structure and transformation of amMSA helps advance our knowledge of sodium aluminate hydrate crystallization in several ways. (i) A key insight into the transformation can be inferred from the presence of tetrahedral Al after physical mixing of AlOOH with NaOH·H2O, which indicates that the octahedral to tetrahedral transformation is spontaneous when an interface between those phases is present. Following dissolution of AlOOH, which goes to completion and thus eliminates the AlOOH-solution interface, a second chemically-distinct interface is formed from the precipitation of the tetrahedral aluminate solution species. This subsequent interface is composed of tetrahedrally-coordinated Al. (ii) Given the presence of mu-oxo bonds in the amorphous tetrahedral phase as demonstrated by the v2 band in the Raman spectra in Fig. 7, the concentration or activity of water is dependent on both the equilibrium between the monomeric and dimeric aluminates in solution, and polycondensation of solution state aluminates into the amorphous tetrahedral phase. Additional work to understand the precipitation of solution-state species will seek to link the dependence of water activity to the mechanisms of polycondensation and oligomerization mechanisms. (iii) Lastly, the precipitation of the amorphous phase after heating for 2 days is much faster than the month-long precipitation processes observed in a neutron pairwise distribution study of a similar composition prepared via dissolution of aluminum wire in concentrated sodium hydroxide solutions.16 The solution prepared via dissolution of aluminum wire in concentrated sodium hydroxide remained stable for weeks to months at room temperature,16 but in the current system studied at 80 °C the precipitation of the amorphous phase occurs at a faster rate, resulting in phase separation of the polymerized amorphous tetrahedral phase.
4. Conclusions
The in situ27Al MAS NMR experiments, coupled with supporting ex situ X-ray diffraction, Raman spectroscopy and 27Al 3QMAS NMR results, identify the emergence of a metastable amorphous sodium aluminate precipitate in which the Al-coordination is tetrahedral. For the compositional space studied here, the amorphous phase mediates the change in coordination from tetrahedral solution-state aluminates to the octahedral aluminate phase in the final phase of the reaction. The presence of a solid amorphous intermediate suggests that the coordination change through which NSA is formed occurs not in the bulk solution but either within the amorphous solid or at its interface. Thus the observation of the intermediate amorphous tetrahedral phase is the first observation of Al species in significant quantities beyond end member species in caustic Na2O : Al2O3 : H2O systems (e.g., Al(OH)4− and gibbsite in (re)crystallization processes, and MSA and NSA in solid-state recrystallization processes).21,41Conflicts of interest
The authors declare no conflicts of interest at the time of submission.Acknowledgements
This research was supported by IDREAM (Interfacial Dynamics in Radioactive Environments and Materials), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES). XRD, Raman spectroscopy, and NMR spectroscopy were performed using facilities at the Environmental Molecular Science Laboratory (EMSL, grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research at Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram national laboratory operated for DOE by Battelle Memorial Institute operating under Contract No. DE AC05-76RL0-1830. The 14.1 T NMR spectrometer was acquired with support from the BES Chemical Sciences, Geosciences, & Biosciences (CSGB) Division. Dave Bazak (PNNL) is thanked for helpful discussions regarding the use of ssnAKE to model quadrupolar line shapes with finite spinning models. The authors thank the anonymous peer reviewers for constructive comments that significantly improved the manuscript.References
- H. R. Watling, P. M. Sipos, L. Byrne, G. T. Hefter and P. M. May, Raman, IR, and, 27Al MAS-NMR Spectroscopic Studies of Sodium (Hydroxy)Aluminates, Appl. Spectrosc., 1999, 53(4), 415–422, DOI:10.1366/0003702991946875.
- P. Sipos, The Structure of Al(III) in Strongly Alkaline Aluminate Solutions—A Review, J. Mol. Liq., 2009, 146(1–2), 1–14, DOI:10.1016/j.molliq.2009.01.015.
- J.-P. Jolivet, C. Chanéac, D. Chiche, S. Cassaignon, O. Durupthy and J. Hernandez, Basic Concepts of the Crystallization from Aqueous Solutions: The Example of Aluminum Oxy(Hydroxi)des and Aluminosilicates, C. R. Geosci., 2011, 343(2), 113–122, DOI:10.1016/j.crte.2010.12.006.
- M. Weinberger, M. Schneider, V. Zabel, D. Müller and W. Geßner, Nonanatrium-Bis(Hexahydroxoaluminat)-Trihydroxid-Hexahydrat (Na9[Al(OH)6]2(OH)3 · 6H2O) – Kristallstruktur, NMR-Spektroskopie und thermisches Verhalten, Z. Anorg. Allg. Chem., 1996, 622(19), 1799–1805, DOI:10.1002/zaac.19966221027.
- V. Zabel, M. Schneider, M. Weinberger and W. Gessner, Nonasodium Bis(Hexahydroxoaluminate) Trihydroxide Hexahydrate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 747–749, DOI:10.1107/S0108270195015277.
- S. Ma, S. Zheng, Y. Zhang and Y. Zhang, Phase Diagram for the Na2O-Al2O3-H2O System at 130 °C, J. Chem. Eng. Data, 2007, 52(1), 77–79, DOI:10.1021/je060244k.
- Y. Zhang, Y. Li and Y. Zhang, Phase Diagram for the System Na2O-Al2O3-H2O at High Alkali Concentration, J. Chem. Eng. Data, 2003, 48(3), 617–620, DOI:10.1021/je025611g.
- Y. Zhang, S. Zheng, H. Du, S. Wang and Y. Zhang, Solubility of Al2O3 in the Na2O-Al2O3-H2O-CH3OH System at (30 and 60) °C, J. Chem. Eng. Data, 2010, 55(3), 1237–1240, DOI:10.1021/je900611g.
- G. Qiu and N. Chen, Phase Study of the System Na2O-Al2O3-H2O, Can. Metall. Q., 1997, 36(2), 111–114, DOI:10.1016/S0008-4433(96)00044-4.
- S. T. Mergelsberg, M. Dembowski, M. E. Bowden, T. R. Graham, M. Prange, H.-W. Wang, X. Zhang, O. Qafoku, K. M. Rosso and C. I. Pearce, Cluster Defects in Gibbsite Nanoplates Grown at Acidic to Neutral PH, Nanoscale, 2021, 13(41), 17373–17385, 10.1039/D1NR01615F.
- J. Z. Hu, X. Zhang, N. R. Jaegers, C. Wan, T. R. Graham, M. Hu, C. I. Pearce, A. R. Felmy, S. B. Clark and K. M. Rosso, Transitions in Al Coordination during Gibbsite Crystallization Using High-Field 27Al and 23Na MAS NMR Spectroscopy, J. Phys. Chem. C, 2017, 121(49), 27555–27562, DOI:10.1021/acs.jpcc.7b10424.
- M. Henry, J. P. Jolivet and J. Livage, Aqueous Chemistry of Metal Cations: Hydrolysis, Condensation and Complexation, 1992, pp. 153–206. DOI:10.1007/BFb0036968.
- X. Zhang, W. Cui, J. Z. Hu, H. W. Wang, M. P. Prange, C. Wan, N. R. Jaegers, M. Zong, H. Zhang and C. I. Pearce, et al., Transformation of Gibbsite to Boehmite in Caustic Aqueous Solution at Hydrothermal Conditions, Cryst. Growth Des., 2019, 19(10), 5557–5567, DOI:10.1021/acs.cgd.9b00468.
- T. R. Graham, J. Z. Hu, X. Zhang, M. Dembowski, N. R. Jaegers, C. Wan, M. Bowden, A. S. Lipton, A. R. Felmy and S. B. Clark, et al., Unraveling Gibbsite Transformation Pathways into LiAl-LDH in Concentrated Lithium Hydroxide, Inorg. Chem., 2019, 58(18), 12385–12394, DOI:10.1021/acs.inorgchem.9b02000.
- M. Dembowski, M. P. Prange, M. Pouvreau, T. R. Graham, M. E. Bowden, A. N'Diaye, G. K. Schenter, S. B. Clark, A. E. Clark and K. M. Rosso, et al., Inference of Principal Species in Caustic Aluminate Solutions through Solid-State Spectroscopic Characterization, Dalton Trans., 2020, 49(18), 5869–5880, 10.1039/D0DT00229A.
- H. W. Wang, T. R. Graham, E. Mamontov, K. Page, A. G. Stack and C. I. Pearce, Countercations Control Local Specific Bonding Interactions and Nucleation Mechanisms in Concentrated Water-in-Salt Solutions, J. Phys. Chem. Lett., 2019, 10(12), 3318–3325, DOI:10.1021/acs.jpclett.9b01416.
- Y. Wang, D. Song, Y. Zhou, C. Cheng, Y. Zhang, C. I. Pearce, Z. Wang, S. B. Clark, J. Zhu and K. M. Rosso, et al., Molecular Examination of Ion-Pair Competition in Alkaline Aluminate Solutions Using In Situ Liquid SIMS, Anal. Chem., 2021, 93(2), 1068–1075, DOI:10.1021/acs.analchem.0c04070.
- R. O. Grishchenko and A. L. Emelina, Synthesis and Thermochemical Characteristics of Na2O·Al2O3·2.5H2O, Russ. J. Phys. Chem. A, 2013, 87(1), 1–5, DOI:10.1134/S0036024413010081.
- S. F. Dec, G. E. Maciel and J. J. Fitzgerald, Solid-State 23Na and 27Al MAS NMR Study of the Dehydration of Na2O·Al2O3·3H2O, J. Am. Chem. Soc., 1990, 112(25), 9069–9077, DOI:10.1021/ja00181a006.
- M. Weinberger, M. Schneider, W. H. Geßner and D. Müller, Die Kristallstruktur Des Natriumoxohydroxoaluminathydrates Na2[Al2O3(OH)2] · 1,5 H2O, Z. Anorg. Allg. Chem., 1995, 621(4), 679–684, DOI:10.1002/zaac.19956210430.
- T. R. Graham, R. Gorniak, M. Dembowski, X. Zhang, S. B. Clark, C. I. Pearce, A. E. Clark and K. M. Rosso, Solid-State Recrystallization Pathways of Sodium Aluminate Hydroxy Hydrates, Inorg. Chem., 2020, 59(10), 6857–6865, DOI:10.1021/acs.inorgchem.0c00258.
- T. R. Graham, M. Dembowski, J. Z. Hu, N. R. Jaegers, X. Zhang, S. B. Clark, C. I. Pearce and K. M. Rosso, Intermediate Species in the Crystallization of Sodium Aluminate Hydroxy Hydrates, J. Phys. Chem. C, 2020, 124(23), 12337–12345, DOI:10.1021/acs.jpcc.0c00205.
- W. Gessner, M. Weinberger, D. Müller, L. P. Ni and O. B. Chaljapina, Zur Kenntnis Der Kristallinen Phasen in Den Systemen M12O-Al2O3-H2O (M1 = K, Na), Z. Anorg. Allg. Chem., 1987, 547(4), 27–44, DOI:10.1002/zaac.19875470404.
- J. Z. Hu, M. Y. Hu, Z. Zhao, S. Xu, A. Vjunov, H. Shi, D. M. Camaioni, C. H. F. Peden and J. A. Lercher, Sealed Rotors for in Situ High Temperature High Pressure MAS NMR, Chem. Commun., 2015, 51(70), 13458–13461, 10.1039/C5CC03910J.
- R. A. Peterson, E. C. Buck, J. Chun, R. C. Daniel, D. L. Herting, E. S. Ilton, G. J. Lumetta and S. B. Clark, Review of the Scientific Understanding of Radioactive Waste at the U.S. DOE Hanford Site, Environ. Sci. Technol., 2018, 52(2), 381–396, DOI:10.1021/acs.est.7b04077.
- D. L. Herting, J. G. Reynolds and W. B. Barton, Conversion of Coarse Gibbsite Remaining in Hanford Nuclear Waste Tank Heels to Solid Sodium Aluminate [NaAl(OH)4·1.5H2O], Ind. Eng. Chem. Res., 2014, 53(36), 13833–13842, DOI:10.1021/ie5014212.
- X. Zhang, P. L. Huestis, C. I. Pearce, J. Z. Hu, K. Page, L. M. Anovitz, A. B. Aleksandrov, M. P. Prange, S. Kerisit and M. E. Bowden, et al., Boehmite and Gibbsite Nanoplates for the Synthesis of Advanced Alumina Products, ACS Appl. Nano Mater., 2018, 1(12), 7115–7128, DOI:10.1021/acsanm.8b01969.
- X. Zhang, W. Cui, K. L. Page, C. I. Pearce, M. E. Bowden, T. R. Graham, Z. Shen, P. Li, Z. Wang and S. Kerisit, et al., Size and Morphology Controlled Synthesis of Boehmite Nanoplates and Crystal Growth Mechanisms, Cryst. Growth Des., 2018, 18(6), 3596–3606, DOI:10.1021/acs.cgd.8b00394.
- C. V. Chandran, C. E. A. Kirschhock, S. Radhakrishnan, F. Taulelle, J. A. Martens and E. Breynaert, Alumina: Discriminative Analysis Using 3D Correlation of Solid-State NMR Parameters, Chem. Soc. Rev., 2019, 48(1), 134–156, 10.1039/C8CS00321A.
- H. Stephen and T. Stephen, Solubilities of Inorganic and Organic Compounds, Vol. 1, Macmillan, New York, 1963 Search PubMed.
- D. S. Raiford, C. L. Fisk and E. D. Becker, Calibration of Methanol and Ethylene Glycol Nuclear Magnetic Resonance Thermometers, Anal. Chem., 1979, 51(12), 2050–2051, DOI:10.1021/ac50048a040.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, SsNake: A Cross-Platform Open-Source NMR Data Processing and Fitting Application, J. Magn. Reson., 2019, 301, 56–66, DOI:10.1016/j.jmr.2019.02.006.
- H. Jacobs and U. Metzner, Ungewöhnliche H-Brückenbindungen in Natriumhydroxidmonohydrat: Röntgen- und Neutronenbeugung an NaOH · H2O bzw. NaOD · D2O, Z. Anorg. Allg. Chem., 1991, 597(1), 97–106, DOI:10.1002/zaac.19915970113.
- D. Balzar and H. Ledbetter, Voigt-Function Modeling in Fourier Analysis of Size- and Strain-Broadened X-Ray Diffraction Peaks, J. Appl. Crystallogr., 1993, 26(1), 97–103, DOI:10.1107/S0021889892008987.
- J. Brus, S. Abbrent, L. Kobera, M. Urbanova and P. Cuba, Chapter Two – Advances in 27Al MAS NMR Studies of Geopolymers, ed. Webb, Annual Reports on NMR Spectroscopy, Academic Press, 2016, vol. 88, pp. 79–147. DOI:10.1016/bs.arnmr.2015.11.001.
- M. Haouas, F. Taulelle and C. Martineau, Recent Advances in Application of 27Al NMR Spectroscopy to Materials Science, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 94-95, 11–36, DOI:10.1016/j.pnmrs.2016.01.003.
- M. Kenisarin and K. Mahkamov, Salt Hydrates as Latent Heat Storage Materials: Thermophysical Properties and Costs. Solar energy materials and solar cells, North-Holland, Amsterdam, 2016, 35, 255–286, DOI:10.1016/j.solmat.2015.10.029.
- P. Sipos, G. Hefter and P. M. May, 27Al NMR and Raman Spectroscopic Studies of Alkaline Aluminate Solutions with Extremely High Caustic Content - Does the Octahedral Species Al(OH)63- Exist in Solution?, Talanta, 2006, 70(4), 761–765, DOI:10.1016/j.talanta.2006.02.008.
- A. Medek, J. S. Harwood and L. Frydman, Multiple-Quantum Magic-Angle Spinning NMR: A New Method for the Study of Quadrupolar Nuclei in Solids, J. Am. Chem. Soc., 1995, 117(51), 12779–12787, DOI:10.1021/ja00156a015.
- L. Frydman, Fundamentals of Multiple-Quantum Magic-Angle Spinning NMR on Half-Integer Quadrupolar Nuclei, Encyclopedia of Nuclear Magnetic Resonance, 2002, 9, 262–274, DOI:10.1002/chin.200332288.
- T. R. Graham, M. Dembowski, E. Martinez-Baez, X. Zhang, N. R. Jaegers, J. Hu, M. S. Gruszkiewicz, H. W. Wang, A. G. Stack and M. E. Bowden, et al., In Situ 27Al NMR Spectroscopy of Aluminate in Sodium Hydroxide Solutions above and below Saturation with Respect to Gibbsite, Inorg. Chem., 2018, 57(19), 11864–11873, DOI:10.1021/acs.inorgchem.8b00617.
Footnote |
| † Electronic supplementary information (ESI) available: Composition calculations, additional characterization of the boehmite starting material, 27Al MAS NMR experiments at 7.05 T in which the excitation pulse length is nutated, 27Al MAS NMR spectroscopy at 14.1 T demonstrating the lack of pentahedral aluminate intermediates and the conserved tetrahedral line shape during the crystallization of amMSA into NSA, SEM micrographs, additional Raman spectra, and additional 1H and 23Na MAS NMR spectra. See DOI: https://doi.org/10.1039/d2qi01642g |
| This journal is © the Partner Organisations 2022 |