
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A conjugated diosma-octacyclic complex and its mixed-valence singly reduced state†
Yu Xuan
Hu‡
a,
Qianqian
Deng‡
bc,
Ya-Ping
Ou‡
d,
Xiaofei
Yang
a,
Jing
Zhang
a,
Emily K.
Garrett
 e,
Jun
Zhu
*bc,
Sheng Hua
Liu
*a and
František
Hartl
*e
e,
Jun
Zhu
*bc,
Sheng Hua
Liu
*a and
František
Hartl
*e
aKey Laboratory of Pesticide and Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: chshliu@mail.ccnu.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
cFujian Provincial Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: jun.zhu@xmu.edu.cn
dKey Laboratory of Functional Metal-Organic Compounds of Hunan Province, Key Laboratory of Functional Organometallic Materials of Hunan Province College, College of Chemistry and Material Science, Hengyang Normal University, Hengyang, P. R. China
eDepartment of Chemistry, University of Reading, Whiteknights, Reading RG6 6DX, UK. E-mail: f.hartl@reading.ac.uk
First published on 30th September 2022
Abstract
In this work two dicationic diosma-octacyclic complexes have successfully been synthesized and fully characterized. One of them, with aromatic osmapentalene termini linked by the non-aromatic osmafuran moiety to the rigid naphthalenediolate bridge, represents the first example of a fused, fully conjugated dimetalla-octacyclic complex. A reference complex with more flexible biphenolate in the bridging position was also prepared. Their redox and electronic properties were investigated by combined methods of cyclic voltammetry and UV-vis-NIR–IR spectroelectrochemistry, supported by density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations. The rigid octacyclic complex forms a stable singly reduced mixed-valence species with the spin density localized at one of the osmapentalene termini.
Introduction
Analysis and understanding of unconventional aromaticity in organometallic compounds (Hückel, Craig and Möbius types) has long been in the center of interest for both experimental chemists and theoreticians. The chemistry of metallaaromatics has been developing rapidly and new challenges have been identified.1–10 Recently, polycyclic metallaaromatic compounds such as metallanaphthalenes,11–14 metallanaphthalynes,15,16 metalla-anthracene,17,18 metallafluoranthenes,19 metallaanthracynes,20,21 and carbolong complexes22–31 have widely been studied due to excellent thermodynamic stability and outstanding performance involving charge transfer,32 NIR luminescence22 photothermal effect,33–35 photovoltaics,36,37 photoactive therapy38 and catalysis.39To date, most of these complexes contain only a single transition metal in the polycyclic skeleton. Whilst abundant transition metal–organic dinuclear complexes have yielded unusually brilliant results in catalysis, magnetism, electronic communication, and optical materials, metallaaromatic complexes containing two metal centres (including dimetallabenzenes) have been less developed.40,41
In 2014, Xia and co-workers synthesised metal-bridged tricyclic aromatic complex 2 (monocationic, a BF4− salt) through the nucleophilic addition of phenol to cationic osmapentalyne 1 (Scheme 1).42 The aromaticity of the red-highlighted core in 2 obtained support from the calculated negative values of nucleus-independent chemical shifts (NICS) for each of the three fused five-membered metalla-rings. In this work, the tricyclic, reportedly osmaaromatic42 moieties in 2 are interconnected by a conjugated bridge to form dicationic dinuclear complexes 3 and 4 (Scheme 1). Flexible 4,4′-biphenol was used as the nucleophilic reagent to form the osmafuran-appended biphenyl bridge in 3. Then, 2,6-naphthalenediol was chosen to prepare octacyclic complex 4 with a rigid bridge core. To the best of our knowledge, metallaaromatic compounds with an octacyclic skeleton are unprecedented in the literature, regarding both monometallic and dimetallic systems. Aromaticity of the molecular rings in both 3 and 4 was investigated by the recently developed electron density of delocalized bonds (EDDB) method43,44 (instead of the popular NICS calculations45–47) and the anisotropy of induced current density (AICD) analyses.48,49 Their redox and electronic properties and the nature of the singly reduced mixed-valence state were explored by cyclic voltammetry, in situ UV-vis and IR spectroelectrochemistry at variable temperature, and quantum mechanical calculations.
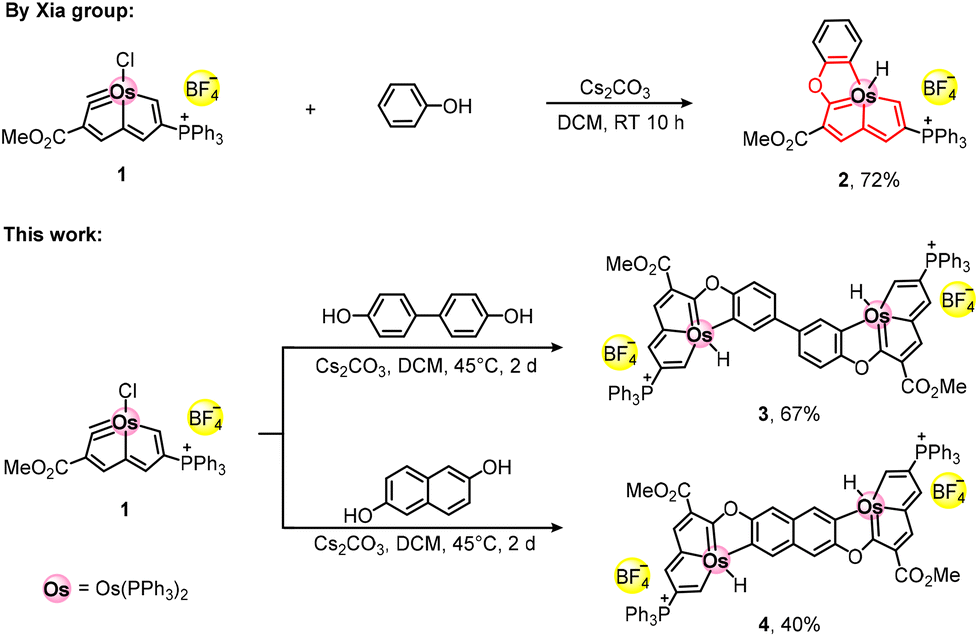 | ||
| Scheme 1 Syntheses of 2–4. | ||
Results and discussion
The synthetic routes to dinuclear 3 and 4 are shown in Scheme 1. Commercially available 4,4′-biphenol and slightly more than two equivalents of osmapentalyne 1 were refluxed in the presence of Cs2CO3 in dichloromethane for 2 days to yield 3 in 67% yield. Complex 4 was synthesized from 1 and 2,6-naphthalenediol under the same reaction conditions and isolated in 40% yield. In the solid state, both 3 and 4 are stable and can be stored in air for one year without any signs of decomposition. The composition and purity of dications 3 and 4 were revealed by their HRMS/ESI+ signals (Fig. S17 and S18, ESI†) and elemental analyses, respectively. The calculated mass of each molecular ion complies with the experimental value.The structures of 3 and 4 are presented in Fig. 1 and 2, respectively, and in Fig. S1 and S2 (ESI†). Crystallographic data are summarized in Tables S1–S3 (ESI†). Single crystals of 3 suited for X-ray diffraction were grown by slow diffusion of n-hexane into the dichloromethane solution of the complex. In the crystal, 3 is centrosymmetric and each osmium atom is shared by three five-membered rings. The sum of the angles in the five-membered ring Os1–C1–C2–C3–C4 is 540.0°, which is an ideal value indicating its perfect planarity. The same applies for the five-membered rings Os1–C4–C5–C6–C7 and Os1–C7–O1–C9–C8, where the deviation from the ideal value for ring planarity is very small, viz. 539.9° and 539.8°, respectively. The small torsion angles in the three osmacycles are illustrated in Fig. S1(a) (ESI†). The sum of the angles in the phenolate rings (C8–C13) is 719.5°, which is again close to the ideal value of a planar six-membered ring. The slight deviation from planarity of the polycyclic skeleton in the crystal of 3 seen in Fig. 1(b and c) has its origin in the twist and S-shaped distortion of the biphenyl core due to the flexible inter-ring C12–C12′ bond. However, the C12–C12′ bond length of 1.378 Å is significantly smaller compared to the value of 1.494 Å for the inter-ring C–C σ-bond in biphenyl and comparable with intra-ring distances in benzene (1.397 Å),50 proving strong π-conjugation in the biphenolate bridge in 3.
 | ||
| Fig. 1 (a) Crystal structure of 3·4CH2Cl2 (thermal ellipsoids set at 50% probability); (b) front view; (c) side view. Hydrogen atoms, BF4− counterions, CH2Cl2 molecules and phenyl substituents in PPh3 have been omitted for clarity. Selected bond lengths (Å) and angles (°) are presented in Table S2 and Fig. S2(a) (ESI†). | ||
 | ||
| Fig. 2 (a) Crystal structure of 4-BPh4·2CH2Cl2 (thermal ellipsoids set at 50% probability); (b) front view; (c) side view. Hydrogen atoms, anions, CH2Cl2 molecules and phenyl substituents in PPh3 have been omitted for clarity. Selected bond lengths (Å) and angles (°) are presented in Table S3 and Fig. S2(b) (ESI†). | ||
Diverse attempts to grow single crystals of dicationic 4 with BF4− counterions failed. Consequently, the counterions had to be exchanged for BPh4− by reacting 4 with Na[BPh4] in methanol. Suitable single crystals of 4-BPh4 were obtained by slow diffusion of n-hexane into the dichloromethane solution of the complex. The crystal structure of dication 4-BPh4 is also centrosymmetric. Each ring of the octacyclic skeleton exhibits excellent planarity, while sum of the ring angles in each half reaching 539.3°, 539.9°, 540.0° and 720.0° (Fig. 2). Differently from 3, the planar octacyclic structure of 4-BPh4 with the rigid naphthalene core is undistorted (Fig. 2b and c). The values of the small torsion angles in the three osmacycles are given in Fig. S1(b) (ESI†).
The three Os–C bonds in the five-membered rings of 3 (2.060–2.124 Å) are in average slightly longer compared with 2,42 but still falling within the range reported for planar Möbius aromatic osmapentalene (1.926–2.139 Å).22–30,51 The Os–C8 bond to the biphenolate linker is slightly longer (2.154 Å). Interestingly, all four Os–C bonds in 4-BPh4 (2.048–2.130 Å) containing the naphathalenediolate linker are shorter compared with 3, which reflects increased conjugation in the rigid octacyclic system, potentially facilitating electronic communication between the redox-active osmapentalene halves (see the Cyclic voltammetry and DFT parts hereinafter).
Complexes 3, 4 and 4-BPh4 were further characterized by nuclear magnetic resonance (NMR) spectroscopy (Fig. S6–S15, ESI†). In the 1H NMR spectra, the resonances in the high-field region (δ −3.33 ppm for 3 and δ −2.92 ppm for 4) are assigned to OsH. The low-field signals at δ 12.00 ppm for 3 and δ 12.22 ppm for 4 belong to OsCH, which complies with the chemical shifts of OsCH reported for aromatic osmapentalenes (11.93–12.46 ppm).42,51 The 13C{1H} NMR spectra of 3 and 4 exhibit low-field signals at δ 257.5 ppm for 3 and 257.0 ppm for 4, which are assigned to the OsCH resonance. Compared to 4, the 13C{1H} NMR spectrum of 4-BPh4 reveals a new quartet signal at δ 164.5 ppm assigned to BC in the BPh4− counterion.
Density functional theory (DFT) calculations of models 2′′, 3′′, and 4′′ (see Computational details in ESI†) have appreciably reproduced the experimental 1H NMR chemical shifts of OsCH (see Fig. S16 in ESI† for details). The downfield chemical shifts of the H1, H3 and H5 nuclei, ranging from 8.54 to 12.22 ppm, comply with the aromaticity of the two metallapentalene rings.22,23,42 The difference between the experimental and calculated OsH values is about 3 ppm; this is understandable, as the hydride is directly bound to the 5d transition metal, and its magnetic shielding can strongly be affected by spin–orbit (SO) effects.48
The planarity, high stability in air and the OsCH downfield shifts in the 1H NMR spectra of 3 and 4 indicate that these diosmium complexes are aromatic in the osmapentalene rings. To probe the aromaticity of the whole octacyclic system and compare the results with the data in the previous report by Xia and co-workers,42 electron density of delocalized bonds (EDDB)43,44 was used to investigate the aromaticity of these compounds instead of the alternative popular indices such as nucleus-independent chemical shifts (NICS),45,46 as the latter calculations feature certain limitations in evaluating the aromaticity of metallacycles.52 The strong local paramagnetic currents around heavy atoms have been reported to affect the value of NICS. Hence, NICS might not be a reliable criterion in evaluating ring aromaticity in osmapentalene derivatives53 and metal cluster systems.54,55 Note that the EDDB analysis is an advanced electronic criterion for aromaticity, proposed by Szczepanik et al., which has shown superiority in the quantification of electronic delocalization in aromatic rings.43,44 The EDDB analysis can efficiently separate the σ- and π-contributions from bonds in the molecules, and can also specifically investigate the electronic delocalization of specific loops in complex systems. With the popularity of corresponding computing programs, the EDDB analysis is often performed to quantitatively assess the strength of aromaticity in an increasing number of aromaticity studies.56–59 Therefore, the EDDB analysis has been used to evaluate the aromaticity of 2′, 3′ and 4′ (see Computational details in ESI†) in comparison with the tricyclic part of 2′ reported by Xia and co-workers42 (Fig. 3). The calculations show that the π-EDDB value for the peripheral 11-membered metal-bridged tricyclic aromatic system is 5.850 e. The corresponding values for the whole series 2′, 3′ and 4′ are slightly smaller (Fig. 3a). Detailed analyses of the π-EDDB values for each individual ring are presented in Fig. 3b. The π-EDDB values determined for the two five-membered metalla-rings of the osmapentalene moiety in 2′, 3′ and 4′ are very similar to those in the limited tricyclic system, indicating that the osmapentalene rings in 2′, 3′ and 4′ are aromatic. In contrast, the π-EDDB value (0.857 e) calculated for the osmafuran ring in the previously reported tricyclic system42 is slightly larger than those (0.687–0.697 e) for the osmafuran ring in 2′, 3′ and 4′ fused with the additional benzene ring of the bridge (Fig. 3b). As the π-EDDB values (0.687–0.857 e) for the osmafuran ring in all these complexes are much smaller than that (2.483 e) of furan (Fig. S20†), together with no distinguishable ring current of the osmafuran ring in the ACID plots (Fig. 4), we can draw the conclusion that the osmafuran rings in all these complexes, including the previously reported42 tricyclic species, is nonaromatic. In addition, the π-EDDB value (2.815 e) for the benzene ring in 4′ is much smaller than those in 2′ and 3′. Similarly, the EDDB and π-EDDB value (3.144 e) for the benzene ring in naphthalene is also significantly reduced in comparison with that (5.327 e) of benzene (Fig. S20†). The much lower π-EDDB value for the naphthalene linker in 4′ (Fig. 3) is understandable due to its mobile π-sextet in the Clar structures.60–62 For comparison, the overall EDDB values obtained from all contributing molecular orbitals in the 11-membered ring system, and the separate overall EDDB values for each ring are summarised in Fig. S19 (ESI†), leading to the same conclusions.
 | ||
| Fig. 3 EDDB analysis of the ring aromaticity in 2′, 3′ and 4′. (a) π-EDDB values for the 11-membered triple ring system. (b) Separate π-EDDB values for each ring, including the additional benzene ring in the bridge fused to the osmafuran moiety. | ||
 | ||
| Fig. 4 AICD plots of (a) 3′ and (b) 4′ visualizing π-ring current contributions. Current density vectors are plotted onto the AICD isosurface of 0.030 to indicate diatropic ring currents. The magnetic field vector is orthogonal with respect to the ring plane and points upwards. The aromatic rings exhibit clockwise diatropic circulations. | ||
In line with the EDDB calculations, AICD analyses48,49 of both mononuclear 2′ (Fig. S21, ESI†) and dinuclear 3′ and 4′ (Fig. 4) also demonstrate the nonaromaticity of the osmafuran rings and aromaticity of the other rings. Specifically, the clockwise diatropic ring currents are shown in the two osmapentalene rings as well as the central biphenyl a naphthalene rings in 3′ and 4′, respectively. In contrast, no distinct ring currents could be identified in the inserted osmafuran rings. This also applies for tetracyclic 2′. In summary, fully conjugated 4 cannot be classified as a so far unprecedented fully aromatic dimetalla-octacyclic compound. Different from the previously reported aromatic osmafurans,63 the osmafuran ring in 4 represents a non-aromatic linker between the aromatic osmapentalene termini and the naphthalene bridge.
The electronic absorption spectra of 2–4 are shown in Fig. 5. The absorption maximum of 2 at λ = 464 nm (with a shoulder at 520 nm) is close to the value of 461 nm reported by Xia and co-workers.42 The corresponding visible absorption of dinuclear 3 and 4 is slightly shifted to a lower energy and gains intensity. An absorption band between 350–400 nm is also observed for all three complexes, its intensity following the same trend, from weakly absorbing 2 to strongly absorbing 4.
 | ||
| Fig. 5 UV-vis absorption spectra of 2–4 measured in CH2Cl2 at T = 293 K. | ||
The electronic absorption of dinuclear 3 and 4 in the visible-near-UV region and the nature of the participating molecular orbitals, including the HOMO and LUMO (Fig. 6) examined in the electrochemical studies (vide infra), have been clarified by DFT and TD-DFT calculations conducted on their optimized models. The relevant data are presented in Tables S4–S6 (ESI†). The conjugated tetracyclic halves in 3 manifest a good planarity, but the large dihedral angle (35.8°) ascertained in the model structure further diminishes their electronic communication. In contrast, the diosma-octacyclic skeleton of optimized 4 is fully planar.
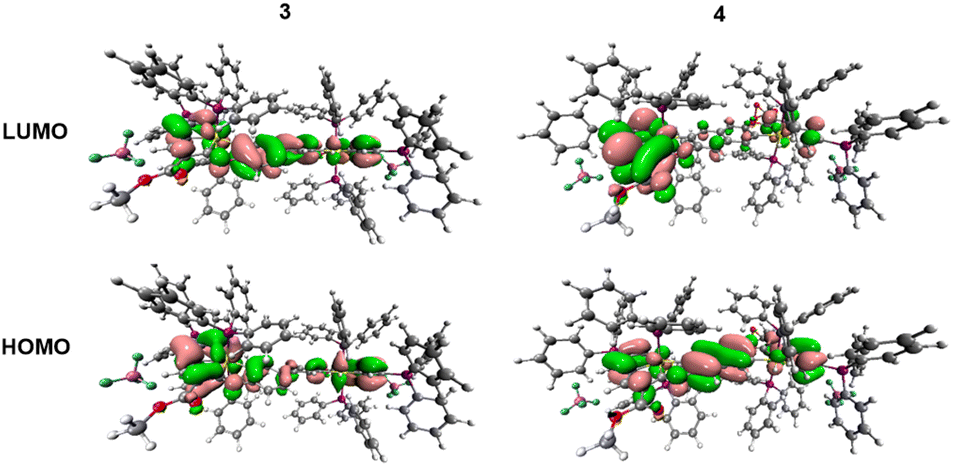 | ||
| Fig. 6 HOMO and LUMO of 3 (left) and 4 (right). | ||
The visible electronic absorption of planar 4 has been adequately reproduced by the TD-DFT results. The broad asymmetric band at 530 nm (at T = 293 K) encompasses two close-lying intense electronic transitions attributed to combined HOMO → LUMO and HOMO−1 → LUMO excitations corresponding to transfers of π-electron density from the naphthalenediolate bridge and/or one of the osmapentalene moieties to the other osmapentalene moiety (interpreted as LMCT/MMCT). The strong electronic absorption of 4 at 395 nm is dominated by a charge transfer from one osmapentalene unit to the triphenylphosphonium substituent, HOMO−1 → LUMO+2, and a charge transfer within the bis(osmapentalene) and central naphthalene π-system, HOMO−3 → LUMO, resembling the HOMO → LUMO excitation in the visible region.
For 3 the TD-DFT data have predicted strong absorption at 498 nm assigned as the HOMO → LUMO excitation within the bis(osmapentalene)-biphenolate backbone π-system, with a limited charge-transfer character. The higher-lying absorption in the visible region has a similar, even more delocalized nature due to the strong contribution from the HOMO−3 → LUMO excitation. On the other hand, the near-UV absorption is dominated by the HOMO−2 → LUMO+1 excitation that involves the biphenyl core and one of the osmapentalene moieties and can therefore be interpreted as a charge-transfer transition (LMCT). Although the TD-DFT data comply appreciably with the experimental near-UV-vis absorption of 3, some prudence needs to be taken, as the biphenolate twist angle in the optimized model structure is larger than unveiled in the crystal structure of the complex (Fig. 1).
The redox behaviour and electron-transfer properties of conjugated complexes 2–4 were essentially explored by cyclic voltammetry (CV) at variable temperature (Fig. 7, Fig. S3 in ESI,† and Table 1). Mononuclear 2 shows reversible reduction at E1/2 = −2.04 V at ambient temperature (Fig. S3a†), but its oxidation remains totally irreversible even at T = 233 K (Fig. 7(a)). Dinuclear 3 and 4 follow this behaviour. Differently from reference 2, however, they exhibit two separate reversible reduction steps (Fig. 7(b) and (c), respectively). The HOMO–LUMO gap (Eg) of 2.07 eV has been estimated64 from the cyclic voltammogram of complex 3. This value is nearly identical with 2.08 eV determined64 from the lowest electronic absorption of 3 (in CH2Cl2/n-Bu4NPF6, see below), which is shown by TD-DFT to encompass largely (86%) the HOMO → LUMO excitation (Table S4, ESI†). For 4 the difference of the optical band gap from the CV-based value of 2.14 eV remains limited to ca. −0.05 eV, which complies with the very close energies of the HOMO−1 and HOMO involved in the lowest-energy optical excitation (Tables S4 and S6, ESI†). The potential difference ΔE1/2 between the successive reduction waves (obtained by deconvolution) increases from 80 mV for 3 to 130 mV for 4. The LUMO of 4 resides largely on one of the osmapentalene termini while the HOMO is strongly delocalized over the naphthalenediolate bridge and both osmapentalene termini (Table S5, ESI†). This observation is signalling some weak electronic coupling between the Os centres in parent 3, which increases in rigid diosma-octacyclic 4. However, the presence of the non-aromatic osmafuran rings in the conjugated dinuclear compounds is the likely reason for the small ΔE1/2 value even in the latter case. The reversibility of the close-lying cathodic steps reveals the formation of stable mixed-valence (M-V) complexes in the 1e−-reduced state, which may, however, undergo redox disproportionation. This assumption is further supported by UV-vis spectroelectrochemistry (vide infra).
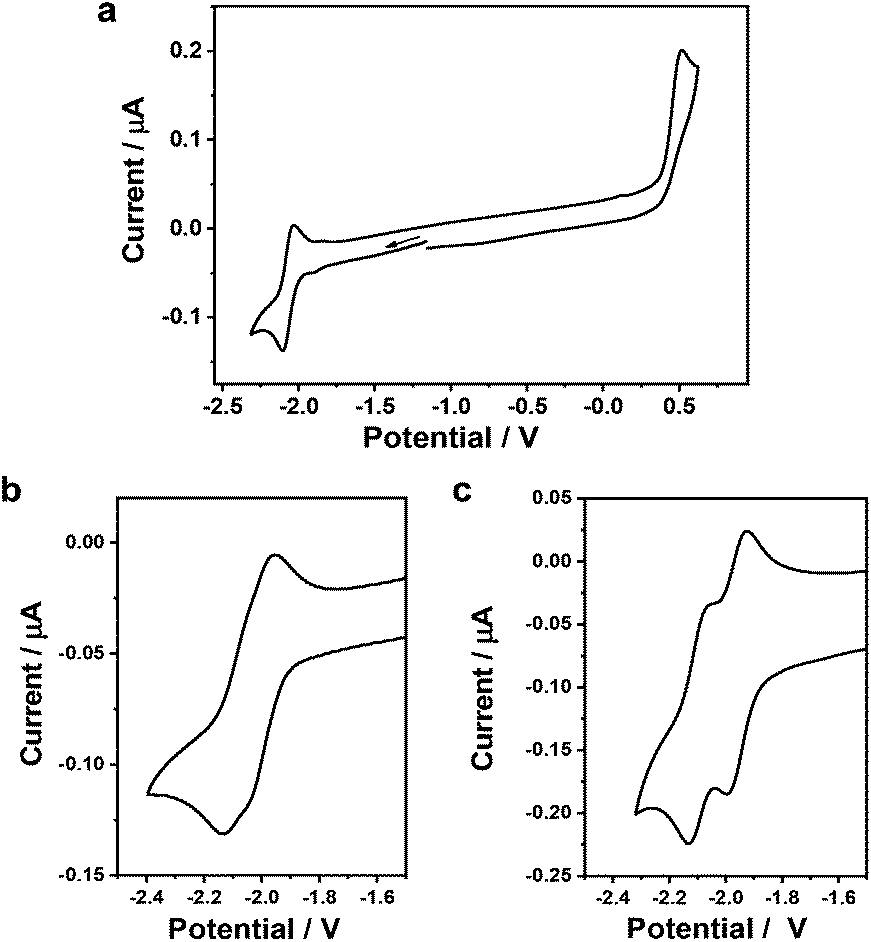 | ||
| Fig. 7 Cyclic voltammograms of 1 mM 2 (a), 3 (b) and 4 (c) in CH2Cl2/n-Bu4NPF6 at v = 100 mV s−1, T = 233 K. The potential scales correspond to the standard ferrocenium/ferrocene redox couple. | ||
| Osmapentalene | E 1/2 (red1), Volt | E 1/2 (red2), Volt | E p,a (oxid.), Volt |
|---|---|---|---|
| a Experimental conditions: dichloromethane/n-Bu4NPF6 and T = 233 K. The electrode potentials are referenced against the standard ferrocenium/ferrocene (Fc+/Fc) couple. | |||
| 2 | −2.04 | n/a | 0.57 |
| 3 | −2.03 | −2.11 | 0.34 |
| 4 | −1.96 | −2.09 | 0.43 |
The DFT description of singly reduced radical 4′′− (with the PPh3 groups in 4− replaced by PH3 for successful geometry optimization) has indeed exposed the α-HOSO and α-LUSO being localized on the opposite osmapentalene units (Table S7, ESI†) and the spin density being distributed mainly over one osmapentalene fragment (Fig. 8). This striking asymmetry (Fig. S5, ESI†), in combination with the moderate ΔE1/2 value (Table 1), permits to assign 4− as a class I rather than class II M-V system, with trapped valences according to the Robin–Day classification;65 although, most M-V systems have been based on distinct metal oxidation states.66–70 Recording of the electronic absorption spectrum of 4− was then attempted to identify a low-lying intervalence charge-transfer (IVCT) absorption, which would otherwise verify the class II M-V assignment.
 | ||
| Fig. 8 Spin density distribution in mixed-valence 4′′−. Contour values: ±0.04 (e bohr−3)1/2. | ||
UV-vis and IR spectroelectrochemical experiments with 2–4 to monitor their electrochemical reduction were conducted in non-coordinating dichloromethane at T = 233 K, using an OTTLE cell. UV-vis spectral changes recorded during the cathodic electron transfer steps are depicted in Fig. 9.
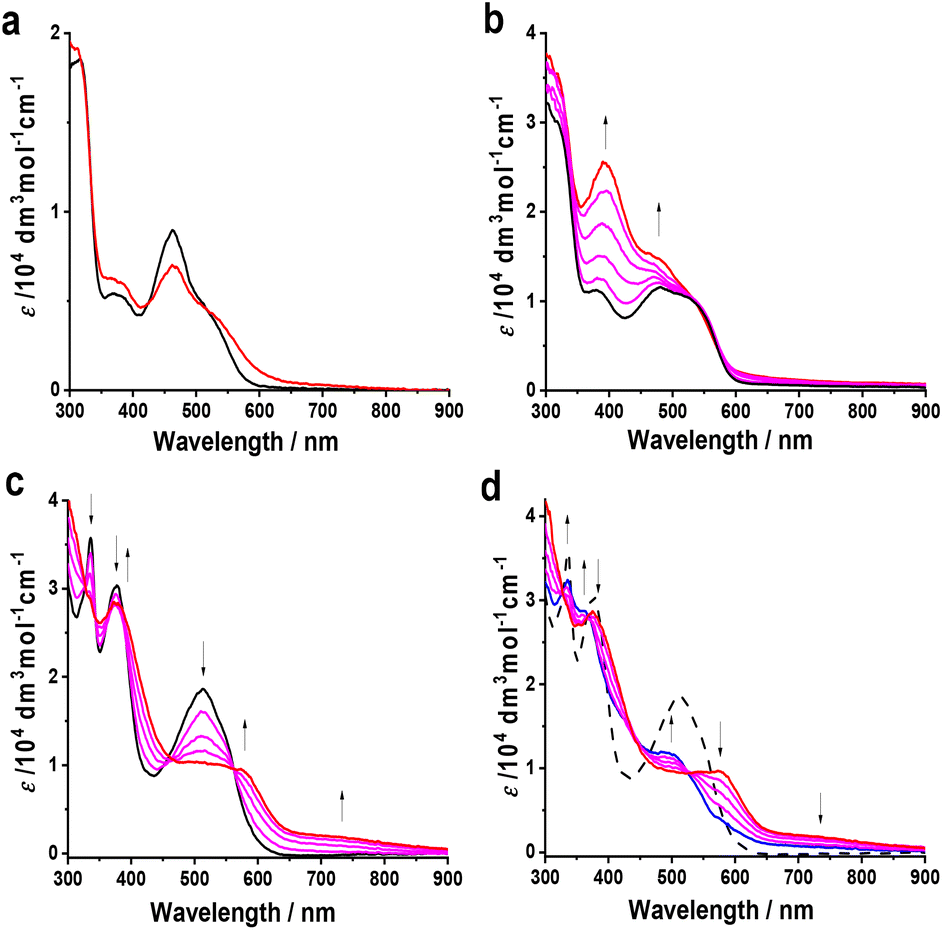 | ||
| Fig. 9 Spectral changes in the UV-vis-NIR absorption region recorded during the electrochemical reduction in a cryostatted OTTLE cell: 2 → 2− (a), 3 → {3}2− (b), 4 → 4− (c), 4− → {4}2− (d). Conditions: CH2Cl2/n-Bu4NPF6, T = 233 K, cparent = 2 mmol dm−3. | ||
The 1e− reduction of 2 in the chilled electrolyte at −2.04 V proved to be reversible only in the initial phase, in contrast to the reversible cathodic CV response (Fig. 7 and Table 1). The prominent absorption band at 463 nm was gradually decreasing, which was accompanied by the growth of new low-energy absorption between 550–650 nm tailing to 800 nm. Having reached ca. 50% conversion (as shown in Fig. 9(a)), the low-energy absorption of 2− began to disappear; subsequent reoxidation at a strongly positively shifted anodic counterwave did not lead to the recovery of parent 2. The secondary reduction product, denoted as {2}−, has not been identified.
On the contrary, the initial 1e− reduction of 4 to 4− was fully reversible at T = 233 K, as manifested by the persistent isosbestic points in the UV-vis absorption spectra (Fig. 9(c)) and smooth recovery of the parent absorption at a relatively small overpotential during the reverse anodic step. The strong absorption band of 4 at 508 nm was replaced by a half-reduced maximum at 570 nm and a broad absorption of low intensity between 650–900 nm. These characteristic features resemble the transient absorption observed for mononuclear 2− (Fig. 9(a)) prior to its irreversible transformation to {2}−, which complies with the localized valence of 4− as manifested by DFT calculations (Fig. 8). The parallel monitoring of the reduction of 4 to 4− in the IR fingerprint region revealed a small red shift of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) absorption of the ester group on the osmapentalene skeleton from 1718 to 1710 cm−1, accompanied by a significant gain of the absorption intensity (Fig. S4, ESI†). The apparent asymmetry of the new ν(CO) band at 1710 cm−1 reveals the presence of the underlying, diminished band at 1718 cm−1 also in valence-localized 4− where only one of the osmapentalene termini gets singly reduced. As demonstrated by Fig. 8, the affected ester group in 4− hardly bears spin density, in line with the limited decrease in the ν(CO) wavenumber. The consecutive cathodic step at −2.09 V (Table 1) converted 4− to a poorly known secondary species, {4}2−, showing a new absorption band at 492 nm on the foot of the strong near-UV absorption (Fig. 9(d)). Like {2}−, the reoxidation of {4}2− was found in the thin-layer cyclic voltammogram strongly positively shifted beyond the parent reduction wave and reproduced neither singly reduced 4− nor parent 4.
O) absorption of the ester group on the osmapentalene skeleton from 1718 to 1710 cm−1, accompanied by a significant gain of the absorption intensity (Fig. S4, ESI†). The apparent asymmetry of the new ν(CO) band at 1710 cm−1 reveals the presence of the underlying, diminished band at 1718 cm−1 also in valence-localized 4− where only one of the osmapentalene termini gets singly reduced. As demonstrated by Fig. 8, the affected ester group in 4− hardly bears spin density, in line with the limited decrease in the ν(CO) wavenumber. The consecutive cathodic step at −2.09 V (Table 1) converted 4− to a poorly known secondary species, {4}2−, showing a new absorption band at 492 nm on the foot of the strong near-UV absorption (Fig. 9(d)). Like {2}−, the reoxidation of {4}2− was found in the thin-layer cyclic voltammogram strongly positively shifted beyond the parent reduction wave and reproduced neither singly reduced 4− nor parent 4.
The stepwise electrochemical reduction of 3 was not resolved sufficiently due to the small ΔE1/2 value (Table 1) to permit the complete generation of singly reduced 3−. As shown in Fig. 9(b), the characteristic electronic absorption of 3− between 600–900 nm (cf.4− in Fig. 9(c)) only appears at the onset of the reduction wave at −1.73 V. The concomitant redox disproportionation of 3− results in the direct observation of the secondary reduction product, {3}2− (Fig. 9(b)), showing similar absorption features between 350–600 nm as {4}2− (Fig. 9(d)). The UV-vis spectroelectrochemical results have revealed that 3− and 4− are electronically close and contain the same chromophores. The twisted 4,4′-biphenolate and coplanar naphathalenediolate linkers in the parent complexes markedly control the weak to moderate electronic communication between the osmapentalene termini.
The electronic absorption of stable 4− in the visible region was analysed by TD-DFT calculations performed on its optimized model 4′′− (see Fig. S5 in ESI†) having the six PPh3 groups replaced by PH3. The data are summarized in Table 2 and Table S7 (ESI†). The weak absorption recorded between 650–900 nm (Fig. 9(c)) is dominated by the α-HOSO → α-LUSO+2 transition within the π-orbital system delocalized over the reduced osmapentalene moiety, also including a charge transfer to the phosphonium ring substituent. There is no evidence for a low-lying intense IVCT transition that would comply with the assignment of model 4′′− as a class II M-V complex, as discussed above at the cyclovoltammetric response of 4. The medium-intensity absorption band of 4− at 570 nm represents a π-electronic excitation close to the weak lowest-energy absorption, with the central naphthalene rings of α-LUSO+6 being more involved. The α-HOSO → α-LUSO transition (Table S7†) with an apparent IVCT character is shown to contribute significantly only to the higher-lying optical excitation around 450–500 nm. In general, the absence of a clear IVCT absorption band in the NIR region and the complex nature of the visible electronic absorption of 4− (represented by model 4′′−) do not meet the criteria for an intermediate class II M-V system marked by a low activation energy for facile interconversion of the distinct valences (electron-hopping) on the timescale of 10−14 s. Even though the rigid central naphthalenediolate linker in 4 may mediate a stronger electronic interaction between the osmapentalene termini compared to slightly twisted dinuclear 3, the singly reduced M-V radical species with the spin density localized at a distinct osmapentalene site (Fig. 8) can more reliably be classified as a class I valence-trapped complex.
| Excitation (percentage) | f | λ max (calc.)/nm | λ max (exp.)/nm (ε/104 × M−1 cm−1) |
|---|---|---|---|
| α-HOSO → α-LUSO+2 (82%) | 0.001 | 719 | ∼710 (0.195) |
| α-HOSO−3 → α-LUSO (28%) | 0.0001 | 663 | |
| α-HOSO → α-LUSO+6 (19%) | 0.092 | 474 | 570 (0.955) |
| α-HOSO → α-LUSO+6 (19%) | 0.091 | 453 | |
| β-HOSO → β-LUSO+ 2 (23%) | 0.023 | 427 | 440 (1.31) |
| α-HOSO → α-LUSO (55%) | |||
| α-HOSO → α-LUSO+6 (18%) | 0.197 | 423 | |
| β-HOSO → β-LUSO+2 (53%) | |||
| β-HOSO−8 → β-LUSO (15%) | 0.071 | 394 | 377 (2.82) |
| β-HOSO−2 → β-LUSO (15%) | |||
| α-HOSO−9 → α-LUSO (16%) | 0.028 | 391 | |
| β-HOSO−9 → β-LUSO (16%) | |||
| α-HOSO−1 → α-LUSO (11%) | 0.094 | 386 | |
| β-HOSO−2 → β-LUSO (11%) |
Conclusions
In summary, two dicationic conjugated diosma-octacyclic complexes 3 and 4 were successfully synthesized by a nucleophilic addition reaction of osmapentalyne with 4,4′-biphenol and 2,6-naphthalenediol, respectively. The downfield proton chemical shifts, AICD analysis and calculated EDDB values confirmed the aromaticity of the terminal osmapentalene units and the bridge core. In contrast, the osmafuran ring linking these aromatic regions is shown to be nonaromatic. Complex 4 is the first reported conjugated dimetalla-octacyclic compound. Cyclic voltammetry and DFT calculations reveal a moderate electronic communication between the osmapentalene termini mediated by the rigid naphthalenediolate core. The rare singly reduced mixed-valence species, 4−, was characterised by UV-vis and IR spectro-electrochemistry as a Robin–Day class I valence-trapped complex. This work has not only enriched the family of metallaaromatic (osmapentalene) complexes by novel structures, but also clarified their peculiar electronic nature, redox behaviour, and ability to undergo electronic communication between polycyclic redox centres, opening a vast new playground for exploring such phenomena in this field by experiment and theory in the years ahead.Author contributions
Funding acquisition: Sheng Hua Liu, František Hartl; syntheses: Yu Xuan Hu; investigation: Yu Xuan Hu, Qianqian Deng, Xiaofei Yang, Jing Zhang, Emily K. Garrett, Yaping Ou, František Hartl; supervision: Jun Zhu, Sheng Hua Liu, František Hartl: visualization: Yu Xuan Hu, Jing Zhang, František Hartl; writing – original draft: Yu Xuan Hu, František Hartl; writing – review & editing: Jun Zhu, Sheng Hua Liu, František Hartl.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21772054 and 22175069) and the Plan 111 Project (B17019). Funding was also raised by Spectroelectrochemistry Reading, a spin-out company of the University of Reading.References
- D. L. Thorn and R. Hoffman, Delocalization in Metallocycles, Nouv. J. Chim., 1979, 3, 39–45 CAS
.
- G. P. Elliott, W. R. Roper and J. M. Waters, Metallacyclohexatrienes or ‘Metallabenzenes.’ Synthesis of Osmabenzene Derivatives and X-ray Crystal Structure of [Os(CSCHCHCHCH)(CO)(PPh3)2], J. Chem. Soc., Chem. Commun., 1982, 811–813 RSC
- B. J. Frogly and L. J. Wright, Recent Advances in Metallaaromatics Chemistry, Chem. – Eur. J., 2018, 24, 2025–2038 CrossRef PubMed
- J. R. Bleeke, Metallabenzenes, Chem. Rev., 2001, 101, 1205–1228 CrossRef CAS PubMed
- J. Chen, H. H. Y. Sung, I. D. Willianms, Z. Lin and G. Jia, Synthesis and Characterization of a Rhenabenzyne Complex, Angew. Chem., Int. Ed., 2011, 50, 10675–10678 CrossRef CAS PubMed
- J. Chen and G. Jia, Recent Development in the Chemistry of Transition Metal-containing Metallabenzenes and Metallabenzynes, Coord. Chem. Rev., 2013, 257, 2491–2521 CrossRef CAS
- D. Chen, Y. Hua and H. Xia, Metallaaromatics Chemistry: History and Development, Chem. Rev., 2020, 120, 12994–13086 CrossRef CAS
- Z. Huang, Y. Zhang, W.-X. Zhang, J. Wei, S. Ye and Z. Xi, A Tris-spiro Metalla-aromatic System Featuring Craig-Möbius Aromaticity, Nat. Commun., 2021, 12, 1319–1325 CrossRef CAS
- S. Gupta, S. Su, Y. Zhang, P. Liu, D. J. Wink and D. Lee, Ruthenabenzene: A Robust Precatalyst, J. Am. Chem. Soc., 2021, 143, 7490–7500 CrossRef CAS PubMed
- J. Zhu, Open Questions on Aromaticity in Organometallics, Commun. Chem., 2020, 3, 161–163 CrossRef CAS
- M. Paneque, C. M. Posadas, M. L. Poveda, N. Rendón, V. Salazar, E. Oñate and K. Mereiter, Formation of Unusual Iridabenzene and Metallanaphthalene Containing Electron-Withdrawing Substituents, J. Am. Chem. Soc., 2003, 125, 9898–9899 CrossRef CAS PubMed
- M. Talavera, S. Bolaño, J. Bravo, J. Castro, S. García-Fontán and J. M. Hermida-Ramón, Formation of Indanone from an Iridanaphthalene Complex, Organometallics, 2013, 32, 4058–4060 CrossRef CAS
- M. Talavera, A. Peña-Gallego, J. L. Alonso-Gómez and S. Bolaño, Metallaaromatic Biaryl Atropisomers, Chem. Commun., 2018, 54, 10974–10976 RSC
- M. Talavera, J. Bravo, J. Castro, S. García-Fotán, J. M. Hermida-Ramón and S. Bolaño, Electronic Effects of Substituents on the Stability of the Iridanaphthalene Compound [IrCp*{=C(OMe)CH=C(o,-C6H4)(Ph)}(PMe3)]PF6, Dalton Trans., 2014, 43, 17366–17374 RSC
- G. He, J. Zhu, T. B. Wen, I. D. Williams, Z. Lin and G. Jia, A Metallanaphthalyne Complex from Zinc Reduction of a Vinylcarbyne Complex, Angew. Chem., Int. Ed., 2007, 46, 9065–9068 CrossRef CAS PubMed
- B. Liu, H. Xie, Q. Zhao, J. Chen, T. B. Wen, Z. Cao and H. Xia, Selective Synthesis of Osmanaphthalene and OsmaNaphthalyne by Intramolecular C-H Activation, Angew. Chem., Int. Ed., 2009, 48, 5461–5464 CrossRef CAS PubMed
- B. J. Frogly and L. J. Wright, A Metallaanthracene and Derived Metallaanthraquinone, Angew. Chem., Int. Ed., 2017, 56, 143–147 CrossRef
- Y. García-Rodeja and I. Fernández, Influence of the Transition-Metal Fragment on the Reactivity of Metallaanthracenes, Chem. – Eur. J., 2017, 23, 6634–6642 CrossRef PubMed
- Y. X. Hu, J. Zhang, X. Wang, Z. Lu, J. Yin, H. Xia and S. H. Liu, One-pot Syntheses of Irida-polycyclic Aromatic Hydrocarbons, Chem. Sci., 2019, 10, 10894–10899 RSC
- M.-X. Zhang, Z. Xu, T. Lu, J. Yin and S. H. Liu, A Visible-Light-Induced Strategy to Construct Osmanaphthaynes, Osmaanthracyne, and Osmaphenanthryne, Chem. – Eur. J., 2018, 24, 14891–14895 CrossRef CAS
- W. Ruan, T.-F. Leung, C. Shi, I. D. Williams, Z. Lin and G. Jia, Facile Synthesis of Polycyclic Metallaarynes, Chem. Sci., 2018, 9, 5994–5998 RSC
- C. Zhu, S. Li, M. Luo, J. Zhu, T. Wen, P. v. R. Schleyer and H. Xia, Stabilization of Anti-aromatic and Strained Five-membered Rings with a Transition Metal, Nat. Chem., 2013, 5, 698–703 CrossRef CAS
- C. Zhu, J. Zhu, X. Zhou, Q. Zhu, Y. Yang, T. Wen and H. Xia, Isolation of an Eleven-Atom Polydentate Carbon-Chain Chelate Obtained by Cycloaddition of a Cyclic Osmium Carbyne with an Alkyne, Angew. Chem., Int. Ed., 2018, 57, 3154–3157 CrossRef CAS
- C. Zhu, J. Wu, S. Li, Y. Yang, J. Zhu, X. Lu and H. Xia, Synthesis and Characterization of a Metallacyclic Framework with Three Fused Five-membered Rings, Angew. Chem., Int. Ed., 2017, 56, 9067–9071 CrossRef CAS PubMed
- C. Zhu, C. Yang, Y. Wang, G. Lin, Y. Yang, X. Wang, J. Zhu, X. Chen, X. Lu, G. Liu and H. Xia, CCCCC Pentadentate Chelates with Planar Möbius Aromaticity and Unique Properties, Sci. Adv., 2016, 2, e1601031 CrossRef PubMed
- C. Zhu and H. Xia, Carbolong Chemistry: A Story of Carbon Chain Ligands and Transition Metals, Acc. Chem. Res., 2018, 51, 1691–1700 CrossRef CAS PubMed
- Z. Lu, Q. Zhu, Y. Cai, Z. Chen, K. Zhuo, J. Zhu, H. Zhang and H. Xia, Access to Tetracyclic Aromatics with Bridgehead Metals via Matalla-click reactions, Sci. Adv., 2020, 6, eaay2535 CrossRef CAS PubMed
- Q. Zhuo, J. Lin, Y. Hua, X. Zhou, Y. Shao, S. Chen, Z. Chen, J. Zhu, H. Zhang and H. Xia, Multiyne Chains Chelating Osmium via Three Metalcarbon σ Bonds, Nat. Commun., 2017, 8, 1912–1918 CrossRef
- M. Luo, Y. Hua, K. Zhuo, L. Long, X. Lin and H. Xia, Carbolong Chemistry: Planar CCCCX-type (X = N, O, S) Pentadentate Chelates by Formal [3+1] Cycloadditions of Metalla-azirines with Terminal Alkynes, CCS Chem., 2020, 2, 758–763 Search PubMed
- Q. Zhuo, H. Zhang, Y. Hua, H. Kang, X. Zhou, X. Lin, Z. Chen, J. Lin, K. Zhuo and H. Xia, Constraint of a Ruthenium-Carbon Triple Bond to a Five-Membered Ring, Sci. Adv., 2018, 4, eaat0336 CrossRef PubMed
- S. Chen, L. Peng, Y. Liu, X. Cao, Y. Zhang and H. Xia, Conjugated Polymers based on Metallaaromatic Building Blocks, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203701119 CrossRef CAS PubMed
- R. Li, Z. Lu, Y. Cai, F. Jiang, C. Tang, W. Hong and H. Xia, Switching of Charge Transport Pathways via Delocalization Changes in Single-Molecule Metallacycles Junctions, J. Am. Chem. Soc., 2017, 139, 14344–14347 CrossRef CAS PubMed
- Z. Lu, Q. Lin, Y. Cai, S. Chen, J. Chen, W. Wu, X. He and H. Xia, Cylindrical NIR-Responsive Metallopolymer Containing Möbius Metalla-aromatics, ACS Macro Lett., 2018, 7, 1034–1038 CrossRef CAS PubMed
- H. Zhang, H. Zhao, K. Zhuo, Y. Hua, J. Chen, X. He, W. Weng and H. Xia, “Cabolong” Polymers with Near Infrared Triggered, Spatially Resolved and Rapid Self-Healing Properties, Polym. Chem., 2019, 10, 386–394 RSC
- Y. Chen, L. Yang, W. Zheng, P. Ouyang, W. Weng, X. He and H. Xia, Dynamic Polymer Network System Mediated by Radically Exchangeable Covalent Bond and Carbolong Complex, ACS Macro Lett., 2020, 9, 344–349 CrossRef CAS PubMed
- J. Wang, J. Li, Y. Zhou, C. Yu, H. Xia and H.-L. Wang, Tuning an Electrode Work Function Using Organometallic Complexes in Inverted Perovskite Solar Cells, J. Am. Chem. Soc., 2021, 143, 7759–7768 CrossRef CAS PubMed
- L. Liu, S. Chen, Y. Qu, Y. Tan, H. Xia and F. He, Nanographene-Osmapentalynes Complexes as a Cathode Interlayer in Organic Solar Cells Enhance Efficiency over 18%, Adv. Mater., 2021, 2101279 CrossRef CAS
- N. Lu, Z. Deng, J. Gao, C. Liang, H. Xia and P. Zhang, An Osmium-peroxo plex for Photoactive Therapy of Hypoxic Tumors, Nat. Commun., 2022, 13, 2245–2255 CrossRef CAS PubMed
- F. Cui, Y. Hua, Y. Lin, J. Fei, L. Gao, X. Zhao and H. Xia, Selective Difunctionalization of Unactivated Aliphatic Alkenes Enabled by a Metal-Metallaaromatic Catalytic System, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS PubMed
- J. Wei, Y. Zhang, Y. Chi, L. Liu, W.-X. Zhang and Z. Xi, Aromatic Dicupra[10]annulenes, J. Am. Chem. Soc., 2016, 138, 60–63 CrossRef CAS PubMed
- C. Yu, M. Zhong, Y. Zhang, J. Wei, W. Ma, W.-X. Zhang, S. Ye and Z. Xi, Butadienyl Diiron Complexes: Nonparnar Metalla-aromatics Involving σ-Type Orbital Overlap, Angew. Chem., Int. Ed., 2020, 59, 19048–19053 CrossRef CAS PubMed
- C. Zhu, Q. Zhu, J. Fan, J. Zhu, X. He, X.-Y. Cao and H. Xia, A Metal-Bridged Tricyclic Aromatic System: Synthesis of Osmium Polycyclic Aromatic Complexes, Angew. Chem., Int. Ed., 2014, 53, 6232–6236 CrossRef CAS
- D. W. Szczepanik, M. Andrzejak, K. Dyduch, E. Zak, M. Makowski, G. Mazur and J. Mrozek, A Uniform Approach to the Description of Multicenter Bonding, Phys. Chem. Chem. Phys., 2014, 16, 20514–20523 RSC
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawelek, T. M. Krygowski, H. Szatylowicz and M. Solà, The Electron Density of Delocalized Bonds (EDDB) Applied for Quantifying Aromaticity, Phys. Chem. Chem. Phys., 2017, 19, 28970–28981 RSC
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed
- Z. F. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-Independent Chemical Shifs (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Which NICS Aromaticity Index for Planar π Rings Is Best, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed
- R. Herges and D. Geuenich, Delocalization of Electrons in Molecules, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Anistropy of
the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed
- G. B. Robertson, Nature, 1961, 191, 593–594 CrossRef CAS
- Q. Zhu, C. Zhu, Z. Deng, G. He, J. Zhu and H. Xia, Synthesis and Characterization of Osmium Polycyclic Aromatic Complexes via Nucleophilic Reactions of Osmapentalyne, Chin. J. Chem., 2017, 35, 628–634 CrossRef CAS
- M. A. Iron, A. C. B. Lucassen, H. Cohen, M. E. van der Boom and J. M. L. Martin, A computational Foray into the Formation and Reactivity of Metallabenzenes, J. Am. Chem. Soc., 2004, 126, 11699–11710 CrossRef CAS PubMed
- C. F. Nejad, J. Vícha and A. Ghosh, Relativity or Aromaticity? A First-principles Perspective of Chemical Shifts in Osmabenzene and Osmapentalene Derivatives, Phys. Chem. Chem. Phys., 2020, 22, 10863–10869 RSC
- Z. Badri, S. Pathak, H. Fliegl, P. Rashidi-Ranjbar, R. Bast, R. Marek, C. Foroutan-Nejad and K. Ruud, All-metal Aromaticity: Revisiting the Ring Current Model Current among Transition Metal Clusters, J. Chem. Theory Comput., 2013, 9, 4789–4796 CrossRef CAS
- C. Foroutan-Nejad, Is NICS a Reliable Aromaticity Index for Transition Metal Clusters, Theor. Chem. Acc., 2015, 134, 8–17 Search PubMed
- G. Mahmoudi, F. A. Afkhami, A. Castiñeiras, I. García-Santos, A. Gurbanov, F. I. Zubkov, M. P. Mitoraj, M. Kukulka, F. Sagan, D. W. Szczepanik, I. A. Konyaeva and D. A. Safin, Quasi-aromatic Möbius Metal Chelates, Inorg. Chem., 2018, 57, 4395–4408 CrossRef CAS PubMed
- D. W. Szczepanik, M. Solà, T. M. Krygowski, H. Szatylowicz, M. Andrzejak, B. Pawelek, J. Dominikowska, M. Kukulka and K. Dyduch, Aromaticity of Acenes: the Model of Migrating π-Circuits, Phys. Chem. Chem. Phys., 2018, 20, 13430–13436 RSC
- D. W. Szczepanik and M. Solà, Electron Delocalization in Plabar Metallacycles: Hückel or Möbius Aromatic?, ChemistryOpen, 2019, 8, 219–227 CrossRef CAS PubMed
- K. Xiao, Y. Zhao, J. Zhu and L. Zhao, Hyperconjugative Aromaticity and Protodeauration Reactivity of Polyaurated Indoliums, Nat. Commun., 2019, 10, 5639–5648 CrossRef CAS
- L. Lin, Q. Zhu, A. M. Rouf and J. Zhu, Probing the Aromaticity and Stability of Metallatricycles by DFT Calculations: Toward Clar Structure in Organometallic Chemistry, Organometallics, 2020, 39, 80–86 CrossRef CAS
- J. Wu and J. Zhu, The Clar Structure in Inorganic BN Analogues of Polybenzenoid Hydrocarbons: Does it Exist or Not?, ChemPhysChem, 2015, 16, 3806–3813 CrossRef CAS PubMed
- J. Zhu, C. Dahlstrand, J. R. Smith, S. Villaume and H. Ottosson, On the Importance of Clar Structures of Polybenzenoid Hydrocarbons as Revealed by the π-Contribution to the Electron Localization Function, Symmetry, 2010, 2, 1653–1682 CrossRef CAS
- X.-Y. Cao, Q. Zhao, Z. Lin and H. Xia, The Chemistry of Aromatic Osmacycles, Acc. Chem. Res., 2014, 47, 341–354 CrossRef CAS PubMed
- A. Shaffiee, M. M. Saleh and M. Yahaya, Determination of HOMO and LUMO of [6,6]-Phenyl C61-butyric Acid 3-Ethylthiophene Ester and Poly (3-octyl-thiophene-2,5-diyl) through voltametry characterization, Sains Malays., 2011, 40, 173–176 Search PubMed
- M. B. Robin and P. Day, Mixed Valence Chemistry-a Survey and Classification, Adv. Inorg. Chem., 1967, 10, 247–422 CrossRef CAS
- C.-J. Yao, Y.-W. Zhong and J. Yao, Charge Delocalization in a Cyclometalated Bisruthenium Complex Bridged by a Noninnocent 1,2,4,5-Tetra(2-pyridyl)benzene Ligand, J. Am. Chem. Soc., 2011, 133, 15697–15706 CrossRef CAS PubMed
- G. E. Pieslinger, P. Alborés, L. D. Slep and L. M. Baraldo, Class III Delocalization in a Cyanide-Bridged Trimetallic Mixed-Valence Complex, Angew. Chem., Int. Ed., 2014, 53, 1293–1296 CrossRef CAS PubMed
- X. Ma, C.-S. Lin, X.-Q. Zhu, S.-M. Hu, T.-L. Sheng and X.-T. Wu, An Unusually Delocalized Mixed-Valence State of a Cyanidometal-Bridged Compound Induced by Thermal Electron Transfer, Angew. Chem., Int. Ed., 2017, 56, 1605–1609 CrossRef CAS PubMed
- Y.-Y. Yang, X.-Q. Zhu, S.-M. Hu, S.-D. Su, L.-T. Zhang, Y.-H. Wen, X.-T. Wu and T.-L. Sheng, Different Degrees of Electron Delocalization in Mixed Valence Ru-Ru-Ru Compounds by Cyanido-/Isocyanido-Bridge Isomerism, Angew. Chem., Int. Ed., 2018, 57, 14046–14050 CrossRef CAS PubMed
- Y. X. Hu, J. Zhang, F. Zhang, X. Wang, J. Yin, F. Hartl and S. H. Liu, Electronic Properties of Oxidized Cyclometalated Diiridium Complexes: Spin Delocalization Controlled by the Mutual Position of the Iridium Centers, Chem. – Eur. J., 2020, 26, 4567–4575 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and syntheses; NMR spectra and HR-MS data; X-ray crystallographic data; complete cyclic voltammograms; IR spectroelectrochemistry; additional DFT, TD-DFT and EDDB data. CCDC 2024859 and 2024860. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi01523d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |