
The exploration of new infrared nonlinear optical crystals based on the polymorphism of BaGa4S7†
 a
Hongwei
Yu
*a
a
Hongwei
Yu
*a
Abstract
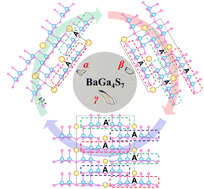
Balancing the key performance metrics, such as a large second harmonic generation (SHG) response and a wide band gap, is an extremely important but intractable challenge for the development of infrared (IR) nonlinear optical (NLO) materials. Herein, we report two new polymorphs of BaGa4S7, β- and γ-BaGa4S7, both of which crystallize in the noncentrosymmetric and polar space groups, Pmc21 (No. 26) for β-BaGa4S7 and Cm (No. 8) for γ-BaGa4S7. They show an interesting structural evolution from AA′ [Ga6S16]∞ chains to AA′A′A [Ga6S16]∞ chains to AAA′AAA′ [Ga6S16]∞ chains as their structures show a transition from the high-temperature phase to the low-temperature phase. More importantly, they both exhibit excellent NLO properties. In particular, β-BaGa4S7 exhibits the best balance along with a large phase-matching SHG response (1.2 × AgGaS2) and a wide band gap (3.94 eV). These indicate that β-BaGa4S7 will be a promising IR NLO crystal for high-power laser application. The research also provides an insight into exploring new IR NLO crystals based on structural rearrangement.
- This article is part of the themed collections: Inorganic Chemistry Frontiers Emerging Investigator Series 2022–2023 and 2022 Inorganic Chemistry Frontiers HOT articles
 Please wait while we load your content...
Please wait while we load your content...

