DOI:
10.1039/D2QI00876A
(Research Article)
Inorg. Chem. Front., 2022,
9, 4101-4110
Controlled synthesis of a porous single-atomic Fe–N–C catalyst with Fe nanoclusters as synergistic catalytic sites for efficient oxygen reduction†
Received
25th April 2022
, Accepted 19th June 2022
First published on 21st June 2022
Abstract
Single-atomic Fe–Nx sites have been widely accepted as active sites for the oxygen reduction reaction (ORR), while the roles played by other symbiotic Fe moieties (such as Fe clusters) are still contentious. Synthesis of Fe–N–C catalysts possessing both Fe–Nx sites and Fe clusters and investigation of their catalytic mechanism are essential but challenging. Herein, the controlled synthesis of a model catalyst is successfully achieved using Fe(II)–phenanthroline (Phen) complexes as the only precursor. Through a solid-phase preparation process, Fe–Phen complexes are synthesized on the surface of silica that is used as a hard template for introducing porosity into the carbon structure. The high density of Fe centers facilitates the simultaneous generation of single atomic Fe–Nx sites and Fe clusters, severe aggregation of which is impeded by the silica template. The as-prepared catalyst delivers an efficient ORR performance in an alkaline environment. Combining with computational analysis, the synergistic catalytic mechanism between the Fe–Nx sites and Fe clusters is revealed that the neighboring Fe clusters can increase the adsorption energy of OOH* on the Fe atom of Fe–Nx sites and lower the energy barrier for the formation of the OOH intermediate, thus accelerating the catalytic process. This study provides insights into the future design and synthesis of efficient Fe–N–C catalysts.
Introduction
The ever-increasing consumption of fossil fuels and pollution of the environment have aroused extensive research interest in exploiting renewable and sustainable energy conversion-storage devices, including fuel cells and metal–air batteries.1–5 As a major component in fuel cells and metal–air batteries, the cathode oxygen reduction reaction (ORR) suffers from sluggish kinetics that traditionally requires platinum group metal catalysts to accelerate it.6–9 The limited reserves and high cost however deeply impede their large-scale commercial application. Developing efficient non-noble metal electrocatalysts for catalyzing the ORR is thus highly necessary.
Among the numerous catalysts investigated based on non-precious metals, the catalysts composed of iron, nitrogen, and carbon (Fe–N–C) have been considered the most promising alternatives owing to their superior ORR performance.10–16 To achieve an efficient 4e− pathway (O2 → H2O), it is of vital importance to understand the active sites of these Fe–N–C materials. As the formation of active sites is closely related to the precursors used, multiple active sites have been suggested for their ORR activity, such as Fe-containing moieties includingsingle Fe sites (nitrogen-coordinating Fe sites, Fe–Nx)17–24 and crystalized iron species (metal, carbide, nitrides, and oxide),25–28 doped nitrogen species (pyridinic N, graphitic N, and pyrrolic N)29–34 and carbon defects.35,36 However, there is a continuing debate over the nature of active sites in these Fe–N–C catalysts. Growing evidence has indicated experimentally and theoretically that Fe–Nx sites are the dominant active centers.37,38 In this perspective, efforts are dedicated to the preparation of catalysts with dense single iron sites.39,40 As the synthesis of Fe–N–C materials involves a pyrolysis process, the simultaneous formation of multiple Fe species (such as Fe clusters, Fe clusters/FexC, FeOx/FeyC, or FeNx nanoparticles, etc.) usually occurs due to the different precursors adopted, which makes it complex to interpret the ORR behaviors of Fe–N–C catalysts.41,42 When oxygen atoms are not involved, the most common accompanying impurities are Fe clusters or Fe clusters/FexC clusters/nanocrystals due to the agglomeration of Fe atoms during the heat treatment and the possible reaction between the outer surfaces of agglomerated Fe with the C atoms in contact. Some reported studies lean toward the viewpoint that these Fe or Fe/FexC moieties are catalytically inactive for the ORR due to their low stability in an acidic environment or inaccessibility to O2 when protected/isolated by carbon layers37,40,43,44 and should be suppressed during the synthesis while maximizing the Fe–Nx sites. On the other hand, the involvement of Fe clusters or Fe/FexC moieties in the catalytic process of the ORR has also been reported.45–51 The catalyst composed of pea-pod-like CNTs with encapsulated Fe nanoparticles is reported with enhanced ORR activity due to the electron transfer from Fe particles to CNTs.52 Gewirth and coworkers identified the Fe particles encapsulated by N-doped carbon as active species and responsible for the activity of the ORR catalyst.45 The high ORR performance of a Fe–N–C catalyst that is devoid of Fe–N coordination is believed by Mukerjee's group to correspond to the N-doped carbon-covered Fe/FexC nanoparticles, which imparts a synergistic effect on N-doped carbon, enabling the stabilization of the peroxide intermediate and 4e− reduction of oxygen. Besides the abovementioned research studies, the Fe clusters/particles have also been found to affect the Fe–Nx sites around them, not only serving as active species. Wan and coworkers suggest that the Fe/Fe3C nanoparticles can boost the activity of Fe–Nx.46 The Fe nanocluster is also believed to play an important role in enhancing the ORR activity of atomic Fe sites.47 The coexistence of M nanoparticles and MN4 (M = Fe and Co) reported by the Sun group offers efficient ORR activity due to the changed adsorption/desorption strength of ORR intermediates.53 These pioneering studies infer that a synergistic effect might appear between the coexistent single atomic Fe sites and Fe clusters/nanoparticles in Fe–N–C materials when used for catalyzing the ORR process, which can provide a good opportunity for fabricating efficient Fe–N–C catalysts. To further confirm this deduction, the synthesis of a new Fe–N–C catalyst possessing both single atomic Fe sites and Fe species through a different strategy and the investigation of its catalytic mechanism are essential but still challenging.
Herein, a model Fe–N–C catalyst with single atomic Fe–Nx sites and Fe nanoclusters embedded in a porous N-doped carbon matrix is successfully fabricated through a solid-phase preparation process followed by pyrolysis. The complexes assembled from Fe(II) and phenanthroline (Phen) are selected and used solely as carbon, iron, and nitrogen sources with consideration of two points: (1) maximizing the transformation of Fe–N bonding in Fe–Phen complexes to atomic Fe–Nx sites; (2) generating Fe clusters due to the high density of Fe centers. Silica nanoparticles are added for two purposes: (1) for using as the hard template for introducing porosity into the carbon structure; (2) for impeding severe aggregation of Fe centers during the heat treatment. After removal of SiO2, the prepared catalyst has been confirmed to have abundant atomic Fe–Nx sites and Fe nanoclusters and a large specific surface area, which contribute to its high activity toward the ORR. DFT calculations indicate that it is beneficial for the Fe atom of Fe–N4 sites to bond with the OOH intermediate when a neighboring Fe nanocluster exists, thus increasing the adsorption energy of OOH and lowering the energy barrier for the formation of the OOH intermediate, resulting in an efficient ORR performance.
Experimental
Materials
All chemical reagents were directly used without any purification. Anhydrous iron(II) acetate (Fe(CH3COO)2), 1,10-phenanthroline and hydrofluoric acid (HF) were purchased from Energy Chemical, and SiO2 powder was supplied by Sigma-Aldrich.
Preparation of the Fe–Phen@SiO2 precursor
The Fe–Phen@SiO2 precursor was prepared by mechanical grinding. A mixture of Fe(CH3COO)2 (0.0348 g, 0.2 mmol), 1,10-phenanthroline (0.1082 g, 0.6 mmol), and the appropriate amount of SiO2 powder was added into an agate grinding jar, and ground at 700 rpm for 120 min. Then, the prepared Fe–Phen@SiO2 precursor was scraped from the grinding jar and collected for further use. The names Fe–Phen, Fe–Phen@SiO2-6, Fe–Phen@SiO2-7, and Fe–Phen@SiO2-8 correspond respectively to precursors with 0 g, 0.0720 g, 0.0840 g, and 0.0960 g of SiO2.
Preparation of the Fe(0)/FeNx-NC catalyst
0.2 g of the as-prepared Fe–Phen@SiO2 precursor was put into the crucible and heated at 800 °C for 3 h with a heating rate of 5 °C min−1 under a N2 atmosphere, which was then soaked in 5% HF solution for 24 h, washed with deionized water to neutral, collected by centrifugation, and dried at 80 °C overnight to obtain the final product. The catalysts Fe-NC, Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7 and Fe(0)/FeNx-NC-8 correspond respectively to the precursors Fe–Phen, Fe–Phen@SiO2-6, Fe–Phen@SiO2-7, and Fe–Phen@SiO2-8.
Characterization
Phase purity and crystallinity were evaluated by powder X-ray diffraction (PXRD) with Cu Kα radiation (λ = 0.15418 nm). The morphology and microstructure of the synthesized catalysts were observed by scanning electron microscopy (SEM, JEOL-7900) and transmission electron microscopy (TEM, JEM-2100F). X-ray photoelectron spectroscopy (XPS) was applied to investigate the surface chemical states using an ESCA250Xi spectrometer and the binding energy was calibrated according to the graphite C 1s peak. Raman spectra were recorded on a LABRAM HR EVO Raman microscope spectrometer at an excitation wavelength of 532 nm to analyze the graphitization degree of the samples and the structural information of the carbon matrix. The N2 adsorption–desorption isotherms and corresponding pore size distributions were acquired from a JW-BK200 surface area analyzer. The content of Fe in the samples was measured by an Agilent 710 inductively coupled plasma optical emission spectrometer (ICP-OES). The Fe K-edge X-ray absorption near edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS) were investigated at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) in fluorescence mode using a fixed-exit Si (111) double crystal monochromator. The incident X-ray beam was monitored in an ionization chamber filled with N2, and X-ray fluorescence detection was performed using a Lytle-type detector filled with Ar. The EXAFS raw data was background-subtracted, normalized and Fourier transformed by the standard procedures with the IFEFFIT package.
Electrochemical measurements
All electrochemical tests were carried out on a Gamry electrochemical workstation (INTERFACE 1000 T) through a three-electrode cell configuration with a glassy carbon rotating disk electrode (RDE, 5 mm in diameter) as the working electrode, Ag/AgCl electrode (KCl saturated) as the reference electrode, and a graphite rod as the counter electrode. All measured potentials were transformed to the reversible hydrogen electrode (RHE) in terms of the Nernst equation: ERHE = EAg/AgCl + 0.059 pH + EθAg/AgCl. For the preparation of the catalyst ink, 4 mg of the as-prepared catalyst was dispersed in a solution of 970 μL of ethanol and 30 μL of Nafion (5 wt%) by ultrasonication for 60 min. To prepare the working electrode, 15 μL of homogeneous catalyst ink was pipetted onto the GC electrode with a loading equivalent to 0.300 mg cm−2. Linear sweep voltammograms (LSVs) were tested in the O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1. Cyclic voltammetry (CV) was performed in both O2- and N2-saturated 0.1 M KOH solution at the same scan rate. The electron transfer number (n) was calculated using the Koutecky–Levich (K–L) equation:
where J is the measured current density, JK and JL are the kinetically limited and diffusion-limited current densities respectively, ω is the angular velocity of the disk, n is the electron transfer number, F is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the bulk concentration of O2 (C0 = 1.2 × 10−6 mol cm−3 in 0.1 M KOH), D0 is the diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm2 s−1 in 0.1 M KOH), and V is the kinematic viscosity of the electrolyte (V = 0.01 cm2 s−1 in 0.1 M KOH).
485 C mol−1), C0 is the bulk concentration of O2 (C0 = 1.2 × 10−6 mol cm−3 in 0.1 M KOH), D0 is the diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm2 s−1 in 0.1 M KOH), and V is the kinematic viscosity of the electrolyte (V = 0.01 cm2 s−1 in 0.1 M KOH).
Based on the measurement data of the rotating ring disk electrode (RRDE), the yield of HO2− and the electron transfer number (n) can be calculated according to the equation:
where
Id is the disk current density,
Ir is the ring current density, and
N is the ring collection efficiency (
N = 0.37).
Density functional theory (DFT) calculations
The Vienna ab initio simulation package (VASP) was applied to perform DFT calculations with the Perdew–Burke–Ernzerhof (PBE) functional and projector augmented wave (PAW) method.54 The kinetic cut-off energy and the Brillouin zone were optimized to be 450 eV and the 3 × 3 × 1 k-point mesh respectively. The ionic relaxation was calculated until the force on each atom was less than 0.03 eV Å−1. The single Fe atom anchored graphene (named FeN4) model was constructed by bonding a single Fe atom with four N atoms in the graphene, and the Fe3 cluster was placed surrounding the FeN4 moiety (named Fe3–FeN4) to simulate the interaction between Fe clusters and FeN4 coordination. Different Fe3–FeN4 configurations were considered to search for the stable structures as the calculation model. For the ORR process in alkaline solution, the Gibbs free energy was calculated based on the standard four-electron reaction mechanism:55
| * + O2 + H2O + e− → *OOH + OH− |
| *O + H2O + e− → *OH + OH− |
The Gibbs free energy of the adsorbed species was calculated as:
| ΔG = ΔEads + ΔEZPE − TΔSads |
where Δ
Eads is the adsorption energy, Δ
EZPE is the zero-point energy difference between adsorbed and gaseous species, and
TΔ
Sads is the corresponding entropy difference between these two states (
T was set to be 298 K).
Results and discussion
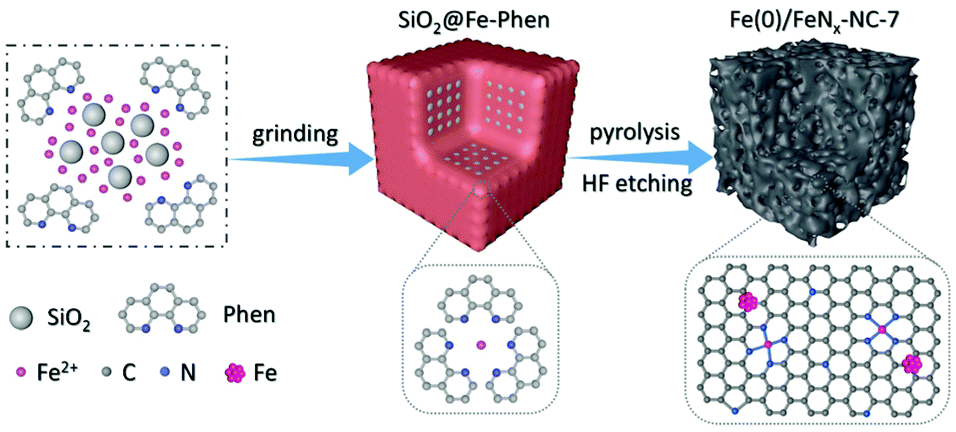
The overall synthetic process of Fe(0)/FeNx-NC catalysts is shown in Fig. 1. The nanosized SiO2 was utilized as the hard template to introduce porosity into the carbon structure (Fig. S1a and S2†). After grinding together with Fe(CH3COO)2 and 1,10-phenanthroline (Phen), the white color of SiO2 turned to dark red (Fig. S1b†), the characteristic color of Fe–Phen complexes, indicating the successful synthesis of Fe–Phen complexes via the solid chemical reaction. As revealed by scanning electron microscopy (SEM) (Fig. S3†), the Fe–Phen complexes had been coated on the surface of the SiO2 template. The prepared Fe–Phen@SiO2 precursors were then heated at 800 °C for 3 h under a N2 atmosphere, during which the Fe–Phen complexes were transformed into the carbon matrix that is decorated with single atomic Fe–Nx sites and Fe nanoclusters. Finally, porous Fe(0)/FeNx-NC catalysts were prepared after removing the SiO2 template by HF etching (Fig. S1c†). As Fe–Phen complexes are the precursors capable of producing single atomic Fe–Nx sites, it is reasonable to disperse them on the carbon substrate to prepare the single atom ORR catalyst. However, no extra carbon substrate is used in this study to increase the density of Fe–Phen complexes. Under high density of Fe–Phen complexes, migration and agglomeration of Fe center would happen during the pyrolysis process, facilitating the simultaneous generation of Fe nanoclusters, further growth of which can be restricted by the confinement effect of SiO2 template as revealed in Fig. 2 and 3a.
 |
| | Fig. 1 Schematic illustration of the preparation process of the Fe(0)/FeNx-NC-7 catalyst. | |
 |
| | Fig. 2 (a and b) TEM images of Fe-NC; (c and d) TEM images, (e and f) HRTEM images, (g and h) HAADF-STEM images (single Fe atoms and Fe nanoclusters highlighted by yellow and blue circles respectively) and (i–m) elemental mapping of C (red), N (green), O (cyan), and Fe (yellow) of Fe(0)/FeNx-NC-7. | |
 |
| | Fig. 3 (a) PXRD patterns, (b) Raman spectra, (c) N2 adsorption–desorption isotherms and (d) corresponding pore size distribution of Fe-NC, Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7 and Fe(0)/FeNx-NC-8. | |
The detailed structural features of the as-prepared Fe(0)/FeNx-NC catalysts were characterized by transmission electron microscopy (TEM) in comparison with the Fe-NC material obtained without the addition of the SiO2 template. As shown in Fig. 2a and b, a mass of anomalous nanoparticles was formed and embedded in the non-porous carbon matrix of Fe-NC. The lattice fringes of the nanoparticles show interplanar distances of 2.37 Å, corresponding to the (110) crystal plane of Fe3C, which is further confirmed by XRD results in Fig. 3a. In contrast, obvious porosity is observed in the TEM images of Fe(0)/FeNx-NC catalysts after SiO2 was introduced with no large aggregated Fe species (Fig. 2c–f and Fig. S4†). In the high-resolution TEM (HRTEM) images, clear lattice fringes with interplanar distances of 3.51 Å (Fe(0)/FeNx-NC-6), 3.42 Å (Fe(0)/FeNx-NC-7) and 3.50 Å (Fe(0)/FeNx-NC-8) can be attributed to the (002) crystal planes of carbon. By applying HRTEM and atomic-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 2g and h), the existence of Fe single atoms and Fe clusters in Fe(0)/FeNx-NC-7 is revealed from the bright dots that are highlighted by yellow and blue circles respectively. It clearly proves the effectiveness of this strategy in fabricating abundant single atomic Fe–Nx sites with the simultaneous generation of Fe nanoclusters in the carbon matrix. The elemental mappings further performed on Fe(0)/FeNx-NC-7 reveal the homogeneous distribution of participating elements (C, O, N, and Fe) (Fig. 2i and m). As indicated by the TEM characterization, the target material designed with atomically dispersed Fe–Nx sites and Fe nanoclusters in carbon has been successfully prepared.
Further characterization of the microstructures was carried out for Fe(0)/FeNx-NC catalysts. As shown in the powder X-ray diffraction (PXRD) patterns in Fig. 3a, there are two diffraction peaks observed at 2θ of 25.1° and 43.1° for Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7 and Fe(0)/FeNx-NC-8, which can be attributed to the (002) and (101) crystal planes of carbon. No characteristic peaks corresponding to Fe species are detected in the PXRD patterns, indicating that severe aggregation of the Fe centers of Fe–Phen complexes did not happen during the pyrolysis, in sharp contrast to the Fe-NCs that present distinct diffraction peaks of the Fe3C phase (JCPDS-75-0910). This great difference originates from the absence of the SiO2 template, further confirming a positive role played by SiO2 in reducing the aggregation of Fe centers during the heat treatment. The detailed structural features of carbon in the as-prepared samples were investigated via Raman spectra (Fig. 3b). The two distinct peaks located at 1346 and 1592 cm−1 are respectively assigned to the D-band and G-band, referring to the disordered and graphitic structure in carbon. The ID/IG values calculated for the Fe(0)/FeNx-NC catalysts are much lower than that of Fe-NC, suggesting that defects are greatly reduced after introducing SiO2 as a template. It is probably due to the more compact and continuous arrangement of the formed Fe–Phen molecules on the SiO2 surface under the force of mechanical grinding that results in a lower defect degree in the final carbon structure. Comparing the three Fe(0)/FeNx-NC catalysts, Fe(0)/FeNx-NC-7 presents a little lower ID/IG value than its counterparts, indicating a higher graphitization degree for Fe(0)/FeNx-NC-7, which would be beneficial for the catalysis of the ORR. The importance of an appropriate Fe–Phen/SiO2 ratio for deriving a superior catalyst in this study is also confirmed by the N2 adsorption–desorption results. As illustrated in Fig. 3c, Fe-NC shows basically no adsorption of N2 in the pressure (P/P0) range of 0–1.0, while much higher adsorption capacity is obtained on Fe(0)/FeNx-NC catalysts with distinct hysteresis loops. The calculated specific surface areas of Fe-NC, Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7 and Fe(0)/FeNx-NC-8 are 16.14, 320.41, 456.58 and 418.81 m2 g−1, respectively. The much higher surface areas of Fe(0)/FeNx-NC catalysts compared to that of Fe-NC prove the successful introduction of porosity by adopting SiO2 as a template. Corresponding pore size follows the order Fe(0)/FeNx-NC-6 > Fe(0)/FeNx-NC-8 > Fe(0)/FeNx-NC-7 (Fig. 3d). The reasons lie in the fact that compared to Fe(0)/FeNx-NC-7, relatively excessive Fe–Phen complexes were added in the case of Fe(0)/FeNx-NC-6, which impeded better dispersion of the SiO2 template. The agglomeration of SiO2 and its incorporation in Fe–Phen complexes not only reduce the possible porosity and surface area but also generate larger pores in the carbon structure. As for the preparation of Fe(0)/FeNx-NC-8, the relatively deficient Fe–Phen complexes led to thinner coating or partial discontinuity of its coating on the surface of SiO2, resulting in the possible collapse of a small fraction of the pores after SiO2 removal. Therefore, Fe(0)/FeNx-NC-7 shows the smallest pore size owing to the optimal ratio of Fe–Phen complexes and SiO2 that provides more uniform dispersion of SiO2 nanoparticles and even coating of Fe–Phen complexes on their surface. In the catalytic reaction process, the larger surface area and higher porosity are conducive to mass transfer and accessibility of the active sites, thus helping enhance the overall ORR catalytic performance of Fe(0)/FeNx-NC-7.
The elemental composition and surface chemical state of Fe(0)/FeNx-NC catalysts were analyzed by X-ray photoelectron spectroscopy (XPS). Four elements C, N, O, and Fe show their characteristic peaks in the survey spectra (Fig. S5a†). In the C 1s high-resolution XPS spectra, the fitted peaks at 284.3, 285.1, 287.3, and 289.6 eV are respectively assigned to the C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CO, and OC–O bonding (Fig. S5b†). Deconvolution of the Fe 2p spectra reveals two pairs of 2p3/2/2p1/2 doublets for Fe2+ (710.1/722.8 eV) and Fe3+ (713.5/726.2 eV), accompanied by two satellite peaks at 718.9 and 730.0 eV (Fig. 4a). The reversible redox reaction between Fe3+/Fe2+ plays a critical role in the demonstrated ORR activity of Fe(0)/FeNx-NC catalysts. Four peaks at 398.1 eV (pyridinic N), 399.1 eV (Fe–N), 399.9 eV (pyrrolic N), and 400.8 eV (graphitic N) are analyzed from the N 1s spectra (Fig. 4b), which confirms the formation of Fe–Nx sites in the structure of Fe(0)/FeNx-NC catalysts. Furthermore, Fe K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were applied to investigate the local bonding environments of Fe in Fe(0)/FeNx-NC-7. Fig. 4c shows the normalized XANES spectra of Fe(0)/FeNx-NC-7 in comparison with Fe foil and Fe2O3. As revealed, the energy absorption threshold of Fe(0)/FeNx-NC-7 is located between Fe foil and Fe2O3, indicating the existence of positively charged Feδ+ stabilized by N atoms in Fe(0)/FeNx-NC-7. The corresponding Fourier transformation of the Co K-edge from EXAFS spectra is shown in Fig. 4d. Two main peaks at about 1.74 and 2.32 Å can be attributed to the Fe–N and Fe–Fe scattering paths respectively, which are further confirmed in the wavelet transform (WT) contour plots. In comparison with Fe foil (Fig. 4f) and Fe2O3 (Fig. 4g), the two signals focused at 5.2 and 6.7 Å−1 in the plot of Fe(0)/FeNx-NC-7 (Fig. 4e) are assigned to the Fe–N and Fe–Fe paths respectively, demonstrating its essential microscopic features of atomic-level Fe coupled with Fe clusters in the carbon structure. These results suggest the evolution of single-atom Fe sites from the Fe–Phen complexes with the simultaneous generation of Fe clusters, which is consistent with the TEM analysis.
N, CO, and OC–O bonding (Fig. S5b†). Deconvolution of the Fe 2p spectra reveals two pairs of 2p3/2/2p1/2 doublets for Fe2+ (710.1/722.8 eV) and Fe3+ (713.5/726.2 eV), accompanied by two satellite peaks at 718.9 and 730.0 eV (Fig. 4a). The reversible redox reaction between Fe3+/Fe2+ plays a critical role in the demonstrated ORR activity of Fe(0)/FeNx-NC catalysts. Four peaks at 398.1 eV (pyridinic N), 399.1 eV (Fe–N), 399.9 eV (pyrrolic N), and 400.8 eV (graphitic N) are analyzed from the N 1s spectra (Fig. 4b), which confirms the formation of Fe–Nx sites in the structure of Fe(0)/FeNx-NC catalysts. Furthermore, Fe K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were applied to investigate the local bonding environments of Fe in Fe(0)/FeNx-NC-7. Fig. 4c shows the normalized XANES spectra of Fe(0)/FeNx-NC-7 in comparison with Fe foil and Fe2O3. As revealed, the energy absorption threshold of Fe(0)/FeNx-NC-7 is located between Fe foil and Fe2O3, indicating the existence of positively charged Feδ+ stabilized by N atoms in Fe(0)/FeNx-NC-7. The corresponding Fourier transformation of the Co K-edge from EXAFS spectra is shown in Fig. 4d. Two main peaks at about 1.74 and 2.32 Å can be attributed to the Fe–N and Fe–Fe scattering paths respectively, which are further confirmed in the wavelet transform (WT) contour plots. In comparison with Fe foil (Fig. 4f) and Fe2O3 (Fig. 4g), the two signals focused at 5.2 and 6.7 Å−1 in the plot of Fe(0)/FeNx-NC-7 (Fig. 4e) are assigned to the Fe–N and Fe–Fe paths respectively, demonstrating its essential microscopic features of atomic-level Fe coupled with Fe clusters in the carbon structure. These results suggest the evolution of single-atom Fe sites from the Fe–Phen complexes with the simultaneous generation of Fe clusters, which is consistent with the TEM analysis.
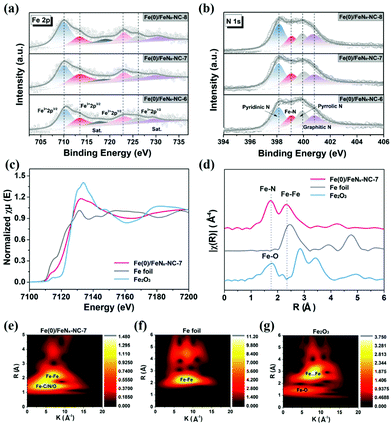 |
| | Fig. 4 (a) Fe 2p and (b) N 1s XPS spectra of Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7, and Fe(0)/FeNx-NC-8; (c) Fe K-edge XANES of Fe(0)/FeNx-NC-7 in comparison with Fe foil and Fe2O3; (d) Fourier transform of the EXAFS spectra of Fe(0)/FeNx-NC-7, Fe foil and Fe2O3; wavelet transform of Fe K-edge EXAFS for (e) Fe(0)/FeNx-NC-7, (f) Fe foil and (g) Fe2O3. | |
Given the intrinsic activity of Fe–Nx in the presence of neighboring Fe clusters, the ORR performance of the as-prepared Fe(0)/FeNx-NC catalysts was investigated in 0.1 M KOH solution using a rotating disk electrode (RDE) and rotating ring-disk electrode (RRDE). As shown in Fig. 5a, the cyclic voltammetry (CV) curves manifest well-defined cathodic peaks in O2-saturated electrolytes that are associated with the oxygen reduction process, while no peaks appear when the electrolyte was N2-saturated. The reduction peak of Fe(0)/FeNx-NC-7 catalyst (0.90 V) is more positive than that of commercial 20% Pt/C (0.87 V), Fe(0)/FeNx-NC-6 (0.86 V), and Fe(0)/FeNx-NC-8 (0.87 V), indicating its superior performance toward the ORR. The ORR performance was further studied by linear sweep voltammograms (LSVs). Fig. 5b shows the ORR polarization curves recorded at the rotating rate of 1600 rpm. As observed, the Fe(0)/FeNx-NC-7 exhibits an activity with a half-wave potential (E1/2) of 0.86 V and a diffusion-limited current density (JL) of 5.11 mA cm−2, which is higher than commercial 20% Pt/C (E1/2 = 0.84 V, JL = 5.05 mA cm−2), Fe(0)/FeNx-NC-6 (E1/2 = 0.83 V, JL = 4.56 mA cm−2), Fe(0)/FeNx-NC-8 (E1/2 = 0.84 V, JL = 4.81 mA cm−2), Fe-NC (E1/2 = 0.80 V, JL = 4.52 mA cm−2) and comparable to many recently reported electrocatalysts (Fig. 5c and Table S1†). The corresponding Tafel plots of these samples are presented in Fig. 5d, in which the calculated Tafel slopes are respectively 91.6, 86.2, 90.3 and 98.8 mV dec−1 for Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7, Fe(0)/FeNx-NC-8 and Fe-NC. The smaller Tafel slope of Fe(0)/FeNx-NC-7 reveals its faster catalytic kinetics toward the ORR than its counterparts. To investigate the catalysis mechanism of the as-prepared catalysts, RDE measurements were performed at different rotating speeds from 400 to 2025 rpm (Fig. 5e and Fig. S6†). The limiting current densities are observed to increase with the rotating speed due to the improved mass transport at a higher rotating speed. The corresponding Koutecky–Levich (K–L) plots of Fe(0)/FeNx-NC-7 with parallel slopes and good linearity in the potential range of 0.5–0.7 V (inset of Fig. 5e) evidence that the reduction of oxygen on Fe(0)/FeNx-NC-7 follows the first-order reaction kinetics of dissolved oxygen. The average electron transfer number (n) calculated based on the K–L equation is about 3.97, suggesting the four-electron pathway (O2 + 2H2O + 4e− → 4OH−). The ORR catalytic pathway was further investigated by monitoring the formation of peroxide species (H2O2) based on RRDE measurements. As illustrated in Fig. 5f and Fig. S7,† the Fe(0)/FeNx-NC-7 presents a much lower H2O2 yield (≤12.3%) than Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-8, and Fe-NC in the potential range of 0.2–0.8 V. The corresponding electron-transfer number evaluated (3.72–3.91) reveals that the process from O2 to OH− on Fe(0)/FeNx-NC-7 is mainly through the four-electron pathway, which is in good agreement with the K–L results, confirming that the superior ORR process of Fe(0)/FeNx-NC-7 is related to its faster dynamics.
 |
| | Fig. 5 (a) CV curves in N2- (dotted line) and O2- (solid line) saturated 0.1 M KOH solution, (b) LSVs curves at 1600 rpm, (c) half-wave potential and limiting current density and (d) Tafel plots of Fe-NC, Fe(0)/FeNx-NC-6, Fe(0)/FeNx-NC-7, Fe(0)/FeNx-NC-8 and 20% Pt/C; (e) LSV curves of Fe(0)/FeNx-NC-7 recorded at different rotation rates in O2-saturated 0.1 M KOH solution (inset: the K–L plots); (f) RRDE tests of Fe(0)/FeNx-NC-7 at 1600 rpm in O2-saturated 0.1 M KOH solution (inset: the H2O2 yield and electron transfer number from RRDE measurements). | |
To understand the contribution of atomic Fe–Nx sites and Fe nanoclusters in Fe(0)/FeNx-NC-7 to its ORR activity, SCN− and H2O2 poisoning experiments were carried out in O2-saturated 0.5 M H2SO4 solution. As shown in Fig. 6a, a significant decline in the ORR activity is observed from the LSV curve of Fe(0)/FeNx-NC-7 when SCN− ions were introduced. However, after the electrode was put back in 0.1 M KOH solution, the ORR performance gradually recovered in 20 min (Fig. 6b). This phenomenon is due to the blocking of Fe–Nx sites by SCN− ions in the acidic environment (strong interaction between Fe centers and SCN− ions) and the subsequent release under the alkaline conditions (dissociation of SCN−), well demonstrating the important role played by Fe–Nx sites in the catalysis of the ORR process. The reason that the recovered performance of Fe(0)/FeNx-NC-7 is a little lower than that in its initial state is probably due to the loss of part of the Fe clusters in the acidic environment. The ORR contribution of Fe clusters was further investigated by H2O2 poisoning experiment as metallic Fe is more easily dissolved in the peroxide-containing electrolyte. Fig. 6c compares the LSV curves of Fe(0)/FeNx-NC-7 before and after the addition of 2 mM H2O2 in 0.5 M H2SO4 solution. An obvious decline in the ORR performance can be observed with a ∼70 mV shift of E1/2, suggesting that the existence of Fe nanoclusters is beneficial for boosting the overall ORR performance of Fe(0)/FeNx-NC-7.
 |
| | Fig. 6 (a) SCN− poisoning experiments represented by RRDE polarization curves at 1600 rpm in 0.5 M H2SO4 for Fe(0)/FeNx-NC-7; (b) LSV curves of SCN− poisoned Fe(0)/FeNx-NC-7 in 0.1 M KOH at different times; (c) H2O2 poisoning experiments represented by RRDE polarization curves at 1600 rpm in 0.5 M H2SO4 for Fe(0)/FeNx-NC-7; (d) chronoamperometric responses of Fe(0)/FeNx-NC-7 and 20% Pt/C for 10 h (inset: methanol tolerance of Fe(0)/FeNx-NC-7 and 20% Pt/C with 4.4% volume methanol in 0.1 M KOH solution). | |
To evaluate the potential of Fe(0)/FeNx-NC-7 for real applications, two important aspects of the methanol crossover effect and long-term durability were tested by chronoamperometry measurement at the potential of 0.85 V. After 4.4% volume methanol was quickly injected into 0.1 M KOH solution at 300 s, 20% Pt/C shows a sharp and unrecoverable current drop of ∼40%, while limited influence on the current is observed for Fe(0)/FeNx-NC-7 (inset of Fig. 6d), suggesting its excellent methanol tolerance. The chronoamperometry response in a constant ORR process was further recorded for Fe(0)/FeNx-NC-7 to explore its durability. As shown in Fig. 6d, 88% of its initial current density can be retained after 10 h of test, higher than that of 20% Pt/C (78%), verifying the good durability of Fe(0)/FeNx-NC-7, which was further demonstrated by the long-time CV cycling measurement (Fig. 7a). After 3000 CV cycles, the half-wave potential only shows a slight shift of 2 mV with a 3.4% reduction of current density. TEM and XPS characterization studies were further performed to reveal the microstructural changes in Fe(0)/FeNx-NC-7 after this CV cycling test. The porous feature of the carbon structure is observed to be unchanged from the TEM image (Fig. 7b), whereas subtle differences have been detected by the XPS spectra. In the Fe 2p spectrum, the deconvoluted doublet of Fe2+ is found to shift to higher binding energy after CV cycling test, indicating the electrochemical oxidation of the Fe2+ during the CV cycling (Fig. 7c). Fitting the N 1s spectrum shows that the ratio of Fe–N and pyridinic N decreases with an increase of pyrrolic N after 3000 CV cycles, which suggests that part of the Fe–N and pyridinic N bonding was damaged during the continuous redox process (Fig. 7d and Fig. S8†). That might be the reason for the slight decrease in ORR performance after the CV cycling test.
 |
| | Fig. 7 (a) LSV curves of Fe(0)/FeNx-NC-7 before and after 3000 CV cycles in 0.1 M KOH solution; (b) TEM image of Fe(0)/FeNx-NC-7 after 3000 CV cycles; (c) Fe 2p and (d) N 1s XPS spectra of Fe(0)/FeNx-NC-7 before and after 3000 CV cycles. | |
As revealed by tremendous studies, atomic Fe–Nx sites should be responsible for the high ORR activity of the Fe–N–C catalyst, especially the Fe–N4 configuration. In this work, enhanced activity was obtained when atomic Fe–Nx sites and Fe nanoclusters co-exist in the carbon structure. To elucidate the mechanism of these two kinds of Fe sites for catalyzing the ORR, DFT calculations were carried out using the VASP code, investigating the effect of Fe species on the theoretical energy diagrams and electronic structure of the catalyst. According to the structure of Fe(0)/FeNx-NC-7, the Fe3-FeN4 configuration is constructed by placing a Fe3 cluster around the Fe–N4 moiety to simulate the possible interaction between Fe clusters and Fe–N4 coordination (Fig. S9a and b†). Fig. 8a displays the Gibbs free energy diagrams along the ORR reaction steps for the Fe–N4 and Fe3-FeN4 model together with the optimized adsorption states of ORR intermediates. For these two configurations, the *OOH formation step is the most sluggish step with the highest uphill energy barriers. The Gibbs free energy barrier of the rate-determining step on Fe3-FeN4 (0.22 eV) is lower than that of Fe–N4 (0.46 eV) at U = 1.23 V, which accounts for the higher ORR activity of Fe(0)/FeNx-NC-7. To gain a deeper understanding of the catalytic behavior, the local density of states (LDOS) of Fe–N4 and Fe3-FeN4 were further investigated as shown in Fig. 8b. The hybridization states between the d orbital of a single Fe atom and the p orbital of N atoms at around 0.1 and 3.2 eV are lower for Fe3-FeN4 than that for Fe–N4, indicating a weaker interaction between the single Fe atom and N atoms in Fe3-FeN4. The charge density distributions in Fig. 8c, d and Fig. S9c, d† also demonstrate that the electron-deficient state around the single Fe atom of Fe3-FeN4 is attenuated in contrast to that of Fe–N4, corresponding to the lower oxidation state of Fe. These results indicate that the additional Fe cluster in the Fe3-FeN4 configuration is able to provide extra electrons to N-doped graphene, resulting in the electronic localization of the single Fe atom. Naturally, the single Fe atom in Fe3-FeN4 can supply more electrons for bonding with the OOH intermediate as reflected in the charge density difference results of OOH adsorbed Fe–N4 and Fe3-FeN4 (Fig. 8e and f). The enhanced electron interactions will increase the adsorption energy of OOH on the single Fe atom of Fe3-FeN4, thus leading to the low energy barrier for the formation of the OOH intermediate. Consequently, high ORR activity is obtained on Fe(0)/FeNx-NC-7.
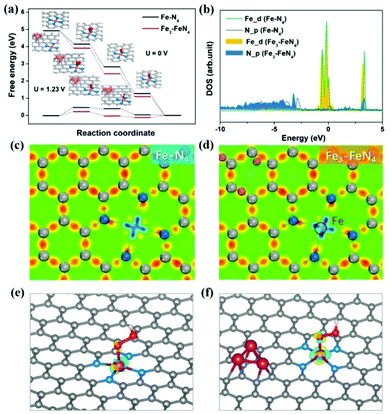 |
| | Fig. 8 (a) Potential Gibbs free energy diagrams for the ORR pathway on Fe–N4 and Fe3-FeN4 at U = 0 V (up) and U = 1.23 V (down) with the optimized configurations for intermediates adsorption; (b) the difference of DOS between Fe–N4 and Fe3-FeN4 systems; charge density distribution of (c) Fe–N4 and (d) Fe3-FeN4 systems; three-dimensional charge density difference of OOH adsorbed (e) Fe–N4 and (f) Fe3-FeN4. Electron accumulation and depletion are in yellow and blue, respectively. The isosurfaces are all set to 0.1 eV Å−3. | |
Conclusions
In summary, we designed and synthesized a model Fe–N–C catalyst with atomic Fe–Nx sites and Fe nanoclusters by using the mechanical grinding strategy followed by pyrolysis. The Fe–Phen complexes assembled by grinding a mixture of iron(II) acetate and Phen are directly used as carbon, metal, and nitrogen sources. As the sole element source, Fe–Phen complexes are expected to generate atomic Fe–Nx sites and Fe clusters at the same time due to their high density of Fe centers and Fe–N bonds. Also added to the starting materials are silica nanoparticles as hard templates to introduce porosity into the final carbon structure for better mass transfer and exposure of active sites, which have been confirmed to impede severe agglomeration of Fe centers during the pyrolysis process. As suggested by the results of electrochemical tests, the synthesized Fe(0)/FeNx-NC-7 catalyst exhibits a high overall ORR performance that is even superior to the commercial 20% Pt/C. DFT calculations indicated that the neighboring Fe nanoclusters of atomic Fe–Nx sites can increase the electron density of a single Fe atom, which drastically decreased the reaction energy barrier for *OOH formation and thus boosted the ORR activity of Fe(0)/FeNx-NC-7. These findings would provide insights into the future design and synthesis of efficient Fe–N–C catalysts for the ORR.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21875285, 22171288, and 22005340), the Key Research and Development Projects of Shandong Province (2019JZZY010331), the Natural Science Foundation of Shandong Province (ZR2020MB017) and the PetroChina Innovation Foundation (2019D-5007-0411).
Notes and references
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Oxygen electrocatalysts in metal-air batteries: from aqueous to nonaqueous electrolytes, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- Z. Jintao, X. Zhenhai and D. Liming, Carbon-based electrocatalysts for advanced energy conversion and storage, Sci. Adv., 2015, 1, 1500564 CrossRef PubMed.
- H.-F. Wang and Q. Xu, Materials Design for Rechargeable Metal-Air Batteries, Matter, 2019, 1, 565–595 CrossRef CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Modulating the local coordination environment of single-atom catalysts for enhanced catalytic performance, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- H.-Y. Wang, C.-C. Weng and Z.-Y. Yuan, Insights into efficient transition metal-nitrogen/carbon oxygen reduction electrocatalysts, J. Energy Chem., 2021, 56, 470–485 CrossRef.
- H. A. Gasteiger and N. M. Markovic, Just a Dream—or Future Reality?, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, MOF-derived electrocatalysts for oxygen reduction, oxygen evolution and hydrogen evolution reactions, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- Y. Wang, X. Zheng and D. Wang, Design concept for electrocatalysts, Nano Res., 2022, 15, 1730–1752 CrossRef CAS.
- L. Lin, Q. Zhu and A.-W. Xu, Noble-metal-free Fe-N/C catalyst for highly efficient oxygen reduction reaction under both alkaline and acidic conditions, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- W. Wang, Q. Jia, S. Mukerjee and S. Chen, Recent Insights into the Oxygen-Reduction Electrocatalysis of Fe/N/C Materials, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Fe–N–C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- X. Wang, H. Zhang, H. Lin, S. Gupta, C. Wang, Z. Tao, H. Fu, T. Wang, J. Zheng, G. Wu and X. Li, Directly converting Fe-doped metal–organic frameworks into highly active and stable Fe-N-C catalysts for oxygen reduction in acid, Nano Energy, 2016, 25, 110–119 CrossRef CAS.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Cascade anchoring strategy for general mass production of high-loading single-atomic metal-nitrogen catalysts, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- X. Zhang, X. Han, Z. Jiang, J. Xu, L. Chen, Y. Xue, A. Nie, Z. Xie, Q. Kuang and L. Zheng, Atomically dispersed hierarchically ordered porous Fe–N–C electrocatalyst for high performance electrocatalytic oxygen reduction in Zn-Air battery, Nano Energy, 2020, 71, 104547 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- Y. Zhou, Y. Yu, D. Ma, A. C. Foucher, L. Xiong, J. Zhang, E. A. Stach, Q. Yue and Y. Kang, Atomic Fe Dispersed Hierarchical Mesoporous Fe–N–C Nanostructures for an Efficient Oxygen Reduction Reaction, ACS Catal., 2020, 11, 74–81 CrossRef.
- Z. Zhang, J. Sun, F. Wang and L. Dai, Efficient Oxygen Reduction Reaction (ORR) Catalysts Based on Single Iron Atoms Dispersed on a Hierarchically Structured Porous Carbon Framework, Angew. Chem., Int. Ed., 2018, 57, 9038–9043 CrossRef CAS PubMed.
- Y. Wang, Y.-J. Tang and K. Zhou, Self-Adjusting Activity Induced by Intrinsic Reaction Intermediate in Fe-N-C Single-Atom Catalysts, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS PubMed.
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Single-Atom Fe-Nx-C as an Efficient Electrocatalyst for Zinc-Air Batteries, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS.
- X. Wang, Y. Jia, X. Mao, D. Liu, W. He, J. Li, J. Liu, X. Yan, J. Chen, L. Song, A. Du and X. Yao, Edge-Rich Fe-N4 Active Sites in Defective Carbon for Oxygen Reduction Catalysis, Adv. Mater., 2020, 32, 2000966 CrossRef CAS PubMed.
- B. Wang, X. Wang, J. Zou, Y. Yan, S. Xie, G. Hu, Y. Li and A. Dong, Simple-Cubic Carbon Frameworks with Atomically Dispersed Iron Dopants toward High-Efficiency Oxygen Reduction, Nano Lett., 2017, 17, 2003–2009 CrossRef CAS PubMed.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Identification of durable and non-durable FeNx sites in Fe–N–C materials for proton exchange membrane fuel cells, Nat. Catal., 2020, 4, 10–19 CrossRef.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. V. Voorhis, J. T. Miller and Y. Surendranath, A pyridinic Fe-N4 macrocycle models the active sites in Fe/N-doped carbon electrocatalysts, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Mullen, 3D nitrogen-doped graphene aerogel-supported Fe3O4 nanoparticles as efficient electrocatalysts for the oxygen reduction reaction, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- M. Li, L. Fan, Z. Xiao, L. Zhang, Z. Wang, Z. Kang, H. Guo, F. Dai, X. Lu and D. Sun, Micelles of Mesoporous Silica with Inserted Iron Complexes as a Platform for Constructing Efficient Electrocatalysts for Oxygen Reduction, ACS Appl. Mater. Interfaces, 2020, 12, 54720–54731 CrossRef CAS PubMed.
- R. Zhong, Y. Wu, Z. Liang, W. Guo, C. Zhi, C. Qu, S. Gao, B. Zhu, H. Zhang and R. Zou, Fabricating hierarchically porous and Fe3C-embeded nitrogen-rich carbon nanofibers as exceptional electrocatalysts for oxygen reduction, Carbon, 2019, 142, 115–122 CrossRef CAS.
- H. Tan, Y. Li, J. Kim, T. Takei, Z. Wang, X. Xu, J. Wang, Y. Bando, Y. M. Kang, J. Tang and Y. Yamauchi, Sub-50 nm Iron-Nitrogen-Doped Hollow Carbon Sphere-Encapsulated Iron Carbide Nanoparticles as Efficient Oxygen Reduction Catalysts, Adv. Sci., 2018, 5, 1800120 CrossRef PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- S. K. Singh, K. Takeyasu and J. Nakamura, Active Sites and Mechanism of Oxygen Reduction Reaction Electrocatalysis on Nitrogen-Doped Carbon Materials, Adv. Mater., 2019, 31, 1804297 CrossRef PubMed.
- L. Chai, L. Zhang, X. Wang, L. Xu, C. Han, T.-T. Li, Y. Hu, J. Qian and S. Huang, Bottom-up synthesis of MOF-derived hollow N-doped carbon materials for enhanced ORR performance, Carbon, 2019, 146, 248–256 CrossRef CAS.
- L. Liu, G. Zeng, J. Chen, L. Bi, L. Dai and Z. Wen, N-doped porous carbon nanosheets as pH-universal ORR electrocatalyst in various fuel cell devices, Nano Energy, 2018, 49, 393–402 CrossRef CAS.
- N. Wang, B. Lu, L. Li, W. Niu, Z. Tang, X. Kang and S. Chen, Graphitic Nitrogen Is Responsible for Oxygen Electroreduction on Nitrogen-Doped Carbons in Alkaline Electrolytes: Insights from Activity Attenuation Studies and Theoretical Calculations, ACS Catal., 2018, 8, 6827–6836 CrossRef CAS.
- J. Liu, P. Song and W. Xu, Structure-activity relationship of doped-nitrogen (N)-based metal-free active sites on carbon for oxygen reduction reaction, Carbon, 2017, 115, 763–772 CrossRef CAS.
- X. Yan, Y. Jia and X. Yao, Defects on carbons for electrocatalytic oxygen reduction, Chem. Soc. Rev., 2018, 47, 7628–7658 RSC.
- A. Zhu, L. Qiao, P. Tan, Y. Ma, W. Zeng, R. Dong, C. Ma and J. Pan, Iron-nitrogen-carbon species for oxygen electro-reduction and Zn-air battery: Surface engineering and experimental probe into active sites, Appl. Catal., B, 2019, 254, 601–611 CrossRef CAS.
- J. H. Kim, Y. J. Sa, H. Y. Jeong and S. H. Joo, Roles of Fe-Nx and Fe-Fe3C@C Species in Fe-N/C Electrocatalysts for Oxygen Reduction Reaction, ACS Appl. Mater. Interfaces, 2017, 9, 9567–9575 CrossRef CAS PubMed.
- Z. Liu, F. Sun, L. Gu, G. Chen, T. Shang, J. Liu, Z. Le, X. Li, H. B. Wu and Y. Lu, Post Iron Decoration of Mesoporous Nitrogen-Doped Carbon Spheres for Efficient Electrochemical Oxygen Reduction, Adv. Energy Mater., 2017, 7, 1701154 CrossRef.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Chemical vapour deposition of Fe-N-C oxygen reduction catalysts with full utilization of dense Fe-N4 sites, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Zinc-Mediated Template Synthesis of Fe-N-C Electrocatalysts with Densely Accessible Fe-Nx Active Sites for Efficient Oxygen Reduction, Adv. Mater., 2020, 32, 1907399 CrossRef CAS PubMed.
- C. Tang and Q. Zhang, Can metal–nitrogen–carbon catalysts satisfy oxygen electrochemistry?, J. Mater. Chem. A, 2016, 4, 4998–5001 RSC.
- H. Tan, J. Tang, J. Kim, Y. V. Kaneti, Y.-M. Kang, Y. Sugahara and Y. Yamauchi, Rational design and construction of nanoporous iron- and nitrogen-doped carbon electrocatalysts for oxygen reduction reaction, J. Mater. Chem. A, 2019, 7, 1380–1393 RSC.
- Y. J. Sa, D. J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T. Y. Kim and S. H. Joo, A General Approach to Preferential Formation of Active Fe-Nx Sites in Fe-N/C Electrocatalysts for Efficient Oxygen Reduction Reaction, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- M.-X. Chen, M. Zhu, M. Zuo, S.-Q. Chu, J. Zhang, Y. Wu, H.-W. Liang and X. Feng, Identification of Catalytic Sites for Oxygen Reduction in Metal/Nitrogen-Doped Carbons with Encapsulated Metal Nanoparticles, Angew. Chem., Int. Ed., 2020, 59, 1627–1633 CrossRef CAS PubMed.
- J. A. Varnell, E. C. Tse, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Identification of carbon-encapsulated iron nanoparticles as active species in non-precious metal oxygen reduction catalysts, Nat. Commun., 2016, 7, 12582 CrossRef CAS PubMed.
- W.-J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L.-J. Zhang, J.-Q. Wang, J.-S. Hu, Z. Wei and L.-J. Wan, Understanding the High Activity of Fe-N-C Electrocatalysts in Oxygen Reduction: Fe/Fe3C Nanoparticles Boost the Activity of Fe-Nx, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- X. Ao, W. Zhang, Z. Li, J. G. Li, L. Soule, X. Huang, W. H. Chiang, H. M. Chen, C. Wang, M. Liu and X. C. Zeng, Markedly Enhanced Oxygen Reduction Activity of Single-Atom Fe Catalysts via Integration with Fe Nanoclusters, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS PubMed.
- X. Zhang, X. Guo, Q. Wang, R. Zhang, T. Xu, P. Liang and X. Huang, Iron-based clusters embedded in nitrogen doped activated carbon catalysts with superior cathodic activity in microbial fuel cells, J. Mater. Chem. A, 2020, 8, 10772–10778 RSC.
- Z. Li, L. Wei, W.-J. Jiang, Z. Hu, H. Luo, W. Zhao, T. Xu, W. Wu, M. Wu and J.-S. Hu, Chemical state of surrounding iron species affects the activity of Fe-Nx for electrocatalytic oxygen reduction, Appl. Catal., B, 2019, 251, 240–246 CrossRef CAS.
- X. Sun, P. Wei, S. Gu, J. Zhang, Z. Jiang, J. Wan, Z. Chen, L. Huang, Y. Xu, C. Fang, Q. Li, J. Han and Y. Huang, Atomic-Level Fe-N-C Coupled with Fe3C-Fe Nanocomposites in Carbon Matrixes as High-Efficiency Bifunctional Oxygen Catalysts, Small, 2020, 16, 1906057 CrossRef CAS PubMed.
- M. Liu, J. Lee, T. C. Yang, F. Zheng, J. Zhao, C.-M. Yang and L. Y. S. Lee, Synergies of Fe Single Atoms and Clusters on N-Doped Carbon Electrocatalyst for pH-Universal Oxygen Reduction, Small Methods, 2021, 5, 2001165 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Iron encapsulated within pod-like carbon nanotubes for oxygen reduction reaction, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- S.-H. Yin, J. Yang, Y. Han, G. Li, L.-Y. Wan, Y.-H. Chen, C. Chen, X.-M. Qu, Y.-X. Jiang and S.-G. Sun, Construction of Highly Active Metal-Containing Nanoparticles and FeCo-N4 Composite Sites for the Acidic Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2020, 59, 21976–21979 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- C. Chen, Y. Tuo, Q. Lu, H. Lu, S. Zhang, Y. Zhou, J. Zhang, Z. Liu, Z. Kang, X. Feng and D. Chen, Hierarchical trimetallic Co-Ni-Fe oxides derived from core-shell structured metal-organic frameworks for highly efficient oxygen evolution reaction, Appl. Catal., B, 2021, 287, 119953 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Digital photos, SEM images, TEM images, XPS survey spectra, LSV curves, H2O2 yields, computational models and calculations. See DOI: https://doi.org/10.1039/d2qi00876a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Ling
Zhang‡
a,
Xuting
Li
a,
Hao
Mei
a,
Mengfei
Li
a,
Zhanning
Liu
*a,
Ling
Zhang‡
a,
Xuting
Li
a,
Hao
Mei
a,
Mengfei
Li
a,
Zhanning
Liu