
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium site occupancy and phosphate speciation in natrophosphate are invariant to changes in NaF and Na3PO4 concentration†
Trent R.
Graham
 *a,
Emily T.
Nienhuis
a,
Jacob G.
Reynolds
b,
Jose
Marcial
a,
John S.
Loring
a,
Kevin M.
Rosso
a and
Carolyn I.
Pearce
ac
*a,
Emily T.
Nienhuis
a,
Jacob G.
Reynolds
b,
Jose
Marcial
a,
John S.
Loring
a,
Kevin M.
Rosso
a and
Carolyn I.
Pearce
ac
aPacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: trenton.graham@pnnl.gov
bWashington River Protection Solutions, LLC, Richland, Washington 99352, USA
cDepartment of Crop and Soil Sciences, Washington State University, Pullman, Washington 99164, USA
First published on 7th July 2022
Abstract
Recent interest in the crystal structure of natrophosphate (Na7FPO4·19H2O) has sought to better understand the propensity of this phase to vary in composition through either alteration of the fluoride/phosphate ratio or subspeciation of phosphate (PO43−) into hydrogen phosphate (HxPO43−x). To address questions brought up in the analysis of the solubility of natrophosphate in multicomponent NaF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Na3PO4:H2O systems relevant to fundamental studies of select geological deposits and industrial processes pertinent to radioactive waste, this work deploys X-ray diffraction, scanning electron microscopy, Raman spectroscopy, Fourier transform infra-red spectroscopy, solid-state multinuclear (1H, 31P, 19F, and 23Na) Magic Angle Spinning Nuclear Magnetic Resonance (MAS NMR) spectroscopy and 23Na multiple quantum MAS NMR spectroscopy. Scrutiny of the integrated microscopy, diffraction and spectroscopy results indicate that the crystalline structure and stoichiometry of natrophosphate are invariant to changes in the fluoride/phosphate ratio of the solution and that formation of hydrogen phosphate can be neglected. This improved knowledge of the chemical composition of natrophosphate crystallites will aide in the application of solubility models relevant to geological studies and radioactive waste processing.
:Na3PO4:H2O systems relevant to fundamental studies of select geological deposits and industrial processes pertinent to radioactive waste, this work deploys X-ray diffraction, scanning electron microscopy, Raman spectroscopy, Fourier transform infra-red spectroscopy, solid-state multinuclear (1H, 31P, 19F, and 23Na) Magic Angle Spinning Nuclear Magnetic Resonance (MAS NMR) spectroscopy and 23Na multiple quantum MAS NMR spectroscopy. Scrutiny of the integrated microscopy, diffraction and spectroscopy results indicate that the crystalline structure and stoichiometry of natrophosphate are invariant to changes in the fluoride/phosphate ratio of the solution and that formation of hydrogen phosphate can be neglected. This improved knowledge of the chemical composition of natrophosphate crystallites will aide in the application of solubility models relevant to geological studies and radioactive waste processing.
1. Introduction
The local polyhedral coordination and solvation environment of cations are of fundamental importance in understanding solution structure and ion–ion assembly,1–3 but experimental techniques often measure ensembles of these species in solution-state systems.4–6 One strategy to analyze sub ensembles of complex electrolyte species such as polyatomic ion aggregates is to unravel the spectroscopic contributions of distinct individual species through the use of model salt hydrates that preserve relevant chemical motifs at the molecular level.7,8 Our group has pioneered this approach for alkali aluminate salts relevant to nuclear waste processing,9–12 and this work expands the strategy of using model salt hydrates to study polyatomic ion aggregates of relevance to geochemistry and radioactive waste to a new ternary electrolyte compositions by investigating the crystal structure of natrophosphate (Na7FPO4·19H2O).The unit-cell structure of natrophosphate is shown in Fig. 1A, and the crystal structure contains two unique sodium ion (Na+) sites in which Na+ occurs in two distinct coordination environments.13–15 The first Na+ site drawn in Fig. 1B encapsulates a fluoride ion (F−) in a supramolecular ion assembly of six, edge linked Na+ octahedra comprising the polyatomic super-octahedral ion aggregate, Na6(H2O)18F5+. The local environment around the Na+ within the super-octahedral ion aggregate is Na(H2O)5Fδ+, where the partial (δ) charge on the ion-pair is due to the association of an F− ion with 5 other Na+ ions. The second unique site of Na+ shown in Fig. 1B is associated with discrete, monomeric phosphate (PO43−) ions, and the phosphate ions link the supramolecular Na–F ion complexes in a complex polyatomic aggregate Na(H2O)(PO4)25−. Note that the tetrahedral Na+ in site 2 is partially occupied with an occupancy factor of 0.5, and water exists in the unoccupied sites.16 Based on the summation of these ion-aggregate motifs, the crystal structure of natrophosphate can be formulized as Na6F(H2O)18(NaH2O)(PO4)2.16
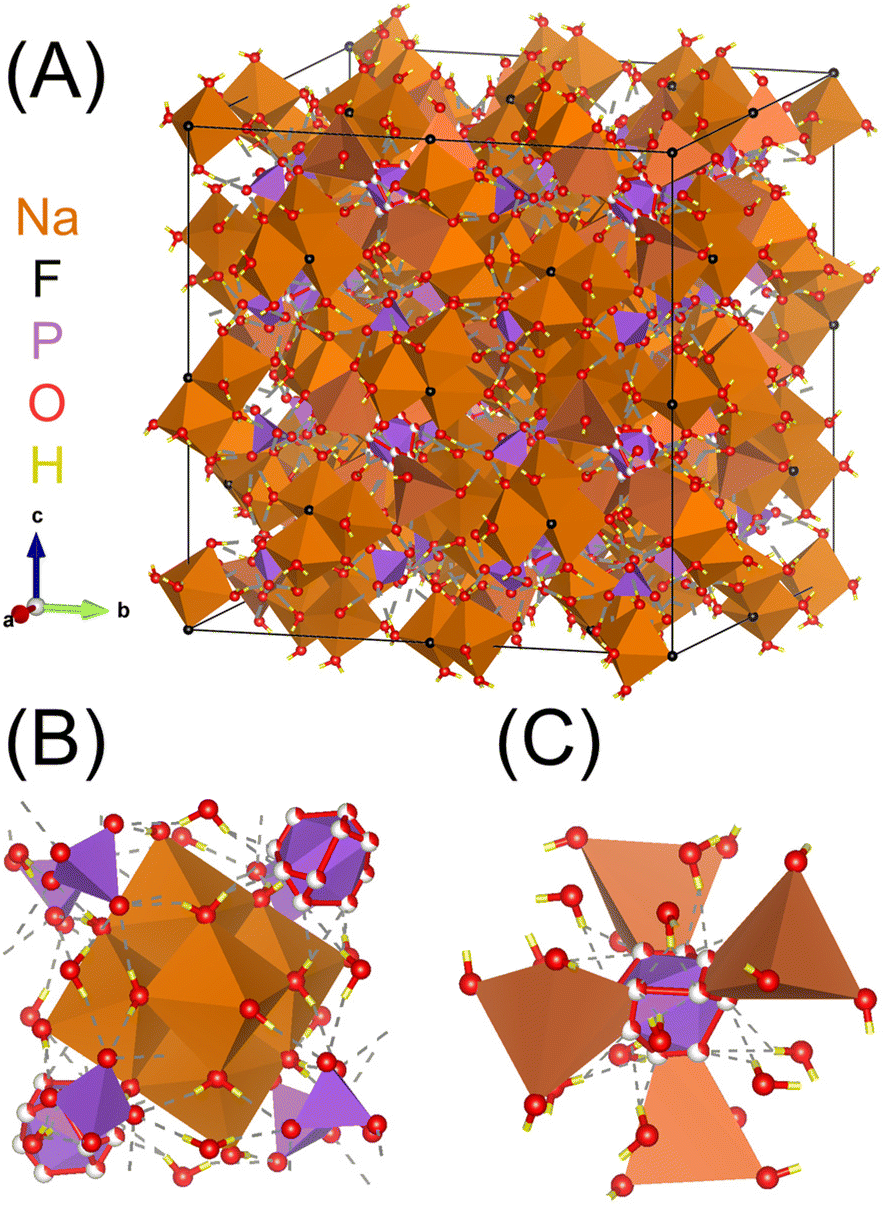 | ||
| Fig. 1 (A) Unit cell of natrophosphate13 and (B) truncated crystal structures highlighting the local chemical environment of the octahedral Na+ in site 1 and the (C) local chemical environment of the tetrahedral Na+ in site 2. Note that partial occupancy of the oxygens is depicted with partially filled spheres, and that the tetrahedral Na+ in site 2 is also partially occupied.16 Based on the size of the unit cell of natrophosphate, the complexity of the hydrogen bond network of water, and partial occupancy of sodium, the structure of natrophosphate is quantitively classified17 as structurally very complex based on information-based parameters.18,19 | ||
Beyond the interest in fundamental chemistry of the disparate environments of Na+ ions in this crystal structure, natrophosphate is of interest because it occurs in alkaline rocks,14,18–20 and is present in radioactive waste stored at the Hanford Site in Richland, WA, United States.20–23 Natrophosphate is one of several fluoride or phosphate-containing salts found in the waste. The fluoride ions are partitioned in solid phase salts between natrophosphate, sodium fluoride (NaF) and kogarkoite (Na3FSO4). The quantity of each salt depends on the relative abundance of PO43−, F− and sulfate (SO42−) in each waste tank.21–24 Likewise, PO43− are partitioned in solid phase salts between natrophosphate, sodium phosphate dodecahydrate (Na3PO4·0.25NaOH·12H2O) and nastrophite (NaSr(PO4)·9H2O).24–26
Previous studies of the solubility of natrophosphate in multicomponent NaF:Na3PO4:H2O systems have suggested that the natrophosphate composition changes based on solution compositions and that the crystalline phase is a solid-solution.27 Likewise, the composition of natrophosphate has been hypothesized to be variable due to partial replacement of the PO43− groups in the structure with HxPO43−x (x = 1 or 2).13,19 Substitution of hydrogen into phosphates containing mono and trivalent cations to occurs in other complex phosphates precipitated from acidic solutions,28 which is explained since hydrogen phosphate and dihydrogen phosphate are the predominant species at low pH conditions.29 If occurring in natrophosphate crystals, the formation of the HxPOx3−x would necessitate a reduction in Na+ in site 2 to satisfy the balance of the charge, specifically resulting in the postulated stoichiometry of natrophosphate occurring as Na7−xHxFPO4·19H2O. A third hypothesis presented regarding the composition of natrophosphate is that the crystalline structure and stoichiometry are invariant with respect to F/HxPO43−x ratio and that formation of hydrogen phosphate can be neglected in compositionally pure NaF:Na3PO4:H2O ternary systems with respect to changes in solution composition.30 Modeling the solubility of natrophosphate solubility has been difficult.31 A changing natrophosphate composition with changing solution composition would explain this difficulty.
The primary contribution of this paper is to fill the knowledge gap regarding whether natrophosphate is (i) a solid-solution of NaF and Na3PO4 hydrate (ii) a complex phosphate in which PO43− ions speciate into HxPO43−x ions, or (iii) if natrophosphate is indeed of a single crystalline form. This is done through multimodal experimental characterization of natrophosphate crystals precipitated at two Na3PO4/NaF compositions in the NaF:Na3PO4:H2O ternary system. The techniques include X-ray diffraction, scanning electron microscopy, Raman spectroscopy, Fourier transform infra-red spectroscopy, solid-state multinuclear (1H, 31P, 19F, and 23Na) Magic Angle Spinning Nuclear Magnetic Resonance (MAS-NMR) spectroscopy and 23Na multiple quantum MAS-NMR spectroscopy. The microscopy, diffraction and spectroscopy results are consistent with the notion that the chemical composition of Natrophosphate is independent of the NaF and NaPO3 concentrations in solution and that formation of HxPO43−x ions does not occur in significant quantities or vary with changes in NaF and NaPO3 concentrations in solution.
2. Experimental methods
2.1 Crystallization of natrophosphate
Natrophosphate was crystallized by cooling sodium fluoride and sodium phosphate solutions with varied phosphate to fluoride ratios. Specifically, to prepare natrophosphate from a sodium fluoride-rich solution (F-rich, mole ratio of NaF/Na3PO4 = 2), a solution of sodium fluoride was prepared by dissolving 1.6795 g of sodium fluoride (NaF, >98%, Sigma-Aldrich) into 40 g of water (18 MΩ cm) and a separate solution of sodium phosphate was prepared by dissolving 3.2788 g sodium phosphate tribasic (Na3PO4·12H2O, >96%, Sigma-Aldrich) into 20 g of deionized water. The solutions were separately heated on a hotplate to a temperature of 70 °C measured by thermometer (Thermco® precision ground glass thermometer) and then mixed with vigorous stirring. After mixing for 30 seconds, the stir-bar was removed, and the solution was stored overnight at 20 °C. To prepare natrophosphate from a fluoride-poor solution (F-poor, mole ratio of NaF/Na3PO4 = 0.25), an NaF solution was prepared by the dissolution of 0.8398 g of NaF salt into 20 g of deionized water and an Na3PO4 solution was prepared by the dissolution of 13.1152 g of Na3PO4·12H2O salt in 80 g of deionized water. The solutions were separately heated on a hotplate to a temperature of 70 °C measured by thermometer and then mixed with vigorous stirring. After mixing for 30 seconds, the stir-bar was removed, and the solution was stored for 16 hours at 20 °C. For both solution compositions, the Natrophosphate crystals were separated from the solution via vacuum filtration after 16 hours, and ground with an Agate mortar and pestle (Cole-Parmer) prior to characterization.2.2 Powder X-ray diffraction
Powder X-ray diffractograms of natrophosphate synthesized in F-rich or F-poor solution compositions were acquired on a Rigaku Smartlab SE instrument, with a Cu sealed anode X-ray source (λ = 1.54056 angstroms) which was operating at 40 kV and 44 mA, with a θ–2θ goniometer, and a 1-dimensional semiconductor detector. The acquisition speed was 1.8° min−1 and the step size was 0.01°. Rietveld fits of the powder XRD patterns were carried out using Topas v6 (Bruker AXS) and the natrophosphate structure published by Genkina and Khomyakov32 with the atomic coordinates not refined. Peak shapes were calculated using the fundamental parameters approach, with instrumental factors established with patterns obtained from NIST silicon (SRM 640D) and LaB6 (SRM 660A) powders. Line broadening from finite crystallite size was modeled using the double-Voigt approach.332.3 Scanning electron microscopy
Scanning electron micrographs were acquired on a Helios NanoLab SEM (FEI, Hillsboro, Oregon) by collection of secondary electrons with an Everhart–Thornley detector. The dwell time was 200 ns, the frame rate was 317.4 ms, and 128 frames were integrated for each micrograph. The electron beam voltage was 5.0 kV and the current was 0.17 nA. The contrast of the micrographs was optimized in ImageJ (v 1.53c).2.4 Raman spectroscopy
Raman spectra were acquired with a Horiba LabRam HR spectrometer installed on an inverted optical microscope (Nikon Ti-E) with a 632.8 nm HeNe laser light source and a 40× objective. Data was collected via a backscattering geometry setup in which the scattered light was returned through the same 40× objective, transmitted through a beam splitter, dispersed through a 1800 g mm−1 grating and detected by an EM-CCD detector. The spectral window was between wavenumbers of 100–4000 cm−1. A select region of the spectrum spanning between 350 and 1100 cm−1 where diagnostic bands of PO43− vibrations occur was processed via application of a spline function and then pure Lorentzian line shapes were used to fit the data. Regression of the Lorentzian line shape parameters was performed in Excel (Microsoft, version 2108) using the generalized reduced gradient method through which the line shape parameters were optimized to minimize the sum of the squared residuals between the data and the fit.2.5 Fourier transform infra-red spectroscopy
FTIR spectra were collected on a Bruker Vertex80 V under the rough vacuum of the spectrometer with a DTGS detector and a standard globar source with a multi bounce diamond ATR cell. The background was the clean diamond cell, and the sample was the solid was pressed against the diamond. The resolution was 4 cm−1, the number of background scans was 512, and the number of scans to acquire spectra of the sample was 128. A select region of the spectrum spanning between 850 and 1100 cm−1 where diagnostic bands of PO43− vibrations occur was processed via application of a spline function and then pure Gaussian line shapes were used to fit the data. Regression of Gaussian line shape parameters was performed in Excel.2.6 Multinuclear MAS NMR spectroscopy
1H, 19F, 31P, and 23Na MAS NMR spectroscopy were performed on an NMR spectrometer (Bruker) energized at a field strength of 14.0954 T. At 14.0954 T, the Larmor frequencies of 1H, 19F, 31P, and 23Na are 600.130, 564.686, 242.938, and 132.294 MHz, respectively. The solid-state MAS NMR spectra of the natrophosphate crystals were acquired with a 2.5 mm HXY probe operating in double resonance mode. The samples were placed within 2.5 mm zirconia rotors using Vespel drive and bottom caps. MAS NMR spectra were then collected with a MAS spin rate of 32 kHz.Single pulse, direct excitation 1H MAS NMR spectra of natrophosphate were acquired using a π/4 pulse width corresponding to a 1.4 μs pulse length (89.3 kHz), which was calibrated using a separate sample composed of solid adamantane (C10H16, ≥99%, Sigma-Aldrich). The chemical shift of the 1H resonance of natrophosphate is calibrated to the 1H resonances of the protons of adamantane, which corresponds to two overlapping resonances and was assigned to δ = 1.8 ppm. The 1H MAS NMR spectra were acquired with a sweep width of 793.476 ppm, a collection of 32 transients, an acquisition time of 24.9858 ms enumerated with 23796 time domain points, and a recycle delay between transients (d1) of 10 s. Post-acquisition processing of the 1H NMR spectra was done in Mestrenova (version 14.01-23559, released 2019-06-07, Mestrelab Research S.L.). Exponential line broadening of 5 Hz was applied. The 1H NMR spectra were fit using a single purely Lorentzian line shape. Regression of Lorentzian line shape parameters was performed in Excel.
Single pulse, direct excitation 19F MAS NMR spectra were measured with a π/4 pulse width (55.6 kHz) corresponding to a duration of 2.25 μs which was calibrated via a 19F pulse nutation experiment on a separate reference of sodium fluoride (NaF, ≥99%, Sigma-Aldrich). The 19F chemical shift of the resonance of natrophosphate is referenced to the 19F chemical shift of the resonance of NaF (δ = −224.2 ppm, with respect to CFCl3 occurring at δ = 0 ppm).3419F MAS NMR spectra were acquired with a sweep width of 1967.79 ppm, a collection of a single transient on a fully-relaxed sample, and an acquisition time of 9.9495 ms enumerated with 22110 time domain points. Post-acquisition processing of the 19F MAS NMR spectra was performed in Mestrenova, where the 19F MAS NMR spectra were zero-filled to 256k points, and then exponential line broadening of 1 Hz was applied. The 19F NMR spectra were fit using a single purely Lorentzian line shape. Regression of Lorentzian line shape parameters was performed in Excel.
Single pulse, direct excitation 31P MAS NMR spectra were obtained with a 1.56 μs excitation pulse (80.1 kHz), which was equivalent to a π/4 pulse width for an aqueous 85 wt% phosphoric acid solution (H3PO4 in H2O, ≥99.99% trace metal basis, Sigma-Aldrich) and the 31P resonance of this solution was also used as a 31P chemical shift reference (δ = 0 ppm). A sweep width of 980.072 ppm, an acquisition time of 49.2366 ms enumerated with 23446 time domain points, a collection of 16 transients, and a d1 of 120 s were used for the single pulse direct excitation 31P MAS NMR spectra. Acquisition of a series of spectra in which the d1 time was varied and subsequent analysis indicated that 120 s was sufficient to avoid saturation effects. Post-acquisition processing of the spectra was performed in Mestrenova, where the spectra were zero-filled to 32k points, and 5 Hz of exponential line broadening was applied. The 31P NMR spectra were fit using two purely Lorentzian line shapes. Regression of Lorentzian line shape parameters was performed in Excel.
Single pulse, direct excitation 23Na MAS NMR spectra were obtained with a 0.45 μs excitation pulse (55.6 kHz), which was equivalent to a π/20 pulse width for a 1 M sodium chloride (NaCl, ≥99%, Sigma-Aldrich) solution prepared in H2O. The 23Na resonance of this solution was also used as the 23Na chemical shift reference (δ = 0 ppm). A sweep width of 7874.24 ppm, an acquisition time of 17.8992 ms enumerated with 44748 time domain points, a collection of 512 transients, and a d1 of 0.5 s were used for the single pulse direct excitation 23Na MAS NMR spectra. Acquisition of a d1 nutation array and subsequent analysis indicated that 0.5 s was sufficient to avoid saturation effects. Post-acquisition processing of the spectra was performed in Mestrenova, where 2 Hz of exponential line broadening was applied. The 23Na MAS NMR spectra were fit in ssNAKE (v 1.3) using the finite MAS, quadrupole model, where the quadrupolar line shape parameters were regressed by minimizing the residual between the data and the fit using the Powell minimization method.35
2.7 Multiple quantum MAS NMR spectroscopy
23Na MQMAS NMR spectra were acquired at a field strength of 14.1 T using the aforementioned Bruker, 2.5 mm MAS probe with a z-filter, 23Na 3QMAS pulse sequence (mp3qzqf) at a spinning rate of 32 kHz. For all samples, the power level for the excitation pulse and conversion pulse was −20 dB, the power level for the selective pulse was 5.23 dB, the time between the second and third pulse (D4) was 20 us, the acquisition time was 14.9424 ms and the spectral width for F1 (evolution) dimension was 32 kHz. Pulse widths were optimized and the P1, P2, and P3 pulse lengths were 4.8, 2.5, and 25.0 µs. The delay between scans was 0.5 seconds, 108 transients were collected, with 768 increments. The width in the F2 dimension was 357.9199 ppm. Acquisition utilized States-Time Proportional Phase Incrementation (TPPI) processing, and the number of processed increments was truncated to 350 from the original 768. The 2D MQMAS spectrum was sheared in Topspin v(3.5.7).3. Results and discussion
The results and discussion of this manuscript are organized as follows. First, X-ray diffraction and scanning electron microscopy were used to ascertain the purity and analyze the crystal morphology of the natrophosphate formed from F-rich or F-poor solution conditions. Then, vibrational spectra of natrophosphate were acquired with Raman spectroscopy and FTIR spectroscopy to analyze the vibrational modes of the PO43− ion to ascertain the lack of formation of hydrogen phosphate or dihydrogen phosphate. Next, the isotope specificity of multinuclear MAS NMR spectroscopy was leveraged to interrogate the bulk resonance from the water in the hydrating salt, the chemical environment of F− ions in the Na5F4+ subunit, and the PO4− speciation with 1H, 19F, and 31P MAS NMR spectroscopy, respectively. 23Na MAS NMR and 23Na MQMAS NMR spectroscopy were respectively used to directly quantify the relative site abundance of Na ions in the two sites of natrophosphate and ascertained that the chemical composition of natrophosphate is invariant across the experimental conditions.3.1 Crystal structure and morphology of natrophosphate synthesized from F-rich and F-poor conditions
X-ray diffractograms of the natrophosphate crystals synthesized from F-rich and F-poor solution compositions are shown in Fig. 2A. All significant reflections obtained experimentally can be attributed to the natrophosphate phase, indicating that there are no significant crystalline impurities. The procedure to refine the XRD diffractogram in TOPAS necessitated the use of preferential orientation to improve the fit of the diffractogram. Based on the full-width-at-half-maxima of the reflections, analysis of the refinements conducted in TOPAS indicates that for natrophosphate crystallized in F-rich and F-poor conditions that the crystallite sizes are greater than 250 nm. The refined unit cell parameter for the F-rich sample was a = 27.7757(2) Å, and the refined parameter for the F-poor sample was a = 27.7635(2) Å. The small magnitude of the difference between the two samples is likely due to sample packing in the XRD cell, which results in some offset in sample height. The refined unit cell parameter in this work is in good agreement with a recent study of the natrophosphate mineral (a = 27.712(2) Å).19 A summary of the goodness of fit and R values for the powder XRD is shown in Table 1. While Rietveld refinement is a technique to measure site occupancies, many other groups in literature have analyzed natrophosphate with X-ray diffraction and subsequential refinements.13,19,32 Despite this, because the X-ray scattering contribution of light atoms, such as H+, are low, the conclusiveness of refinements of XRD data in discriminating HPO42− from PO43− in the natrophosphate salt is limited.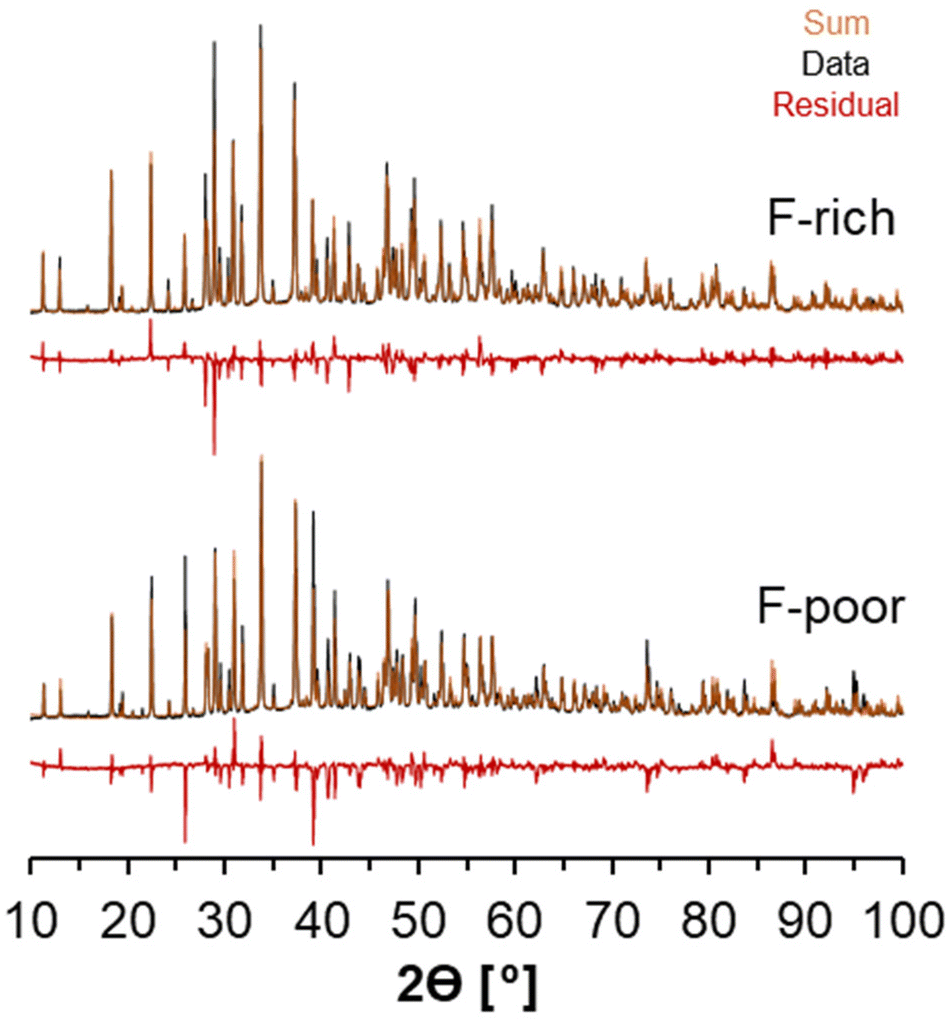 | ||
| Fig. 2 X-ray diffractograms of natrophosphate synthesized in F-rich and F-poor conditions and the corresponding Rietveld refinements are shown. Note that the intensity of the reflections and the residual between the data and the Rietveld refinement are shown with square root intensity units. The diffractograms and residuals are vertically offset. The residual between the data and the fit diffractogram for both samples is attributed to preferential orientation. | ||
| Sample | a [angstroms] | Size [nm] | GOF | Bragg-R | R p | Rwp |
|---|---|---|---|---|---|---|
| F-Poor | 27.764 | >250 | 8 | 10% | 9% | 13% |
| F-Rich | 27.777 | >250 | 7 | 9% | 8% | 12% |
Note that analysis of subsequent X-ray diffraction experiments of the same batches of natrophosphate after 2 years of storage at 20 °C in a sealed cryogenic vial (2 mL, Corning, internal thread/washer/self-stand) under N2 in an N2-filled glovebox indicated that the residual could be reduced through finer homogenization of the sample. The diffractograms are shown in the ESI.† In addition, inspection of this subsequent XRD results indicated that after 2 years of storage in an N2-filled glovebox, additional reflections were found in trace amounts, which are attributed to trace amounts of sample decomposition. While the decomposition of natrophosphate is outside the scope of this study, the reflections could not be fit by a single phase of sodium orthophosphate, protonated sodium phosphate, sodium carbonate, nor sodium fluoride, nor hydrates of these phases.
The large crystallite size determined through refinement of the XRD results was then scrutinized through scanning electron microscopy. In Fig. 3A–D, representative scanning electron micrographs of natrophosphate synthesized from F-rich and F-poor solutions conditions are shown with higher magnification images of select regions outlined in yellow. The crystals of natrophosphate synthesized from either solution condition are observed to be greater than 50 microns in diameter with euhedral surfaces. Adsorbed to the euhedral surfaces are smaller crystals of natrophosphate generated from homogenizing the salt with a mortar and pestle. The XRD and SEM results indicate that the natrophosphate synthesized from F-rich and F-poor conditions are similar in that natrophosphate is the only crystalline phase and that the produced salt is euhedral with crystals between 5 and 50 microns in size. As shown in the ESI,† scanning electron microscopy energy-dispersive X-ray spectroscopy analysis of the natrophosphate samples indicate that the compositions of the samples are within error of each other and also within error of the nominal composition of natrophosphate. Therefore, vibrational spectroscopy and MAS NMR spectroscopy was performed to define the molecular structure of the constituents of the natrophosphate crystals.
 | ||
| Fig. 3 Scanning electron micrograph of natrophosphate synthesized in (A and C) an F-rich solution composition and (B and D) an F-poor solution composition. Note that in parts A and B, the region highlighted in yellow indicates that area selected for imaging at higher magnification. The micrographs obtained at higher magnification are shown in parts C and D. | ||
3.2 Molecular level description of natrophosphate synthesized from F-rich and F-poor conditions
The Raman spectra of natrophosphate synthesized from F-rich and F-poor sodium phosphate solutions are shown in Fig. 4 over a spectral range of 350 to 1100 cm−1. This region contains a series of diagnostic bands that can be attributed to the Raman active modes of the phosphate anions in natrophosphate. Specifically, the Raman active modes of the phosphate anion include the symmetric stretching mode of PO43− (v1), the out-of-plane bending mode of PO43− (v2), the antisymmetric stretching mode of PO43− (v3), and the in-plane bending mode of PO43− (v4).36,37 The lack of variability in the phosphate bands as a function of F− and PO43− concentrations in the mother liquor suggests that variability in phosphate speciation, such as the production of hydrogen phosphate or dihydrogen phosphate anions, does not occur under either of the synthesis conditions. Formation of hydrogen phosphate is expected to produce a Raman band of moderate intensity assigned to v2-POH near 850 cm−1 based on hydrated salts that contain the HPO42− motif.38 The full spectral width spanning between 100–4000 cm−1 of the Raman spectra acquired for these salts is available in Table 2, and the similarity of the natrophosphate crystals prepared under F-rich and F-poor conditions extends across the full spectral window. The Raman spectra acquired from natrophosphate synthesized in F-rich and F-poor solution compositions are in good agreement both in the peak maxima of the Raman band, the full-width-at-half-maxima (fwhm) and the relative intensity as shown in the ESI.† Avdontceva et al. hypothesized that protonated phosphate species formed in their sample, but there is no peak at 850 cm−1 in the Raman spectra they published either.19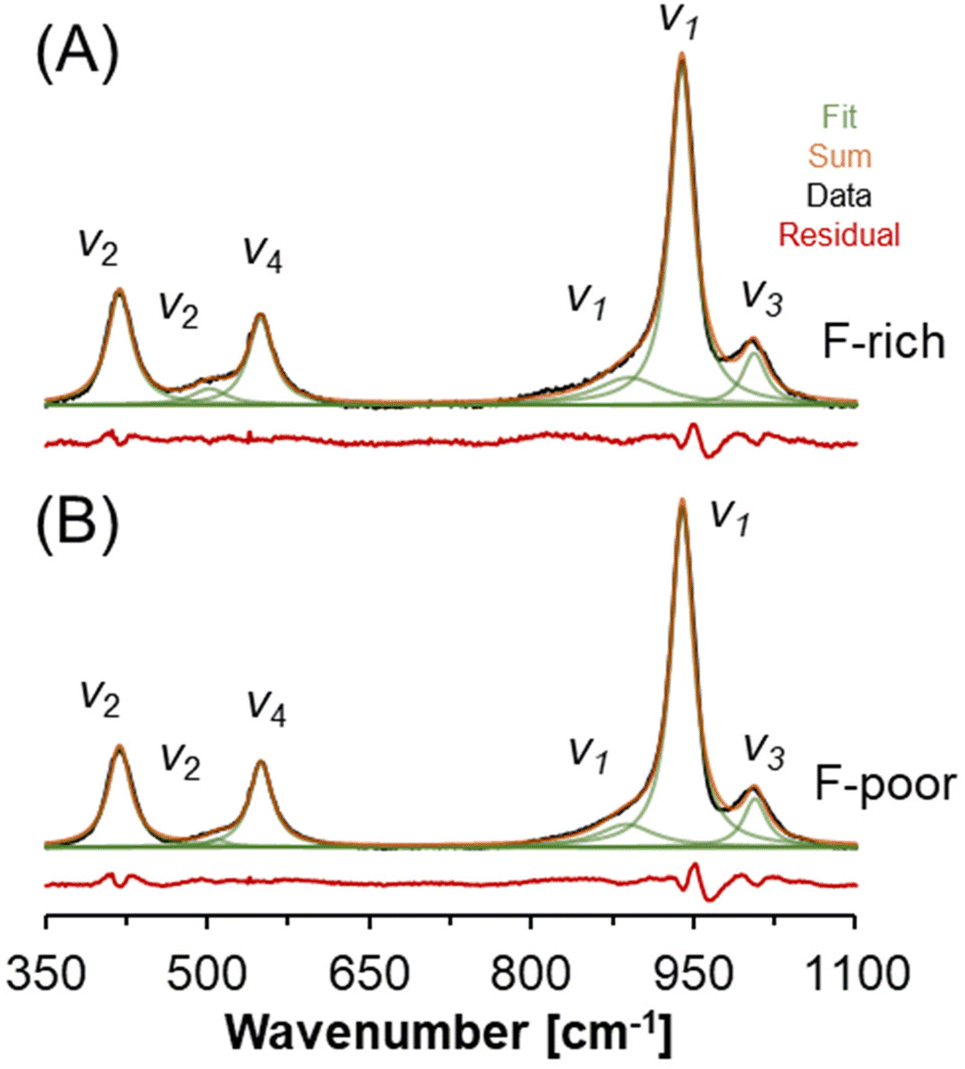 | ||
| Fig. 4 Raman spectroscopy of natrophosphate crystallized from the (A) F-rich and (B) F-poor solution conditions. The vibrational modes of the PO43− ion are annotated. | ||
| F-Poor | F-Rich | Assignment36,37 | ||||
|---|---|---|---|---|---|---|
| Wavenumber [cm−1] | fwhm [cm−1] | Intensity [%] | Wavenumber [cm−1] | fwhm [cm−1] | Intensity [%] | |
| 418.8 | 25.6 | 15.7 | 418.8 | 27.5 | 16.5 | v 2 |
| 509.6 | 23.4 | 1.1 | 502.1 | 40.3 | 3.6 | v 2 |
| 549.6 | 25.7 | 13.6 | 549.3 | 27.8 | 13.2 | v 4 |
| 887.7 | 63.2 | 8.7 | 890.2 | 74.9 | 10.8 | Shoulder of v1 |
| 939.6 | 24.9 | 53.2 | 939.1 | 26.2 | 48.5 | v 1 |
| 1006.7 | 25.9 | 7.8 | 1006.0 | 27.1 | 7.5 | v 3 |
A select region of the FTIR spectra acquired from natrophosphate synthesized in either F-rich or F-poor solutions is shown in Fig. 5. Two FTIR-active modes fall within this region: (i) the symmetric stretching vibration of PO43− (v1) near 950 cm−1 and (ii) the asymmetric stretching vibration of PO43− (v3) near 1020 cm−1.39 The splitting of these two modes into multiple bands has been attributed to crystalline field effects,39 and at least three Gaussian components are needed to fit the spectra in this region. The similarity between the FTIR spectra of the natrophosphate synthesized in either condition is evident both from the total spectrum and the line shape parameters of the deconvoluted components. If HxPO43−x ions were present in significant quantities, additional FTIR bands at about 1200 cm−1 and 860 cm−1 would be expected based on infrared studies of H3PO4 solutions that were titrated with NaOH,40 density functional theory based calculations of infrared spectra,41 and Born-Oppenheimer molecular dynamics simulations followed by calculation of infrared spectra from trajectories.42 The line shape parameters of the deconvoluted FTIR spectra are shown in Table 3.
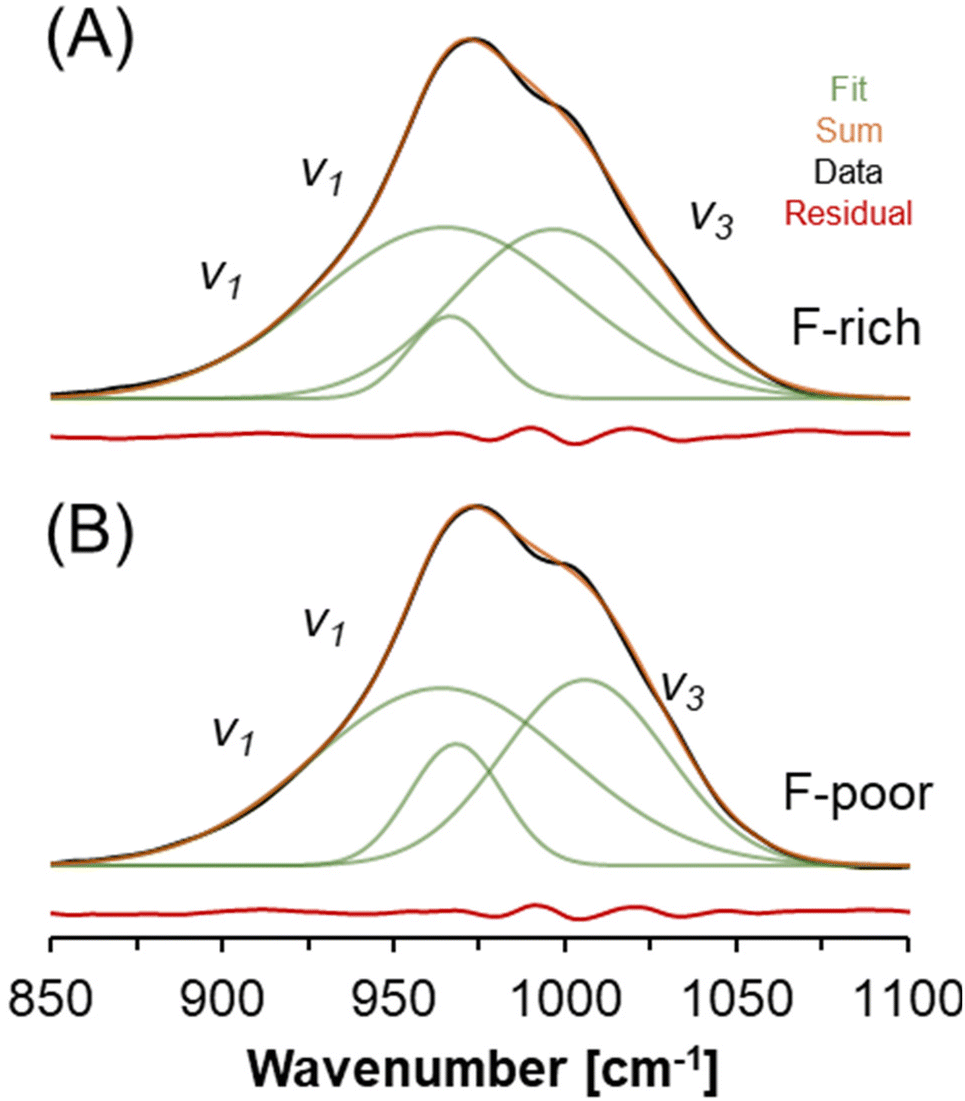 | ||
| Fig. 5 FTIR spectroscopy of natrophosphate crystallized from the (A) F-rich and (B) F-poor solution conditions. The vibrational modes of the PO43− ion are annotated. | ||
| F-Poor | F-Rich | Assignment39 | ||||
|---|---|---|---|---|---|---|
| Wavenumber [cm−1] | fwhm [cm−1] | Intensity [%] | Wavenumber [cm−1] | fwhm [cm−1] | Intensity [%] | |
| 964.0 | 37.4 | 52.0 | 965.1 | 38.0 | 52.6 | v 1 |
| 968.3 | 13.5 | 12.8 | 966.6 | 12.0 | 7.9 | v 1 |
| 1006.0 | 24.1 | 35.1 | 996.8 | 28.7 | 39.5 | v 3 |
Multinuclear MAS NMR spectroscopy was conducted to characterize the local chemical environment of H2O, F−, PO43− and Na+ ions in natrophosphate crystals grown in F-rich or F-poor solution conditions via1H, 19F, 31P and 23Na, respectively. The 1H MAS NMR spectra of the natrophosphate samples is shown in Fig. 6A. The 1H resonance of natrophosphate crystallized from either solution condition displays a single resonance, indicating that the protons of water are undergoing chemical exchange on the millisecond timescale. The resulting ensemble resonance is well-fit by a single Lorentzian function with a peak maximum of approximately 6 ppm and a fwhm of 4000 Hz. Both the peak maxima and the fwhm of the 1H resonance are similar between the two samples. Based on the reported crystal structure of natrophosphate, there are 7 unique water sites. However, a confounding complexity is that the tetrahedral sodium site has a partial occupancy factor of 0.5. It is likely that the water proximal to the tetrahedral sodium (versus an unoccupied tetrahedral sodium site) would have a perturbed chemical shift. Therefore, there are well over 7 proton resonances, with the exact number contributing to this distribution depending on the extent of effects arising from the next nearest neighbor of water, and the 2nd nearest neighbor effects from water, with respect to tetrahedral sodium site occupancies and vacancies.
 | ||
| Fig. 6 Multinuclear single pulse direct excitation MAS NMR of natrophosphate synthesized from an F-rich or F-poor solution condition. (A) 1H MAS NMR spectra, (B) 19F MAS NMR spectra, (C) and (D) 23Na MAS NMR spectra are shown. The line fit parameters are reported in the Supporting Information. | ||
19F MAS NMR spectra of natrophosphate crystallized in F-rich and F-poor solution conditions are shown in Fig. 6B. A single resonance is observed for both samples in the 19F MAS NMR spectra, which is well-fit by a Lorentzian function centered at approximately −222.7 ppm and both the location and the fwhm of the resonance are similar between the two samples. The fwhm of the 19F MAS NMR resonances of the two samples is about 270 Hz. The presence of a single 19F resonance is in good agreement with the XRD-derived crystal structure of natrophosphate,13 where only a single F− site was refined.
31P MAS NMR spectra of natrophosphate crystallized in solutions that were F-rich or F-poor are shown in Fig. 6C. The 31P MAS NMR spectra of natrophosphate crystallized under either solution condition exhibits two resonances. The predominant resonance occurs at 8.3 ppm with a second resonance occurring at about 7.6 ppm. The signal is well-fit by two purely Lorentzian line shapes, with respective fwhm of about 270 and 140 Hz, respectively. The presence of two resonances in the 31P MAS NMR spectra is in agreement with the XRD-derived crystal structure of natrophosphate.13 Notably, the XRD-derived crystal structure of natrophosphate13 also indicates that the relative abundance of PO43− in the two sites are 3 to 1, and that ratio is in excellent agreement with the relative integrals of the 31P MAS NMR resonances.
Similarly, the 23Na MAS NMR spectra shown in Fig. 6D, of natrophosphate synthesized in F-rich or F-poor solutions contains two resonances, which is in agreement with the two sites present in the XRD-derived crystal structure of natrophosphate.13 The relative abundance of sodium in site 1 proximal to F and site 2 proximal to PO43− is invariant to perturbation of F−/PO43− ratio in the mother liquor, and the relative integrated signal intensity of 6 to 1 matches the expected site abundance based on the XRD-derived crystal structure of natrophosphate.13 Notably, hydrogen phosphate is not present in appreciable amounts since its presence would necessitate a reduction in the relative abundance of the resonance at 4 ppm, attributed to tetrahedral Na+ proximal to the PO43− ion, to maintain the balance of charge. The isotropic chemical shift of the 23Na MAS NMR resonance of the predominant line is about 8 ppm, while the isotropic chemical shift of the minor resonance is about 4.4 ppm. The quadrupolar coupling coefficient of the two resonances are 0.9 and 0.4, respectively, for a fixed asymmetry parameter of a value of 0. As shown in the ESI,† the lack of archetypical quadrupolar line shapes was also found at 7.05 T, which is consistent with 23Na quadrupolar coupling coefficients with magnitudes less than 1 MHz. The Lorentzian line shape parameters for the 1H, 19F, and 31P MAS NMR spectra and the quadrupolar line shape parameters for the 23Na MAS NMR spectra are tabulated in the ESI.†
Formation of HxPO43−x, if present, would be expected to appear as a separate resonance at a chemical shift near 15 ppm, based on hydrogen phosphate salts with 1H MAS NMR.43 Note that while it is possible that chemical exchange in the intermediate time regime relative to the NMR timescale could lead to 1HPO42− being undetectable in 1H MAS NMR spectroscopy, based on the integration of Raman and FTIR spectroscopy alongside 23Na and 31P MAS NMR results, there is an absence of spectroscopic features that could be attributed to hydrogen phosphate in the natrophosphate crystals synthesized under either F-rich or F-poor conditions. Note that in this manuscript, absolute quantification (spin counting) of spin ½ nuclei was not performed because the two phosphate and two sodium sites were resolved, and their relative abundances were directly deducible from the spectra. Spin counting potentially introduces uncertainties due to differences in spin dynamics (arising from phenomena such as magnetic susceptibility differences, inequivalent reflected and transmitted powers, and inequivalent relaxation dynamics, etc.) between the sample of interest and standards.44
To further scrutinize the 23Na MAS NMR resonances, 23Na multiple quantum (MQ)MAS NMR spectroscopy was performed. MQMAS NMR spectroscopy is used to generate NMR spectra of greater resolution than one dimensional spectra for quadrupolar nuclei by correlating multiple quantum coherences with their conversion into single-quantum coherences.45–50 Given that relative signal intensity of sites in MQMAS spectra is dependent on the quadrupolar coupling parameter of the site,45,46 MQMAS spectra are often used qualitatively to validate the lack of poorly resolved resonances that would be overlapping in one dimensional NMR spectra. The 23MQMAS NMR spectra shown in Fig. 7 confirm that only two Na+ sites are present in natrophosphate synthesized with either F-poor or F-rich solution compositions. The MQMAS NMR results therefore validate the use of only two resonances used in Fig. 6D to fit the 23Na MAS NMR spectra. The NMR results in tandem indicate that the local chemical environment of H2O, F−, PO43− and Na+ ions are conserved in natrophosphate crystals grown in F-rich or F-poor solution conditions.
 | ||
| Fig. 7 23Na Multiple Quantum (MQ) MAS NMR of natrophosphate synthesized from an (A) F-rich solution composition and an (B) F-poor solution composition. The f1 and f2 projections are the summation of the 23Na MQMAS NMR spectrum. Two Na sites are observable, confirming that the natrophosphate crystal structure contains only two sodium sites. | ||
4. Implications
Salts have been hypothesized to precipitate most rapidly when the local chemical structures of the solution state species are topologically similar to the structures of the solid phase species.51 natrophosphate is one of several fluoride or phosphate-containing salts found in Hanford nuclear waste. natrophosphate competes with sodium fluoride (NaF) and Kogarkoite (Na3FSO4) for fluoride. The quantity of each salt in different Hanford tanks depends on the relative inventory of phosphate, fluoride and sulfate.21–24 Likewise, natrophosphate competes with sodium phosphate dodecahydrate (Na3(PO4)·0.25NaOH·12H2O) and Nastrophite (NaSr(PO4)·9H2O) for phosphate.24–26 Natrophosphate can grow up to 500 micron-sized crystals in as little as four days.52 Sodium phosphate dodecahydrate grows even faster, going from a solid free liquid to a dense gel in literally the blink of an eye as solutions cool on a countertop.53 In one study, sodium phosphate dodecahydrate precipitated even though natrophosphate was the stable phase; the sodium phosphate dodecahydrate then slowly recrystallized into natrophosphate.52 While quantifying the kinetics of crystallization of natrophosphate as a function of solution composition and temperature remains an active area of research, we note that the crystallization of many of these salts is rapid relative to aluminum hydroxide minerals also present in the radioactive waste.54,55 Hawthorne has hypothesized that crystals precipitate most rapidly when the liquid phase species look most like the solid phase species,51 therefore the local structure around ions in natrophosphate is compared to solution structures.The structure of hydrated salts can indicate how water interacts with ions in the salt when they are dissolved in aqueous solution.8,56 There are 19 water molecules in the chemical formula of natrophosphate, and 1.9 water molecules per ion. Despite there being many water molecules in natrophosphate, none of them coordinate fluoride. Fluoride binds water stronger than many other monovalent ions in aqueous solution, but not so strongly that it irrotationally binds the water molecules. Others have predicted that inner-sphere ion pairs are formed in solution between ions with similar hydration energy, even when both ions individually form strong interactions with water when the other ion is not present.57–59 Collins Law58,59 thus predicts that smaller alkali ions like Li+ or Na+ form inner-sphere ion-pairs with fluoride. More importantly with the present study, this is consistent with the observation that fluoride is not coordinated with water in natrophosphate, NaF solid, or Kogarkoite (the three fluoride-bearing salts found in Hanford waste). We hypothesize that Na+ and F− also form inner-sphere ion-pairs in multicomponent Hanford waste liquids. Buchner et al.57,60 indicated that only a small amount of Na+ and F− form inner-sphere ion pairs in dilute aqueous solution, but Hanford waste has greater than five moles of Na+ per liter, providing ample Na+ for ion-pairing.
Hawthorne and Schindler61 developed rules for the location of water molecules in salts that have water in their crystal structure. Water molecules incorporate into the structure of solids when there is a large charge density discrepancy between the cation and anion.61 Phosphate has four oxygen and a negative three charge, which means that there is net negative 0.75 valence units per oxygen. Each phosphate oxygen is generally bound to about three cations surrounding each phosphate oxygen, giving an average of 0.25 valence units of charge donated to each cation.62 The Na+ in natrophosphate, nastrophite, and sodium phosphate dodecahydrate is coordinated by six electronegative elements. With the one positive charge divided by six there is an average of 0.17 charge units per ligand. There is thus a relatively large discrepancy between the ideal charge donated from phosphate to each cation (0.25 valence units, v.u.) and the ideal charge received by Na+ per each cation (0.17 v.u.). When there is a large charge-density discrepancy between electronegative and electropositive element like this, Hawthorne and Schindler61 noted that water is incorporated into crystals to mediate the charge density discrepancy because water has high flexibility in how it accepts and donates charge. This is consistent with the structure of natrophosphate, Nastrophite, and sodium phosphate dodecahydrate, where nearly all phosphate–oxygens are coordinated by water, meaning that water molecules are in-between the Na+ and phosphate ions.16,63,64
5. Conclusion
Despite varying the PO43− to F− concentrations in the ternary NaF:Na3PO4:H2O system, little variation in the molecular structure of natrophosphate was found with powder X-ray diffraction, vibrational spectroscopy, and multinuclear MAS NMR. The results are consistent with the notion that natrophosphate is a classical salt of fixed composition. The 23Na MAS NMR results in particular directly interrogated the relative sites abundances of Na+ ions in Na6(H2O)18F5+ and Na(H2O)(PO4)25− sites and would have detected, if present, changes in the F− to PO43− ratio and hydrogen phosphate formation. This improved understanding of the discrete chemical environments of the ion–ion aggregates in the natrophosphate crystal structure enables this salt to be used to study ion–ion interactions between fluoride, phosphate and sodium in well-defined ionic clusters and also enables further study of the solubility of sodium fluoride and sodium phosphate solutions pertinent to select geological deposits and radioactive waste processing.
Conflicts of interest
The authors declare no conflicts of interest at the time of submission.Acknowledgements
This research was supported by IDREAM (Interfacial Dynamics in Radioactive Environments and Materials), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Science (BES). Scanning electron microscopy, X-ray diffraction, Raman spectroscopy and nuclear magnetic resonance spectroscopy were performed using facilities at the Environmental Molecular Science Laboratory (EMSL, grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research at Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram national laboratory operated for DOE by Battelle Memorial Institute operating under Contract No. DE AC05-76RL0-1830. The 14.1 T NMR spectrometer was acquired with support from the BES Chemical Sciences, Geosciences & Biosciences (CSGB) Division. PNNL colleagues are thanked for the following: (i) Sarah Burton for assistance in setting up the 7.05 T NMR spectrometer, (ii) Nancy Washton, Dave Bazak and Ying Chen for helpful discussions regarding the analysis and interpretation of NMR data, and (iii) Mark Bowden for assistance in collecting and fitting the XRD data.References
- M. J. Servis, E. Martinez-Baez and A. E. Clark, Hierarchical phenomena in multicomponent liquids: simulation methods, analysis, chemistry, Phys. Chem. Chem. Phys., 2020, 22, 9850–9874, 10.1039/D0CP00164C.
- R. Andersson, F. Årén, A. A. Franco and P. Johansson, Ion Transport Mechanisms via Time-Dependent Local Structure and Dynamics in Highly Concentrated Electrolytes, J. Electrochem. Soc., 2020, 167, 140537, DOI:10.1149/1945-7111/abc657.
- M. Soniat, G. Pool, L. Franklin and S. W. Rick, Ion association in aqueous solution, Fluid Phase Equilib., 2016, 407, 31–38, DOI:10.1016/j.fluid.2015.05.001.
- B. P. Fingerhut, J. Schauss, A. Kundu and T. Elsaesser, Aqueous Contact Ion Pairs of Phosphate Groups with Na+, Ca2+ and Mg2+ – Structural Discrimination by Femtosecond Infrared Spectroscopy and Molecular Dynamics Simulations, Z. Phys. Chem., 2020, 234, 1453–1474, DOI:10.1515/zpch-2020-1614.
- S. Kumar and R. Nussinov, Relationship between Ion Pair Geometries and Electrostatic Strengths in Proteins, Biophys. J., 2002, 83, 1595–1612, DOI:10.1016/S0006-3495(02)73929-5.
- D. M. Sherman and M. D. Collings, Ion association in concentrated NaCl brines from ambient to supercritical conditions: results from classical molecular dynamics simulations, Geochem. Trans., 2002, 3, 102, DOI:10.1186/1467-4866-3-102.
- S. B. Pillai, R. J. Wilcox, B. G. Hillis, B. P. Losey and J. D. Martin, Understanding the Water-in-Salt to Salt-in-Water Characteristics across the Zinc Chloride: Water Phase Diagram, J. Phys. Chem. B, 2022, 126, 2265–2278, DOI:10.1021/acs.jpcb.1c10530.
- H. L. Friedman and L. Lewis, The coordination geometry of water in some salt hydrates, J. Solution Chem., 1976, 5, 445–455, DOI:10.1007/BF00650462.
- T. R. Graham, M. Dembowski, J. Z. Hu, N. R. Jaegers, X. Zhang, S. B. Clark, C. I. Pearce and K. M. Rosso, Intermediate Species in the Crystallization of Sodium Aluminate Hydroxy Hydrates, J. Phys. Chem. C, 2020, 124, 12337–12345, DOI:10.1021/acs.jpcc.0c00205.
- T. R. Graham, R. Gorniak, M. Dembowski, X. Zhang, S. B. Clark, C. I. Pearce, A. E. Clark and K. M. Rosso, Solid-State Recrystallization Pathways of Sodium Aluminate Hydroxy Hydrates, Inorg. Chem., 2020, 59, 6857–6865, DOI:10.1021/acs.inorgchem.0c00258.
- M. P. Prange, T. R. Graham, R. Gorniak, M. Pouvreau, M. Dembowski, H.-W. Wang, L. L. Daemen, G. K. Schenter, M. E. Bowden, E. T. Nienhuis, K. M. Rosso, A. E. Clark and C. I. Pearce, Theory-Guided Inelastic Neutron Scattering of Crystalline Alkaline Aluminate Salts Bearing Principal Motifs of Solution-State Species, Inorg. Chem., 2021, 60, 16223–16232, DOI:10.1021/acs.inorgchem.1c02006.
- M. Dembowski, M. P. Prange, M. Pouvreau, T. R. Graham, M. E. Bowden, A. N’Diaye, G. K. Schenter, S. B. Clark, A. E. Clark, K. M. Rosso and C. I. Pearce, Inference of principal species in caustic aluminate solutions through solid-state spectroscopic characterization, Dalton Trans., 2020, 49, 5869–5880, 10.1039/D0DT00229A.
- Y. L. Kapustin, A. V. Bykova and V. I. Bukin, Natrophosphate, a new mineral, Int. Geol. Rev., 1972, 14, 984–989, DOI:10.1080/00206817209475790.
- E. Tillmanns and W. H. Baur, A new type of polycation in heptasodiumfluoridebisarsenate-19-hydrate, Naturwissenschaften, 1970, 57, 242–242, DOI:10.1007/BF01010263.
- O. V. Petersen, T. Fockenberg, P. C. Toft and M. Rattay, Natrophosphate from the Aris phonolites, Windhoek, Namibia, Neues Jahrb. Mineral., Monatsh., 1997, 1997, 511–517, DOI:10.1127/njmm/1997/1997/511.
- W. H. Baur and E. Tillmanns, Salt hydrates. X. The crystal structure determinations of heptasodium fluoride bisphosphate 19-hydrate and heptasodium fluoride bisarsenate 19-hydrate and the computer simulation of the isomorphous vanadate salt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2218–2224, DOI:10.1107/S0567740874006777.
- S. Krivovichev, Topological complexity of crystal structures: quantitative approach, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 393–398, DOI:10.1107/S0108767312012044.
- S. V. Krivovichev, Structural complexity of minerals: information storage and processing in the mineral world, Mineral. Mag., 2013, 77, 275–326, DOI:10.1180/minmag.2013.077.3.05.
- M. Avdontceva, S. Krivovichev and V. Yakovenchuk, Natrophosphate, Arctic Mineral and Nuclear Waste Phase: Structure Refinements and Chemical Variability, Minerals, 2021, 11, 186, DOI:10.3390/min11020186.
- R. A. Peterson, E. C. Buck, J. Chun, R. C. Daniel, D. L. Herting, E. S. Ilton, G. J. Lumetta and S. B. Clark, Review of the Scientific Understanding of Radioactive Waste at the U.S. DOE Hanford Site, Environ. Sci. Technol., 2018, 52, 381–396, DOI:10.1021/acs.est.7b04077.
- S. D. Bolling, J. G. Reynolds, T. M. Ely, J. S. Lachut, M. E. Lamothe and G. A. Cooke, Natrophosphate and kogarkoite precipitated from alkaline nuclear waste at Hanford, J. Radioanal. Nucl. Chem., 2020, 323, 329–339, DOI:10.1007/s10967-019-06924-9.
- J. G. Reynolds, G. A. Cooke, D. L. Herting and R. W. Warrant, Salt Mineralogy of Hanford High-Level Nuclear Waste Staged for Treatment, Ind. Eng. Chem. Res., 2013, 52, 9741–9751, DOI:10.1021/ie400822r.
- J. G. Reynolds, H. J. Huber, G. A. Cooke and J. A. Pestovich, Solid-phase zirconium and fluoride species in alkaline zircaloy cladding waste at Hanford, J. Hazard. Mater., 2014, 278, 203–210, DOI:10.1016/j.jhazmat.2014.05.097.
- J. G. Reynolds, J. S. Page, G. A. Cooke and J. Pestovich, A scanning electron microscopy study of bismuth and phosphate phases in bismuth phosphate process waste at Hanford, J. Radioanal. Nucl. Chem., 2015, 304, 1253–1259, DOI:10.1007/s10967-015-3980-1.
- J. G. Reynolds, G. A. Cooke and D. L. Herting, Sodium strontium phosphate nonahydrate (NaSrPO4·9H2O) found in Hanford nuclear waste, J. Radioanal. Nucl. Chem., 2020, 326, 435–443, DOI:10.1007/s10967-020-07296-1.
- R. W. Warrant and G. A. Cooke, Characterization of the solid waste in the Hanford waste tanks using a combination of XRD, SEM and PLM, Adv. X-Ray Anal., 2003, 43, 251–256 Search PubMed.
- O. N. Roslyakova, M. R. Petrov and M. I. Zhikharev, The NaF-Na3PO4-H2O system at 25 °C, Russ. J. Inorg. Chem., 1979, 24, 115–116 Search PubMed.
- L. N. Komissarova, M. G. Zhizhin and A. A. Filaretov, Complex phosphates containing mono- and trivalent cations, Russ. Chem. Rev., 2002, 71, 619–650, DOI:10.1070/RC2002v071n08ABEH000728.
- D. R. Migneault and R. K. Forcé, Dissociation constants of phosphoric acid at 25 °C and the ion pairing of sodium with orthophosphate ligands at 25 °C, J. Solution Chem., 1988, 17, 987–997, DOI:10.1007/BF00649742.
- D. L. Herting and J. G. Reynolds, The composition of natrophosphate (sodium fluoride phosphate hydrate), Environ. Chem. Lett., 2016, 14, 401–405, DOI:10.1007/s10311-016-0574-2.
- A. R. Felmy and G. T. MacLean, Development of an Enhanced Thermodynamic Database for the Pitzer Model in ESP: The Fluoride and Phosphate Components, Richland, Washington, p. 99352, 2001 Search PubMed.
- E. A. Genkina and A. P. Khomyakov, Refinement of the structure of natural natrophosphate, Kristallografiya, 1992, 37, 1559–1560 Search PubMed.
- D. Balzar and H. Ledbetter, Voigt-function modeling in Fourier analysis of size- and strain-broadened X-ray diffraction peaks, J. Appl. Crystallogr., 1993, 26, 97–103, DOI:10.1107/S0021889892008987.
- A. Sadoc, M. Body, C. Legein, M. Biswal, F. Fayon, X. Rocquefelte and F. Boucher, NMR parameters in alkali, alkaline earth and rare earth fluorides from first principle calculations, Phys. Chem. Chem. Phys., 2011, 13, 18539, 10.1039/C1CP21253B.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, ssNake: A cross-platform open-source NMR data processing and fitting application, J. Magn. Reson., 2019, 301, 56–66, DOI:10.1016/j.jmr.2019.02.006.
- R. L. Frost, A. López, R. Scholz, F. M. Belotti and Y. Xi, A vibrational spectroscopic study of the anhydrous phosphate mineral sidorenkite Na3Mn(PO4)(CO3), Spectrochim. Acta, Part A, 2015, 137, 930–934, DOI:10.1016/j.saa.2014.08.029.
- K. D. Litasov and N. M. Podgornykh, Raman spectroscopy of various phosphate minerals and occurrence of tuite in the Elga IIE iron meteorite, J. Raman Spectrosc., 2017, 48, 1518–1527, DOI:10.1002/jrs.5119.
- K. A. Syed, S.-F. Pang, Y. Zhang, G. Zeng and Y.-H. Zhang, Micro-Raman Observation on the HPO42− Association Structures in an Individual Dipotassium Hydrogen Phosphate (K2HPO4) Droplet, J. Phys. Chem. A, 2012, 116, 1558–1564, DOI:10.1021/jp2110743.
- W. Jastrzębski, M. Sitarz, M. Rokita and K. Bułat, Infrared spectroscopy of different phosphates structures, Spectrochim. Acta, Part A, 2011, 79, 722–727, DOI:10.1016/j.saa.2010.08.044.
- J. Baril, J.-J. Max and C. Chapados, Titrage infrarouge de l’acide phosphorique, Can. J. Chem., 2000, 78, 490–507, DOI:10.1139/v00-038.
- M. Klähn, G. Mathias, C. Kötting, M. Nonella, J. Schlitter, K. Gerwert and P. Tavan, IR Spectra of Phosphate Ions in Aqueous Solution: Predictions of a DFT/MM Approach Compared with Observations, J. Phys. Chem. A, 2004, 108, 6186–6194, DOI:10.1021/jp048617g.
- J. VandeVondele, P. Tröster, P. Tavan and G. Mathias, Vibrational Spectra of Phosphate Ions in Aqueous Solution Probed by First-Principles Molecular Dynamics, J. Phys. Chem. A, 2012, 116, 2466–2474, DOI:10.1021/jp211783z.
- J. P. Yesinowski and H. Eckert, Hydrogen environments in calcium phosphates: proton MAS NMR at high spinning speeds, J. Am. Chem. Soc., 1987, 109, 6274–6282, DOI:10.1021/ja00255a009.
- R. J. Smernik and J. M. Oades, The use of spin counting for determining quantitation in solid state 13C NMR spectra of natural organic matter, Geoderma, 2000, 96, 101–129, DOI:10.1016/S0016-7061(00)00007-0.
- A. Medek, J. S. Harwood and L. Frydman, Multiple-Quantum Magic-Angle Spinning NMR: A New Method for the Study of Quadrupolar Nuclei in Solids, J. Am. Chem. Soc., 1995, 117, 12779–12787, DOI:10.1021/ja00156a015.
- L. Frydman, Fundamentals of Multiple-Quantum Magic-Angle Spinning NMR on Half-Integer Quadrupolar Nuclei, Encycl. Nucl. Magn. Res., 2002, 9, 262–274, DOI:10.1002/chin.200332288.
- S. E. Ashbrook, J. McManus, K. J. D. MacKenzie and S. Wimperis, Multiple-quantum and cross-polarized 27Al MAS NMR of mechanically treated mixtures of kaolinite and gibbsite, J. Phys. Chem. B, 2000, 104, 6408–6416, DOI:10.1021/jp000316t.
- C. Vinod Chandran, C. E. A. Kirschhock, S. Radhakrishnan, F. Taulelle, J. A. Martens and E. Breynaert, Alumina: discriminative analysis using 3D correlation of solid-state NMR parameters, Chem. Soc. Rev., 2019, 48, 134–156, 10.1039/c8cs00321a.
- C. D. Pilgrim, J. R. Callahan, C. A. Colla, C. A. Ohlin, H. E. Mason and W. H. Casey, 27Al MQMAS of the δ-Al13-Keggin, Dalton Trans., 2017, 46, 2249–2254 RSC.
- C. V. Chandran, J. Cuny, R. Gautier, L. Le Pollès, C. J. Pickard and T. Bräuniger, Improving sensitivity and resolution of MQMAS spectra: A 45Sc-NMR case study of scandium sulphate pentahydrate, J. Magn. Reson., 2010, 203, 226–235, DOI:10.1016/j.jmr.2009.12.021.
- F. C. Hawthorne, A bond-topological approach to theoretical mineralogy: crystal structure, chemical composition and chemical reactions, Phys. Chem. Miner., 2012, 39, 841–874, DOI:10.1007/s00269-012-0538-4.
- J. G. Reynolds and D. L. Herting, Waste Management '16 Proceedings, 2016 Search PubMed.
- M. E. LaMothe, Determination of Phosphate Hydrate Transition Temperatures, M.S. Thesis, Washington State University, Pullman, WA, 2017, https://rex.libraries.wsu.edu/esploro/outputs/graduate/Determination-of-phosphate-hydrate-transition-temperatures/99900525160101842#file-0 Search PubMed.
- R. F. Scotford and J. R. Glastonbury, Effect of temperature on the rates of dissolution of gibbsite and boehmite, Can. J. Chem. Eng., 1971, 49, 611–616, DOI:10.1002/cjce.5450490510.
- C. Skoufadis, D. Panias and I. Paspaliaris, Kinetics of boehmite precipitation from supersaturated sodium aluminate solutions, Hydrometallurgy, 2003, 68, 57–68, DOI:10.1016/S0304-386X(02)00165-2.
- E. Sokolova and F. C. Hawthorne, The Crystal Chemistry of the [M3ϕ11–14] Trimeric Structures: from Hyperagpaitic Complexes to Saline Lakes, Can. Mineral., 2001, 39, 1275–1294, DOI:10.2113/Gscanmin.39.5.1275.
- R. Buchner, W. Wachter and G. Hefter, Systematic Variations of Ion Hydration in Aqueous Alkali Metal Fluoride Solutions, J. Phys. Chem. B, 2019, 123, 10868–10876, DOI:10.1021/acs.jpcb.9b09694.
- K. Collins, Ions from the Hofmeister series and osmolytes: effects on proteins in solution and in the crystallization process, Methods, 2004, 34, 300–311, DOI:10.1016/j.ymeth.2004.03.021.
- K. D. Collins, The behavior of ions in water is controlled by their water affinity, Q. Rev. Biophys., 2019, 52, e11, DOI:10.1017/S0033583519000106.
- R. Buchner, G. T. Hefter and J. Barthel, Dielectric relaxation of aqueous NaF and KF solutions, J. Chem. Soc., Faraday Trans., 1994, 90, 2475, 10.1039/FT9949002475.
- F. C. Hawthorne and M. Schindler, Understanding the weakly bonded constituents in oxysalt minerals, Z. Kristallogr. – Cryst. Mater., 2008, 223, 41–68, DOI:10.1524/zkri.2008.0003.
- F. C. Hawthorne, Structure and chemistry of phosphate minerals, Mineral. Mag., 1998, 62, 141–164, DOI:10.1180/002646198547512.
- E. Tillmanns and W. H. Baur, On the crystal chemistry of salt hydrates. VII. The crystal structures of pseudo trisodium orthoarsenate dodecahydrate and the isomorphous phosphate and vanadate salts, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2124–2132, DOI:10.1107/S0567740871005454.
- S. Takagi, M. Mathew and W. E. Brown, The structure of sodium strontium phosphate nonahydrate, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1408–1413, DOI:10.1107/S0567740882006049.
Footnote |
| † Electronic supplementary information (ESI) available: SEM-EDS derived compositional measurements, additional X-ray diffraction studies, Raman and FTIR spectra between 100–4000 cm−1 and also tables of regressed line shape parameters. See DOI: https://doi.org/10.1039/d2qi00868h |
| This journal is © the Partner Organisations 2022 |