
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroxyalkyl-substituted double-decker silsesquioxanes: effective separation of cis and trans isomers†
Anna
Władyczyn
a,
Anna
Gągor
 b,
Katarzyna
Ślepokura
a and
Łukasz
John
*a
b,
Katarzyna
Ślepokura
a and
Łukasz
John
*a
aFaculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: lukasz.john@uwr.edu.pl
bInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, 2 Okólna, 50-422 Wrocław, Poland
First published on 9th June 2022
Abstract
In this work, we report the preparation, isolation, and characterization of a family of double-decker silsesquioxane architectures starting from the previously reported DDSQ-Me2H2 precursor, a mixture of cis and trans isomers. Using such a starting material results in a subsequent mixture of isomers when hydrosilylation reactions using DDSQ-Me2H2 and hydroxyalkyl-substituted alkenes catalyzed by Karstedt catalyst are performed. We describe a procedure to sequentially crystallize the two isomers from one another, affording a protocol potentially applicable to other closely related derivatives. The ability to access pure cis and trans isomers of double-decker silsesquioxanes may result in the preparation of new materials and polymers with well-defined properties. All resulting silsesquioxanes were characterized by multinuclear (1H, 13C, and 29Si) NMR spectroscopy, FT-IR, and HR-MS. Additionally, we report five crystal structures of geometrical isomers, including two cis forms described in the literature for the first time. This is a breakthrough in itself and sets a new strategy for obtaining such exiting organosilicon-based systems.
1. Introduction
Double-decker silsesquioxanes (DDSQs) occupy an indisputable place among the group of functionalized siloxane-based compounds. They throw light on the development of molecular and macromolecular organosilicon chemistry concepts.1 This attractive group of silicon-containing species has been extensively studied due to their outstanding physicochemical properties. Double-decker silsesquioxanes have shown excellent potential for various applications, including such intriguing areas as materials engineering, smart and self-healing polymers, coordination chemistry, catalysis, and others.2–19 The structure of a typical double-decker silsesquioxane is different from that of the symmetric cubic T8 polyhedral oligomeric silsesquioxanes (POSSs). It constitutes an open cage network including two connected, via oxygen bridges, decks of cyclosiloxane rings located on top of one another with eight phenyl groups attached to Si atoms (Fig. 1).20 This group of silsesquioxanes includes the two following structures: an open (Fig. 1, C) architecture and a closed framework (Fig. 1, A and B with one and two reactive group(s) on each lateral arm, respectively). Difunctional double-decker silsesquioxanes (Fig. 1, A) are routinely obtained as isomer mixtures, with geometric cis and trans isomer formation corresponding to the different spatial arrangements of the labile functional groups and inert groups on the double-decker silsesquioxane core.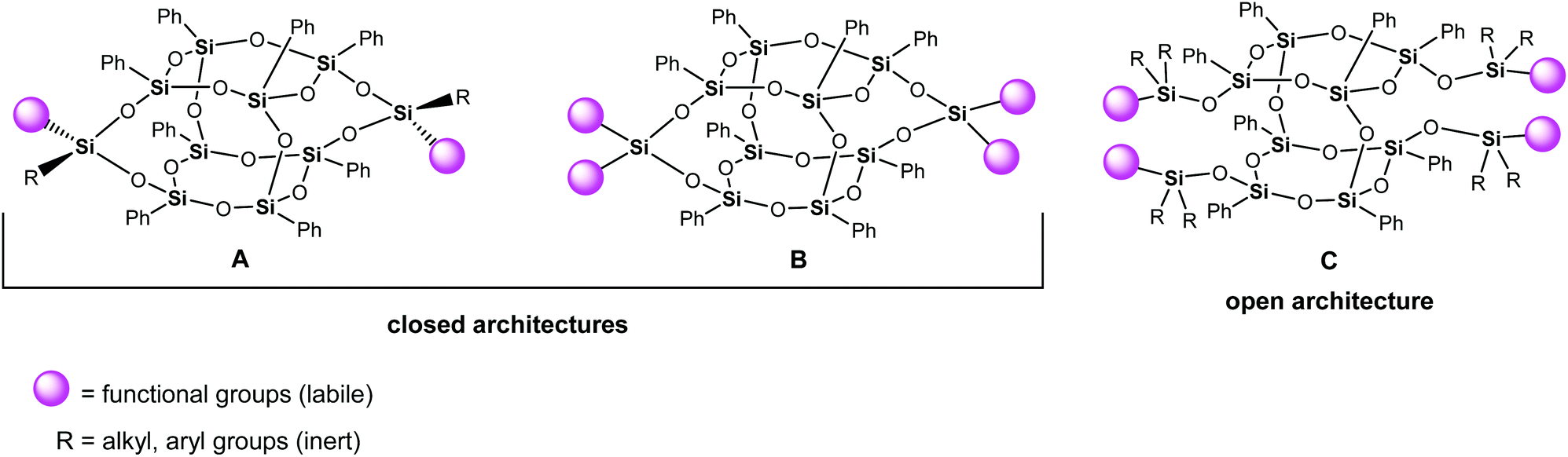 | ||
| Fig. 1 Possible architectures of the double-decker silsesquioxanes. | ||
Among the broad palette of the functionalized double-decker silsesquioxanes, several articles are devoted to forming a mixture containing cis and trans geometrical isomers in the case of difunctionalized silsesquioxane (Fig. 1, A) derivatives.21–23 This limits the number of available pure crystalline double-decker silsesquioxane reactive monomers. Unfortunately, these isomers, regardless of the nature of the external organic arms, have identical or similar retention times during chromatographic purification, which makes them difficult to separate. The identical drawback is also observed in polyhedral oligomeric silsesquioxanes chemistry. For instance, Laine et al. pointed out that molecules containing different numbers of phenyl substituents exhibited the same Rf values.24 Even though the distinction may seem unnoticed, it is worth noticing that polyhedral oligomeric silsesquioxane species can be regarded as oligomers rather than discrete molecular compounds.25 This is one of the reasons for looking for non-chromatographic separation methods.
Since polyhedral oligomeric silsesquioxane species can be regarded as oligomers, scientists have become inspired to find more or less effective synthetic approaches for novel di-substituted double-decker silsesquioxane monomers, including recrystallization, coprecipitation, formation of soluble coordination entities, and others. One of the attempts consists of coupling isobutyltrichlorosilane leading to the mixture of sodium salts. Unfortunately, only hydrolyzable cis and trans derivatives of closed double-decker silsesquioxanes were found; no further analysis was reported.24
Moreover, during this approach, other silsesquioxanes, except for the expected mixture of cis and trans isomers, such as mono-substituted cage-type polyhedral oligomeric silsesquioxane (T7) and double-decker silsesquioxane with different substitution on lateral silicon atoms, are observed, which additionally complicates the reaction.22 A similar strategy, based on the mixture of suitable sodium salts, led to diazido-functionalized double-decker silsesquioxane, obtained in the reaction of double-decker octasilsesquioxane-tetraol tetrasodium salt and (3-chloropropyl) methyldichlorosilane. Further azidation of the resulting dichloropropyl derivative gave an isomeric mixture of diazido double-decker silsesquioxane, not separable by column chromatography. Finally, careful recrystallization of the mixture solution was performed to obtain a pure crystalline trans isomer.23 The above-mentioned examples show how difficult it is to separate a mixture of geometric isomers. If it is successful, this process requires long-term and often breakneck procedures that end with the separation of the trans isomer, with the cis isomer that remains in the mixture, and only due to the easier crystallization of a “more symmetrical” (center of symmetry in trans isomer) derivative.
A recent increase in the demand for well-defined and thermally stable systems has highlighted the importance of silsesquioxane-based architectures.41–43 This paper describes a new approach to obtaining and separating cis and trans geometrical isomers of selected double-decker silsesquioxanes. More precisely, our interest was focused on various alkyl organic arms containing terminal hydroxy group(s) attached to double-decker silsesquioxane core, which have great potential in further modifications and formation of the hybrid building nano-blocks. We also report on crystal structures of cis isomers of the resulting double-decker silsesquioxanes. This is a breakthrough in itself and has never been done, according to our best knowledge.
2. Experimental
2.1. General procedures and chemicals
All the reactions and operations that required an inert atmosphere of N2 or Ar were performed using a standard Schlenk apparatus and vacuum line techniques. Solvents for the synthesis: THF (tetrahydrofurane) and toluene; were purified using the Solvent Purification Systems (Inert, PureSolv EN 1–7 Base). Catalyst species were removed after reaction by filtration through Celite® 545 (Sigma-Aldrich) pad. Solvents for standard workup (methanol, hexane, acetone) were purchased from VWR International and ChemPur and were used as received. All the chemicals were obtained from commercial sources and used without further purification: TetraSilanolPhenyl POSS (Hybrid Plastics Inc.), dichloromethylsilane (>97% Sigma Aldrich), Karstedt catalyst (platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution) (in xylene, Pt ∼ 2%, Sigma-Aldrich), ethylene glycol vinyl ether (97%, Sigma Aldrich), allyl alcohol (99%, Acros Organics), 3-allyloxy-1,2-propanediol (99%, Sigma-Aldrich), 4-allyl-2-methoxyphenol (99%, Sigma-Aldrich), triethylamine (Et3N) (>99%, Sigma Aldrich).2.2. Methods
1H and 13C NMR spectra were recorded with a Bruker Avance III 500 MHz spectrometer. 29Si NMR spectra were recorded with a Bruker Avance III 600 MHz and a Bruker Avance III 500 MHz spectrometer. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) and are referenced to residual peaks of deuterated NMR solvents. Chemical shift values for 29Si{1H} NMR spectra were referenced to TMS or DSS (2,2-dimethylsilapentane-5-sulphonate). Assignments are based on COSY and HSQC correlation experiments. Coupling constants (J) are reported in Hz. Standard abbreviations s, d, t, q, and m refer to singlet, doublet, triplet, quartet, and multiplet. High-resolution mass spectra were recorded using a Bruker apex ultra FTMS and Bruker microTOF-Q spectrometer using the electrospray technique. Elemental analyses (C, H, Si) were performed using a Vario EL III element analyzer (Hanau, Germany).Single crystal X-ray diffraction data for trans-1, trans-2, trans-3, cis-31 and cis-32 crystals were collected on an Agilent Technologies Gemini Ultra or Rigaku XtaLAB Synergy R, DW system at 100 K and 300 K. The data collection and processing utilized CrysAlis suite of programs. The structures were solved using direct methods (SHELXS97) and also using the dual-space algorithm (SHELXT program) and refined by full-matrix least-squares on F2 (SHELXL program). Most of the non-hydrogen atoms were refined with anisotropic displacement parameters, and hydrogen atoms were placed at their calculated positions. For the details of disorder refinements, see the ESI.† Graphics were made using the Diamond 4.4.0 (Crystal Impact). Crystallographic data for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, no. 2158320–2158323† and 2158968. The complex dielectric permittivity, ε* = ε′ − iε′′, was measured between 80 and 300 K using an Agilent E4980A precision LCR meter in the frequency range of a 135 Hz–2 MHz. The overall experimental uncertainties were less than 5%. The dielectric experiment was performed on a polycrystalline sample in a pressed pellet with geometrical parameters (S = 20 mm2, d = 0.5 mm). The silver electrodes were painted on both opposing faces.
2.3. Syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CAr), 1431 (Si–CH3), 1133 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1175.06 {calcd for [M + Na]+ 1175.06}. Elemental analyses calcd for C50H48O14Si10: C, 52.05; H, 4.19; Si, 24.34; found C 52.06; H 4.18; Si, 24.36.
CAr), 1431 (Si–CH3), 1132 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1291.14 {calcd for [M + Na]+ 1291.15}. Elemental analyses calcd for C56H60O16Si10: C, 52.96; H, 4.76; Si, 22.12; found C, 52.86; H 4.80; Si, 22.20.
CAr), 1461 (Si–CH3), 1134 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1351.15 {calcd for [M + Na]+ 1351.17}. Elemental analyses calcd for C58H64O18Si10 C, 52.38; H, 4.85; Si, 21.12; found C, 52.26; H, 4.92; Si, 21.08.
CAr), 1431 (Si–CH3), 1132 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1439.24 {calcd for [M + Na]+ 1439.22}. Elemental analyses calcd for C62H72O20Si10: C, 52.51; H, 5.12; Si, 19.80; found C, 52.46; H 5.13; Si, 19.87.
CAr), 1431 (Si–CH3), 1133 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1175.06 {calcd for [M + Na]+ 1175.06}. Elemental analyses calcd for C50H48O14Si10: C, 52.05; H, 4.19; Si, 24.34; found C 52.06; H 4.18; Si, 24.36.
CAr), 1431 (Si–CH3), 1132 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1291.14 {calcd for [M + Na]+ 1291.15}. Elemental analyses calcd for C56H60O16Si10: C, 52.96; H, 4.76; Si, 22.12; found C, 52.86; H 4.80; Si, 22.20.
CAr), 1461 (Si–CH3), 1134 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1351.15 {calcd for [M + Na]+ 1351.17}. Elemental analyses calcd for C58H64O18Si10 C, 52.38; H, 4.85; Si, 21.12; found C, 52.26; H, 4.92; Si, 21.08.
CAr), 1431 (Si–CH3), 1132 (br, Si–O–Si). HRMS (ESI+, TOF/CH3Cl): m/z: 1439.24 {calcd for [M + Na]+ 1439.22}. Elemental analyses calcd for C62H72O20Si10: C, 52.51; H, 5.12; Si, 19.80; found C, 52.46; H 5.13; Si, 19.87.
3. Results and discussion
Following the latest literature reports on double-decker silsesquioxanes, it can be easily noted that in the case of difunctionalized double-decker silsesquioxane derivatives (Fig. 1, A), the authors describe the preparation of a mixture of geometric isomers. If a monocrystal is likely to be obtained, the trans form usually crystallizes, and the cis isomer remains in the mixture of the products, which is further difficult to separate. In the case of cis isomers, the number of crystal structures is negligible.26,27–29 They constitute the examples in which the organic arms attached to the double-decker silsesquioxane core have aromatic groups that stabilize the side substituents, among other possible interactions, by π–π stacking. In turn, there are no reports on derivatives containing alkyl side groups. This inspired us to undertake research in an attempt to obtain well-defined cis-type isomers that would contain alkyl substituents possessing hydroxyl groups, which would be easy to modify in further reactions to obtain new nano-blocks for complex hybrid materials (Fig. 2).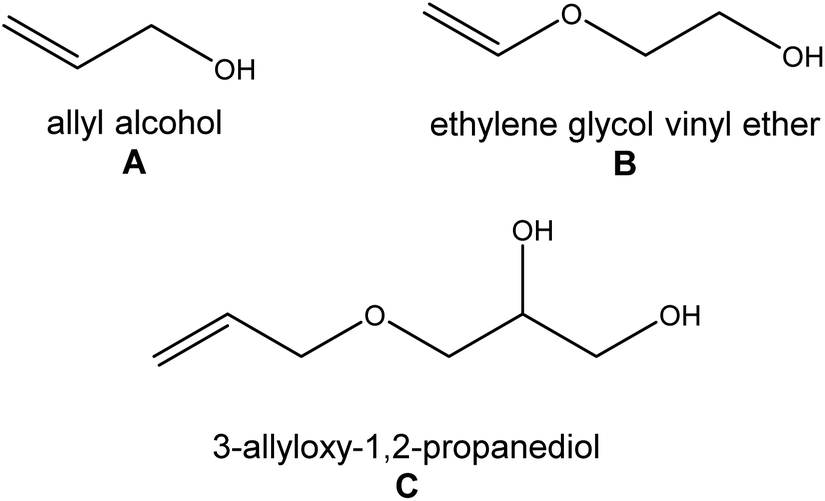 | ||
| Fig. 2 Hydroxyalkyl-substituted alkenes used in hydrosilylation reactions. | ||
Moreover, the selection of organic arms was also dictated by creating weak intermolecular interactions possibilities (e.g., hydrogen bonds, hydrophobic interactions, π–π stacking, C–H⋯π interactions, etc.). The presence of weak interactions between phenyl substituents, sidearms, and solvent molecules in the crystal stabilize architectures formed in the solid state.
We considered DDSQ-Me2H2 (1) as a starting substrate to further modifications. This compound is routinely obtained in the reaction between DDSQ-(OH)4 or its sodium salt and methyldichlorosilane.30–32 However, it is commonly known that in most synthetic protocols, a significant excess of silane is used, and on the other hand, yields are around or even below 50%.
We slightly modified its synthesis, focusing on substrates’ ratios and raising the yield, mixing substrates in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 molar ratio that resulted in 86% overall yield. The final product was characterized by NMR (1H, 13C, and 29Si), FT-IR spectroscopy, and HR-MS spectrometry (see ESI, Fig. S1–S5†). The results state that the difunctional 1 was successfully obtained. 29Si NMR spectrum (Fig. 3) indicates five signals (δ = −32.28, −77.30, −78.59, −78.79, and −78.99 ppm) which is expected for the mixture of isomers.33 Although both isomers have the same chemical environment in the SiO3Ph unit, it should not be excluded that their 29Si resonance signals are not isochronous for isomers. It can also be found in the literature that geometric isomerism influences the shifts of signals on the 29Si NMR spectrum.23,28,33,34
:6 molar ratio that resulted in 86% overall yield. The final product was characterized by NMR (1H, 13C, and 29Si), FT-IR spectroscopy, and HR-MS spectrometry (see ESI, Fig. S1–S5†). The results state that the difunctional 1 was successfully obtained. 29Si NMR spectrum (Fig. 3) indicates five signals (δ = −32.28, −77.30, −78.59, −78.79, and −78.99 ppm) which is expected for the mixture of isomers.33 Although both isomers have the same chemical environment in the SiO3Ph unit, it should not be excluded that their 29Si resonance signals are not isochronous for isomers. It can also be found in the literature that geometric isomerism influences the shifts of signals on the 29Si NMR spectrum.23,28,33,34
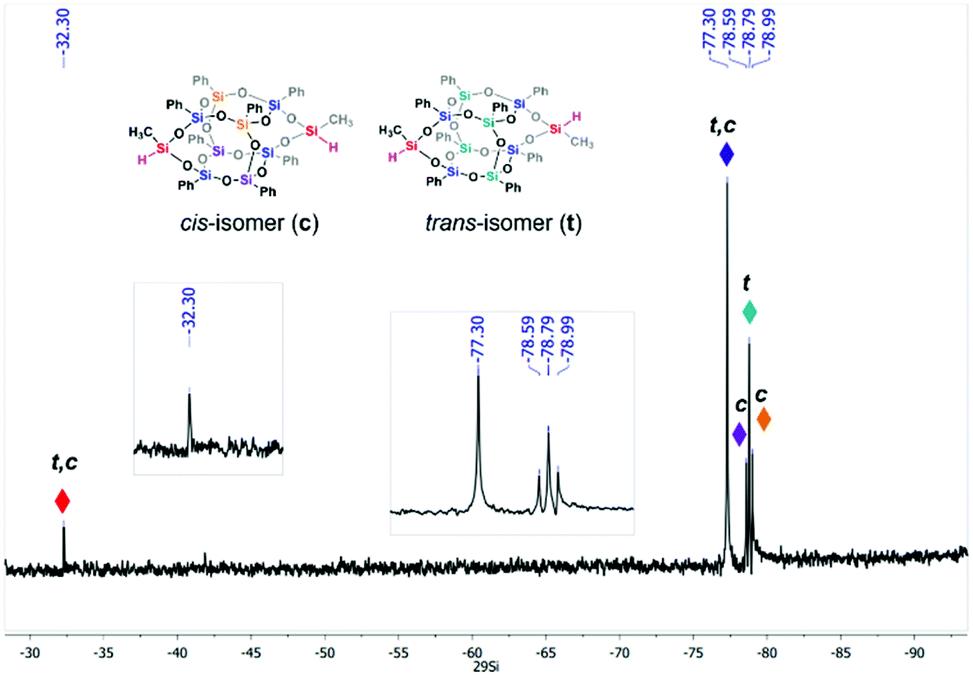 | ||
| Fig. 3 29Si NMR spectrum of cis/trans mixture of 1. | ||
Furthermore, the X-ray quality single crystals of trans-1·C7H8 were obtained from toluene saturated solution. As a result, the trans-1 crystal structure was determined (see ESI, Table S1†). For this isomer, to date, only a crystal structure that does not form a solvate has been published.33 Therefore, a valid justification here seems to be that the crystal structure obtained for trans-1·C7H8 is stabilized by a solvent molecule (toluene) in the crystal lattice by C–H⋯π interactions.36 To confirm this, we also investigated the effect of the solvent in further extensions of 1via hydrosilylation by modifying the lateral hydroxyalkyl arms. Our model shows that the structure has three conformers (depending on the arrangement of lateral Si atoms with methyl groups, Si–Me). Diffraction data analysis reveals that trans-1·C7H8 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . There are two crystallographically independent molecules in a unit cell, both centrosymmetric. One of them is presented in Fig. 4A. Symptomatic value for the C–Si–Si–C pseudo-torsion angle (180°) indicates obtaining the trans isomer, but there is a disorder in the Si–Me group in both molecules. The double-decker silsesquioxane core, as presented in Fig. 4B, arranges itself in three positions that differ in Si–Si(lateral)–Si angle, which causes the disorder. The lengths of the Si–O bonds are consistent with the literature data, with an average value of 1.6 Å.26,33,35 As shown in Fig. 4C, there are C–H⋯π interactions between toluene molecules and phenyl substituents from one layer (blue) and second layer of 1 (green).
. There are two crystallographically independent molecules in a unit cell, both centrosymmetric. One of them is presented in Fig. 4A. Symptomatic value for the C–Si–Si–C pseudo-torsion angle (180°) indicates obtaining the trans isomer, but there is a disorder in the Si–Me group in both molecules. The double-decker silsesquioxane core, as presented in Fig. 4B, arranges itself in three positions that differ in Si–Si(lateral)–Si angle, which causes the disorder. The lengths of the Si–O bonds are consistent with the literature data, with an average value of 1.6 Å.26,33,35 As shown in Fig. 4C, there are C–H⋯π interactions between toluene molecules and phenyl substituents from one layer (blue) and second layer of 1 (green).
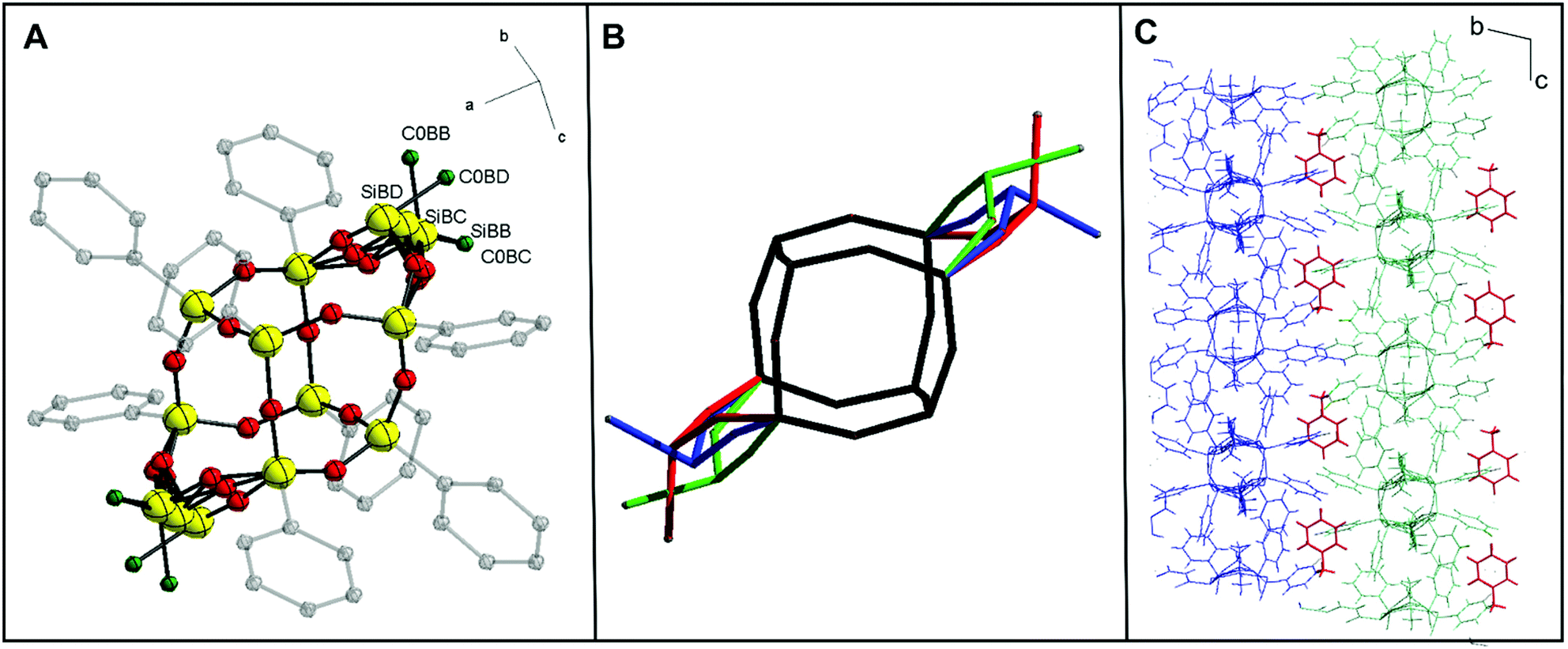 | ||
| Fig. 4 (A) Molecular structure of trans-1·C7H8, illustrating disordered “lateral” part of the molecule. Color code: C (grey), Si (yellow), O (red). Hydrogen atoms are mitted for clarity. Displacement ellipsoids (for Si and O atoms) are drawn at 50% probability. (B) Schematic representation of three possible conformations. (C) A fragment of the crystal packing. | ||
In the case of 1, due to the high similarity of the flank substituents, the mixture of the geometrical isomers is cumbersome for separation. That is why 1 was applied as an isomeric mixture for further modifications of the inorganic core. Herein, 1 was used to obtain 2–4 double-decker silsesquioxanes via hydrosilylation reactions. Those reactions were conducted on hydroxyalkyl-substituted alkenes not blocked with TMS or by using epoxides, thus avoiding the stage of deprotection of the blocking group.37–40
The syntheses of difunctionalized by hydroxyalkyl substituents double-decker silsesquioxanes are depicted with Scheme 1. In general, all derivatives were synthesized in the reaction between 1 and allyl alcohol (A; in the case of 2), ethylene glycol vinyl ether (B; 3), and 3-allyloxy-1,2-propanediol (C; 4) (Fig. 2), respectively, using a 1:5 molar ratio, in anhydrous toluene at 95 °C, under dinitrogen atmosphere, in the presence of Karstedt catalyst.
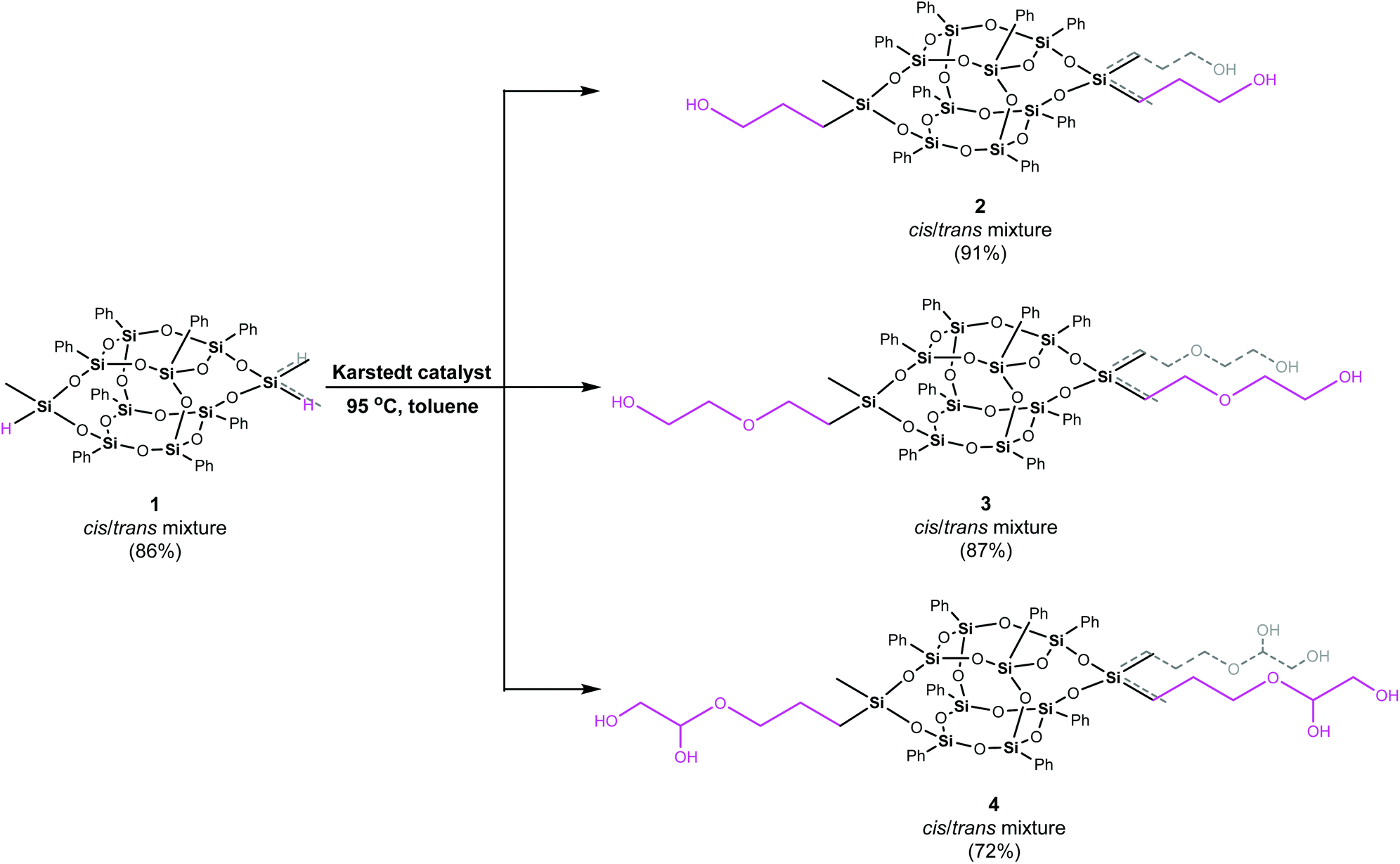 | ||
| Scheme 1 Synthesis of 2–4. | ||
It is worth mentioning that in these reactions, 2.5 equivalents of alcohol per one lateral silicon atom were used, which was a breaking point. The applied excess of alcohol causes no O-silylation by-products. Moreover, this synthetic strategy does not work for unsaturated amines. It was observed that the dehydrogenative silylation occurs for them.
Before the first hydroxyalkyl double-decker silsesquioxane derivative was characterized by physicochemical methods, an uncomplicated and effective separation of the two geometric isomers was carried out (Scheme 2). The crude product was dried and recrystallized in methanol in the first step.
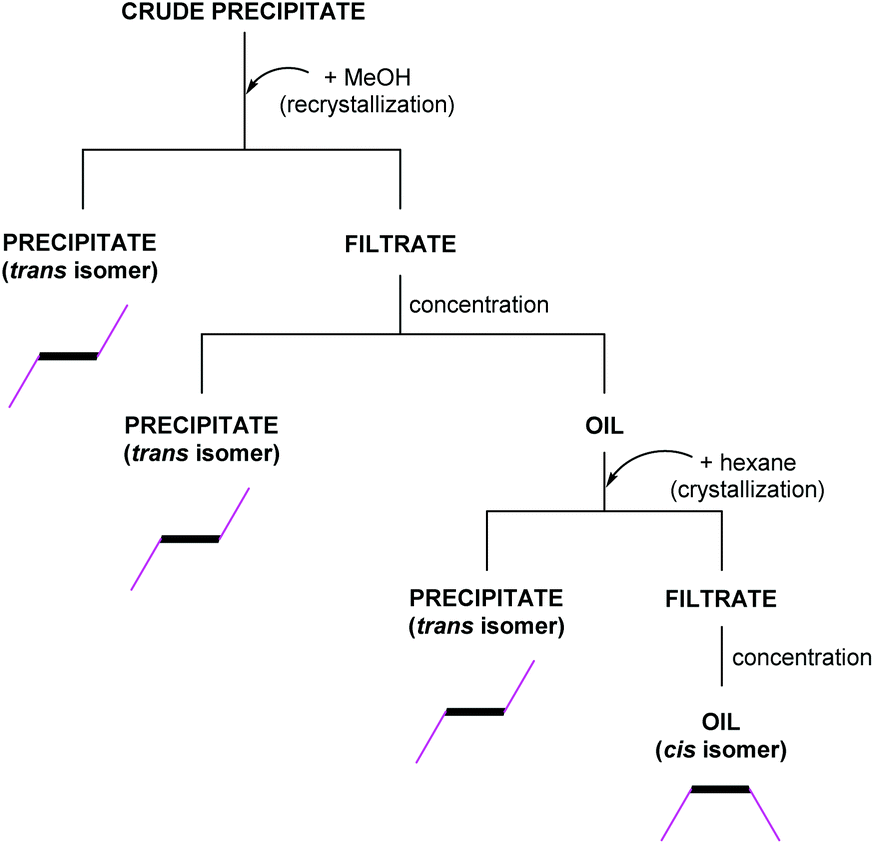 | ||
| Scheme 2 Flow chart of the fractional crystallization leading to the separation of geometrical isomers. | ||
The precipitate did not completely dissolve in the alcohol, and it was confirmed that the trans isomer remained in the solid. Next, the filtrate was concentrated using a rotary evaporator to an oil. Hexane was then added to the oil, causing precipitation of the additional amount of trans isomer. Finally, the remaining filtrate was concentrated again to oil, and the oil was analyzed. The analysis showed the presence of the cis isomer and traces of unreacted functionalized alcohol. An analogous separation strategy can be applied to all derivatives described in this paper. The omission of purification by chromatographic methods is an advantage of using the above-mentioned fractional crystallization.
Fractional crystallization of 2 led to pure trans-2 and the cis/trans mixture. The trans-2 crystallizes in an orthorhombic system (P212121 space group; Fig. 5A). The Si–O bond lengths are in the ranges of 1.59–1.63 Å (average value: 1.61 Å), which stays in agreement with the previously reported double-decker silsesquioxane-type structures.23,24,27 The pseudo-torsion angle C–Si–Si–C (177°) confirms the formation of the trans-2 regioisomer. The Si–Me distances in the “lateral” fragment equal 1.796(2) Å (SiA–C0A) and 1.859(3) Å (SiB–C0B), which is within the length range for already reported systems.27,33,35Trans-2 molecules are involved with the strong O–H⋯O interactions with each other (D⋯A distances: 3.08(6), 2.80(3), 2.65(4), 2.65(4) Å), which results in the formation of a hydrogen-bonded polymer (Fig. 5B).
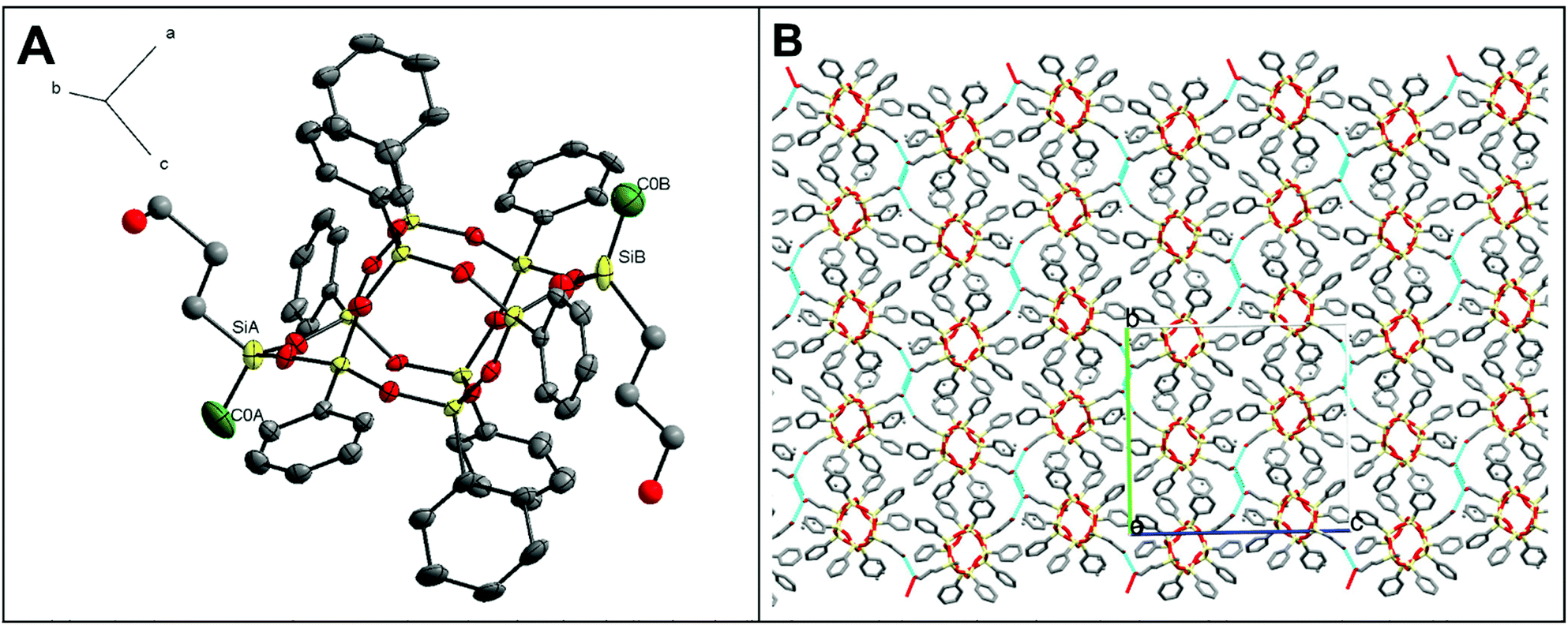 | ||
| Fig. 5 (A) Molecular structure of trans-2. Color code: C (grey), Si (yellow), O (red), C from methyl group (green). For the clarity of the picture, disordered fragments of molecule and hydrogen atoms are omitted. All displacement ellipsoids are drawn at 30% probability. Hydroxyalkyl fragments are represented using “ball and stick” (radius 0.3 Å) style. (B) Fragments of crystal packing with hydrogen interactions are marked with blue dots. | ||
Furthermore, the structure of 2 was unambiguously confirmed by multinuclear (1H, 13C, 29Si) NMR spectroscopy, infrared spectroscopy, and high-resolution mass spectrometry. The chemical shifts of 29Si NMR signals were within the expected region for phenyl-substituted closed double-decker silsesquioxane at δ = −78.03, −78.74, and −79.11 ppm for cis-2, and at −78.59 and −79.56 ppm for trans-2 isomer.
In turn, chemical shifts of the lateral Si nuclei are localized at δ = −17.23 and −17.41 ppm for cis and trans form, respectively. Furthermore, the characteristic multiplets in the aromatic and alkyl regions in the 1H NMR spectrum indicated the phenyls bounded to Si atoms forming a closed inorganic framework and organic side arms attached to flank silicon nuclei (ESI, Fig. S6†). Additionally, in the FTIR spectrum of 2, the Si–O–Si stretching gave strong absorption at 1132 cm−1, whereas the presence of hydroxyl groups was observed at 3435 cm−1. The mass spectrum {HRMS (ESI+, TOF/CH3Cl): m/z (%): 1292.19 (calcd 1292.15 [M + Na]+)} confirmed the formation of a closed frame structure composed of eight silicon atoms with attached phenyl groups and two silicon atoms in the lateral positions containing terminal hydroxypropyl side-chains and methyl groups.
Since silsesquioxane 2 could not be separated in the form of a crystalline cis derivative, it was decided to further modify the organic arm attached to lateral silicon atoms of the inorganic core. The idea of this object was that in the resulting 3, introducing an additional ether oxygen atom can force the other weak interactions in the crystal lattice. This simple trick made it possible to isolate the trans-3 and cis-3 stereoisomers of double-decker silsesquioxane species in the monocrystalline forms. Their structures were confirmed via complex X-ray analyses (see ESI, Tables S3 and S4†). A view of two isomers is presented in Fig. 6. In the case of cis-3, two solvates were obtained: cis-3·(CHCl3)·0.25(C2H5OH) (cis-33, ESI, Fig. S25†) with trichloromethane and ethanol, and cis-3·0.5(CH3OH) (cis-31, ESI, Fig. S25†). Both possess the same space group (triclinic, P at 100 K) with an ordered double-decker silsesquioxane core of C1 symmetry. In both solvates, methyl groups attached to silicon atoms point towards the same direction with a small C0A–SiA–SiB–C0B torsion angles of −3.5° in cis-31 (−5° in cis-33), confirming the presence of pure cis isomer (Fig. 6B). Si–O skeletons are rigid and adopt almost identical molecular conformation with geometry characteristic of other phenyl-substituted double-decker silsesquioxanes.35 At 100 K, in cis-31, both side chains are disordered over two inequivalent positions with 0.5/0.5 and 0.6/0.4 occupancy. Van der Waals forces solely design the long-range crystal architecture. Therefore, with increasing temperature (cis-3·0.5(CH3OH) (cis-32, ESI, Table S4†), molecular dynamic comes into play and modifies the crystal structure. Around 220 K, global symmetry grows to monoclinic (C2/m), double-decker silsesquioxane adopts Cs symmetry, and the C0A–Si0A–Si0B–C0B torsion angle is reduced to 0°. Both side chains are disordered due to the mirror plane within two equivalent positions. The dielectric relaxation process observed around 220 K confirms the changes in the disorder of side chains (the details concerning the dielectric analysis are presented in ESI, Fig. S26 and S27†). Additionally, the crystal is characterized by high temperature-induced dynamics, which manifests in large values of atoms’ displacement parameters.
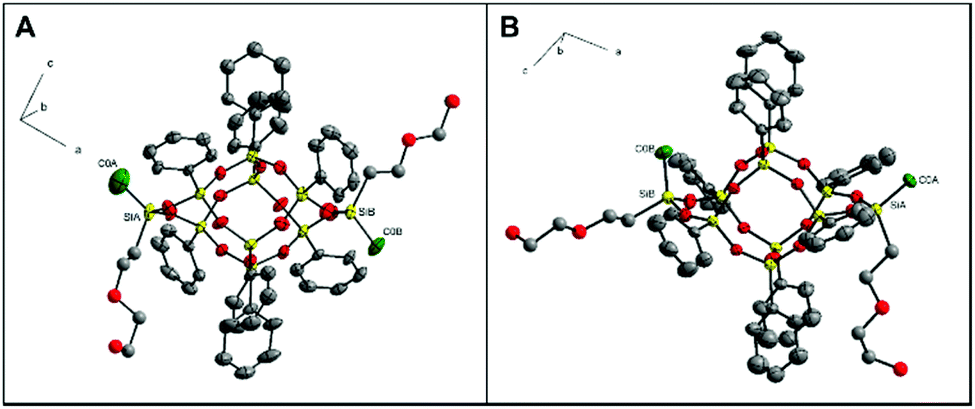 | ||
| Fig. 6 (A) Molecular structure of trans-3, and (B) cis-32 (room-temperature phase). Color code: C (grey), Si (yellow), O (red), methyl group (green). Hydrogen atoms and disorder of Si(CH3)(CH2CH2OCH2CH2OH) fragments in trans-3 and cis-32 are omitted for clarity. All displacement ellipsoids are draw at 30% probability. Hydroxyalkyl fragments are represented using “ball and stick” (radius 0.3 Å) style. | ||
The presence of trichloromethane stabilizes the side chains in the crystal structure of cis-33. Solvents aggregate in (a,c) layers and act as spacers between double-decker silsesquioxane molecules. In cis-33, the b lattice parameter of 14.27 Å is remarkably longer than 13.95 Å in cis-31. This affects both, the distances between molecules and the nature of interactions between them. One hydroxyl group is involved in O–H⋯Cl interactions (with O⋯Cl approx. 3.85 Å), whereas the other acts as a donor in the interchain O–H⋯O hydrogen bond (O⋯O distance of 2.90 Å) forming polymeric architectures extending towards [110] direction. Fig. 7 shows the crystal packing (without solvates) of cis-32 with a highlighted polymeric configuration.
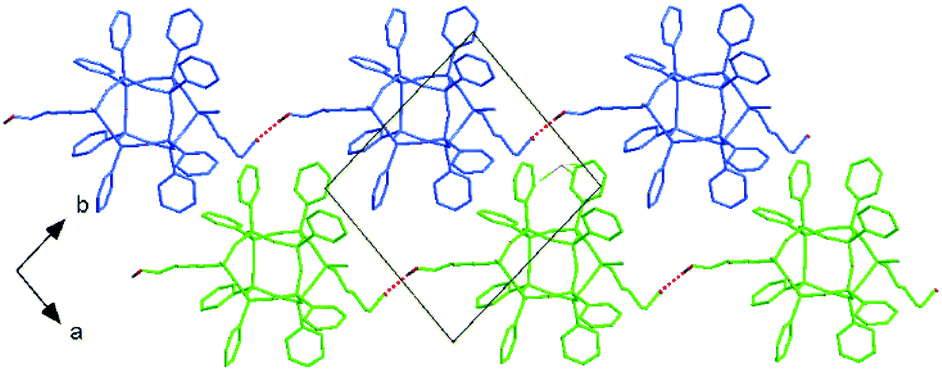 | ||
| Fig. 7 Crystal packing of cis-32 with the hydrogen-bonded polymeric architecture. Chains of cis-32 propagate along the [110] direction. | ||
In the case of the trans-3 (Fig. 6A), even though the crystals were obtained by slow evaporation from the methanol/toluene mixture, this structure does not form a solvate. Based on the crystallographic data of the analogous compounds (e.g., trans-1, and others published previously, CSD ref. codes: BEHPOV, NIWLAJ, NODWUA, TASBAT, TIYVAB, TIYVEF, UFEMAW, XETWUR5,27–29,33,35), it can be suspected that the trans isomer possesses Ci symmetry; however, it crystallizes in the non-centrosymmetric P212121 space group, like trans-2. Both Si(CH3)(CH2CH2OCH2CH2OH) fragments were found to be disordered and were refined in two positions each, with site occupation factors equal to 0.7 and 0.3. As a result, some of the molecules in the crystal are rotated 180° compared to others. Therefore, for structure clarity, these groups/atoms are omitted in Fig. 6A. Typically for trans geometry, two methyl groups attached to lateral silicon atoms point differently. The torsion angle between C0A–SiA–SiB–C0B is −175°. The angle distortion from ideal 180°, which was noted for these types of structures,27,33,35 may result from the presence of relatively long alkyl substituents. The overlay of Si–O skeletons (along with methyl groups) for cis-31 and trans-3 isomers is shown in Fig. 8.
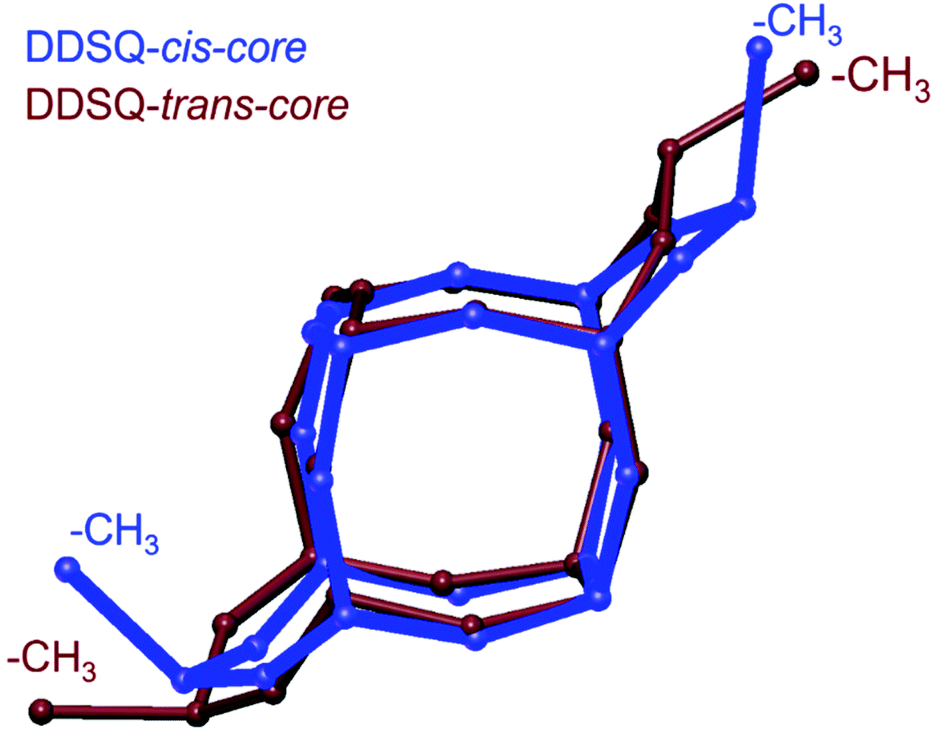 | ||
| Fig. 8 Overlay of cis-31 (dark-blue) and trans-3 (brown) isomers’ cores. The major occupied atoms were selected for the trans-3 model. The atoms of the cis-3:trans-3 structures were superimposed as follows: Si1:Si3, Si1(+x, 1 − y, +z), Si2:Si5, O4:O16, O1:O9, O3:O14, Si4:Si2. | ||
Similarly to 2, for 3, in the solution sets of assigned to the Si nuclei chemical shifts at δ = −20.05 (Si–Me), −77.45 (![[S with combining low line]](https://www.rsc.org/images/entities/i_char_0053_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/i_char_0069_0332.gif) –O–Si–Ph), −78.88, −78.94 (Si–O––Ph), and −79.26 ppm (–O–Si–Ph) for cis-3, and at −19.55 (Si–Me), −78.59 (–O–Si–Ph), −79.60 (Si–O––Ph) ppm for isomer trans-3 were observed. The HR-MS spectrum {HRMS (ESI+, TOF/CH3Cl): m/z (%): 1352.21 (calcd 1352.18 [M + Na]+)} also confirmed difunctionalized double-decker silsesquioxane framework (Fig. 1, A) possessing –CH2CH2OCH2CH2OH side arms.
–O–Si–Ph), −78.88, −78.94 (Si–O––Ph), and −79.26 ppm (–O–Si–Ph) for cis-3, and at −19.55 (Si–Me), −78.59 (–O–Si–Ph), −79.60 (Si–O––Ph) ppm for isomer trans-3 were observed. The HR-MS spectrum {HRMS (ESI+, TOF/CH3Cl): m/z (%): 1352.21 (calcd 1352.18 [M + Na]+)} also confirmed difunctionalized double-decker silsesquioxane framework (Fig. 1, A) possessing –CH2CH2OCH2CH2OH side arms.
Another modification of the side arms by 3-allyloxy-1,2-propanediol did not lead to the crystallization of further derivatives. Moreover, the spectroscopic characterization of 4 in solution is much more demanding. Due to a chiral carbon atom in organic side arms attached to lateral Si nuclei, the 1H NMR spectra manifest a diastereoisotopic effect. The protons of the methylene group are not equivalent (see signals 7a and 7b in Fig. S18†) due to their vicinity to the stereogenic center. Therefore, it was feasible to observe the separation of their signals. Signals assigned to the Si nuclei have chemical shifts at δ = −78.64 and −79.57 ppm match to Si–O––Ph and –O–Si–Ph for trans-4, whereas a chemical shift of Si–Me is observed at δ = −17.52 ppm, which confirms the presence of pure trans isomer. Chemical shifts at δ = −78.63 (Si–O––Ph), −79.44 (–O–Si–Ph) −79.69 (Si–O––Ph), 17.53 (–Me) correspond to cis isomer. In turn, the mass spectrum {HRMS (ESI+, TOF/CH3Cl): m/z (%): 1439.24 (calcd 1439.22 [M + Na]+)} confirmed the formation of a A-type (Fig. 1, A) double-decker silsesquioxane structure.
4. Conclusions
This work describes a crucial issue related to the preparation of difunctionalized double-decker silsesquioxane derivatives, which are routinely obtained in the form of a mixture of cis and trans geometric isomers. A problem often highlighted in the literature is that if the authors describe attempts to separate the mixture, isolating the trans isomer in a crystalline form is usually possible. However, the cis remains in the difficult to separate mixture of the products. Additionally, it is also in vain to look for the reports regarding the crystal structures of the cis double-decker silsesquioxane forms. The present work shows new synthetic strategies for obtaining well-defined cis and trans isomers containing hydroxyalkyl lateral arms attached to siloxane core. In this manner, we have proof that the modification of the organic arm attached to lateral silicon atoms of the inorganic core by introducing an additional ether oxygen atom can force the other weak interactions in the crystal lattice, resulting in the successful isolation of two stereoisomers in the monocrystalline forms. On the one hand, such derivatives have never been reported. On the other, presented effective protocols can be applied in the further modifications of the double-decker silsesquioxanes leading to cis isomers isolation, so few are described in the literature. We are currently exploring further reactions of the resulting hydroxyalkyl derivatives, which may shed more light on creating attractive hybrid materials in various applications, including biomaterials. Investigations along these lines are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Centre, Poland (Grant No. 2020/39/B/ST4/00910). Furthermore, the authors would like to express their gratitude to Dr Anna Piecha-Bisiorek, D.Sc. from the Faculty of Chemistry, University of Wrocław, Poland, for her cooperation on dielectric studies and to Prof. Tadeusz Lis for his expertise in the X-ray analysis.References
- K. Yoshida, T. Hattori, N. Ootake, R. Tanaka and H. Matsumoto, Silsesquioxane-Based Polymers: Synthesis of Phenylsilsesquioxanes with Double-Decker Structure and Their Polymers, in Silicon Based Polymers, ed. F. Ganachaud, S. Boileau and B. Boury, Springer, Dordrecht, 2008, pp. 205–211 Search PubMed.
- J. Duszczak, K. Mituła, A. Santiago-Portillo, L. Soumoy, M. Rzonsowska, R. Januszewski, L. Fusaro, C. Aprile and B. Dudziec, Double-Decker Silsesquioxanes Self-Assembled in One-Dimensional Coordination Polymeric Nanofibers with Emission Properties, ACS Appl. Mater. Interfaces, 2021, 13, 22806–22818 CrossRef CAS PubMed.
- R. Sodkhomkhum and V. Ervithayasuporn, Synthesis of Poly (Siloxane/Double-Decker Silsesquioxane) via Dehydrocarbonative Condensation Reaction and Its Functionalization, Polymer, 2016, 86, 113–119 CrossRef CAS.
- P. Żak, L. Delaude, B. Dudziec and B. Marciniec, N-Heterocyclic Carbene-Based Ruthenium-Hydride Catalysts for the Synthesis of Unsymmetrically Functionalized Double-Decker Silsesquioxanes, Chem. Commun., 2018, 54, 4306–4309 RSC.
- H.-L. Au-Yeung, S. Y.-L. Leung and V. W.-W. Yam, Supramolecular Assemblies of Dinuclear Alkynylplatinum(II) Terpyridine Complexes with Double-Decker Silsesquioxane Nano-Cores: The Role of Isomerism in Constructing Nano-Structures, Chem. Commun., 2018, 54, 4128–4131 RSC.
- J. Guan, K. Tomobe, I. Madu, T. Goodson, III, K. Makhal, M. T. Trinh, S. C. Rand, N. Yodsin, S. Jungsuttiwong and R. M. Laine, Photophysical Properties of Functionalized Double Decker Phenylsilsesquioxane Macromonomers: [PhSiO1.5]8[OSiMe2]2 and [PhSiO1.5]8[O0.5SiMe3]4. Cage-Centered Lowest Unoccupied Molecular Orbitals Form Even When Two Cage Edge Bridges Are Removed, Verified By Modeling and Ultrafast Magnetic Light Scattering Experiments, Macromolecules, 2019, 52, 7413–7422 CrossRef CAS.
- J. Guan, Z. Sun, R. Ansari, Y. Liu, A. Endo, M. Unno, A. Ouali, S. Mahbub, J. C. Furgal, N. Yodsin, S. Jungsuttiwong, D. Hashemi, J. Kieffer and R. M. Laine, Conjugated Copolymers That Shouldn't Be, Angew. Chem., Int. Ed., 2021, 60, 11115–11119 CrossRef CAS PubMed.
- B. Zhao, H. Ding, S. Xu and S. Zheng, Organic-Inorganic Segmented Polyurethanes Simultaneously Having Shape Recovery and Self-Healing Properties, ACS Appl. Polym. Mater., 2019, 1, 3174–3184 CrossRef CAS.
- M. Rzonsowska, K. Mituła, J. Duszczak, M. Kasperkowiak, R. Januszewski, A. Grześkiewicz, M. Kubicki, D. Głowacka and B. Dudziec, Unexpected and frustrating transformations of double-decker silsesquioxanes, Inorg. Chem. Front., 2022, 9, 379–390 RSC.
- J. Guan, J. J. R. Arias, K. Tomobe, R. Ansari, M. de F. V. Marques, A. Rebane, S. Mahbub, J. C. Furgal, N. Yodsin, S. Jungsuttiwong, D. Hashemi and J. Kieffer, Unconventional conjugation via vinylMeSi(O-)2siloxane bridges may imbue semiconductiong properties in [vinyl(Me)SiO(PhSiO1.5)8OSi(Me)vinyl-Ar] double-decker copolymers, ACS Appl. Polym. Mater., 2020, 2, 3894–3907 CrossRef CAS.
- B. Zhao, K. Wei, L. Wang and S. Zheng, Poly(hydroxy urethane)s with double decker silsesquioxanes in the main chains: Synthesis, shape recovery, and reprocessing properties, Macromolecules, 2020, 53, 434–444 CrossRef CAS.
- Y. Liu, N. Takeda, A. Ouali and M. Unno, Synthesis, characterization, and functionalization of tetrafunctional double-decker siloxanes, Inorg. Chem., 2019, 58, 4093–4098 CrossRef CAS PubMed.
- Y. Liu, M. Kigure, K. Koizumi, N. Takeda, M. Unno and A. Ouali, Synthesis of tetrachloro, tetraiodo, and tetraazido double-decker siloxanes, Inorg. Chem., 2020, 59, 15478–15486 CrossRef CAS PubMed.
- T. Uchida, Y. Egawa, T. Adachi, N. Oguri, M. Kobayashi, T. Kudo, N. Takeda, M. Unno and R. Tanaka, Synthesis, structures, and thermal properties of symmetric and Janus “latern cage” siloxanes, Chem. – Eur. J., 2019, 25, 1683–1686 CrossRef CAS PubMed.
- B.-D. Barry, J. E. Dannatt, A. K. King, A. Lee and R. E. Maleczka, A general diversity oriented synthesis of asymmetric double-decker shaped silsesquioxanes, Chem. Commun., 2019, 55, 8623–8626 RSC.
- J. Duszczak, K. Mituła, R. Januszewski, P. Żak, B. Dudziec and M. Marciniec, Highly efficient route for the synthesis of a novel generation of tetraorganofunctional double-decker type of silsesquioxanes, ChemCatChem, 2019, 11, 1086–1091 CAS.
- Y. Du and H. Liu, Cage-like silsesquioxanes-based hybrid materials, Dalton Trans., 2020, 49, 5396–5405 RSC.
- M. Soldatov and H. Liu, Hybrid porous polymers based on cage-like organosiloxanes: synthesis, properties and applications, Prog. Polym. Sci., 2021, 119, 101419 CrossRef CAS.
- W. Mingyue, C. Hong, K. S. Joshy and W. Fuke, Progress in the synthesis of bifunctionalized polyhedral oligomeric silsesquioxane, Polymers, 2019, 11, 2098 CrossRef PubMed.
- B. Dudziec and M. Marciniec, Double-decker silsesquioxanes: current chemistry and applications, Curr. Org. Chem., 2017, 21, 2794–2813 CAS.
- M. A. Hoque, Y. Kakihana, S. Shinke and Y. Kawakami, Polysiloxanes with Periodically Distributed Isomeric Double-Decker Silsesquioxane in the Main Chain, Macromolecules, 2009, 42, 3309–3315 CrossRef CAS.
- V. Ervithayasuporn, R. Sodkhomkhum, T. Teerawatananond, C. Phurat, P. Phinyocheep, E. Somsook and T. Osotchan, Unprecedented formation of cis- and trans-di[(3-chloropropyl)isopropoxysilyl]-bridged double-decker octaphenylsilsesquioxanes, Eur. J. Inorg. Chem., 2013, 3292–3296 CrossRef CAS.
- V. Ervithayasuporn, X. Wang and Y. Kawakami, Synthesis and characterization of highly pure-azido-functionalized polyhedral oligomeric silsesquioxanes (POSS), Chem. Commun., 2009, 34, 5130–5132 RSC.
- J. Jung, H. J. C. Furgal, T. Goodson III, T. Mizumo, M. Schwartz, K. Chou, J.-F. Vonet and R. M. Laine, 3-D Molecular Mixtures of Catalytically Functionalized [vinylSiO1.5]10/[vinylSiO1.5]12. Photophysical Characterization of Second Generation Derivatives, Chem. Mater., 2012, 24, 1883–1895 CrossRef.
- M. Janeta, Ł. John, J. Ejfler and S. Szafert, Novel organic-inorganic hybrids based on T8 and T10 silsesquioxanes: synthesis, cage-rearrangement and properties, RSC Adv., 2015, 5, 72340–72351 RSC.
- Y. Morimoto, K. Watanabe, N. Ootake, J. Inagaki, K. Yoshida and K. Ohguma, Silsesquioxane derivative and production process for the same, US Pat, 7449539B2, 2008 Search PubMed.
- L. M. J. Moore, J. J. Zavala, J. T. Lamb, J. T. Reams, G. R. Yandek, A. J. Guenthner, T. S. Haddad and K. B. Ghiassi, Bis-phenylethynyl polyhedral oligomeric silsesquioxanes: new high-temperature, processable thermosetting materials, RSC Adv., 2018, 8, 27400–27405 RSC.
- D. F. Vogelsang, J. E. Dannatt, B. W. Schoen, R. E. Maleczka, Jr. and A. Lee, Phase Behavior of cis–trans Mixtures of Double-Decker Shaped Silsesquioxanes for Processability Enhancement, ACS Appl. Nano Mater., 2019, 2, 1223–1231 CrossRef CAS.
- P. Żak, B. Dudziec, M. Kubicki and B. Marciniec, Silylative Coupling versus Metathesis—Efficient Methods for the Synthesis of Difunctionalized Double-Decker Silsesquioxane Derivatives, Chem. – Eur. J., 2014, 20, 9387–9393 CrossRef PubMed.
- V. Vij, G. Yandek, S. Ramirez, J. M. Mabry and T. S. Haddad, Polym. Prepr., 2012, 53, 513 CAS.
- K. Wei, L. Wang and S. Zheng, Organic-inorganic polyurethanes with 3,13-dihydroxyoctaphenyl double-decker silsesquioxane chain extender, Polym. Chem., 2013, 4, 1491–1501 RSC.
- K. Koppe, J. Eichhorn, B. Jeffery, A. Davis, W. Mitchell, P. Miskiewicz and T. Nonaka, WO 2020/182636A1, 2020.
- M. Walczak, R. Januszewski, M. Majchrzak, M. Kubicki, B. Dudziec and B. Marciniec, Unusual cis and trans architecture of dihydrofunctional double-decker shaped silsesquioxane and synthesis of its ethyl bridged π-conjugated arene derivatives, New J. Chem., 2017, 41, 3290–3296 RSC.
- B. W. Schoen, D. Holmes and A. Lee, Identification and quantification of cis and trans isomers in aminophenyl double-decker silsesquioxanes using 1H-29Si gHMBC NMR, Magn. Reson. Chem., 2013, 51, 490–496 CrossRef CAS PubMed.
- T. Tanaka, Y. Hasegawa, T. Kawamori, R. Kunthom, N. Takeda and M. Unno, Synthesis of Double-Decker Silsesquioxanes from Substituted Difluorosilane, Organometallics, 2019, 38, 743–747 CrossRef CAS.
- E. Arunan and H. S. Gutowsky, The rotational spectrum, structure and dynamics of a benzene dimer, J. Chem. Phys., 1993, 98, 4294–4296 CrossRef CAS.
- Z. Niu, D. Tian, L. Yan, X. Ma, C. Zhang and X. Hou, Synthesis of 3,13-diglycidyloxypropyloctaphenyl double-decker polyhedral oligomeric silsesquioxane and the thermal reaction properties with thermosetting phenol-formaldehyde resin, J. Appl. Polym. Sci., 2020, 137, 49376–49386 CrossRef CAS.
- L. Wang, C. Zhang and S. Zheng, Organic-inorganic poly(hydroxyether of bisphenol A) copolymers with double-decker silsesquioxane in the main chains, J. Mater. Chem., 2011, 21, 19344–19352 RSC.
- J. Cao, H. Fan, B.-G. Li and S. Zhu, Synthesis and evaluation of double-decker silsesquioxanes as modifying agent for epoxy resin, Polymer, 2017, 124, 157–167 CrossRef CAS.
- C.-H. Lin, W.-B. Chen, W.-T. Whang and C.-H. Chen, Characteristics of Thermosetting Polymer Nanocomposites: Siloxane-Imide-Containing Benzoxazine with Silsesquioxane Epoxy Resins, Polymers, 2020, 12, 2510 CrossRef CAS PubMed.
- S.-E. Zhu, W.-J. Yang, Y. Zhou, W.-H. Pan, C.-X. Wei, A. C. Y. Yuen, T. B. Y. Chen, G. H. Yeoh, H.-D. Lu and W. Yang, Chem. Eng. J., 2022, 442, 136367 CrossRef CAS.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- L. Liu, M. Zhu, Z. Ma, X. Xu, J. Dai, Y. Yu, S. Mohsen Seraji, H. Wang and P. Song, Chem. Eng. J., 2022, 440, 135645 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 29Si NMR, FT-IR, HR-MS spectra, X-ray crystallographic data, and dielectric studies. CCDC 2158320–2158323. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi00577h |
| This journal is © the Partner Organisations 2022 |