
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimeric iodine(I) and silver(I) cages from tripodal N-donor ligands via the [N–Ag–N]+ to [N–I–N]+ cation exchange reaction†
Essi
Taipale
 a,
Jas S.
Ward
*a,
Giorgia
Fiorini
a,
Daniel L.
Stares
b,
Christoph A.
Schalley
b and
Kari
Rissanen
*a
a,
Jas S.
Ward
*a,
Giorgia
Fiorini
a,
Daniel L.
Stares
b,
Christoph A.
Schalley
b and
Kari
Rissanen
*a
aUniversity of Jyvaskyla, Department of Chemistry, P.O. Box 35, Survontie 9B, 40014 Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi
bInstitut für Chemie und Biochemie, Organische Chemie, Freie Universität Berlin, Arnimallee 20, 14195 Berlin, Germany
First published on 31st March 2022
Abstract
The directionality of the [N–I–N]+ halogen bond makes iodine(I) ions impeccable tools in the design and construction of [N–I–N]+ halogen-bonded assemblies. The synthesis of dimeric iodine(I) cages with imidazole-derived N-donor tripodal ligands is described, as well as their corresponding silver(I) precursors. The addition of elemental iodine to the parent two-coordinate Ag(I) complexes produces iodine(I) complexes with three-center four-electron (3c–4e) [N–I–N]+ bonds. Complex formation via this cation exchange was confirmed by 1H and 1H–15N HMBC NMR studies in solution, and additionally by electrospray ionisation and ion mobility mass spectrometry analysis (MS) in the gas phase. The structural analysis of the single crystal X-ray structures of 11 silver(I) cages and the computationally modelled iodine(I) cages, along with MS analysis, revealed structural similarities between the two different capsular assemblies. In addition to strong electrostatic interactions, C–H⋯F hydrogen bonds were determined to have a directing effect on the silver(I) cage formation and binding of suitably sized anions into the cavities in the solid state.
Introduction
The term clathrate was first devised by H. M. Powell in 1948 when the first definition of a solid-state cage-like supramolecular structure came to life. According to Powell, a clathrate was a kind of a solid-state inclusion compound “in which two or more components are associated without an ordinary chemical union, but through the complete enclosure of one set of molecules in a suitable structure formed by another”.1 These structures are generally further divided into two categories: the first are clathrates, viz lattice inclusion compounds, in which guests are trapped into a host matrix, generally speaking, a crystal lattice; the second are molecular host–guest inclusion compounds, in which the guest is trapped inside a larger, often macro- or multimacrocyclic, host molecule or a self-assembled concave entity comprising multiple components, e.g., molecular capsules and cages.1Molecular hosts and capsular assemblies have attracted much interest since Rebek's first hydrogen-bonded supramolecular capsule in 1993.2 The possibility of encapsulating various guests in these cages has yielded a plethora of applications,3 ranging from biomedical applications and selective encapsulation,4,5 to stabilisation of reactive compounds using supramolecular capsular entities and catalysis in confined spaces.6,7 To date, various host–guest capsular assemblies have been reported using molecules such as resorcin[4]arenes,8–11 calix[4]-arenes,12–14 and pyridine[4]arenes.15,16 The self-assembled capsular assemblies comprise two or more suitable preorganised molecules spontaneously forming capsular entities with either metal coordination,17–21 or hydrogen bonding.22,23
Ever since the 1990s, halogen bonding has developed into a true competitor for hydrogen bonding and other non-covalent interactions to the point that, to date, it is one of the most studied interactions in supramolecular chemistry.24–27 Halogen bonding was only recently defined by IUPAC as a net attractive interaction between the positive regions of the electrostatic potential associated with a halogen atom and a Lewis base.28 Perhaps the controversial nature of the halogen bond led to its late definition, however, halogen bonding has been deftly utilised in the formation of supramolecular assemblies, amongst other non-covalent interactions, for its strength, tunability, and directionality, which provides the necessary tools for sufficient control of the self-assembly processes. Despite its utility, the use of halogen bonding in the formation of discrete molecular capsules is rare, with only a few examples previously reported using the classical halogen bond.29–31
More recently, halogen bonding has also received interest in the form of halogen(I), especially iodine(I), cations.32–34 The halogen(I) cations, also known as halenium ions (X+), can be considered as extremely polarised halogen atoms which are capable of forming symmetric three-center four-electron (3c–4e) [L–X–L]+ bonds and constructing a halogen(I) complex by simultaneously interacting with two Lewis bases (L; commonly aromatic amines).35–38 The [N–X–N]+ 3e–4e bonds are found to be among the strongest halogen bonds, which in addition to their high directionality and robust nature, has made them an interesting supramolecular synthon in modern-day crystal engineering.
Iodine(I) ions are commonly used in organic chemistry as halogenating agents39–42 and their inherent reactivity makes them harder to isolate and structurally characterise. Recently, a number of heteroleptic or unconventional iodine(I) complexes have been prepared and characterised, mainly in the solid state.37,38,43–48 So far, only a few supramolecular capsular or macrocyclic49 assemblies incorporating [N–I–N]+ halogen bonds have been reported,50–53 which are obtained using the analogous silver(I) metallocages as the starting point for the iodine(I) cages, obtained via the [N–Ag–N]+ to [N–I–N]+ cation exchange reaction.54
Herein, we report the solid-state structures of 11 parent silver(I) tripodal metallocages, and a comprehensive theoretical, gas, and solution-state study confirming the formation of iodine(I) [N–I–N]+ cages. This report serves as further proof of the indisputable importance of halogen(I) ions as supramolecular synthons, and not just in simple monodentate ligand-complexes, but also in more sophisticated capsular assemblies.
Results and discussion
Synthesis of the silver(I) and iodine(I) cages
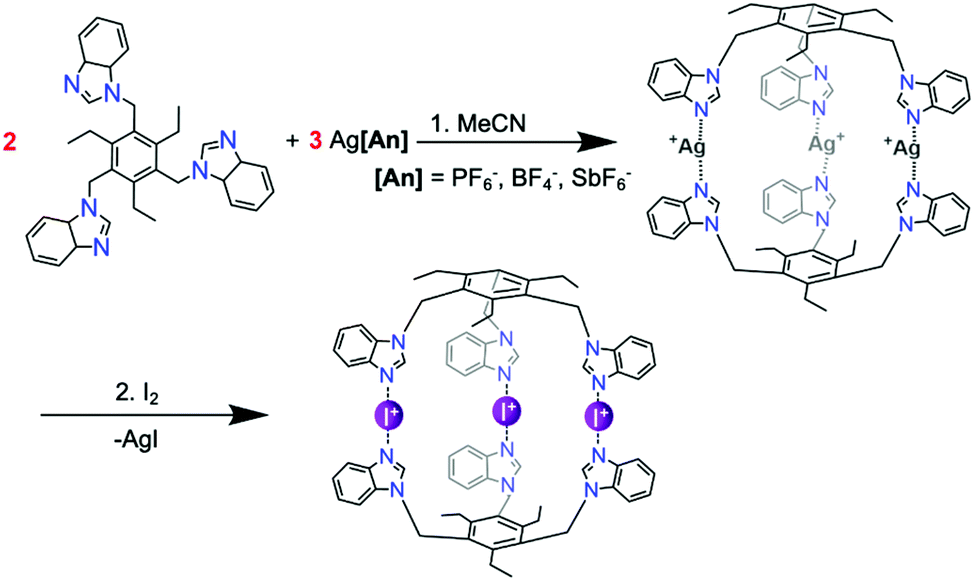
The imidazole- and benzoimidazole-based N-donor tripodal ligands 1–6 with a trialkylbenzene backbone were chosen for the capsular assembly (Scheme 1). | ||
| Scheme 1 The structures of N-donor tripodal ligands 1–6 used in the formation of the iodine(I) cages. | ||
Ligands 1–6 were straightforwardly synthesised, with only 6 not being previously described in the literature. Imidazole, benzoimidazole, or dimethyl-benzoimidazole moieties are attached to the core benzene ring via methylene-linkers, making the ligands flexible and large enough to accommodate guests inside. The ligands must be able to first bind silver(I), and then iodine(I), cations leading to two distinct complexes, which in many cases prefer slightly different conformations with dissimilar bond lengths and angles. The syntheses of the ligands are straightforward and mostly follow literature procedures, with a few adaptions stated in the ESI.†55
The most common route for iodine(I) complex formation is through the [N–Ag–N]+ → [N–I–N]+ cation exchange reaction.54 All cage reactions were performed in either acetonitrile or DMSO dictated by the lower solubility of some of the complexes. In the first step, presented in Scheme 2, two equivalents of ligand are mixed with three equivalents of a silver(I) salt (here AgPF6, AgBF4, or AgSbF6). Many different weakly coordinating anions can be used, as it has been shown that the counteranions do not influence the linear centrosymmetric geometry of the [N–I–N]+ halogen bond neither in solution nor in solid state.56 In the second step, elemental iodine (I2) is added to the parent silver(I) cage solution and stirred for an hour. Due to the more complex structure of the cages, the reaction requires more time for completion than for the previously reported small-ligand iodine(I) complexes.45 Upon completion of the reaction, the silver iodide (AgI) precipitates from the solution leaving the cationic tris-iodine(I) cage with the three anions in the solution.
 | ||
| Scheme 2 The synthesis of the iodine(I) cage of tripodal ligand 4 through its analogous silver(I) metallocage. | ||
Crystallography of the silver(I) cages
The X-ray structures of ligands 1–5 have been previously reported,21,57–60 as well as the [X-⊂1-Ag-1][X]2 cages with various counter-anions.17,20,61,62 For 1 and 2, other topologies including polymeric structures have been reported,17,61 which is not the case for the larger ligands 3–6. Very likely, the additional benzene rings point away from the capsule surface (Fig. 1) and thus do not hamper capsule formation, while they likely interfere with polymerization. Consequently, encountering polymeric structures was expected and one [1-Ag-]n[OTf]n structure was successfully characterized with X-ray crystallography (Fig. S3†). In addition, cages [BF4-⊂3-Ag-3][BF4]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and [BF4-⊂4-Ag-4][BF4]221 have been X-ray structurally characterised. In total, 11 single crystal X-ray structures of the analogous silver(I) cages were determined (Fig. S1–S13†), two of them polymorphs to previously reported structures.17,20 Unlike the halogen(I) 3c–4e bonds, the [N–Ag–N]+ environment is easily distorted by the solvent or anion coordination to the silver(I) cations and angles of as small as 149° have previously been reported.44 Further demonstrating this, the silver(I) cages were determined to have varying N–Ag bond lengths from 2.068(12) Å to 2.170(7) Å and N–Ag–N angles ranging from 163.1(2)° to 180.0(5)°. These bond lengths are similar to previously observed ones with smaller monodentate versions of these ligands.45 However, in the silver(I) cage formation the N–Ag–N angles are more linear when compared to the simple mononuclear complexes where the anion to silver(I) cation interactions can cause distortion from linearity.45
18 and [BF4-⊂4-Ag-4][BF4]221 have been X-ray structurally characterised. In total, 11 single crystal X-ray structures of the analogous silver(I) cages were determined (Fig. S1–S13†), two of them polymorphs to previously reported structures.17,20 Unlike the halogen(I) 3c–4e bonds, the [N–Ag–N]+ environment is easily distorted by the solvent or anion coordination to the silver(I) cations and angles of as small as 149° have previously been reported.44 Further demonstrating this, the silver(I) cages were determined to have varying N–Ag bond lengths from 2.068(12) Å to 2.170(7) Å and N–Ag–N angles ranging from 163.1(2)° to 180.0(5)°. These bond lengths are similar to previously observed ones with smaller monodentate versions of these ligands.45 However, in the silver(I) cage formation the N–Ag–N angles are more linear when compared to the simple mononuclear complexes where the anion to silver(I) cation interactions can cause distortion from linearity.45
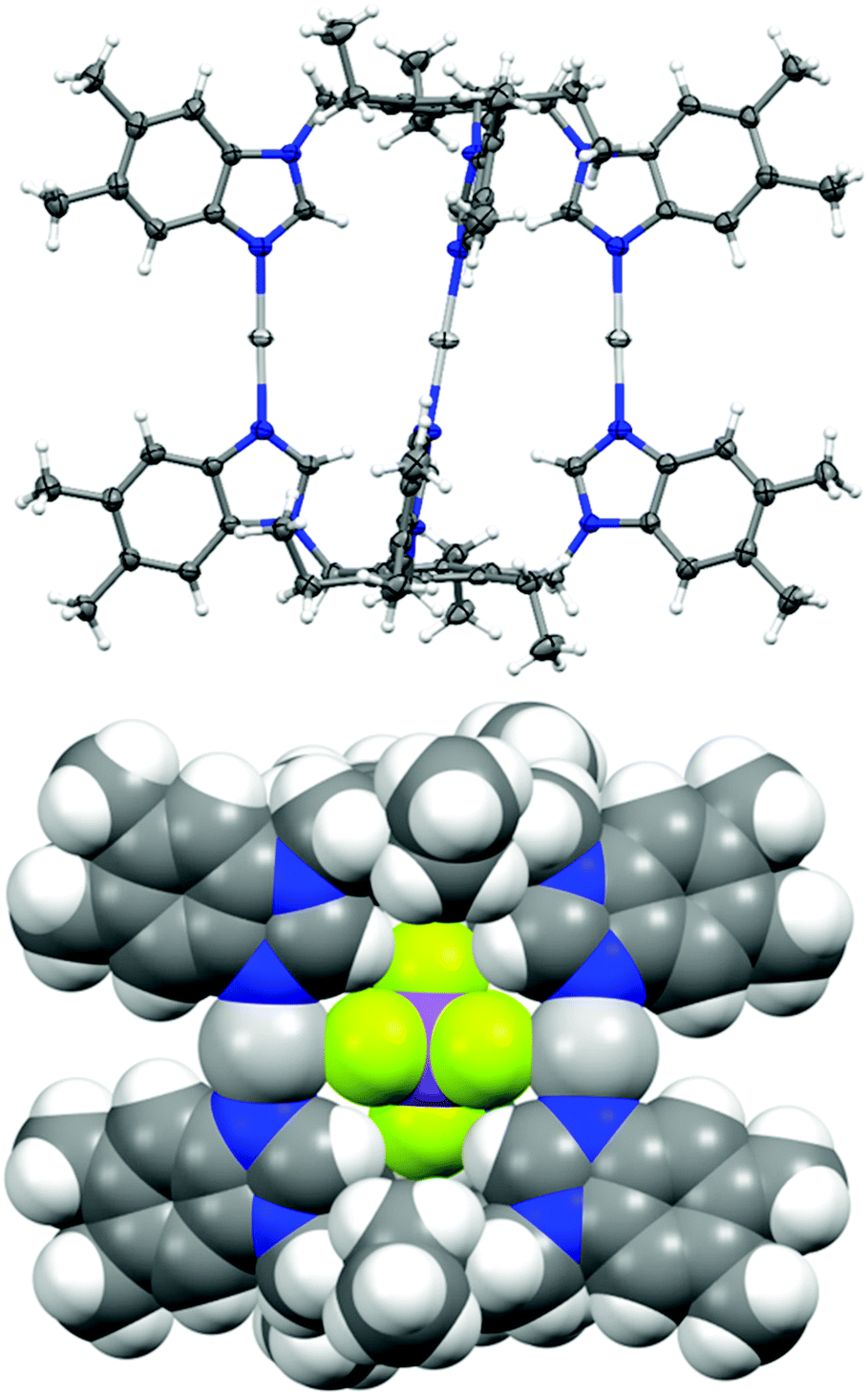 | ||
| Fig. 1 The thermal ellipsoid (top) and spacefill (bottom) representations of the X-ray crystal structure of [SbF6-⊂6-Ag-6][SbF6]2. Solvents and anions omitted for clarity except for the encapsulated SbPF6− anion in the bottom image. Thermal displacement ellipsoids are drawn at the 50% probability level. The N–Ag–N bond lengths and angles were determined to be between 2.091(4)–2.095(4) Å and 172.3(2)–173.0(2)°. | ||
The cages were designed to encapsulate small guests inside the cavites of the cages. In nine out of 11 cases, the electron density map of the X-ray crystallographic analysis indicated that one of the counterions was positioned inside the cages. However in the case of [SbF6-⊂1-Ag-1][SbF6]2 and [SbF6-⊂3-Ag-3][SbF6]2 the electron density inside the cavity was diffuse, indicating a heavily disordered SbF6− anion, and due to this they could not be modelled adequately, and were accounted for using SQUEEZE in the final model.63Fig. 1 depicts, as an example, the encapsulation of a hexafluoroantimonate(V) anion in [SbF6-⊂6-Ag-6][SbF6]2 as a result of the CH⋯anion and cation⋯anion interactions between the ligand, the Ag(I) cations and the SbF6− anion.
Moreover, the complexes observed in the solid state generally encapsulate one anion leaving the two remaining anions outside the cage. In addition, tandem MS experiments support the anion also to be present inside the cages in solution and the gas phase. When PF6- is encapsulated, the collision energies, at which 50% of the cage ions are still intact is significantly higher as compared to the same cage ions carrying a triflate anion attached to the outside (Fig. S45-S47†).
The anion interactions inside the cage were estimated by determining the shortest distances between the anionic and cationic species for structures [BF4-⊂3-Ag-3][BF4]2 and [PF6-⊂3-Ag-3][PF6]2, [BF4-⊂5-Ag-5][BF4]2 and [PF6-⊂5-Ag-5][PF6]2, and also for [PF6-⊂6-Ag-6][PF6]2 and [SbF6-⊂6-Ag-6][SbF6]2, in which the anion resides inside the cage cavity. Many factors are to be taken into account when discussing interactions strengths, and the short distances alone do not always indicate strong interactions. Furthermore, the interaction lengths can be affected by packing in the crystal structure. However, the interaction lengths and other analyses on the crystal structures are here discussed to shed light on the type of interactions involved in the anion encapsulation.
The shortest silver(I) to fluorine (Ag⋯F) constact distances were found to be between 2.66(1) Å–2.802(9) Å, which is significantly shorter than the sum of the van der Waals radii for these atoms (vdW(Ag⋯F) = 3.19 Å). Similarly, the C–H⋯F hydrogen bonds were found to be shorter than the sum of the van der Waals radii of the respective atoms (vdW(F⋯H) = 2.67 Å) varying between 2.33 Å–2.66 Å. However, for cages with the shortest Ag⋯F contacts, the hydrogen-bond distances (C–H⋯F) were found to be the longest, indicating that these distances rely heavily on the orientation of the anion inside the cage cavity.
The anion-to-cage interactions were further studied with Hirshfeld surface analysis (Fig. S14–S19†) using CrystalExplorer.64,65 In each case, large bright red spots can be observed on the Hirshfeld surface of the anions in the regions where the fluorines are in close contact with the silver(I) cations confirming the interactions to be shorter than the van der Waals radii. Slightly smaller, though still fairly bright red spots can also be seen for the C–H⋯F interactions. The 2D fingerprint plots of the Hirshfeld surface analysis provide information on the unique crystal packing in each case, at the same time easily revealing similarities between the structures. The fingerprint plot depicts the relationship of the distance to the nearest atom center interior to the surface (di) and to the exterior to the surface (de).64 The fingerprint plots (Fig. S14–S19†) display the Ag⋯F and C–H⋯F interactions as a pair of spikes at the bottom-left part of the plot. The percentage contributions to the close intermolecular contacts can also be determined from the 2D plots. For all cages, the contribution of the Ag⋯F contacts was determined to be between 9–17% (largest for [BF4-⊂5-Ag-5][BF4]2), whereas the C–H⋯F hydrogen bonds contributed 82–91% to the overall interactions. The electrostatic interactions are generally stronger than hydrogen bonding, and in here, they act in unison. Therefore, even though the Hirshfeld surface depicts Ag⋯F contacts to be the closest, the hydrogen bonds most certainly play a role in the stabilisation of the anion. In some cases, a small percentage (<4%) of other interactions (F⋯N and F⋯C) were observed to affect the anion binding. The results from the Hirshfeld surface analysis indicate a multitude of stabilising interactions directed toward the anion. For example, Fig. 2 depicts the X-ray crystal structure of [SbF6-⊂6-Ag-6][SbF6]2 with the calculated Hirshfeld surface for the SbF6− anion inside the cage, where the punctures in the surface display strong cation–anion (Ag+⋯F6Sb−) interactions. Additionally, the strong interactions are supported by the non-disordered nature of the SbF6− anion with well-defined thermal movement inside the cage due to the supramolecular interactions.66,67
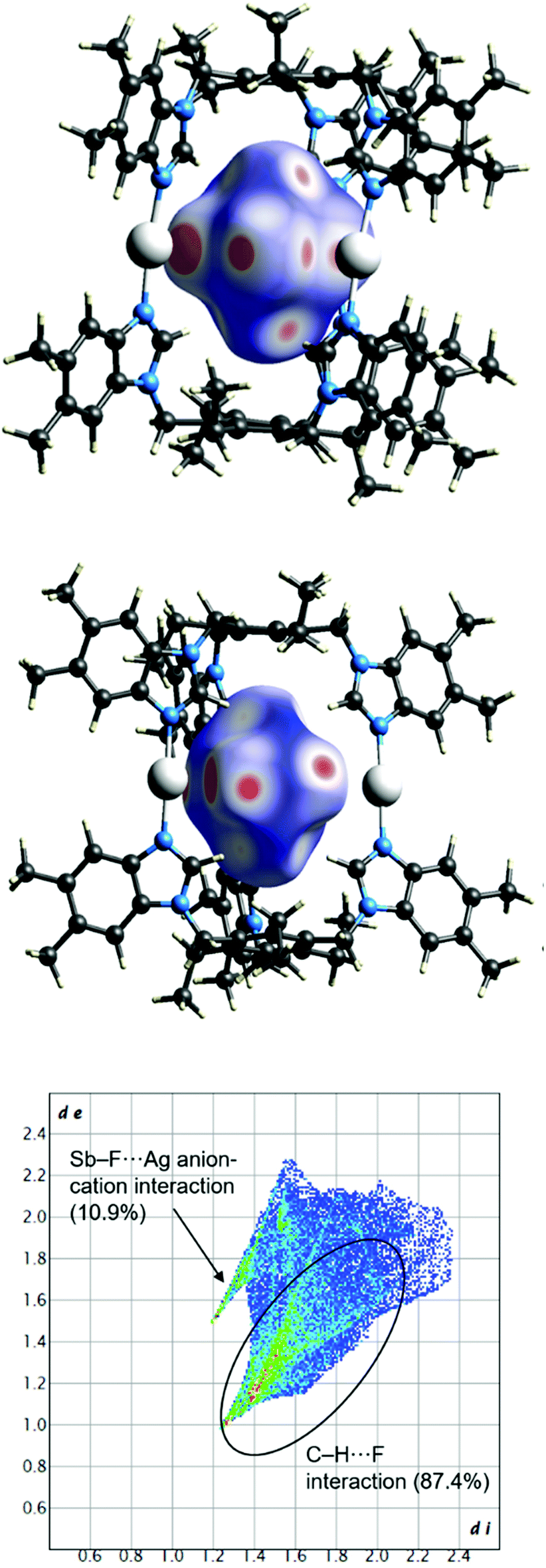 | ||
| Fig. 2 Full fingerprint regions of the hexafluoroantimonate anion in the complex [SbF6-⊂6-Ag-6][SbF6]2 and two views of the intermolecular contacts to the Hirshfeld surface of the anion. Other interactions contributing to the anion binding: Sb–F⋯C (1.7%). In the fingerprint plot, di represents the closest internal distance from a given point on the Hirshfeld surface, and de is the closest external contact (in Å). | ||
Characterisation of the iodine(I) cages
Despite numerous silver(I) cages, to date, only the solid-state structure of dimeric [N–I–N]+ halogen-bonded cage made from 1 has been reported.53 The nucleophilic nature of the iodine(I)38 cation renders the interactions with anions very weak, and the inability to get diffraction quality single crystals of the iodine(I) cages in this study highlights the challenge in obtaining solid-state crystal structures of larger [N–I–N]+ halogen(I) assemblies with multiple 3c–4e bonds.50,51 Unfortunately, also in this study, the crystallisation of the [N–I–N]+ cages proved to be unsuccessful. During the crystallisation, conceivably the inherent anisotropic nature of the 3c–4e [N–I–N]+ bond results in conflicting anion interactions, with the repulsive nature of the I+⋯F interactions and the attractive endohedral cavity,53 making the anion encapsulation and the crystallisation process of the iodine(I) cages more problematic. Therefore, the iodine(I) cages were modelled with fixed N–I bond lengths of 2.25 Å and 180° angles for the most accurate description based on the known coordination geometry of the [N–I–N]+ halogen bond (Fig. 3 and Fig. S20–S25†).56,68,69 The resulting calculated iodine(I) cage structures are very similar to the analogous silver(I) crystal structures (see below discussion and Fig. S12†), apart from the slight distortion of the silver(I) cages in the solid state due to packing effects.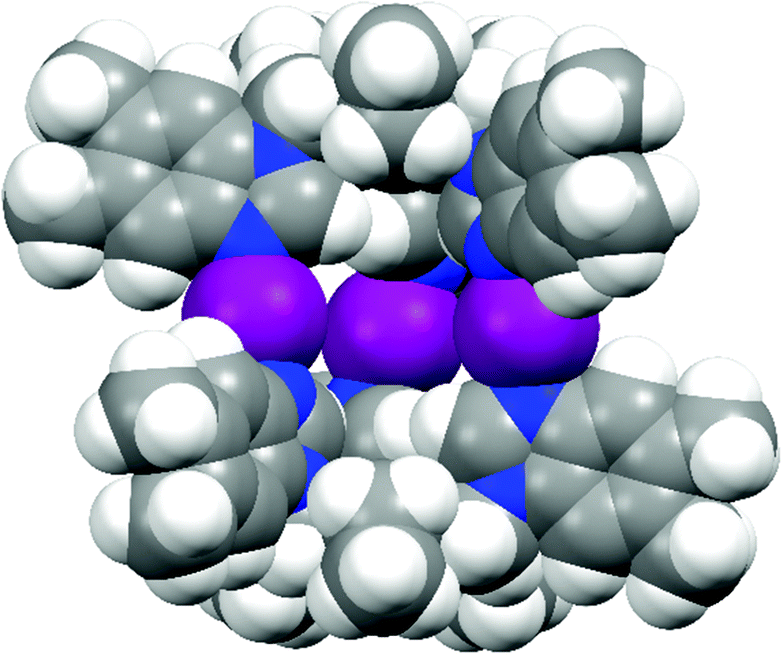 | ||
| Fig. 3 The spacefill representation of the calculated structure of [PF6-⊂6-I-6][PF6]2 iodine(I) cage. | ||
The cage composition was further confirmed in the gas phase with electrospray ionisation mass spectrometry (ESI-MS) and ion mobility mass spectrometry (IM-MS) analysis51,70 (Fig. S26–S49†). For ligand 4, the formation of the cages was verified by the detection of the ions [PF6–⊂4–Ag–4]2+ and [PF6–⊂4–I–4]2+ at m/z values of 787 and 815, respectively. Additionally, the cages were observed with other anions in the gas phase, such as I− and PF2O2−. In ion mobility mass spectrometric experiments, the silver(I) and iodine(I) cages appear with very similar, narrow-width arrival time distributions confirming that the overall cage structures have not changed during the Ag(I)-to-I(I) ion exchange reactions. The IM-MS provided evidence of the cages being close in their sizes, though the iodine(I) cages are slightly bigger than the corresponding silver(I) cages. All silver(I) and iodine(I) cages depicted a narrow peak width in the arrival time distribution indicating a discrete, well-defined structure (Fig. 4). Also, the iodine(I) and silver(I) cages show closely related fragmentation patterns in collision-induced dissociation (CID) experiments (Fig. S45–S49†). The dissociation of the mass-selected doubly charged [PF6–⊂L–Ag–L]2+ and [PF6–⊂L–I–L]2+ ions leads to a singly charged [LAg]+ and [LI]+ ions, respectively. The corresponding singly charged [LAg2PF6]+ and [LI2PF6]+ fragments are, however, not observed, which indicates a subsequent, rapid loss of AgPF6 or IPF6 ion pairs leading again to a second [LAg]+ and [LI]+ fragment, respectively. The fact that these ion pair losses do not occur similarly from the parent ions directly is in agreement with an encapsulated PF6− counterion, which can only escape in the form of the ion pairs when the cage has already fragmented. This assumption is also in line with the observation of 2+, but no 1+ or 3+ charge states for the intact cages. The fragmentation was studied at different collision voltages to obtain a survivor yield curve for both the silver(I) and the iodine(I) cages (Fig. S49†). The 50% survivor yields are obtained at collision energies of 17 V for [PF6–⊂L–I–L]2+ and of 26 V for [PF6–⊂L–Ag–L]2+. Thus, the iodine(I) cage is somewhat less stable in the gas phase than its silver(I) analogue. In conclusion, not only the sizes of the two cages are in the same range, but also the ionisation behavior (only +2 charge states) and the fragmentation patterns. This is straightforward evidence that both cages have analogous capsular structures.
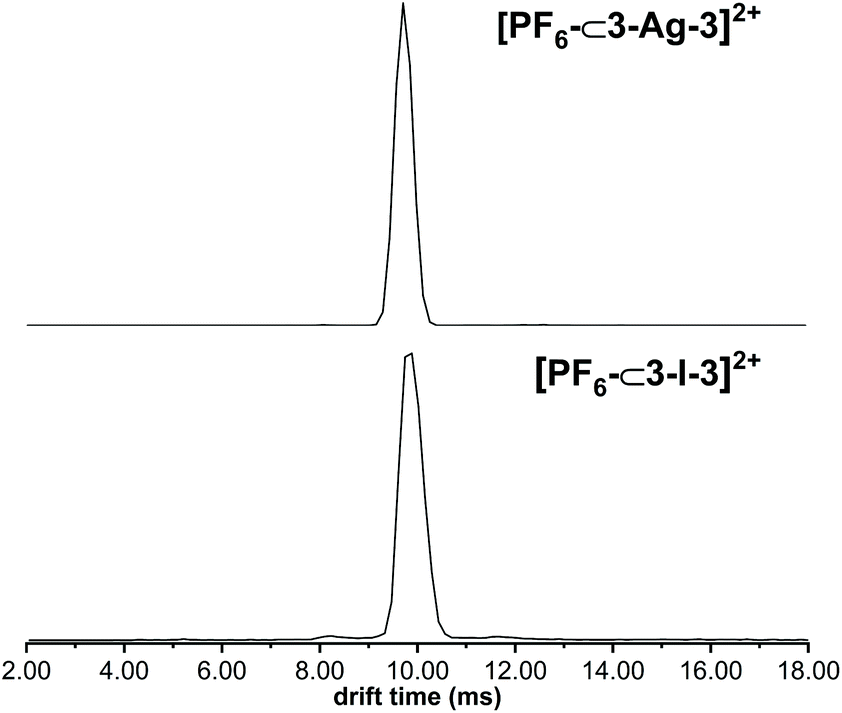 | ||
| Fig. 4 ATDs of [PF6–⊂3–Ag–3]2+ (top) and [PF6–⊂3–I–3]2+ (bottom). The arrival time of both are similar and the distributions narrow. | ||
The formation of the silver(I) and iodine(I) cages in solution was studied with 1H NMR and 1H–15N HMBC measurements (Fig. 5 and Fig. S50–S59†). Ligand 1 was not included in the NMR study since similar experiments have been previously reported.53 Earlier studies have shown that the formation of the silver(I) cage results in a downfield change in the 1H NMR chemical shifts of the ligand,43–45,53 most prominently observed for the imidazole C(2) proton (Ha, singlet, Fig. 1, top). Changes in the 1H NMR chemical shift range were between Δδ 0.16 and Δδ 0.48 ppm when going from the uncomplexed ligands to the silver(I) complexes. The cation exchange reaction from Ag+ to I+ results in a further downfield shift, which for these ligands, ranges from Δδ 0.16 to Δδ 1.21 ppm, resulting in notable shift differences between the free ligands and the iodine(I) cages of up to almost 1.7 ppm. It has been previously observed that the solubility of the complex is often lower for the I+ complex than for the Ag+ complex, and greatly reduced compared to the respective free ligand.45 A complete set of measurements in CD3CN could be performed only for two ligands (4 and 5) out of the five systems, as the solubility of some of the silver(I) and iodine(I) cages required the use of DMSO-d6, and the poor solubility of the silver(I) complexes, as well as interactions with the competing DMSO, results in an incomplete conversion of the silver(I) cage to the iodine(I) cage, as confirmed by 1H NMR spectroscopy. This is most clearly observed for ligand 2, while being much less pronounced for 3 and 6. However, the majority of the silver(I) cages fully convert to the iodine(I) cages. The conversion from the ligands to the silver(I) complexes were further studied with 1H–15N HMBC measurements (Fig. S51, S53, S55, S57, and S59†). Even though the 1H NMR spectra could be obtained for all the silver(I) and iodine(I) cages, in most cases the decreased solubility of the iodine(I) cages would cause them to precipitate out of solution during the time required for satisfactory quality HMBC acquisitions to be collected, and therefore, a full set of 15N NMR resonances were obtained for only one iodine(I) cage, [PF6–⊂2–I–2][PF6]2 (Fig. S51†). However, as demonstrated before,45 upon coordination to silver(I), or iodine(I), the resonance of the coordinating nitrogen (N1) is found to change markedly downfield due to the deshielding effect upon complexation. In contrast, the non-coordinating nitrogen (N2) is shielded and experiences only minor changes in the opposite direction. Due to low solubility, the signal of the coordinating nitrogen (N1) is in some cases undetectable, making the N2 value ever more important, and giving valuable information about the complexation. The silver(I) complexation induces a coordination shift around −50 ppm in the 15N NMR resonance of N1 for all ligands, whereas the resonance of N2 only changes by around +5 ppm.
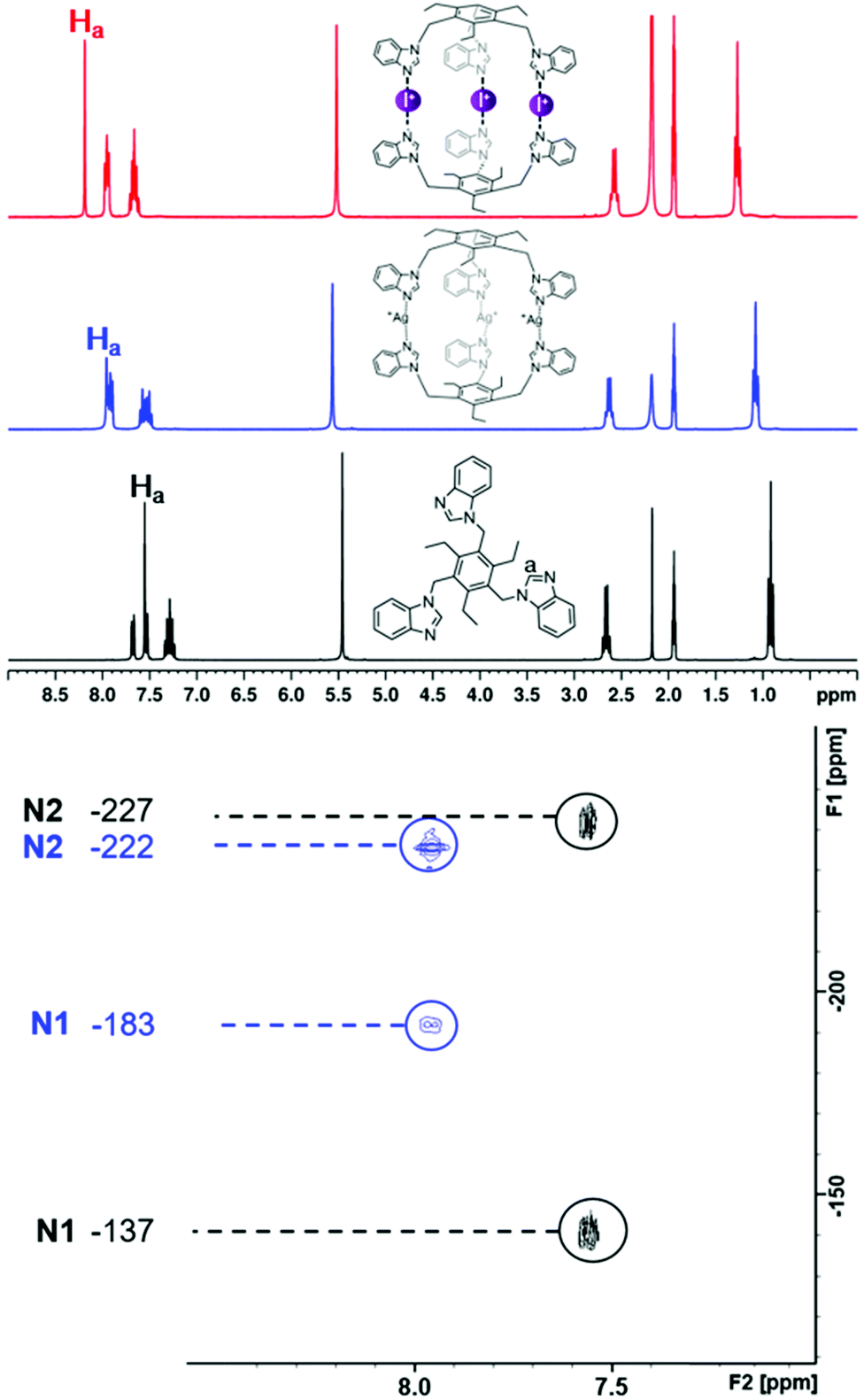 | ||
| Fig. 5 The 1H (top) and 1H–15N HMBC* (bottom) NMR spectra of the uncomplexed ligand 4 (black), the [PF6-⊂4-Ag-4][PF6]2 silver(I) complex (blue), and the [PF6-⊂4-I-4][PF6]2 iodine(I) complex (red), where Ha is the proton between the coordinating (N1) and non-coordinating (N2) nitrogens. All values are in ppm and referenced to CD3CN (1H) and CD3NO2 (15N) (500 MHz, 303 K). *Due to poor solibility, the 1H–15N HMBC NMR spectra of the iodine(I) complex could not be obtained. | ||
Conclusions
In summary, we have presented comprehensive evidence of dimeric imidazole-based iodine(I) cage formation in the solution and gas phase. The formation of the iodine(I) cages occurs via the Ag+ to I+ cation exchange reaction from the analogous silver(I) cages. In addition to NMR and MS experiments, the silver(I) cages were also crystallised and further studied in the solid state using X-ray crystallography. The resulting endohedral cavity of the self-assembled dimeric cages yields an anion encapsulation with strong supramolecular interactions. The silver(I) crystal structure of [PF6-⊂6-Ag-6][PF6]2 and the calculated structure of [PF6-⊂6-I-6][PF6]2 along with the ion mobility-MS analysis, clearly display structural similarities amongst the two well-defined structures with the silver(I) cage having slightly shorter Ag–N bond lengths (2.068(12)–2.170(7) Å) and less linear N–Ag–N angles (163.1(2)°–180.0(5)°) than expected for the iodine(I) cage. This is strongly reflected in the almost identical arrival time distributions determined in ion mobility mass spectrometric experiments, which reveal only a very slight size increase when the silver ions are replaced by iodine(I). The flexibility of the cages enabled them to host anions of varying sizes, forming complexes with strong supramolecular interactions in each case. In addition to anion–cation interactions, a surprisingly large contribution to the anion stabilisation was made by the C–H⋯F hydrogen bonds.For all the complexes, [PF6–⊂L–Ag–L]2+ ions were observed in the mass spectra, which indicates a stonger interaction of one anion compared to the remaining two. This would suggest that the anion encapsulation occurs for each complex in the gas phase. Furthermore, this would also support the occurrence of the anion encapsulation in solution. Even though the destabilizing interactions in the [N–I–N]+ cages rendered the crystallization attempts ineffective, the directionality of the [N–I–N]+ halogen bond makes iodine(I) ions excellent tools in supramolecular cage design and crystal engineering. The work presented here serves as further proof of the indisputable importance of halogen(I) ions as supramolecular synthons, aiding the design of more complex supramolecular capsular assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Academy of Finland (K. R. grant no. 317259), Finnish Cultural Foundation Central Fund (J. S. W. grant number 00201148), the Magnus Ehrnrooth Foundation (J. S. W.), European Union NOAH project (D. L. S, H2020-MSCA-ITN project ref. 765297, the Core Facility BioSupraMol (supported by the Deutsche Forschungsgemeinschaft, DFG) and the University of Jyväskylä, Finland for financial support.Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd, Chichester, United Kingdom, 2nd edn, 2009 Search PubMed.
- R. Wyler, J. de Mendoza and J. Rebek Jr., A synthetic cavity assembles through self–complementary hydrogen bonds, Angew. Chem., Int. Ed. Engl., 1993, 32, 1699 CrossRef.
- C. M. A. Gangemi, A. Pappalardo and G. T. Sfrazzetto, Applications of supramolecular capsules derived from resorcin[4]arenes, calix[n]arenes and metallo-ligands: From biology to catalysis, RSC Adv., 2015, 5, 51919 RSC.
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Biomedical and biochemical applications of self-assembled metallacycles and metallacages, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS PubMed.
- K. Harris, D. Fujita and M. Fujita, Giant hollow MnL2n spherical complexes: Structure, functionalisation and applications, Chem. Commun., 2013, 49, 6703 RSC.
- A. Galan and P. Ballester, Stabilization of reactive species by supramolecular encapsulation, Chem. Soc. Rev., 2016, 45, 1720 RSC.
- M. Yoshizawa, M. Tamura and M. Fujita, Diels-Alder in aqueous molecular hosts: Unusual regioselectivity and efficient catalysis, Science, 2006, 312, 251 CrossRef CAS PubMed.
- K. Twum, K. Rissanen and N. K. Beyeh, Recent advances in halogen bonded assemblies with resorcin[4]arenes, Chem. Rec., 2021, 21, 386 CrossRef CAS PubMed.
- N. K. Beyeh and K. Rissanen, Dimeric resorcin[4]arene capsules in the solid state, Isr. J. Chem., 2011, 51, 769 CrossRef CAS.
- H. Mansikkamäki, M. Nissinen and K. Rissanen, C-Methyl resorcin[4]arene packing motifs with alkyl ammonium salts: From molecular capsules to channels and tubes, CrystEngComm, 2005, 7, 519 RSC.
- R. Puttreddy, N. K. Beyeh, E. Kalenius, R. H. A. Ras and K. Rissanen, 2-Methylresorcinarene: A very high packing coefficient in a mono-anion based dimeric capsule and the X-ray crystal structure of the tetra-anion, Chem. Commun., 2016, 52, 8115 RSC.
- I. Thondorf, F. Broda, K. Rissanen, M. Vysotsky and V. Böhmer, Dimeric capsules of tetraurea calix[4]arenes. MD simulations and X-ray structure, a comparison, J. Chem. Soc., 2002, 2, 1796 Search PubMed.
- M. Chas, G. Gil-Ramírez and P. Ballester, Exclusive self-assembly of a polar dimeric capsule between tetraurea calix[4]pyrrole and tetraurea calix[4]arene, Org. Lett., 2011, 13, 3402 CrossRef CAS PubMed.
- D. Ken and J. Rebek, Synthesis and assembly of self-complementary calix[4jarenes, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 12403 CrossRef PubMed.
- A. Kiesilä, N. K. Beyeh, J. O. Moilanen, R. Puttreddy, S. Götz, K. Rissanen, P. Barran, A. Lützen and E. Kalenius, Thermodynamically driven self-assembly of pyridinearene to hexameric capsules, Org. Biomol. Chem., 2019, 17, 6980 RSC.
- A. Kiesilä, L. Kivijärvi, N. K. Beyeh, J. O. Moilanen, M. Groessl, T. Rothe, S. Götz, F. Topić, K. Rissanen, A. Lützen and E. Kalenius, Simultaneous endo and exo complex formation of pyridine[4]arene dimers with neutral and anionic guests, Angew. Chem., Int. Ed., 2017, 56, 10942 CrossRef PubMed.
- W.-Y. Sun, J. Fan, T.-A. Okamura, J. Xie, K.-B. Yu and N. Ueyama, Self-assembly of frameworks with specific topologies: Construction and anion exchange properties of M3L2 architectures by tripodal ligands and silver(I) salts, Chem. – Eur. J., 2001, 7, 2557 CrossRef CAS.
- C.-Y. Su, Y.-P. Cai, C.-L. Chen, F. Lissner, B.-S. Kang and W. Kaim, Self-assembly of trigonal-prismatic metallocages encapsulating BF4− or CuI32− as anionic guests: Structures and mechanism of formation, Angew. Chem., Int. Ed., 2002, 41, 3371 CrossRef CAS PubMed.
- V. Amendola, M. Boiocchi, B. Colasson, L. Fabbrizzi, M.-J. Rodriguez Douton and F. Ugozzoli, A metal-based trisimidazolium cage that provides six C-H hydrogen-bond-donor fragments and includes anions, Angew. Chem., Int. Ed., 2006, 45, 6920 CrossRef CAS PubMed.
- H.-K. Liu, X. Huang, T. Lu, X. Wang, W.-Y. Sun and B.-S. Kang, Discrete and infinite 1D, 2D/3D cage frameworks with inclusion of anionic species and anion-exchange reactions of Ag3L2 type receptor with tetrahedral and octahedral anions, Dalton Trans., 2008, 3178 RSC.
- S. Bhattacharya and B. K. Saha, Conformation driven complexation of two analogous benzimidazole based tripodal ligands with Ag(I) resulting in a trigonal prism and a coordination polymer, J. Chem. Sci., 2016, 128, 207 CrossRef CAS.
- H. Mansikkamäki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902 RSC.
- H. Mansikkamäki, M. Nissinen, C. A. Schalley and K. Rissanen, Self-assembling resorcinarene capsules: Solid and gas phase studies on encapsulation of small alkyl ammonium cations, New J. Chem., 2003, 27, 88 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The halogen bond, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Halogen bonding in supramolecular chemistry, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed.
- M. H. Kolář and P. Hobza, Computer modeling of halogen bonds and other σ-hole interactions, Chem. Rev., 2016, 116, 5155 CrossRef PubMed.
- H. Wang, W. Wang and W. J. Jin, σ-Hole bond vs π-hole bond: A comparison based on halogen bond, Chem. Rev., 2016, 116, 5072 CrossRef CAS PubMed.
- G. R. Desiraju, P. Shing Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Definition of the halogen bond (IUPAC recommendations 2013), Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- C. B. Aakeröy, A. Rajbanshi, P. Metrangolo, G. Resnati, M. F. Parisi, J. Desper and T. Pilati, The quest for a molecular capsule assembled via halogen bonds, CrystEngComm, 2012, 14, 6366 RSC.
- Y.-J. Zhu, Y. Gao, M.-M. Tang, J. Rebek Jr. and Y. Yu, Dimeric capsules self-assembled through halogen and chalcogen bonding, Chem. Commun., 2021, 57, 1543 RSC.
- N. K. Beyeh, F. Pan and K. Rissanen, A halogen-bonded dimeric resorcinarene capsule, Angew. Chem., Int. Ed., 2015, 54, 7303 CrossRef CAS PubMed.
- J. A. Creighton, I. Haque and J. L. Wood, The iododipyridinium ion, Chem. Commun., 1966, 229 RSC.
- J. A. Creighton, I. Haque and J. L. Wood, The iododipyridinium ion, Chem. Commun., 1966, 892 RSC.
- I. Haque and J. L. Wood, The vibrational spectra and structure of the bis(pyridine)iodine(I), bis(pyridine)bromine(I), bis(γ-picoline)iodine-(I) and bis(γ-picollne)bromine(I) cations, J. Mol. Struct., 1968, 2, 217 CrossRef CAS.
- A.-C. C. Carlsson, M. Uhrbom, A. Karim, U. Brath, J. Gräfenstein and M. Erdélyi, Solvent effects on halogen bond symmetry, CrystEngComm, 2013, 15, 3087 RSC.
- L. Turunen and M. Erdélyi, Halogen bonds of halonium ions, Chem. Soc. Rev., 2020, 49, 2688 RSC.
- A. C. Reiersølmoen, S. Battaglia, S. Oien-Odegaard, A. K. Gupta, A. Fiksdahl, R. Lindh and M. Erdélyi, Symmetry of three-center, four-electron bonds, Chem. Sci., 2020, 11, 7979 RSC.
- S. Yu, P. Kumar, J. S. Ward, A. Frontera and K. Rissanen, A “nucleophilic” iodine in a halogen-bonded iodonium complex manifests an unprecedented I+⋯Ag+ interaction, Chem, 2021, 7, 948 CAS.
- J. Barluenga, J. M. González, M. A. García-Martín, P. J. Campos and G. Asensio, An expeditious and general aromatic iodination procedure, J. Chem. Soc., Chem. Commun., 1992, 1016 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, Efficient reagents for the synthesis of 5-, 7-, and 5,7-substituted indoles starting from aromatic amines: Scope and limitations, J. Org. Chem., 1996, 61, 5804 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Control of the iodination reaction on activated aromatic residues in peptides, Chem. Commun., 2000, 1307 RSC.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Bis(pyridine)iodonium tetrafluoroborate (IPy2BF4): A versatile oxidizing reagent, Chem. – Eur. J., 2004, 10, 4206 CrossRef CAS PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Asymmetric [N-I-N]+ halonium complexes, Chem. Commun., 2020, 56, 8428 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Iodonium complexes of the tertiary amines quinuclidine and 1-ethylpiperidine, Dalton Trans., 2021, 50, 8297 RSC.
- E. Taipale, M. Siepmann, K.-N. Truong and K. Rissanen, Iodine(I) and silver(I) complexes of benzoimidazole and pyridylcarbazole derivatives, Chem. – Eur. J., 2021, 27(69), 17412–17419 CrossRef CAS PubMed.
- J. S. Ward, A. Frontera and K. Rissanen, Nucleophilic iodonium interactions (NIIs) in 2-coordinate iodine(I) and silver(I) complexes, Chem. Commun., 2021, 57, 5094 RSC.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Halogen bonding helicates encompassing iodonium cations, Angew. Chem., Int. Ed., 2019, 58, 9012 CrossRef CAS PubMed.
- J. S. Ward, A. Frontera and K. Rissanen, Utility of three-coordinate silver complexes toward the formation of iodonium ions, Inorg. Chem., 2021, 60, 5383 CrossRef CAS PubMed.
- S. Yu, E. Kalenius, A. Frontera and K. Rissanen, Macrocyclic complexes based on [N⋯I⋯N]+ halogen bonds, Chem. Commun., 2021, 57, 12464–12467 RSC.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, [N⋯I+⋯N] Halogen-bonded dimeric capsules from tetrakis(3-pyridyl)ethylene cavitands, Angew. Chem., Int. Ed., 2016, 55, 14033 CrossRef CAS PubMed.
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Surprising solvent-induced structural rearrangements in large [N⋯I+⋯N] halogen-bonded supramolecular capsules: An ion mobility-mass spectrometry study, Chem. Sci., 2018, 9, 8343 RSC.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Nano-sized I12L6 molecular capsules based on the [N⋯I+⋯N] halogen bond, Chem, 2017, 3, 861 CAS.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Tetrameric and dimeric [N⋯I+⋯N] halogen-bonded supramolecular cages, Chem. – Eur. J., 2017, 23, 11714 CrossRef CAS PubMed.
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdélyi, Symmetry of [N–X–N]+ halogen bonds in solution, Chem. Commun., 2012, 48, 1458 RSC.
- Y. Yuan, Z.-L. Jiang, J.-M. Yan, G. Gao, A. S. C. Chan and R.-G. Xie, A convenient and effective synthesis of tris-bridged tricationic azolophanes, Synth. Commun., 2000, 30, 4555 CrossRef CAS.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topic, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Counterion influence on the N-I-N halogen bond, Chem. Sci., 2015, 6, 3746 RSC.
- W.-Y. Sun, J. Fan, J. Hu, K.-B. Yu and W.-X. Tang, Molecular structure and 1H, 13C NMR assignments of 1,3,5-tris(imidazol-1-ylmethyl)-2,4,6-trimethylbenzene, J. Chem. Crystallogr., 2000, 30, 115 CrossRef CAS.
- S. N. L. Andree, A. S. Sinha and C. B. Aakeröy, Structural examination of halogen-bonded co-crystals of tritopic acceptors, Molecules, 2018, 23, 163 CrossRef PubMed.
- C. E. Willans, S. French, K. M. Anderson, L. J. Barbour, J.-A. Gertenbach, G. O. Lloyd, R. J. Dyer, P. C. Junk and J. W. Steed, Tripodal imidazole frameworks: Reversible vapour sorption both with and without significant structural changes, Dalton Trans., 2011, 40, 573 RSC.
- C. B. Aakeröy, M. Smith and J. Desper, Finding a single-molecule receptor for citramalic acid through supramolecular chelation, Can. J. Chem., 2015, 93, 822–825 CrossRef.
- J. Fan, W.-Y. Sun, T. Okamura, J. Xie, W.-X. Tang and N. Ueyama, First example of a dumbbell-like architecture containing M3L2 cages and terephthalate anions, New J. Chem., 2002, 26, 199 RSC.
- J. Fan, H.-F. Zhu, T. Okamura, W.-Y. Sun, W.-X. Tang and N. Ueyama, Discrete and infinite cage-like frameworks with inclusion of anionic and neutral species and with interpenetration phenomena, Chem. – Eur. J., 2003, 9, 4724 CrossRef CAS PubMed.
- A. L. Spek, PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, Substituent effects on the [N-I-N]+ halogen bond, J. Am. Chem. Soc., 2016, 138, 9853 CrossRef CAS PubMed.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, Symmetric halogen bonding is preferred in solution, J. Am. Chem. Soc., 2012, 134, 5706 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek Jr., The 55% solution: A formula for molecular recognition in the liquid state, Chem. – Eur. J., 1998, 4, 1016 CrossRef CAS.
- K. Rissanen, Crystallography of encapsulated molecules, Chem. Soc. Rev., 2017, 46, 2638 RSC.
- E. Kalenius, M. Groessl and K. Rissanen, Ion mobility–mass spectrometry of supramolecular complexes and assemblies, Nat. Rev. Chem., 2019, 3, 4 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2126429–2126441. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d1qi01532j |
| This journal is © the Partner Organisations 2022 |