
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards degradable polyethylene: end-functionalised polyethylene (PE-X) and PE-I/LDPE blends from iron-catalysed chain growth of ZnEt2 with ethylene†
Richard
von Goetze
a,
Ahmad
Aljaber
a,
Koon-Yang
Lee
 c,
Gavin
Hill
b,
Christopher
Wallis
ab and
George J. P.
Britovsek
*a
c,
Gavin
Hill
b,
Christopher
Wallis
ab and
George J. P.
Britovsek
*a
aDepartment of Chemistry, Imperial College London, MSRH, 82 Wood Lane, White City Campus, London W12 0BZ, UK. E-mail: g.britovsek@imperial.ac.uk
bPolymateria Ltd, Innovation-Hub, 80 Wood Lane, London, W12 0BZ, UK
cDepartment of Aeronautics, Imperial College London, Exhibition Road, South Kensington Campus, London SW7 2AZ, UK
First published on 1st November 2022
Abstract
A series of end-functionalised polyethylene materials (PE-X) has been prepared via the catalysed chain growth (CCG) reaction of diethyl zinc with ethylene, catalysed by a bis(imino)pyridine iron catalyst activated by MAO. This CCG catalyst system enables the in situ formation of long alkyl chain zinc species Zn(PE)2, which are subsequently quenched to form PE-X. Quenching with oxygen results in PE-OH, but the functionalisation appears to be limited to approximately 80% due to the formation of mixed [ZnR(OR)]n clusters. Functionalisation with sulfur leads to polysulfides, PE-Sk-PE, whereby k is affected by temperature. Functionalisation with iodine leads to PE-I with high conversion, but the degree of functionalisation appears to be chain length dependant. PE-I has been blended with LDPE, either through solution mixing or via melt blending to give PE-I/LDPE blends with different chain lengths. Characterisation of the PE-I/LDPE blends has been carried by IR spectroscopy and thermal analysis (DSC). Surface analysis by FIB-SEM and EDX analysis up to 6 μm into the surface has shown a uniform distribution of PE-I within the LDPE matrix. The propensity of alkyl iodides to undergo photolytic cleavage makes these PE-I/LDPE materials interesting candidates for degradable PE.
Introduction
Polyethylene (PE) is currently the most widely used synthetic polymer on the market and accounts for almost one third of the total global plastics production, which translates to approximately 90 Mt per annum.1–3 A wide range of different types of PE are available, all with polymer properties designed for specific applications. Many of these properties cannot easily be obtained by other polymers at a reasonable cost, including polymers from bio-derived or renewable resources. The unique and tuneable properties of PE-based materials and their versatility, combined with their low cost and ease of production, have made PE the most successful polymer on the market and the material of choice for many applications today and for some time to come.The main classes of polyethylene, LDPE, LLDPE, HDPE and UHMWPE, are prepared using different catalyst systems and, in the case of LLDPE through incorporation of co-monomers.4 Post-functionalisation of polyethylene can be carried out to modify certain polymer properties using a range of methods,5 for example through oxidation or chlorination of surface C–H bonds to prepare oxygenated or chlorinated PE.6 An alternative strategy to introduce specific functionality into polyolefins like PE is through melt-blending with end-functionalised polyethylene. In this context, well-defined functionalised polymers, so called smart materials, have become increasingly important targets in recent years.7–10
Commercially available end-functionalised alkanes are only up to C18. A convenient method to generate longer polyethylene chains with reactive chain ends (PE-X) is via reversible coordinative chain transfer polymerisation.9,11 This method uses a polymerisation catalyst (M1) in combination with a chain transfer agent (M2) and makes use of a very fast and reversible exchange of the growing polymer chains (P1 and P2) between the catalyst and a transfer agent (see eqn. (1)). A range of transition metal catalysts can be used, whereby transfer agents are typically based on main group metals such as trialkyl aluminium,12–23 dialkyl magnesium,24–27 or dialkyl zinc reagents.12,28–33
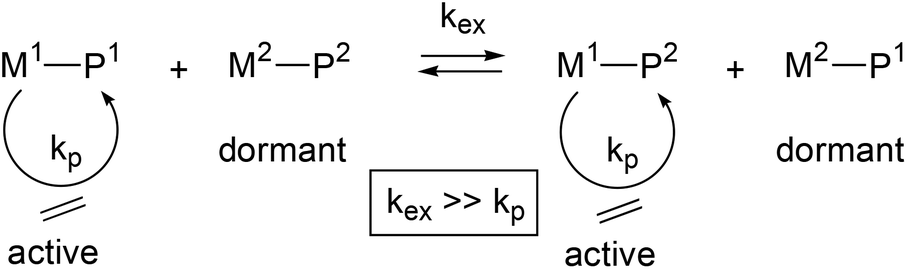 | (1) |
A specific case where all metal alkyl chains are equally engaged in reversible coordinative chain transfer polymerisation is termed Catalysed Chain Growth (CCG).28 CCG leads to a controlled growth of all alkyl chains on the main group metal (M2) and results in M(PE)n with a Poisson distribution of PE chain lengths.34 Cleavage of the M–C bond in M(PE)n can be achieved in a number of ways. Hydrolysis (or protonolysis with HX) will generate linear alkanes PE-H and Zn(OH)2 (or ZnX2), whereas the use of an additional catalyst that enables a fast termination via β-H transfer can generate a Poisson distribution of linear 1-alkenes.12,35 The high reactivity of main group M–C bonds can also be exploited to generate end-functionalised polyethylenes PE-X, where X is for example a halide, hydroxide or amine functionality. End-functionalised PE-X can then be used for the preparation of diblock copolymers.8,22,36–41 Oxidative workup of the M–C bonds to generate PE-OH has been reported for aluminium,21,23,42 magnesium,43,44 and zinc alkyls.36–41,45,46 The end-functionalisation of polyethylene using magnesium alkyls as the chain transfer agent in combination with a neodymium polymerisation catalyst [(C5Me5)2NdCl2Li(OEt2)2], first reported by Mortreux,24 has been extensively investigated by Boisson and D'Agosto and co-workers. In these CCG reactions, Mg(PE)2 is used to generate various end-functionalised PE-X.43,44,47–50
Here we report our studies on the iron-based CCG reaction using ZnEt2 to prepare Zn(PE)2 with defined chain lengths and conversion to a range of end-functionalised PE-X materials. While the reactivity of Zn(PE)2 in many ways resembles that of Mg(PE)2, key advantages of the Fe/Zn system are the ease of synthesis and handling of the iron-based catalyst, the use of highly abundant and cheap first row metals iron and zinc, and crucially, the absence of any alkene by-products, which is often seen in other systems. A range of functionalised PE-X materials has been prepared in this study, where X is either from Group 16 (PE-OH and PE-Sk-PE, where k = 1–8) or Group 17 (PE-I, PE-Br) (see Scheme 2).
A further aim in this work has been the preparation of polymer blends between PE-I and LDPE. Such blends could improve the degradability of polyethylene because alkyl iodides are prone to homolysis of the C–I bond under UV irradiation,51–53 generating alkyl radicals which in the presence of air can form alkyl peroxy radicals,54 and these alkylperoxy radicals are key intermediates in the degradation of polyethylene.55 Furthermore, the low entanglement molecular weight for polyethylene of Me ∼1250 g mol−1,56 suggests chain lengths of approximately 90 carbons will be sufficient to ensure efficient entanglement of PE-I within the polyethylene matrix and avoid any leaching of the PE-I additive.
Results and discussion
Synthesis and characterisation of Zn(PE)2
The preparation of Zn(PE)2 was carried out using our previously described method from ZnEt2 and [(2,6-diacetylpyridinebis(2,6-diisopropylanil))FeCl2] (1) in combination with MAO as the activator, as shown in Scheme 1.28 The extremely fast alkyl exchange reaction between iron and zinc ensures the formation of Zn(PE)2 with excellent selectivity and low dispersity. By varying the reaction time and the ratio between Zn(Et)2 and 1, the chain length of the resulting PE chains can be controlled (see Table 1). Upon hydrolysis, a Poisson distribution of alkanes is obtained with a low dispersity of molecular weights.28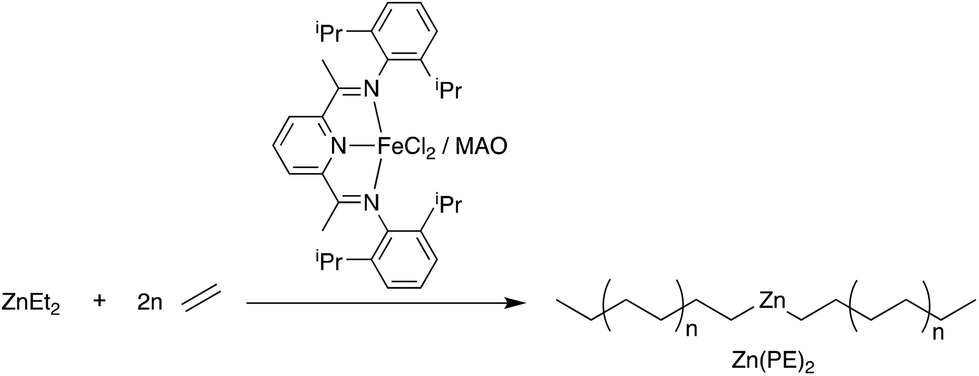 | ||
| Scheme 1 Catalysed chain growth of ZnEt2 using the catalyst system [(2,6-diacetylpyridinebis(2,6-diisopropylanil))FeCl2] (1) and MAO. | ||
| Run | ZnEt2 [mmol] | ZnEt2 Equiv. | Activity [g mmol−1 h bar−1] | λ | Cnb |
|---|---|---|---|---|---|
| Conditions: catalyst 1 (10 μmol), MAO (100 eq.), toluene solvent (30 mL), room temperature, 1 barg ethylene, 1 hour reaction time.a λ = mean number of ethylene units inserted, determined by GC analysis.b Cn = mean chain length, determined by 13C-NMR analysis.c No exchange to zinc is observed. | |||||
| 1 | 0 | 0 | PEc | –– | –– |
| 2 | 2.5 | 250 | PEc | –– | –– |
| 3 | 3.0 | 300 | 1380 | 31 | 64 |
| 4 | 3.5 | 350 | 1420 | 21 | 44 |
| 5 | 4.0 | 400 | 820 | 14 | 30 |
| 6 | 4.5 | 450 | 600 | 11 | 24 |
| 7 | 5.0 | 500 | 690 | 8 | 18 |
| 8 | 5.5 | 550 | 482 | 7 | 16 |
| 9 | 6.0 | 600 | 500 | 4 | 10 |
In the absence of the chain transfer agent ZnEt2, the catalyst system 1/MAO polymerises ethylene with very high activity and produces high molecular weight polyethylene (run 1 in Table 1).57 Small amounts of ZnEt2 (<250 equiv., run 2) have little effect on the polymerisation reactions and only at Zn/Fe ratios of >300 equiv., catalysed chain growth to form Zn(PE)2 is observed. While larger Zn/Fe ratios will produce more Zn(PE)2, this results in shorter chains within a given time and also lowers the catalytic activity, probably due to competitive binding of ZnEt2versus ethylene to the catalyst.58 Zn(PE)2 can be isolated from the reaction mixture by filtration and characterised by NMR and IR spectroscopy (see Fig. S1–3†). The characteristic triplet for the ZnCH2 moiety is found at 0.30 ppm in the 1H NMR and at 16.3 ppm in the 13C NMR spectrum, as well as the in-plane rocking vibration at 628 cm−1, typical for ZnCH2 units.59 The high reactivity of the Zn–C bond can be exploited to prepare a range of end-functionalised PE-X compounds, as illustrated in Scheme 2
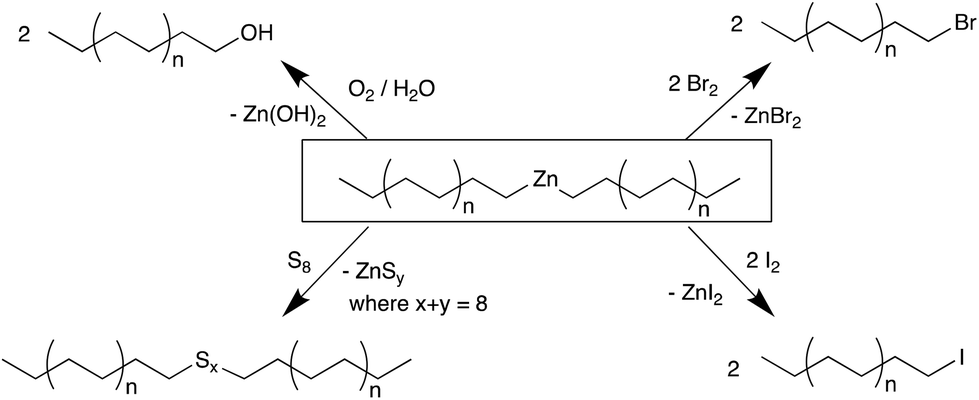 | ||
| Scheme 2 Overview of reactions of Zn(PE)2 with Group 16 and 17 elements. | ||
Hydrolysis of Zn(PE)2 results in a distribution of linear alkanes. The Poisson distribution of the n-alkane oligomers allows quantitative 13C NMR analysis to be used to estimate the mean chain length Cn by comparing integrals for CH3 with those for CH2 signals (Table 1, last column). Further analysis can be achieved by GC-FID. The average molecular weight is generally too low for accurate GPC analysis, whereas analysis by GC-FID will only capture part of the total distribution, because the longer alkanes are only partially soluble and insufficiently volatile for GC analysis, as illustrated in Fig. 1. Only the first part of the total distribution is therefore quantifiable, and these values can be used as inputs for the Poisson equation (eqn. (2)) to extract λ, the mean number of inserted ethylene units (L is the amount of catalyst used). The total amount of each alkane of chain length n obtained at the end of the reaction can then be calculated.60,61 For example, the calculated value of λ = 14 for run 5 in Table 1 suggests an average chain length of 30 carbons which is very close to the value of 30 carbons determined by 13C NMR spectroscopy (see Table 1 for other values). Comparison of the average chain lengths determined by GC and NMR analysis shows excellent agreement (Fig. 2). This fitting method allows the entire product distribution to be determined from a subset of experimental data obtained by GC analysis.
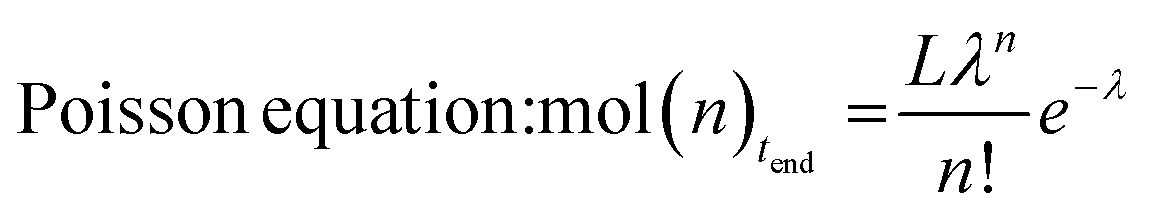 | (2) |
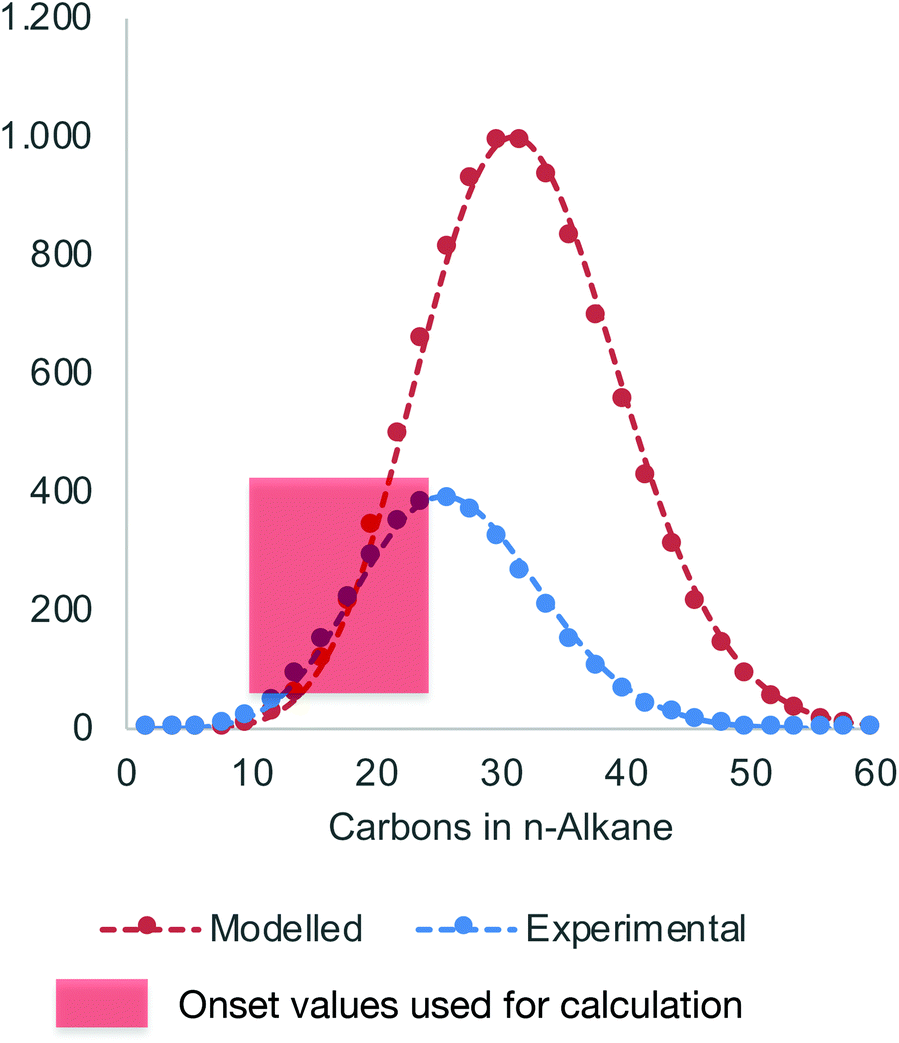 | ||
| Fig. 1 Experimentally determined n-alkane distribution by GC-FID (blue) versus calculated distribution based on the Poisson equation (red, data from run 5 in Table 2). | ||
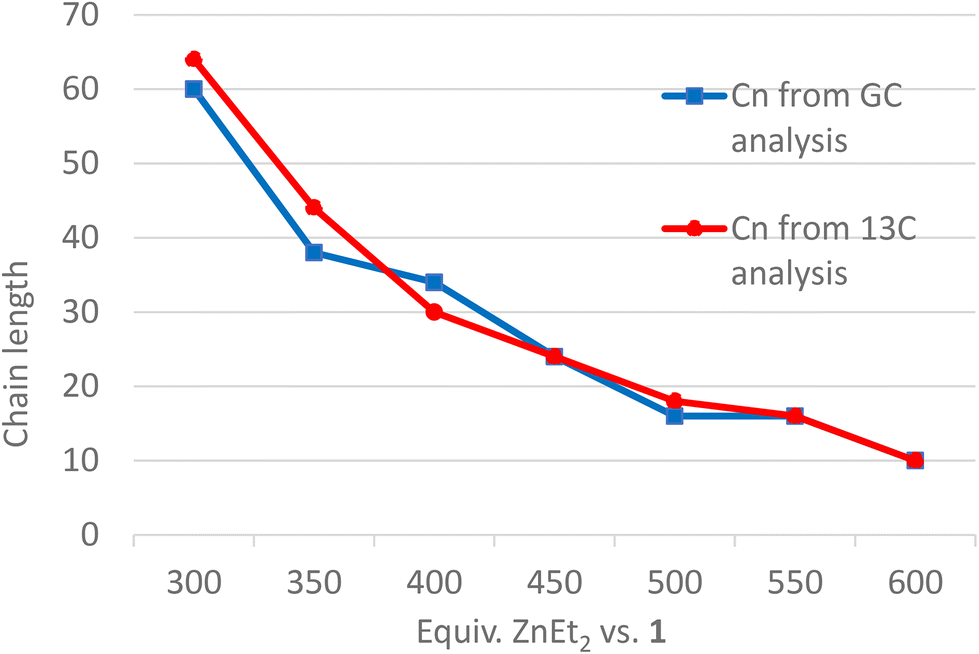 | ||
| Fig. 2 Zn(PE)2 chain length (Cn) as a function of Zn/Fe ratio. | ||
There appears to be no convenient nomenclature to describe mixtures of compounds with long alkyl chains, where the length of the chains follows a Poisson distribution, other than quoting an Mn value. In order to describe the chain length in our product mixtures, we will henceforth use the italicised n value (n) to describe an average chain length, i.e. CnH2n±2 to describe a mixture of alkanes with an average chain length n. We will use the same nomenclature for other related compounds, i.e. Zn(C30H61)2 will result in C30H62 upon hydrolysis, or C30H61I upon reaction with iodine. C30H61I is thus a mixture of alkyl iodides with chain lengths clustered around C30, whereas C30H61I would be the pure compound.
End-functionalisation with oxygen
Initial experiments with dipentylzinc as a model compound for Zn(PE)2 and synthetic air (80% N2/20% O2, dry) resulted in a mixed alkyl alkoxide complex, [Zn(OPent)Pent]n, according to NMR analysis (see Fig. S4 and 5†), presumable a tetramer (n = 4) as seen in related examples.62,63 Hydrolysis of the zinc cluster resulted in pentanol with 78% selectivity, the remainder being pentane. A reaction with pure oxygen instead of air gave similar results but was subsequently avoided for safety reasons. Subsequently, CCG reactions using ZnEt2 and catalyst 1 (Zn/Fe = 300) have been carried out at 1 bar ethylene pressure and the product Zn(PE)2 was reacted with air prior to hydrolysis (see Table 2). Reactions were carried out using different amounts of catalyst, which did affect the activity somewhat. The products in both reactions have a similar average chain length, analysed as C68H137OH according to 13C NMR with 65–74% OH-functionalisation, the remainder of the product mixture being linear alkanes.| Run | Yield [g] | Zn/Fe ratio | Activity [g mmol−1 h bar−1] | λ | Cnd | Funct.(%) |
|---|---|---|---|---|---|---|
| a Conditions: catalyst 1 (10 μmol), MAO (100 eq.), toluene solvent (30 mL), room temperature, 1 bar ethylene, 1 hour reaction time, quenched with air b Conditions: catalyst 1 (40 μmol), MAO (100 eq.), toluene solvent (120 mL), room temperature, 1 bar ethylene, 1 hour reaction time, quenched with air c λ = mean number of ethylene units inserted, determined by GC analysis d Cn = mean chain length, determined by quantitative 13C-NMR analysis | ||||||
| 1a | 3.84 | 300 | 384 | 33 | 68 | 74 |
| 2b | 19.9 | 300 | 497 | 33 | 68 | 65 |
The formation of end-functionalised PE-OH from the reaction of Zn(PE)2 with oxygen has been investigated previously using Ziegler–Natta catalysts and metallocenes,45,46,64 titanium guanidate catalysts,41 and also with the Fe/Zn catalyst system use here.36–40 In all these reports, the degree of OH-functionalisation is less than 80%, with the remainder being alkanes (PE-H) formed upon hydrolysis. The incomplete reaction of zinc alkyls with oxygen is likely due to the formation of mixed zinc alkyl alkoxide “ZnR(OR)” clusters, as reported by Lewinski.62,65,66 For example, the reaction of ZnMe2 with oxygen has been reported to generate [Zn(OMe)2(Zn(OMe)Me)6], which upon hydrolysis would only yield 57% MeOH. It is possible that for ZnR2 with longer alkyl chains, similar [Zn(OR)xRy] clusters are generated which upon hydrolysis will generate different yields of ROH.62,67,68 We can thus conclude that end-functionalisation of Zn(PE)2 to generate PE-OH appears to have an upper limit of approximately 80% selectivity, because this functionalisation suffers from an inherent problem due to the formation of stable mixed alkyl/alkoxide zinc clusters upon reaction with oxygen.
End-functionalisation with sulfur
The incorporation of sulfur into polymers, for example the vulcanisation of rubber, has been an important aspect in polymer chemistry for a long time, and is currently finding renewed interest for other polymers.69–71 The reaction of main group alkyls with elemental sulfur has been known for some time and generally results in the formation of dialkyl polysulfides R-Sk-R.72 Boisson et al. have shown that Mg(PE)2 reacts with sulfur to generate PE-Sk-PE polysulfide polymers, where k = 1–8. Previous studies on the reaction between ZnEt2 with sulfur in equimolar amounts resulted in the formation of [Zn(SEt)Et]n clusters such as [Zn(SEt)Et]10,73 similar as for the reaction with oxygen discussed in the previous section. Increasing the ratio of sulfur to zinc leads to the formation of [(ZnS)Zn(SEt)2]n clusters, together with the formation of SEt2 and S2Et2.74Initial reactions of dipentylzinc with sulfur in an equimolar ratio showed the formation of C5H11-Sk-C5H11 (where the ratio of k = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3+ was 21:42:37), together with unreacted dipentylzinc (see Fig. S6†). The reaction of excess sulfur (4 equiv.) with Zn(PE)2 resulted in the formation of PE-Sk-PE polymers with 71% functionalisation, the remainder being linear alkanes formed upon hydrolysis. Analysis by 1H NMR reveals a ratio between PE-S-PE, PE-S2-PE and PE-S3+-PE of 11:42:47 (see Fig. 3). Considering an average chain length of C22 in this case, an average value of k = 4 could be calculated from elemental analysis (see Table S1†). Analysis of PE-Sk-PE by Raman spectroscopy (Fig. S8†) has shown a characteristic C–S vibration at 744 cm−1.75 When PE-Sk-PE was heated in toluene at 80 °C for one hour a reduction of k was observed, specifically in the amount of PE-Sk-PE with k ≥ 3 (see Fig. S7†), in line with the general thermal instability of polysulphides with k > 3.76
:2:3+ was 21:42:37), together with unreacted dipentylzinc (see Fig. S6†). The reaction of excess sulfur (4 equiv.) with Zn(PE)2 resulted in the formation of PE-Sk-PE polymers with 71% functionalisation, the remainder being linear alkanes formed upon hydrolysis. Analysis by 1H NMR reveals a ratio between PE-S-PE, PE-S2-PE and PE-S3+-PE of 11:42:47 (see Fig. 3). Considering an average chain length of C22 in this case, an average value of k = 4 could be calculated from elemental analysis (see Table S1†). Analysis of PE-Sk-PE by Raman spectroscopy (Fig. S8†) has shown a characteristic C–S vibration at 744 cm−1.75 When PE-Sk-PE was heated in toluene at 80 °C for one hour a reduction of k was observed, specifically in the amount of PE-Sk-PE with k ≥ 3 (see Fig. S7†), in line with the general thermal instability of polysulphides with k > 3.76
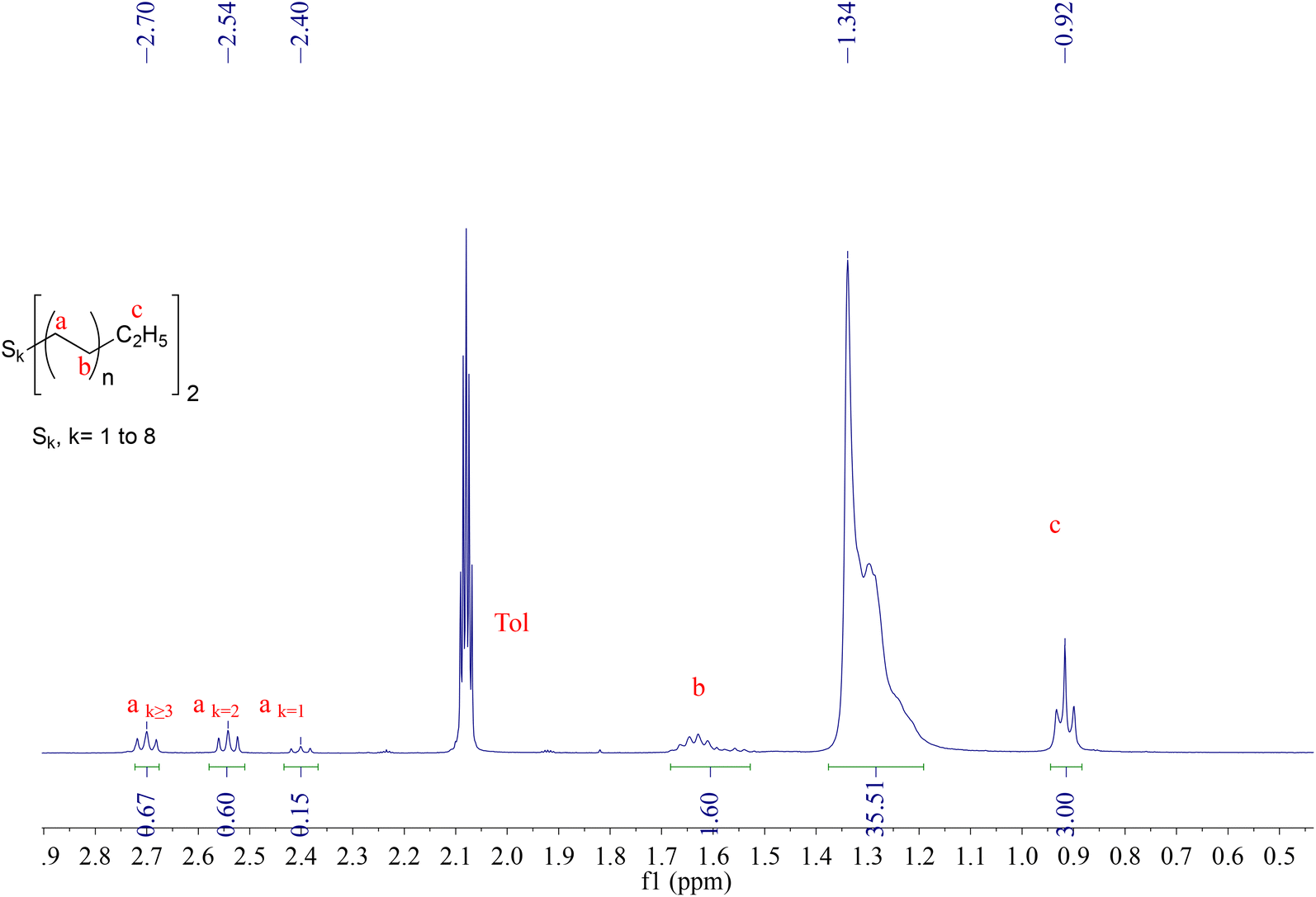 | ||
| Fig. 3 1H-NMR spectrum in d8-toluene at 298K of PE-Sk-PE (k = 1–8) obtained from the reaction of Zn(PE)2 with 4 equiv. sulfur. Zn(PE)2 = Zn(C22H45)2 in this case. | ||
End-functionalisation with iodine and bromine
The reaction of Zn(PE)2 with an excess I2 results in 1-iodoalkanes (PE-I), together with ZnI2. A range of 1-iodoalkanes with various average chain lengths has been prepared, a selection is shown in Table 3. The products have been analysed by GC, 1H, 13C NMR and IR spectroscopy, as well as thermal analysis (Fig. S10–15†). Analysis of long chain 1-iodoalkanes by GC is reliable for short chain lengths up to C20 and fitting of the GC results to the Poisson formula allows full characterisation of the distribution of chain lengths to be determined (see Fig. S14†). Average chain length values have been determined by 13C NMR analysis (see Fig. S11†). IR spectroscopic analysis can also be used for relatively short alkyl iodides, using the ratio of ν(C–H) signal at 3000 cm−1 relative to the characteristic ν(C–I) stretch at 603 cm−1 (see Fig. S15–17†).| Run | Eq. Zn | Activity [g mmol−1 h bar−1] | Chain lengtha (Cn) | PE-Ib (%) |
|---|---|---|---|---|
| Conditions for the polymerization reactions: catalyst 1 (10 μmol), MAO (100 eq.), toluene solvent (50 mL), 30 °C, 1 barg ethylene, 1 hour reaction time.a Cn = average chain length, determined by 13C NMR analysis.b Iodo-functionality yield determined by 1H NMR analysis. | ||||
| 1 | 300 | 1460 | 70 | 89 |
| 2 | 350 | 1220 | 46 | 96 |
| 3 | 450 | 950 | 40 | 97 |
| 4 | 500 | 900 | 30 | 98 |
| 5 | 650 | 640 | 12 | 100 |
Thermal analysis was caried out on a series of four longer chain PE-I samples with different chain lengths, ranging from C48 to C88 alkyl iodides. Analysis by DSC shows two endothermic processes (Fig. 4), one centred around 60–70 °C and another around 110–120 °C. The former signal is more dominant for the shorter chain alkyl iodides, whereas the latter becomes the main transition for the longer chain products. Melting points for linear alkyl iodides CnH2n+1I of different chain lengths follow a hyperbolic relationship (Fig. S20†), approaching the melting point of HDPE for n → ∞, and the melting points of pure C48H97I and C88H177I are 91 °C and 110 °C, respectively.77,78 The two transitions seen here are due to the fact that we have mixtures of linear alkyl iodides with different chain lengths that lead to the formation of solid solutions before complete melting of the material takes place. Similar complex behaviour has been reported for mixtures of alkanes and perfluorinated alkanes.79,80
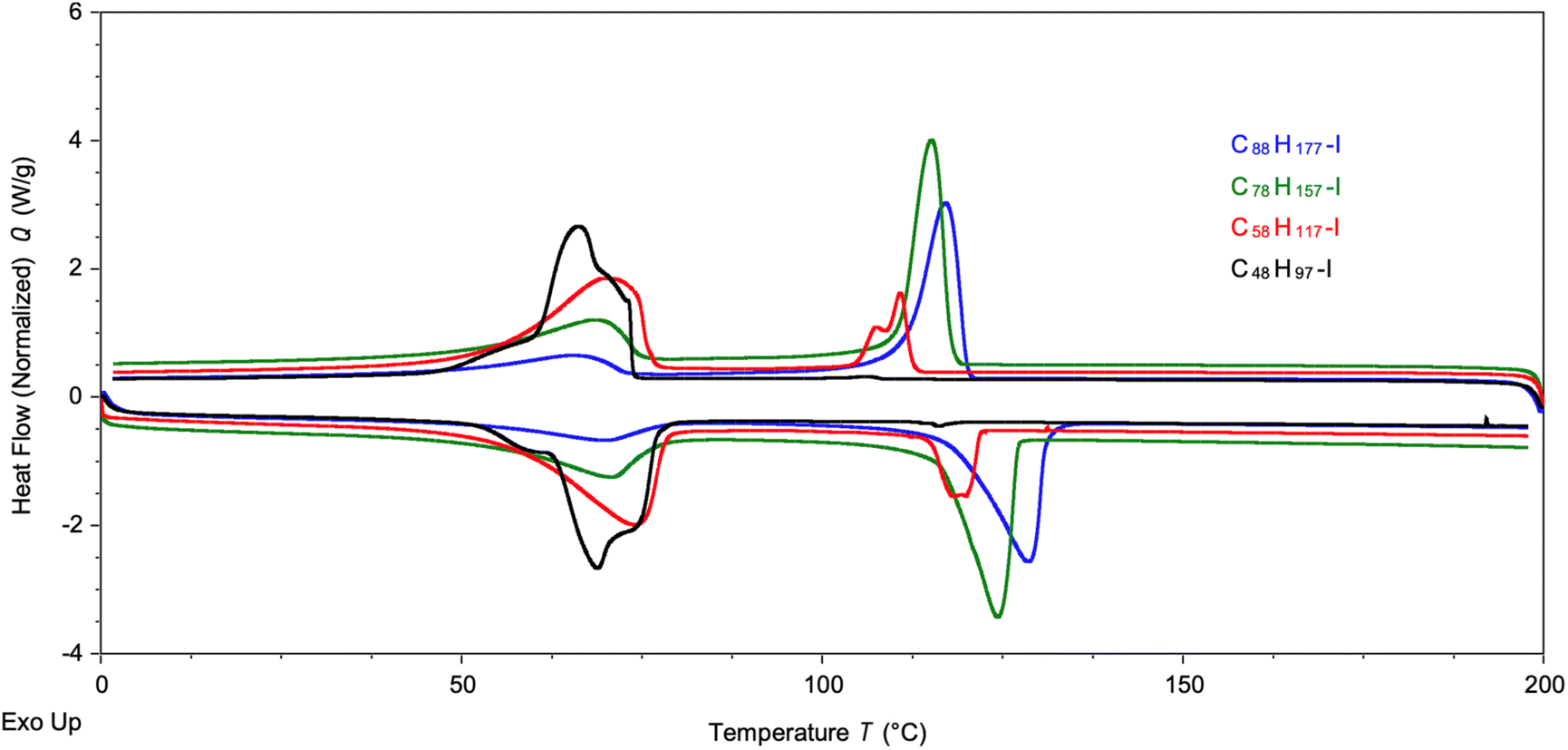 | ||
| Fig. 4 DSC analysis of four PE-I samples of different chain length. Measurements shown were taken during the second heating cycle. | ||
Whilst selectivities for the formation of PE-I are typically very high (>90%), the functionalisation appears to depend to some degree on the alkyl chain length, something that was also noted by Boisson in catalysed chain growth reactions of magnesium alkyls followed by iodine quenching.48 For example, the degree of functionalisation is quantitative for C20H41I, but decreases to 97% for C30H61I and 89% for C70H141I, the remainder being the corresponding alkanes (see Fig. S12†). This may be due to the reduced solubility of Zn(PE)2 with increasing chain length, or alternatively, encapsulation of the reactive zinc centres with increased PE chain length could result in restricted access to the metal centre and reduced reactivity with I2. Unreacted zinc-carbon bonds are eventually cleaved upon hydrolysis, resulting in the formation of alkanes.
Analogous to the reaction of Zn(PE)2 with iodine, reactions with bromine result in the formation of PE-Br. Upon addition of Br2 to a solution of Zn(PE)2 in toluene, C32H65Br was obtained with 67% functionalisation, the remainder being C32H66 (see Fig. S18 and 19†). The reason for the lower functionalisation is not clear at this stage, but it should be noted that compared to iodine, the addition of bromine is considerably more reactive and may have led to some decomposition of alkyl zinc species.
Synthesis and characterisation of PE-I/LDPE polymer blends
A series of polymer blends has been prepared by incorporating PE-I into LDPE (SABIC N0W2021). The polymer blends were prepared by two different methods, either by solution mixing or melt blending. Solution mixing involved dissolving LDPE in hot toluene,81 followed by the addition of PE-I and precipitation of the resulting PE-I/LDPE polymer blend in methanol. Melt blending of PE-I and LDPE was carried out via co-extrusion at 130 °C (see ESI†). TGA analysis has shown that PE-I and LDPE are thermally stable up to 200 °C (Fig. S21†). LDPE was blended with C18H37I, C30H61I and C70H141I at 10 wt% loading. Analysis of the polymer blends was carried out by IR spectroscopy based on the characteristic signals at 1163 cm−1 for ω(CH2I) and 603 cm−1 for ν(C–I) (see Fig. S15†). Despite dilution in the LDPE matrix the characteristic IR signals can be clearly observed (Fig. 5 for solution mixing and S22† for melt blending). Further characterisation was caried out by DSC analysis, which shows superposition of the DSC profiles for LDPE and PE-I (Fig. S23†).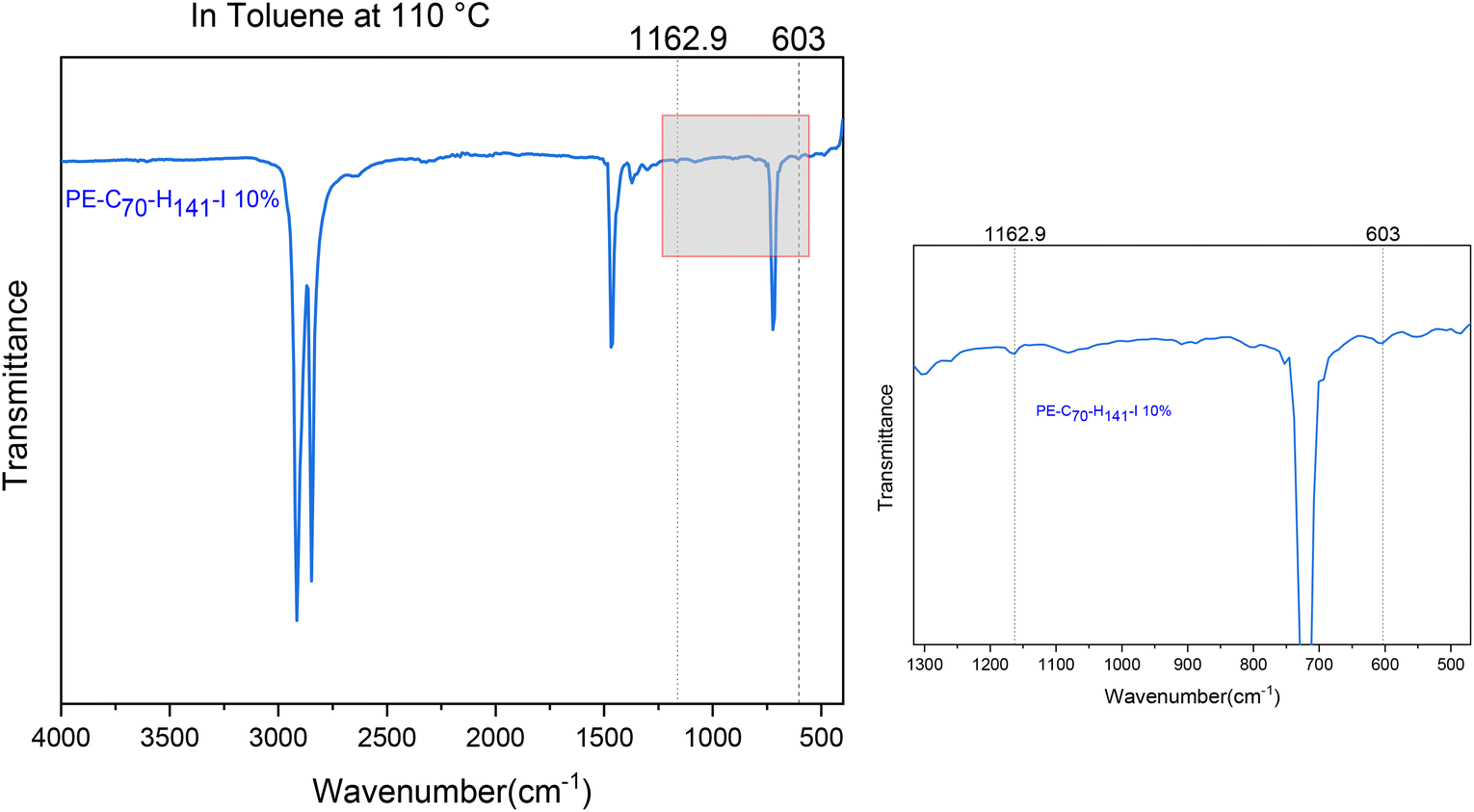 | ||
| Fig. 5 FT-IR analysis of C70H141I/LDPE polymer blend obtained via solution blending in toluene. | ||
In order to establish the dispersion of PE-I throughout the PE-I/LDPE polymer blend, surface analysis has been caried out on the material obtained through solution blending, using FIB-SEM and EDX analysis.82–84 The SEM image in Fig. 6 shows a magnification of the PE-I/LDPE polymer surface. A square hole of approximately 10 × 10 × 10 μm was cut by FIB micro machining using a gallium ion beam (see ESI for details†). EDX measurements were caried out at approximately 1 μm depth intervals up to 6 μm from the surface, as indicated in Fig. 6. Iodine was detected in all samples from its characteristic Lα signal at 3.937 keV, and there was no indication of a concentration gradient along this trajectory, indicating a random dispersion of PE-I, at least within 6 μm from the polymer surface.
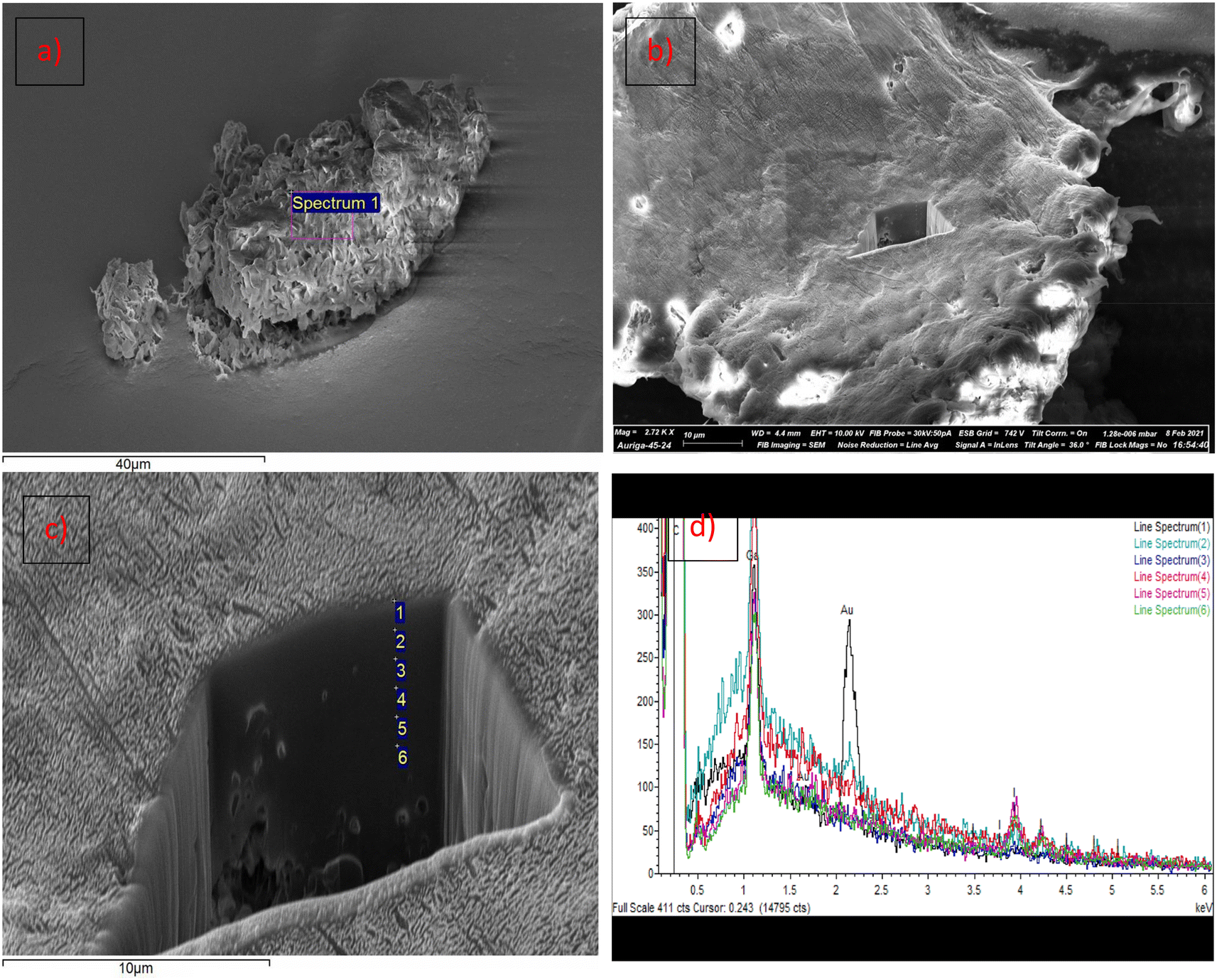 | ||
| Fig. 6 (a) SEM image of PE-I/LDPE particle. (b) FIB-SEM image of PE-I/LDPE surface with square hole cut using FIB micro machining. (c) EDX analysis at ∼1 μm depth intervals. (d) EDX spectra taken at surface (spectrum 1) and at ∼1 μm depth intervals (SEM samples were Au-plated for conductivity). | ||
In conclusion, we have shown that the CCG reaction of ZnEt2 with ethylene, catalysed by the iron-based catalyst system [(2,6-diacetylpyridinebis(2,6-diisopropylanil))FeCl2] (1) and MAO gives access to ZnPE2, whereby the PE chain length can be controlled by the Zn/Fe ratio and the reaction time. The reaction of ZnPE2 with a variety of electrophiles leads to the formation of end-functionalised polyethylene PE-X. The selectivity for the formation of PE-OH appears to be limited to approximately 80%, probably due to the formation of zinc cluster [Zn(OR)R]n intermediates. The reaction of ZnPE2 with elemental sulfur results in the formation of polysulfides PE-Sk-PE. Polysulfides with k > 3 are thermally unstable and heating of PE-Sk-PE results in the elimination of sulfur and a lowering of k. The reaction of ZnPE2 with iodine to generate PE-I is generally highly selective and is the preferred functionalisation method. Blends of PE-I and LDPE can be prepared by solution mixing or by melt blending through extrusion. These novel PE-I/LDPE polymer blends have been characterised by IR spectroscopy and DSC analysis. Surface analysis using SEM-EDX has indicated a uniform distribution of PE-I within 6 μm from the PE-I/LDPE surface. Degradation studies of these novel PE-I/LDPE materials are currently underway and will be reported in due course.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Polymateria Ltd for funding. Dr Mahmoud Ardakani is thanked for SEM and EDX measurements. We thank SABIC for a generous donation of LDPE (SABIC N0W2021) and AA is grateful for a scholarship from SABIC.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 768–771 CrossRef PubMed.
- M. Gahleitner, L. Resconi and P. Doshev, MRS Bull., 2013, 38, 229–233 CrossRef CAS.
- D. Jeremic, in Ullmann's Encyclopedia of Industrial Chemistry, 2014, pp. 1–42 Search PubMed.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722–9744 CrossRef CAS PubMed.
- C. M. Plummer, L. Li and Y. Chen, Poly. Chem., 2020, 11, 6862–6872 RSC.
- J. B. Williamson, W. L. Czaplyski, E. J. Alexanian and F. A. Leibfarth, Angew. Chem., Int. Ed., 2018, 57, 6261–6265 CrossRef CAS PubMed.
- N. M. Franssen, J. N. Reek and B. de Bruin, Chem. Soc. Rev., 2013, 42, 5809–5832 RSC.
- P. D. Goring, C. Morton and P. Scott, Dalton Trans., 2019, 48, 3521–3530 RSC.
- L. R. Sita, Angew. Chem., Int. Ed., 2009, 48, 2464–2472 CrossRef CAS PubMed.
- Y. J. Zhang, H. Y. Li, J. Y. Dong and Y. L. Hu, Prog. Chem., 2014, 26, 110–124 CAS.
- A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836–3857 CrossRef CAS PubMed.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef CAS PubMed.
- I. Haas, W. P. Kretschmer and R. Kempe, Organometallics, 2011, 30, 4854–4861 CrossRef CAS.
- R. Leino, H. J. G. Luttikhedde, P. Lehmus, C.-E. Wilén, R. Sjöholm, A. Lehtonen, V. J. Seppälä and J. H. Näsman, Macromolecules, 1997, 30, 3477–3483 CrossRef CAS.
- L. Rocchigiani, V. Busico, A. Pastore and A. Macchioni, Organometallics, 2016, 35, 1241–1250 CrossRef CAS.
- J. S. Rogers and G. C. Bazan, Chem. Commun., 2000, 1209–1210 RSC.
- K. Nomura, U. Tewasekson and Y. Takii, Organometallics, 2015, 34, 3272–3281 CrossRef CAS.
- J. Obenauf, W. P. Kretschmer and R. Kempe, Eur. J. Inorg. Chem., 2014, 1446–1453 CrossRef CAS.
- S. K. T. Pillai, W. P. Kretschmer, M. Trebbin, S. Förster and R. Kempe, Chem. – Eur. J., 2012, 18, 13974–13978 CrossRef CAS PubMed.
- E. C. Samsel and D. C. Eisenberg, Ethyl Corporation, EP0574854, 1993.
- R. S. A. Meyer, J. S. Scholtyssek and G. A. Luinstra, Macromol. Mater. Eng., 2015, 300, 218–225 CrossRef CAS.
- C. J. Han, M. S. Lee, D.-J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923–8925 CrossRef CAS.
- W. P. Kretschmer, T. Bauer, B. Hessen and R. Kempe, Dalton Trans., 2010, 39, 6847–6852 RSC.
- J.-F. Pelletier, A. Mortreux, X. Olonde and K. Bujadoux, Angew. Chem., Int. Ed. Engl., 1996, 35, 1854–1856 CrossRef CAS.
- T. Chenal, X. Olonde, J.-F. Pelletier, K. Bujadoux and A. Mortreux, Polymer, 2007, 48, 1844–1856 CrossRef CAS.
- X. Olonde, A. Mortreux, F. Petit and K. Bujadoux, J. Mol. Catal., 1993, 82, 75–82 CrossRef CAS.
- R. Ribeiro, R. Ruivo, H. Nsiri, S. B. S. Norsic, F. D'Agosto, L. Perrin, C. Boisson, F. D'Agosto, L. Perrin and C. Boisson, ACS Catal., 2016, 6, 851–860 CrossRef CAS.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson, P. J. Maddox and M. van Meurs, Angew. Chem., Int. Ed., 2002, 41, 489–491 CrossRef CAS PubMed.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS PubMed.
- M. Van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, J. Am. Chem. Soc., 2005, 127, 9913–9923 CrossRef CAS PubMed.
- A. Xiao, L. Wang, Q. Liu, H. Yu, J. Wang, J. Huo, Q. Tan, J. Ding, W. Ding and A. M. Amin, Macromolecules, 2009, 42, 1834–1837 CrossRef CAS.
- W. Zhang, J. Wei and L. R. Sita, Macromolecules, 2008, 41, 7829–7833 CrossRef CAS.
- O. H. Hashmi, M. Visseaux and Y. Champouret, Polym. Chem., 2021, 12, 4626–4631 RSC.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson, P. J. Maddox and M. van Meurs, Angew. Chem., Int. Ed., 2002, 41, 489–491 CrossRef CAS PubMed.
- R. Cariou and J. W. Shabaker, ACS Catal., 2015, 5, 4363–4367 CrossRef CAS.
- H. Kaneyoshi, Y. Inoue and K. Matyjaszewski, Macromolecules, 2005, 38, 5425–5435 CrossRef CAS.
- F. He, P. Li, A. Wu, T. Xu, Z. Fu, L. Zhu and Z. Fan, Polyolefins J., 2017, 4, 191–200 CAS.
- T. Li, W. J. Wang, R. Liu, W. H. Liang, G. F. Zhao, Z. Li, Q. Wu and F. M. Zhu, Macromolecules, 2009, 42, 3804–3810 CrossRef CAS.
- P. Li, Z. Fu and Z. Fan, J. Appl. Polym. Sci., 2015, 132, 1–10 Search PubMed.
- Y. Zhao, X. Shi, H. Gao, L. Zhang, F. Zhu and Q. Wu, J. Math. Chem., 2012, 22, 5737–5745 RSC.
- S. Rutkowski, A. Zych, M. Przybysz, M. Bouyahyi, P. Sowinski, R. Koevoets, J. Haponiuk, R. Graf, M. R. Hansen, L. Jasinska-Walc and R. Duchateau, Macromolecules, 2016, 50, 107–122 CrossRef.
- C. J. Han, M. S. Lee, D. J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923–8925 CrossRef CAS.
- J. Mazzolini, E. Espinosa, F. D'Agosto and C. Boisson, Polym. Chem., 2010, 1, 793–800 RSC.
- T. Chenal, M. Drelon, B. Marsh, F. F. Silva, M. Visseaux and A. Mortreux, Catal. Sci. Technol., 2020, 10, 6809–6824 RSC.
- T. Shiono, K. Yoshida and K. Soga, Makromol. Chem., Rapid Commun., 1990, 11, 169–175 CrossRef CAS.
- T. Shiono, K. Yoshida and K. Soga, Stud. Surf. Sci. Catal., 1990, 56, 285–299 CrossRef CAS.
- J. E. O. Mazzolini, I. Mokthari, Ŕ. Briquel, O. Boyron, F. E. Delolme, V. Monteil, D. Bertin, D. Gigmes, F. D'Agosto and C. Boisson, Macromolecules, 2010, 43, 7495–7503 CrossRef CAS.
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311–9313 CrossRef CAS PubMed.
- W. N. Ottou, S. Norsic, F. D’Agosto and C. Boisson, Macromol. Rapid Commun., 2018, 39, 1800154 CrossRef PubMed.
- B. H. Staudt, J. Wagner and P. Vana, Macromolecules, 2018, 51, 8469–8476 CrossRef CAS.
- P. L. Ross and M. V. Johnston, J. Phys. Chem., 2002, 99, 16507–16510 CrossRef.
- P. L. Ross and M. V. Johnston, J. Phys. Chem., 2002, 99, 4078–4085 CrossRef.
- P. J. Kropp and R. L. Adkins, J. Am. Chem. Soc., 2002, 113, 2709–2717 CrossRef.
- G. Heimann, H.-J. Benkelberg, O. Böge and P. Warneck, Int. J. Chem. Kinet., 2002, 34, 126–138 CrossRef CAS.
- A. Ammala, S. Bateman, K. Dean, E. Petinakis, P. Sangwan, S. Wong, Q. Yuan, L. Yu, C. Patrick and K. H. Leong, Prog. Polym. Sci., 2011, 36, 1015–1049 CrossRef CAS.
- V. M. Litvinov, M. E. Ries, T. W. Baughman, A. Henke and P. P. Matloka, Macromolecules, 2013, 46, 541–547 CrossRef CAS.
- G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef CAS PubMed.
- B. Nagel and W. Brüser, Z. Anorg. Allg. Chem., 1980, 468, 148–152 CrossRef CAS.
- C. T. Young, R. von Goetze, A. K. Tomov, F. Zaccaria and G. J. P. Britovsek, Top. Catal., 2020, 63, 294–318 CrossRef CAS.
- G. V. Schulz, Makromol. Chem., 1950, 5, 83–94 CrossRef CAS.
- S. Jana, R. J. F. Berger, R. Fröhlich, T. Pape and N. W. Mitzel, Inorg. Chem., 2007, 46, 4293–4297 CrossRef CAS PubMed.
- C. H. Bamford and D. M. Newitt, J. Chem. Soc., 1946, 688 RSC.
- W. Wang, L. P. Hou, T. Y. Zhang and C. C. Liu, J. Polym. Res., 2018, 25, 145 CrossRef.
- J. Lewinski, W. Marciniak, J. Lipkowski and I. Justyniak, J. Am. Chem. Soc., 2003, 125, 12698–12699 CrossRef CAS PubMed.
- M. Kubisiak, K. Zelga, W. Bury, I. Justyniak, K. Budny-Godlewski, Z. Ochal and J. Lewinski, Chem. Sci., 2015, 6, 3102–3108 RSC.
- L. Makolski, V. Szejko, K. Zelga, A. Tulewicz, P. Bernatowicz, I. Justyniak and J. Lewinski, Chemistry, 2021, 27, 5666–5674 CrossRef CAS PubMed.
- J. Maury, L. Feray, S. Bazin, J. L. Clement, S. R. A. Marque, D. Siri and M. P. Bertrand, Chem. – Eur. J., 2011, 17, 1586–1595 CrossRef CAS PubMed.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105 RSC.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS.
- J. F. Boscato, J. M. Catala, E. Franta and J. Brossas, Tetrahedron Lett., 1980, 21, 1519–1520 CrossRef CAS.
- D. Zeng, M. J. Hampden-Smith and E. N. Duesler, Inorg. Chem., 1994, 33, 5376–5377 CrossRef CAS.
- M. D. Nyman, M. J. Hampden-Smith and E. N. Duesler, Inorg. Chem., 1996, 35, 802–803 CrossRef CAS PubMed.
- D. Lin-Vien, N. Colthup, W. Fateley and J. G. Graselli, Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, San Diego, 1991 Search PubMed.
- P. D. Clark and C. O. Oriakhi, Energy Fuels, 1992, 6, 474–477 CrossRef CAS.
- M. F. Bolotnikov and Y. A. Neruchev, Russ. J. Phys. Chem. A, 2007, 81, 1198–1202 CrossRef CAS.
- G. R. Somayajulu, Int. J. Thermophys., 1990, 11, 555–572 CrossRef CAS.
- J. Visjager, T. A. Tervoort and P. Smith, Polymer, 1999, 40, 4533–4542 CrossRef CAS.
- E. P. Gilbert, Phys. Chem. Chem. Phys., 1999, 1, 1517–1529 RSC.
- S. L. Wong, N. Ngadi and T. A. T. Abdullah, Appl. Mech. Mater., 2014, 695, 170–173 Search PubMed.
- W. Brostow, B. P. Gorman and O. Olea-Mejia, Mater. Lett., 2007, 61, 1333–1336 CrossRef CAS.
- M. Kato, T. Ito, Y. Aoyama, K. Sawa, T. Kaneko, N. Kawase and H. Jinnai, J. Polym. Sci., Part B: Polym. Lett., 2007, 45, 677–683 CrossRef CAS.
- E. Beach, M. Keefe, W. Heeschen and D. Rothe, Polymer, 2005, 46, 11195–11197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic procedures, NMR, details for DSC and SEM analysis. See DOI: https://doi.org/10.1039/d2py01123a |
| This journal is © The Royal Society of Chemistry 2022 |