
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-catalyzed synthesis of a nano-capsule and its application as a heterogeneous RCMP catalyst and nano-reactor†
Xin Yi
Oh‡
 ,
Jit
Sakar‡
,
Ning
Cham
,
Gerald Tze Kwang
Er
,
Houwen Matthew
Pan
and
Atsushi
Goto
*
,
Jit
Sakar‡
,
Ning
Cham
,
Gerald Tze Kwang
Er
,
Houwen Matthew
Pan
and
Atsushi
Goto
*
School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 62 Nanyang Drive, 637459 Singapore. E-mail: agoto@ntu.edu.sg
First published on 21st October 2022
Abstract
A novel polymeric nano-capsule bearing quaternary ammonium iodide (QAI) groups on both the outer and inner surfaces of the shell was synthesized via self-catalyzed polymerization-induced self-assembly (PISA). Because QAI works as a catalyst of reversible complexation mediated living radical polymerization (RCMP), the obtained nano-capsule was exploited as a dual RCMP catalyst based on the outer and inner QAI groups. Benefitting from the outer QAI groups, the nano-capsule served as a supported heterogeneous RCMP catalyst with good recyclability to generate polymers outside the nano-capsule. Benefitting from the inner QAI groups, the nano-capsule served as a nano-reactor to generate polymers inside the nano-capsule. The nano-capsule served as a substrate-sorting nano-reactor based on the selective diffusivity of small molecules and polymers through the shell by their sizes. Namely, large molecules (polymers) once generated in the nano-reactor are not permeable through the shell, enabling the entrapment of the generated polymers in the nano-capsule. A homopolymer, an amphiphilic block copolymer, and a multi-polarity and multi-elemental block copolymer were synthesized and entrapped in the nano-capsule.
Introduction
Polymer nano-capsules (vesicles) are widely used as nano-reactors,1–3 sensors,4,5 and delivery containers.6,7 Polymerization-induced self-assembly (PISA) is a technique to generate nano-assemblies such as vesicles in situ during the polymerization.8–34 PISA can generate assemblies at high solid contents, which is an attractive feature. In PISA, a solvophilic macroinitiator is extended via polymerization to form an amphiphilic block copolymer. As the second block segment grows, the block copolymer becomes insoluble in the polymerization solvent, forming nano-assemblies during the polymerization. PISA involves living polymerizations such as living radical polymerizations (also termed reversible deactivation radical polymerizations) including reversible addition–fragmentation chain transfer (RAFT) polymerization, atom transfer radical polymerization (ATRP), and nitroxide mediated polymerization.8–30 Our research group also used reversible complexation mediated polymerization (RCMP) for conducting PISA.31–34RCMP35–38 uses a polymer–iodide (polymer–I) as a dormant species and an iodide anion (I−) as a catalyst. I− is used in the form of a quaternary ammonium iodide (QAI) (R4N+I−). The polymer–I dormant species coordinates the I− catalyst to form a halogen bonding complex (polymer–I⋯I−). The complex subsequently reversibly generates a propagating radical (polymer˙) (Scheme 1). An advantage of RCMP is the use of no metals or odorous compounds and tolerance to a range of monomers.
 | ||
| Scheme 1 Reversible activation in RCMP. | ||
Our research group previously synthesized QAI-containing monomers such as [2-(methacryloyloxy)ethyl]dimethylhexylammonium iodide (C6MAI) (Scheme 2) and used them for attaining self-catalyzed RCMP.39 The monomers contain a polymerizable methacrylate moiety and also a catalytic QAI moiety. Because the monomers contain the catalytic moiety, the polymerization (RCMP) can proceed without adding extra catalysts.
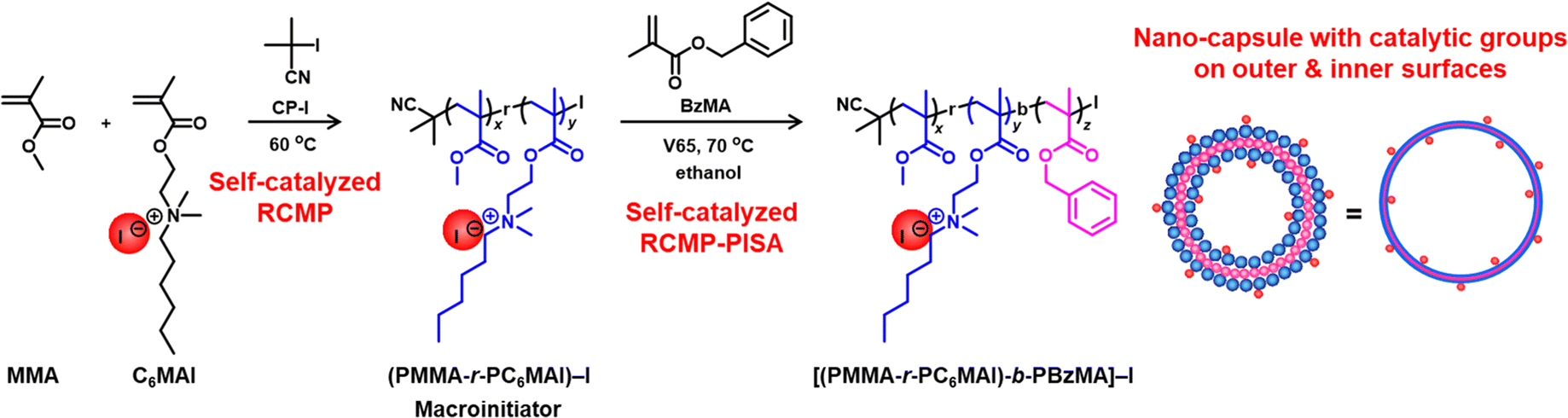 | ||
| Scheme 2 Self-catalyzed synthesis of the nano-capsule via RCMP-PISA. | ||
Herein, we report a combination of self-catalyzed RCMP with PISA for synthesizing a nano-capsule (vesicle) without adding an extra catalyst. As Scheme 2 shows, we used C6MAI as a monomer and a catalyst and conducted self-catalyzed RCMP for synthesizing a QAI-containing macroinitiator. Subsequently, we used the macroinitiator as an initiator and a catalyst and conducted self-catalyzed RCMP-PISA for synthesizing a nano-capsule. This is the first self-catalyzed synthesis of a nano-assembly (vesicle) in the fields of living radical polymerization and PISA.
Importantly, the obtained nano-capsule bears QAI groups on both outer and inner surfaces of the shell (Scheme 2). Hence, by exploiting the QAI groups on the outer surface, we tried to use the nano-capsule as a supported heterogeneous catalyst for RCMP. Heterogeneous catalysts have been extensively studied in polymerizations such as coordination polymerizations (supported Ziegler–Natta catalysts),40 ATRP (supported transition-metal catalysts),41–49 and RAFT polymerizations (supported photocatalysts).50–52 An advantage of heterogeneous catalysts is the recyclability of the catalysts, which is beneficial for sustainability. The present catalytic nano-capsule for RCMP is purely organic (with no inorganic support), which may be an attractive feature for applications where inorganic impurities are unwanted.
Also importantly, owing to the presence of QAI groups on the inner surface, the nano-capsule can also serve as a nano-reactor of RCMP. Namely, monomers can not only polymerize outside the nano-capsule via outer surface catalysis (as a supported catalyst), but also diffuse into the inner cavity and polymerize inside the nano-capsule via inner surface catalysis (as a nano-reactor). Such dual catalysis outside and inside the nano-capsule is novel in the field of living radical polymerization. Catalytic nano-reactors have been used for the syntheses of small molecules1–3,8,53–55 and polymers,56,57 offering, e.g., faster reaction rates, better compatibility with solvents, and multiple reactions occurring in one pot.58–61 Recently, substrate-sorting nano-reactors have been developed based on the selective diffusivity of substrates through the shells.62,63 In the present work, we synthesized polymers in a nano-reactor. While monomers (small molecules) can diffuse into the nano-reactor, polymers (large molecules) generated in the nano-reactor are not permeable through the shell, which enabled us to entrap polymers in a nano-reactor even in their good solvents. For demonstration, we entrapped various homopolymers and copolymers in a nano-reactor in a stable manner in their good solvents. Because the shell permeability mainly depended on the molecular size but not the molecular polarity, we were also able to use multiple monomers with different polarities altogether and entrap a mixed-polar multi-functional random copolymer. Although the present paper focuses on the proof of principle, the potential applications of polymers entrapped in a nano-capsule (nano-reactor) would include non-leaking polymer adsorbents and encapsulation of phase change materials (PCM) to prevent PCM from leaking and modulate the phase change temperature during the phase change processes,64–66 for example. Fig. 1 shows the monomers used in the present work.
 | ||
| Fig. 1 Monomers used in this work. | ||
Results and discussion
Synthesis of the macroinitiator ((PMMA-r-PC6MAI)–I)
A mixture of methyl methacrylate (MMA) (19.3 equiv., main monomer), C6MAI (1 equiv., co-monomer and catalyst), and 2-iodo-2-methylpropionitrile (CP–I, initiating dormant species) was heated at 60 °C for 1 h (Scheme 2 and Table 1). C6MAI contains both methacrylate and QAI groups and acts as a monomer and a polymerization catalyst. Hence, the polymerization proceeded without an extra catalyst. We intentionally stopped the polymerization for a short period of time (1 h) and at low monomer conversions (30% for MMA and 41% for C6MAI) in order to retain the chain-end iodide. The obtained polymer was purified by reprecipitation in a hexane/ethanol mixture (7/3 (v/v)). The number-average molecular weight (Mn) and dispersity (Đ = Mw/Mn) of the purified (PMMA-r-PC6MAI)–iodide ((PMMA-r-PC6MAI)–I) random copolymer were 1700 and 1.08, respectively, where Mw is the weight-average molecular weight, PMMA is poly(methyl methacrylate), and PC6MAI is poly([2-(methacryloyloxy)ethyl]dimethylhexylammonium iodide) (Table 1). These values are PMMA-calibrated gel permeation chromatography (GPC) values. From the 1H NMR spectrum of the purified (PMMA-r-PC6MAI)–I (Fig. S1 in the ESI†), we calculated the molar compositions of the MMA and C6MAI units to be 79% and 21%, respectively. The original amounts (19.3 equiv. and 1 equiv.) and conversions (30% and 41%) of MMA and C6MAI suggest that the polymer compositions before the purification were 93% and 7% for MMA and C6MAI, respectively. During the reprecipitation, MMA-rich polymers seemed to be removed more, which would result in the observed increase in the C6MAI composition (21%) after the purification. Using the Mn value (1700) and molar compositions (79% and 21% for the MMA and C6MAI units, respectively), we estimate the degree of polymerization (DP) to be approximately 9 and 2 (8.6 and 2.3) for the MMA and C6MAI units, respectively. Because the Mn value is not an absolute value but a PMMA-calibrated GPC value, these DP values are viewed as rough estimate. We use these estimated DP values in the present paper. Thus, we obtained (PMMA9-r-PC6MAI2)–I. In the present paper, the subscript in the polymer denotes the DP of each monomer. The exact chain-end fidelity of (PMMA9-r-PC6MAI2)–I was difficult to estimate with 1H NMR, because the peaks of the ω-terminal chain-end units of MMA and C6MAI adjacent to the chain-end iodide overlapped with other peaks (Fig. S1 in ESI†).| [MMA]0/[C6MAI]0/[CP–I]0 (equiv.) | T (°C) | t (h) | Conv.a (%) (MMA/C6MAI) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Đ | DPc (MMA/C6MAI) |
|---|---|---|---|---|---|---|
| a Monomer conversions were determined by 1H NMR. b PMMA-calibrated GPC (THF eluent) values of the purified polymer. c The DP values of the MMA and C6MAI units were calculated from the Mn value and the polymer compositions obtained from the 1H NMR spectrum of the purified polymer. | ||||||
| 19.3/1/1 | 60 | 1 | 30/41 | 1700 | 1.08 | 9/2 |
Self-catalyzed PISA
Using the obtained (PMMA9-r-PC6MAI2)–I as a macroinitiator, we carried out PISA. We heated a mixture of benzyl methacrylate (BzMA) (100 equiv., monomer), (PMMA9-r-PC6MAI2)–I (1 equiv., macroinitiator), 2,2′-azobis(2,4-dimethylvaleronitrile) (V65) (0.5 equiv., azo initiator), and ethanol (70 wt% of total mixture, solvent) at 70 °C (Scheme 2 and Table 2). The total fraction of the macroinitiator (2.7 wt%) and monomer (BzMA) (27.3 wt%) was 30 wt%, and hence the solid (polymer) content expected at a full (100%) monomer conversion was 30 wt%. V65 was added to increase the polymerization rate. Azo initiators are used to decrease the deactivator concentration and hence effectively increase the polymerization rate in RCMP37 and other living radical polymerizations.67| [BzMA]0/[(PMMA9-r-PC6MAI2)–I]0/[V65]0 (equiv.)a | t (h) | Conv.b (%) |
M
nc (Mn,theod) |
Đ | Hydrodynamic size in DLS (d(DLS))e (nm) | DLS size distribution indexf |
|---|---|---|---|---|---|---|
| a Solvent was ethanol (70 wt%). b Monomer (BzMA) conversion determined by 1H NMR. c PMMA-calibrated GPC values (THF eluent). d Theoretical Mn values calculated according to ([BzMA]0/[macroinitiator]0) × (monomer conversion) + (molecular weight of macroinitiator (1700)). e Intensity-average DLS peak-top value (in the water/ethanol mixture (98.6/1.4 (v/v))). f The intensity-average size distribution index in DLS defined as [(standard deviation)/(peak-top value)]2 (in the water/ethanol mixture (98.6/1.4 (v/v))). | ||||||
| 100/1/0.5 | 0.33 | 20 | 5000 (5200) | 1.41 | — | — |
| 1 | 33 | 6100 (7500) | 1.38 | — | — | |
| 1.25 | 40 | 7500 (8700) | 1.22 | — | — | |
| 1.5 | 52 | 8400 (11000) |
1.22 | — | — | |
| 2 | 62 | 12000 (13000) |
1.19 | 220 | 0.139 | |
The polymerization mixture turned cloudy at 1 h, suggesting the formation of a self-assembly as the PBzMA segment became long enough to be insoluble in ethanol (solvent), where PBzMA is poly(benzyl methacrylate). An aliquot of the polymerization mixture was taken out at 0.33, 1, 1.25, 1.5, and 2 h and diluted with CDCl3 for NMR analysis (to determine the monomer conversion) and with tetrahydrofuran (THF) for GPC analysis. The monomer conversion reached 62% for 2 h (Table 2 and Fig. 2a). The GPC chromatogram at 2 h (Fig. 2b) shows that a large fraction of the macroinitiator chains smoothly extended to block copolymer chains, suggesting a high block efficiency. The slight tailing in the GPC chromatogram would be ascribed to dead polymer chains that should be generated via radical–radical termination because of radical polymerization and new polymer chains that should be generated via the decomposition of V65 (azo compound). The Mn value increased with an increase in the monomer (BzMA) conversion and the Đ value was 1.19–1.41 during the polymerization (Table 2 and Fig. 2c), suggesting that the C6MAI units in the macroinitiator worked as effective catalysts in the PISA.
 | ||
| Fig. 2 (a) Plot of ln([M]0/[M]) vs. time (t), (b) GPC chromatograms, and (c) plots of Mn and Mw/Mnvs. monomer conversion for the BzMA/(PMMA9-r-PC6MAI2)–I/V65 system (70 °C): [BzMA]0/[(PMMA9-r-PC6MAI2)–I]0/[V65]0 = 100/1/0.5 (equiv.) (Table 2). The weight fractions were 27.3 wt% for BzMA, 2.7 wt% for (PMMA9-r-PC6MAI2)–I, and 70 wt% for ethanol. | ||
At 2 h, the DP of PBzMA is estimated to be 62 from the original amount of BzMA (100 equiv. to the macroinitiator) and the conversion of BzMA (62%). We use this estimated DP value in the present paper and denote the block copolymer as (PMMA9-r-PC6MAI2)-b-PBzMA62. The self-assembly obtained at 2 h was studied using transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Fig. 3). For the TEM analysis, the polymerization mixture was diluted with ethanol (50 times), added dropwise on a TEM grid, and dried under vacuum. The TEM image (image A in Fig. 3a) suggests the generation of a vesicle. For the DLS analysis, the polymerization mixture was diluted with water (50 times). The DLS analysis (Fig. 3a and Table 2) shows that the hydrodynamic size (intensity-average DLS peak top value) of the assembly was 220 nm (in a water/ethanol (98.6/1.4 (v/v)) mixture). The size (diameter = 220 nm) of the assembly was larger than the two-fold (ca. 37 nm) of the contour length (ca. 18 nm = 0.25 nm × 73 (= 9 + 2 + 62) total monomer units) of the block copolymer, supporting that the generated assembly was not a micelle but a vesicle. The size of the assembly shown in the TEM image (image A in Fig. 3a) is somewhat smaller than that determined using DLS, because the assembly was dry in the TEM analysis and swollen in the DLS analysis (in the solvent). The solid content at 2 h was 19.6 wt% (2.7 wt% of the macroinitiator and 16.9 wt% of the PBzMA segment generated during the PISA). Thus, we successfully obtained a (PMMA9-r-PC6MAI2)-b-PBzMA62 nano-capsule with a relatively high solid content via self-catalyzed PISA.
 | ||
| Fig. 3 DLS curves in water (water/ethanol mixture (98.6/1.4 (v/v)) (dashed line) and in THF (THF/ethanol mixture (98.6/1.4 (v/v)) (solid line) for (a) (PMMA9-r-PC6MAI2)-b-PBzMA62 and (b) (PMMA9-r-PC6MAI2)-b-(PBzMA-r-PEGDMA)62 nano-capsules. TEM images for A = (PMMA9-r-PC6MAI2)-b-PBzMA62 (assembly generated in ethanol), B = (PMMA9-r-PC6MAI2)-b-(PBzMA-r-PEGDMA)62 (assembly generated in ethanol), and C = (PMMA9-r-PC6MAI2)-b-(PBzMA-r-PEGDMA)62 (assembly retained in THF). | ||
Synthesis of crosslinked nano-capsules
Crosslinking is useful to fix self-assembly structures.68,69 Our research groups previously crosslinked self-assemblies using a small amount of a divinyl monomer as a co-monomer in the hydrophobic segment during RCMP-PISA (not self-catalyzed RCMP-PISA).34 In the present work, in order to fix the nano-capsule structure, we co-used ethylene glycol dimethacrylate (EGDMA) as a crosslinkable divinyl monomer as well as BzMA in self-catalyzed RCMP-PISA (Scheme S1 in the ESI†). A small amount of EGDMA (6.25% molar fraction of EGDMA and 93.75% molar fraction of BzMA) was used to crosslink the polymer at a later stage of polymerization, i.e., after the assembly (nano-capsule) was formed.We carried out the same polymerization as above (Table 2) but with EGDMA (Table 3) for 2 h. Both BzMA and EGDMA are methacrylates. We assume that the two monomers have the same reactivity and that the total monomer conversion was the same as that using BzMA only (62% (Table 2)). Thus, we denote the obtained polymer as (PMMA9-r-PC6MAI2)-b-(PBzMA-r-PEGDMA)62. As the TEM image (image B in Fig. 3b) shows, a vesicle was generated. Because the crosslinking occurred in the core-forming inner layer (BzMA and EGDMA) (pink layer in Scheme 1 and Scheme S1 in the ESI†) (not the shell-forming outer layer) of the bilayer, the capsules were not crosslinked in an inter-capsule manner, as observed in the TEM image (images B and C in Fig. 3b).
| [BzMA]0/[EGDMA]0/[(PMMA9-r-PC6MAI2)–I]0/[V65]0 (equiv.)a | t (h) | Hydrodynamic size in DLS (d(DLS))b (nm) | DLS size distribution indexc |
|---|---|---|---|
| a Solvent was ethanol (70 wt%). b Intensity-average DLS peak-top value (in the water/ethanol mixture (98.6/1.4 (v/v))). c The intensity-average size distribution index in DLS defined as [(standard deviation)/(peak-top value)]2 (in the water/ethanol mixture (98.6/1.4 (v/v))). | |||
| 93.75/6.25/1/0.5 | 2 | 234 | 0.139 |
The DLS analysis (Fig. 3b and Table 3) showed that the hydrodynamic size (DLS peak top) of the assembly in a water/ethanol (98.6/1.4 (v/v)) mixture was 234 nm, which is close to that (220 nm) of the non-crosslinked vesicle obtained using BzMA only (Fig. 3a and Table 2).
We added THF to the non-crosslinked (Fig. 3a) and crosslinked (Fig. 3b) vesicles. The non-crosslinked vesicle decomposed to single polymer chains with a hydrodynamic size of 7 nm (in a THF/ethanol (98.6/1.4 (v/v)) mixture), because the PBzMA segment is soluble in THF. In contrast, the crosslinked vesicle was stable in THF (THF/ethanol (98.6/1.4 (v/v)) mixture) with a hydrodynamic size of 191 nm, which is close to that (234 nm) in water (water/ethanol (98.6/1.4 (v/v)) mixture). The TEM analysis confirmed that the assembly structure (vesicle structure) was unchanged in water (image B in Fig. 3b) and THF (image C in Fig. 3b). Thus, we successfully obtained the crosslinked vesicle.
Polymerizations of MMA using a catalytic nano-capsule
The obtained crosslinked nano-capsule (Table 3) contains QAI (C6MAI) units on both the outer and inner surfaces of the nano-capsule (Scheme 2). The QAI unit can work as an RCMP catalyst, motivating us to use this crosslinked nano-capsule as a catalytic nano-capsule. We exploited the outer and inner QAI groups in a dual manner (as a supported catalyst and a nano-reactor catalyst) to conduct RCMP outside and inside the nano-capsule. We previously studied micrometer-sized beads with 300–900 μm diameters bearing catalysts on the outer surfaces as supported RCMP catalysts70 (which had no cavity (non-capsules)).First, we used the outer QAI groups. We studied RCMP outside the nano-capsule for exploiting the nano-capsule as a supported RCMP catalyst. A mixture of MMA (52 wt% of total reaction mixture, 100 equiv., monomer), CP–I (1 equiv., initiator), the crosslinked nano-capsule (13 wt% of total reaction mixture, 0.5 equiv. of the C6MAI unit, catalyst), and toluene (35 wt% of total reaction mixture, solvent) was heated at 70 °C (Table 4 (entry 1) and Fig. 4 (circles)). After 24 h, the monomer (MMA) conversion reached 42%. The polymerization took place both inside and outside the nano-capsule because MMA and CP–I were present (diffused) both inside and outside. Here we focus on the polymerization outside the nano-capsule. The polymer generated outside the nano-capsule had an Mn value of 5100, which is close to the theoretical value (4200), and a Đ value of 1.22. In order to further decrease the Đ value, we added a small amount of I2 (0.0125 equiv. to CP–I) (Table 4 (entry 2) and Fig. 4 (squares)). The Đ value slightly decreased to 1.20 for 24 h (monomer conversion = 28% and Mn = 3200). I2 is a good deactivator of polymer˙ and its addition can ensure a sufficiently high deactivation rate.37
 | ||
| Fig. 4 Plots of (a) ln([M]0/[M]) vs. time (t) and (b) Mn and Mw/Mnvs. monomer conversion for the MMA/CP–I/(catalytic nano-capsule)/I2/AIBN systems (70 °C). The reaction conditions are given in Table 4. The symbols are indicated in the figure. | ||
| Entry | [MMA]0/[CP–I]0/[I2]0/[AIBN]0 (equiv.)a | Catalytic nano-capsule | T (°C) | t (h) | Conv.b (%) |
M
n (Mn,theoc) |
Đ |
|---|---|---|---|---|---|---|---|
| a 52 wt% MMA, 13 wt% catalytic nano-capsule, and 35 wt% solvent (toluene) for entries 1 and 2 and 56 wt% MMA, 6 wt% catalytic nano-capsule, and 38 wt% solvent (toluene) for entry 3. b Monomer conversions determined by 1H NMR. c Theoretical Mn values calculated according to ([MMA]0/[CP–I]0) × (monomer conversion). | |||||||
| 1 | 100/1/0/0 | 13 wt% (0.5 equiv. of the C6MAI unit) | 70 | 24 | 42 | 5100 (4200) | 1.22 |
| 2 | 100/1/0.0125/0 | 13 wt% (0.5 equiv. of the C6MAI unit) | 70 | 24 | 28 | 3200 (2800) | 1.20 |
| 3 | 100/1/0.0125/0.25 | 6 wt% (0.2 equiv. of the C6MAI unit) | 70 | 9 | 91 | 9800 (9100) | 1.23 |
While a low-dispersity polymer was successfully obtained, the polymerization was relatively slow (Table 4 (entry 2) and Fig. 4 (squares)). To increase the polymerization rate, we added a small amount (0.25 equiv.) of an azo initiator, i.e., 2,2′-azobis(2-methylpropionitrile) (AIBN) (Table 4 (entry 3) and Fig. 4 (triangles)). With the addition of AIBN, the polymerization rate increased by a factor of approximately 15 (Fig. 4a), even though the amount of the crosslinked nano-capsule was approximately halved (6 wt% for Table 4 (entry 3) vs. 13 wt% for Table 4 (entry 2)). The monomer (MMA) conversion reached 91% for 9 h with keeping a small Đ value (1.23). These results (Table 4 and Fig. 4) demonstrate that the crosslinked nano-capsule successfully worked as a heterogeneous catalyst for RCMP of MMA (outside the nano-capsule).
Polymerizations of several monomers using a catalytic nano-capsule
Besides MMA, we studied RCMPs of other methacrylates, i.e., BzMA and poly(ethylene glycol) methyl ether methacrylate (PEGMA) (molecular weight = 300), and other families of the monomer, i.e., styrene (St) and acrylonitrile (AN) using the catalytic nano-capsule (Table 5). Relatively low-dispersity polymers (Đ = 1.34–1.48) were obtained with 75–89% monomer conversions for 1–8 h. The results suggest a wide monomer scope of the catalytic nano-capsule.| Entry | Monomer | [Monomer]0/[CP–I]0/[Azo initiator]0 (equiv.)a,b | Catalytic nano-capsule | T (°C) | t (h) | Conv.c (%) |
M
nd (Mn,theoe) |
Đ |
|---|---|---|---|---|---|---|---|---|
| a 38 wt% monomer, 9 wt% catalytic nano-capsule, and 53 wt% toluene (solvent) for entries 1 and 3, and 58 wt% monomer, 6 wt% catalytic nano-capsule, and 36 wt% toluene (solvent) for entry 2, and 58 wt% monomer, 6 wt% catalytic nano-capsule, and 36 wt% ethylene carbonate (solvent) for entry 4. b Azo initiator = AIBN for entries 1, 3, and 4 and V65 for entry 2. c Monomer conversions determined by 1H NMR. d PMMA-calibrated THF-GPC values for entries 1 and 3 and PMMA-calibrated DMF-GPC values for entries 2 and 4. e Theoretical Mn values calculated according to ([monomer]0/[CP–I]0) × (monomer conversion). | ||||||||
| 1 | BzMA | 100/1/0.25 | 9 wt% (0.8 equiv. of the C6MAI unit) | 70 | 2 | 89 | 14000 (16000) |
1.48 |
| 2 | PEGMA | 100/1/1 | 6 wt% (0.6 equiv. of the C6MAI unit) | 60 | 2 | 89 | 23000 (27000) |
1.35 |
| 3 | St | 100/1/1 | 9 wt% (0.5 equiv. of the C6MAI unit) | 80 | 8 | 75 | 5500 (7800) | 1.34 |
| 4 | AN | 100/1/0.125 | 6 wt% (0.1 equiv. of the C6MAI unit) | 75 | 1 | 86 | 19000 (4600) |
1.48 |
Recycling of the catalytic nano-capsule
The catalytic nano-capsule was able to be recovered from the polymerization solution through precipitation; namely, we used toluene to dilute the reaction mixture and collected the nano-capsule by centrifugation. To demonstrate the recyclability of the catalytic nano-capsule, we used the recovered catalytic nano-capsule in the polymerization of MMA under the same conditions (conditions in Table 4 (entry 3)) in recycled manners. We added I2 in each run, because I2 was washed out from the nano-capsule in the recovery process.In ten polymerization cycles, similar monomer conversions (83–91%) and Mn values (9200–10400) were consistently achieved for the 9 h polymerization (Fig. 5 and Table S1 in the ESI†). The Đ value gradually increased from 1.23 (first cycle) to 1.39 (tenth cycle), suggesting a gradual loss of the catalytic activity. Nevertheless, the Đ value was still relatively small (1.39) even in the tenth cycle. The results show the good recyclability of the catalytic nano-capsule.
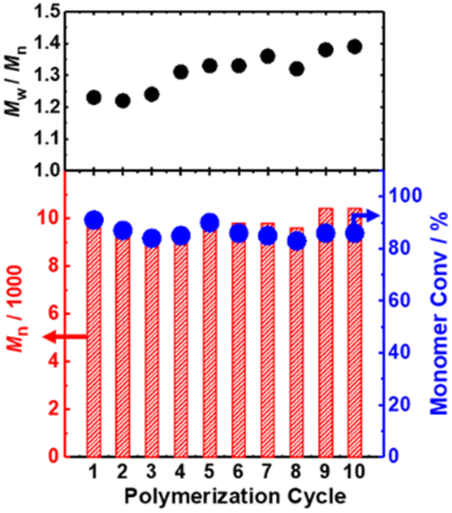 | ||
| Fig. 5 RCMPs of MMA using a catalytic nano-capsule in a recycled manner. The reaction conditions for each cycle were the same as in Table 4 (entry 3): MMA/CP–I/(catalytic nano-capsule)/I2/AIBN system (70 °C) with [MMA]0/[CP–I]0/[I2]0/[AIBN]0 = 100/1/0.0125/0.25 equiv. and [C6MAI unit]0 = 0.2 equiv. | ||
Use of the catalytic nano-capsule as a nano-reactor
Second, we used the nano-capsule as a catalytic nano-reactor. As mentioned, the present nano-capsule bears QAI groups on the inner surface (as well as the outer surface), and hence we conducted RCMP in the cavity of the nano-capsule. We dispersed the catalytic nano-capsule (6 wt% of total reaction mixture) in toluene (38 wt% of total reaction mixture) at room temperature with stirring for 2 h and subsequently added a mixture of MMA (56 wt% of total reaction mixture, 100 equiv.), CP–I (1 equiv.), AIBN (0.25 equiv.), and I2 (0.025 equiv.). We stirred the whole mixture for another 2 h at room temperature to ensure diffusion of the monomer (MMA), initiating dormant species (CP–I) and other species into the cavity of the nano-capsule (in an equilibrated manner inside and outside the nano-capsule) and heated the whole mixture at 70 °C for 6 h. The polymerization took place in both inside and outside the nano-capsule. After the polymerization, we immersed the nano-capsule in excess toluene and subsequently collected the nano-capsule by centrifugation, completely removing small molecules from the cavity. We suppose that the polymer formed outside the nano-capsule was rinsed out while the polymer formed inside the nano-capsule stayed in the cavity because large molecules (polymer chains) are not able to penetrate the nano-capsule shell.The collected nano-capsule was analyzed using DLS and TEM. The DLS analysis showed that the hydrodynamic size (DLS peak top) increased from 191 nm (before the polymerization) to 396 nm (after the polymerization) in THF (solvent) (Fig. S2 in the ESI†). The observed increase in the particle size suggests the increase in osmotic pressure in the cavity and hence the presence of polymers in the cavity. The TEM image shows that the cavity was empty before the polymerization (image C in Fig. 3) and became dark after the polymerization (image D in Fig. 6), demonstrating that the polymerization occurred in the cavity and that the nano-capsule served as a catalytic nano-reactor for RCMP. Notably, as a result, in the present system, PMMA was entrapped in the nano-capsule in its solvophilic medium (hydrophobic MMA/toluene mixture).
 | ||
| Fig. 6 TEM image of the PMMA-loaded catalytic nano-capsule. | ||
Because the nano-capsule was chemically crosslinked, we were not able to take out the PMMA from the nano-capsule or analyze the Mn and Đ values of the PMMA. We also repeatedly washed the polymer-entrapped nano-capsule. The TEM image showed that the cavity remained dark even after repeated washing (Fig. S3 in the ESI†), demonstrating that the polymer chains were impermeable and entrapped in the nano-capsule.
Synthesis of an amphiphilic block copolymer in a nano-reactor
We further used this nano-reactor for synthesizing an amphiphilic block copolymer inside the cavity. We stirred the catalytic nano-capsule (6 wt%) in toluene (36 wt%) for 2 h, added a mixture of PEGMA (58 wt%, 100 equiv.), CP–I (2 equiv.), and V65 (1 equiv.), stirred the whole mixture for 2 h, and heated it at 60 °C for 0.5 h. We intentionally stopped the polymerization at a relatively low monomer conversion of 20% in order to retain the chain-end iodide and obtained PPEGMA–I in the nano-capsule, where PPEGMA is poly(poly(ethylene glycol) methyl ether methacrylate). The polymer generated outside the nano-capsule had an Mn value of 6500 and a Đ value of 1.29. The Mn and Đ values of the polymers generated inside the nano-capsule may or may not be similar to those of the polymers generated outside the nano-capsule, because the catalytic activity, viscosity of medium, concentrations of species, and hence polymerization behaviour may be different inside and outside the nano-capsule. Nevertheless, we assumed that the PPEGMA–I generated in the nano-capsule also had an Mn value of 6500 when we calculated the molar quantity of the PPEGMA–I in the nano-capsule (below), but this molar quantity is viewed as a rough estimate for the mentioned reason. We purified the nano-capsule by centrifugation (removed the PEGMA monomer and other small molecules). The TEM image of the purified nano-capsule (Fig. S4 in the ESI†) showed that the center of the nano-capsule was darker than its shell, suggesting the entrapment of PPEGMA–I in the nano-capsule.The present nano-reactor served as a substrate-sorting nano-reactor based on the selective diffusivity of substrates (small molecules and polymers) through the shell by their sizes. Thus, the nano-reactor could entrap polymer chains (left in Fig. 7a), as mentioned. The polymer-entrapped nano-reactor further enabled a selective reaction in the nano-reactor (right in Fig. 7a); namely, the reaction could occur only inside the nano-reactor but not outside the nano-reactor.
 | ||
| Fig. 7 (a) Schematic illustration of block polymerization in the nano-capsule. (b) TEM image and fluorescence confocal microscopy image of the PPEGMA-b-PNMA-loaded catalytic nano-capsule. | ||
We used the PPEGMA–I in the nano-capsule as a macroinitiator for block polymerization. We dispersed the PPEGMA–I containing nano-capsule (12 wt%, 1 equiv. of PPEGMA–I (assuming Mn = 6500)) in a mixture of 2-naphthyl methacrylate (NMA) (38 wt%, 100 equiv.) and toluene (50 wt%), stirred for 2 h, and heated at 70 °C for 24 h. Polymerization occurred only inside the nano-capsule in which the PPEGMA–I dormant species was present. No polymerization took place outside the nano-capsule because of the absence of alkyl iodide dormant species. Thus, the PPEGMA–I-containing nano-capsule selectively induced the reaction (block polymerization) inside the nano-capsule. After the purification of the nano-capsule, the DLS analysis showed that the hydrodynamic size (DLS peak top) increased from 166 nm (before block polymerization) to 393 nm (after block polymerization) in THF (Fig. S5 in the ESI†). The hydrodynamic size (166 nm) of the PPEGMA–I containing nano-capsule (before block polymerization) was similar to or slightly smaller than that (191 nm) of the empty nano-capsule, because THF is a theta or slightly poor solvent of PPEGMA. THF is a good solvent of hydrophobic poly(2-naphthyl methacrylate) (PNMA). The growth of PNMA in the block copolymer would enable the expansion (swelling) of the polymer chain and hence lead to the observed increase in the hydrodynamic size of the nano-capsule in THF (393 nm). The TEM image (Fig. 7b) shows that the polymer was loaded inside the nano-capsule. PNMA is a fluorescent polymer. The confocal fluorescence microscopy image (Fig. 7b and Fig. S6 in the ESI†) clearly shows discrete fluorescent spots, demonstrating the formation of a PPEGMA-b-PNMA block copolymer in the nano-capsule. The fluorescence spots in Fig. 7b were micrometer-sized (not nanometer-sized), which is because of the microscope resolution.
Synthesis of a multi-elemental polymer in the nano-reactor
For more demonstration (Fig. 8a), we synthesized a multi-elemental copolymer in the nano-capsule. We first synthesized the PPEGMA–I macroinitiator in the nano-capsule as described above. Subsequently, the PPEGMA–I containing nano-capsule (10.3 wt%, 1 equiv. of PPEGMA–I (assuming Mn = 6500)) was dispersed in a mixture of toluene (50 wt%) and several monomers (Fig. 8b), i.e., boron-containing (2-phenyl-1,3,2-dioxaborolan-4-yl)methyl methacrylate (PDBMA) (7.8 wt%, 20 equiv.), silicon-containing trimethylsilyl methacrylate (TMSMA) (0.3 wt%, 20 equiv.), phosphorus-containing 2-(methacryloyloxy)ethyl 2-(trimethylammonio)ethyl phosphate (MPC) (9.4 wt%, 20 equiv.), sulfur-containing 2-(methylthio)ethyl methacrylate (MTEMA) (5.1 wt%, 20 equiv.), fluorine-containing 2,2,3,3,3-pentafluoropropyl methacrylate (PFPMA) (6.9 wt%, 20 equiv.), bromine-containing 3-bromostyrene (Br-St) (5.8 wt%, 20 equiv.), and chlorine-containing 4-chlorostyrene (Cl-St) (4.4 wt%, 20 equiv.), stirred for 2 h, and heated at 70 °C for 24 h, yielding a block copolymer with the first-block PPEGMA segment and the second-block random copolymer segment. The resultant copolymer comprises multiple elements and also comprises amphiphilic (PEGMA), hydrophilic (MPC), hydrophobic (GMMA-PBA, MTEMA, Br-St, and Cl-St), and super-hydrophobic (TMSMA and PFPMA) monomer units with different polarities. | ||
| Fig. 8 (a) Schematic illustration of the block copolymerization of multi-polarity and multi-elemental polymers in the nano-capsule, (b) seven monomers used in block copolymerization, (c) elemental mapping image, and (d) table of elements. 12 elements in red frames in (d) were incorporated in the multi-elemental particle. | ||
We analyzed the purified nano-capsule. The hydrodynamic size (DLS peak top) increased from 164 nm (before block polymerization) to 260 nm (after block polymerization) in THF (Fig. S9 in ESI†), suggesting that the polymer chains inside the nano-capsule became more hydrophobic by the chain growth. The second block segment is a multi-polar random copolymer of the seven monomers. The TEM image (Fig. S10 in ESI†) confirmed the loading of the polymer in the nano-capsule. The scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS) analysis (Fig. 8c and Fig. S11 in the ESI†) confirmed the presence of multiple elements. The SEM-EDS mapping (Fig. 8c) clearly shows the presence of multiple elements on the whole area, as indicated in different colors. The SEM-EDS spectrum on the selected area of the nano-capsule (Fig. S10 in the ESI†) also showed the presence of C, O, F, P, Cl, Br, S, Si, and I. The B (boron) peak was overlapped with the high-intensity C (carbon) peak and was not distinguished. Assuming the presence of B, the result suggests successful random copolymerization of all of the seven monomers in the nano-capsule. The observed I (iodine) results from the C6MAI unit in the catalytic nano-capsule and the chain-end iodide of polymers. N (nitrogen) of C6MAI and MPC (whose peak was overlapped with the high-intensity O (oxygen) and C (carbon) peaks), and H (hydrogen) of all monomer units should be present. Overall, a multi-element particle containing non-metallic 12 elements (SEM-EDS-confirmed C, O, F, P, Cl, Br, S, Si, and I as well as B, N, and H (Fig. 8d)) was obtained. The 12 elements came from the block copolymer and the nano-capsule.
Conclusions
A QAI-containing nano-capsule was successfully synthesized via self-catalyzed RCMP-PISA at a relatively high solid content (19.6 wt%). The obtained nano-capsule was subsequently used as a supported heterogeneous catalyst of RCMP to generate polymers outside the nano-capsule. The heterogeneous catalyst yielded relatively low-dispersity (Đ = 1.23–1.48) polymers at high monomer conversions (75–91%) for several monomers and showed good recyclability in ten polymerization cycles for MMA. The nano-capsule was further used as a catalytic nano-reactor of RCMP to generate polymers inside the nano-capsule. The present nano-reactor served as a substrate-sorting nano-reactor based on the size-selective diffusivity of small molecules and polymers through the shell, enabling the entrapment of the generated polymers. The macroinitiator-entrapped nano-reactor further selectively induced reactions (block polymerizations) in the nano-reactor; namely, the reactions occurred only inside the nano-reactor but not outside the nano-reactor. Thus, we were able to synthesize and entrap a homopolymer, an amphiphilic block copolymer, and a multi-polarity and multi-elemental block copolymer in the nano-capsule. Because the nano-capsule was chemically crosslinked, we were not able to directly analyze the polymers generated in the nano-capsule. Nevertheless, we confirmed the formation of the polymers using TEM and the chain growth in block copolymerization using fluorescence microscopy and SEM-EDS at least in qualitative manners, demonstrating the synthesis and entrapment of the polymers in the nano-reactor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) Investigatorship in Singapore (NRF-NRFI05-2019-0001).References
- S. A. Dergunov, A. T. Khabiyev, S. N. Shmakov, M. D. Kim, N. Ehterami, M. C. Weiss, V. B. Birman and E. Pinkhassik, ACS Nano, 2016, 10, 11397–11406 CrossRef CAS PubMed.
- W. Zhu, Z. Chen, Y. Pan, R. Dai, Y. Wu, Z. Zhuang, D. Wang, Q. Peng, C. Chen and Y. Li, Adv. Mater., 2019, 31, 1800426 CrossRef PubMed.
- M. Wang, Z. Xu, Y. Shi, F. Cai, J. Qiu, G. Yang, Z. Hua and T. Chen, J. Org. Chem., 2021, 86, 8027–8035 CrossRef CAS PubMed.
- B. Hambly, M. Guzinski, F. Perez, B. Pendley and E. Lindner, ACS Appl. Nano Mater., 2020, 3, 6328–6335 CrossRef CAS.
- S. Zhang, R. Geryak, J. Geldmeier, S. Kim and V. V. Tsukruk, Chem. Rev., 2017, 117, 12942–13038 CrossRef CAS PubMed.
- A. Plucinski, Z. Lyu and B. V. K. J. Schmidt, J. Mater. Chem. B, 2021, 9, 7030–7062 RSC.
- S. P. Chali and B. J. Ravoo, Angew. Chem., Int. Ed., 2020, 59, 2962–2972 CrossRef PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef PubMed.
- G. Cheng and J. Pérez-Mercader, Macromol. Rapid Commun., 2019, 40, 1800513 CrossRef PubMed.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- H. Phan, V. Taresco, J. Penelle and B. Couturaud, Biomater. Sci., 2021, 9, 38–50 RSC.
- M. J. Monteiro and M. F. Cunningham, Macromolecules, 2012, 45, 4939–4957 CrossRef CAS.
- S. Pearce and J. Perez-Mercader, Polym. Chem., 2021, 12, 29–49 RSC.
- P. Gurnani and S. Perrier, Prog. Polym. Sci., 2020, 102, 101209 CrossRef CAS.
- J. Lefley, C. Waldron and C. R. Becer, Polym. Chem., 2020, 11, 7124–7136 RSC.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 RSC.
- F. Jasinski, P. B. Zetterlund, A. M. Braun and A. Chemtob, Prog. Polym. Sci., 2018, 84, 47–88 CrossRef CAS.
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid Commun., 2017, 38, 1600528 CrossRef PubMed.
- P. Damsongsang, V. P. Voravee and S. Yusa, New J. Chem., 2021, 45, 12776–12791 RSC.
- C. J. Mable, L. A. Fielding, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2019, 9, 1454–1463 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. L. d. l. Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef PubMed.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605–8615 CrossRef CAS.
- A. Alzahrani, D. Zhou, R. P. Kuchel, P. B. Zetterlund and F. Aldabbagh, Polym. Chem., 2019, 10, 2658–2665 RSC.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Macromolecules, 2013, 46, 4285–4295 CrossRef CAS.
- J. Sarkar, Y. F. Lim and A. Goto, Macromol. Chem. Phys., 2021, 222, 2100349 CrossRef CAS.
- J. Sarkar, K. B. J. Chan and A. Goto, Polym. Chem., 2021, 12, 1060–1067 RSC.
- J. Sarkar, A. W. Jackson, A. M. V. Herk and A. Goto, Polym. Chem., 2020, 11, 3904–3912 RSC.
- J. Sarkar, L. Xiao, A. W. Jackson, A. M. van Herk and A. Goto, Polym. Chem., 2018, 9, 4900–4907 RSC.
- C.-G. Wang, A. M. L. Chong, H. M. Pan, J. Sarkar, X. T. Tay and A. Goto, Polym. Chem., 2020, 11, 5559–5571 RSC.
- A. Goto, T. Suzuki, H. Ohfuji, M. Tanishima, T. Fukuda, Y. Tsuji and H. Kaji, Macromolecules, 2011, 44, 8709–8715 CrossRef CAS.
- A. Goto, A. Ohtsuki, H. Ohfuji, M. Tanishima and H. Kaji, J. Am. Chem. Soc., 2013, 135, 11131–11139 CrossRef CAS PubMed.
- C.-G. Wang, F. Hanindita and A. Goto, ACS Macro Lett., 2018, 7, 263–268 CrossRef CAS PubMed.
- C.-G. Wang, X. Y. Oh, X. Liu and A. Goto, Macromolecules, 2019, 52, 2712–2718 CrossRef CAS.
- L. L. Böhm, Angew. Chem., Int. Ed., 2003, 42, 5010–5030 CrossRef PubMed.
- G. Kickelbick, H.-J. Paik and K. Matyjaszewski, Macromolecules, 1999, 32, 2941–2947 CrossRef CAS.
- D. M. Haddleton, D. J. Duncalf, D. Kukulj and A. P. Radigue, Macromolecules, 1999, 32, 4769–4775 CrossRef CAS.
- Y. Shen, S. Zhu, F. Zeng and R. H. Pelton, Macromolecules, 2000, 33, 5427–5431 CrossRef CAS.
- A. Kanazawa, K. Satoh and M. Kamigaito, Macromolecules, 2011, 44, 1927–1933 CrossRef CAS.
- C. Bernhardt, C. B. Osman, B. Charleux and F. Stoffelbach, Polymer, 2015, 77, 199–207 CrossRef CAS.
- H.-C. Lee, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2016, 7, 7199–7203 RSC.
- H.-C. Lee, M. Fantin, M. Antonietti, K. Matyjaszewski and B. V. K. J. Schmidt, Chem. Mater., 2017, 29, 9445–9455 CrossRef CAS.
- O. S. Taskin, B. Kiskan and Y. Yagci, Polym. Int., 2018, 67, 55–60 CrossRef CAS.
- E. Feiz, M. Mahyari, H. R. Ghaieni and S. Tavangar, Sci. Rep., 2022, 11, 8257 CrossRef PubMed.
- S. Shanmugam, S. Xu, N. N. M. Adnan and C. Boyer, Macromolecules, 2018, 51, 779–790 CrossRef CAS.
- Y. Chu, N. Corrigan, C. Wu, C. Boyer and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15245–15253 CrossRef CAS.
- W. L. Guo, Y. Zhou, B. Duan, W. F. Wei, C. Chen, X. Li and T. Cai, Chem. Eng. J., 2022, 429, 132120 CrossRef CAS.
- B. Saini, S. Singh and T. K. Mukherjee, ACS Appl. Mater. Interfaces, 2021, 13, 51117–51131 CrossRef CAS PubMed.
- N. Qin, A. Pan, J. Yuan, F. Ke, X. Wu, J. Zhu, J. Liu and J. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 12463–12471 CrossRef CAS PubMed.
- C. Carrillo-Carrión, R. Martínez, E. Polo, M. Tomás-Gamasa, P. Destito, M. Ceballos, P. Pelaz, F. López, J. L. Mascareñas and P. d. Pino, ACS Nano, 2021, 15, 16924–16933 CrossRef PubMed.
- G. K. K. Clothier, T. R. Guimarães, M. Khan, G. Moad, S. Perrier and P. B. Zetterlund, ACS Macro Lett., 2019, 8, 989–995 CrossRef CAS PubMed.
- G. K. K. Clothier, T. R. Guimarães, M. Khan, G. Moad and P. B. Zetterlund, Macromolecules, 2022, 55, 1981–1991 CrossRef CAS.
- P. Qu, M. Kuepfert, S. Jockusch and M. Weck, ACS Catal., 2019, 9, 2701–2706 CrossRef CAS.
- M. Kuepfert, E. Ahmed and M. Weck, Macromolecules, 2021, 54, 3845–3853 CrossRef CAS.
- Y. Que, J. Ruan, Y. Xiao, C. Feng, G. Lua and X. Huang, Polym. Chem., 2020, 11, 1727–1734 RSC.
- S. Dutta, N. Kumari, S. Dubbu, S. W. Jang, A. Kumar, H. Ohtsu, J. Kim, S. H. Cho, M. Kawano and I. S. Lee, Angew. Chem., Int. Ed., 2020, 59, 3416–3422 CrossRef CAS PubMed.
- T. Nishimura, S. Hirose, Y. Sasaki and K. Akiyoshi, J. Am. Chem. Soc., 2020, 142, 154–161 CrossRef CAS PubMed.
- Z. Gao, Y. Li, Z. Liu, Y. Zhang, F. Chen, P. An, W. Lu, J. Hu, C. You, J. Xu, X. Zhang and B. Sun, Nano Lett., 2021, 21, 3401–3409 CrossRef CAS PubMed.
- P. F. De Castro and D. G. Shchukin, Chem. – Eur. J., 2015, 21, 11174–11179 CrossRef PubMed.
- H. Hu, Composites, Part B, 2020, 195, 108094 CrossRef CAS.
- Y. Zhao and Y. Luo, Macromol. React. Eng., 2022, 16, 2100049 CrossRef CAS.
- A. Goto and T. Fukuda, Polym. Sci., 2004, 29, 329–385 CAS.
- S. Li, G. Han and W. Zhang, Polym. Chem., 2020, 11, 4681–4692 RSC.
- C. J. Marsden, C. Breen, J. D. Tinkler, T. R. Berki, D. W. Lester, J. Martinelli, L. Tei, S. J. Butler and H. Willcock, Polym. Chem., 2022, 13, 4124–4135 RSC.
- C.-G. Wang, J. J. Chang, E. J. F. Yong, H. Niino, S. Chatani, S. Y. Hsu and A. Goto, Macromolecules, 2020, 53, 51–58 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and additional data. See DOI: https://doi.org/10.1039/d2py01086k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |