
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic carboxylate salt-enabled alternative synthetic routes for bio-functional cyclic carbonates and aliphatic polycarbonates†
Yuya
Watanabe‡
 a,
Shunya
Takaoka‡
a,
Yuta
Haga
a,
Kohei
Kishi
a,
Shunta
Hakozaki
a,
Atsushi
Narumi
a,
Takashi
Kato
b,
Masaru
Tanaka
c and
Kazuki
Fukushima
*abd
a,
Shunya
Takaoka‡
a,
Yuta
Haga
a,
Kohei
Kishi
a,
Shunta
Hakozaki
a,
Atsushi
Narumi
a,
Takashi
Kato
b,
Masaru
Tanaka
c and
Kazuki
Fukushima
*abd
aDepartment of Polymer Science and Engineering, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan. E-mail: k_fukushima@chembio.t.u-tokyo.ac.jp
bDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
cInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dJapan Science and Technology Agency (JST), PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 8th August 2022
Abstract
Simple and efficient synthetic routes for functionalized cyclic carbonates are indispensable for the practical application of side-chain bio-functionalized aliphatic polycarbonates as biodegradable functional biomaterials. In this study, a six-membered cyclic carbonate with a triethylammonium carboxylate has been prepared in one step from 2,2-bis(methylol)propionic acid (bis-MPA). We have demonstrated the suitability of the organic carboxylate salt of the bis-MPA cyclic carbonate for esterification with alkyl bromides via the SN2 mechanism, leading to the formation of functionalized cyclic carbonate monomers. The esterification of the organic carboxylate salt proceeds efficiently when alkyl bromides with α-carbonyl, allyl, and benzyl groups are used. This approach enables a two-step synthesis of functionalized cyclic carbonates from bis-MPA. The organocatalyzed ring-opening polymerization of the resultant functionalized cyclic carbonates is effectively controlled, indicating that the synthetic process involving the organic carboxylate salt does not influence their polymerizability. The ether-functionalized aliphatic polycarbonates obtained from the organic carboxylate salt exhibit good antiplatelet properties, comparable to those of a previously developed blood-compatible aliphatic polycarbonate. The synthetic pathways exploiting organic carboxylate salts enable alternative shortcuts to functionalized cyclic carbonates from bis-MPA.
Introduction
Biodegradable aliphatic polycarbonates have been extensively studied as materials for biomedical applications, electrolytes, and sustainable processes.1–3 Poly(trimethylene carbonate) (PTMC) is a hydrophobic, flexible, and amorphous polymer that can be hydrolyzed in vivo or with the assistance of enzymes.4 Various functionalized PTMC analogs have been developed over the last two decades towards the preparation of biodegradable functional biomaterials for drug delivery applications, antimicrobial agents, and hydrogels.1,5 A combination of organocatalytic ring-opening polymerization (ROP) techniques and substituted cyclic carbonates enabled the production of such analogs.1,62,2-Bis(methylol)propionic acid (bis-MPA; Fig. 1) is most frequently used as a starting material for the synthesis of functionalized six-membered cyclic carbonates such as 1 (Fig. 1).1,5 The controlled ROP of 1 using organocatalysts has been well studied for producing aliphatic polycarbonates 2 (Fig. 1) with different functional groups (FGs).1,5,6 Our research team recently initiated the development of degradable, water-insoluble, and blood-compatible polymers7–9 with potential applications in stent coatings, artificial blood vessels, and antiadhesive materials.10 The first antiplatelet aliphatic polycarbonate (2a; Fig. 1) exhibited high blood compatibility due to the hydration of ether side groups.8 However, its degree of antiplatelet adhesion was smaller than those of clinically applicable blood-compatible polymers, such as poly(2-methacryloyloxyethyl phosphorylcholine)11,12 and poly(2-methoxyethyl acrylate) (PMEA)13 and its analogs.14 In this context, we designed 2b and 2c, which are analogs of 2a with different ether-containing side chains (Fig. 1).
 | ||
| Fig. 1 Synthesis of aliphatic polycarbonates with FGs in the side chains from bis-MPA through key intermediate AC1, which enables a shortcut to functional monomers 1. Representative side chain structures a–c are designed for blood compatibility. | ||
Several routes were developed for the synthesis of cyclic carbonate monomers 1 from bis-MPA.15–18 Its carboxy group is used to install FGs via esterification and amidation with alcohols and amines, respectively, while the two hydroxy groups can be applied to carbonylative cyclization. Because of the poor solubility and competing reactivity of the carboxy and hydroxy groups of bis-MPA, their protection and deprotection are often required,16,17 thus increasing the number of synthetic steps for 1. Bis(pentafluorophenyl)carbonate (PFC) enabled the direct cyclization of bis-MPA as a part of a two-step efficient synthesis of 1.18 However, the cost of PFC and cytotoxicity of liberated pentafluorophenol can be potential issues. Therefore, we revisited the widely used route proposed by Pratt et al.,17 in which MTC-H (Scheme 1) serves as a versatile intermediate to produce 1. Three-step reactions including (de)protection steps are required to obtain MTC-H from bis-MPA in the conventional route.17 We found that the organic carboxylate salt of bis-MPA cyclic carbonate AC1 (Fig. 1) can be formed directly from bis-MPA and converted to MTC-H via protonation (route A in Scheme 1).19 Recently, Tan et al. reported a similar route with a more sophisticated procedure.20
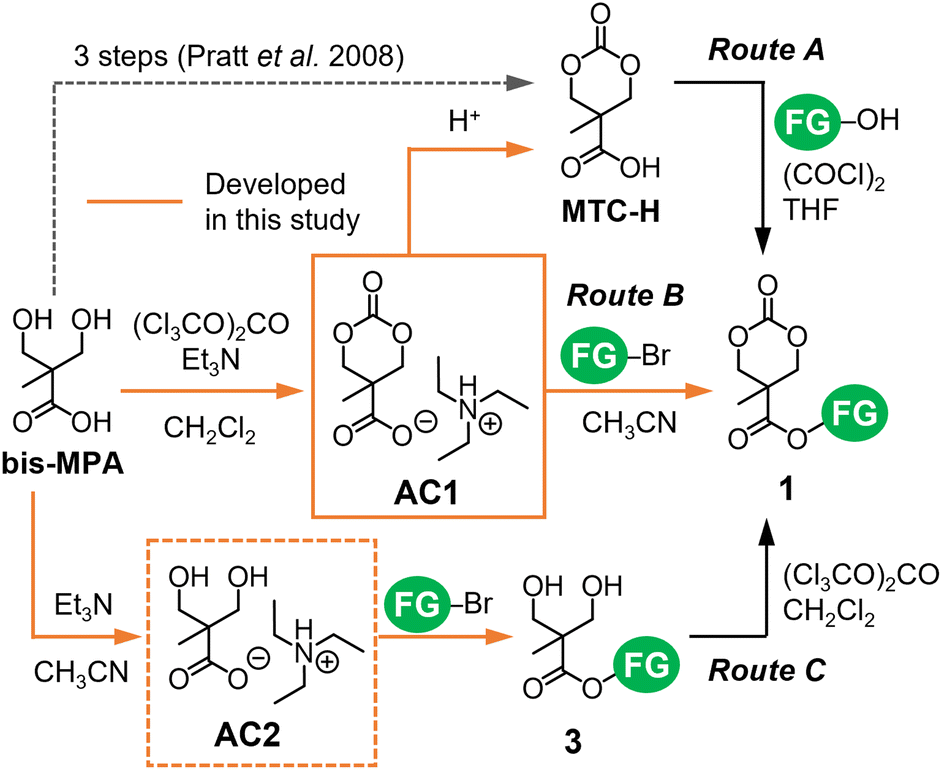 | ||
| Scheme 1 Synthesis of functionalized cyclic carbonates 1 exploiting organic carboxylate salts AC1 and AC2. The orange routes represent the new processes developed in the present study. | ||
In this study, we further explored the shortcut between AC1 and 1 and the utilization of the AC2 intermediate (Fig. 1) during the synthesis of AC1. Coulembier et al. successfully initiated the ROP of β-butyrolactone by the carboxylate anion of MTC-H in the form of an adduct with N-heterocyclic carbene.21 Their results motivated us to use the carboxylate anion of AC1 as a nucleophile for esterification with halogenated alkanes (such as alkyl bromides) containing FGs to obtain 1via the SN2 mechanism (route B in Scheme 1), which has never been examined previously. In a similar manner, we examined the efficiency of AC2 for the formation of bis-MPA esters 3 serving as precursors of 1.
Results and discussion
Direct cyclization of bis-MPA
Based on a previously developed protocol,17 we employed triphosgene as a carbonylative cyclization reagent often used in aprotic low-polarity solvents such as tetrahydrofuran (THF) and CH2Cl2. However, bis-MPA is hardly soluble in these organic solvents. In addition, both hydroxy and carboxy groups can react with triphosgene, which necessitates a benzyl protection step in the conventional approach.17 Inspired by the work of Koga et al.,22 we examined the solubility of acid–base organic salts of bis-MPA using eight commercially available nitrogen bases (Scheme S1†). Although no clear tendency was observed, these salts were soluble in solvents with high dielectric constants when paired bases with high pKa values were used. Accordingly, triethylammonium (TEAH+) salt AC2 (Fig. S1†) exhibited a broad spectrum of solubility in aprotic organic solvents (Table S1†).Next, we studied the direct cyclization of bis-MPA in the presence of 3 equivalents of triethylamine (TEA) in CH2Cl2, in which 1 equivalent of TEA was consumed to form AC2in situ, and the remaining 2 equivalents of TEA were used to scavenge HCl generated from triphosgene. Eventually, 3.5 equivalents of TEA were added for safety. As a result, cyclic carbonate AC1 was successfully formed with quantitative conversion (>99%). Two types of TEAH+ salts were generated in the reaction mixture: AC1 and triethylammonium hydrochloride (TEAH+Cl−). Cyclic carbonate organic salt AC1 remained soluble in THF and CH2Cl2, while TEAH+Cl− precipitated in these solvents. Thus, TEAH+Cl− was effectively separated by filtration, and the filtrate was evaporated to obtain AC1 (Fig. 2). The ratio between the carboxylate and ammonium species was not 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in most cases (Fig. S2 and S3†) owing to the imperfect removal of TEAH+Cl− and the partial evaporation of ammonium species during purification. The latter reflects the dynamic nature of the organic salt form.
:1 in most cases (Fig. S2 and S3†) owing to the imperfect removal of TEAH+Cl− and the partial evaporation of ammonium species during purification. The latter reflects the dynamic nature of the organic salt form.
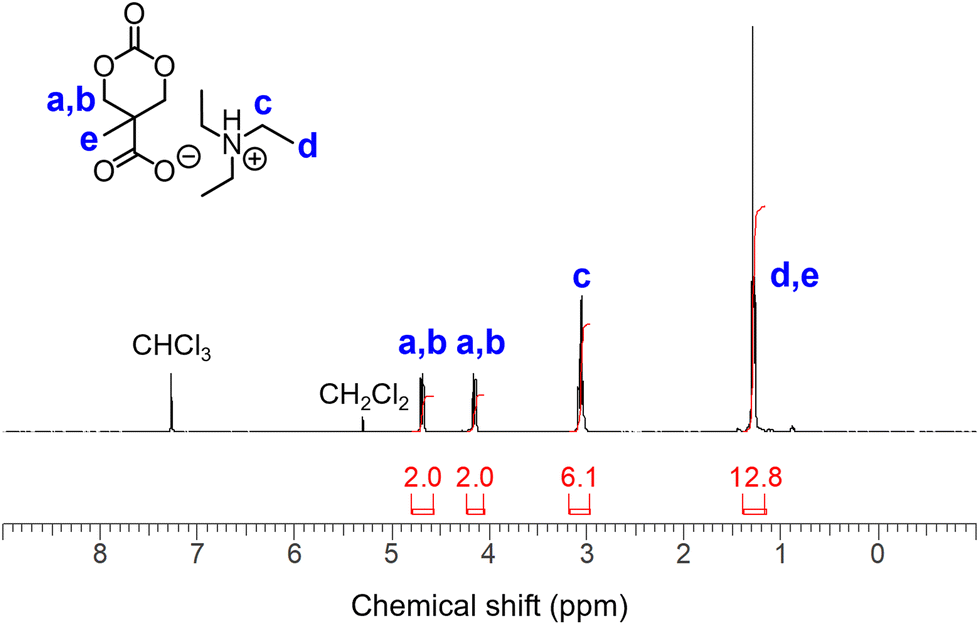 | ||
| Fig. 2 1H NMR spectrum of AC1 (400 MHz, CDCl3). | ||
This successful formation of AC1 suggests that the carboxylate anion of AC2 formed in situ does not react with triphosgene. In turn, the TEAH+ ion deactivates the carboxylate, thus preventing interference in the cyclization reaction. The reactivity of the carboxylate may correlate with solvent polarity as the counter ion is often liable to reside nearby in low-polarity solvents. As demonstrated previously by Tan et al.,20 TEAH+ was easily removed by an ionic exchange resin to yield MTC-H (route A in Scheme 1). The quality of MTC-H produced in this study was comparable to that of MTC-H formed through the conventional route involving the hydrogenolysis of the benzyl-functionalized cyclic carbonate (1d; Fig. 3), which was previously developed by Pratt et al.17 (Fig. S4 and S5†). It is noteworthy that the catalytic transfer hydrogenolysis23 using cyclohexene with Pd/C was also effective for the deprotection of the benzyl ester group of 1d (Scheme S2†).
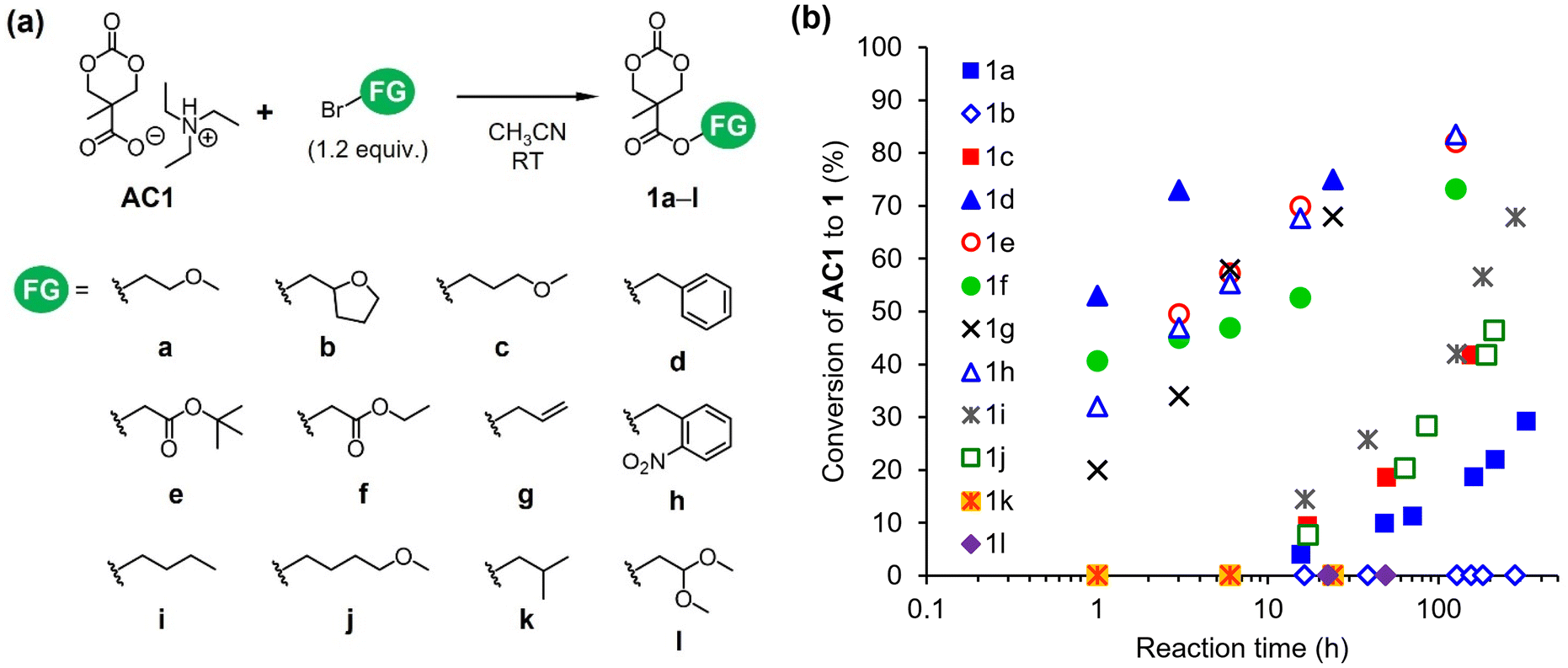 | ||
| Fig. 3 Formation of functionalized cyclic carbonates 1a–l from AC1 through esterification with various alkyl bromides. (a) Synthetic scheme and (b) conversion of AC1 to 1 plotted as a function of reaction time. | ||
The ion-exchange protonation process generates heat owing to the neutralization reaction between the resins and substrates. Because the heat can induce polymerization, it should be carefully managed, especially in a large-scale process.20 Alternatively, approaches for circumventing the cumbersome protonation work-up would be explored.
Esterification of cyclic carbonate organic salt
The carboxylate anion serves as a nucleophile in the SN2 esterification reaction24,25 and an initiator of the ROPs of 4-membered ring lactones.21 Because of their high accessibility and reactivity, we employed alkyl bromides (FG-Br in Scheme 1) as electrophiles for the esterification of AC1 (route B in Scheme 1). In the present study, the nucleophilic attack of the carboxylate anion of AC1 must be directed only towards alkyl bromides rather than ring opening. Hence, we first monitored the temperature, stoichiometry, and concentration for the optimization of reaction conditions using benzyl bromide as an electrophile (Scheme S3†). Acetonitrile was used as a reaction medium due to its ability to promote typical SN2 processes in polar aprotic environments. After optimizing other parameters (Table S2†), the reaction was performed using 1.5 M AC1 and a small excess of alkyl bromide (1.2 equivalents relative to AC1) at 25 °C, which was considered the standard condition.Subsequently, the esterification of AC1 with various alkyl bromides was examined (Fig. 3a). The conversions of AC1 to the desired products 1 at predetermined time points were obtained from the 1H NMR spectra of the reaction mixtures (Fig. S6†). Fig. 3b reveals the existence of two types of FGs with different reactivities toward the carboxylate. The formation of 1d–h proceeded faster than that of the other compounds, leading to higher conversions. The FGs of 1d–h include α-carbonyl, allyl, and benzyl structures. The corresponding alkyl bromides typically exhibit high reactivity as electrophiles in SN2 reactions because the resonance effect of π-conjugation stabilizes the transition state.26 Functionalized monomers 1d–g were isolated, and some were further used for ROP, as described below (Fig. S7–S10†). In contrast, the formation of 1a, 1c, 1i, and 1j required longer reaction times (Fig. 3b). These products possess linear alkyl and methoxy alkyl structures as FGs. Moreover, the conversions of ether-functionalized cyclic carbonates 1a, 1c, and 1j were below 50%. These results imply that the esterification of AC1 is practically useful when highly reactive electrophiles with α-carbonyl, allyl, and benzyl FGs are employed. The transformation of AC1 into 1b, 1k, or 1l was not observed under the standard reaction condition in this study (Fig. 3b). The alkyl bromides with branched and heteroalicyclic structures were less reactive than the other forms, which was likely due to steric effects.26 Overall, the successful esterification of AC1 enabled a two-step synthesis of functionalized cyclic carbonates 1 from bis-MPA.
Cyclization using N,N′-carbonyldiimidazole, as developed by Tan et al. is beneficial for avoiding the use of hazardous phosgene derivatives. However, the successful formation of MTC-H requires a large excess of acetic acid (4–16 equivalents) upon heating, which may be detrimental to the ion-exchange work-up step.20 Recent progress in microflow synthesis may alleviate concerns about the use of hazardous chemicals including phosgene derivatives while exploiting the intense reactivities.27 Advantageously, our approach requires neither heating nor large excess reagents and circumvents cumbersome work-up procedures.
Esterification of bis-MPA organic salt
The esterification of AC1 using alkyl bromides via the SN2 mechanism restricts the FGs of 1. Hence, this chemistry was applied to the TEAH+ salt of bis-MPA AC2 to form bis-MPA esters 3 (Scheme 2). The formation of 3 has been implemented by the esterification of the potassium salt of bis-MPA and alkyl bromides.17,28 This approach requires heating the reaction mixture to approximately 100 °C and prolonged vacuum drying because dimethylformamide was used for the dissolution of the bis-MPA potassium salt. In addition, a trace amount of potassium might affect the polymerizability of the resultant ether-functionalized carbonate monomers, such as 1a, 1b, 1c, and 1j (Fig. 3a). The higher solubility of AC2 enabled the use of more volatile solvents as reaction media (Table S1†). We examined the reaction of AC2 with several alkyl bromides (Scheme 2). Similar to the esterification of AC1, acetonitrile was used as a reaction medium. Owing to the absence of side reactions such as the ring-opening reaction of AC1, the esterification process was conducted at 60 °C. Although the reactivity trend of alkyl bromides was similar to that observed during the esterification of AC1, most conversions of AC2 to corresponding esters 3 were approximately 80%, except for 3k (∼63% conversion). The seven resultant bis-MPA esters were successfully isolated (Fig. S11–S17†). These high conversions were due to the reaction temperature being higher than that for the esterification of AC1 (60 vs. 25 °C). The low isolated yields of 3a and 3c (Scheme 2) were attributed to loss during the isolation step, as some fractions of these compounds were trapped in the water phase during the extraction process. Nevertheless, the efficiency of the esterification of AC2 was comparable to that of the bis-MPA potassium salt, with the additional benefit of using low-boiling-point solvents. Accordingly, the reactions of AC2 were performed at 60 °C, making AC2 advantageous over the potassium salt, which requires a higher reaction temperature (∼100 °C).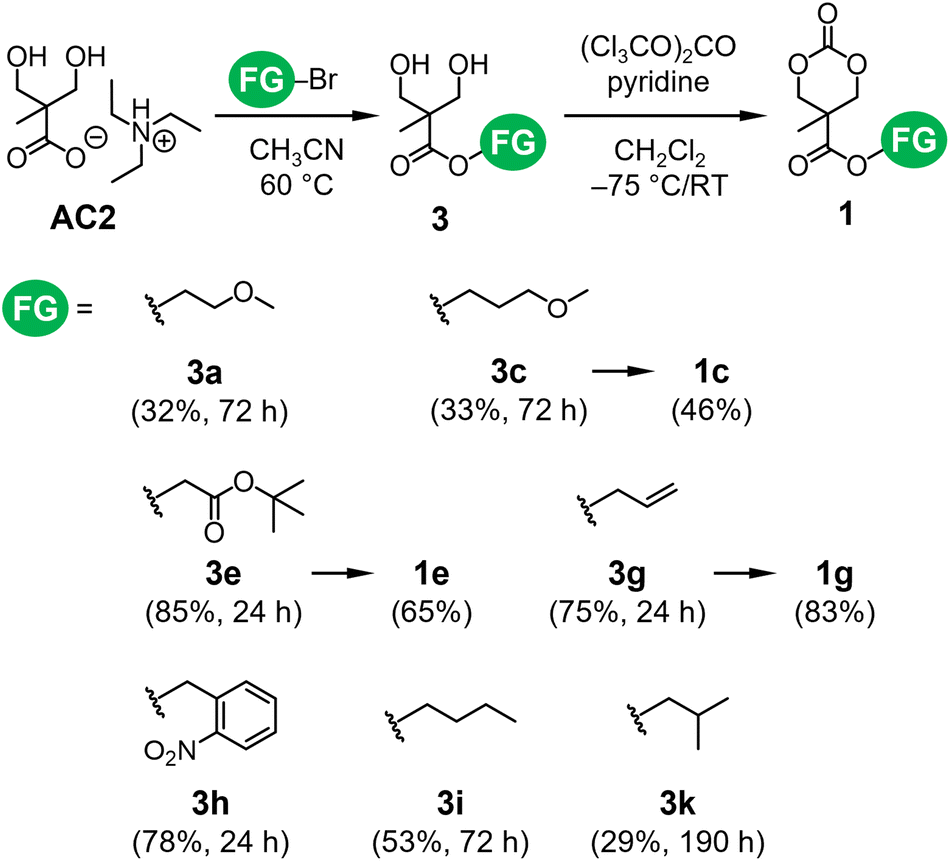 | ||
| Scheme 2 Esterification of AC2 with different alkyl bromides and the subsequent cyclization of 3 to form 1. The values in parentheses denote the product yields and reaction times. | ||
Polymerization of functionalized cyclic carbonates prepared using carboxylate organic salts
In this section, we verified the applicability of selected functionalized cyclic carbonates 1 as monomers for ROP (Scheme 3). Monomer 1b was difficult to prepare through the esterification of AC1 and AC2. Thus, 1b was derived from MTC-H obtained through the protonation of AC1 by ion exchange resins using the previously established method15,17 (route A in Scheme 1; Fig. S18 and S19†). Monomers 1d and 1g were isolated and purified after the reaction of AC1 with alkyl bromides (route B in Scheme 1). Monomers 1c, 1e, and 1g were synthesized from corresponding bis-MPA esters 3 (route C in Scheme 1; Fig. S20–S23†). All ROPs were conducted in CH2Cl2 using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and a thiourea (TU) cocatalyst29,30 to form corresponding polycarbonates 2 (Fig. S24–S28†). Table 1 summarizes the degrees of polymerization (DPs) and molecular weights of resultant functionalized aliphatic polycarbonates 2. These results show that the utilized pathways and reaction intermediates do not affect the polymerizability of functionalized cyclic carbonates 1. Most of the ROP results in this study were not ideal, suggesting insufficient monomer purity, as small unidentified peaks were observed in their NMR spectra (ESI†). As reported previously,7–9 these types of monomers often contain trace amounts of water that can serve as initiators and are difficult to remove completely by column chromatography. In addition, the impurities may include decarbonylated or unreacted diol, which can also serve as initiators. Thus, polymers 2b, 2c, and 2g-i were formed without initiators (Table 1). The size-exclusion chromatography (SEC) traces of the polymers prepared with an alcohol initiator (2d, 2e, and 2g-ii) were somewhat broader than those of other products (Fig. S29†), suggesting the concomitant formation of polymer chains initiated by the impurities and moisture.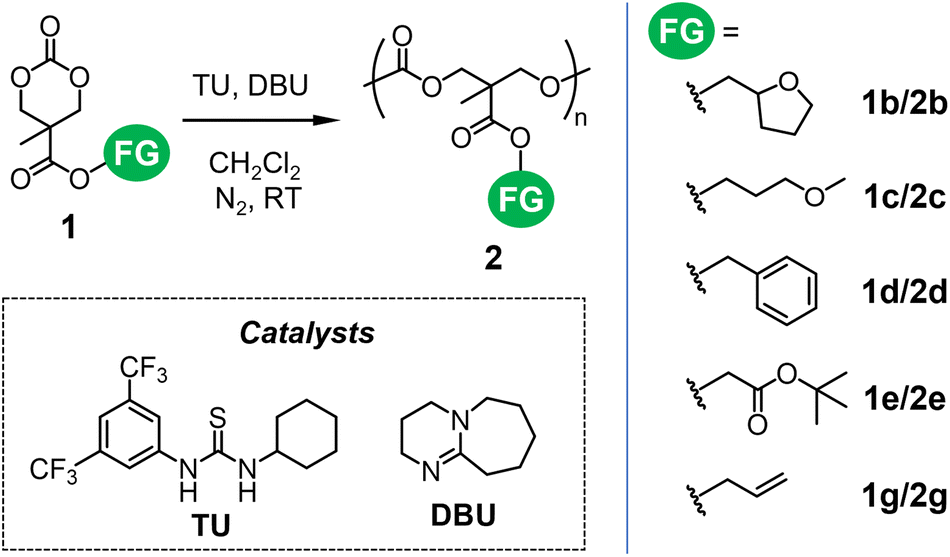 | ||
| Scheme 3 Organocatalytic ROP of functionalized cyclic carbonates 1. | ||
a realized via different routes
| Polymer | Monomer/precursor | [1]/[I]/[TU]/[DBU] | DPb |
M
nc (kg mol−1) |
Đ
Mc |
|---|---|---|---|---|---|
| a [M] = 0.6–1.4 M in CH2Cl2. b Degree of polymerization determined by 1H NMR. c Determined by SEC (THF, 40 °C) with calibration using polystyrene standards. d 1-Pyrenebutanol was used. e Benzyl alcohol was used. | |||||
| 2b | 1b/MTC-H | 100/0/5/5 | 32 | 11 | 1.12 |
| 2c | 1c/3c | 100/0/2/2 | 34 | 14 | 1.09 |
| 2d | 1d/AC1 | 100/2d/5/5 | 57 | 14 | 1.25 |
| 2e | 1e/3e | 100/1e/5/5 | 32 | 10 | 1.26 |
| 2g-i | 1g/3g | 100/0/2/2 | 107 | 24 | 1.12 |
| 2g-ii | 1g/AC1 | 100/2d/5/5 | 40 | 13 | 1.56 |
Evaluation of biocompatibility
Ether-functionalized aliphatic polycarbonates 2b and 2c were obtained with reasonable molecular weights of 11 and 14 kg mol−1, respectively (Table 1), and were not water soluble. Thus, the blood compatibility of 2b and 2c was evaluated by conducting platelet adhesion tests. For this purpose, the polymers were dissolved in chloroform at a concentration of 0.2 wt/vol% and then spin-coated on poly(ethylene terephthalate) (PET) sheets with diameters of 14 mm. A human platelet suspension (4 × 107 cells per cm2) was placed onto the polymer-coated substrates, which were subsequently incubated at 37 °C for 1 h. The numbers and shapes of platelets adhered to the polymer were evaluated by scanning electron microscopy (SEM; Fig. S30†). PET and PTMC were used as positive controls, and 2a, a previously developed blood-compatible aliphatic polycarbonate,8 was used as a negative control. Newly developed polycarbonates 2b and 2c exhibited low numbers of adherent platelets, which were comparable to that for 2a (Fig. 4). Fewer dendritic and spinal-shaped platelets were observed in the SEM images of 2b and 2c than in those of the PET and PTMC surfaces (Fig. S30†), indicating that the adherent platelets on 2b and 2c were not activated. Although the difference was not statistically significant, the number of adhered platelets on 2b was higher than that on 2a (Fig. 4a). This trend was similar to the tendency observed for polyacrylate analogs with the same side chains.31 Kobayashi et al. previously reported that poly(3-methoxypropyl acrylate) exhibited higher blood compatibility than PMEA.32 The antiplatelet adhesion properties of aliphatic polycarbonate analog 2c were comparable to those of 2a (Fig. 4b). However, as compared to PMEA, which is a medically-endorsed antiplatelet polymer, and its analogs, the blood compatibility levels of 2a–2c were not sufficient (Fig. S31†).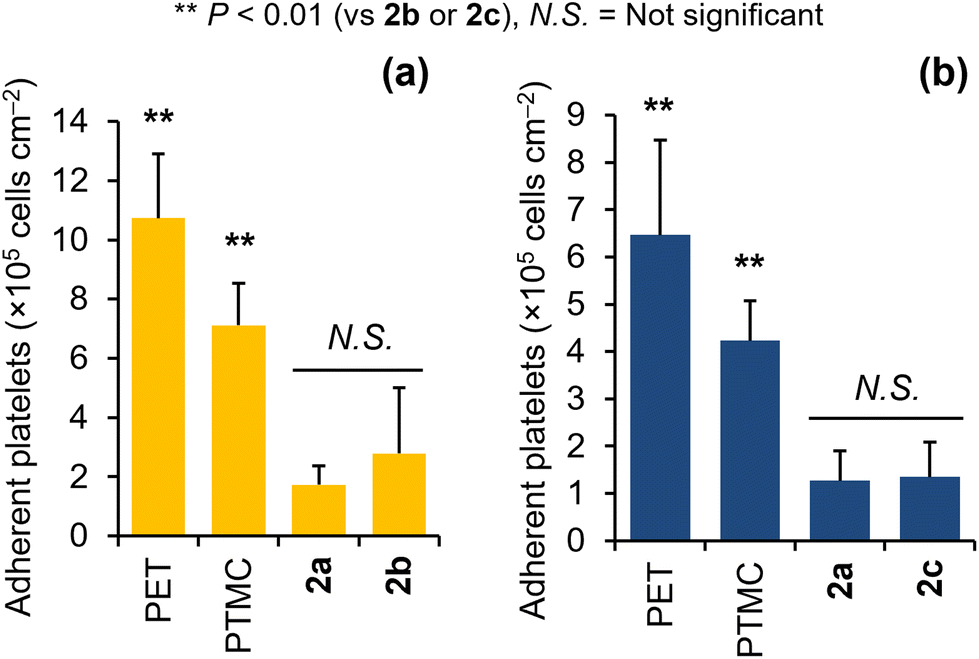 | ||
| Fig. 4 Numbers of adherent platelets on the polymer-coated substrates displayed as mean values with standard deviations. (a) n = 10 (2 substrates × 5 points) per polymer and (b) n = 15 (3 substrates × 5 points) per polymer. Different platelet suspensions were used in the tests for 2b and 2c. | ||
As for the biodegradation of 2a, one would expect the release of 2-methoxyethanol, which is further metabolized into the Substances of Very High Concern (SVHC) compound 2-methoxyacetic acid, causing reproductive malformation.33 Accordingly, the development of alternatives to 2a with comparable antiplatelet adhesion properties is of high significance for researchers investigating biodegradable and biocompatible materials.
Conclusions
In this study, we demonstrated that the ammonium carboxylate organic salts of cyclic carbonate and bis-MPA serve as effective nucleophiles for esterification with alkyl bromides via the SN2 mechanism. The alkyl bromides containing α-carbonyl, allyl, and benzyl structures exhibited high reactivity with the ammonium carboxylate organic salts. The esterification reaction produced functionalized cyclic carbonate monomers with different substituents. Although the efficient two-step synthesis of the functionalized monomers was applicable to limited FGs, esterification via the SN2 mechanism was expanded to the organic salt of bis-MPA. In addition, more diverse FGs could be attached using commercially available modified α-carbonylalkyl and benzyl bromides. The resultant monomers exhibited good polymerizability during oragnocatalyzed ROP, reflecting the absence of any effects of the synthetic pathways on the monomer quality. Moreover, the new ether-functionalized aliphatic polycarbonates possessed excellent blood compatibility. The described synthetic pathways using the organic salts can serve as alternative routes for the facile and efficient synthesis of functionalized cyclic carbonates from bis-MPA. These functionalization methods may also be applicable to aliphatic polycarbonates as sustainable and recyclable materials.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by JSPS KAKENHI (grant numbers JP25870078, JP18K12074, JP19H05715, JP19H05716, and JP19H05720), JST-Mirai Program (grant number JPMJMI21EH), JST COI (grant number JPMJCE1312), Izumi Science and Technology Foundation (grant number H24-J-129), and Iketani Science and Technology Foundation (grant number 0301087-A). We would like to thank Editage for English language editing.Notes and references
- W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS PubMed.
- K. Saito, C. Jehanno, L. Meabe, J. L. Olmedo, D. Mecerreyes, K. Fukushima and H. Sardon, J. Mater. Chem. A, 2020, 8, 13921–13926 RSC.
- R. Miyake, A. Maehara, N. Chanthaset and H. Ajiro, ChemistrySelect, 2022, 7, e202104326 CrossRef CAS.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736–1740 CrossRef CAS.
- K. Fukushima, Biomater. Sci., 2016, 4, 9–24 RSC.
- K. Fukushima, ROP of Cyclic Carbonates, in Organic Catalysis for Polymerisation, ed. A. P. Dove, H. Sardon and S. Naumann, Royal Society of Chemistry, Cambridge, 2019, ch. 7, pp. 274–327 Search PubMed.
- V. Montagna, J. Takahashi, M.-Y. Tsai, T. Ota, N. Zivic, S. Kawaguchi, T. Kato, M. Tanaka, H. Sardon and K. Fukushima, ACS Biomater. Sci. Eng., 2021, 7, 472–481 CrossRef CAS PubMed.
- K. Fukushima, Y. Inoue, Y. Haga, T. Ota, K. Honda, C. Sato and M. Tanaka, Biomacromolecules, 2017, 18, 3834–3843 CrossRef CAS PubMed.
- K. Fukushima, K. Honda, Y. Inoue and M. Tanaka, Eur. Polym. J., 2017, 95, 728–736 CrossRef CAS.
- X. Liu, L. Yuan, D. Li, Z. Tang, Y. Wang, G. Chen, H. Chen and J. L. Brash, J. Mater. Chem. B, 2014, 2, 5718–5738 RSC.
- E. Zhang, B. Song, Y. Shi, H. Zhu, X. Han, H. Du, C. Yang and Z. Cao, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32046–32055 CrossRef CAS PubMed.
- K. Ishihara, J. Biomed. Mater. Res., Part A, 2019, 107, 933–943 CrossRef CAS PubMed.
- M. Tanaka, T. Motomura, M. Kawada, T. Anzai, Y. Kasori, T. Shiroya, K. Shimura, M. Onishi and A. Mochizuki, Biomaterials, 2000, 21, 1471–1481 CrossRef CAS PubMed.
- W. Lee, S.-H. Jeong, Y.-W. Lim, H. Lee, J. Kang, H. Lee, I. Lee, H.-S. Han, S. Kobayashi, M. Tanaka and B.-S. Bae, Sci. Adv., 2021, 7, eabi6290 CrossRef CAS PubMed.
- K. Fukushima, R. C. Pratt, F. Nederberg, J. P. K. Tan, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2008, 9, 3051–3056 CrossRef CAS PubMed.
- T. F. Al-Azemi and K. S. Bisht, Polymer, 2002, 43, 2161–2167 CrossRef CAS.
- R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS PubMed.
- M. Tanaka, K. Fukushima, C. Sato, A. Sasaki and Y. Inoue, Yamagata University, WO2014133102A1, 2014.
- E. W. P. Tan, J. L. Hedrick, P. L. Arrechea, T. Erdmann, V. Kiyek, S. Lottier, Y. Y. Yang and N. H. Park, Macromolecules, 2021, 54, 1767–1774 CrossRef CAS.
- O. Coulembier, S. Moins and P. Dubois, Macromolecules, 2011, 44, 7493–7498 CrossRef CAS.
- K. Koga, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4351–4355 CrossRef CAS.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- A. Kimura, S. Kawauchi, T. Yamamoto and Y. Tezuka, Org. Biomol. Chem., 2014, 12, 6717–6724 RSC.
- F. Cardellini, L. Brinchi, R. Germani and M. Tiecco, Synth. Commun., 2014, 44, 3248–3256 CrossRef CAS.
- P. R. Rablen, B. D. McLarney, B. J. Karlow and J. E. Schneider, J. Org. Chem., 2014, 79, 867–879 CrossRef CAS PubMed.
- N. Sugisawa, Y. Otake, H. Nakamura and S. Fuse, Chem. – Asian J., 2020, 15, 79–84 CrossRef CAS PubMed.
- S. H. Kim, J. P. K. Tan, K. Fukushima, F. Nederberg, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomaterials, 2011, 32, 5505–5514 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- K. Fukushima and K. Nozaki, Macromolecules, 2020, 53, 5018–5022 CrossRef CAS.
- A. Mochizuki, T. Hatakeyama, Y. Tomono and M. Tanaka, J. Biomater. Sci., Polym. Ed., 2009, 20, 591–603 CrossRef CAS PubMed.
- S. Kobayashi, M. Wakui, Y. Iwata and M. Tanaka, Biomacromolecules, 2017, 18, 4214–4223 CrossRef CAS PubMed.
- M. J. Brabec, Encyclopedia of Toxicology, 2nd edn, 2005, pp. 63–66 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00705c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |