
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and self-assembly of cytidine- and guanosine-based copolymers†
Sany
Chea
ab,
Kristin
Schade
a,
Stefan
Reinicke
 a,
Regina
Bleul
c and
Ruben R.
Rosencrantz
*a
a,
Regina
Bleul
c and
Ruben R.
Rosencrantz
*a
aFraunhofer Institute for Applied Polymer Research IAP, Biofunctionalized Materials and (Glyco)Biotechnology, Geiselbergstr. 69, 14476 Potsdam, Germany. E-mail: ruben.rosencrantz@iap.fraunhofer.de
bUniversity of Potsdam, Chair of Polymer Materials and Polymer Technologies, Institute of Chemistry, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany
cFraunhofer Institute for Microengineering and Microsystems IMM, Nanomaterials for Cancer Therapy, Carl-Zeiss-Str. 18-20, 55129 Mainz, Germany
First published on 11th August 2022
Abstract
The base pairing property and the “melting” behavior of oligonucleotides can take advantage to develop new smart thermoresponsive and programmable materials. Complementary cytidine- (C) and guanosine- (G) based monomers were blockcopolymerized using RAFT polymerization technique with poly-(N-(2-hydroxypropyl) methacrylamide) (pHPMA) as the hydrophilic macro chain transfer agent (macro-CTA). C–C, G–G and C–G hydrogen bond interactions of blockcopolymers with respectively C and G moieties have been investigated using SEM, DLS and UV-Vis. Mixing and heating both complementary copolymers resulted in reforming new aggregates. Due to the ribose moiety of the isolated nucleoside-bearing blockcopolymers, the polarity is increased for better solubility. Self-assembly investigations of these bioinspired compounds are the crucial basis for the development of potential future drug delivery systems.
Introduction
The integration of hydrogen bond interactions in polymeric materials results in supramolecular programmable, stimuli responsive architectures with captivating optoelectronic,1,2 mechanical,3,4 self-assembled5–7 and thermal properties,6,8 which is inspired by nature. These noncovalent interactions are crucial in biological systems, for example for stability reasons of secondary, tertiary and quaternary structures of proteins or for the molecular self-assembly of nucleic acids based on the complementary base pairing property. Regarding to Watson–Crick base pairing, the nucleobases adenine (A) and thymine (T) (or uracil in RNA) as well as guanine (G) and cytosine (C) interact. While the interaction of A and T involves 2 hydrogen bonds, G and C interact by 3 hydrogen bonds, which results in a stronger interaction of G–C compared to A–T.9,10 The versatile properties of nucleic acids like DNA or RNA have motivated the synthesis of nucleobase-bearing compounds.7,10,11 DNA/RNA-like polymers result in controlled self-assembled structures with attractive properties like a thermoresponsive, DNA-like melting behaviour.12,13Various nucleobase-containing polymers were prepared by different polymerization methods.7,11,14 For “melting” behaviour investigations, A- and T-functionalized copolymers were prepared using free radical polymerization technique.12 Silyl-protected uridine- and adenosine based (PEG-functionalized) copolymers were prepared using atom transfer radical polymerization (ATRP).15–17 In addition, nucleobase monomer derivatives were used for a templated copper-mediated living radical polymerization on solid support, which was mediated by complementary nucleoside interactions.16,18 However, the ability to coordinate with metal ions might affect ATRP polymerization kinetics of nucleobases. Cu(I), which is involved in ATRP, coordinates purine and pyrimidine derivatives.19 Reversible addition–fragmentation chain transfer (RAFT) mediated polymerization might be a preferable method to isolate nucleobase-containing polymers.11,20 RAFT polymerization enables the synthesis of synthetic polymers with a defined molecular weight, low molar mass dispersity (PDI) and an opportunity for chain growth. It is allows to polymerize a broad spectrum of monomers with high conversions.21–23 In addition, this technique has a high tolerance regarding implementation and is inexpensive compared to competitive methods. In terms of a RAFT-mediated synthesis of nucleobase containing polymers, the choice of the polymerization solvent is significant as it influences the morphology of the polymers.24,25 While syntheses of A- and T-containing polymer architectures have already been described successfully, the synthesis of G-based molecules remains more challenging due to the lower solubility.10
To increase the solubility of nucleobase functionalized derivatives, an extension with water soluble polymer chains is possible. Polyethylene glycol (PEG) is the gold standard when it comes to drug delivery systems. Even though PEG has many advantages like low toxicity, biocompatibility and hydrophilicity, it has its limits when it comes to biodegradability or immunogenicity.26,27 The use of poly-(N-(2-hydroxypropyl) methacrylamide) (pHPMA) as an alternative to PEG has grown interest in recent years.28 pHPMA is a linear, biocompatible and non-immunogenic polymer, which accomplished clinical trials in the past. The predominant application of pHPMA includes the use as potential anticancer therapeutics.29
In this work, we describe the synthesis and characterization of a new class of ribonucleoside-bearing block copolymers. Therefore, methacrylamide-based monomers with cytidine and guanosine moieties (Fig. 1) were synthesized by a two-step synthesis. RAFT-mediated polymerization was applied to isolate blockcopolymers using pHPMA as the macro chain transfer agent (macro-CTA) to increase the hydrophilicity and therefore the solubility. The nucleoside-based blockcopolymers were further investigated in their base-pairing interactions and self-assembly behavior (Scheme 1) by SEM, DLS and UV-Vis.
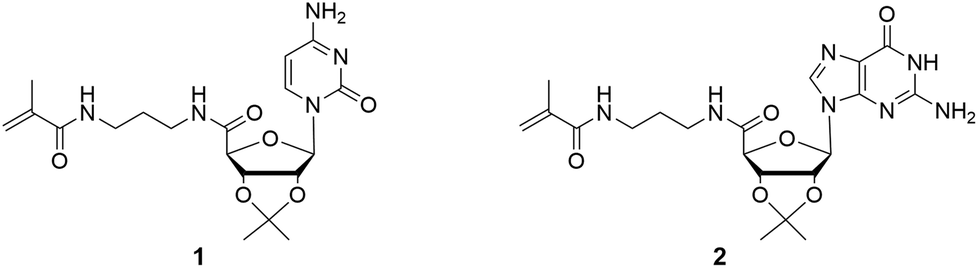 | ||
| Fig. 1 Chemical structures of ribonucleoside methacrylamides, including cytidine (1) and guanosine (2) derivatives. | ||
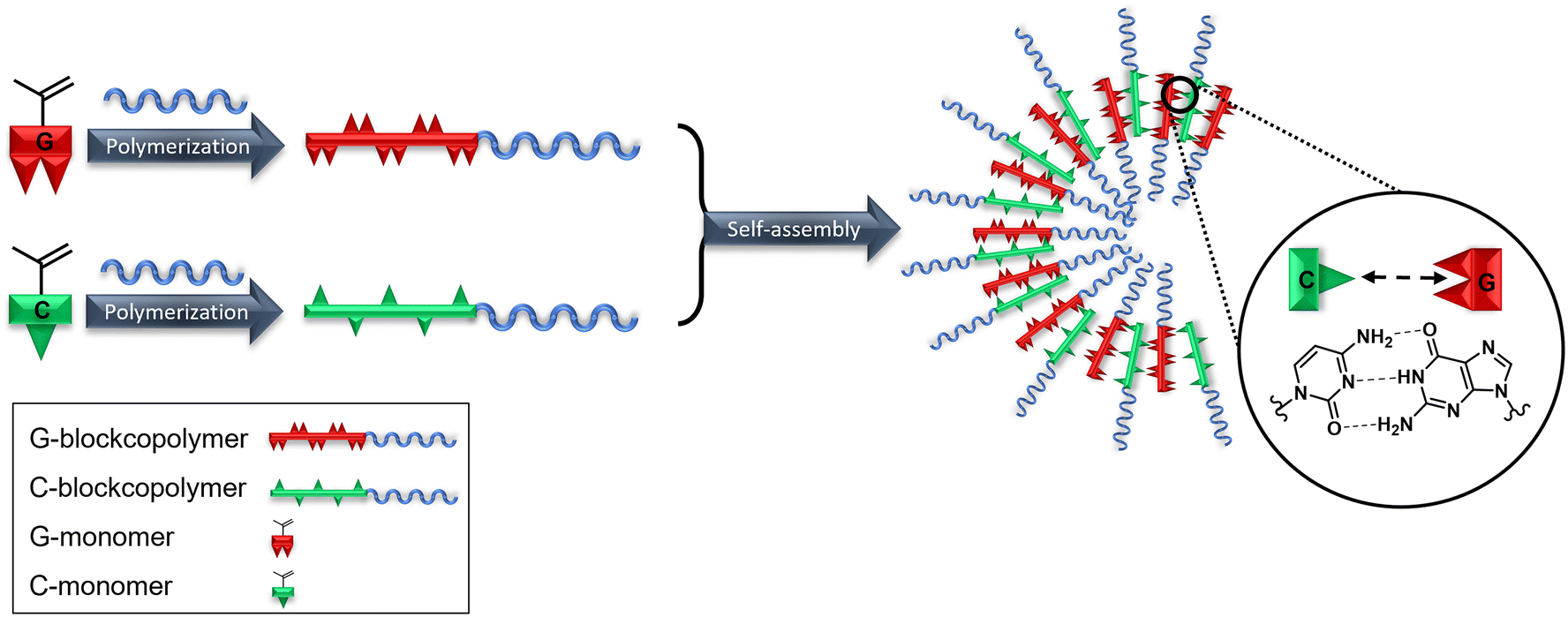 | ||
| Scheme 1 Polymerization and self-assembly of complementary nucleoside (C and G) blockcopolymers. | ||
Results and discussion
Monomer synthesis
The nucleosides were used as the protected 2′,3′-acetonide forms to address the 5′-position. Due to stability reasons, methacrylamide functionalized ribonucleosides were preferred compared to methacrylate derivatives, which can be synthesized enzymatically.16,18 The synthesis of the cytidine- (1) and guanosine-based monomers (2) were prepared via a two-step procedure, including the oxidation of the primary hydroxyl group, followed by an amide coupling with N-(3-aminopropyl)-methacrylamide (Scheme 2). Despite the higher nucleophilicity of the exocyclic –NH2, a protection step of this functionality was not required, unlike the enzymatic esterification route.18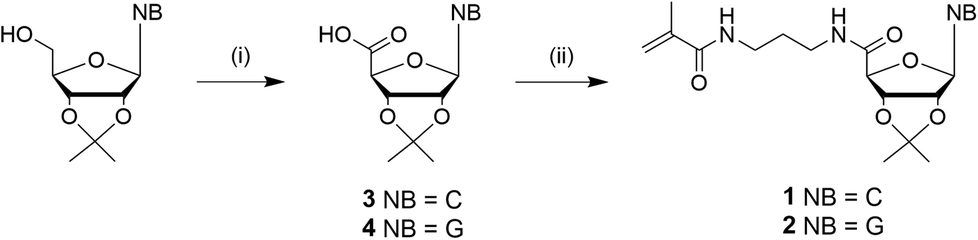 | ||
| Scheme 2 Synthesis of nucleobase (NB) monomer derivatives: (i) TEMPO, BAIB, CH3CN/H2O, rt, overnight (3: 44%, 4: 98%); (ii) APMA*HCl, CDMT, NMM, MeOH, rt, overnight (1: 44%, 2: 52%). | ||
The oxidation of commercially available 2′,3′-isopropylidene cytidine and guanosine to the carboxylic derivatives has been described previously.30 Shortly, the acetal protected ribonucleosides were oxidized with TEMPO and BAIB in the presence of NaHCO3. After filtration of the precipitate, oxidized cytidine (3) was afforded in a yield of 44%, while the yield of oxidized guanosine (4) was quantitative as a white powder. 1 and 2 were obtained after the amide coupling of the 3 and 4 with N-(3-aminopropyl)-methacrylamide hydrochloride using 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) and N-methylmorpholine (NMM) with a yield of 44% and 52% respectively. The chemical structure was confirmed by NMR spectroscopy and ESI-MS analyses. Attempts using DCC, EDC and HATU as coupling agents resulted in lower yields. Coupling of 2-aminoethyl methacrylate hydrochloride instead of N-(3-aminopropyl)-methacrylamide hydrochloride with the stated coupling reagents resulted in an isolation of the methacrylate pendant with undefined byproducts resulting in a significantly lower yield. 2 exhibit a lower solubility compared to 1, but both ribonucleoside methcrylamide-based monomers showed appropriate solubility in non-polar solvents like chloroform and diethyl ether as well in polar solvents like dichloromethane, acetone and dimethylformamide as aprotic solvents and water, methanol and ethanol as protic solvents. This solubility property can be explained by both the formation of hydrogen bond interactions and the hydrophobic parts in one molecule simultaneously. Due to the high solubility of both monomer molecules, the nucleoside monomers were refrained from further deprotection for polymerizations.
As the hydrophilic part of the desired blockcopolymer, poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA) was chosen due to its biocompatibility. The synthesis of N-(2-hydroxypropyl)methacrylamide (HPMA) was described before.31 The HPMA structure was confirmed by 1H NMR spectroscopy analysis after isolation following the published protocol.
Polymerization
RAFT-mediated polymerization ranks among the crucial and well-known polymerization techniques, which involves a radical initiator and the chain transfer agent (CTA). As the selected CTA influences the polymerization efficiency, the choice needs to be done carefully. CTA's consist of a stabilizing Z and a leaving R group.32The dithioester-based CTA 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB) was selected, as it is described for polymerization of methacrylamide-based monomers. After polymerization using RAFT technique, a macro-CTA with the derived CTA end groups was achieved. This macro-CTA can form block copolymers by reacting further with other monomers. Dithiobenzoate (Z-group) as the end-group was confirmed by 1H NMR and UV-Vis analysis.
1 and 2 were homopolymerized using RAFT polymerization with ACVA as the thermal initiator at 75 °C (Fig. 2). Polymerizations of nucleoside-based monomers were conducted with a [M0]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA0]:[I0] ratio of 75:3:1. Solvent mixtures observations of 8:2 DMF/H2O and 9:1 1,4-dioxane/H2O showed different effects depending on the nucleoside type. The choice of solvent mixtures was respectively related to previously described (co-)polymerisations of nucleobase analogues and pHPMA.18,24,25,33–36 Using a 9:1 1,4-dioxane/H2O mixture gave a high conversion (94%) of the G-based polymer (piGPMA), while the conversion of cytidine-based polymer (piCPMA) was lowered to 34% with the same solvent mixture. On the other hand, the conversion of piCPMA was increased to 40% and of the piGPMA was decreased to only 70% in 8:2 DMF/H2O. Polymerizations of nucleoside homopolymers and their monomer conversions were determined by comparing the integrals of the typical C-4 protons of piCPMA (δ 4.43 ppm) and piGPMA (δ 4.50 ppm) with the integrals of the monomer vinyl peaks of iCPMA (δ 5.64 ppm and 5.30 ppm) and iGPMA (δ 6.39 ppm and 5.61 ppm). The theoretical molecular weights (Mn, theory, NMR) were calculated following eqn (2, see ESI†) based on the resulted conversion and are summarized in Table 1.
:[CTA0]:[I0] ratio of 75:3:1. Solvent mixtures observations of 8:2 DMF/H2O and 9:1 1,4-dioxane/H2O showed different effects depending on the nucleoside type. The choice of solvent mixtures was respectively related to previously described (co-)polymerisations of nucleobase analogues and pHPMA.18,24,25,33–36 Using a 9:1 1,4-dioxane/H2O mixture gave a high conversion (94%) of the G-based polymer (piGPMA), while the conversion of cytidine-based polymer (piCPMA) was lowered to 34% with the same solvent mixture. On the other hand, the conversion of piCPMA was increased to 40% and of the piGPMA was decreased to only 70% in 8:2 DMF/H2O. Polymerizations of nucleoside homopolymers and their monomer conversions were determined by comparing the integrals of the typical C-4 protons of piCPMA (δ 4.43 ppm) and piGPMA (δ 4.50 ppm) with the integrals of the monomer vinyl peaks of iCPMA (δ 5.64 ppm and 5.30 ppm) and iGPMA (δ 6.39 ppm and 5.61 ppm). The theoretical molecular weights (Mn, theory, NMR) were calculated following eqn (2, see ESI†) based on the resulted conversion and are summarized in Table 1.
 | ||
| Fig. 2 Chemical structures of ribonucleoside-based homopolymers piCPMA 5 and piGPMA 6. | ||
The presence of the nucleobases might be responsible for the long polymerization time, as the basic aromatic rings (purine and pyrimidine) can act as radical scavengers. The acetonide protecting groups of homopolymers of both ribonucleosides were removed under acidic conditions with trifluoroacetic acid to improve the hydrophilicity due to demanding solubility properties. The deprotection steps were observed by 1H NMR spectroscopy. The reduction of the two shielded singlets of piCPMA (δ 1.47 ppm and 1.29 ppm) and piGPMA (δ 1.49 ppm and 1.31 ppm) indicated a successful removal of the acetonide functional groups. Even after increasing the hydrophilicity by deprotection, the homopolymers 7 and 8 still exhibited low solubility, so the synthesis of the hydrophilic pHPMA 9 as the macromolecular chain transfer agent (macro-CTA) for further copolymerization with nucleoside monomers was decided (Scheme 3).
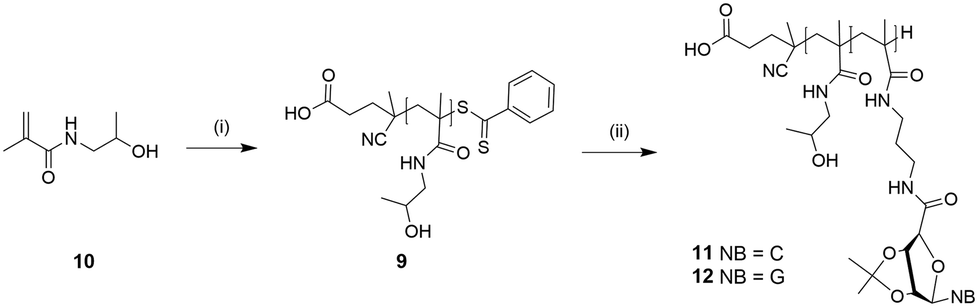 | ||
| Scheme 3 Synthesis of pHPMA 9 and nucleoside-based blockcopolymers pHPMA-b-piCPMA 11 and pHPMA-b-piGPMA 12: (i) ACVA, acetate buffer (pH 5)/EtOH, 70 °C, 24 h; (ii) ACVA, DMF/H2O or 1,4-dioxane/H2O, 75 °C, 24 h. | ||
HPMA macroinitiator was prepared using a modified procedure via RAFT-mediated polymerization.32 The monomer concentration was kept low, as the propagation kinetic constant (KP) of hydrophilic monomers influences positively the transition state of propagation step and can be increased by using water as polymerization solvent and using a decreased monomer concentration.37 The structure of pHPMA was confirmed by 1H NMR spectroscopy. Monomer conversion was at 75%, resulting in a theoretical Mn of 7.8 kDa. DP was determined by comparing the integrals of the phenylic peaks (δ 7.93 and 7.81 ppm) of the end-group with the peaks of the pHPMA backbone (δ 4.69 ppm). In addition, UV-Vis analysis confirmed the attachment of the dithiobenzoyl end group and showed a similar DP like 1H NMR spectroscopy DP determination. Theoretical and actually determined Mn do not go together, which means that the RAFT agent did not get completely consumed.
Block copolymers of nucleosides (pHPMA-b-piCPMA 11 and pHPMA-b-piGPMA 12) were prepared using the RAFT-mediated polymerization technique. As the resulting blockcopolymers were analyzed via UV-Vis spectroscopy to evaluate the hydrogen-bonding interactions of the nucleobases, blockcopolymers were synthesized with a low “livingness” rate. “Livingness” is a feature, which allows the chain extension. It implies, how many “living” chains remain intact for further blockcopolymerizations. A low “livingness” results in high quantities with dead ends led to nucleobase-based polymers without the phenylic Z-group, which may interfere in further UV-Vis spectroscopy analysis. The calculated “livingness” rates were kept low and are 36.0% of 11, whereas of 12 is at 15.6%.
Polymerization of both nucleosides were performed in the solvent system, which worked the best for the homopolymers, respectively: 1 in 8:2 DMF/H2O, 2 in 9:1 1,4-dioxane/H2O. Purine-based 2 monomer lead to higher conversion and therefore higher molecular weight in our case, unlike A-based monomers, which were polymerized via ATRP with possibly complexation of Cu(I) affording lower conversion compared to the pyrimidine counterpart.16 The monomer conversion was specified using eqn (1, see ESI†) by comparing the integrals of the monomer peak (1: δ 5.30 ppm; 2: δ 5.61 ppm) with the nucleoside-based polymer peak (b-piCPMA δ 4.37 ppm or b-piGPMA δ 6.14 ppm). The monomer conversion of 11 was 68%, while 12 was at 78%, summarized in Table 2. The lower PDI of polymer 11 compared to macro-CTA 10 is due to the different purification methods. While 10 was purified by dialysis against H2O, 11 was purified by repeated precipitation, which may lead to fractional precipitation. Due to the poor similarity of the standard used with the polymers and the difficult solubility of them, the values of the SEC analysis are to be regarded as inaccurate and therefore not really reliable. They only give an indication of the comparison of the polymers with each other.
Both nucleoside-based blockcopolymers 11 and 12 revealed low solubilities due to the integrated nucleobases. The blockcopolymers were removed by an acidic deprotection of the acetal functionalities with trifluoroacetic acid (Scheme 4). The successful deprotection was confirmed by the disappearance of the two singlets in the upfield resulting from the acetal protecting groups of 11 (δ 1.46 ppm and 1.28 ppm) and 12 (δ 1.48 and 1.33 ppm) in 1H NMR spectroscopy analysis. Agitating for in total 2 h yielded 13 and 14.
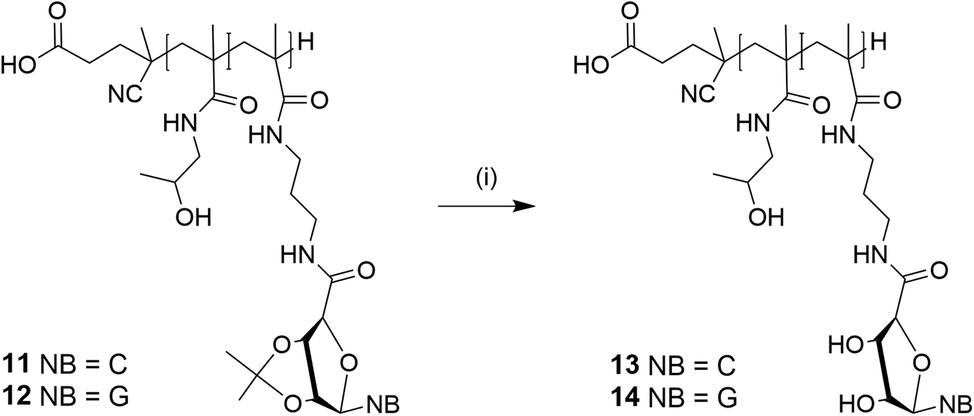 | ||
| Scheme 4 Acidic deprotection of the acetonide functional group of pHPMA-b-piCPMA 11 and pHPMA-b-piGPMA 12: (i) TFA, H2O, rt, 2 h (11: 53%, 12: 81%). | ||
Self-assembly analysis
Due to lower solubilities caused by 3 strong hydrogen binding sites, self-assembly studies of G- and C-based blockcopolymers were less reported compared to A- and T-containing polymers.15,16,20,24,25 To investigate aggregate formation due to hydrogen bonding interactions between the purine and pyrimidine functionalities, SEM and DLS analysis were carried out (Fig. 3). Regarding to SEM images, A- and T-containing polymers showed popcorn-like structures in CHCl3 and 1,4-dioxane,25 whereas our synthesized pHPMA-b-pCPMA 13 polymers form large network structures in aqueous solution. Since a gentle precipitation was already observed in the aqueous solution, the formation of the network structures seen in the SEM image due to cohesion during the drying process is unlikely. These structures result from stronger C–C interactions with a broad size distribution and an average size of 280 nm. This broad size distribution was also detected with DLS with a polydispersity index (PDI) of 0.421. The average hydrodynamic diameter of 13 is around 507 nm. However, SEM images of pHPMA-b-pGPMA 14 revealed small particles due to G–G interactions with an average size of 86 nm. The size distribution was smaller according to DLS with an average hydrodynamic diameter of around 144 nm (PDI = 0.213). Mixing both complementary blockcopolymers results in particle with an average hydrodynamic diameter of around 165 nm and a PDI of 0.3. Heating up to 100 °C for 30 min and cooling down of this mixture led to a narrower size distribution and a smaller average size of 266 nm and a hydrodynamic diameter of 136 nm. This observation might be explained by breaking the strong C–C and G–G hydrogen bond in increased temperatures and re-assembling of C–G interactions when cooling down to room temperature (rt). Sonication of 13, 14 and the mixture of both lead to no morphology change indicating a strong stability like previously reported nucleobase-containing polymers.25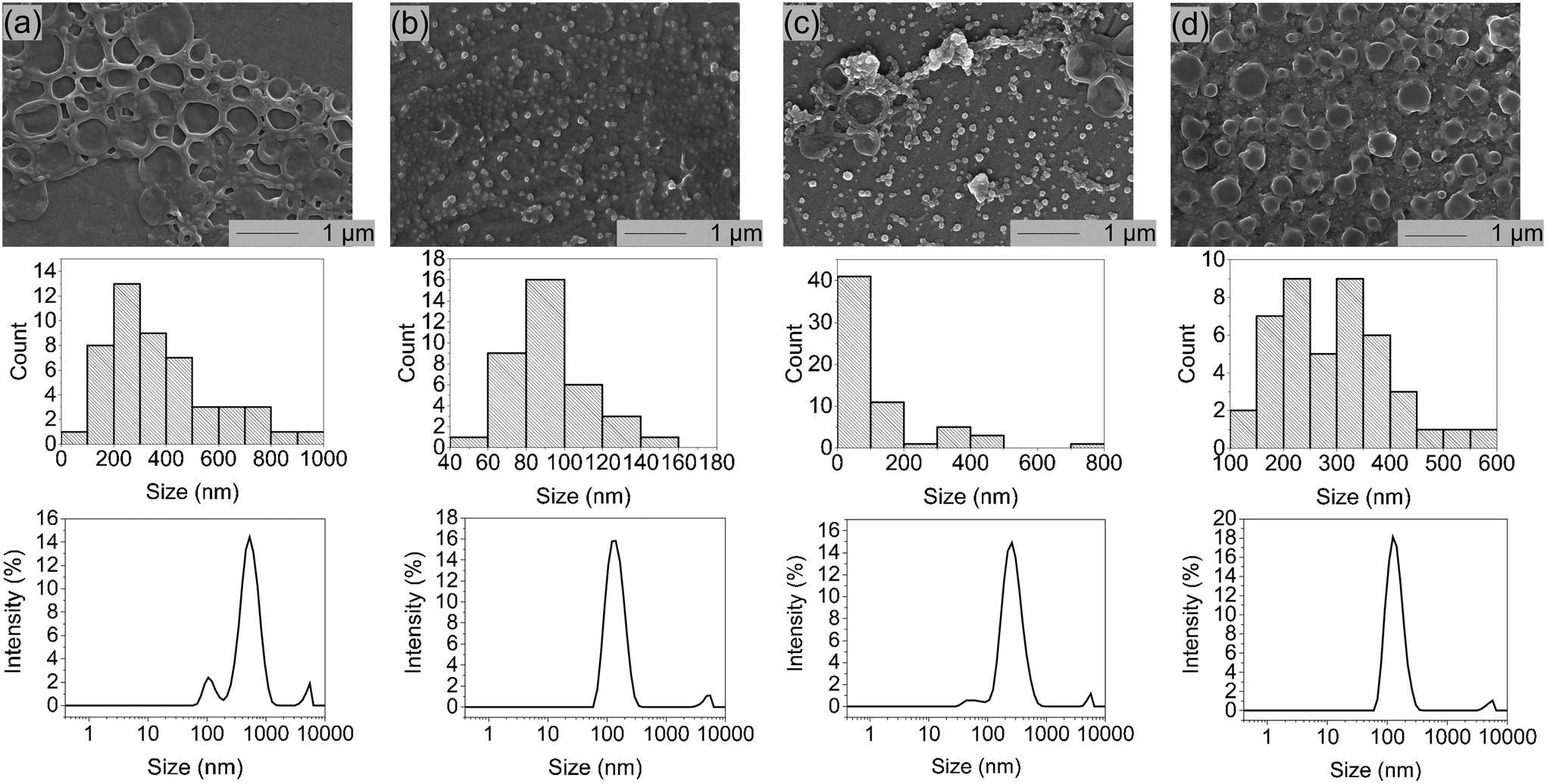 | ||
| Fig. 3 SEM-image, size distribution and hydrodynamic size distribution by DLS of (a) pHPMA-b-pCPMA 13, (b) pHPMA-b-pGPMA 14, (c) mixture of 13 and 14 before heating and (d) mixture of 13 and 14 after heating. | ||
Nucleobases show strong UV absorption due to hydrogen bond and π–π interactions.38 Base-pairing interactions of nucleobase derivatives result in changes in the UV-Vis spectroscopy. To investigate hydrogen bonding interactions of the complementary nucleoside-containing polymers 13 and 14, spectrophotometric measurements were conducted (Fig. 4). The UV absorption spectra of the individual polymers were compared with the spectrum of the mixture after heating. The average values of the individual polymers pHPMA-b-pCPMA 13 and pHPMA-b-pGPMA 14 matches with the absorption values of the mixture of both polymers using same concentrations due to hydrogen bond pre-assembly of the single polymers. After heating the polymer mixture for 30 min at 100 °C, hypochromicity was observed like expected from literature.12 This decrease of absorbance resulted from re-assembly of the complementary C–G interactions after heating and cooling down. Hypochromicity at a wavelength of 260 nm might be an indication for dsDNA-like structures, which show lower absorbance compared to ssDNA. Absorption maxima of both polymers individually at 274 nm for 13 and at 258 nm for 14 were comparable with other previously described amphiphilic blockcopolymers containing T- and A-structures.15,16
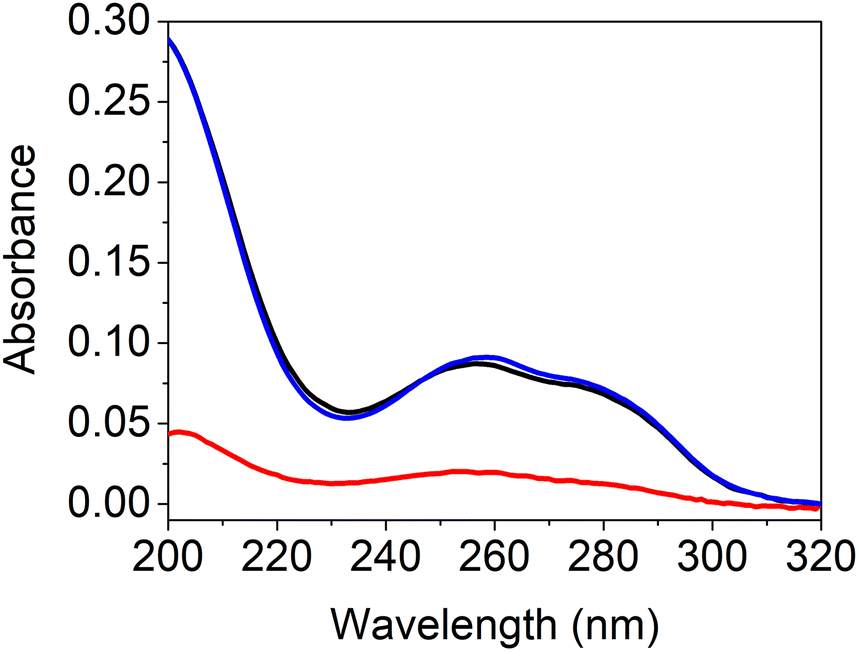 | ||
| Fig. 4 UV-Vis spectrum of the average of individual 13 and 14 (blue), mixture of 13 and 14 before heating (black) and mixture of 13 and 14 after heating (red). | ||
Conclusion
Two complementary nucleoside-based monomers were isolated by a two-step synthesis starting with the oxidation of the primary hydroxyl group which was followed by an amide coupling affording a methacrylamide-based nucleoside monomer. Nucleoside-bearing monomers were homopolymerized using the RAFT polymerization technique. The monomer conversion was depended on the polymerization solvent system. C-based polymer showed a higher monomer conversion using a solvent mixture of DMF/H2O, whereas G-based polymer yielded higher using 1,4-dioxane/H2O. Chain extension with HPMA of both homopolymers were due to high insolubility hampered, even after removal of the protection groups. Therefore, pHPMA was synthesized as the macro-CTA for further polymerizations of both nucleoside-based monomers. Using the RAFT techniques in the solvent system, which works the best for homopolymerization of the nucleosides, two complementary blockcopolymers were isolated with a low solubility. After acidic deprotection, the C–G hydrogen bond interaction between these two blockcopolymers was studied by SEM, DLS and UV-Vis. These analyses revealed strong C–C and G–G interactions within one nucleoside-based polymer type. C-based blockcopolymers aggregate to a network with a broad size distribution, whereas G-based blockcopolymers assemble to smaller particles with a narrower size distribution. Heating the polymer mixtures resulted in breaking these base pairing of the same nucleobase type to form new aggregates by forming C–G interactions after cooling down. Further investigation of complementary blockcopolymers in different sizes and their analysis can lead to a programmable, thermoresponsive material for a targeted drug delivery. Synthesis of well-defined pHPMA as macroCTA under optimised conditions should be considered to keep the PDI low.Experimental
Materials
All reagents and solvents were used without further purification. Na2CO3 (>99.5%), NaHCO3 (>99%), acetic acid (100%), N-methylmorpholine (NMM, >99%) and 1,4-dioxane (>99.8%) were purchased from Carl Roth (Karlsruhe, Germany). Na2SO4 (>99%), NaCl (>99%), dichloromethane (>99.8%), acetone (>99%) and ethanol (EtOH, >99.5%) were received from Chemsolute (Renningen, Germany), sodium acetate (>99%) and trifluoroacetic acid (TFA, 99%) from Acros Organics (Geel, Belgium). Et3N (>99.5%), methacryloyl chloride (97%), 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO, 98%), 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT, 97%), 4-cyano-4-(phenylcarbonothioylthio)-pentanoic acid (CPADB), 4,4′-azobis(4-cyanovaleric acid) (ACVA, >98%) and DMF (>99%) were obtained from Merck KGaA (Darmstadt, Germany). 1-Aminopropan-2-ol (94%) was from Alfa Aesar (Kandel, Germany), 2′,3′-O-isopropylidene cytidine and 2′,3′-O-isopropylidene guanosine from Biosynth (Berkshire, UK). Bis(acetoxy)iodobenzene (BAIB, 97%) and N-(3-aminopropyl)-methacrylamide hydrochloride (APMA, 97%) were purchased from BLD pharm (Shanghai, China), acetonitrile (>99.9%) and THF from VWR (Radnor, US). Deuterated solvents D2O (99.9%) and DMSO-d6 (99.8%) were received from Deutero (Kastellaun, Germany).Characterization techniques
ESI-MS spectra were recorded with a Perkin Elmer Flexar SQ 300 MS Detector. Nuclear magnetic resonance (NMR) spectroscopy was performed with a Bruker AVANCE NEO (400 MHz) spectrometer. Deuterated solvents were used as standards. Chemical shifts are given in the δ-scale in ppm relative to solvent peaks. Multiplicities are displayed with the coupling constants in hertz (Hz).Size exclusion chromatography (SEC) was performed in HPLC grade DMF containing 0.1% LiBr with a flow rate at 1 mL min−1 and calibrated with polystyrene (PS) or poly(methyl methacrylate) (PMMA). Mn,UV–Vis was determined using a Specord 210 spectrophotometer.
Individual polymeric samples (pHPMA-b-pCPMA 13 and pHPMA-b-pGPMA 14) for UV-Vis, dynamic light scattering (DLS), SEM and AFM investigations were prepared by the solvent switch method separately. Blockcopolymers 13 and 14 were dissolved in DMSO with a concentration of 8 mg mL−1. After stirring for 10 min, 7 mL of water was added using a syringe pump with a rate of 1 mL h−1. The solutions were dialyzed against water for 3 days to remove DMSO. Samples have a final concentration of around 1 mg mL−1. Diluted polymer solutions with a final concentration of 10 μg mL−1 were placed in a 10 mm quartz cuvette for UV-Vis and DLS investigations. DLS analysis were conducted with a Malvern Zetasizer Nano ZS, UV-Vis with a Specord 210 spectrophotometer. The SEM images were taken with a GeminiSEM 300 after a drop of the sample had been applied to the sample plate and dried and then sputtered with a 4 nm thick platinum layer. AFM images were acquired using a Bruker Dimension Icon using NanoScope 9.1.
Synthesis of N-(2-hydroxypropyl)methacrylamide (HPMA) 10
1-Aminopropan-2-ol (8.35 mL, 107 mmol) and Na2CO3 (12.6 g, 119 mmol) were added to cold dichloromethane (28 mL). The reaction solution was cooled to −10 °C and freshly distilled methacryloyl chloride (10.6 mL, 110 mmol), diluted in dichloromethane (11 mL) was added dropwise within 35 min. After complete addition, the reaction solution was stirred for additional 20 min at 5 °C and then allowed to warm up to rt. The white precipitate was filtered and washed with dichloromethane (3 × 100 mL). The resulting filtrate was dried over anhydrous Na2SO4, filtered, washed with dichloromethane (3 × 100 mL) and concentrated under vacuo. The concentrate was left in the fridge to crystallize. The resulting crystals were filtrated and washed with cold dichloromethane. After recrystallization in acetone, 10 was isolated as white crystals (9.96 g, 69.6 mmol, 65%); δH (D2O, 400 MHz): 5.72 (1H, t, 4J = 0.8 Hz, H2′), 5.47 (1H, qin, 4J = 1.6 Hz, H2′′), 3.96 (1H, ddt, 3J = 4.8 Hz, 3J = 13.2 Hz, 3J = 6.4 Hz, H4), 3.33 (1H, dd, J = 4.8 Hz, 3J = 13.8 Hz, H3′), 3.25 (1H, dd, J = 6.8 Hz, 3J = 13.8 Hz, H3′′), 1.95 (3H, s, H1), 1.18 (3H, d, 3J = 6.4 Hz, H5) ppm.Synthesis of 2′,3′-O-isopropylidene-5′-carboxynucleosides (iC-COOH 3 and iG-COOH 4)
A solution of 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO) (0.331 g, 2.12 mmol) in acetonitrile (10.1 mL) was added to a reaction solution of 2′,3′-O-isopropylidene nucleoside (10.6 mmol), NaHCO3 (1.78 g, 21.2 mmol) and bis(acetoxy)iodobenzene (BAIB) (6.80 g, 21.2 mmol) in H2O (10.1 mL). The reaction solution was stirred for 1 h at 0 °C and then overnight at rt. The precipitate was filtered, washed with acetone (3 × 50 mL) and diethyl ether (3 × 50 mL) and dried in vacuo to afford oxidized nucleosides.iC-COOH 3: white powder, yield: 44%, δH (D2O, 400 MHz): 8.99 (1H, d, 3J = 7.6 Hz, H5), 6.12 (1H, d, 3J = 7.6 Hz, H6), 5.86 (1H, s, H2), 5.27 (1H, dd, 3J = 2 Hz, 3J = 6 Hz, H3), 5.23 (1H, d, 3J = 6.4 Hz, H2), 4.66 (1H, d, 3J = 2 Hz, H7), 1.59 (3H, s, H1′), 1.43 (3H, s, H1′′) ppm.
iG-COOH 4: white powder, yield: 98%, δH (D2O, 400 MHz): 7.85 (1H, s, H5), 6.15 (1H, s, H4), 5.58 (1H, d, 3J = 5.6 Hz, H2), 5.47 (1H, d, 3J = 5.6 Hz, H3), 4.63 (1H, s, H6), 1.62 (3H, s, H1′), 1.47 (3H, s, H1′′) ppm.
Synthesis of 2′,3′-O-isopropylidene-5′-propylmethacrylamide nucleosides (iCPMA 1 and iGPMA 2)
2′,3′-O-Isopropylidene-5′-carboxynucleoside 3 or 4 (7.27 mmol) was reacted with N-(3-aminopropyl)-methacrylamide hydrochloride (APMA) (1.43 g, 8.02 mmol), 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (1.40 g, 7.98 mmol) and N-methylmorpholine (NMM) (1.76 mL, 16.0 mmol) in methanol (65 mL) overnight at rt. The crude product was purified by a preparative HPLC device (reverse phase C18 silica, gradient of 10% to 20% acetonitrile in H2O) to afford nucleoside monomers.iCPMA 1: white powder, yield: 38%, δH (D2O, 400 MHz): 7.63 (1H, d, 3J = 7.6 Hz, H5), 5.96 (1H, d, 3J = 7.2 Hz, H4), 5.73 (1H, s, H6), 5.68 (1H, s, H11′), 5.44 (1H, s, H11′′), 5.40–5.38 (2H, m, H2, H3), 4.68 (1H, d, 3J = 0.8, H7), 3.29–3.07 (4H, m, H8, H10), 1.92 (3H, s, H12), 1.78–1.61 (2H, m, H9), 1.58 (3H, s, H1′), 1.43 (3H, s, H1′′) ppm; δH (DMSO-d6, 400 MHz): 7.87 (1H, t, 3J = 6 Hz, H5), 7.75–7.73 (2H, m, H9, H13), 7.27 (2H, s, H7), 5.75 (1H, d, 3J = 1.2 Hz, H4), 5.69 (1H, d, 3J = 7.2 Hz, H6), 5.64 (1H, s, H14′), 5.30 (1H, t, J = 1.6 Hz, H14′′), 5.08 (1H, dd, 3J = 2.8 Hz, 3J = 6.4 Hz, H3), 5.02 (1 H, dd, 3J = 1.2 Hz, 3J = 6.2 Hz, H2), 4.35 (1H, d, 3J = 2.8 Hz, H8), 3.17–2.88 (4H, m, H10, H12), 1.85 (3H, s, H15), 1.50 (2H, quin, 3J = 6.8 Hz, H11), 1.47 (3H, s, H1′), 1.29 (3H, s, H1′′) ppm; δC (D2O + DMSO-d6, 100 MHz): 173.58, 158.39, 148.01, 140.75, 122.47, 115.33, 115.31, 99.88, 97.24, 90.11, 85.80, 85.55, 38.39, 37.84, 29.52, 27.23, 25.64, 19.32 ppm; ESI-MS: m/z for C19H27N5O6: [M + H]+ calculated: 422.46, found: 422.25; [M + Na]+ calculated: 444.44, found: 444.24.
iGPMA 2: white powder, yield: 52%, δH (D2O, 400 MHz): 7.89 (1H, s, H5), 6.30 (1H, s, H4), 5.74 (1H, d, 3J = 6.0 Hz, H3), 5.64 (1H, d, J = 0.8 Hz, H10′), 5.53 (1H, d, 3J = 6.0 Hz, H10′′), 5.42 (1H, d, J = 0.7 Hz, H2′′), 3.05–2.83 (4H, m, H7, H9), 1.90 (3H, s, H11), 1.62 (3H, s, H1′), 1.47 (3H, s, H1′′), 1.36 (1H, dp, J = 6.9 Hz, 3J = 13.9 Hz, H8′), 1.21 (1H, dp, J = 7.1 Hz, 3J = 14.2 Hz, 8′′) ppm; δH (DMSO-d6, 400 MHz): 10.57 (1H, bs, H6), 7.82 (1H, s, H5), 7.78 (1H, t, 3J = 6 Hz, H13), 7.54 (1H, t, 3J = 6 Hz, H9), 6.41 (2H, s, H7), 6.14 (1H, d, 3J = 1.6 Hz, H4), 5.61 (1H, s, H14′), 5.43 (1H, dd, 3J = 2.4 Hz, 3J = 6 Hz, H3), 5.29 (1H, t, J = 1.2 Hz, H14′′), 5.25 (1H, dd, 3J = 1.2 Hz, 3J = 6.2 Hz, H2), 4.50 (1H, d, 3J = 2.4 Hz, H8), 2.99–2.78 (4H, m, H10, H12), 1.83 (3H, s, H15), 1.51 (3H, s, H1′), 1.33 (2H, do, J = 6.8 Hz, 3J = 31.4 Hz, H11), 1.33 (3H, s, H1′′) ppm; δC (D2O + DMSO-d6, 100 MHz): 172.27, 171.99, 160.05, 154.54, 151.80, 140.00, 139.73, 121.70, 117.03, 114.78, 90.99, 88.32, 84.44, 84.30, 37.19, 36.98, 28.56, 26.35, 24.89, 18.47 ppm; ESI-MS: m/z for C20H27N7O6: [M + H]+ calculated: 462.49, found: 462.27; [M + Na]+ calculated: 484.47, found: 484.25.
Homopolymerization of nucleoside-based monomers (piCPMA 5 and piGPMA 6)
Cytidine-based monomer 1 (63.0 mg, 150 μmol), CPADB (1.70 mg, 6.00 μmol) and ACVA (0.560 mg, 2.00 μmol; CTA/I molar ratio = 3) were dissolved in a solvent mixture of 8:2 DMF/H2O or 9:1 1,4-dioxane/H2O (437 μL) and purged with N2 for 30 min. The reaction mixture was then placed in a preheated oil bath at 75 °C and reacted for 24 h. The reaction was quenched by exposing to air and cooling to rt. The polymer was isolated purified by repetitive precipitation from cold acetone and dried on high vacuum.
The same procedure was applied to isolate the guanosine-based homopolymers using 2 as the starting material.
piCPMA 5: pinkish powder, monomer conversion: 40% (piCPMADMF) and 34% (piCPMA1,4-dioxane); Mn = 2.1 kDa (piCPMADMF) and 4.1 kDa (piCPMA1,4-dioxane), PDI = 1.3 (SEC-DMF, PS standard); δH (DMSO-d6, 400 MHz): 7.92–7.82 (H5, H9, H13), 5.98 (H7), 5.82 (H4), 5.11 (H6, H8), 4.43 (H2), 3.32–2.99 (H10, H12), 1.47 (H1′), 1.29 (H1′′), 0.94–0.80 (H11) ppm.
piGPMA 6: pinkish powder, monomer conversion: 70% (piGPMADMF) and 94% (piGPMA1,4-dioxane); Mn = 8.4 kDa (piGPMADMF) and 11.4 kDa (piGPMA1,4-dioxane), PDI = 1.3 (SEC-DMF, PS standard); δH (DMSO-d6, 400 MHz): 10.79 (H6), 7.85 (H5), 7.62 (H9, H13, H16), 6.49 (H7), 6.15 (H4), 5.42 (H3), 5.27 (H2), 4.50 (H8), 2.88 (H10, H12), 1.49 (H1′), 1.31 (H1′′), 1.23 (H14), 0.96–0.68 (H11) ppm.
Deprotection of nucleoside-based homopolymers (pCPMA 7 and pGPMA 8)
Cytidine- and guanosine-based homopolymers 5 and 6 were deprotected under acidic conditions, respectively. 5 and 6 (23.4 mg) were stirred for 2 h at rt in H2O (93.6 μL) and trifluoroacetic acid (TFA) (608 μL), followed by reprecipitation into cold THF/Et3N (9:1). Precipitated polymers were centrifuged, washed with THF (3×), acetone (3×) and dichloromethane (3×). Deprotected nucleoside homopolymers were isolated after drying on high vacuum as pale red powders (23.0 mg).
pCPMA 7: yield: 50%; δH (DMSO-d6, 400 MHz): 10.91 (H6), 8.32 (H5), 8.17 (H9, H13), 7.54–7.32 (H16), 6.62 (H7), 5.84 (H4), 4.53 (H2), 4.32 (H3), 4.21 (H8), 3.88 (H1), 2.94 (H10, H12), 1.50 (H14), 0.96–0.66 (H11) ppm.
pGPMA 8: yield: 62%; δH (DMSO-d6, 400 MHz): 8.51 (H5, H6), 8.31 (H9, H13), 7.44 (H16), 6.10 (H7), 5.79 (H4), 4.32 (H2), 4.22 (H3), 4.04 (H8), 3.50 (H1), 2.96 (H10, H12), 1.54 (H14), 0.97–0.80 (H11) ppm.
Polymerization of HPMA (pHPMA 9)
A mixture of HPMA 10 (1.10 g, 7.69 mmol), CPADB (31.0 mg, 110 μmol; target DP = 70), ACVA (10.3 mg, 136.5 μmol; CTA/I molar ratio = 3), ethanol (3 mL) and acetate buffer (7 mL; pH 5) was purged with N2 for 30 min before placing in a preheated oil bath at 70 °C. After 24 h, the reaction was stopped by exposing to air and cooling to rt. Resulting 9 was purified by dialysis against water for 5 days, followed by freeze-drying as a pinkish powder (monomer conversion: 75%, yield: 70%); Mn = 38.7 kDa (by 1H NMR); Mn = 42.8 kDa (by UV-Vis); Mn = 9.1 kDa, PDI = 1.66 (SEC-DMF, PMMA standard); δH (DMSO-d6, 400 MHz): 7.17 (1H, bs, H5), 4.69 (1H, s, H3), 3.67 (1H, s, H1), 2.90 (2H, s, H4), 1.57 (1H, m, H7), 1.02 (3H, s, H6), 0.89 (3H, d, 3J = 64.4 Hz, H2) ppm.Blockcopolymerization of pHPMA-b-piCPMA 11
A mixture of iCPMA 1 (101 mg, 0.240 mmol), pHPMA 9 (40 kDa) (68.5 mg, target DP = 150) and ACVA (0.815 mg, 2.90 μmol, CTA/I molar ratio = 0.55) in 8:2 DMF/H2O (899 μL) was flushed with N2 for 30 min before placing in a preheated oil bath at 75 °C. The reaction mixture was reacted for 24 h and purified by repetitive precipitation from cold acetone, followed by drying under high vacuum. The desired product was yielded as a white powder (monomer conversion: 77%, yield: 76%); δH (DMSO-d6, 400 MHz): 7.74 (H5, H9, H13), 7.18 (H18), 5.75 (H4, H7), 5.10–5.05 (H6, H8), 4.69 (H20), 4.37 (H2), 3.68 (H22), 2.91 (H10, H12, H19), 1.57 (H15, H17), 1.46 (H1′), 1.28 (H1′′), 1.02 (H16), 0.81 (H11, H21) ppm.
Block-copolymerization of pHPMA-b-piGPMA 12
iGPMA 2 (62.0 mg, 0.134 mmol), pHPMA 9 (40 kDa) (11.5 mg, target DP = 150) and ACVA (0.415 mg, 1.50 μmol, CTA/I molar ratio = 0.134) were dissolved in 9:1 1,4-dioxane/H2O (938 μL) and flushed with N2 for 30 min. The reaction mixture was placed in a preheated oil bath at 75 °C and reacted for 24 h. The desired compound was isolated after dialysis against water for 3 days and drying as a white powder (monomer conversion: 77%, yield: 68%); δH (DMSO-d6, 400 MHz): 10.77 (H6), 7.85 (H5), 7.18 (H9, H13, H16), 7.18 (H18), 6.53 (H7), 6.15 (H4), 5.43 (H3), 5.27 (H2), 4.69 (H20), 4.50 (H8), 3.68 (H22), 2.91 (H10, H12, H19), 1.42 (H1′), 1.32 (H1′′), 1.19 (H14), 1.02 (H16), 0.82 (H11, H21) ppm.
Deprotection of pHPMA-b-nucleosides (pHPMA-b-pCPMA 13 and pHPMA-b-pGPMA 14)
pHPMA-b-nucleosides 11 and 12 (20 mg) were deprotected under acidic conditions by agitating with TFA (520 μL) and H2O (80 μL) for 2 h. The reaction mixtures were dialyzed against water for 3 days and dried by freeze-drying.pHPMA-b-pCPMA 13: yield: 53%; Mn = 91.4 kDa (by 1H NMR), Mn = 11.7 kDa, PDI = 1.10 (SEC-DMF, PMMA standard); δH (DMSO-d6, 400 MHz): 9.12 (H6), 8.73–8.44 (H5, H9, H13), 7.15 (H18), 6.15 (H7), 5.81 (H4), 4.48–4.03 (H1, H2, H3, H8, H20), 3.68 (H22), 2.99 (H10, H12, H19), 1.57 (H15, H17), 1.26 (H14), 1.01 (H16), 0.62 (H11, H21) ppm.
pHPMA-b-pGPMA 14: yield: 81%; Mn = 163.2 kDa (by 1H NMR), Mn = 24.7 kDa, PDI = 2.50 (SEC-DMF, PMMA standard); δH (DMSO-d6, 400 MHz): 10.81 (H6), 8.13 (H5, H9, H13), 7.18 (H18), 5.84 (H4), 5.61 (H7), 4.70 (H20), 4.38 (H1), 4.28–4.18 (H3, H8), 3.68 (H22), 2.90 (H10, H12, H19), 1.57 (H15, H17), 1.23 (H14), 1.02 (H16), 0.81 (H11, H21) ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Angela Krtitschka from the University of Potsdam for enabling measurements of NMR spectra and Dr Matthias Hartlieb from the University of Potsdam for scientific advice. In addition, the authors thank Dr Andreas Bohn and Minh Thu Tran from Fraunhofer IAP for taking SEM pictures. Thanks also go to Dr Erik Wischerhoff from Fraunhofer IAP for GPC measurements.This work was financially supported by the Fraunhofer Internal Programs under grant no. Discover 840 205 and by the Fraunhofer Cluster of Excellence Immune-Mediated Diseases CIMD.
R. B. acknowledges additional financial support by the German Federal Ministry of Education and Research (BMBF), grant number 13XP5113.
References
- Q. Li and Z. Li, Molecular Packing: Another Key Point for the Performance of Organic and Polymeric Optoelectronic Materials, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- C. S. Sarap, P. Partovi-Azar and M. Fyta, Optoelectronic Properties of Diamondoid-DNA Complexes, ACS Appl. Bio Mater., 2018, 1, 59–69 CrossRef CAS.
- Y. Feng, L. B. Luo, J. Huang, K. Li, B. Li, H. Wang and X. Liu, Effect of molecular rigidity and hydrogen bond interaction on mechanical properties of polyimide fibers, J. Appl. Polym. Sci., 2016, 133, 1–10 Search PubMed.
- N. V. Medhekar, A. Ramasubramaniam, R. S. Ruoff and V. B. Shenoy, Hydrogen bond networks in graphene oxide composite paper: structure and mechanical properties, ACS Nano, 2010, 4, 2300–2306 CrossRef CAS PubMed.
- H. Lan, F. Wang, M. Lan, X. An, H. Liu and J. Qu, Hydrogen-Bond-Mediated Self-Assembly of Carbon-Nitride-Based Photo-Fenton-like Membranes for Wastewater Treatment, Environ. Sci. Technol., 2019, 53, 6981–6988 CrossRef CAS PubMed.
- P. Song, Z. Xu and Q. Guo, Bioinspired Strategy to Reinforce PVA with Improved Toughness and Thermal Properties via Hydrogen-Bond Self-Assembly, ACS Macro Lett., 2013, 2, 1100–1104 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Nucleobase Containing Synthetic Polymers: Advancing Biomimicry via Controlled Synthesis and Self-Assembly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- F. Jiang, S. Cui, N. Song, L. Shi and P. Ding, Hydrogen Bond-Regulated Boron Nitride Network Structures for Improved Thermal Conductive Property of Polyamide-imide Composites, ACS Appl. Mater. Interfaces, 2018, 10, 16812–16821 CrossRef CAS PubMed.
- M. Meot-Ner, The ionic hydrogen bond, Chem. Rev., 2005, 105, 213–284 CrossRef CAS PubMed.
- S. Sivakova and S. J. Rowan, Nucleobases as supramolecular motifs, Chem. Sci., 2005, 34, 9–21 CAS.
- H. Yang and W. Xi, Nucleobase-Containing Polymers: Structure, Synthesis, and Applications, Polymers, 2017, 9, 666 CrossRef PubMed.
- J.-F. Lutz, A. F. Thünemann and K. Rurack, DNA-like “Melting” of Adenine- and Thymine-Functionalized Synthetic Copolymers, Macromolecules, 2005, 38, 8124–8126 CrossRef CAS.
- H.-W. Yang, A.-W. Lee, C.-H. Huang and J.-K. Chen, Characterization of poly(N-isopropylacrylamide)-nucleobase supramolecular complexes featuring bio-multiple hydrogen bonds, Soft Matter, 2014, 10, 8330–8340 RSC.
- J. Li, Z. Wang, Z. Hua and C. Tang, Supramolecular nucleobase-functionalized polymers: synthesis and potential biological applications, J. Mater. Chem. B, 2020, 8, 1576–1588 RSC.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, Synthesis and assembly behavior of nucleobase-functionalized block copolymers, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS.
- M. Garcia, M. P. Beecham, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Water soluble triblock and pentablock poly(methacryloyl nucleosides) from copper-mediated living radical polymerisation using PEG macroinitiators, Eur. Polym. J., 2015, 66, 444–451 CrossRef CAS.
- A. Marsh, A. Khan, D. M. Haddleton and M. J. Hannon, Atom Transfer Polymerization: Use of Uridine and Adenosine Derivatized Monomers and Initiators, Macromolecules, 1999, 32, 8725–8731 CrossRef CAS.
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Templated polymerizations on solid supports mediated by complementary nucleoside interactions, Polym. Chem., 2015, 6, 1944–1951 RSC.
- P. Amo-Ochoa and F. Zamora, Coordination polymers with nucleobases: From structural aspects to potential applications, Coord. Chem. Rev., 2014, 276, 34–58 CrossRef CAS.
- M. Wang, B. Choi, X. Wei, A. Feng and S. H. Thang, Synthesis, self-assembly, and base-pairing of nucleobase end-functionalized block copolymers in aqueous solution, Polym. Chem., 2018, 9, 5086–5094 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, RAFT polymerization and some of its applications, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Advances in RAFT polymerization: the synthesis of polymers with defined end-groups, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- S. Rosencrantz, J. S. J. Tang, C. Schulte-Osseili, A. Böker and R. R. Rosencrantz, Glycopolymers by RAFT Polymerization as Functional Surfaces for Galectin–3, Macromol, Chem. Phys., 2019, 220, 1900293 CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, Effect of Complementary Nucleobase Interactions on the Copolymer Composition of RAFT Copolymerizations, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS PubMed.
- Y. Kang, A. Pitto-Barry, H. Willcock, W.-D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Exploiting nucleobase-containing materials – from monomers to complex morphologies using RAFT dispersion polymerization, Polym. Chem., 2015, 6, 106–117 RSC.
- J. Ulbricht, R. Jordan and R. Luxenhofer, On the biodegradability of polyethylene glycol, polypeptoids and poly(2-oxazoline)s, Biomaterials, 2014, 35, 4848–4861 CrossRef CAS PubMed.
- T. T. Hoang Thi, E. H. Pilkington, D. H. Nguyen, J. S. Lee, K. D. Park and N. P. Truong, The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation, Polymers, 2020, 12, 298 CrossRef PubMed.
- M. Talelli, C. J. F. Rijcken, C. F. van Nostrum, G. Storm and W. E. Hennink, Micelles based on HPMA copolymers, Adv. Drug Delivery Rev., 2010, 62, 231–239 CrossRef CAS PubMed.
- B. S. Tucker and B. S. Sumerlin, Poly(N-(2-hydroxypropyl) methacrylamide)-based nanotherapeutics, Polym. Chem., 2014, 5, 1566–1572 RSC.
- J. B. Epp and T. S. Widlanski, Facile Preparation of Nucleoside-5′-carboxylic Acids, J. Org. Chem., 1999, 64, 293–295 CrossRef CAS PubMed.
- K. Ulbrich, V. Šubr, J. Strohalm, D. Plocová, M. Jelínková and B. Říhová, Polymeric drugs based on conjugates of synthetic and natural macromolecules, J. Controlled Release, 2000, 64, 63–79 CrossRef CAS PubMed.
- S. Perrier, 50th Anniversary Perspective : RAFT Polymerization—A User Guide, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- K. Zhang, M. Aiba, G. B. Fahs, A. G. Hudson, W. D. Chiang, R. B. Moore, M. Ueda and T. E. Long, Nucleobase-functionalized acrylic ABA triblock copolymers and supramolecular blends, Polym. Chem., 2015, 6, 2434–2444 RSC.
- K. Zhang, S. J. Talley, Y. P. Yu, R. B. Moore, M. Murayama and T. E. Long, Influence of nucleobase stoichiometry on the self-assembly of ABC triblock copolymers, Chem. Commun., 2016, 52, 7564–7567 RSC.
- K. Hatanaka, H. Takeshige and T. Akaike, Synthesis of a New Polymer Containing Uridine and Galactose as Pendent Groups, J. Carbohydr. Chem., 1994, 13, 603–610 CrossRef CAS.
- X. Pan, F. Zhang, B. Choi, Y. Luo, X. Guo, A. Feng and S. H. Thang, Effect of solvents on the RAFT polymerization of N-(2-hydroxypropyl) methacrylamide, Eur. Polym. J., 2019, 115, 166–172 CrossRef CAS.
- M. Sponchioni, L. Morosi, M. Lupi and U. Capasso Palmiero, Poly(HPMA)-based copolymers with biodegradable side chains able to self assemble into nanoparticles, RSC Adv., 2017, 7, 50981–50992 RSC.
- I. Lacík, A. Chovancová, L. Uhelská, C. Preusser, R. A. Hutchinson and M. Buback, PLP-SEC Studies into the Propagation Rate Coefficient of Acrylamide Radical Polymerization in Aqueous Solution, Macromolecules, 2016, 49, 3244–3253 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00615d |
| This journal is © The Royal Society of Chemistry 2022 |