
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in self-immolative linkers and their applications in polymeric reporting systems
Alexander G.
Gavriel
 a,
Mark R.
Sambrook
b,
Andrew T.
Russell
*a and
Wayne
Hayes
*a
a,
Mark R.
Sambrook
b,
Andrew T.
Russell
*a and
Wayne
Hayes
*a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: w.c.hayes@reading.ac.uk
bCBR Division, Defence Science & Technology Laboratory (Dstl), Porton Down, Salisbury, Wiltshire, SP4 0JQ, UK
First published on 18th May 2022
Abstract
Interest in self-immolative chemistry has grown over the past decade with more research groups harnessing the versatility to control the release of a compound from a larger chemical entity, given a specific stimulus. Originally conceived in 1981 to overcome electronic or steric features which may prohibit cleavage of a prodrug linkage; today's self-immolative linkers are widely used, inter alia, within medicinal chemistry, analytical chemistry, and material science. The incorporation of these linkers can be found in small molecules, dendritic and polymeric systems where a controlled release profile is required. This control can be of a binary character, release or not release, or a more nuanced issue of rate of release. In this article, we build upon our previous review in 2011 discussing key literature within the self-immolative field and, in particular, a selection of more recent examples that highlight how this field has matured in the past decade, with relevant earlier literature to provide context.
1 Introduction and key concepts
Over the past two decades, the chemistry of self-immolative systems has received significant attention and the technology has developed sufficiently to permit application in areas as diverse as the pharmaceutical sector,1–3 degrade-on-demand adhesives,4–6 chemosensors7 and the detection or disclosure of toxic compounds.8–10 In 1981 seminal studies conducted by Katzenellenbogen and co-workers on prodrugs led to the term ‘self-immolative connector’ being proposed, and this degradable unit has subsequently been referred to in the literature as a ‘self-immolative spacer’ or ‘self-immolative linker’.11 The chemistry of self-immolative systems has been the subject of several reviews in the past decade, and indeed we published a review a decade ago on the topic of self-immolative linkers in polymeric delivery systems.12–37 In this review, we will build upon our original survey12 to evaluate the most recent developments in self-immolative linkers including those utilised in polymeric systems and will assess the degradation chemistries plus the different macromolecular architectures used. Important advances have also occurred in the more general area of self-reporting materials, not always incorporating a self-immolative linker, and these have been reviewed recently by Mutlu and Barner-Kowollik.38The self-immolative linker (spacer) has been defined by Schmidt and Jullien in their 2015 review as: ‘self-immolative spacers (linkers) are covalent assemblies tailored to correlate the cleavage of two chemical bonds in an inactive precursor’.21 In a similar way, Corso and Gennari used the following definition in 2021: ‘self-immolative (SI) spacers (linkers) are covalent constructs designed to degrade spontaneously in response to specific stimuli’.39
They have developed from traditional protecting group strategies, wherein a reactive functionality in a molecule would be protected, modifying the reactivity at that site. Following whatever synthetic manipulations are desired, the protecting group is cleaved in a selective manner to release the modified molecule. In the present context, the act of protection has been used to modify a particular property of a molecule e.g. colour, fluorescence, biological activity; whereas, the selectivity in deprotection has been utilised in a number of ingenious ways as a means of detecting a specific environmental factor and responding to it e.g. the presence of a hydrolytic enzyme or toxic molecule to release a drug or indicator molecule. The molecules prepared are often referred to as SIEs (Self-Immolative Entities) and in their classical manifestation are composed of three parts, a trigger group (a protecting group), a linker, and a reporter group (it should be noted that in Katzenellenbogen's original work these were referred to as a specifier, a self-immolative connector or link and a drug). In principle, the linker may not be necessary, whereupon the SIE becomes a two-component system of the type that predated Katzenellenbogen's work. However, the field has attracted its own lexicon of terminology and the reader should be aware of synonyms used in the literature such as cargo, effector or reporter groups for the released molecules or atoms. In that case, upon a given specific stimulus (herein referred to as an activation event), the protecting group is activated so that the bond to the reporter group becomes labile and leads to its release (Scheme 1a). However, this approach is not always feasible as the covalent linkage between the trigger and reporter may be resistant to cleavage; for example, by steric bulk surrounding the scissile bond or electronic factors that enhance the stability of the bond.40,41 To overcome this impairment, a linker is incorporated joining the trigger and the reporter moieties. The linker forms a scissile bond to the trigger group and a stable linkage to the reporter; however, upon activation of the trigger and subsequent cleavage, the linker–reporter coupling is rendered labile, resulting in disassembly of the now two-component system and release of the reporter (Scheme 1b). The incorporation of a linker has facilitated further developments in the field that go beyond reactivity and embrace the ability to release multiple reporter groups, vide infra. Prior to our 2011 review, the field had integrated advancements from the polymer field such as dendrons and self-immolative polymers to allow for amplification of detected events (Scheme 1c and d),23,42–44 this reaching impressive levels with dendritic chain reactions (Scheme 1e), potentially delivering exponential growth of the signal from the analyte.27,45 The classic tripartite structure proved adaptable; for example, such that the linker moiety simultaneously functioned as a reporter group, vide infra. From the impressive foundations already present in 2011, many ingenious applications, new dendritic chain reactions, and exciting developments in information storage have been reported.
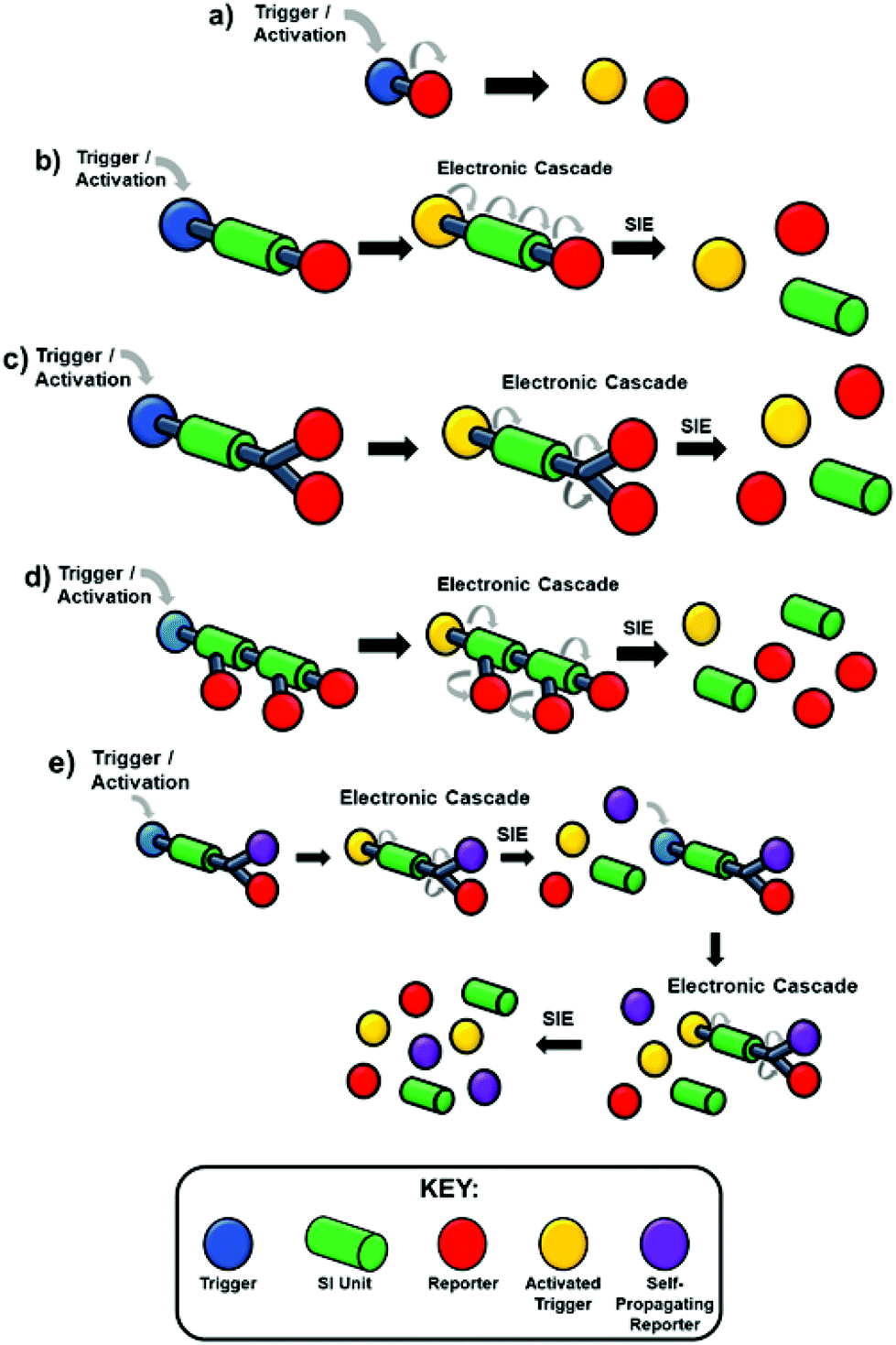 | ||
| Scheme 1 (a) Two-component system, (b) traditional non-amplified self-immolative system, (c) dendritic self-immolative system resulting in an amplified reporting event, (d) polymeric self-immolative system resulting in an amplified reporting event and depolymerisation, (e) dendritic chain reaction whereby the reporter event allows for a further trigger event. | ||
2 Classes of self-immolative elimination pathways
Self-immolative elimination occurs when a molecular system undergoes a spontaneous and irreversible disassembly into its constituent fragments through either an electronic cascade, via an elimination pathway or, alternatively, where self-immolation is achieved through a cyclisation–elimination event. Although these degradation processes differ fundamentally from each other, the pathways that drive self-immolative elimination both lead to: (i) an increase in entropy coupled with (ii) the irreversible formation of thermodynamically stable products (e.g. CO2). The more general area of stimuli responsive materials, where a single trigger site is not always present, has also led to important developments e.g. recyclable polymers,46 and has been recently reviewed by Gillies.34 These will only be covered for examples that include a specific trigger(s) within the structure e.g. an OTBS group.47In order to aid the reader of this review, a specific colour scheme has been employed in the diagrams to highlight the individual components of self-immolative systems herein; trigger (blue), self-immolative linker (green), reporter (red).
2.1 Self-immolation via an electronic cascade process
A significant number of self-immolative systems that undergo an electronic cascade commonly feature an aromatic linker with an electron-donating substituent (such as hydroxyl, amino or thiol residues) that is in conjugation (ortho- or para-) with a leaving group in a benzylic position on the molecule. The presence of an electron-donating group is essential to lowering the energy barrier of dearomatisation within the self-immolative process (thus forming a quinone methide, azaquinone methide or thioquinone methide intermediate). In general, amine groups have been found to be the most effective at promoting rapid cleavage, post activation, though hydroxyls, particularly when deprotonated, have been found to have significant roles, such as when linked to the anomeric position of carbohydrates and when generated from C–B bonds, vide infra.Katzenellenbogen's seminal account reported a model tripartite prodrug that employed the self-immolative linker 4-aminobenzyl alcohol.11 The self-immolative elimination of 4-nitroaniline (used as a model for drug compounds) from 1 was demonstrated, following hydrolysis its scissile amide bond, catalysed by trypsin, with Nα-Boc-Lys serving as the trigger moiety (Scheme 2). The Nα-Boc-Lys protected amide 1 is insufficiently electron releasing in nature (σp+ = −0.6) to facilitate spontaneous disassembly at a rate relevant to its use as prodrug (t1/2 ≈ 40 h in 0.05 M Bistris buffer (pH 6.9) at 25 °C). However, upon trypsin mediated hydrolysis of the amide, a strongly electron-releasing amine (σp+ = −1.31) is revealed and promotes self-immolation through a 1,6-elimination process. Elimination proceeds from the intermediate 2 with concomitant decarboxylation, resulting in the release of 4-nitroaniline 3. In conjunction with release of 3, highly reactive aza-quinone methide species 4 was presumably formed which was readily quenched by water to afford 4-aminobenzyl alcohol 5. Thus, the presence of a nucleophile was required to complete product formation from 2 and the rate of fragmentation was likely increased by the reaction occurring in a polar protic solvent system.48,49 The decomposition of 2 could also be described as solvolysis (or solvolytic cleavage).
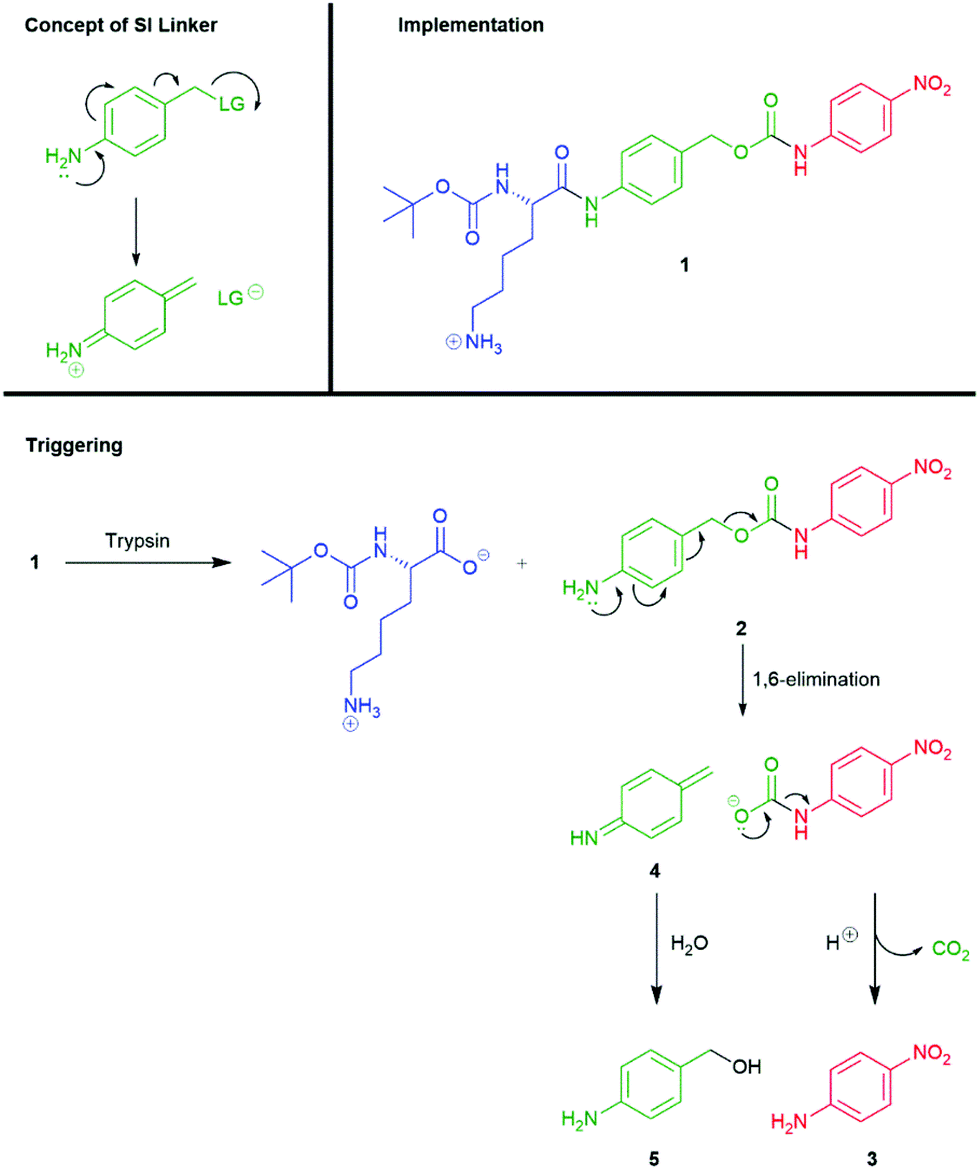 | ||
| Scheme 2 Trypsin mediated release of 4-nitroaniline through self-immolative elimination of a 4-aminobenzylalcohol linker.11 | ||
An interesting point, raised in a footnote to the paper, concerns the fate of 4 when alternative nucleophiles are present; for example, in the case of formation of 4 in a cell, the possibility of quenching of 4 by glutathione was considered. This raises a very important point regarding the fate of such reactive intermediates, generated from the linkers, that is worthy of consideration in the application of a SIE system; some examples of recapture of the reporter group has been observed by Hay, Du and Li, and Phillips.50–52 In addition, reversible reactions of the reactive intermediates from self-immolative processes with DNA bases have reported by Fakhari and Rokita.53 These observations and reports highlight an important aspect of use of self-immolative systems in biological environments – there is a need to understand the biocompatibility and toxicity of such reactive intermediates before these highly innovative, degradable materials can be translated into ‘real-world’ applications. Established tumour imaging applications; for example, by chemiluminescence, have been carried out in animals without significant side effects being reported (vide infra), though this aspect was not the focus of the studies. In the treatment of cancer, selective cell death is desirable and release of reactive intermediates at the target site may not be an issue and possibly beneficial. By contrast, off-site release may be problematical but, to date, detailed findings of such studies have yet to be published.
Since Katzenellenbogen's ground breaking report,11 numerous groups have utilised successfully this type of benzylic spacer including some recent examples from Byun et al., Anami et al., Wei and Safina et al., Gennari and Piarulli.54–58 It has also been demonstrated that the hydroxyl and thiol analogues, 4-hydroxybenzyl alcohol and 4-mercaptobenzyl alcohol, respectively, also undergo 1,6-elimination upon cleavage of a triggering moiety attached to the ring heteroatom.59,60 In a study by Senter on mercaptobenzyl alcohols, following rapid disulfide reduction, the para-derivative 6 fragmented with a t1/2 value of 10 minutes, as compared to a t1/2 of 72 minutes for the ortho-derivative 7, to release mitomycin C (Scheme 3). The meta-derivative 8 proved stable toward release.59 That the rates of fragmentation increased with a change in pH from 6, to 7.2 to 8.0 supports the role of deprotonated thiol (thiophenol pKa 6.6).
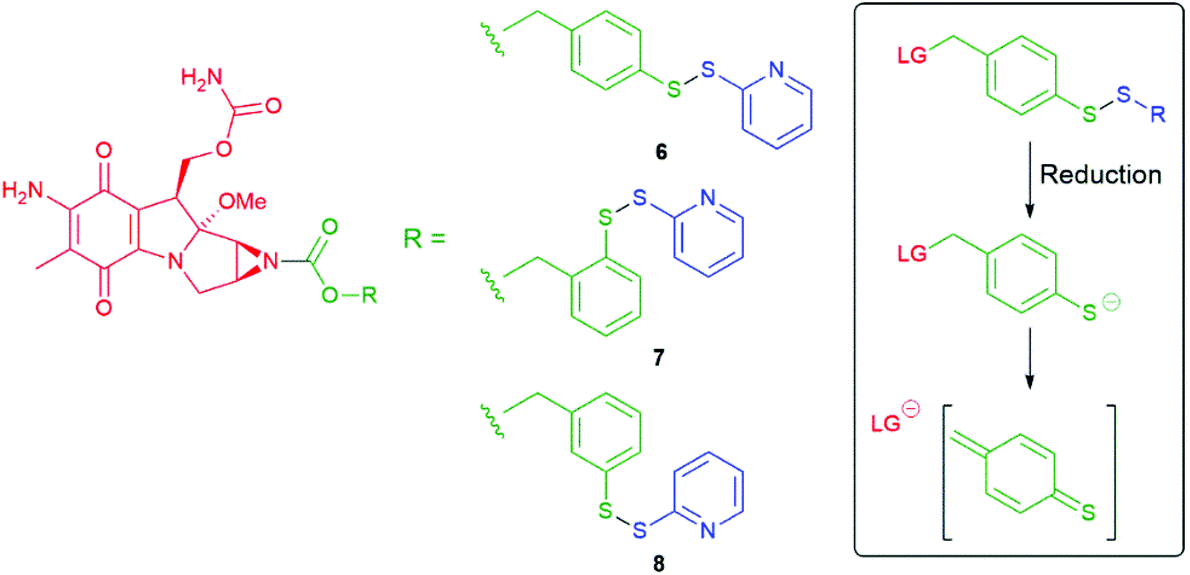 | ||
| Scheme 3 Prodrugs 6–8 for mitomycin C with release triggered by reductive cleavage of a disulfide bond.59 | ||
Shabat and co-workers have reported previously kinetic studies with aminobenzylalcohol linkers that undergo single para/1,6- or ortho/1,4-elimination and observed 1,6-elimination to be more slightly rapid that 1,4-elimination, in agreement with Senter's observations on thiobenzyl alcohol linkers.61 As a result of the increased electronegativity of oxygen with respect to nitrogen, hydroxybenzyl alcohol-based linkers undergo disassembly at relatively reduced rates, although these can be enhanced through the incorporation of additional ring substituents or the use of mild basic conditions to effect deprotonation.49,62–65
In addition, Shabat and co-workers evaluated a pyridine core-based AB and AB2 self-immolative dendron (ABn, where A and B represent an electron-donating group and suitable leaving groups, respectively, and n correlates to the number of leaving groups present).66 Under physiological conditions, the AB system constructed from a pyridine core, a reporter, and an enzyme (penicillin-G-amidase, (PGA)) sensitive trigger unit underwent 1,4-elimination more rapidly than its parent system featuring a benzene core. Furthermore, the pyridine core-based AB2 dendron was also found to release its two reporter units upon activation through 1,6- and 1,4-pyridinone-methide elimination reactions and faster than the analogous benzene system. To promote the elimination the conjugate base must be formed; given the pKa of 3-hydroxypyridine (8.7) vs. phenol (10.0) under aqueous conditions, a larger concentration of the conjugate base is expected in the pyridine system in comparison to the benzene system. This difference in pKa serves to explain the variation in the rates of elimination, though it introduces the potential for variance in aromatic resonance energy to affect rates of elimination vide infra.
Hay et al.50,67 have investigated the influence of ring substituents on the rate of 1,6-elimination of para-hydroxylaminobenzyl alcohol linkers 9, following radiolytic reduction of the corresponding nitro compound 10 to release aniline 11 (Scheme 4). Twelve analogues were analysed and revealed that the presence of electron-donating groups were found to enhance the rate; whereas, electron-withdrawing groups serve to decrease the rate of elimination. The highest effect on self-immolative elimination rate occurs when electron-donating or withdrawing groups were ortho to the benzylic leaving group (R2 in 9 rather than R1), permitting conjugation with the developing cation formed during departure of the reporter group. Indeed, data on the electron-donating substituents e.g. OMe could be fitted to the equation log(Mt1/2) = 0.57σ + 1.30, where Mt1/2 is the maximum half-life and σ represents σp for 2-substituents and σm for 3-substituents.
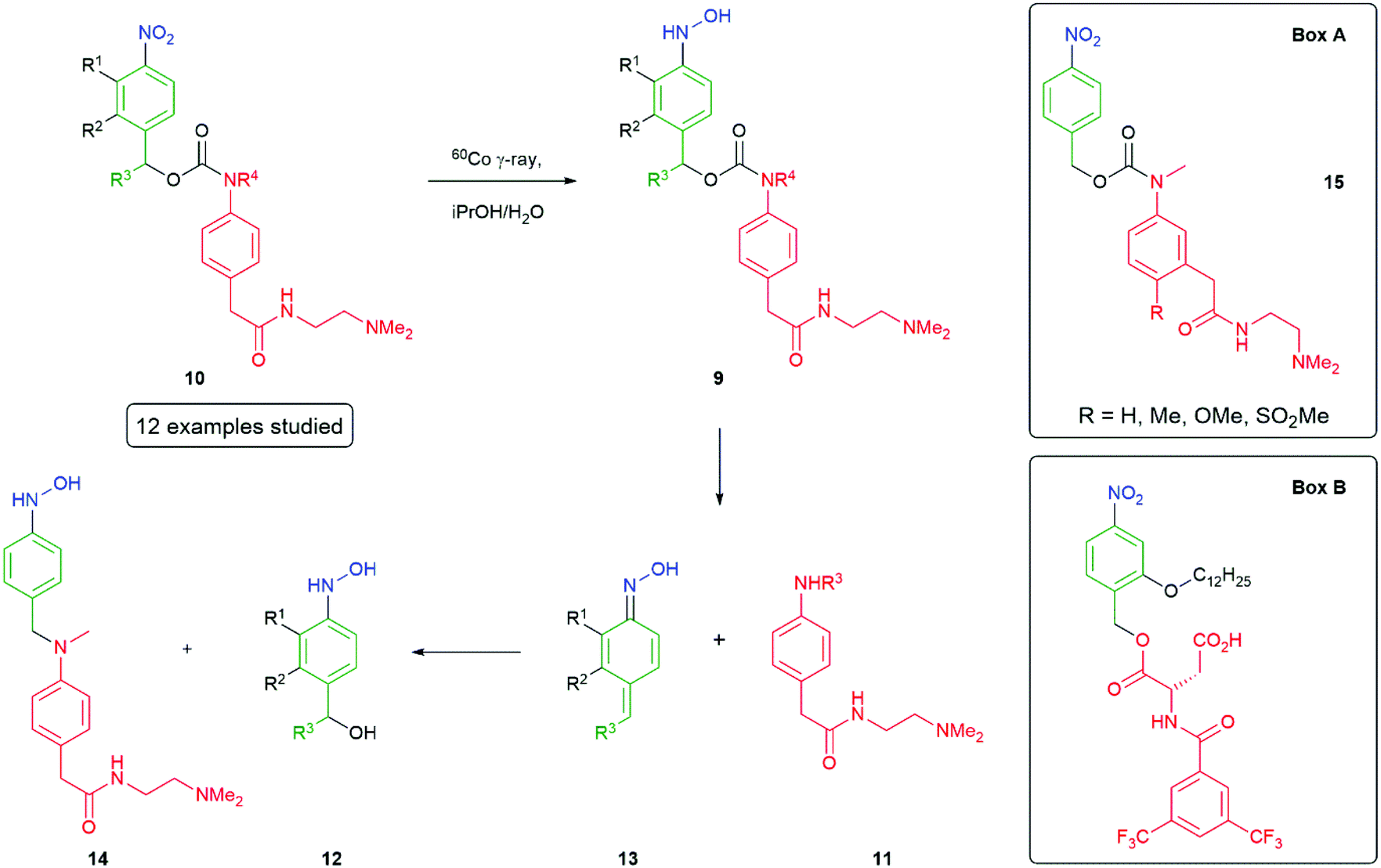 | ||
| Scheme 4 Reductively triggered self-immolative compounds showing the effects of ring substituents on the rate of fragmentation and recapture of the reporter group.50,68 | ||
An increase of elimination rate was also observed when a methyl group was substituted at R3 to create a secondary carbon, consistent with its ability to stabilise a developing positive charge. As noted above, Hay also reported that the extent of release of the reporter group can be compromised by recombination of the reporter group with a precursor to 12, probably the azaquinone methide intermediate 13: a detailed analysis being given for 14. This was found to be pH dependent with greater formation of the reporter group at pH 3 and more recombination at pH 7.4.50 The observation of a reduced fragmentation rate (factor of 2) upon methylation at R4 suggested the leaving group structure may also be important. A study of the four related compounds 15 (Box A) showed different extents of release of reporter group, at the end of the reduction process, dependent upon its structure though a clear connection to pKa of the reporter group remained elusive. Hamachi has reported an interesting application of nitroreductase to trigger a signal-amplifiable, self-assembling 19F NMR-MRI probe, based on the self-immolative molecule shown in Box B.68 Cleverly, this molecule utilises a hydrophobic ortho-alkoxy group that simultaneously facilitates its self-assembly into an NMR silent aggregate (mean diameter of 100 nm by DLS), prior to self-immolation, and increases the rate of that self-immolation, following reduction of the nitro group.
The generation of an aniline by reduction, to initiate 1,6-elimination, has also been examined from the diazo functional group. This has proved of value in drug release in the colon by action of azo reductase e.g. for controlled release of Tofacitinib for the treatment of ulcerative colitis.69,70
Senter et al. have also flagged the importance of the leaving group during an investigation into release of a reporter group linked through an alcohol moiety and have shown the effect on self-immolative ability, elimination rates, and conjugate stability.71 In this study, 4-aminobenzyl ether-based conjugates were studied with the N-protected dipeptide trigger group, benzyloxycarbonylvaline-citrulline (Z-val-cit). Z-val-cit was conjugated with the amino group of aminobenzyl ether derivatives of 1-naphthol 16 and N-acetylnorephedrine 17 (Scheme 5). The amide bond in these derivatives was sensitive to hydrolysis by the protease enzyme Cathepsin B. Upon treatment with Cathepsin B, the peptide bond of conjugate 16 was hydrolysed and release of 1-naphthol occurred through 1,6-elimination from intermediate 18. Under identical reaction conditions, the conjugate 17 featuring N-acetylnorephedrine was also subject to hydrolysis of the amide bond to yield the corresponding aniline 19, but 1,6-elimination of N-acetylnorephedrine did not occur. The failure of 19 to undergo 1,6-elimination was attributed to a higher pKa of the reporter moiety. Stirling has noted the nucleofugality of leaving groups cannot always be correlated to the pKa of their conjugate acid.72 However, in the context of β-elimination reactions with an E1cBR character, Stirling assigns the leaving group rankings (via a logarithmic scale) of PhO− (8.9) and MeO− (6.1), consistent with the above observations. Germane to the upcoming discussion, stirling notes that ‘small variations of structure within a series of leaving groups of the same type, e.g. aryloxy, do, however, show a correlation with pKa’.
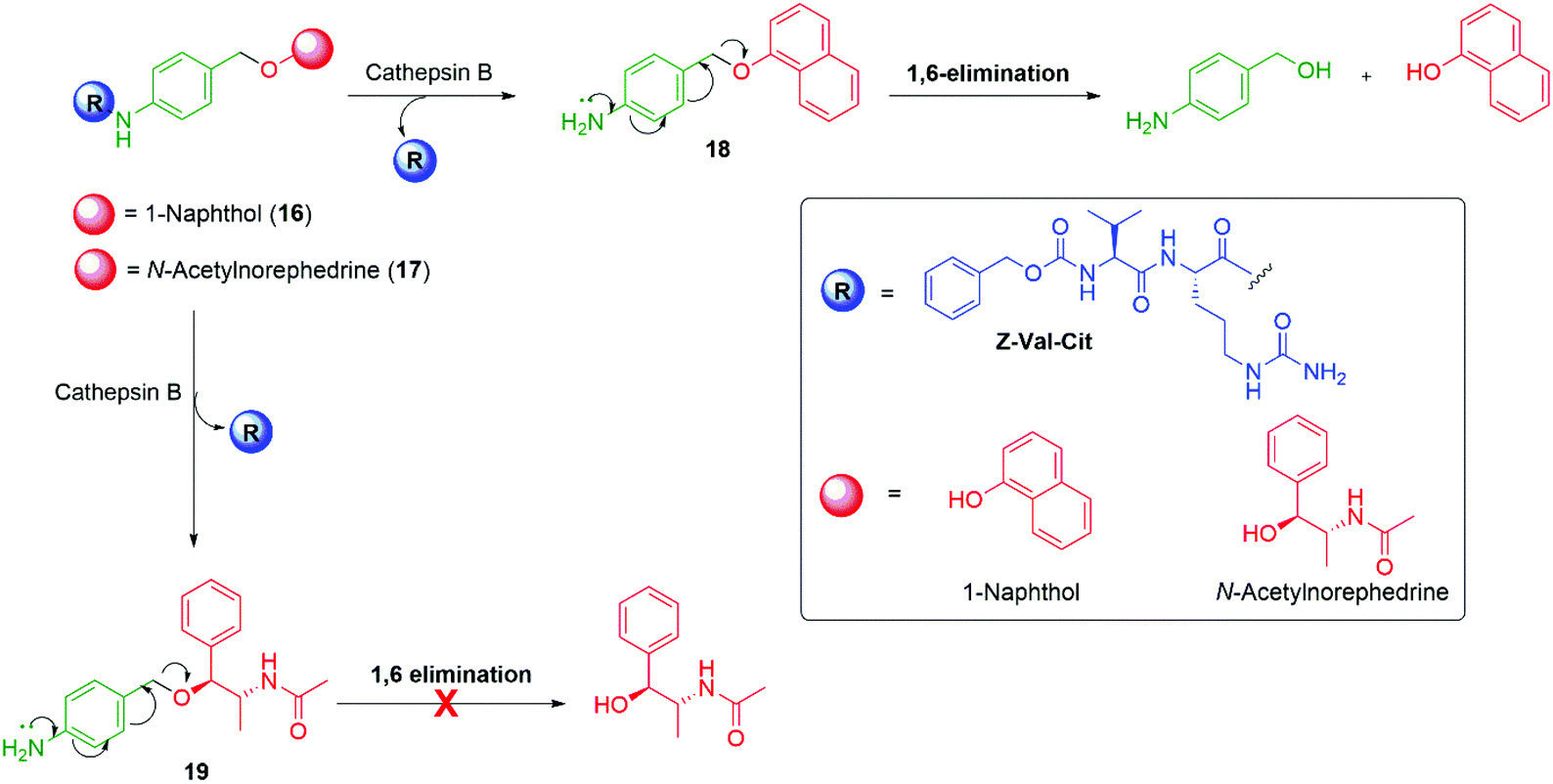 | ||
| Scheme 5 Variation of the self-immolative ability of the 4-aminobenzyl ether linker when coupled to either naphtholic or alcoholic reporter groups.71 | ||
Based upon these studies, the phenolic anticancer drugs etoposide and combretastatin were synthesised with ether 20–21 and carbonate 22–23 linkages to the Z-Val-Cit-p-amidobenzyl self-immolative unit, forming the corresponding tripartite prodrugs (Fig. 1). The phenolic ethers 20–21 proved to be stable in aqueous buffers as well as in human serum, and self-immolative fragmentation occurred only upon treatment with Cathepsin B. In contrast, the carbonates 22 and 23 were found to be unstable in aqueous buffers as well as in human serum. This study highlights that the design of a self-immolative system should consider the possibility of release by other than the desired mechanism. Similar studies were reported by Burke and co-workers,73 and Leu and co-workers74 who described the self-immolative ability of glucuronide hydroxybenzyl ether prodrugs of 10-hydroxycamptothecin.74–76 Recently, Zhang, Pillow and co-workers have investigated the self-immolation of 4-aminobenzylalcohol linkers with phenol-based drugs as reporter groups in antibody drug conjugates.77 They concluded that a case-by-case evaluation was necessary to determine their likely effectiveness. Related to these studies have been the application of self-immolative linkers in protein activity assays that take advantage of the highly reactive (aza)quinone methide78 and quinone methide species.79
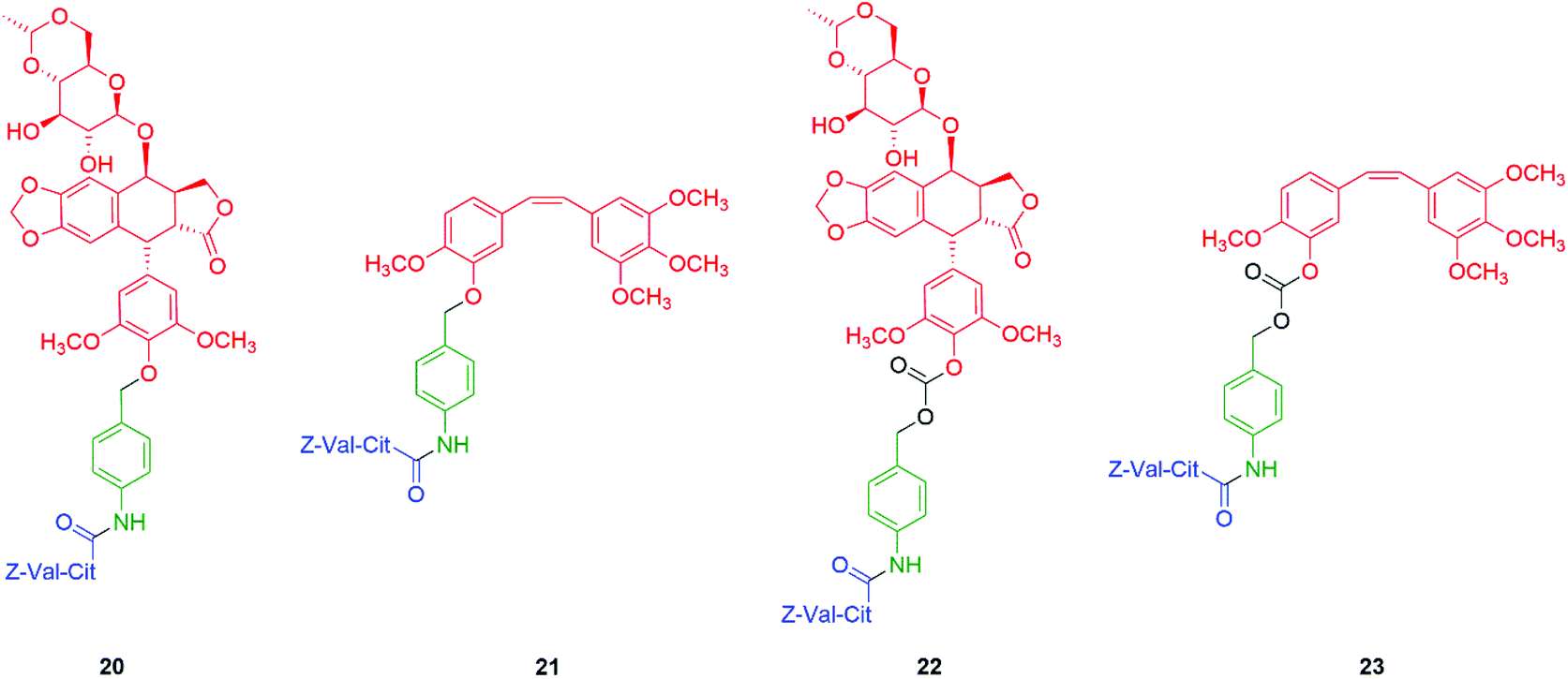 | ||
| Fig. 1 Self-immolative tripartite prodrugs of anticancer agents etoposide and combretastatin A-4 described by Senter et al. in their ether (20–21) and carbonate (22–23) forms, respectively.71 | ||
Building upon these observations, Phillips and co-workers have reported protected 4-aminobenzyalcohol linkers such as 24, substituted by electron donating groups, that allow tunable release of phenols with pKa values ≤11.5, directly connected to the benzylic carbon and under neutral conditions (Fig. 2).49 A particularly reactive conformation, that simultaneously allowed facile interaction of the methoxy lone pairs with the aromatic π-system and, by conjugation, the σ* orbital of the departing C–O bond, was shown to be favoured. Of general significance to SIE triggered decomposition, the effect of solvent polarity was explicitly considered by variation of the ratio of two solvents, MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O; an increased rate of release was observed with increased polarity. That the fragmentation of many prodrugs has utilised enzymatic reactions in water could leave this point less easily recognised.
:H2O; an increased rate of release was observed with increased polarity. That the fragmentation of many prodrugs has utilised enzymatic reactions in water could leave this point less easily recognised.
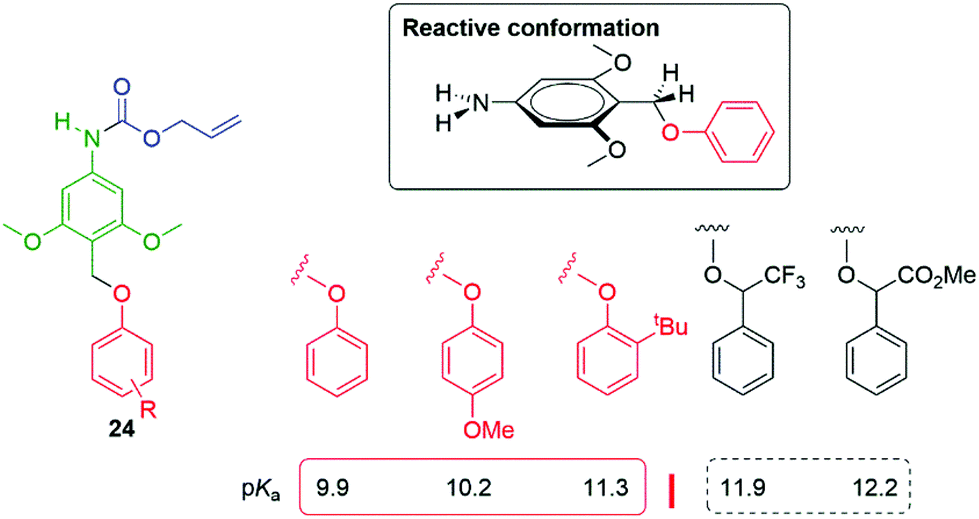 | ||
| Fig. 2 Protected 4-aminobenzyalcohol linker that facilitates phenol release at neutral pH (for pKa ≤ 11.5).49 | ||
Use of a trigger group based on oxidation of a C–B bond has been reported by Chang and co-workers (Scheme 6).80 Specifically, the use of hydrogen peroxide to mediate the oxidation is of medicinal significance as elevated levels in a cell are often correlated to significant disease states such as cancer and neurodegenerative diseases. Earlier work from this group has established the selectivity of this type of trigger for hydrogen peroxide over other reactive oxygen species.81 Chang reported a SIE system 25 that is capable of releasing firefly luciferin with a view to quantifying the level of hydrogen peroxide in a cell. Thus, exposure of boronic acid 25 to intracellular H2O2 releases 26 (firefly luciferin) that, in turn, upon exposure to luciferase, gives oxyluciferin 27 and chemiluminescence (612 nm).
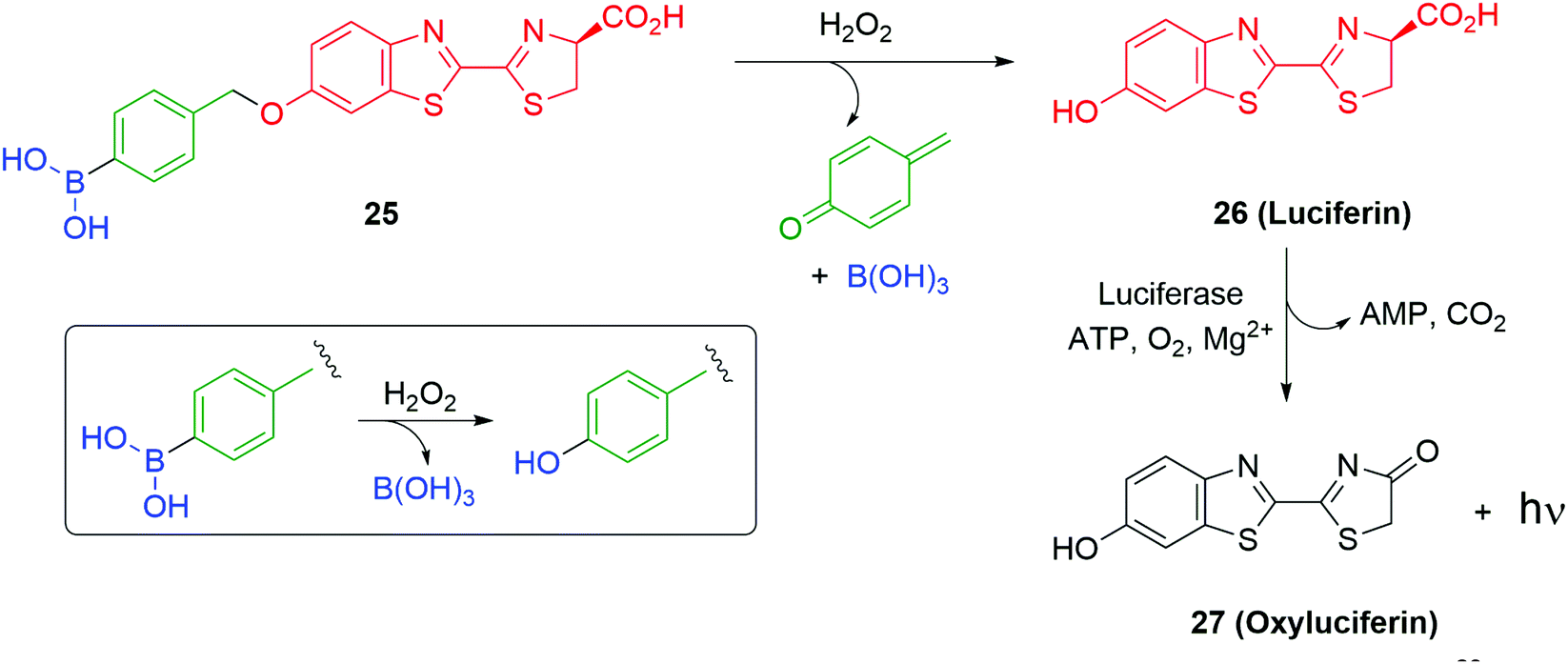 | ||
| Scheme 6 Oxidatively driven SIE for release of luciferin: quantification of intracellular H2O2.80 | ||
Further studies on such SIE systems, by Cohen and co-workers,82 has mapped out several useful design features (Fig. 3). In a comparative experiment, measuring rates of release of the phenol methyl salicylate (pKa 9.8), from three precursors 28–30, the carbonate 29 showed a slightly higher rate of release when compared to the phenol 28 but was subject to some background hydrolysis. Removing the self-immolative linker to give 30 revealed a significantly slower rate of formation; this latter issue highlights the general point that a self-immolative linker offers a consistent substructure within the molecule, with predicable kinetics and attenuated influence from changes in the reporter group structure, even when it is not an enzyme catalysed reaction that is under consideration. Amine-based reporter groups were unsuccessful e.g., 31a unless part of a carbamate (32); a thiophenol (31b) also proved ineffective. At the reaction pH of 7.5, the amine-based reporter groups would be protonated to a very small extent, but this provided insufficient drive to eliminate, presenting an interesting contrast to N-alkylation (see Schemes 9, 33 and Fig. 12). Interestingly, 33, containing a hydroxamic acid reporter group, also failed to self-immolate, perhaps reflecting the acidity of the NH group. The related compound 34 fragmented readily. Boronic acids were generally cleaved more rapidly than their corresponding esters, as well as improving aqueous solubility.
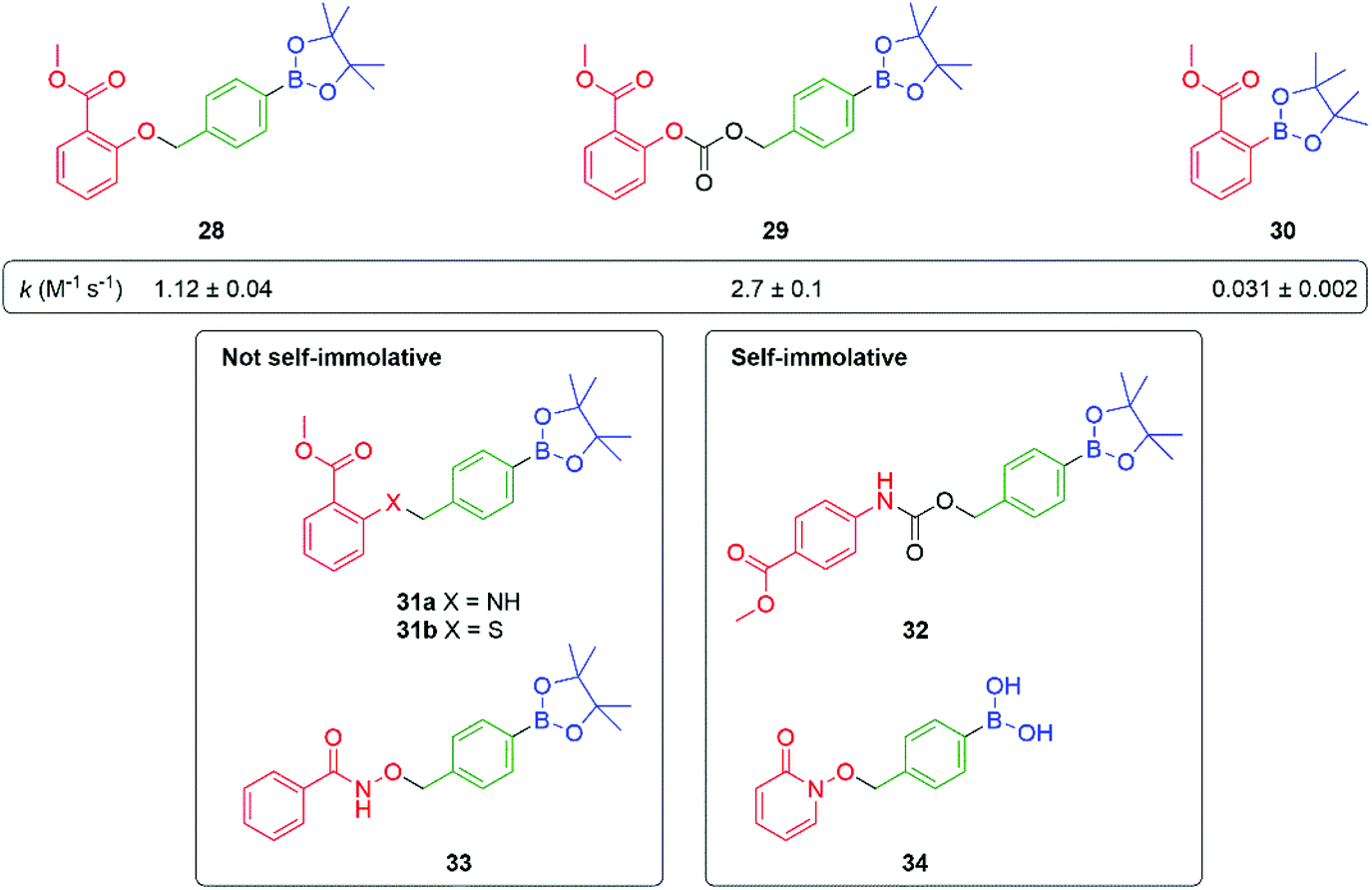 | ||
| Fig. 3 Effectiveness of C–B bond oxidation to trigger release of various reporter groups.82 | ||
An alternative approach to the release of alcohols that addresses the difficulty of self-immolative release of alkyl alcohols has been advanced by Mosey and Floreancig (Scheme 7).83 Building on the body of work reported by Chang and co-workers,80 using a C–B bond oxidation of e.g., 35 to 36 triggered by exposure to hydrogen peroxide, a key α-alkoxy carbamate 37 is released. Of significance is the observation that self-immolation of the linker is slow compared to decarboxylation to give 38 and subsequent breakdown of the resultant tetrahedral intermediate, leading to 39. When 40a and 40b (R1 = R2 = H) are treated with hydrogen peroxide, the corresponding phenol and neopentyl alcohol were released at comparable rates, despite their difference in nucleofugacity. Interestingly, the authors also note the rate enhancement of linker fragmentation that attends the presence of a OMe at R2 in 40 or a methyl group at R1, (cf.Scheme 4 and Fig. 2).
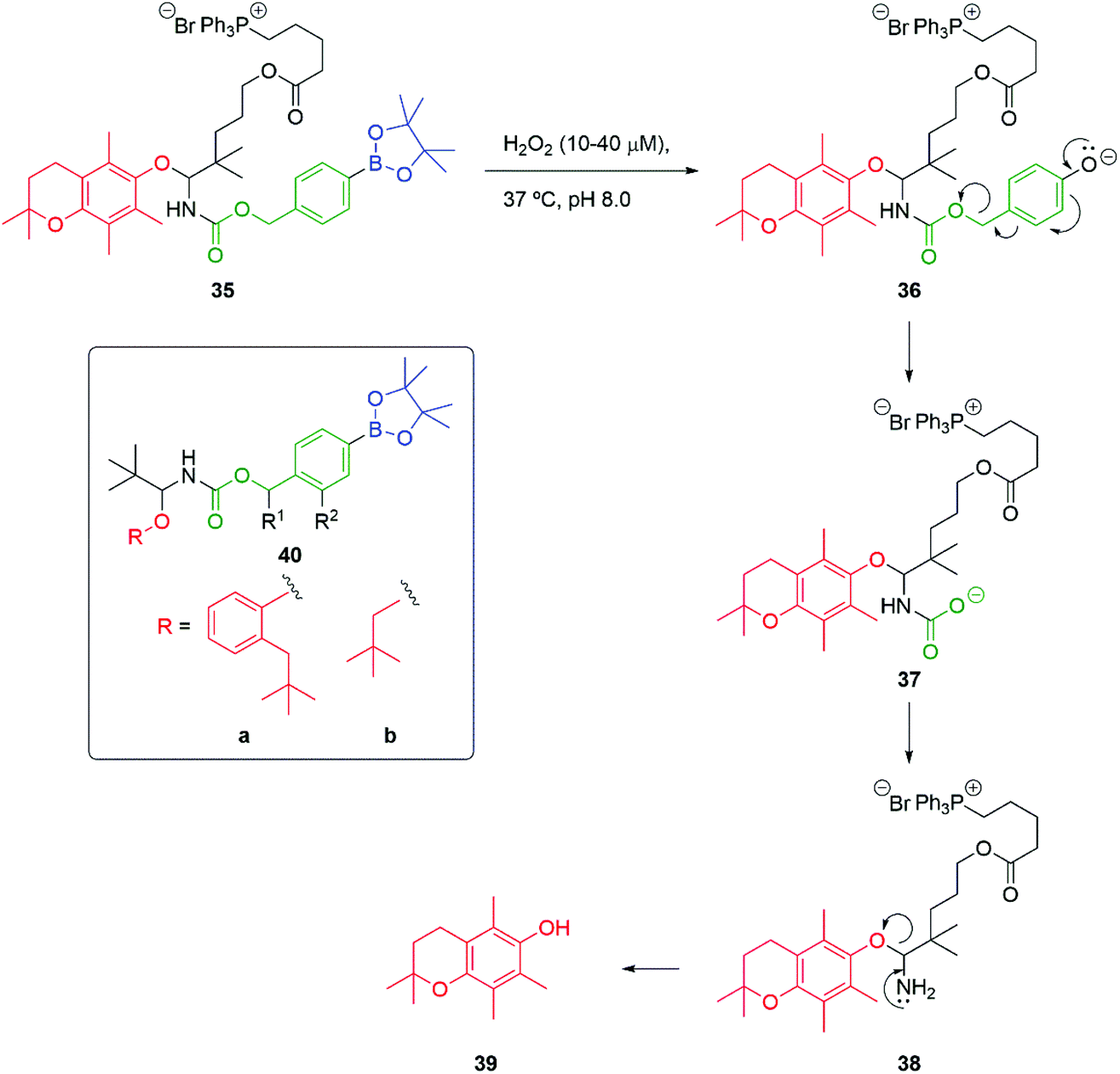 | ||
| Scheme 7 Hydrogen peroxide induced self-immolative release of alcohols under pseudo biological conditions.83 | ||
Overall, compound 35 is an interesting example of the general method; it is designed to deliver an antioxidant reporter group 39 to mitochondria, directed by the triphenylphosphonium group, as initially described by Murphy and co-workers.84,85 It could be considered that the O,N-acetal functional group forms a second part of the linker moiety, as well as affording an attachment point for the directing group. In the latter sense, Santi has applied this type of O,N-acetal in the delivery of SN-38.86 Kolakowski and Jeffrey have shown this type of linker to be effective in drug delivery from antibody–drug conjugates.87 A related approach was exploited independently by Wang and Xian for the delivery of persulfides (vide infra).88,89 In later work, Floreancig and Deiters utilised hydrogen peroxide to trigger disassembly of O,O-acetals.90
In contemporaneous studies, Phillips and co-workers demonstrated release of two alcohols from symmetric or asymmetric acetals of the type present in 41, at pH 7 under the influence of hydrogen peroxide (Scheme 8).52 The amino benzaldehyde acts simultaneously as a reporter group (by colour, see Scheme 77 for the likely origin of the yellow colour) and a second linker group to the alcohol reporters. The observation of the secondary products 42 and 43 formed during fragmentation of 41 relates to the original suggestion by Katzenellbogen11 that the fate of the linker could be influenced by nucleophiles in the system, other than water. Presumably, these arise via the para-quinone methide 44; however, as these are also coloured, fortuitously they do not influence that part of the read-out.
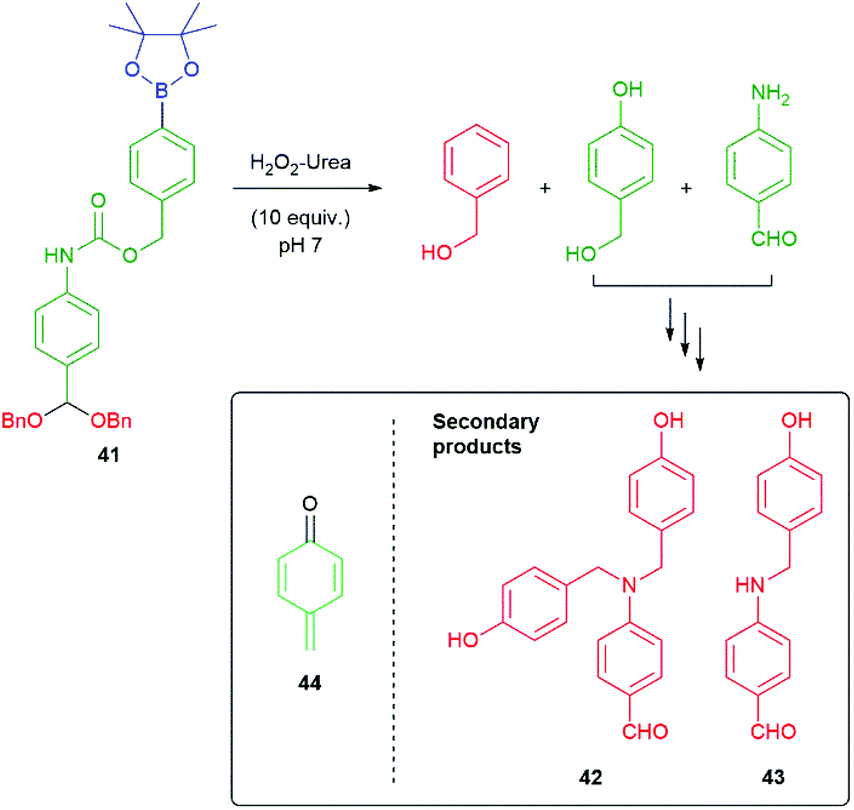 | ||
| Scheme 8 Hydrogen peroxide responsive detector that releases alcohols from an acetal under pseudo biological conditions.52 | ||
The modular approach to synthesis of the detection system allows ready construction of detectors that respond to a range of environmental triggers; for example, UV radiation (45a), fluoride (45b), Pd(0) (45c), PGA (45d) and β-glucuronidase (45e) (see exemplars in Fig. 4).52
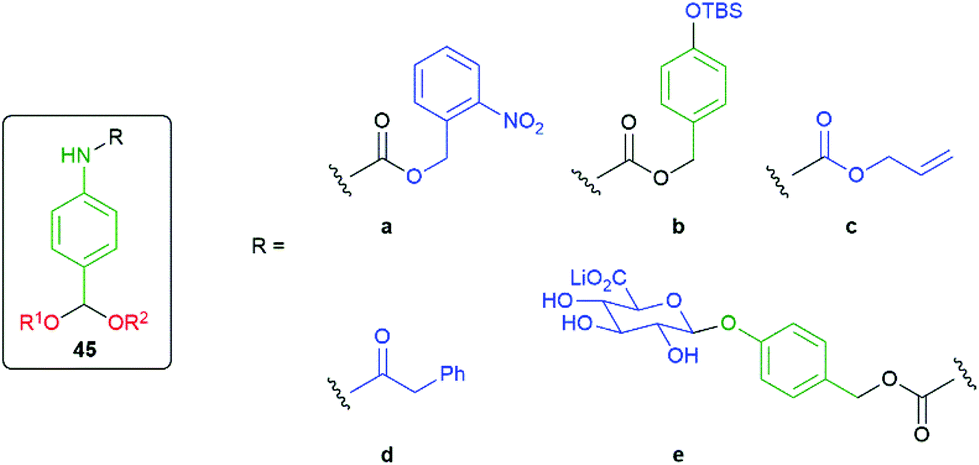 | ||
| Fig. 4 Detectors that respond to a range of environmental triggers.52 | ||
In addition to amine reporter groups being released as carbamates, ammonium ions have also been used to significant advantage. For example, Peng and co-workers have applied this approach to the delivery of nitrogen mustard anticancer agents e.g. mustine 46 (Scheme 9).91–93 Utilising the nitrogen's lone pair in bonding to the linker within 47 simultaneously forms a leaving group and prevents its use in neighbouring group participation, a key step that precedes DNA strand cross-linking, such as that to guanine N-7 illustrated in the boxed structure in Scheme 9. As noted above, intracellular hydrogen peroxide, frequently present in elevated levels in cancer cells, oxidises C–B bonds delivering borate esters such as 48 and thence via hydrolysis to phenols/phenolates 49 that self-immolate to release 46. A related use of ammonium ion reporter groups has been discussed by Taran and Le Gall, vide infra.9 In addition, Staben et al. have described the use of ammonium ions as leaving groups in self-immolative drug delivery systems based upon para-aminobenzyl alcohol and cyclization-based linkers.94
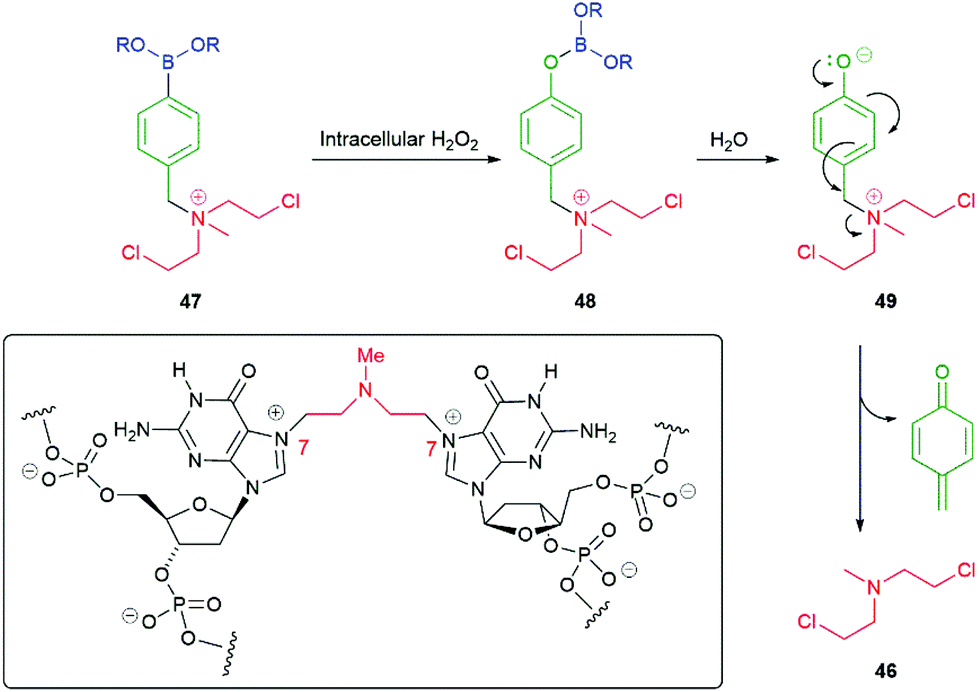 | ||
| Scheme 9 Hydrogen peroxide induced self-immolative release of a nitrogen mustard.91–93 | ||
Gois and co-workers have adapted the C–B bond oxidation method to the targeted delivery of the fluorescent imaging agent 7-hydroxycoumarin 50 to folate-positive cancer cells (MDA-MB-231). Thus, 51 in the presence of glutathione releases 52 which will undergo oxidation and self-immolative cleavage to afford 50 (Scheme 10).95
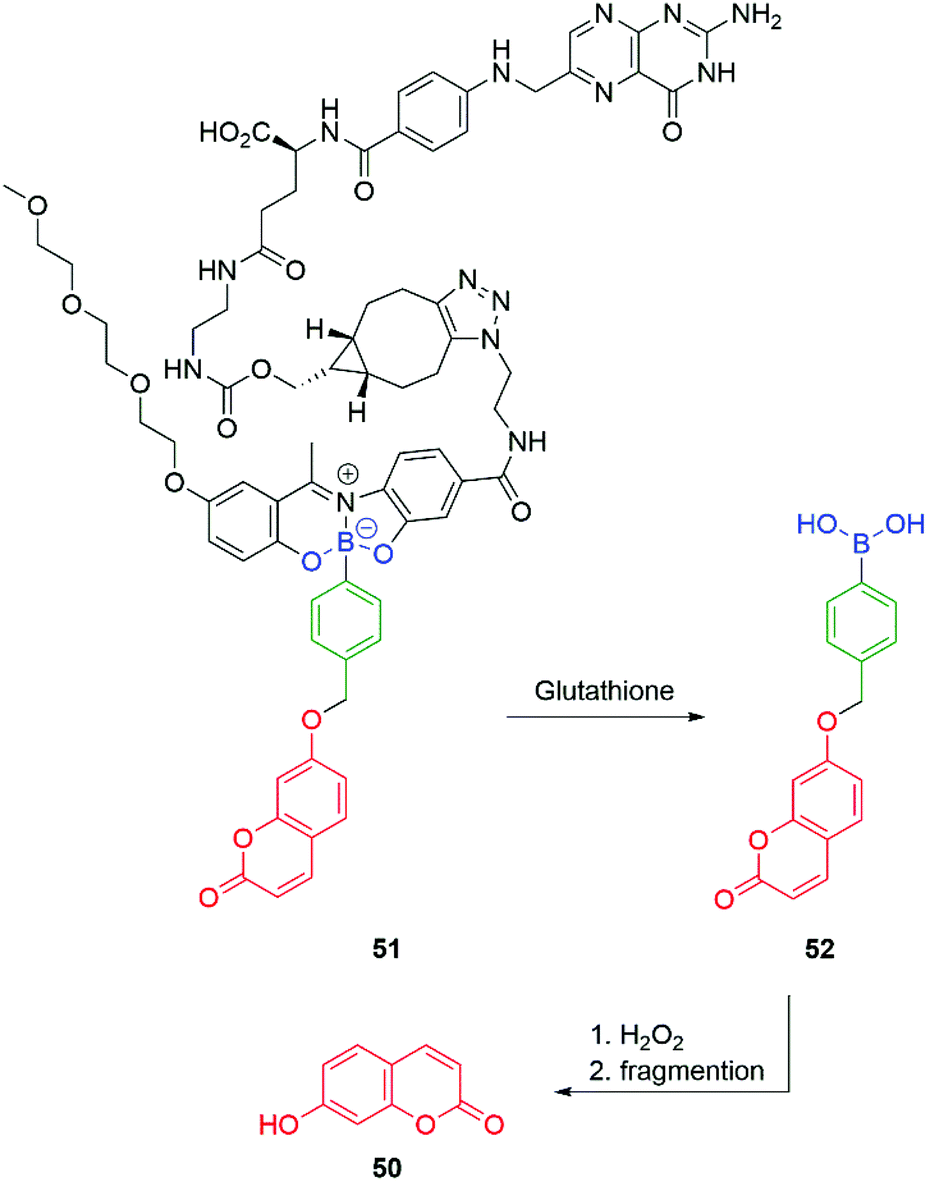 | ||
| Scheme 10 Glutathione/hydrogen peroxide induced self-immolative release of a fluorescent coumarin.95 | ||
Again, the modular approach for assembling the molecules greatly facilitates development of a glutathione triggered release of the cytotoxic drug Bortezomid.95 A model study identified the cleavage products (Scheme 11) and led to a proposal for the mechanism of release of the boronic acid.
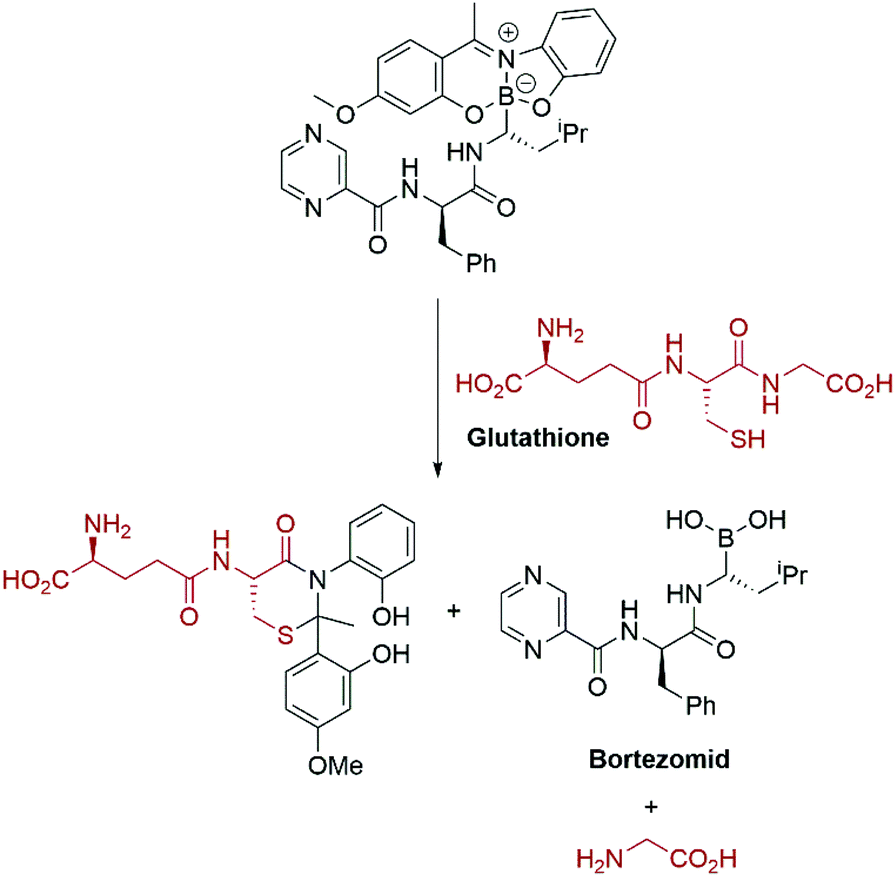 | ||
| Scheme 11 Glutathione mediated release of Bortezomid.95 | ||
Fréchet et al. have prepared a modified version of the biocompatible polymer dextran that is sensitive to the presence of H2O2 (Scheme 12).96 This new polymer, named OxiDEX, was found to be soluble in organic solvents, facilitating formation of microparticles (100–200 nm) capable of carrying a payload. OxiDEX was demonstrated to encapsulate ovalbumin to a protein loading of 1.6 ± 0.1 wt%. In the presence of H2O2, the particles were shown to degrade with a half-life of 36 ± 1 minutes. The OxiDEX particles were found to provide effective antigen presentation.
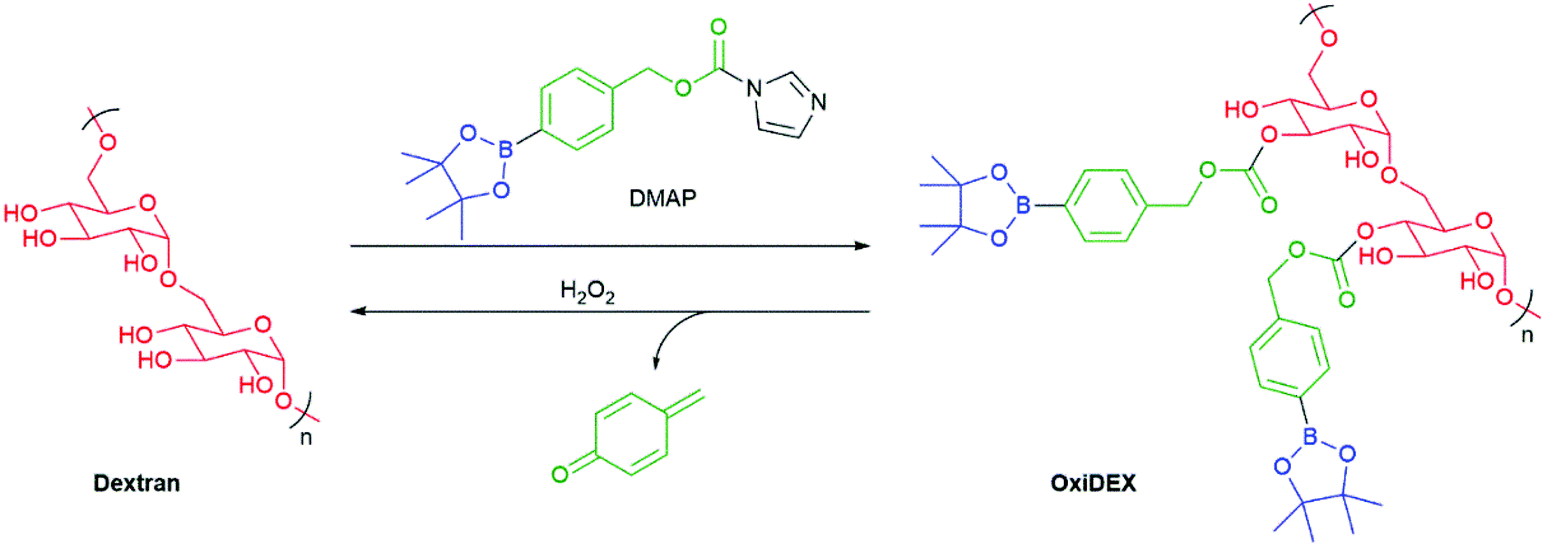 | ||
| Scheme 12 The modified dextran carrier polymer that was sensitive to H2O2 as reported by Fréchet et al.96 | ||
The value of C–B bond oxidation in affecting/investigating function within a cell was further exemplified by Pluth and co-workers by the development of a range of SIEs (53) capable of controlled release of COS and thence, by action of carbonic anhydrase, to accelerated release of H2S (some non-enzymatic background release).97–99 The discovery of H2S as a gasotransmitter implicated in human health and disease has heightened interest in such molecules and 54 was shown to afford protection against oxidative stress in HeLa cells: it's mode of action, via55, is illustrated below (Scheme 13). Release was preferentially triggered by H2O2 but O2− and ONOO− were also found to be competent oxidants; this being less discriminating than in its preferred oxidant than the systems reported by Chang and co-workers.81 An alternative approach to the delivery of sulfur species has been developed by Toscano et al. via a cyclisation linker strategy (see section 2.2).100
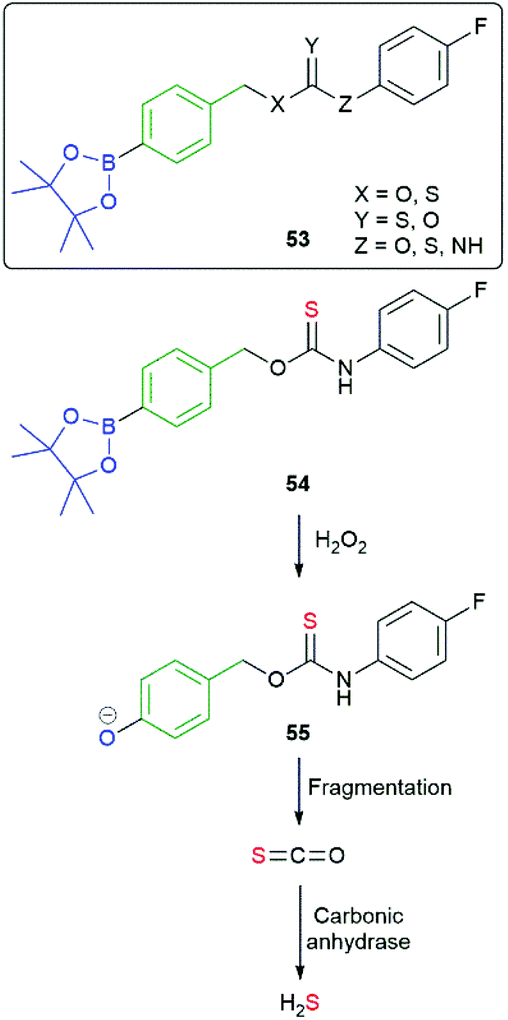 | ||
| Scheme 13 Reactive oxygen species triggered release of COS and thence gasotransmitter H2S.97–99 | ||
Significant efforts have been expended to develop a range of self-immolative molecules, based upon 1,6-elimination, that deliver COS/H2S but are triggered by a range of methods.
As a variation on 54, Chakrapani and co-workers prepared a range of compounds exemplified by 56 (Fig. 5). The key feature of these compounds being the sulfur atom through which the linker is attached to the leaving group.101 The significantly lower C–S as compared to C–O bond energy being considered advantageous during fragmentation. Similar compounds have also been reported by Pluth and their degradation pathways were subject to detailed theoretical analysis.99 Despite the change of linking atom in 56, it fragments efficiently to deliver, initially, COS and thence H2S. The co-released aniline is the methyl ester of the non-steroidal anti-inflammatory drug mesalamine, used in the treatment of colitis. Together, the concept was to co-release a drug (mesalamine) and the cell-protective agent H2S. The correlation of the pKaH+ of the various amines/anilines employed in the study to the rate of release of H2S (via COS) was also considered.
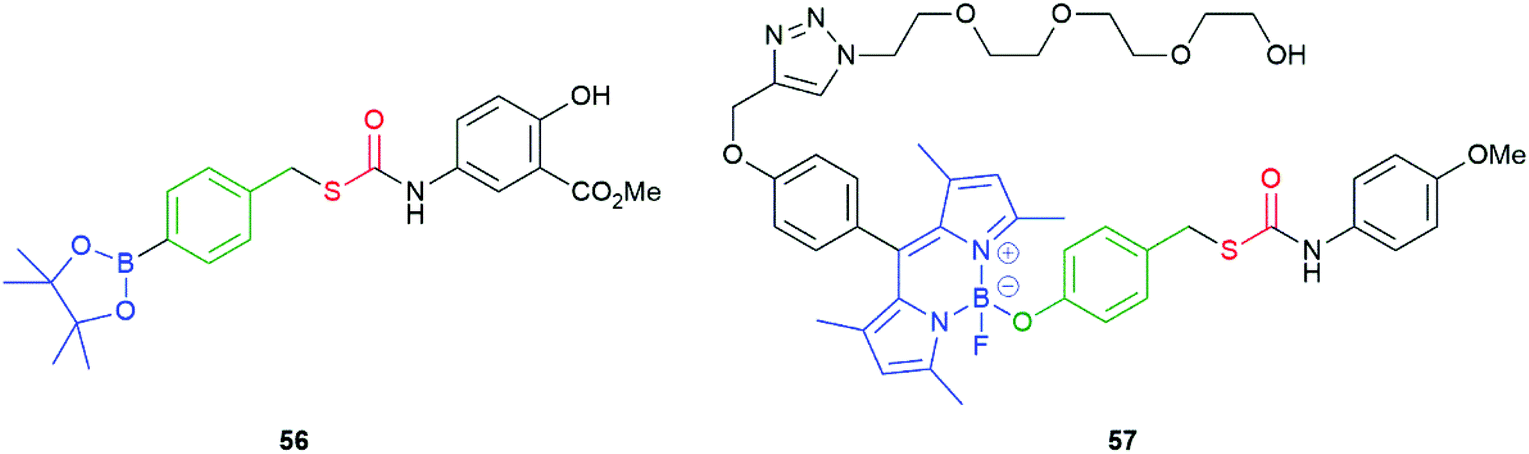 | ||
| Fig. 5 COS/H2S donor compounds 56 and 57, triggered by H2O2 and visible light, respectively.101,102 | ||
An alternate system, triggered by visible light, was also reported by the Chakrapani group.102 Thus, 57 was prepared that featured a BODIPY-based photolabile trigger group (Fig. 5); this group being modified by ‘click’ methodology to append an oligo-ethylene glycol unit so as to improve aqueous solubility. Irradiation of 57 for 2 minutes at 470 nm afforded cleavage of the trigger group, within HeLa cells. Successful cleavage was established by observation of fluorescence at 540 nm. Additionally, compound 57 was found to be non-toxic. Klán and co-workers have reported use of BODIPY-based prodrug systems, but without use of a self-immolative linker.103 Chakrapani and co-workers have also developed hydrogen peroxide triggered nitric oxide donors; whereas, Yuan et al. have reported a hydrogen peroxide triggered H2O2 sensor with NO detection capability.104,105
Developing the 1,6-elimination system in 56 and 57, Chakrapani and co-workers have exploited enzymatic triggering of COS/H2S release by protecting the phenolic OH as a POM (PivaloylOxyMethyl) group to afford compounds 58 (Fig. 6).106 For the examples when X = O, rapid cleavage of the pivalate ester occurred upon exposure to an esterase followed immediately by sequential 1,6-elimination and formation of COS with the rate largely invariant with nature of R. Broadly the same mechanistic observations were made for examples when X = NH; however, some evidence for formation of a short-lived intermediate 58a was presented. Additionally, for the X = NH compounds, the rate of release of COS/H2S was variable with the nature of R; for example, a benzyl group was found to generate the gasotransmitters more slowly than a para-methoxyphenyl group.
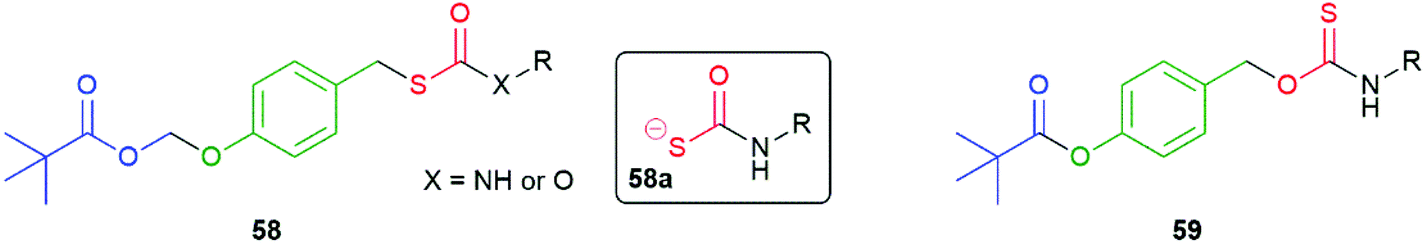 | ||
| Fig. 6 The esterase triggered COS/H2S donors 58 and 59 reported by the Chakrapani and Pluth groups, respectively.106,107 | ||
In a related study, Pluth and co-workers noted the advantage of intracellular cleavage in concentrating the release of H2S where required to solicit its action.107 The relatively slow non-catalysed rate of hydrolysis associated with the sterically demanding pivalate ester in 59 was noted, a particular concern for a phenolic ester (Fig. 6). By contrast, rapid hydrolysis was demonstrated in the presence of porcine liver esterase. The generation of H2S in this manner does not directly consume intracellular nucleophiles, such as glutathione; however, the potential of consuming them by reaction with the para-quinone methide by-product was noted (much as suggested in the original paper by Katzenellenbogen). Compound 59 was noted to be cytotoxic and the cause was attributed to inhibition of mitochondrial respiration.
The abundant intracellular nucleophile cysteine has been utilised by Pluth in a two-step triggering of release of COS/H2S from 60 (Scheme 14).108 Following an initial conjugate addition of thiolate to the acrylate moiety in 60 to afford 61, cyclisation to ε-lactam 62 releases the linker/reporter unit 63 that undergoes rapid 1,6-elimination to release COS. The two-step activation does make 60 vulnerable to trapping of the acrylate moiety by alternative nucleophiles, such as glutathione or N-acetyl cysteine, that can only function in the first step. Competitive hydrolysis by porcine liver esterase was also noted, though the rate of release of COS/H2S was slower than that with cysteine.
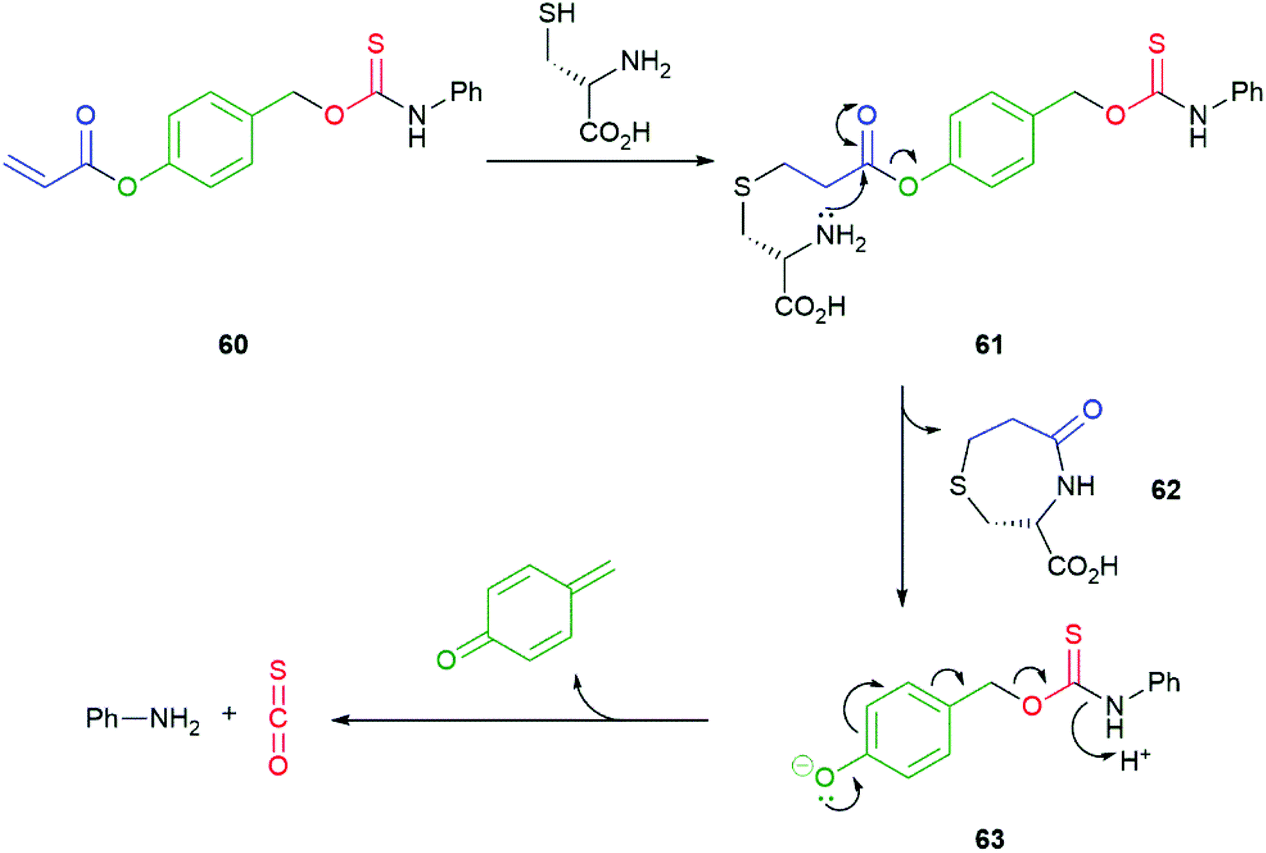 | ||
| Scheme 14 Pluth's cysteine-mediated two-step triggering of COS release from acrylate 60.108 | ||
Recognizing that self-immolation by 1,6-elimination can be initiated by an aniline as well as a phenoxide, reductively triggered self-immolative systems have been reported by Pluth, and by Singh and Chakrapani (Fig. 7).97,109 Compound 64 was designed as an analytical tool for H2S and takes advantage of the well-known reduction of azides by two equivalents of H2S to afford the corresponding amine. The subsequent 1,6-elimination and release of COS, following hydrolysis by carbonic anhydrase, replaces one half of the consumed H2S analyte. The highly fluorescent methylrhodol (λem 480–650 nm) is simultaneously released allowing quantification. A self-immolative polymer, triggered by azide reduction, for amplified release of COS, has recently been reported by Matson and co-workers (see section 4).110 An additional method for release of COS, based upon a trans-cyclooctene trigger/linker system is discussed later on in this section.111
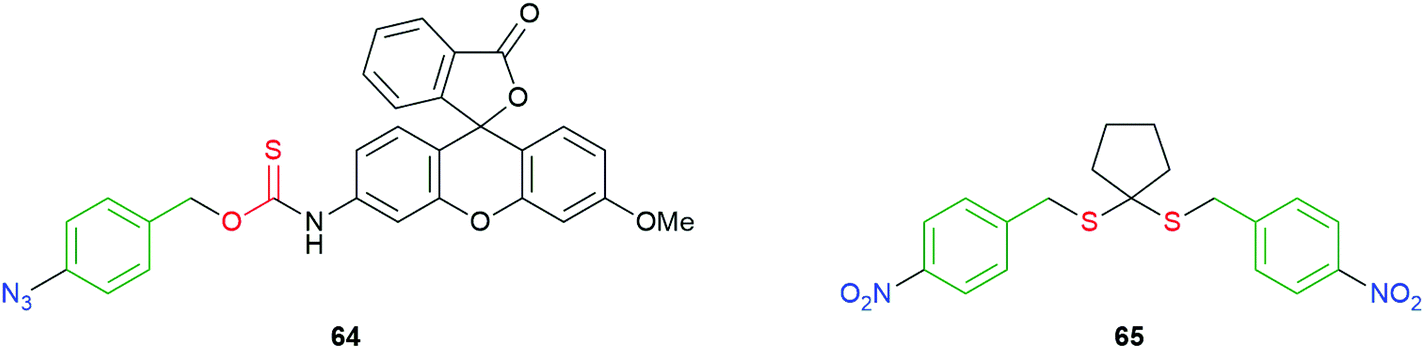 | ||
| Fig. 7 Reductively triggered COS/H2S donors 64 and 65.97,109 | ||
Rather than first releasing COS, Singh and Chakrapani utilised reduction of the nitro groups in molecules such as 65 to release H2S directly, following 1,6-elimination of the so-formed aniline/hydroxylamine (Fig. 7). E. coli nitroreductase (NTR) would be expected to reduce to the nitro group to the corresponding hydroxylamine 65a or amine 65b (see also Scheme 4). Exposure of 65 to NTR was shown to liberate H2S, as was its intracellular release in a variety of bacteria. As NTR is found in bacteria but not mammalian cells, 65 can be seen as a species selective H2S donor.
By adapting the enzymatically triggered species 58 and 59 to sulfonate 66 and analogues, Chakrapani was able to prepare cell-permeable, self-immolative molecules for controlled delivery of SO2: a cellular signalling compound (Scheme 15).112 The cyclopropyl ester was selected according to its typically slower background rate of hydrolysis than simple aliphatic esters. The bond dissociation energy of the C–S bond was assessed to be comparable to a C–O bond; thus, fragmentation during 1,6-elimination and α-elimination of SO2 was anticipated to be facile. Forming the sulfonate ester with, inter alia, a 7-hydroxycoumarin (umbelliferone) facilitated fluorescence monitoring of fragmentation (phenyl sulfonate esters were also effective SO2 donors). As an alternative to aromatic sulfonates, some alkyl sulfonate esters were examined and found to be relatively poor SO2 donors, reflecting their inferior leaving group ability. As expected, the more reactive benzyl and allyl sulfonate esters were subject to significant background hydrolysis at pH 7.4, though no generation of SO2 was observed from the resultant sulfonate. Exposure of 66 to an esterase led to consumption with a half-life of 5 minutes. Umbelliferone was formed at a comparable rate and co-formation of SO2 was established by its reaction with a coumarin-hemicyanine dye.
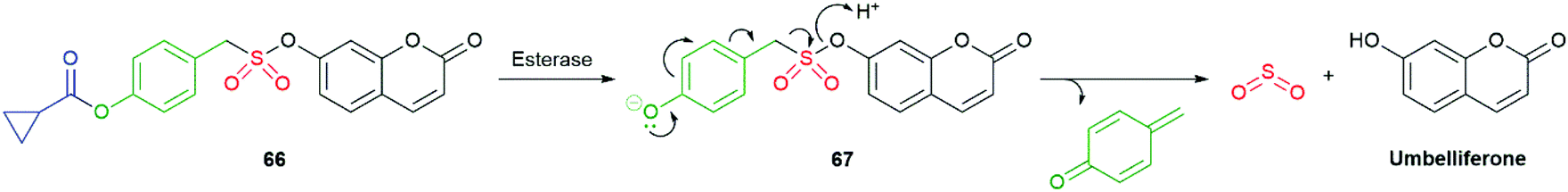 | ||
| Scheme 15 The esterase triggered SO2 donor 66 reported by Chakrapani and co-workers.112 | ||
Interest in this area has also embraced hydropersulfides, such as cysteine and glutathione hydropersulfide, as ubiquitous in mammalian cells with protein-based cysteine hydropersulfides also being significant. Together, such hydropersulfides are considered mediators of biological function. The development of small molecules capable of effecting delivery of hydropersulfides, as reagents for the formation of their biologically significant counterparts, has become a matter of interest and self-immolative linkers have played a role in their design. In particular, the instability of persulfides, especially when an unprotected SH is present, suggests that selective release of such species from a protected form is important. For example, in the style of Mosey and Floreancig's hemiaminal-based linker 35 (Scheme 7), a persulfide donor 68 that liberates an unstable disulfaneylmethanol 69, upon cleavage with an esterase, was developed by Wang (Scheme 16a).88 Once formed, 69 spontaneously undergoes a 1,2-elimination to release a single hydropersulfide species 70 allowing a detailed study of its biological effects. Reaction of 70 with thiols also generates the gasotransmitter H2S. Variation of the structure of R1 allows control of the rate of release; whereas, changes to R can tune its aqueous solubility, e.g.68a (R1 = Et) was found to exhibit protective effects in a murine model of heart myocardial infarction-reperfusion-injury studies. Xian has described a range of self-immolative persulfide donors of which 71 serves as an example, these are triggerable by fluoride/acid, Scheme (15a).89 Of further interest is the conversion of 71 into the potentially esterase sensitive compound 72. Additionally, related H2S and HSNO donors were reported. Overall, these silyl-triggered SIEs appear very flexible synthetically.
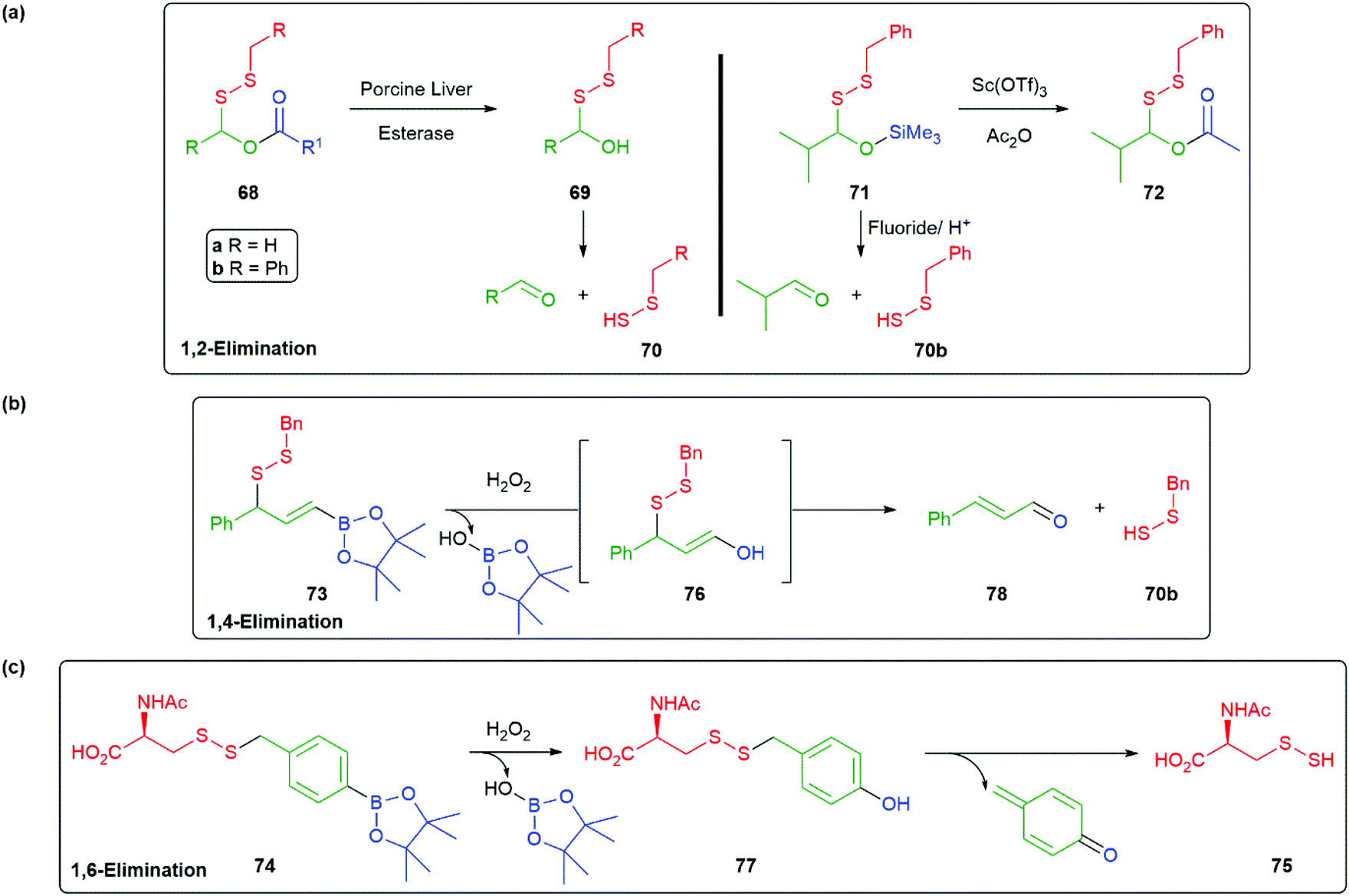 | ||
| Scheme 16 (a) Esterase or fluoride/acid mediated release of a hydropersulfide from 68 or 71via a 1,2-elimination; (b) hydrogen peroxide triggered release of hydropersulfide from 73 by 1,4-elimination; (c) hydrogen peroxide triggered release of hydropersulfide from 74 by 1,6-elimination.88,107,113 | ||
Two related reports have appeared, by Chakrapani and Matson, that target elevated levels of hydrogen peroxide in cells undergoing oxidative stress (vide supra) by oxidative cleavage of the C–B bond in 73 and 74 (Scheme 16b and c).101,113 These reagents utilise 1,4- and 1,6-elimination to liberate 70b and 75 from 76 and 77, respectively. In each case, these compounds were shown to offer protection to cells undergoing oxidative stress. Chakrapani and co-workers noted that the cinnamaldehyde by-product 78 is a Generally Regarded As Safe (GRAS) compound, though it does offer some potential for re-trapping 70b. In each case, the oxidation with hydrogen peroxide was found to be chemoselective for the boronate ester over the persulfide moiety. These and other studies on reactive sulfur species has been expertly reviewed by Bora et al. in 2018.114
Using new versions of Schaap's dioxetane,115–117 such as 79, with improved chemiluminescence properties, Shabat and co-workers have developed a range of self-immolative detectors e.g. for galactosidase 80 and glutathione 81a (Fig. 8).118 A particular advantage of these new dioxetanes is that they maintain high light emission efficiency in aqueous solutions, greatly assisting their application in biological systems. The fragmentation of dioxetane 79 affords a deprotonated, excited state benzoate, as the final emissive species. Attachment of an acrylate/acrylonitrile moiety ortho to the phenolic OH was key to the improved emissive properties. The ortho-chloro substituent lowers the pKa of the phenol, increasing the percentage that is deprotonated at a given pH, a prerequisite for chemiexcitation and chemiluminescence.119 Compound 79 exhibits ϕCL 13.8% (λmax = 525 nm); in the absence of the acrylonitrile moiety, the chemiluminescent quantum yield falls to ϕCL 0.003% (λmax = 470 nm).32 A detailed summary of the chemistry underpinning these dioxetanes has been reported by Gnaim et al.28 Additionally, Shabat and co-workers were able to demonstrate further enhancements to light emission by formation of an inclusion complex between a range of dioxetane probes, e.g.80 and trimethyl β-cyclodextrin; further, some of these were shown to be effective for in vivo imaging.120 Utilising the H2S released by biodegradation of β-lactam antibiotics, following ring opening by a β-lactamase, Spitz and Shabat have developed several H2S sensitive probes e.g.81b that allow screening for antibiotic resistance.121 Wu and Zheng have used a similar trigger to that in 81a-b to release camptothecin (see section 3.1).122
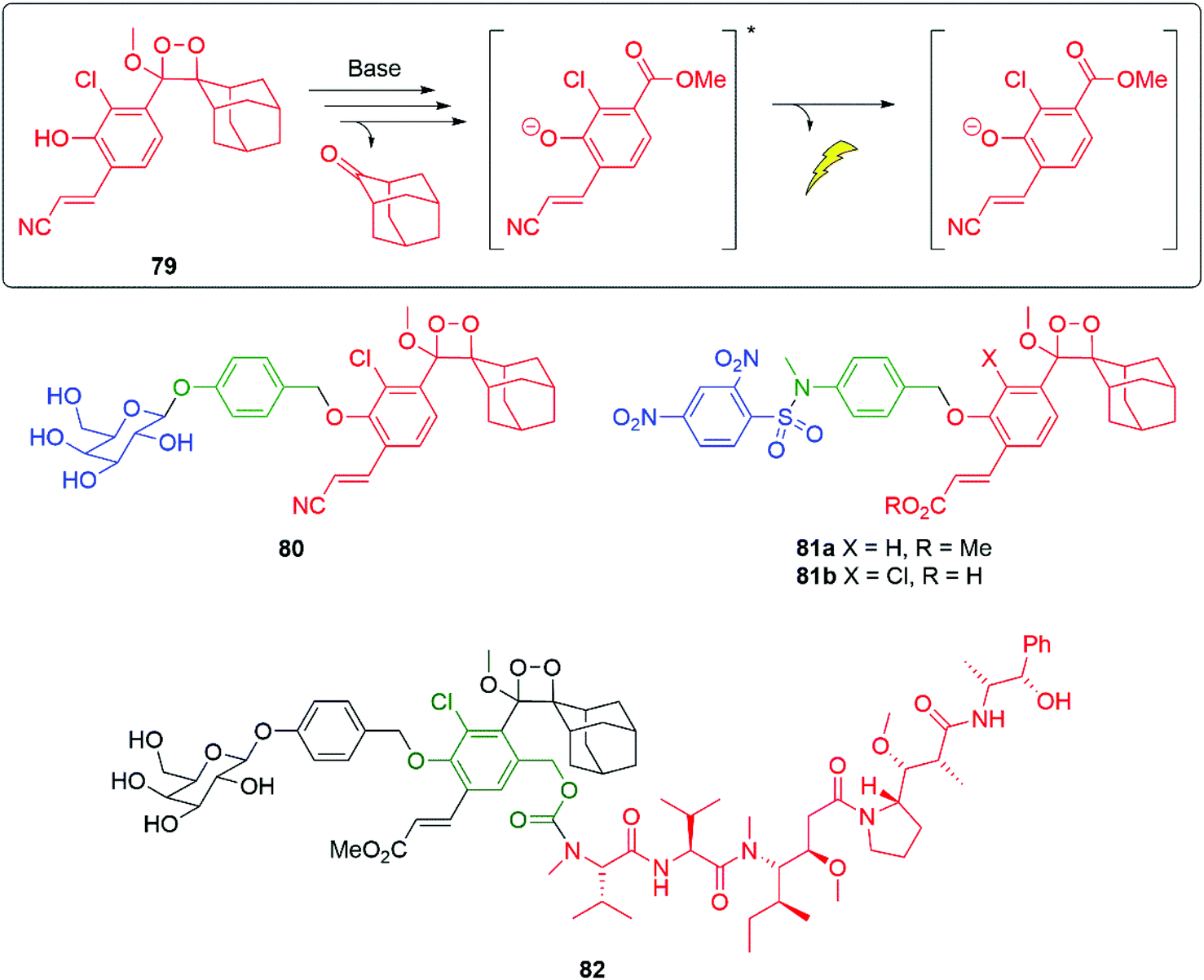 | ||
| Fig. 8 Modified Schaap's dioxetanes for improved chemiluminescence.118 | ||
Developing self-immolative entity 80 into 82, to make the aromatic ring of the chemiluminescent moiety a second linker unit, allows the luminescent reporting to be accompanied by delivery of the chemotherapeutic agent monomethyl auristatin E (Fig. 8).123 Of significance is that such reporting was established to work in vivo in mice bearing CT26-LacZ tumours.
Developing the applications of these new 1,2-dioxetanes has facilitated ultrasensitive self-immolative sensors for the key food pathogens Salmonella and Listeria monocytogenes. The limit of detection of these sensors outperforms common fluorescent probes. The sensors 83 and 84 function by targeting, respectively, a Salmonella esterase and a phosphatidylinositol-specific phospholipase C that is species specific (Fig. 9).124
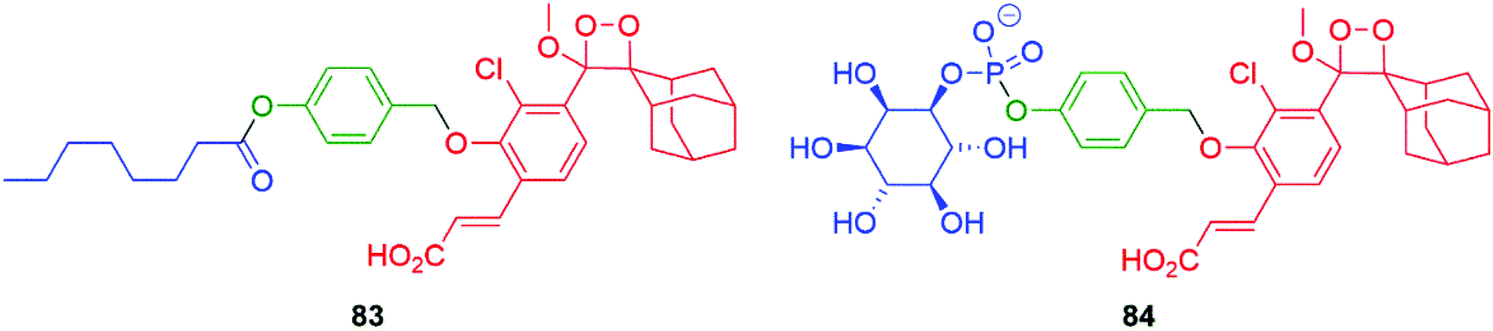 | ||
| Fig. 9 Selective chemiluminescent sensors 83 and 84 for rapid detection of Salmonella and Listeria monocytogenes.124 | ||
Building on earlier work by Renard and Romieu,125,126 connecting the chemiluminescent probe through a self-immolative aniline linker, giving probes of type 85 (see Fig. 10), allows a range of peptide triggers to be attached that can target specific proteases. For example, Brik and Shabat reported that attaching the C-terminal sequence of ubiquitin protein (Ub(1–75)) to the aniline nitrogen through a glycine residue (85a) allows an assay for deubiquitinase activity to be developed.127 Since deubiquitinating enzymes are involved in various human diseases, monitoring of their activity is valuable. Probe 85a was found to be effective at detecting activity from three different deubiquitinases by cleavage of the scissile bond: UCH-L3, UCH-L1 and USP-2, with the first two giving the stronger response. Kitamura, Shabat and Vendrell have shown that appending peptide 85b was found to selectively target granzyme B, a key enzyme in the functioning of natural killer cells; these cells can kill certain types of cancer cells. This probe has allowed imaging of granzyme B activity in mouse models.128 Attaching the peptide sequence in 85c allows targeting of prostate specific antigen proteolytic activity; this has been applied to the forensic detection of human semen on fabrics.129 Bogyo et al. have studied the imaging of the clinically very important disease tuberculosis; this has been by addressed with the peptidic sequence in 85d. This sequence targets the Hip1 protease in Mycobacterium tuberculosis, allowing imaging of such cells either in culture or in human sputum samples.130
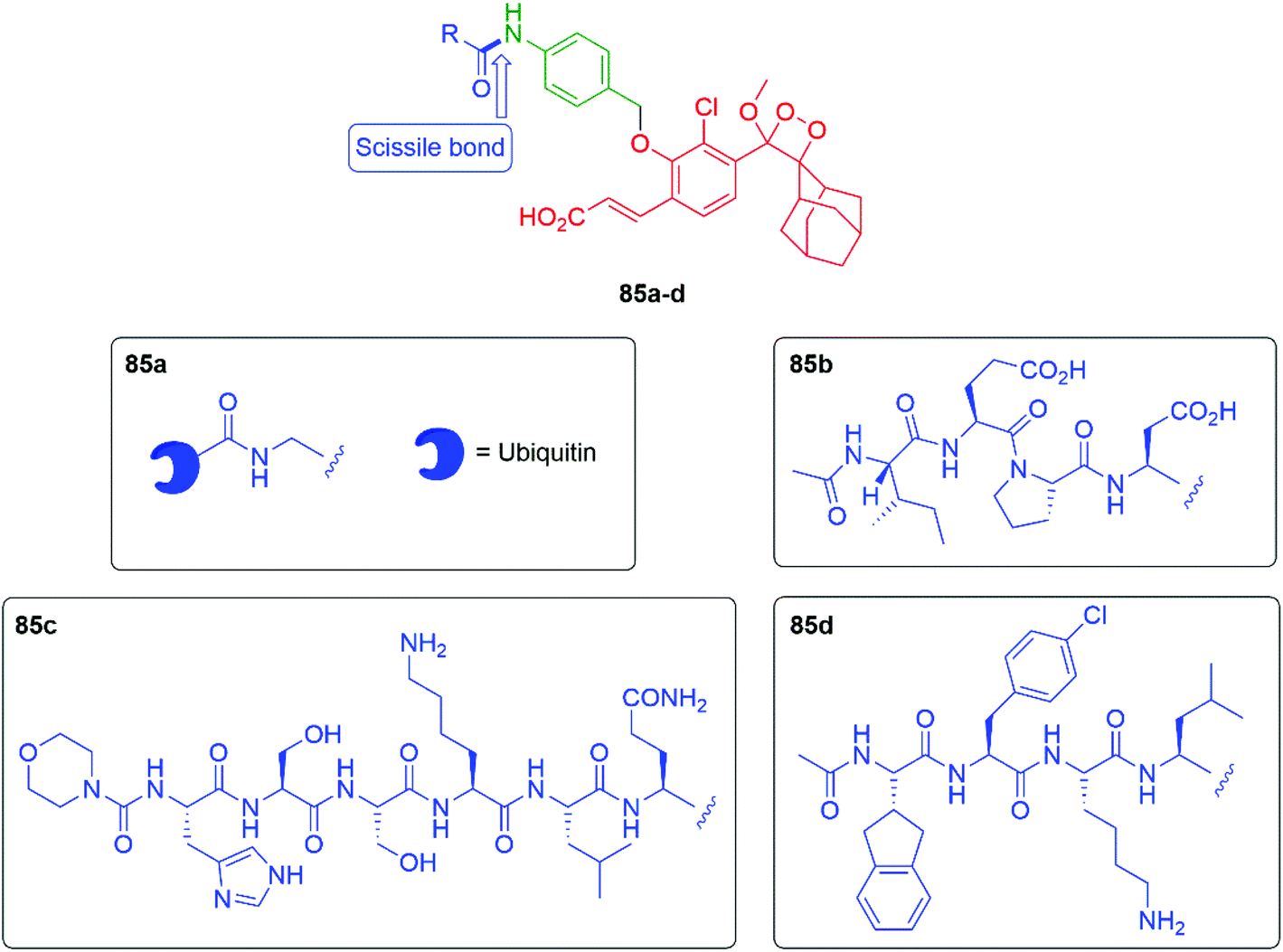 | ||
| Fig. 10 Chemiluminescent probes 85a–d that target of protease activity in four different contexts.127–130 | ||
Using solid-phase methods, a general approach to the synthesis of these self-immolative, chemiluminescent probes that are triggered by proteases, has been described by Ponomariov et al.131
As an alternative to targeting proteases, conjugation of the nitrogen of the aniline linker to a substrate for the copper dependent enzyme tyrosinase allows selective oxidation from sensor 86 to ortho-quinone 87 (Scheme 17).132 When 87 forms in the presence of thiols, for example, glutathione then cleavage occurs to 88 and sets in train disassembly of the linker and ultimately chemiluminescence from an excited state of 89. The electrophilicity of ortho-quinones toward thiols is known and has been examined in a biological context by Fellmann, Doudna and Francis.133 Such conjugate addition is expected to form a catechol, allowing the trigger to cleave. The presence of the 2-dimethylaminoethyl unit facilitates cell permeability. The typically high levels of tyrosinase expressed in melanoma cells makes this probe of significance in cancer imaging. Similar metabolic activity of tyrosinase has been shown as effective at triggering self-immolation by Ma and Zhang, in turn acting as sensors for that enzyme.134,135
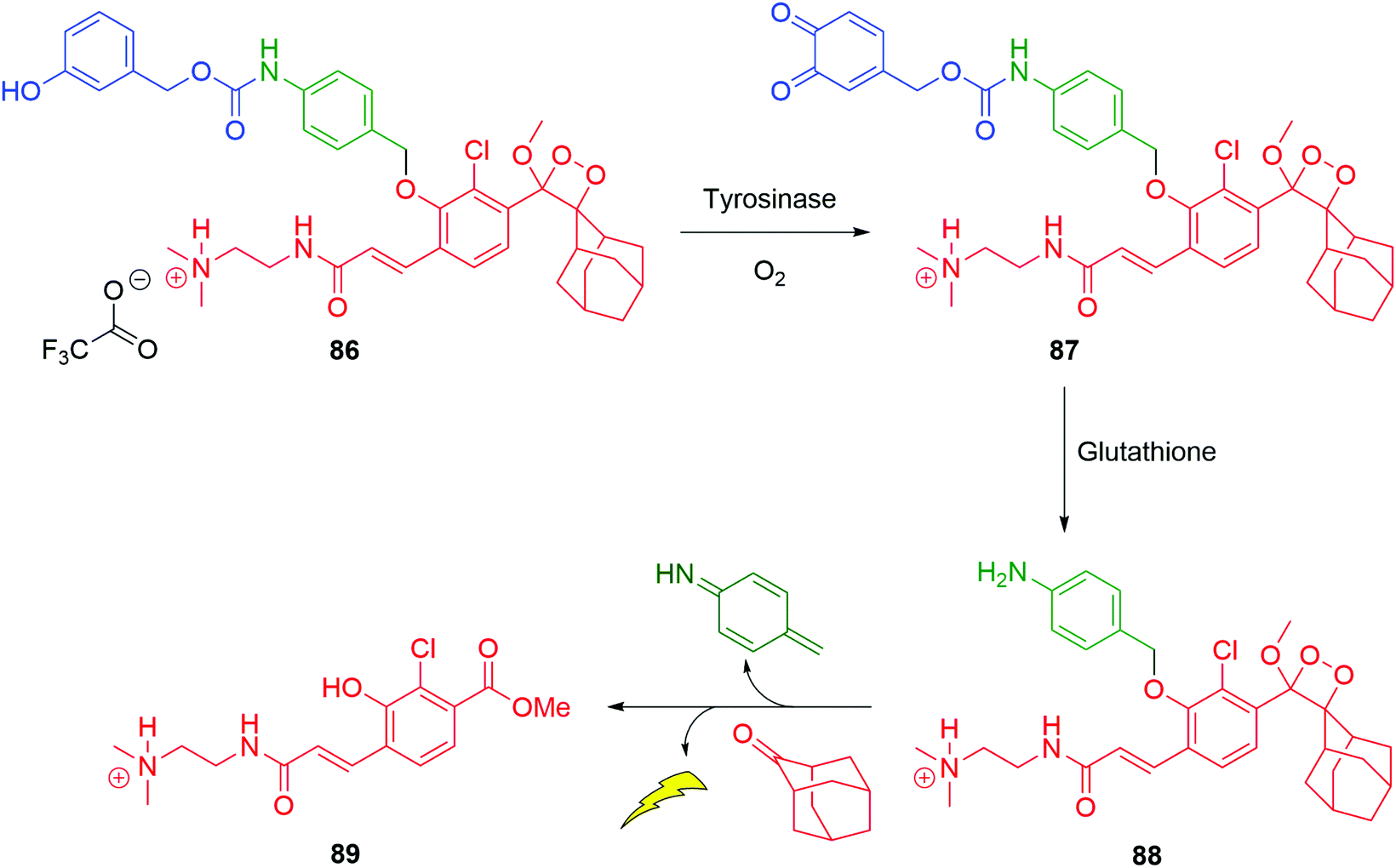 | ||
| Scheme 17 Satchi-Fainaro, Shabat and Sessler's sensor 86 for a chemiluminescent response to the simultaneous presence of tyrosinase and thiols.132 | ||
Further studies by Shabat and co-workers have seen deployment of chemiluminescent 1,2-dioxetanes in a polymeric framework (see section 4.1).43
Hou et al. have developed a ratiometric self-immolative probe 90 for β-galactosidase that changes from blue (475 nm) to green (545 nm) fluorescence upon triggering to release the 4-amino-1,8-naphthalimide fluorophore 91 (Scheme 18).136 They report an interesting observation with respect to the substituent R on the linker, finding that the rate limiting step for the probe was enzymatic hydrolysis and thus the primary contribution of the substituent was to affect the Michaelis–Menten constant (Km) of the substrate for the β-galactosidase, the nitro substituted compound having the lowest value.
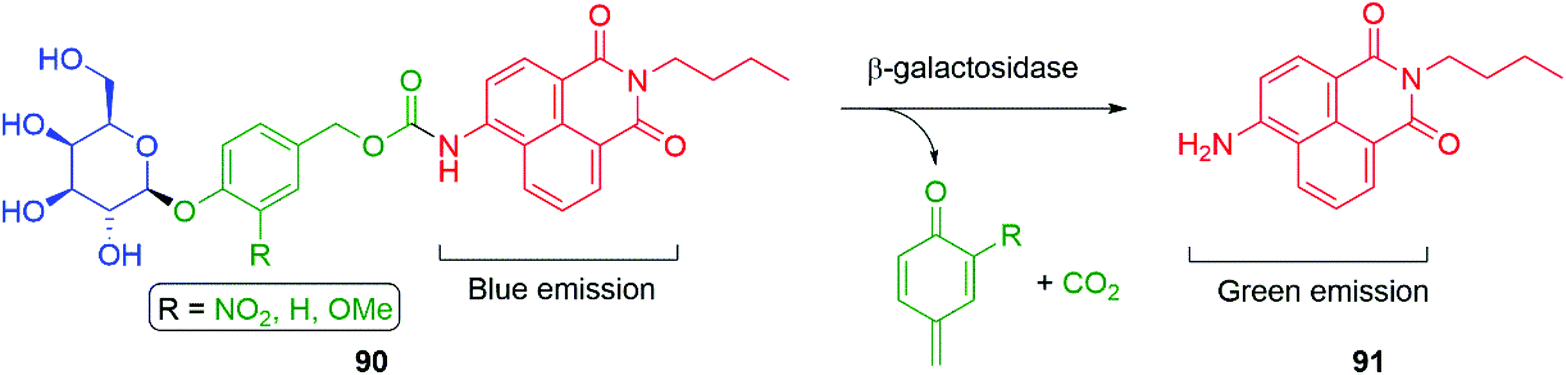 | ||
| Scheme 18 The β-galactosidase probes 90 developed by Hou and co-workers.136 | ||
Probing redox biology is important in understanding cellular function. Yang has introduced an innovative method for sensing hydrogen peroxide based upon sequential Payne and then Dakin reactions; hydrogen peroxide is converted into a reagent suitable for rapid Dakin oxidative conversion of an aromatic aldehyde into a phenol (Scheme 19).137 When applied to pro-fluorescent agents 92 or 93, the presence of hydrogen peroxide triggers 1,6-elimination and release of their strongly fluorescent reporter groups at a rapid rate. It was found that Cl3CCN, 92 and 93 were non-toxic to RAW264.7 macrophages and were successful in imaging hydrogen peroxide activity in RAW264.7 cells. Shabat and Yang successfully extended this methodology to chemiluminescence by preparing probe 94 and demonstrating its effectiveness in imaging live cells and real-time monitoring of rat brain.138
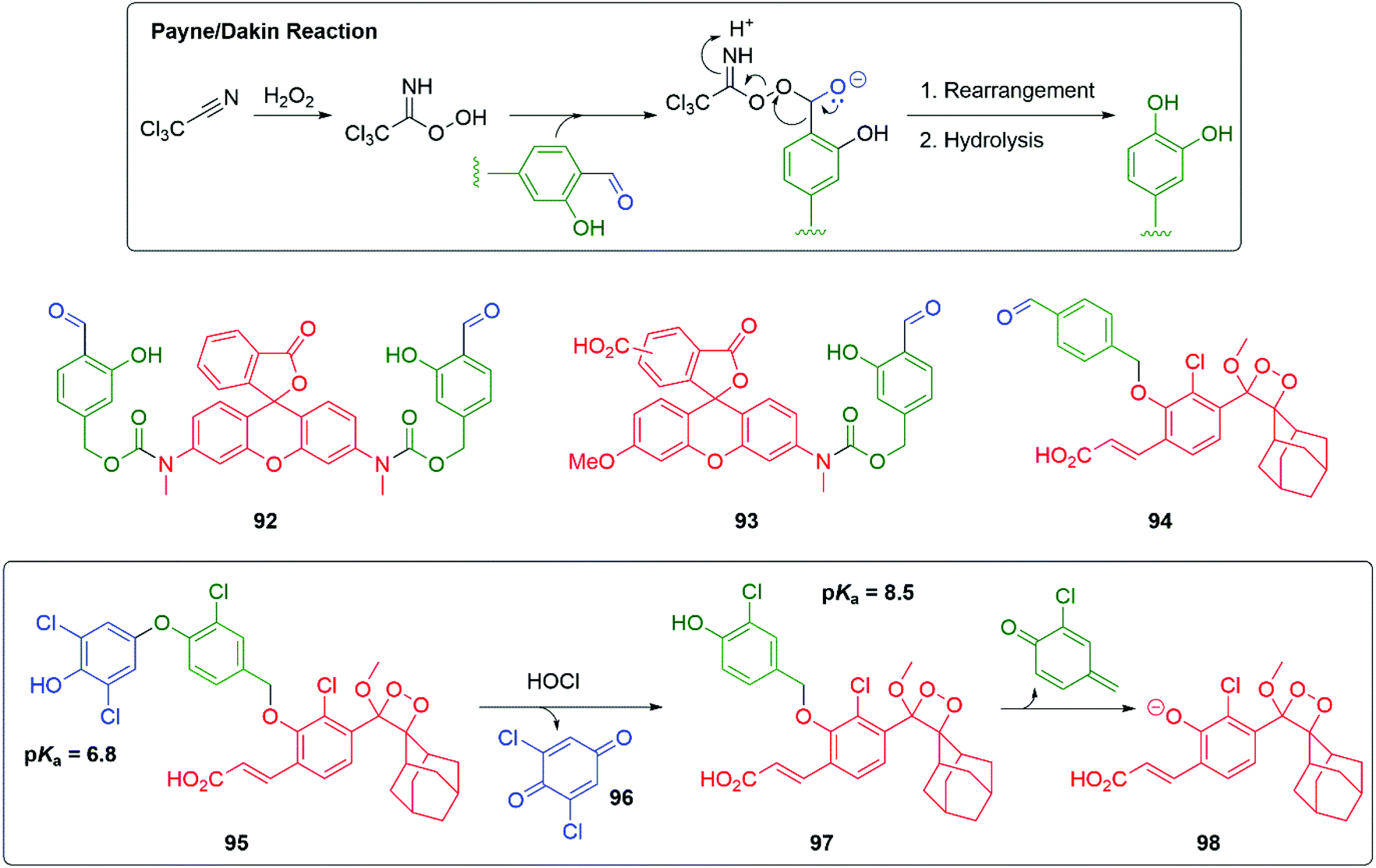 | ||
| Scheme 19 Yang and Shabat's sensors for oxidative activity the cell triggered by H2O2 or HOCl.137–139 | ||
By varying the trigger group and a subtle modification to the linker, Shabat and Yang have extended this work to the sensing and real-time imaging of HOCl in mice (Scheme 19).139 Thus, in 95 a phenoxy trigger group has been attached to the linker; the two ortho-chloro substituents on the trigger (pKa 6.8) ensured that at physiological pH greater than half of the phenol will be ionised, enhancing the nucleophilicity of the aromatic ring. It would also resist chlorination of this ring by HOCl. When exposed to HOCl, oxidative cleavage occurred with loss of quinone 96 and release of the linker reporter unit 97; this will fragment by 1,6-elimination leading, after fragmentation of 98, to chemiluminescence. The addition of a chloro substituent to the linker resisted further chlorination and lowered the pKa of the phenol.
Antibiotic resistance is one of the major challenges in medicine and medicinal chemistry. The role of β-lactamases is central to that challenge and probes that can offer an easily applied diagnostic tool to a given sample, for their presence, is of considerable importance. Spitz and Shabat have reported such a probe 99 for carbapenemase activity (Scheme 20).140 In the presence of a carbapenemase e.g. SPM-1 (a metallo-β-lactamase), the β-lactam ring of 99 undergoes hydrolysis to give 100, setting up an enamine-driven 1,8-elimination to give the chemiluminescent dioxetane 98 discussed above.
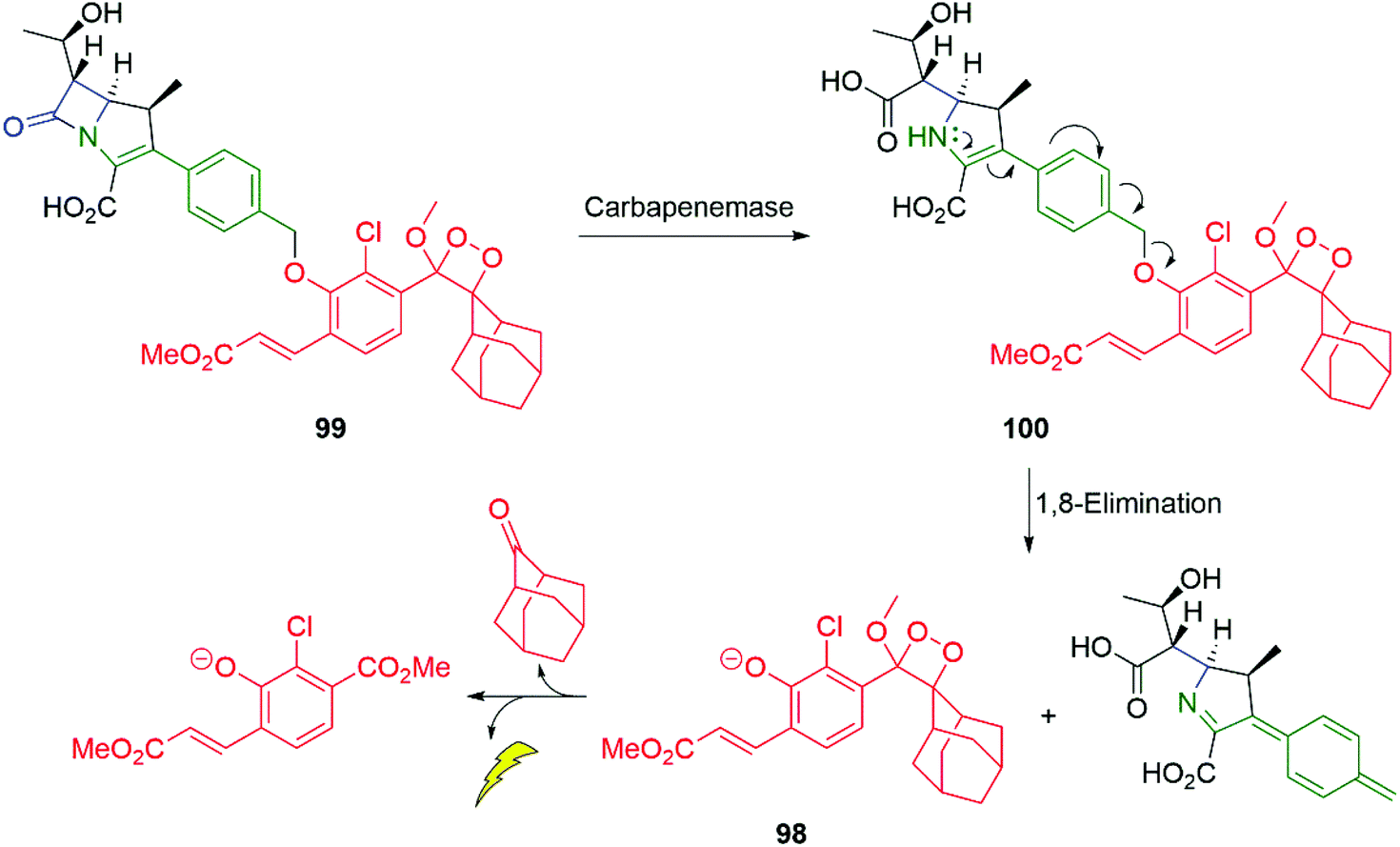 | ||
| Scheme 20 Spitz and Shabat's probe 99 for carbapenemase activity.140 | ||
Formaldehyde is an important one carbon source for biological systems, frequently stored as part of a folate molecule. The low concentrations and transient nature of formaldehyde in biological systems has resulted in an interest in developing probes for its detection. Building on Chang's extensive studies on the self-immolative aza-Cope rearrangement strategy to trigger a fluorescent response to the presence of formaldehyde, chemiluminescent probe 101 was developed by Chang and Shabat (Scheme 21).141–144 Upon exposure to formaldehyde, the primary amine of the Chang trigger condenses with it to give the corresponding imine 102 that undergoes an aza-Cope rearrangement to afford 103. Hydrolysis of 103 delivers ketone 104 that undergoes β-elimination to release the dioxetane 98 that undergoes decomposition with release of a photon at 540 nm. To improve imaging in vivo imaging probe 105 was developed that emits radiation at 700 nm.
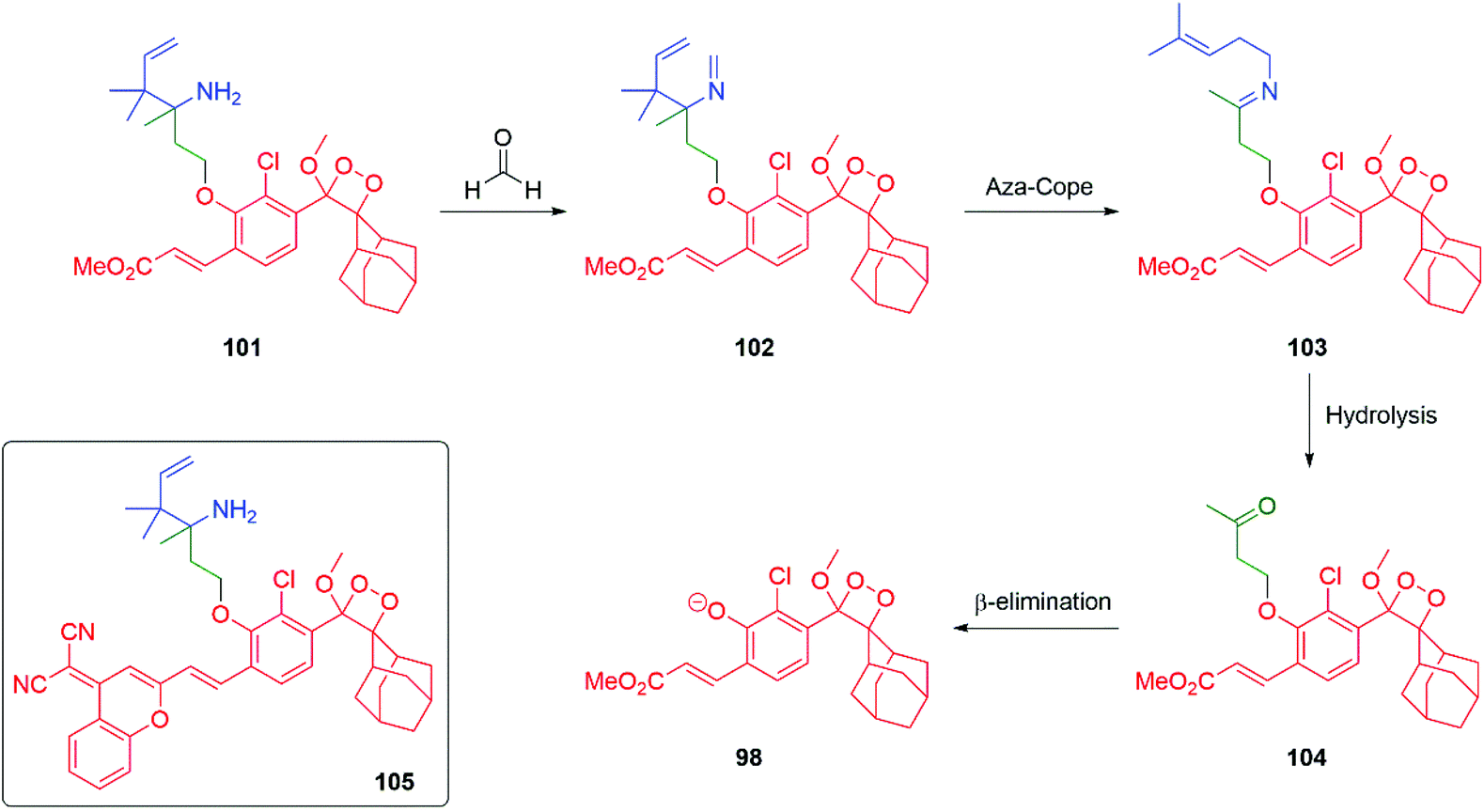 | ||
| Scheme 21 Chang and Shabat's aza-Cope driven probe 101 for folate-derived formaldehyde.144 | ||
By contrast, Ma has demonstrated that imine formation by oxidation of an amine, followed by hydrolysis and β-elimination of acrolein, to release a self-immolative phenol, can serve as a selective sensor for monoamine oxidase A (MAO-A) over MAO-B.145
The Staudinger reduction of α-azido-ether groups to generate unstable N,O-acetal moieties that collapse with the release of a reporter group have been important, inter alia, in nucleic acid detection studies and have been reviewed by Winssinger, Seitz and Kool.146–148 The utility of the N,O-acetal functional group in SIEs has been illustrated in Scheme 7. The development of templated Staudinger azide reduction into a SIE technology for release of functional molecules has been reported independently in 2013 by Sadhu and Winssinger (Scheme 22), and by Ito and Abe (Scheme 23).149,150 In particular, the bioorthogonality of the azide group was seen as important in allowing the method's implementation within cells. Winssinger employed a strategy of synthesising a peptide nucleic acid then condensing it with the SIE to afford oxime 106 (Scheme 22). Subsequent reduction of the azide in 106 generates the corresponding aniline 107, that then permits degradation to liberate doxorubicin as the reporter group. The alternative reporter groups rhodamine methylurea and estradiol were also examined. Model azide reductions indicated Staudinger reduction and fragmentation of the first linker stage to be very fast; whereas, the second linker self-immolated at a rate related to the pKa of the reporter group, with some attenuation for the carboxyl linker.151 However, even the slowest system examined, doxorubicin, was found to have a t1/2 of 21 minutes. Further studies by Winssinger have utilised templated azide photoreduction, mediated by a Ru(II) complex, in live cells.152 The method was found to give very high turnover in the reduction and high sensitivity in nucleic acid sensing.
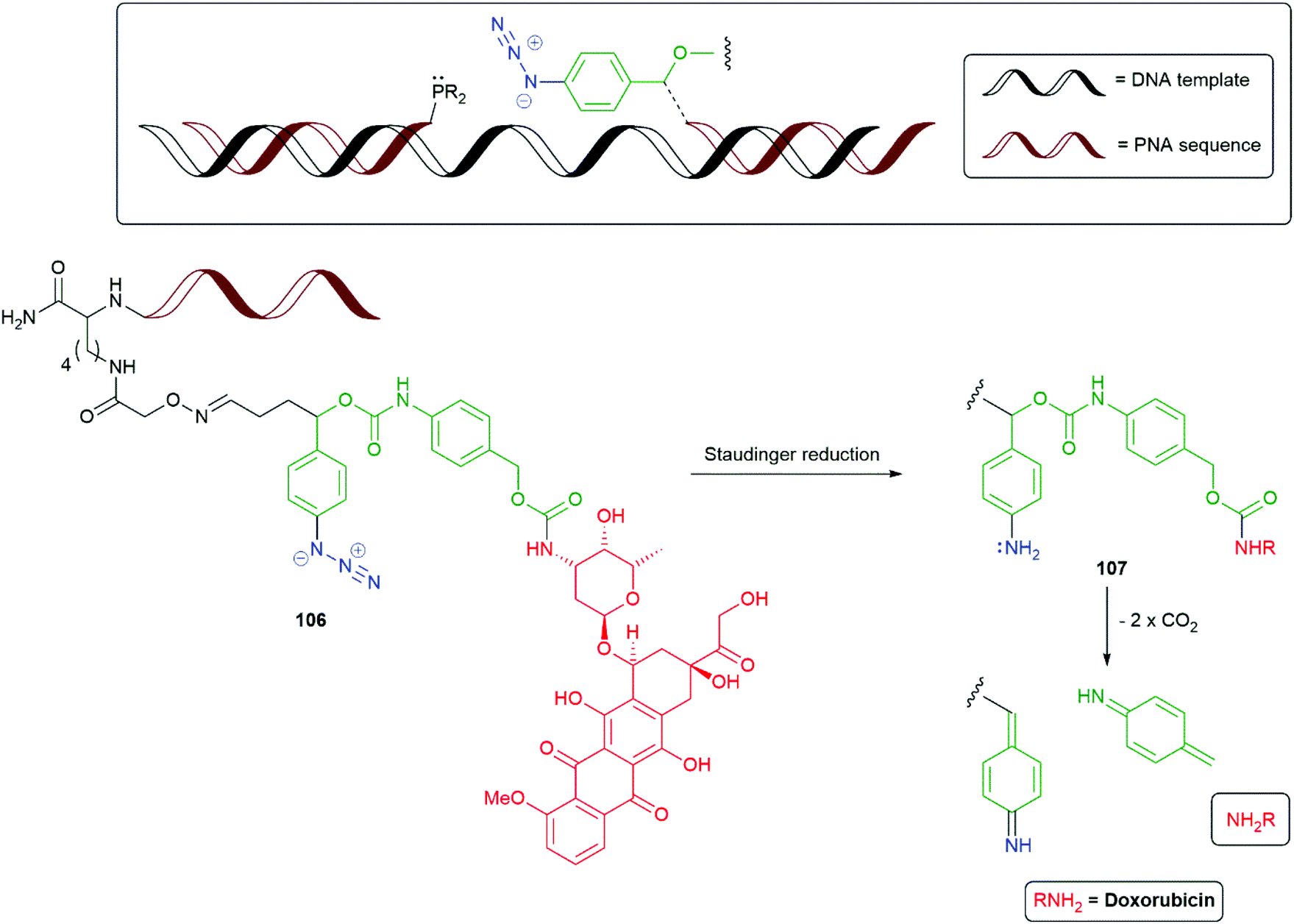 | ||
| Scheme 22 The templated Staudinger azide reduction triggered SIE 106 reported by Winssinger and co-workers.149 | ||
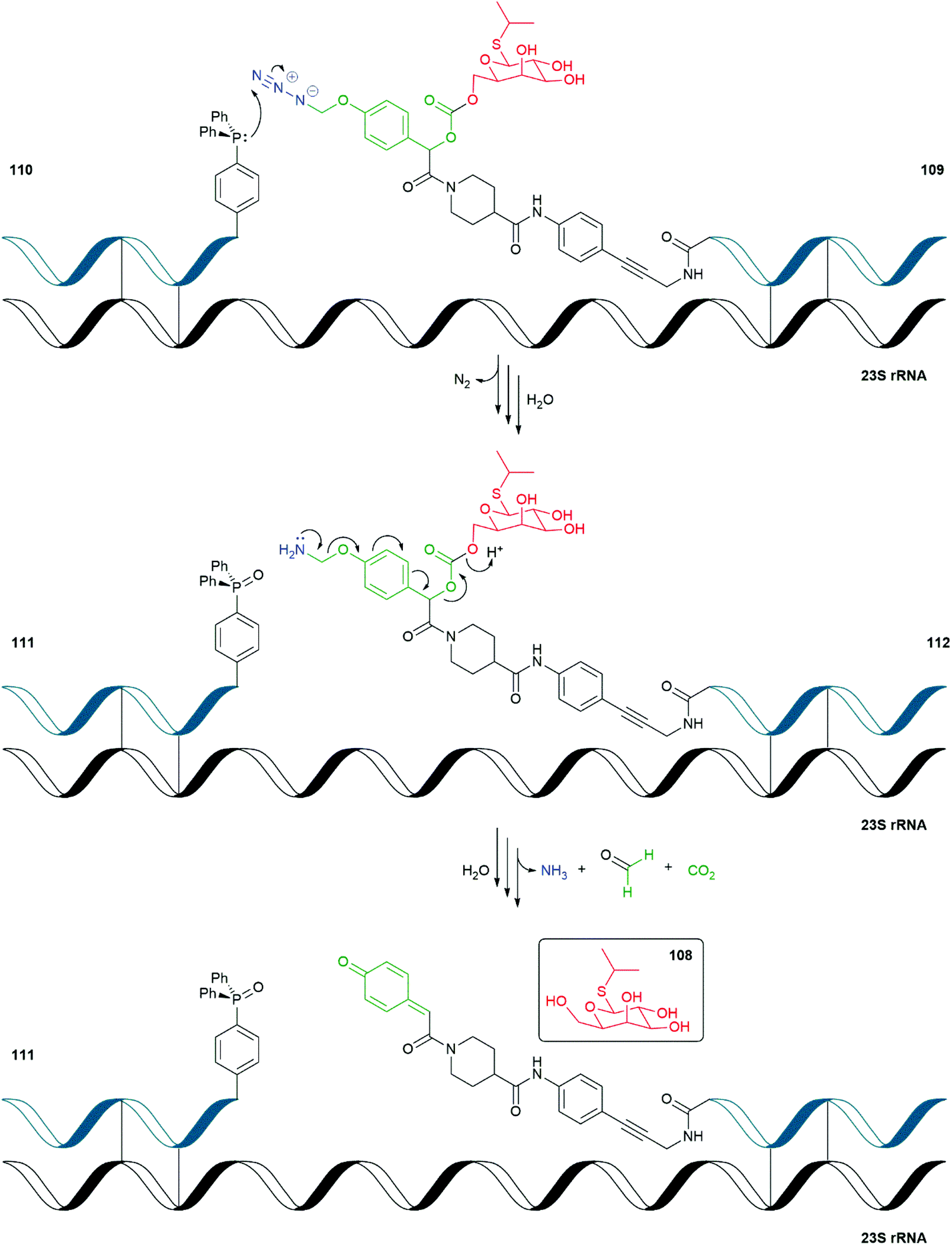 | ||
| Scheme 23 The RNA templated self-immolative system 109 reported by Ito and Abe.150 | ||
Ito and Abe have described an RNA-templated SIE decomposition that releases isopropyl-β-D-thiogalactoside (IPTG) 108 within an E. coli cell (Scheme 23).150 IPTG, when present in a bacterial cell, induces expression of plasmid-based genes that are under the control of the lac operator by binding to the lac repressor. In their study, Ito and Abe have used the Staudinger reduction of azide triggered para-hydroxymandelic acid-based SIE 109 by RNA bound triphenylphosphine 110 affording, after hydrolysis, the corresponding phosphine oxide 111 and linker system 112. Upon loss of formaldehyde imine and 1,6-elimination of the mandelate unit, decarboxylation releases IPTG 108. Additional DNA-templated FRET systems were also reported.
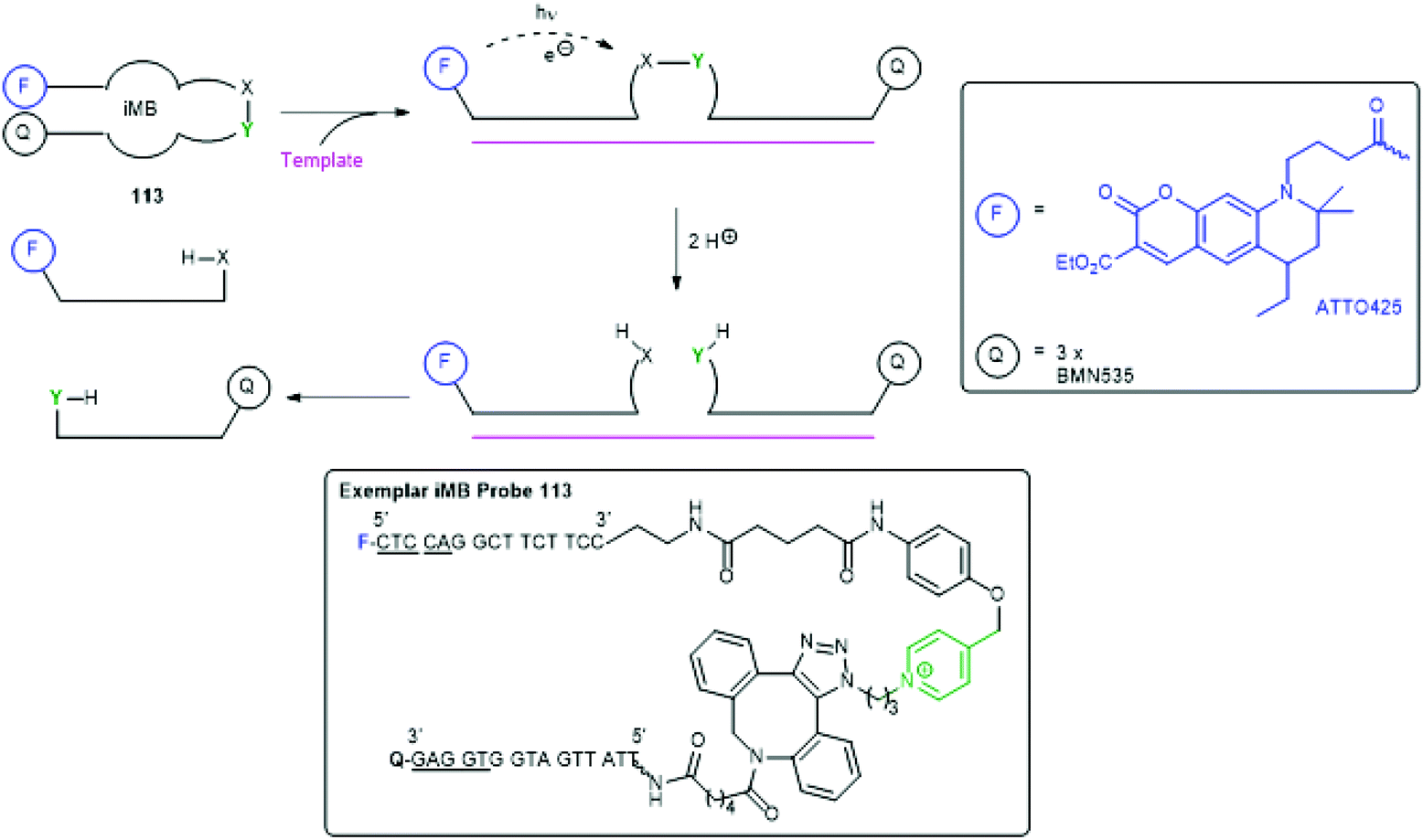
Recently Seitz and co-workers have presented an interesting, templated SIE method (an immolative Molecular Beacon iMB) that allows amplification of a fluorescence signal, facilitating detection of RNA/DNA target sequences at pM levels (Scheme 24).153 It involves a novel and unusual situation in which the trigger and reporter group are the same, activation of which photosensitises cleavage of a remote linker. Thus, an immolative molecular beacon 113 was prepared that uses a pentamer stem (underlined nucleotides) to give a hairpin structure that brings the fluorescent reporter group (ATTO425) into proximity with a quencher group (three copies of BMN535). Upon exposure to the target RNA/DNA template, the hairpin opens allowing fluorescence upon exposure to 455 nm light. Simultaneously, this photosensitises cleavage of the N-alkylpicolinium-phenol ether linker, mediated by ascorbic acid. Each fragment of the cleaved probe has a lower binding energy for the template than the probe itself, allowing dissociation, continued fluorescence and reuse of the template.
 | ||
| Scheme 24 The immolative molecular beacon (iMB) 113 reported by Seitz and co-workers.153 | ||
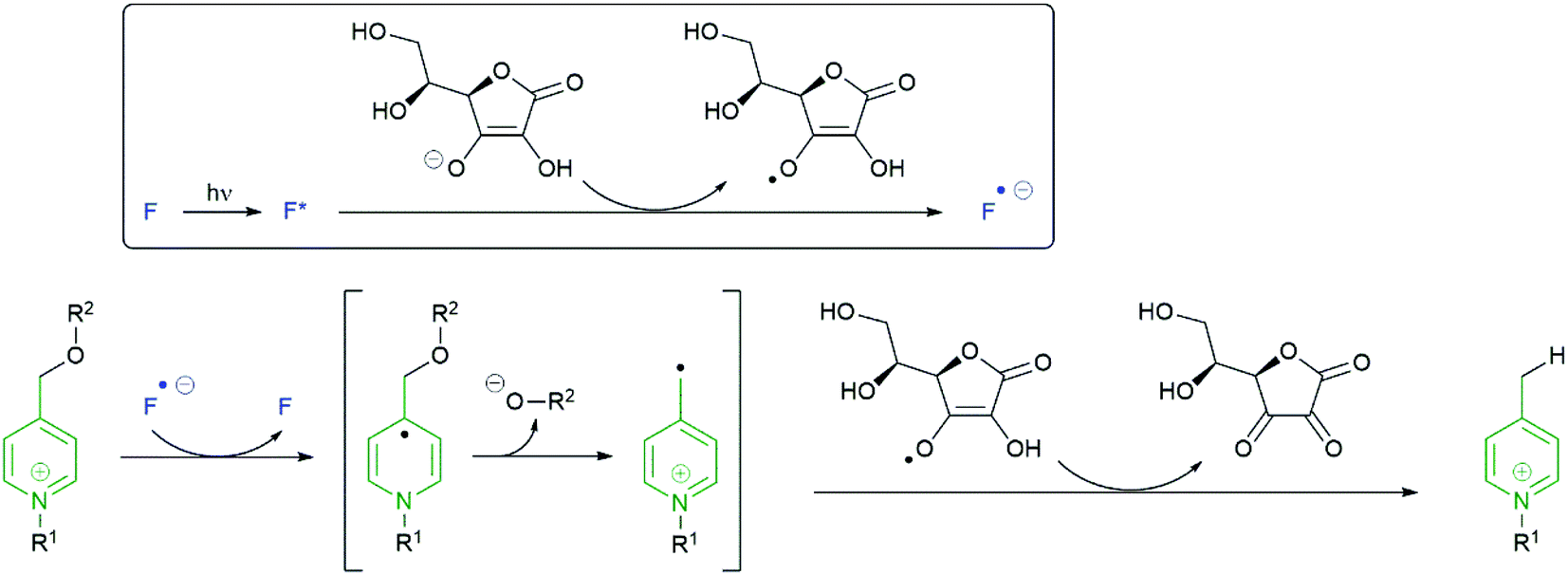
The mechanism illustrated in Scheme 25 has been proposed153 to account for reductive cleavage of the N-alkylpicolinium-phenol ether (NAP) linker.
 | ||
| Scheme 25 The proposed mechanism for cleavage of the NAP-phenyl ether linker 113.153 | ||
Building upon earlier work on light activation of 114, Schmidt, Jullien and co-workers studied light activation (365 ± 25 nm) of SIEs of type 115 to underpin a detailed assessment of the kinetics of disassembly of representative self-immolative systems, such as 116, and the relationship their of structure to the observed rates (Fig. 11).151,154 The results emphasise the speed and utility of photochemical triggering, a technique that has found a number of applications in the field, e.g.Schemes 47 and 57. The data reported are of general importance; for example, the relative rates of decarboxylation of carbamates and carbonates, two commonly employed parts of SIE design, are determined. Also, quantitative data on the effect of electron withdrawing and electron donating groups on the linker add further useful design information. The authors have drawn attention to the idea of a self-immolative linker as a ‘molecular clock’ and report further quantitative information on the importance of pH on their disassembly. The significance of such considerations is given useful focus in the recent results reported155 by Bradley and co-workers that show reaction-condition-dependent stability of a self-immolative (SI) linker. This phenol-based linker–reporter system proved isolable under some conditions yet disassembled expeditiously under others (see Schemes 35 and 36). A broader consideration of the kinetic aspects of SIE disassembly, across many systems, was covered in their recent review.21
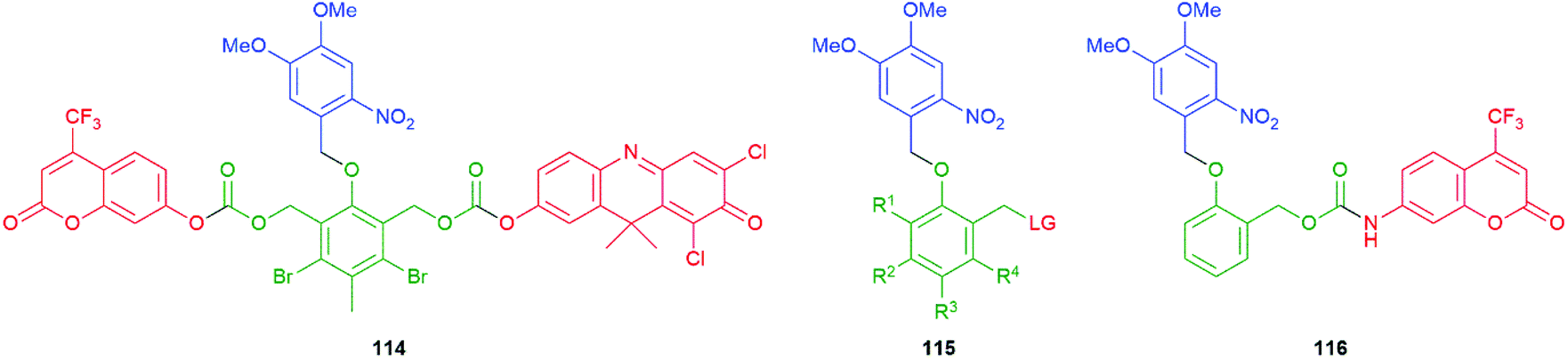 | ||
| Fig. 11 The photoactivatable self-immolative systems 114–116 subject to kinetic studies by Schmidt, Jullien and co-workers.151,154 | ||
An interesting application of photo-triggered self-immolative linkers has been reported by Zhou (Scheme 26).156 The system 117 conjugates a new self-immolative linker/reporter group to a carrier peptide or protein; this peptide/protein has an appropriately placed primary amine e.g. a lysine residue, the ε-amino group of which then becomes critical to fragmenting the system once photo-decaging has taken place. The method is based upon earlier work on the fragmentation of DNA by primary amines at C4′-oxidized abasic sites (referred to as C4APs) and has focused upon the biocompatibility of both the initial system and the breakdown products, with the added advantage that the carrier could assists with important functions such as cell penetration. Thus, irradiation of 117 at 365 nm led to quantitative cleavage of its two ortho-nitrobenzyl trigger groups to afford 118. Equilibria with ring-opened forms of 118 facilitates formation of imine 119 using the intramolecular ε-amino group of a lysine side-chain. Elimination of the carbamate, possibly via the enamine form of 119, releases the doxorubicin reporter group. Cyclisation of 120 with stepwise addition and re-elimination of water gives 121 as the final product. Release of doxorubicin was found to reach 85% within 1 h, following irradiation for 5 min at 365 nm, when H3-V35C was used as the carrier peptide. The conjugate was also shown to be transported into the cytoplasm of HeLa cells and release its doxorubicin upon irradiation, only then did the drug move to the nucleus. The new linker was also shown to be compatible with antibody directed methods.
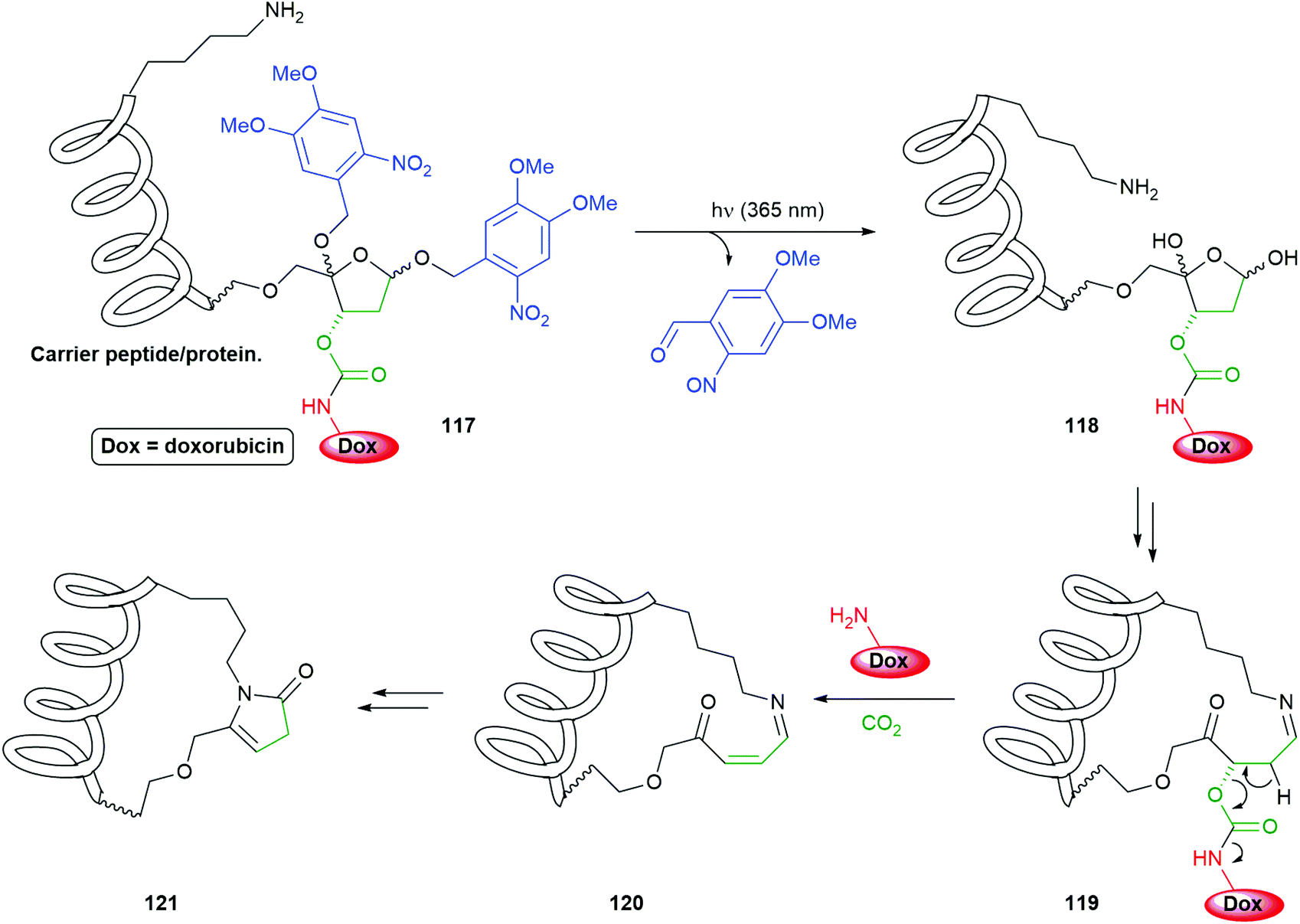 | ||
| Scheme 26 Zhou's PC4AP (photocaged C4′-oxidized Abasic site) self-immolative linker attached to a carrier peptide to form a delivery system for doxorubicin.156 | ||
Though an amine-containing reporter group (doxorubicin) is illustrated in Scheme 26, alcohol bearing reporter groups are also usable.
The design of linkers with varying rates of release has been important to the advancements in the field. In respect of linkers based upon an aromatic core, in addition to the use of judiciously placed donor substituents on the ring, employing an aromatic ring of lower resonance energy can also be useful; for instance, Shabat's pyridine linker66 (vide supra) and the use of polycyclic aromatics. Indeed, Gillies has utilised the facile thermal reversibility of a furan Diels–Alder reaction, coupled with furan's electron rich 2 and 5 positions, to facilitate fragmentation of a self-immolative polyglyoxylate polymer, see section 4.4. Some measure of relative aromaticity can be garnered, inter alia, from aromaticity indices; for example, those proposed by Hess and Schaad which relate a number of heterocycles to benzene as 100%. Thus, pyridine (82%), pyrrole (37%), furan (12%) can be seen as having lower aromaticity.157
Phillips has shown that naphthyl linkers can be effective, if designed to give 1,6-elimination.158 As part of this study, the contributions of resonance energy of a particular aromatic were considered and evaluated against a benzene ring as standard (Scheme 27). The results were applied to the fragmentation rates of oligo-linkers 122–124, formed after triggering of the corresponding boronate esters. The naphthyl linker disassembled a little slower than a methoxy bearing benzene ring; however, it still represents a valuable, alternative approach to varying fragmentation rates. Solvent polarity effects were also considered with more polar solvent mixtures encouraging faster fragmentation rates. The naphthyl and methoxybenzene linkers 123 and 124 were able to function effectively in a significantly less polar medium than 122.
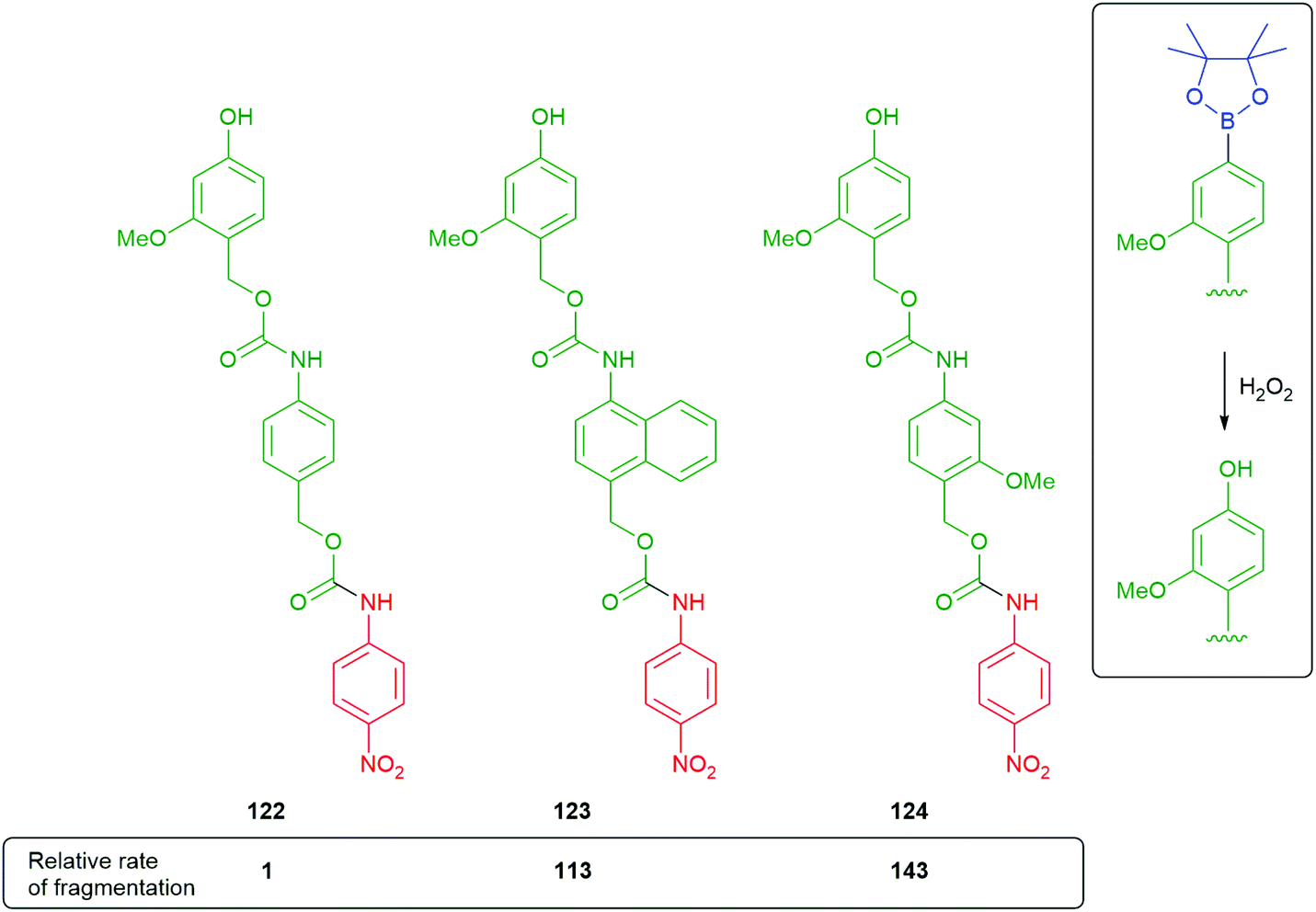 | ||
| Scheme 27 Comparative rates of fragmentation of modified, aromatic self-immolative linkers 122–124.158 | ||
Self-immolative systems that operate, post-triggering, via an electronic cascade have been utilised in innovative synthetic processes. For example, Goggins et al. have described the use of a self-immolative electronic cascade process to permit signal transduction and amplification via an enzymatically-triggered ligand release and accelerated catalysis process.159 The pro-ligand enzyme substrate 125 was designed to selectively self-immolate in the presence of an alkaline phosphatase enzyme to release a ligand 126 that can bind to a metal pre-catalyst [(cp*IrCl2)2] to afford 127, thus accelerating the rate of a transfer hydrogenation reaction (Scheme 28). Using SI proligands to facilitate a change in the properties of metals may prove quite generally significant (vide infra).
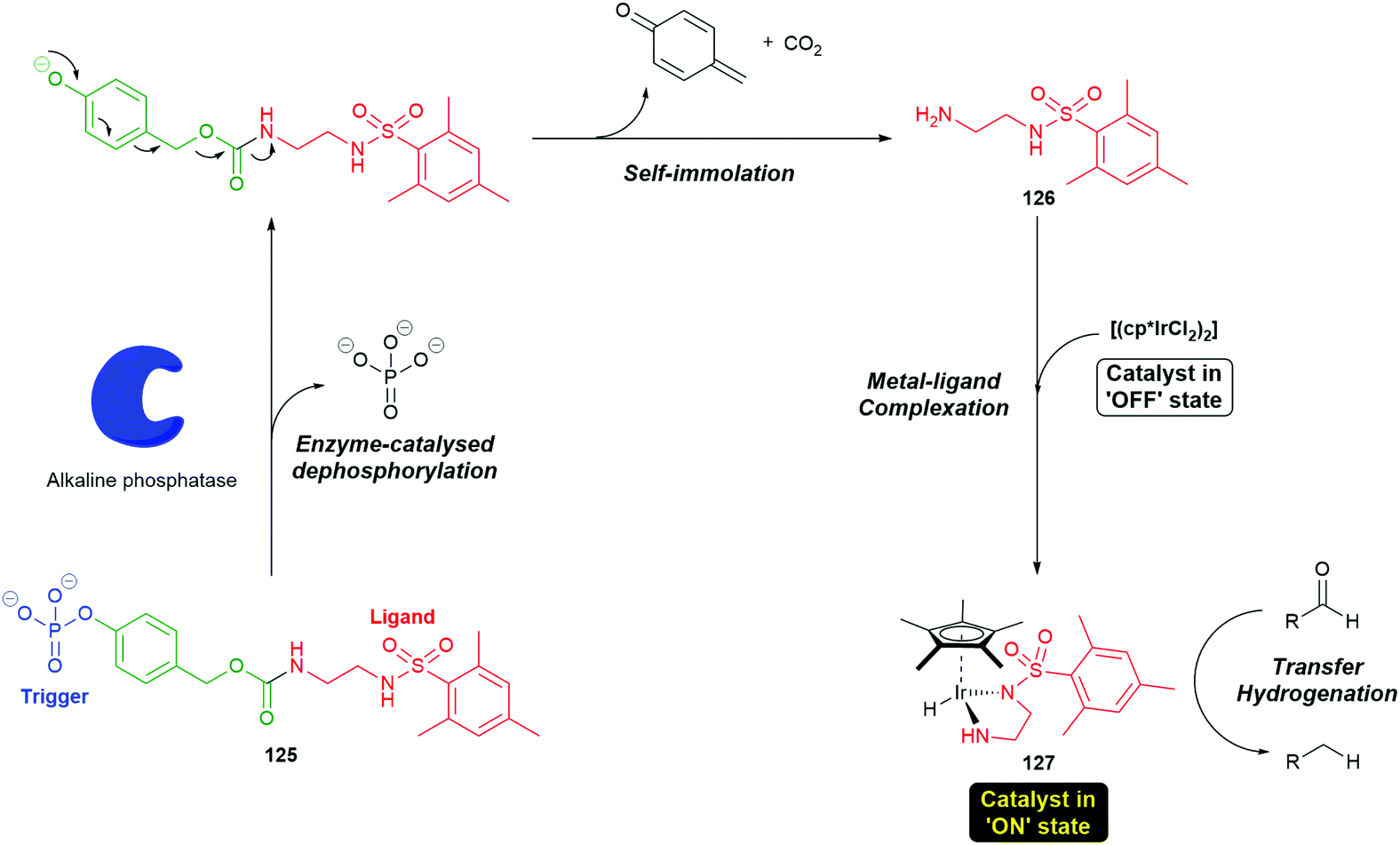 | ||
| Scheme 28 The self-immolative electronic cascade process to permit signal transduction and amplification via an enzymatically triggered ligand release from the proligand system 125 and accelerated catalysis process.159 | ||
Using an esterase driven cleavage of a SIE, Gale and co-workers have described a very interesting means by which the magnetic properties of a complex can be profoundly changed, from antiferromagnetic to paramagnetic (Scheme 29).160 A proligand was designed into the Fe3+ μ-oxo-bridged dimer complex 128 that, upon exposure to a porcine liver esterase, triggered cleavage of the pivalate ester to afford 129; concomitant 1,6-elimination gave 130. The facility with which elimination of the phenol occurs probably reflects the predominantly aqueous solution in which the reaction takes place, as noted by Phillips (vide supra).49 By bonding of the newly generated phenolic ligand in 130 it spontaneously splits into two equivalents of a monomeric, distorted octahedral, high-spin Fe3+ complex 131 that displays paramagnetic properties. This change in magnetic properties can support, inter alia, magnetic resonance imaging experiments.
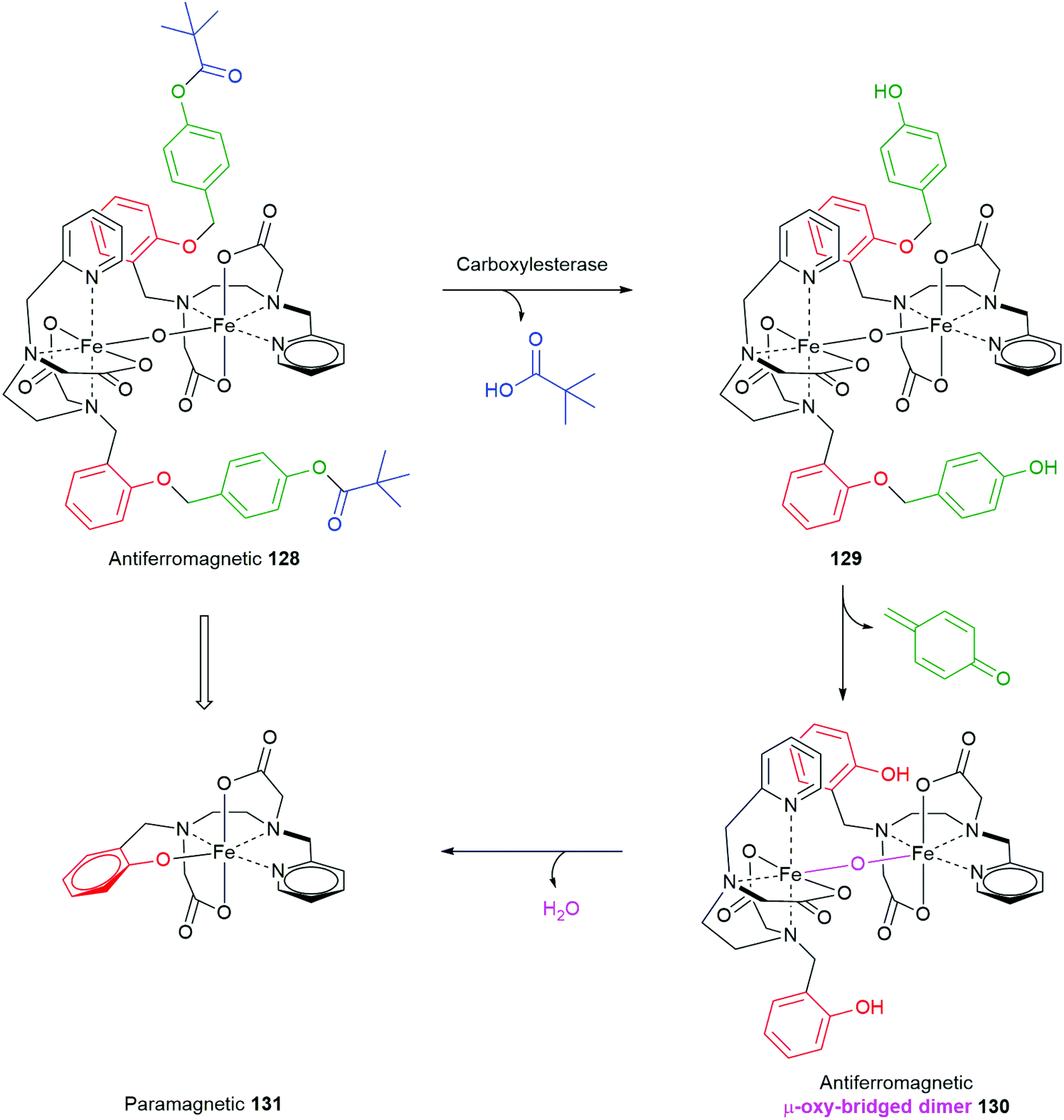 | ||
| Scheme 29 Gale's porcine liver esterase sensitive SIE that drives conversion of an antiferromagnetic complex 128 into a paramagnetic complex 131.160 | ||
An alternative approach to the targeted delivery of nitrogen mustards to that described by Peng and co-workers (Scheme 9) has been reported by Shabat et al. An elegant prodrug system based on a 7-hydroxycoumarin fluorophore-linker was developed which allowed for an efficient 1,8-elimination cascade (Scheme 30).161 The success of a 1,8-elimination across a coumarin mirrors that across cinnamyl alcohol systems but contrasts with that attempted across a naphthalene ring system.162,163 The prodrug 132 was comprised of a dipeptide, Z-Phe-Lys, the 4-aminobenzyl alcohol linker, an ethylenediamine-derived cyclisation spacer, coumarin-based fluorophore-linker and the anticancer agent, melphalan. Exposure of the prodrug 132 to cathepsin B, an enzyme found in elevated levels in cancer cells and tumour endothelial cells, resulted in cleavage of the trigger group to reveal the aniline moiety 133 capable of 1,6-elimination to yield a secondary amine that in turn cyclises to afford a urea and the free ionised hydroxycoumarin species 134. The phenolate species 134 fragmented via a 1,8-elimination process, resulting in the release of melphalan and the coumarin-quinone-methide 135, which is then subject to hydration in the aqueous medium to afford the highly fluorescent coumarin derivative 136. This design facilitates monitoring of fragmentation by fluorescence.
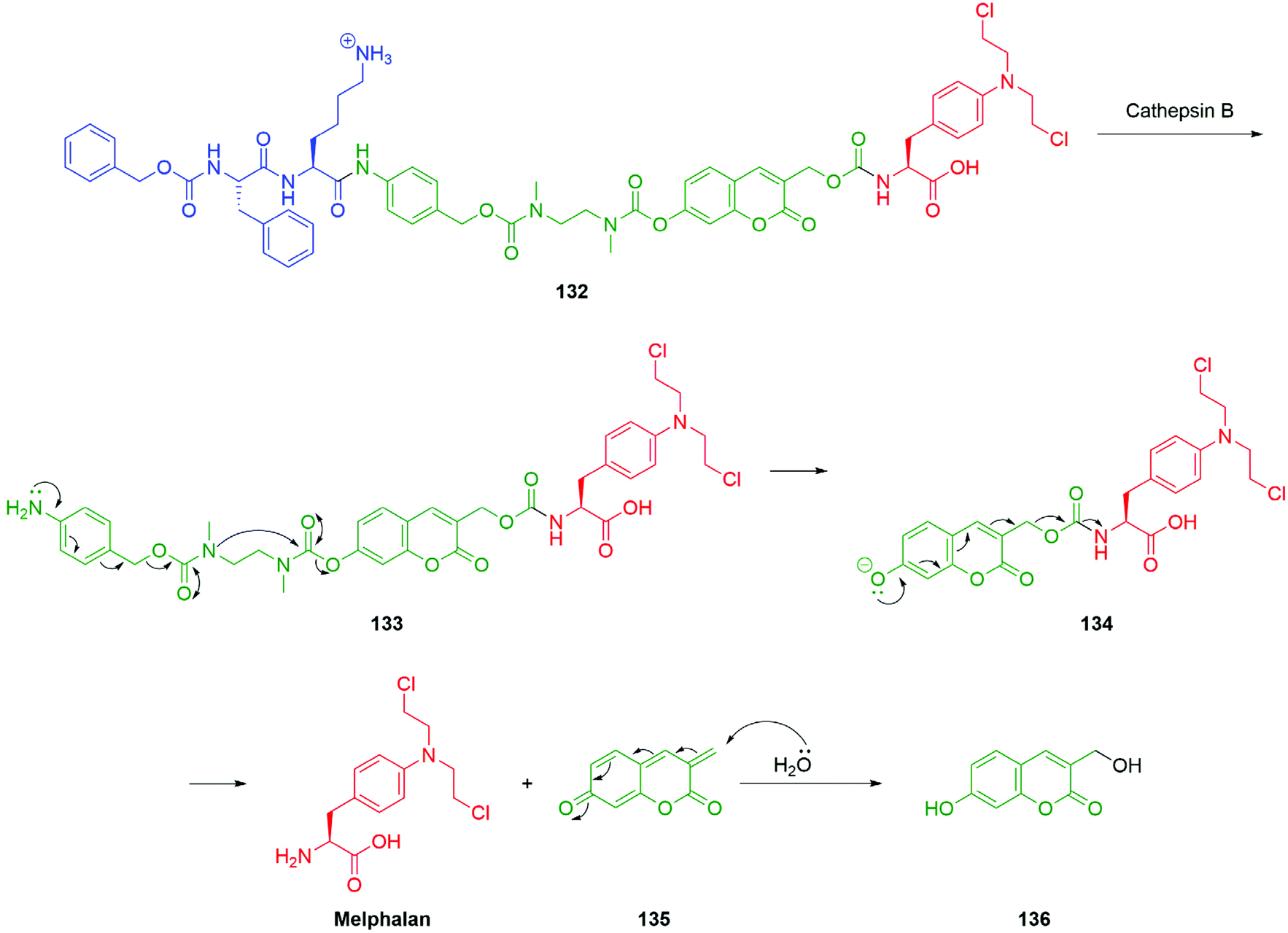 | ||
| Scheme 30 Melphalan release upon exposure of prodrug 132 to cathepsin B featuring 7-hydroxycoumarin fluorophore-linker as reported by Shabat and co-workers.161 | ||
Inspired by the work of Bertrand and Gesson on acid sensitive triazolylmethylcarbamate linkers,164 Blencowe et al.165 demonstrated the base sensitivity of triazolylmethylcarbamate self-immolative systems 137, capable of a 1,4-elimination cascade with a pivaloyloxymethyl (POM) trigger (Scheme 31). It was highlighted that the rate of self-immolative elimination was tuneable by the degree and nature of substitution at the triazole α-methine position (R1 and R2), with the fastest rate of self-immolation when R1 and R2 were gem-dimethyl groups. Self-immolation was triggered in a mixture of NaOCD3 and MeOD3 with formaldehyde generated as a by-product, affording the triazole anion 138. Under these experimental conditions, the triazole anion 138 initiates a 1,4-elimination to allow for release of the carbamic anion which subsequently underwent a decarboxylation event resulting in the formation of the deuterated benzylamine 139. The so-formed triazafulvene 140 was trapped by excess base to afford 141. Acid mediated degradation of polymer bonded triazoles have also been described.166
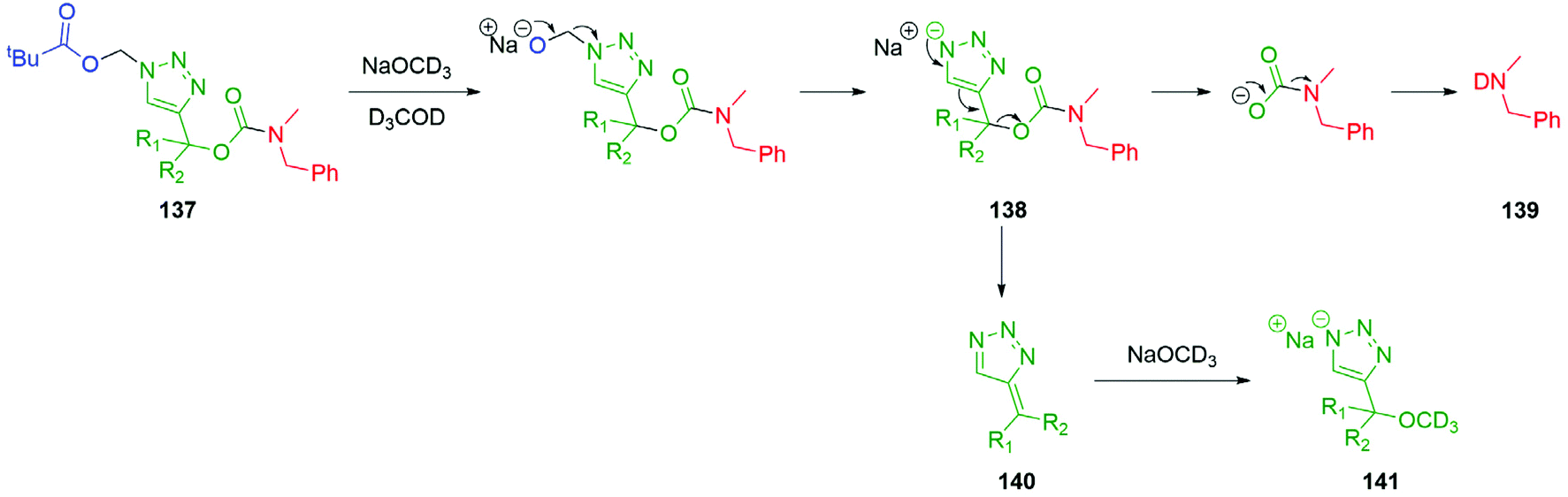 | ||
| Scheme 31 Base mediated disassembly of self-immolative triazole 137via 1,4-elimination reported by Blencowe et al.165 | ||
Roberts and Stevens further developed the 1,2,3-triazolyl-based self-immolative scaffold to afford a dynamic, pH sensitive switch that allows controlled delivery of the reporter group. A double linker system was prepared that combined the triazole moiety with a 1,2-diamine cyclisation stage to facilitate the use of carbamate trigger moieties.48 Removal of the trigger from 142 with a Pd(0) catalyst and morpholine as stoichiometric nucleophile yielded the free amine 143. Cyclization to a urea, with concomitant elimination of formaldehyde, gave triazolide anion 144. The triazolide anion then underwent a pH dependent 1,4-elimination; under basic conditions, release the reporter moiety together with carbon dioxide and triazafulvene 140 occurred. In turn, 140 undergoes a Michael addition with the excess morpholine to give 145 (Scheme 32). Alternatively, upon the addition of acid, the triazolide anion protonates, thus pausing the self-immolation. Treatment with base was observed to restart the paused 1,4-elimination. The 1,4-elimination was found to be faster in the presence of D2O, in accord with other fragmentations discussed above.
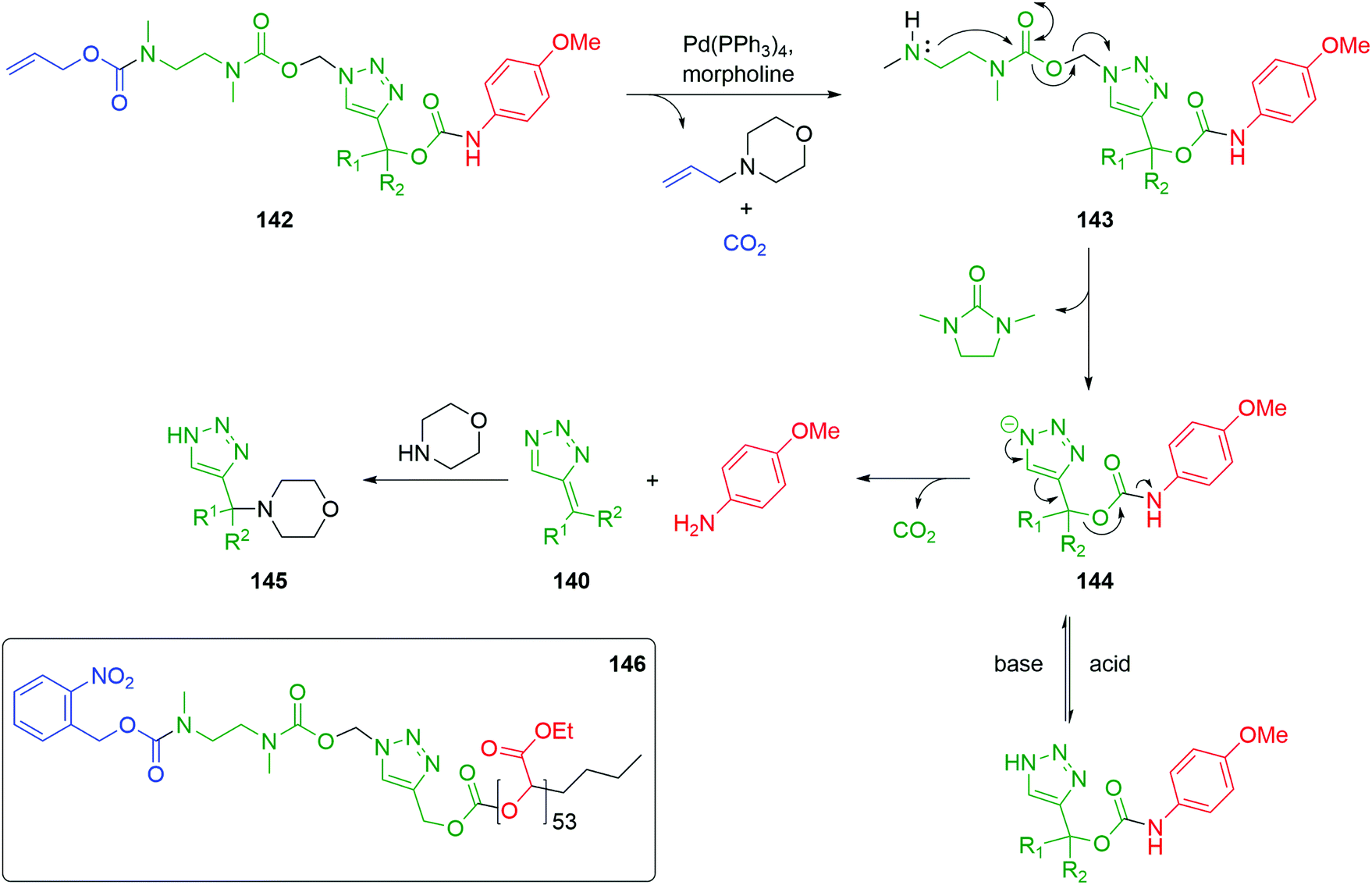 | ||
| Scheme 32 Dynamic pH responsive self-immolative triazole 142 with an ethylenediamine linker permitting the use of carbamate trigger groups as described by Roberts and Stevens.48 | ||
Subsequently, the authors have applied this methodology to ‘click’ their self-immolative unit to a polymer with pendant alkyne groups in order to develop a material with tuneable properties.167 Further work, with Gillies, has seen the method used to effect post-polymerisation modification of self-immolative polymers (SIPs), allowing depolymerisation168 under selective conditions e.g. Pd(0) catalysts or UV light (365 nm) e.g.146, for related SIPs, see section 4.4.
The effectiveness of ammonium ions as leaving groups in self-immolative processes has been exemplified in drug delivery by Peng, using hydrogen peroxide triggering (Scheme 9).91–93 Pillow et al. have used both glutathione and enzymatic triggering to release drugs, in ammonium ion form, from antibody–drug conjugates.94 By contrast, Taran and Le Gall have used the generation of an ammonium ion (147) by alkylation of 148 as a means of triggering its self-immolative fragmentation (Scheme 33).9 By this means, the FRET quenching apparent in 148 is removed in 149, allowing quantitative detection of a variety of alkylating agents by fluorescence upon excitation of coumarin 149 at 365 nm. This method is particularly interesting as one of the early examples of an electrophilically triggered SIE and, as with Seitz's iMB, has the rare property that the reporter group and trigger are in the same moiety. The self-immolative linker features an ortho-nitrophenol, to enhance the acidity of the phenolic OH; an alternative role for the nitro group, in enzyme catalysed reactions, was discussed by Hou and co-workers (Scheme 18).136
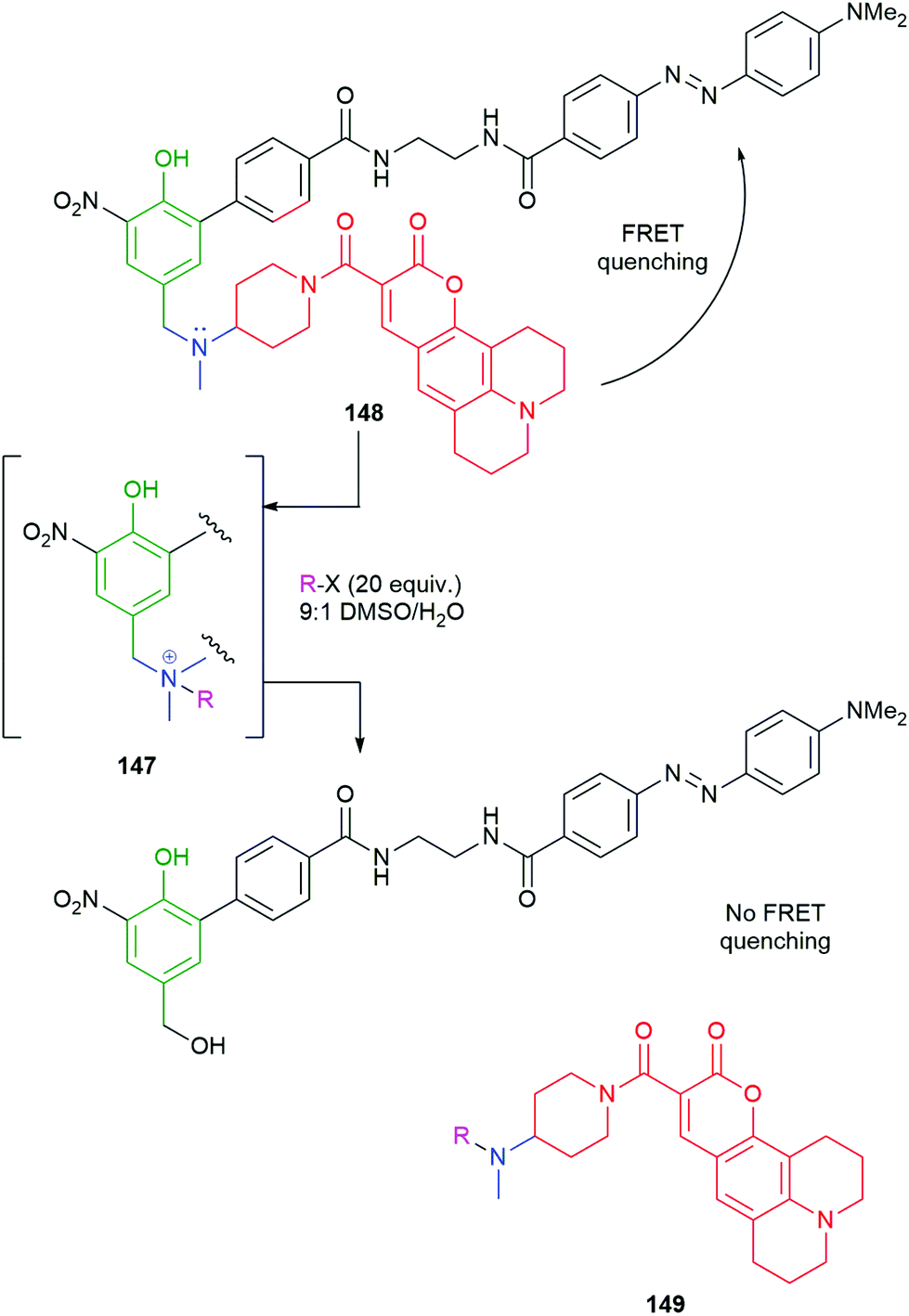 | ||
| Scheme 33 Taran and LeGall's FRET-based detection system 148 for electrophilic alkylating agents.9 | ||
The utility of the related iminium ions, as leaving groups in SIEs, has also been used in an elegant DNA ‘walking’ mechanism of a 2,6-disubstituted phenol system by Fakhiri and Rokita (see section 3).53
Acton et al. reported a self-immolative system triggered by non-acidic electrophilic species such as methyl, allyl and benzylic halides.8 Inspired by work from Kunz169 and Verducci,170 2-(diphenylphosphino)ethyl group (DPPE) was conjugated with N-methyl-4-nitroaniline via a carbamate to afford 150 (Scheme 34). Subjecting phosphine 150 to a Type II SN2 alkylation resulted in formation of the corresponding phosphonium salt 151, thus increasing the acidity of the α-methylene protons and activating the trigger towards base mediated elimination. Addition of N,N-diisopropylethylamine (DIPEA) to a solution of 151 in acetonitrile led to 1,2-elimination, releasing the nitroaniline reporter moiety, confirmed visually by the distinct yellow colour of the solution (Scheme 34). A one-pot procedure proved equally effective. Recent optimisation studies by Gavriel et al., to probe both electronic and steric effects of the reporter unit, identified the self-immolative system 152 as the best balance of stability and reactivity.171
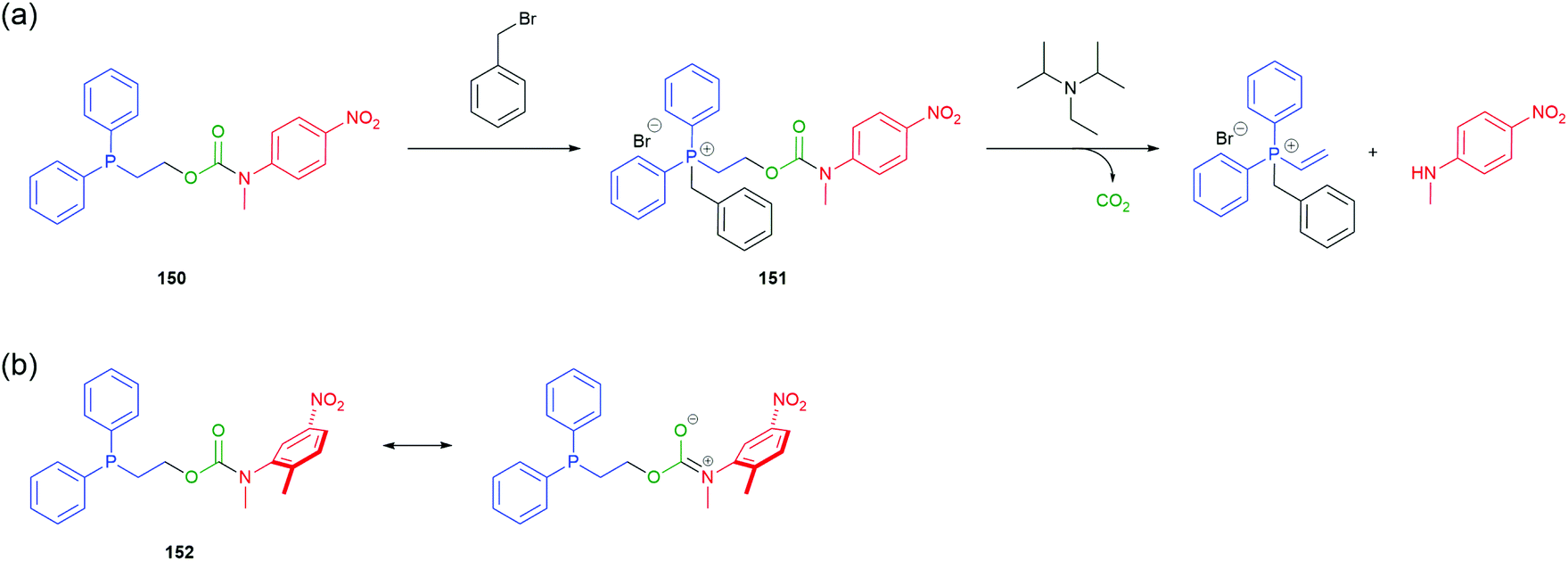 | ||
| Scheme 34 (a) Self-immolative system 150 for disclosure of electrophilic alkylating agents, as described by Acton et al.;8 (b) optimisation of 150 to afford compound 152.171 | ||
Bradley and co-workers have recently adapted a self-immolative electronic cascade process, that affords a quinone methide intermediate, to give an elegant protecting group strategy and demonstrated its application to solid-phase peptide synthesis.155 In this approach, the side chain amine of a lysine residue is protected in the form of a modified benzylic carbamate that uses a 4-demethoxyveratryl alcohol unit that bears a para-vinyl ether (referred to as the vinyl ether benzyloxycarbonyl (VeZ) protecting group), for example 153 (Scheme 35). The vinyl unit of the VeZ protecting group allows a thermally induced triggering event that is orthogonal to the standard range of protecting groups used in peptide synthesis. Thus, treatment of 153 with tetrazine 154, induces a tandem inverse electron-demand Diels–Alder reaction, retro-Diels–Alder affording, after tautomerism, 155. An eliminative rearomatisation released the linker–reporter unit 156; indeed, this elimination could be regarded as an additional linker unit (vide infra).172,173 The stability of 156 proved condition dependent, with isolation being possible.
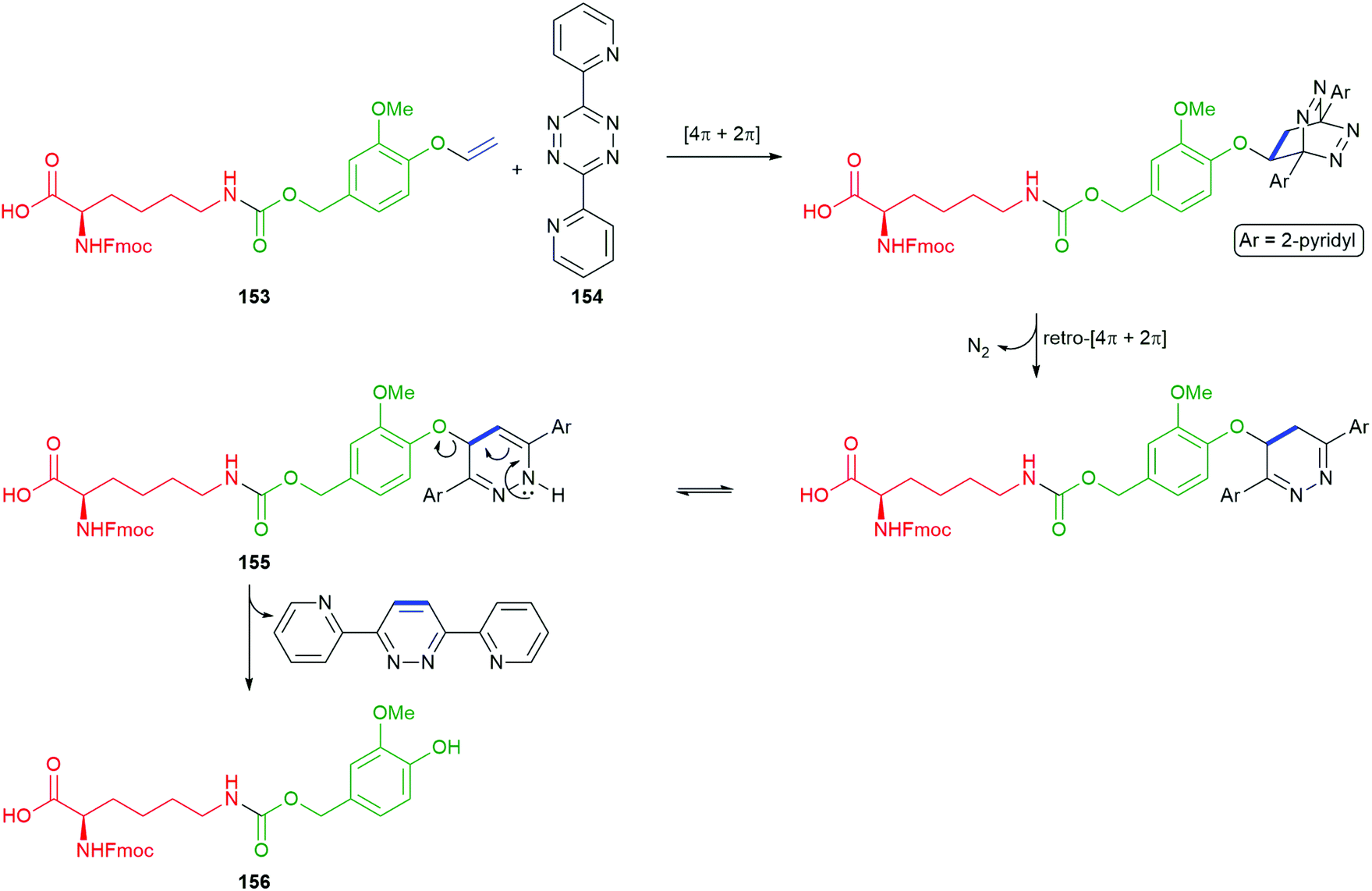 | ||
| Scheme 35 The thermally induced cleavage of the trigger moiety of the vinyl ether benzyloxycarbonyl (VeZ) protecting group on 153 with tetrazine 154 reported by Bradley and co-workers.155 | ||
Consistent with Phillips’ observations on the fragmentation of phenolic systems (Fig. 2),49 plus those of Roberts and Stevens48 on the fragmentation of triazole carbamate systems (Scheme 32), an increase in the percentage of water in the solvent assisted the rate 1,6-elimination of 156. Thus, the reaction conditions of 50% water/DMF mixture (buffered to pH 5 using sodium citrate), in conjunction with exposure to microwave radiation, allowed for an accelerated Diels–Alder reaction and a facile self-immolative deprotection step: these conditions could be employed when generating peptides on solid-supports (Scheme 36). Fmoc-Lys(VeZ)-OH was applied to the synthesis of two cyclic peptides, melanotan II (MTII) and a constrained BAD BH3 peptide analogue – both of which are examples of side-chain to side-chain lactam-bridge peptides. Additionally, the approach has proved effective at drug release from prodrug systems.174
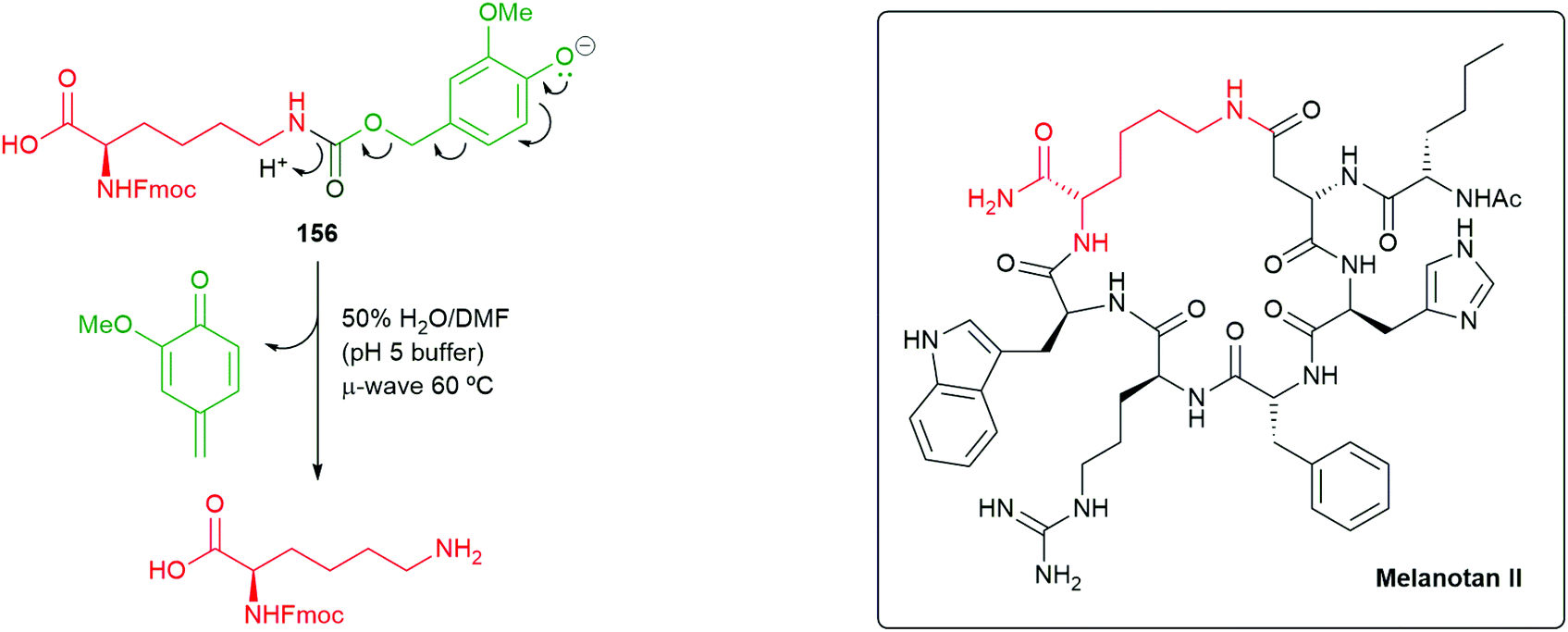 | ||
| Scheme 36 Fragmentation of the conjugate base of SIE linker 156 and a target peptide melanotan II.155 | ||
The use of thermal triggering of SIE systems, using Diels–Alder reactions, has also proved advantageous in SIE polymer systems (see section 4.4).175
The method has proved effective at release of doxorubicin (for its structure see Schemes 22 and 39) from an amphiphilic methyl methacrylate/PEG copolymer 157 in HEK273 T and PC3 cells (Scheme 37).176 In view of the vinyl ether unit, the polymer was best prepared by RAFT. When in water, the copolymer formed nanoparticles of 35 nm diameter with PEG at the surface and four doxorubicin moieties in the hydrophobic core.
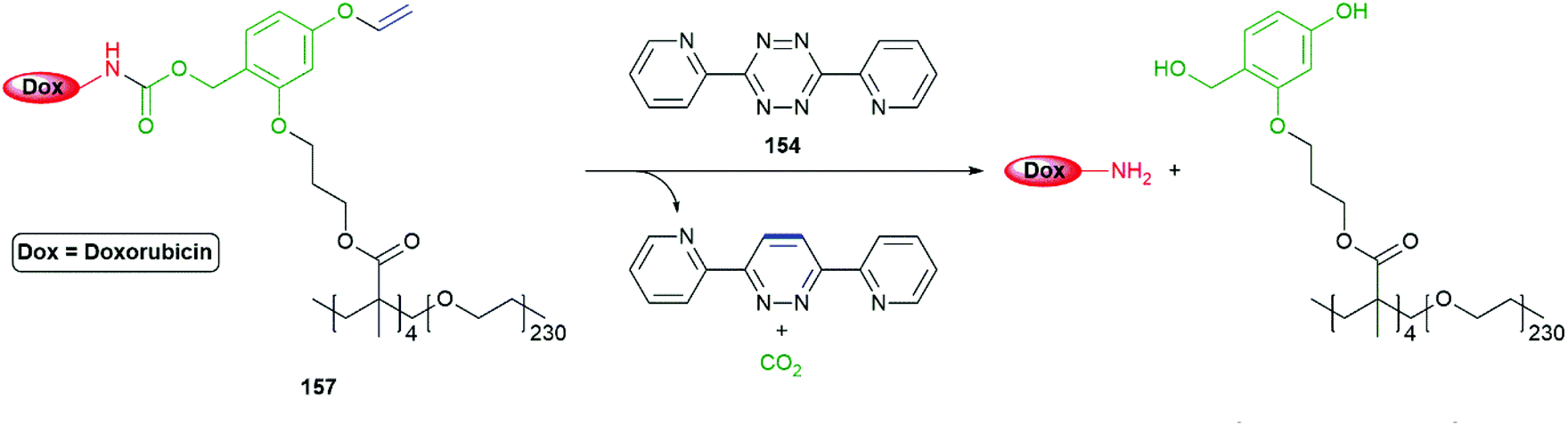 | ||
| Scheme 37 VeZ protecting group methodology adapted to controlled release of doxorubicin from an amphiphilic copolymer, initiated by biocompatible tetrazine 157.176 | ||
This work was developed from reports of effective bioconjugation and protein activation of tetrazines with trans-cyclooctene derivatives.177–179 Robillard developed this facile ligation method into an interesting self-immolative method termed ‘click to release’ (Scheme 38).180 Of particular interest is the innovative way that the triggering event simultaneously completes construction of a self-immolative unit. The nature of the R group on the tetrazines, e.g.158–159 being found to exert a significant effect of its rate of reaction, alkyl substituents enhancing reactivity over their aromatic counterparts. Thus, trans-cyclooctene 160 (X = O) undergoes the previously discussed tandem inverse electron-demand Diels–Alder reaction, retro-Diels–Alder, under physiologically compatible conditions, affording 161. Dihydropyridazine 161 undergoes tautomerism to give 162, completing the self-immolative unit. Once formed, 162 undergoes 1,4-elimination to release the reporter group (a detailed analysis of this process was reported), CO2 and 163 that, in turn, isomerises to aromatic pyridazine 164. The significance of water in the reaction medium was again noted.
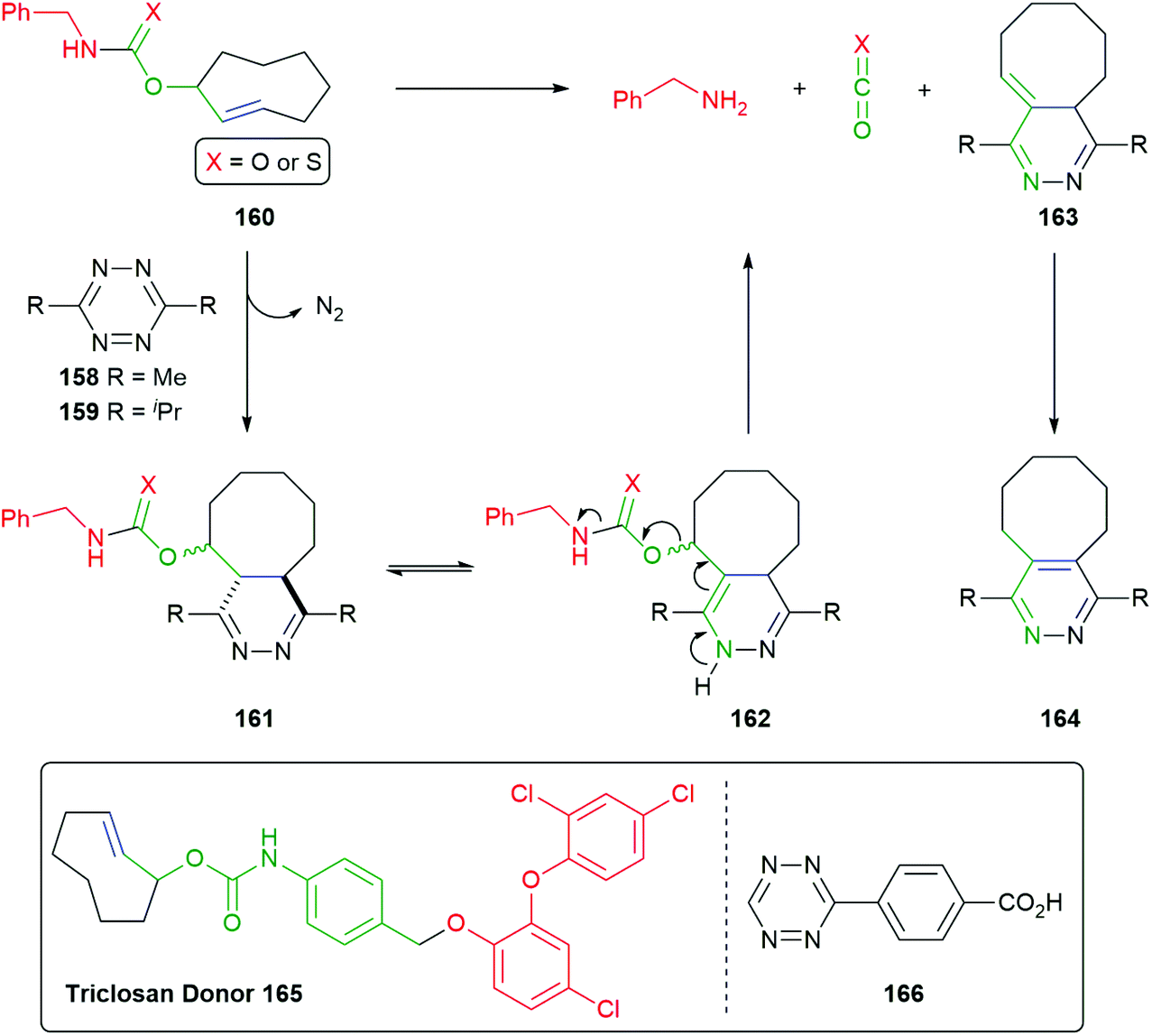 | ||
| Scheme 38 ‘Click to release’ reaction of tetrazines with trans-cyclooctenes 160 and 165.180 | ||
For the case when X = S, Pluth has cleverly adapted the approach to delivery of COS, mediated by tetrazine 159, and thence by catalysis with carbonic anhydrase to gasotransmitter H2S.111
The use of the trans-cyclooctene-based methodology has also been adapted to the release of oxygen-connected reporter groups by inserting an aniline-based linker and exemplified by preparation of the Triclosan donor 165 by Bernardes (Scheme 38).181 Release of this antibacterial agent was demonstrated within live E-Coli cells by reaction with tetrazine 166: restoration of triclosan's activity was observed.
Further, Robillard has substituted the benzylamine reporter group in 160 for doxorubicin in 167 and demonstrated its release in A431 cell culture upon treatment with 158, as well as noting the method's potential for use in antibody drug conjugates (Scheme 39).180 The delivery of doxorubicin by this approach has been further extended by Mejia Oneto et al. to target soft tissue sarcoma by using an injectable polymeric reagent 168; specifically, an alginate hydrogel modified with tetrazines (Scheme 39).182
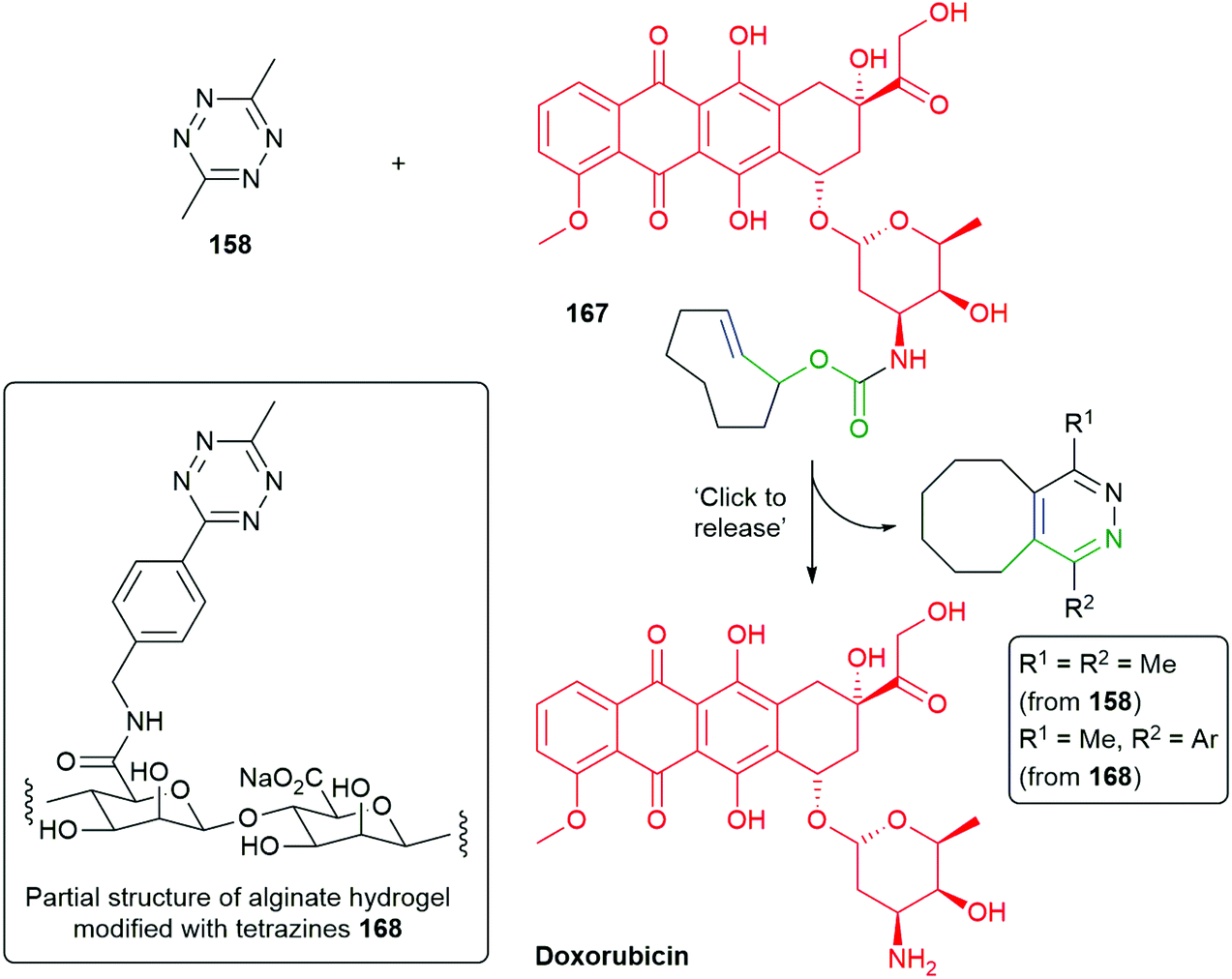 | ||
| Scheme 39 ‘Click to release’ of doxorubicin system 167, modified with trans-cyclooctene, by tetrazines 158 or 168.180,182 | ||
Further impressive advances in the application of trans-cyclooctene, tetrazine methodology in the anticancer area have been published.183,184 Very recently, Fan and Chen have reported a novel approach to on-demand pyroptosis via tetrazine mediated activation of a cytosine base editor, blocked by a trans-cyclooctene.185
An interesting variation on the method is the use of a 1,3-dipolar cycloaddition of an aromatic azide e.g. hydrogelator 169 to a trans-cyclooctene 170 to give an unstable adduct 171 that fragments to diazonium species 172 with concomitant ring contraction to deliver, after hydrolysis, an aniline 173 (Scheme 40). This aniline undergoes 1,6-elimination to yield dipeptide 174; in so doing, a gel–sol transition takes place raising the potential of such systems to be employed in inter alia a drug delivery role e.g. for doxorubicin.186 This reaction has also proved successful in prodrug systems e.g. for doxorubicin and profluorescence compounds.187,188 Such a change of physical properties of a gelator has also been used as part of a dendritic chain reaction (see section 3.3).
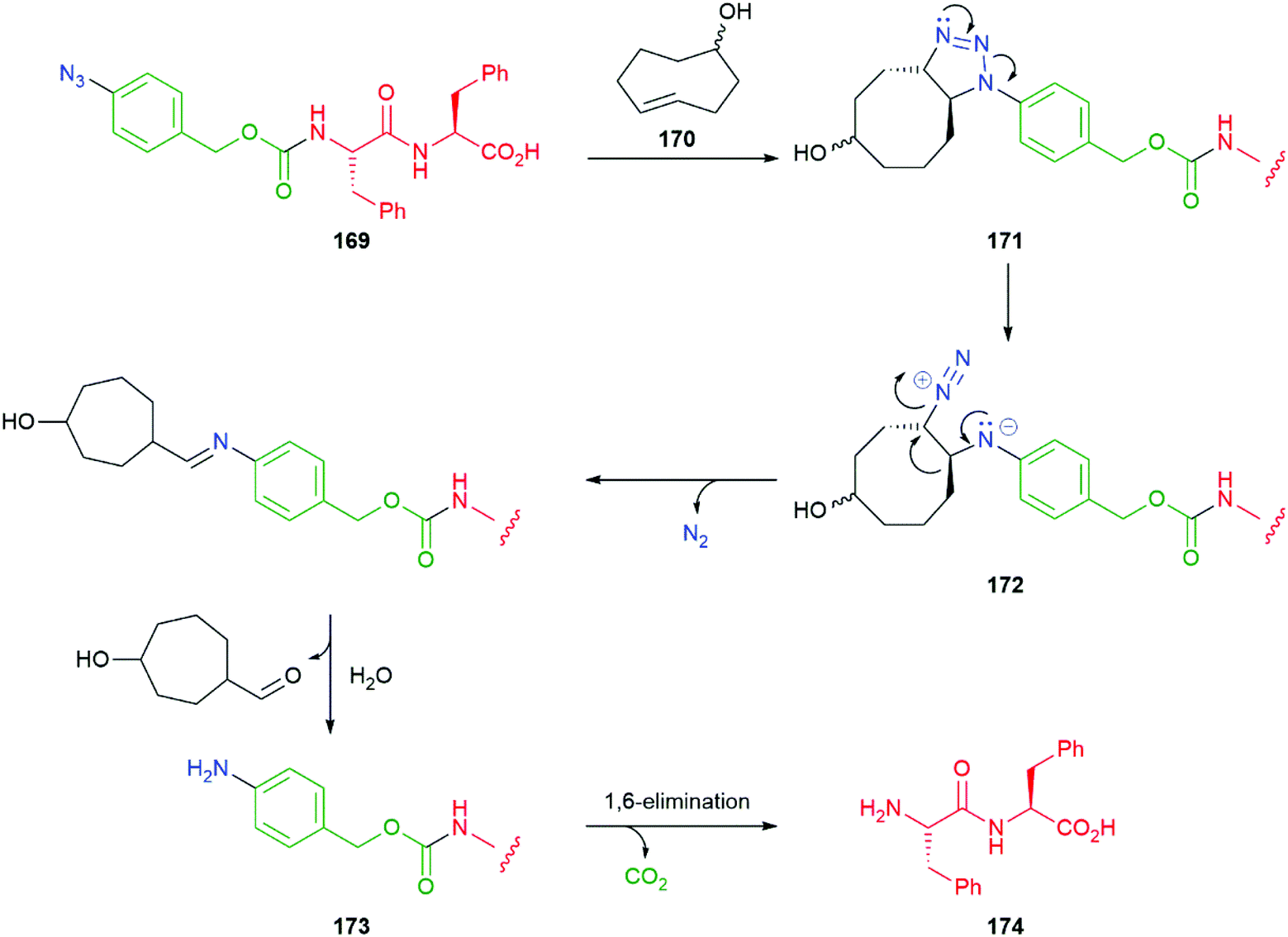 | ||
| Scheme 40 1,3-Dipolar cycloaddition of 169 to a trans-cyclooctene 170 triggers a gel–sol transition.186 | ||
Reduction of azides to initiate 1,6-elimination and targetted release of reporters by cyclisation–elimination, triggered by saccharide azides, has also been achieved by Staudinger reduction.189,190
Alternative reactive alkenes have also been used in ‘click to release’ reactions of tetrazenes; for example, Franzini has demonstrated the effectiveness of 7-oxa/azabenzonorbornadienes e.g.175 (Scheme 41).191,192
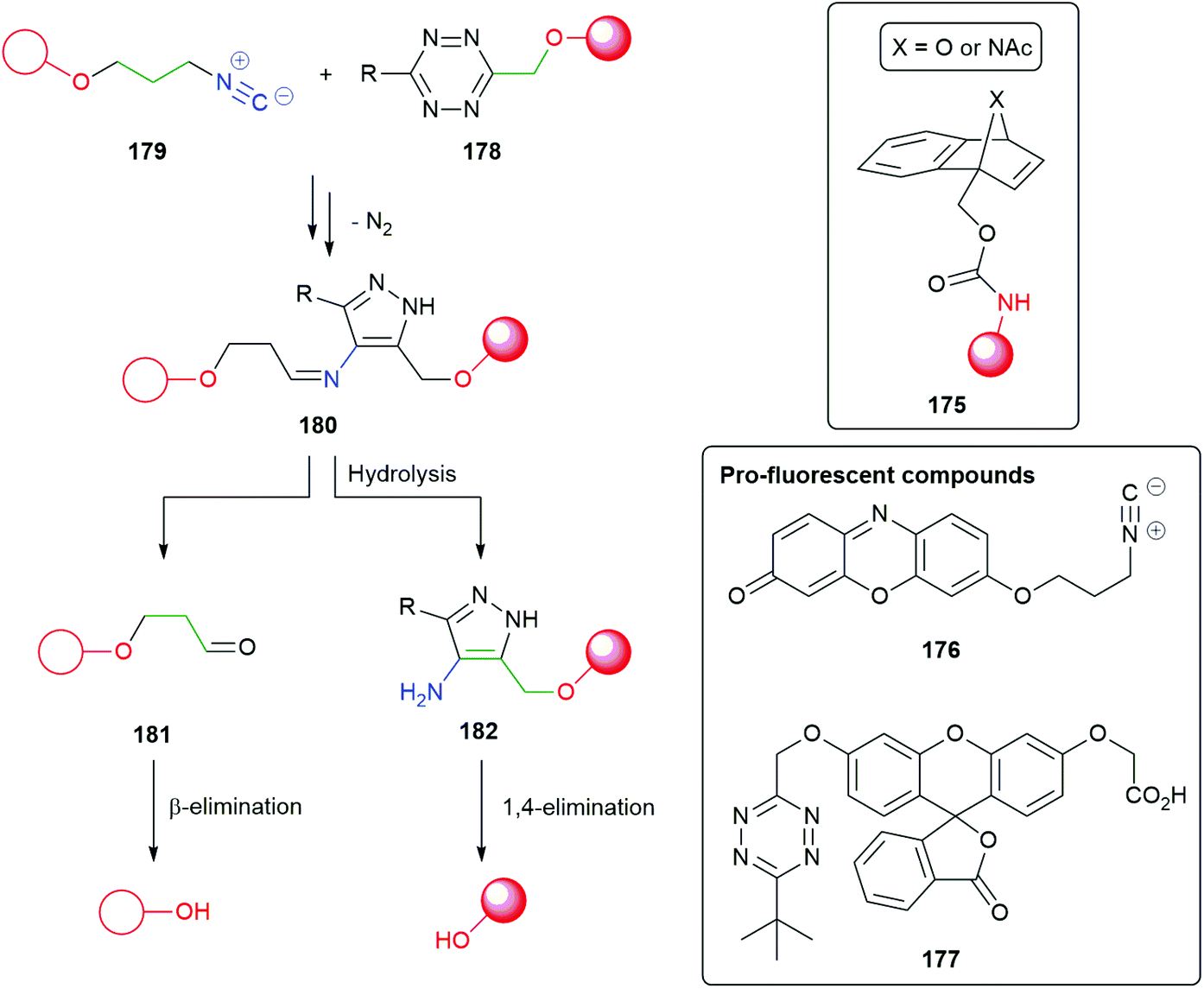 | ||
| Scheme 41 Franzini's isonitrile and norbornadiene triggered release systems from and with tetrazines.191–194 | ||
Developing work by Imming et al. and Leeper et al., Franzini has also reported a distinctive method for dual reporter group release from a combination of tetrazines and isonitriles and found this to work in vivo, using zebra fish embryos (Scheme 41).193–196 The capacity for dual release offers an interesting alternative to the use of dendrons and can be used, for example, to deliver two different, synergistic drugs, in theranostic applications or to release two distinct fluorescent molecules (see pro-fluorescent 176 and 177). Thus, treatment of tetrazines 178 with isonitriles 179 results in a tandem cheletropic reaction, retro-[4π + 2π] cycloaddition to afford pyrrazole 180. Hydrolysis of 180 liberates aldehyde 181 that undergoes β-elimination to deliver one reporter group; whereas, 182 undergoes 1,4-elimination to give the second reporter group.
Related results have been reported for release of sulfonamide groups by Houk and Liang using cycloaddition of sydnonimines onto the strained alkyne, dibenzoazacyclooctyne. An orthogonality of delivery was noted between this system and the benzonorbornadiene-terazine system of type 175, mentioned above.197
2.2 Self-immolation via cyclization elimination
Self-immolative linkers that utilize cyclization for disassembly via activation of a latent nucleophile (e.g. amine,198 phenol,199 and thiol groups200,201) followed by an intramolecular exo-trig type cyclization mechanism are driven by (i) an increase in entropy coupled with (ii) the irreversible formation of thermodynamically stable products (e.g. CO2). For example, this degradation pathway was demonstrated by Amir and Shabat with the release of para-nitrophenol from a first-generation diethylene tri-amine-based dendron 183 (Scheme 42) using ethylenediamine as the linker.202 The amide linkage is stable under physiological conditions; however, the selection of the amide side chain of penicillin G affords an elegant solution for mild cleavage of an otherwise stable functional group. Thus, cleavage of the two trigger groups by the enzyme penicillin-G-amidase affords the nucleophilic amine species 184 that participates in a favourable 5-exo-trig-cyclisation to liberate para-nitrophenol (the reporter unit in this model study) with concomitant formation of imidazolidinone 185. The cyclisation–elimination approach to self-immolative linkers, either alone or in tandem with other linker types, has been applied in a number of ways in recent years and below we illustrate some of these.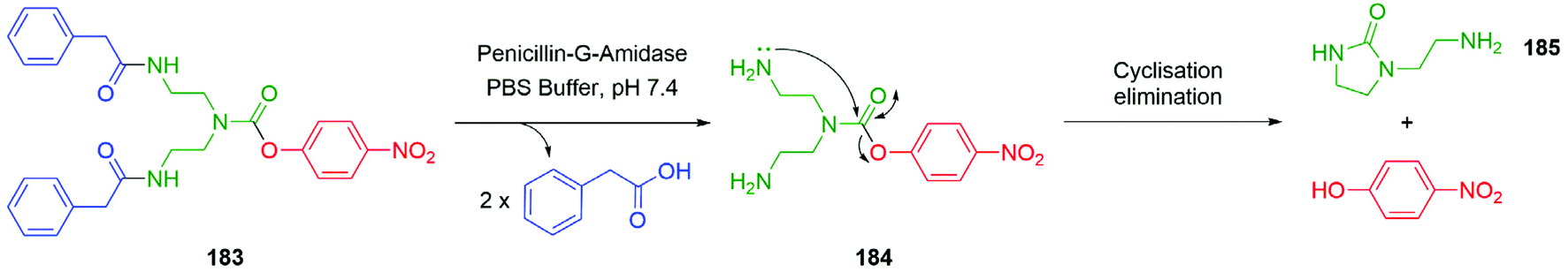 | ||
| Scheme 42 Pencillin-G-Amidase triggered self-immolative system 183 featuring a cyclisation linker as described by Shabat and co-workers.202 | ||
The biological significance of controlled release of COS/H2S and persulfides has been discussed above (Schemes 13, 14, 16, 38 and Fig. 5–7). Additionally, Toscano notes the recent discovery of the use of tRNA bound Cys-SSH in protein synthesis.100 Toscano and co-workers have thus prepared a range of self-immolative molecules of which compounds 186a–d were found to be effective for persulfide or COS/H2S delivery (Scheme 43). They are triggered by deprotonation of its terminal nitrogen, at physiological pH, to give free amines 187 or by nucleophilic attack by thiols to give persulfides 188 and thence COS/H2S. The predictable cyclisation rate (pH dependent) of the linker affords a suitable release profile for hydropersulfide 189, with ureas 190 as by-product. The rate of release of the persulfide 189 is slowed at lower by pH, or with the absence of a terminal methyl group (186b and 186d) or a longer linker (186c–d). The steric bulk presented by the gem-dimethyl group in the N-acetyl penicillamine unit of 186 is significant. The analogue of 186 derived from N-acetylcysteine methyl ester releases hydropersulfide anion 191 that reattacks the S–S bond of the starting material to deliver trisulfide 192 as a significant by-product. In the presence of thiols, in competition with cyclisation to cyclic ureas 190, attack on the persulfides 186 occurs to give 188 and 193, and thence release of COS, with subsequent hydrolysis, catalysed by carbonic anhydrase, to deliver H2S (see also Scheme 13). Though a minimal pathway with 186a, slower cyclising compounds e.g.186c–d led to this becoming a major pathway. Thus, linker design is crucial to achieving a desired mode of action. In addition, 188 also reacts with N-acetylcysteine to afford polysulfides. Artaud and Galardon have also described a pH sensitive persulfide donor molecule based on penicillamine.203
 | ||
| Scheme 43 The pH/thiol triggered SIEs 186 for release of a hydropersulfide 189 or COS/H2S reported by Toscano and co-workers.100 | ||
The cleavage of disulfide bonds in 194 by cellular thiols has been described by Feng and co-workers as means of controlled release of sorafenib 195via a cyclisation–elimination event to yield 1,3-oxathiolan-2-one 196 as the by-product via the thiol intermediate 197 (Scheme 44).204 The drug release system 194 was prepared from multi-armed PEG-NH2 adding to acrylate 198 to deliver an amphiphilic polymer that exhibited good aqueous solubility and self-assembled into micellar-like nanoparticles (hydrodynamic diameter ca. 150 nm). Sorafenib could be released in vitro by treatment with dithiothreitol. Dose dependent cytotoxicity was observed toward HeLa and HepG2 cells, with evidence for improved intracellular localization; in addition, in vivo activity was reported. A similar cyclisation–elimination, but onto a carbonate group, was reported by Low for the delivery of camptothecin.205
 | ||
| Scheme 44 The PEG-based SIE 194 for intracellular delivery of sorafenib as described by Feng and co-workers.204 | ||
As part of their studies on traceless linkers to quaternary ammonium ions, Pillow et al. have used a glutathione sensitive persulfide trigger to initiate a tandem cyclisation–elimination, 1,6-elimination sequence to release tubulysin from tripartite antibody–linker–drug conjugate 199in vitro (Fig. 12).94
 | ||
| Fig. 12 Pillow's glutathione triggered antibody-double linker-drug conjugate 199.94 | ||
Perez has developed a SIE that combines targeting of doxorubicin delivery with a folate unit and a cyclisation–elimination linker that was triggered by intracellular cleavage of a persulfide bond to release the drug.206
The ethylenediamine-carbamate module has been shown to be highly versatile, and has been used in combination with self-immolative triggers that are activated upon the application of different stimuli (e.g. enzymatic action,64,201,207 irradiation with UV,208 or near-infrared light209). However, as noted by Dal Corso, Gennari and co-workers, the cyclisation can prove to be rate-limiting and ultimately affect prodrugs’ therapeutic efficacy.210 In 2020, Dal Corso and Gennari proposed a suitable modification of the ethylenediamine-carbamate resulting in a proline-derived self-immolative linker with an enhanced rate of cyclisation whilst retaining the desirable features of the ethylenediamine linker (Scheme 45).210 The reactivity of the proline-derived linker in 200 was compared directly to the ethylenediamine linker in 201 in their trifluoroacetic acid (TFA)-salt pro-drug forms. The half-life for cyclisation of 201 was 10.6 hours; whereas, 200 was found to release the reporter moiety with a half-life of only 1.6 hours. In this system, the cyclic amine attacks the carbamate connected to the exocyclic amine thus forming a bicyclic urea.
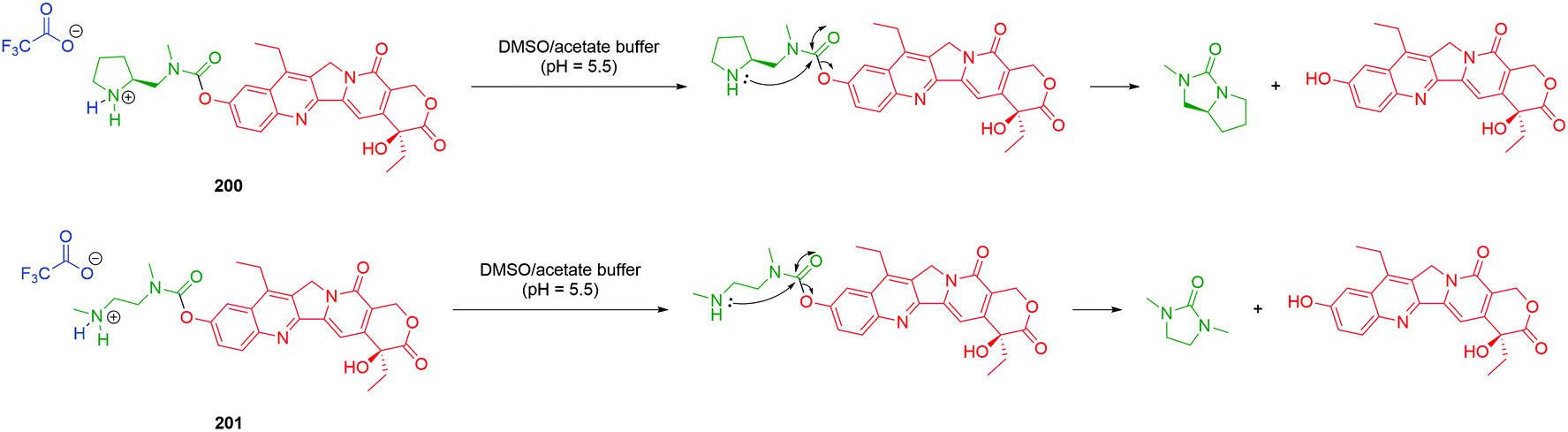 | ||
| Scheme 45 The proline-derived self-immolative linker in 200 as described by Dal Corso and Gennari et al.210 | ||
Subsequently, this was extended to a trifunctional linker system 202 which functions by non-sequential, dual activation wherein the presence of a protease enzyme, though sufficient to remove the dipeptide trigger, with concomitant 1,6-elimination to give 203, is insufficient to give rapid cyclisation to release the camptothecin reporter group (Scheme 46).39 This is thought to arise because of extensive protonation of the proline-derived amine, preventing its nucleophilic cyclisation onto the carbamate in 203. By contrast, the combined presence of both a protease and a phosphatase converts 202 into 204 which undergoes rapid cyclisation–elimination forming bicyclic urea 205 and releasing the reporter group. This nuanced approach allows for the general idea of selecting for environments in which two analytes must be present e.g. a cell type that contains two enzymes; this should allow for more selective targeting of a reporter group.
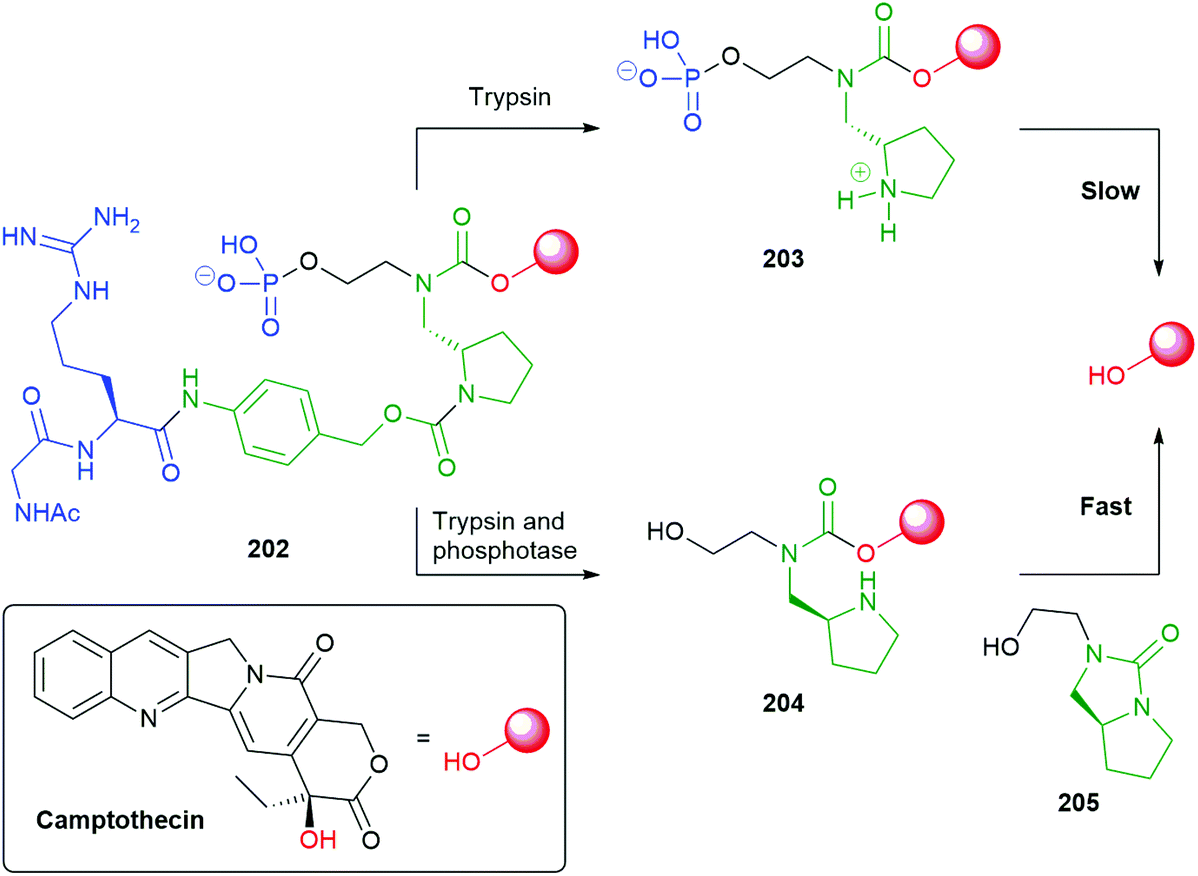 | ||
| Scheme 46 Dal Corso and Gennari's dual triggered SIE for camptothecin delivery 202.39 | ||
In a related study, the rates of cyclisation of variously substituted 4-aminobutyric acids were examined by Gillies as potential self-immolative linkers.211
Schnermann has developed a number of drug delivery systems, based around a cyanine caging group, of which the antibody–cyanine–drug conjugate 206 serves as an example (Scheme 47). These are triggered by irradiation with near-IR light (780 nm), affording good tissue penetration of the light and lower risk of tissue damage than UV radiation.212–216 Formation of singlet oxygen allows cleavage of the indicated double bonds via the corresponding dioxetanes 207. Once cleaved to compounds 208 and 209, the enamine moiety becomes susceptible to hydrolysis releasing self-immolative linker–reporter unit 210 that undergoes cyclisation–elimination to afford cyclic urea 211 and release of the duocarmycin reporter group. It should be noted that these systems also allow for fluorescence monitoring of the systems distribution in vivo.
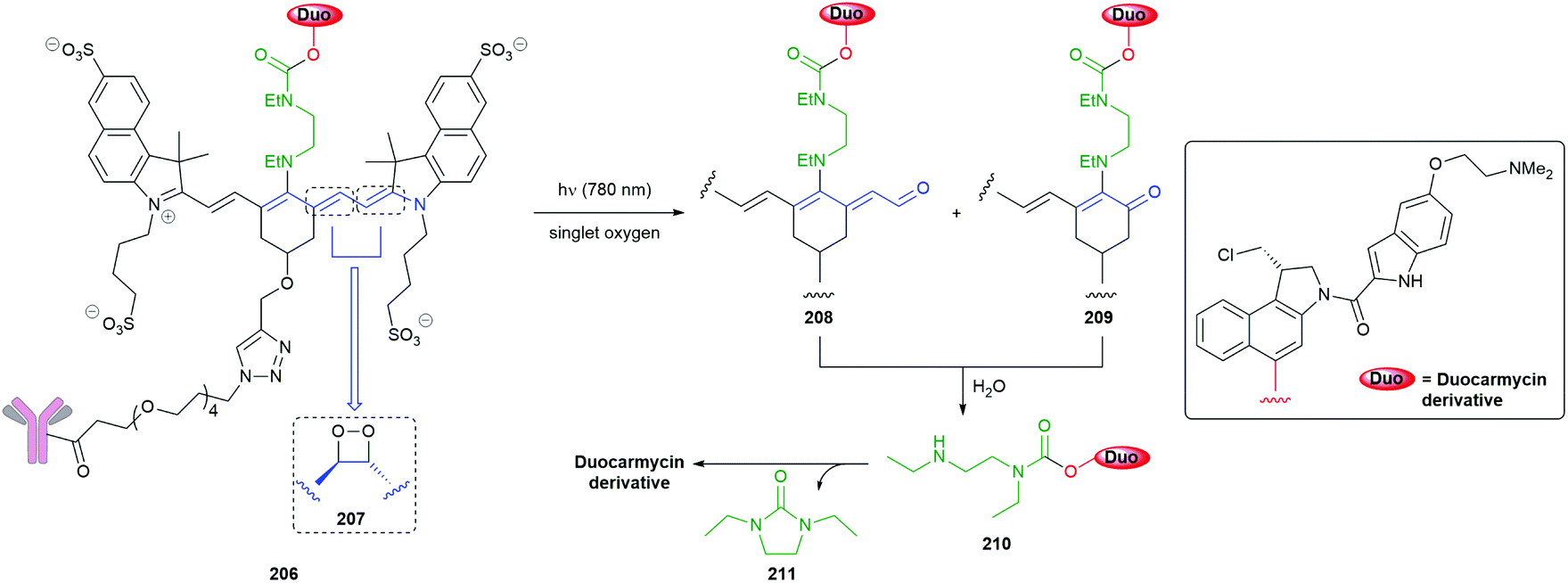 | ||
| Scheme 47 Schnermann's near-IR triggered drug delivery system 206 for duocarmycin.213 | ||
The E/Z-isomerism of ortho-hydroxycinnamates of type 212 leads to spontaneous cyclisation from 213 to coumarin 214 with release of the reporter group (Scheme 48). Such self-immolation may be regarded as an example of a reagentless form of activation in that no formal trigger group is present and the only ‘reagent’ is a UV or near IR two-photon excitation. However, this approach does allow for double targeting, as exemplified by Wang and Zhang's theranostic pro-prodrug 215 where reduction of the aromatic nitro group by nitroreductase directs release toward cells over expressing this enzyme e.g. hypoxic solid tumours, after which external irradiation (365 nm) effects E/Z isomerism and release of the gemcitabine reporter group.217 Concomitant with this is formation of a strongly fluorescent coumarin reporter from the self-immolative linker. A related double activation strategy has also been exemplified by Zeng and Wu (see Scheme 52).
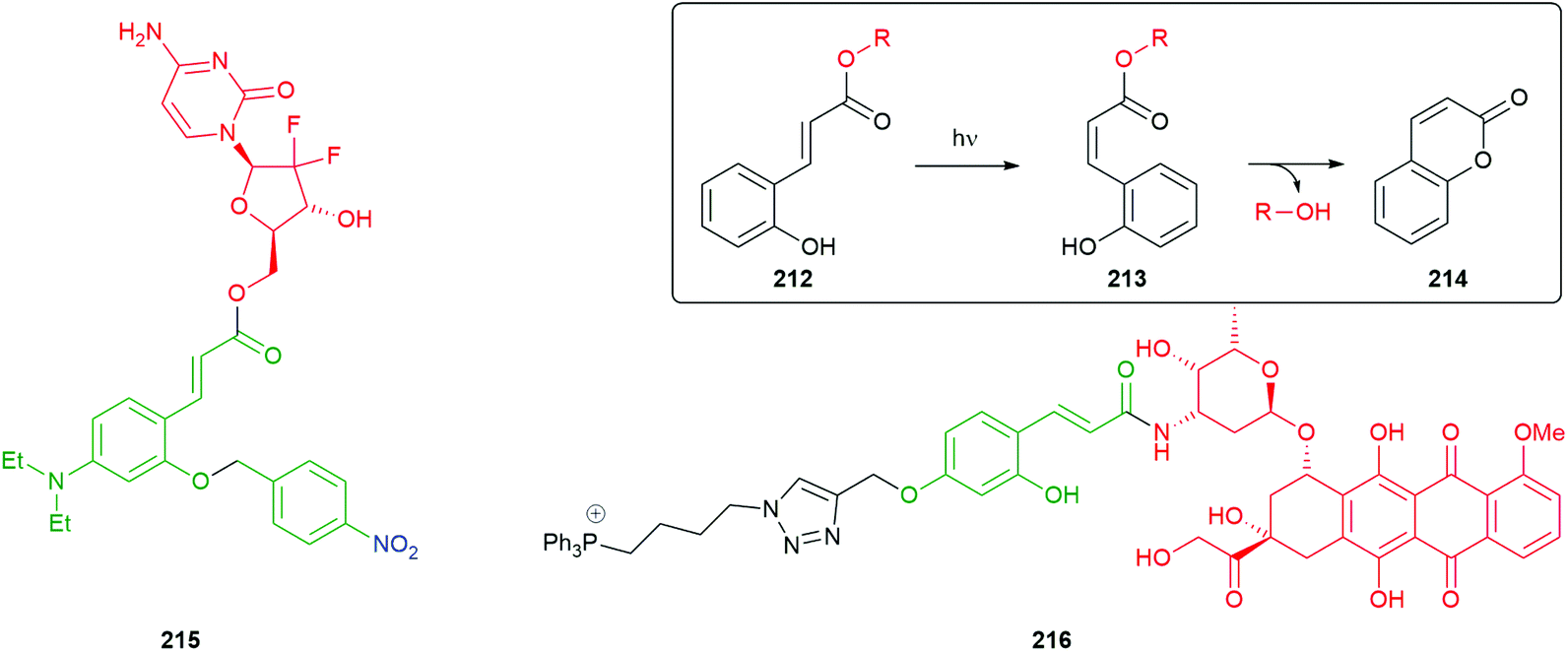 | ||
| Scheme 48 Drug delivery platforms 215 and 216 based around photoisomerism of cinnamate linkers.217,218 | ||
Sasmal et al. have recently reported drug delivery platform 216 based upon a key cinnamate photoactivation.218 In the same way as Mosey and Floreancig (Scheme 7), Sasmal et al. have employed Murphy's triphenylphosphonium directing group to target the mitochondria of the relevant cell type, and the success of this approach was evident in the fluorescence microscopy studies following treatment of HeLa cells with 216 (92% of cellular uptake into the mitochondria within 2 h). Use of one (365 nm) or two-photon irradiation (700 nm)219 of 216 proved successful in releasing the doxorubicin reporter group.
An alternative platform for effective intramolecular cyclisation systems have been realised by the trimethyl-lock linkers, these take advantage of a Thorpe–Ingold effect in tandem with repulsion from an adjacent aromatic methyl group: recently, these have been thoroughly reviewed by Klahn.220–223 Jullien and Schmidt have reported valuable kinetic data of their rates of cyclisation, utilising a photoactivatable self-immolative system 217 that, upon trigger activation, results in a 6-exo-trig-cyclisation (218), favoured by steric hindrance, resulting in lactonization of the linker to give 219 in ca. 3 minutes liberating the fluorescent reporter 220 (Scheme 49).199 This sterically induced effect occurs on account of the unfavourable interactions between the methyl residue at the meta-position on the phenolic core and geminal methyl groups at the benzylic position of the alkyl chain. Further data is reported on a phenol-carbamate cyclisation linker. They note the general point that cyclisation-based linkers tend to self-immolate less rapidly than elimination-based linkers.
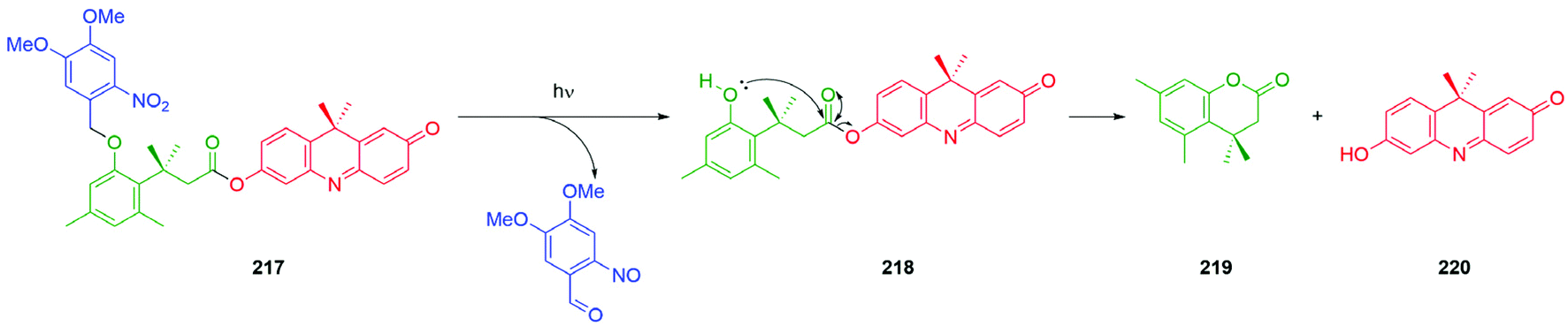 | ||
| Scheme 49 Near-UV triggered self-immolative system 217 cable of undergoing a trimethyl-lock accelerated cyclisation event resulting in the release of a fluorescent reporter moiety as described by Jullien, Schmidt and co-workers.199 | ||
Wang and co-workers have used the trimethyl-lock linker as an esterase-triggered source of the gasotransmitter H2S (Scheme 50).224 Treatment of 221a–b with porcine liver esterase gives rapid cleavage of the trigger group. The resultant phenol 222 undergoes facile cyclisation, possibly following protonation to the thioacid, to give δ-lactone 219 and the reporter group, H2S. By variation of the ester group, the rate of hydrolysis could be tuned (221at1/2 13.0 ± 2.4; 221bt1/2 28.7 ± 1.5). Additionally, the absence of the aromatic methyl groups could significantly slow the rate of the cyclisation reaction. Together these allow variation in the rate of delivery of the H2S gasotransmitter. Zheng and Wang have also modified this approach to deliver hydrogen persulfide using 223a–b (Scheme 50).225 By altering the trigger group to a phosphate in 224, triggering can occur using alkaline phosphatase.
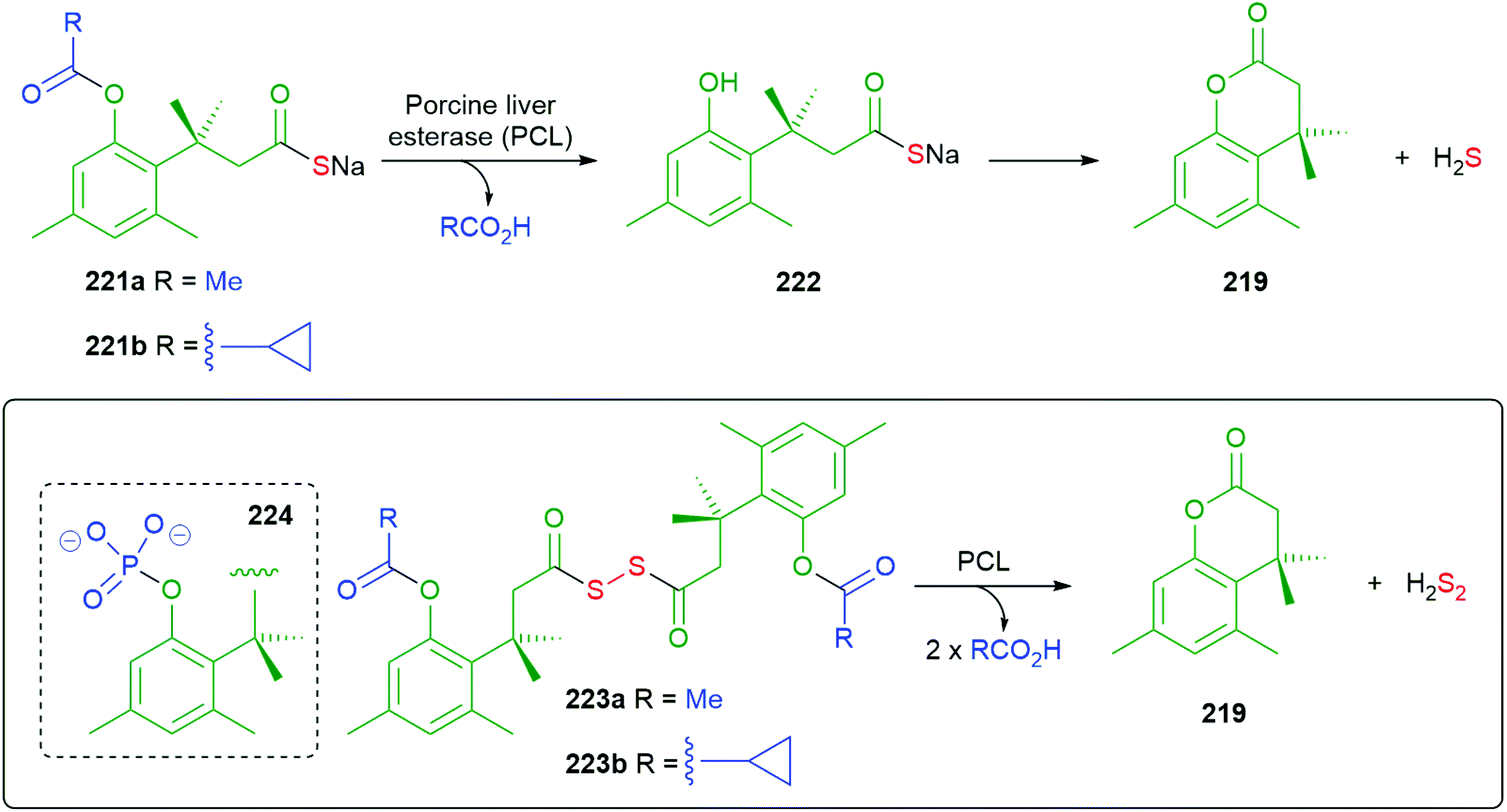 | ||
| Scheme 50 The esterase/alkaline phosphatase triggered trimethyl-lock linker for controlled release of H2S and H2S2 reported by Wang, Zheng and co-workers.224,225 | ||
By coupling the ability of some 1,4-benzoquinones to function as substrates for DT-diaphorase, being reduced to the corresponding hydroquinone, a combined trigger–linker of the trimethyl-lock type has been used by Cheng as part of a delivery system 225 for N-azidoacetylmannosamine (Scheme 51).226 The trigger–linker and reporter group are connected by a second, N,N′-dimethylethylene diamine, linker; this linker was established to give negligible non-specific hydrolysis. Thus, following reduction of 225 by DT-diaphorase, 226 undergoes cyclisation to eliminate δ-lactone 227. The N,N′-dimethylethylene diamine linker in 228 then cyclises to urea 190 to deliver N-azidoacetylmannosamine 229; the latter is known to be incorporated into cell surface saccharides, offering a partner for subsequent ‘click’ chemistry modifications. The overexpression of DT-diaphorase in multiple cancer cell lines makes this of particular interest in the study of cancer biology; for example, 225 was shown to deliver 229 in Bxps-3 cells (pancreatic cancer cells).
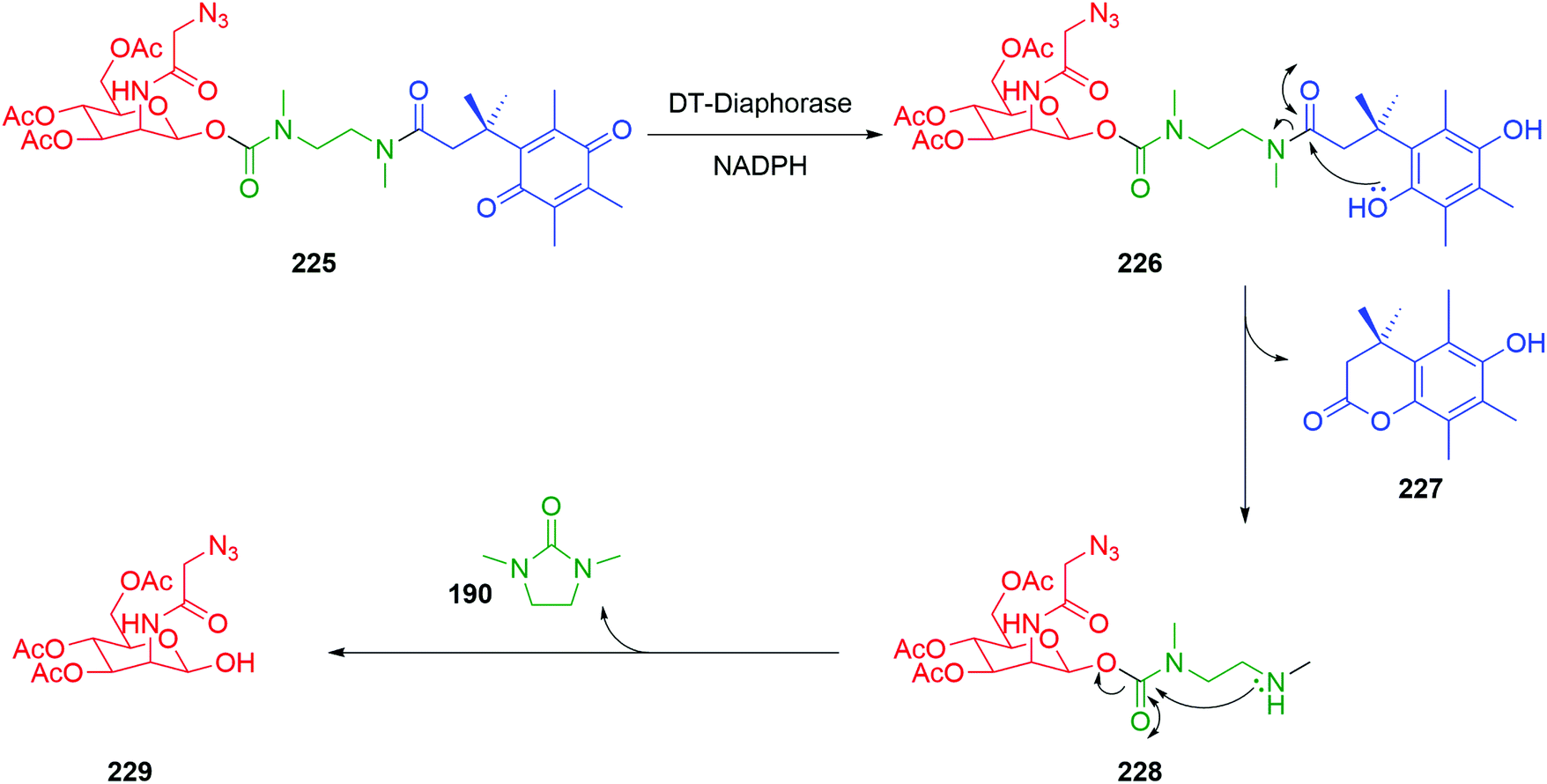 | ||
| Scheme 51 Cheng's DT-diaphorase triggered SIE 225 for release of N-azidoacetylmannosamine.226 | ||
In 2015, Zeng and Wu used the combined trigger–linker of the trimethyl-lock type with an aniline-based 1,6-elimination linker to give a camptothecin delivery system 230 (Fig. 13).227 In this system, an additional feature is the ability to self-indicate fragmentation by the loss of fluorescence quenching (photoinduced electron transfer – PeT – from the camptothecin to the quinone). Simultaneously, this also functions as diagnostic for DT-diaphorase activity, with the fluorescence intensity being shown to reflect the levels of the enzyme. This strategy has been adapted to a dendritic platform (see section 3).228
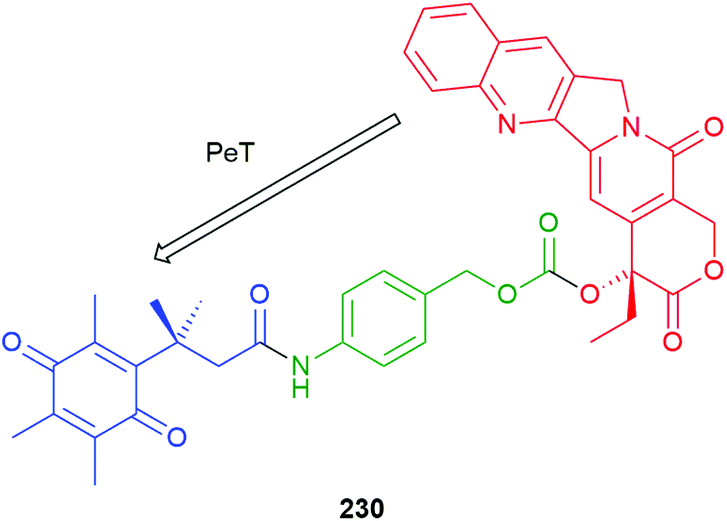 | ||
| Fig. 13 Zeng and Wu's theranostic SIE 230 for camptothecin delivery.227 | ||
In 2018, Wu and Zeng developed a selective methotrexate delivery system 231, in a liposome carrier; as it requires two triggering stimuli, it may be designated as a pro-prodrug (Scheme 52).229 First, reduction/cyclisation of the DT-diaphorase sensitive 1,4-benzoquinone trigger/linker in 231 leads to selective cleavage in cells overexpressing that enzyme. Concomitant with this, activation of fluorescence (475 nm) of the coumarin moiety in 232 occurs, facilitating assessment of the enzyme's levels. Subsequent irradiation by one-photon (visible – 400–450 nm) or two-photon (near infrared) light cleaves the coumarin to release the dihydrofolate reductase inhibitor, methotrexate. Critically, in the absence of DT-Diaphorase, cleavage of the coumarin under irradiation at 400–450 nm is blocked. Such double triggering offers the potential of detection and selective cytotoxicity toward cancerous cells overexpressing DT-diaphorase e.g. HeLa and A549.
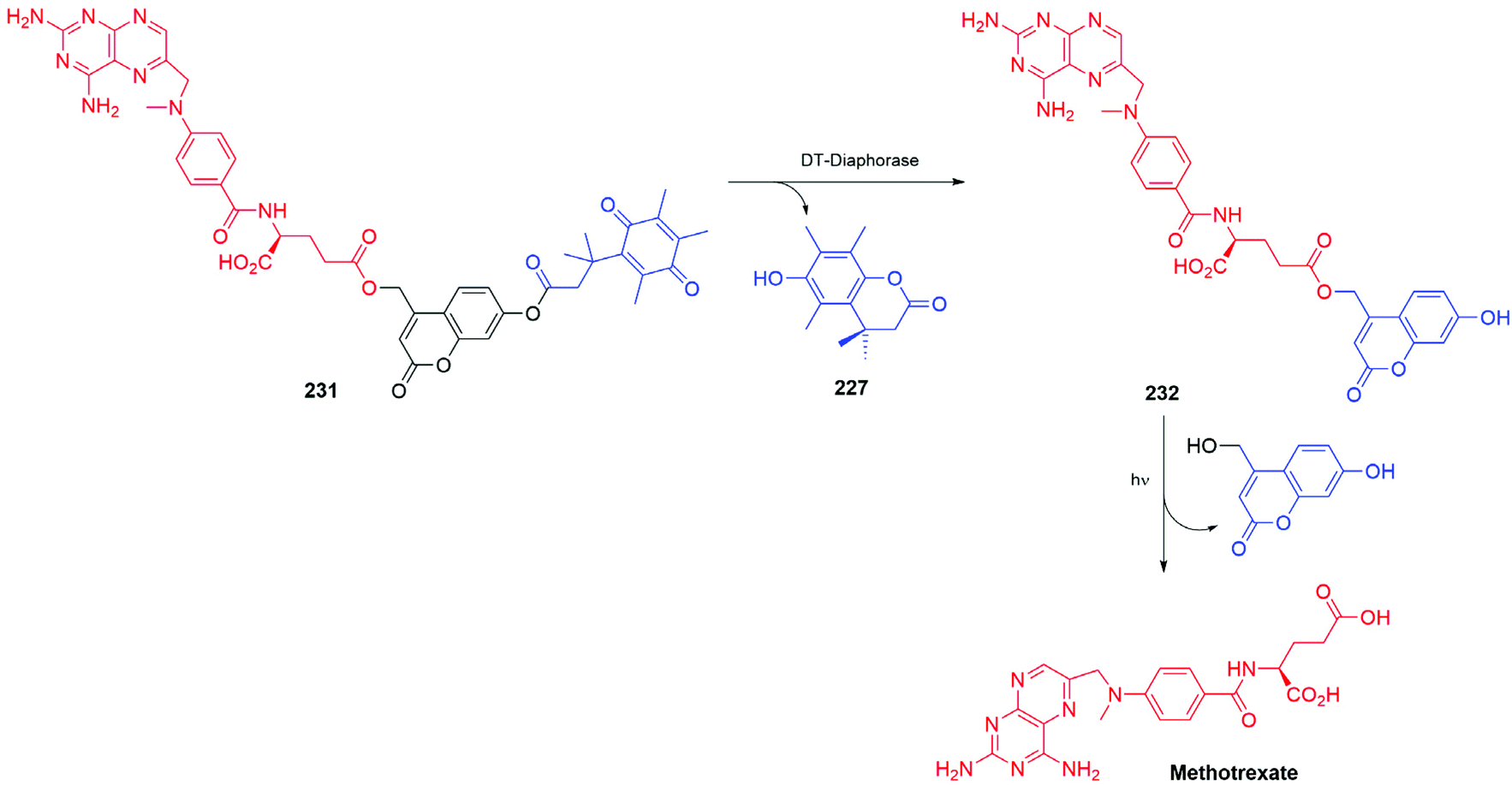 | ||
| Scheme 52 Wu and 's pro-prodrug system 231 for selective targeting of cancer cells.229 | ||
A practically interesting in vivo imaging application of the 1,4-benzoquinone trigger/linker has been reported by a collaboration between Kwak, Sessler, Shabat and Kim, utilising an optimised chemiluminescent reporter group described by Sigman and Shabat, comparison was made between the pro-chemiluminescence 233 and pro-fluorescence probes 234 shown in Fig. 14.230,231 Amongst the advantages attributed to chemiluminescence is good penetration depths into tissue, higher signal-to-noise ratios than fluorescence and reduced interference from autofluorescence. Upon exposure to DT-diaphorase (NQO1), the chemiluminescent probe 233 was observed to rapidly begin emission of green light (≤90 s, monitored at 515 nm), undergoing rapid reduction, cyclisation, 1,6-elimination and fragmentation of the dioxetane without an observed intermediate. In vitro, this probe was found to differentiate between A549 cells (elevated NQO1 levels) and H596/WI-38 cells (low NQO1 levels). The fluorescence-based probe 234, developed by McCarley, was also effective;232,233 however, chemiluminescence was found to deliver a higher signal-to-noise ratio in reporting on NQO1 activity in A549 cells. In vivo imaging of NQO1 activity in A549-tumour-bearing mice, by chemiluminescence, was effective whilst no signal was seen with H596 derived xenografts.
 | ||
| Fig. 14 Chemiluminescence 233 and fluorescence-based 234 trimethyl-lock linker probes for in vitro and in vivo imaging of NQO1 activity.230–233 | ||
Ji and Miller have developed a self-immolative system 235 for the selective transport of the antibiotic ciprofloxacin into bacterial cells, utilizing the microbial iron acquisition system (Fig. 15).234 The targeting is achieved by a siderophore (desferrioxamine B) that is attached to a succinate trigger; in turn, this connects to a trimethyl-lock linker and, finally, the reporter group. As a cautionary note, whilst displaying antibiotic activity, it proved less active than ciprofloxacin alone, raising the probability that the enzymatic cleavage of the trigger was inefficient. Linkers were originally introduced to ease the problem of enzyme access to a scissile bond, but here a problem remains.
 | ||
| Fig. 15 The siderophore directed SIE 235 for delivery of ciprofloxacin reported by Ji and Miller.234 | ||
In an ingenious adaptation of the Julia olefination, Wang and Wang have reported an esterase sensitive prodrug 236 for delivery of the potential gasotransmitter SO2 (Scheme 53).235 A range of molecules related to 236 were prepared, with the glycinate ester shown below facilitating rapid hydrolysis and enhanced aqueous solubility. The substituents on the linker facilitates rearrangement via237 and 238 to give 239, the gem-dimethyl precludes β-elimination of the ester prior to hydrolysis, then, critically, stabilises the antiperiplanar transition state for fragmentation of 239 to release SO2. Compound 236 was shown to release SO2 in HeLa cells.
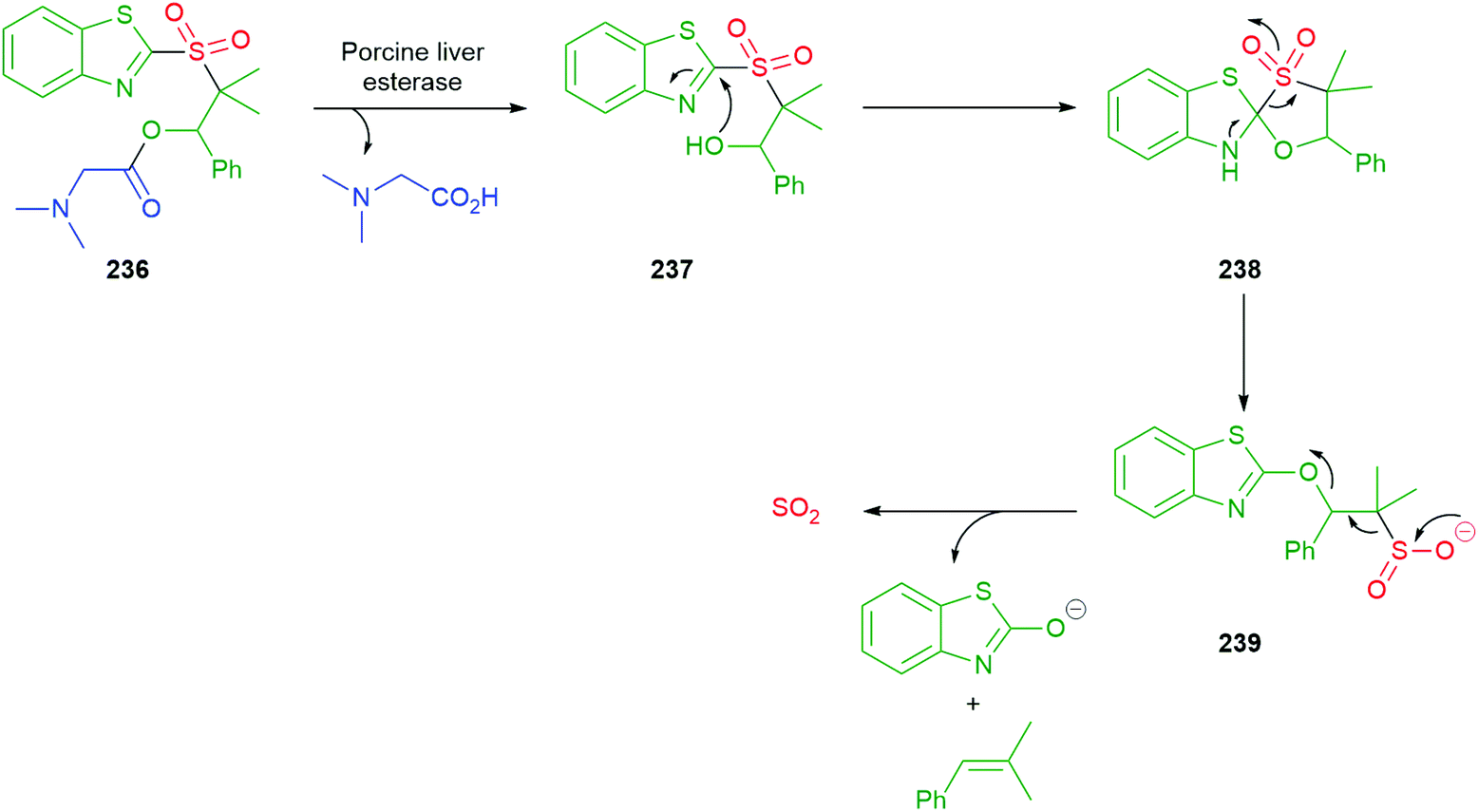 | ||
| Scheme 53 Wang and Wang's esterase triggered SO2 release SIE 236 based on a modified Julia reaction.235 | ||
Linkers featuring a phosphate/phosphonate unit have proved to be of significant value, in both the cyclisation–elimination mode and from para-aminobenzylalcohol236 linkers where they may readily replace the role played by carbonates or carbamates in connecting to the reporter group (see Scheme 50 for use as a trigger group). These groups have found an early implementation in prodrugs; for example, they have been employed in the form of ester hydrolysis triggered phosphonamidates for the delivery of tenofovir237 and Propofol,238 building upon earlier technology reported by McGuigan et al. (Scheme 54).239–243 Thus, following administration of Tenofovir alafenamide 240, hydrolysis to carboxylate 241 is thought to occur and that spontaneously cyclises to 242; in turn, this hydrolyses twice to give first 243 then release of the drug Tenofovir (NB the usual colour scheme has been abandoned as trigger, linker and reporter groups overlap). This approach was adapted by Wei et al. to give prodrugs 244 and 245 for the phenolic reporter groups Propofol and HSK3486, respectively: compounds with a variety of uses including starting and maintaining anaesthesia. This approach offers a general platform for the delivery of phenolic drugs.
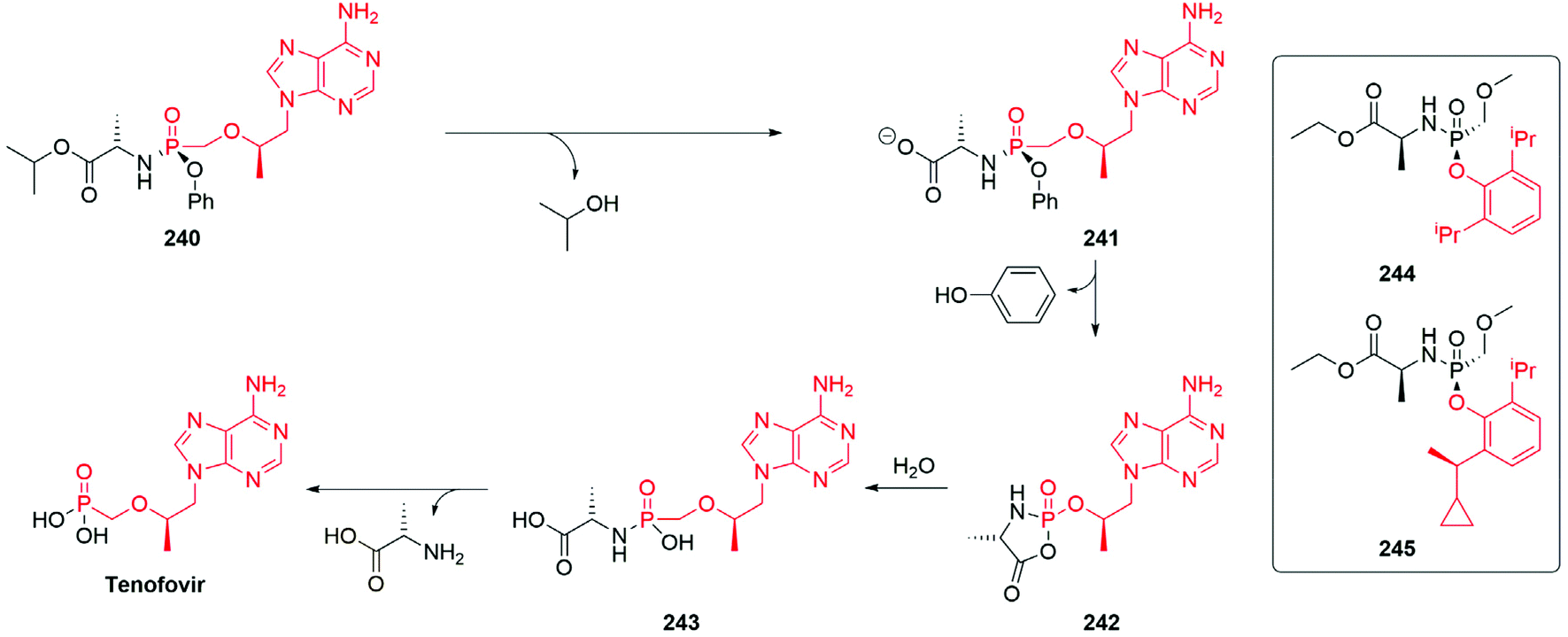 | ||
| Scheme 54 The release of Tenofovir from prodrug Tenofovir alafenamide 240.237,238 | ||
Berkman and co-workers have reported a number of pH triggered phosphoramidate-based systems for the delivery of amines, with general acid catalysis of cleavage of the P–N bond being noted (Scheme 55). The pKa of the amine reporter group was found to be important in determining the rate of hydrolysis.2441H and 31P NMR studies on 246 revealed cleavage of 247 proceeding via a two-step mechanism, passing through an acylphosphate intermediate 248 to give 249.245 Further work on a series of compounds related to 250 confirmed the importance of the nature of the amine reporter group and established the significance of the carboxylic acid (or other acid group), its pKa and its position relative to the phosphoramidate in labializing the P–N bond.246 In studies on the rate of hydrolysis of 250, it was shown to vary significantly over the pH range 3–7.4.246
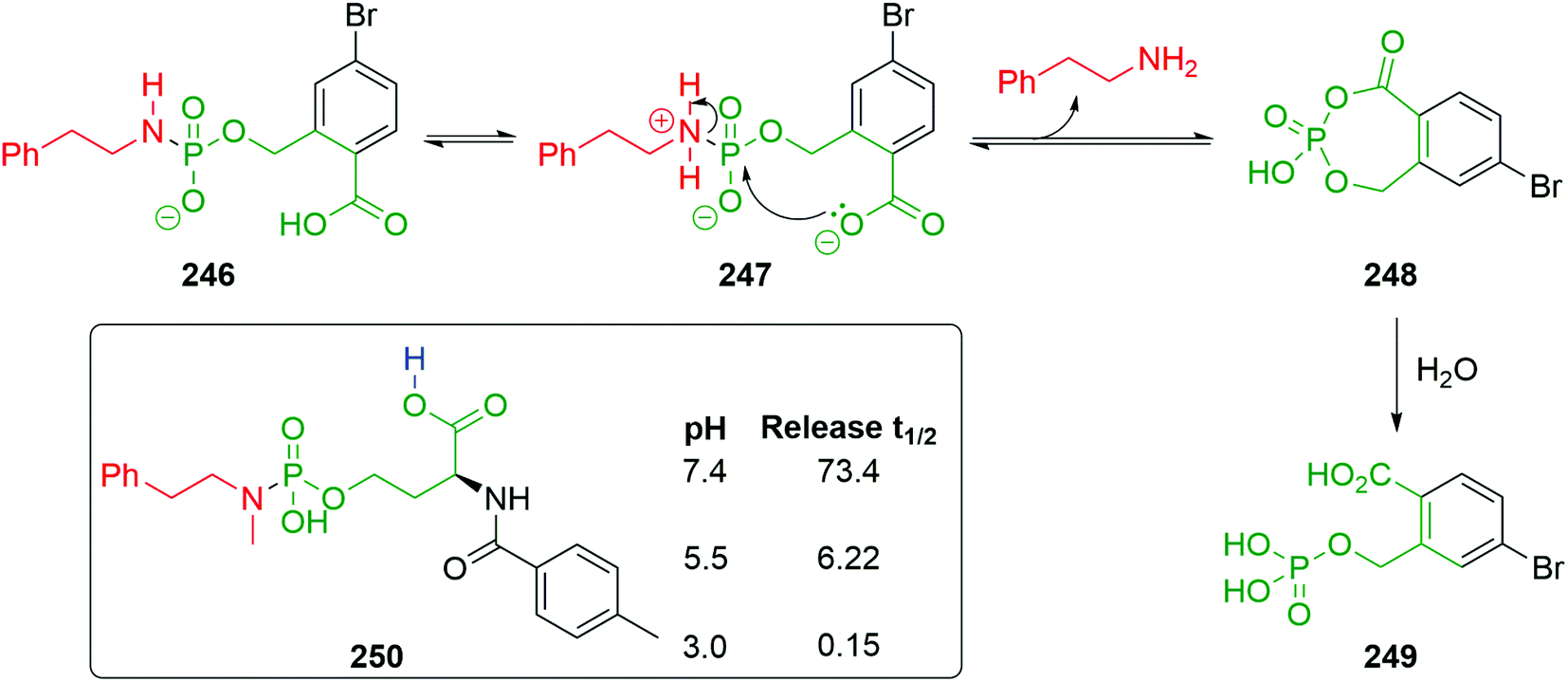 | ||
| Scheme 55 The pH triggered phosphoramidate-based systems for the delivery of amines reported by Berkman and co-workers.244–246 | ||
Building upon this work, a SIE 251 for the delivery of the antimitotic agent monomethyl auristatin E has been reported (Scheme 56).247 Inclusion of an N,N′-ethylenediamine cyclisation linker, coupled with a 1,6-elimination linker in 251 achieved consistent rates of hydrolysis, regardless of varying steric hindrance of the reporter group, the proposed mechanism of fragmentation of 251 to yield the antimitotic agent and the phosphate 252via intermediates 253–255 is illustrated in Scheme 56. The presence of an azide in 251 makes it adaptable to a number of purposes through ‘click’ chemistry e.g. the potential for incorporation into an antibody–drug conjugate. This methodology has also been applied to controlled release of L-Dopa.248
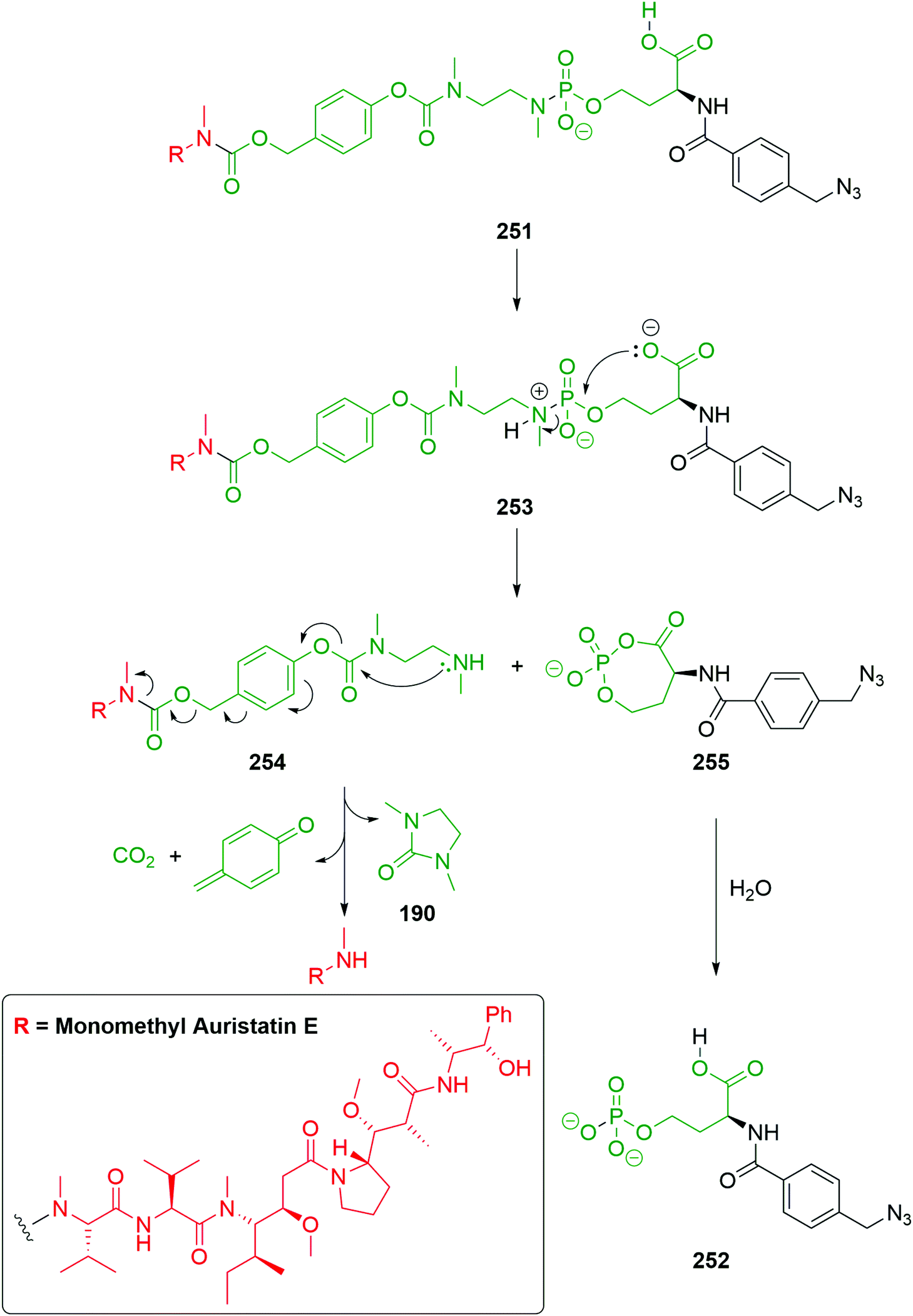 | ||
| Scheme 56 The pH triggered release of monomethyl auristatin E from the SIE 251.247 | ||
Recently, Baszczyňski and Procházková have described a set of UV light triggered, phosphate-based self-immolative entities that facilitate the release of two reporter groups, exploiting that particular advantage of using a phosphate over, say, a carbamate (Scheme 57).249 The release of more than one reporter group has mainly been achieved by the use of dendritic and polymeric systems (vide infra) though, for example, Phillips has described an acetal-based system for achieving this (Scheme 9, and Fig. 4). The rate of release from 256 proved tunable by a combination the Thorpe–Ingold effect on the cyclisation step and variation of the pKa of the reporter group. The first reporter to be released occurs via an SI mechanism (257 to 258a–b, then hydrolysis to 259a–b); whereas, the second release to 260 occurs by direct hydrolysis. Phenols were found to be good leaving groups; whereas, alkanols were retained on the phosphate. A particularly rapid release of phenols was observed when a gem-dimethyl group was present on the linker (256, R1 = R2 = Me).
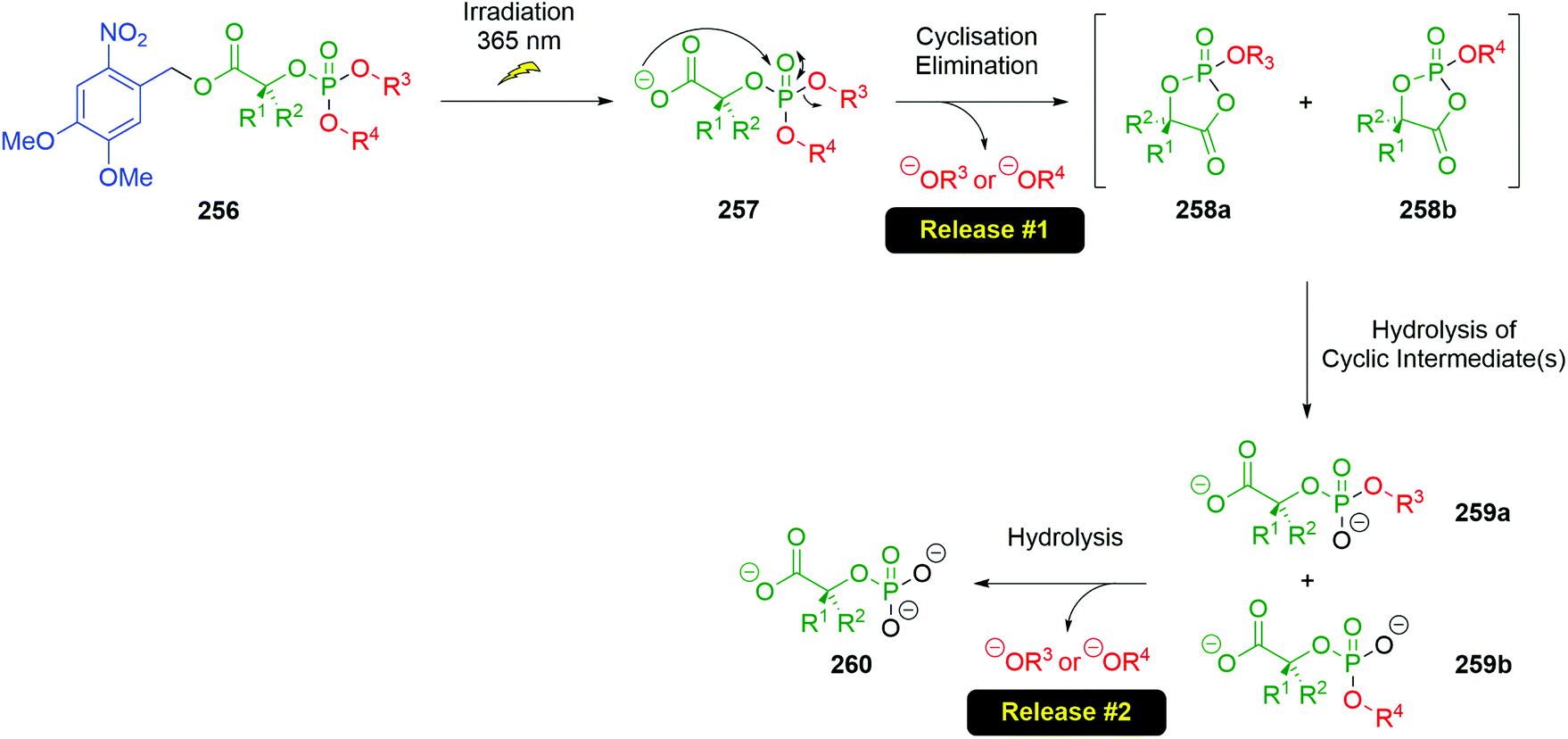 | ||
| Scheme 57 Baszczyňski and Procházková's phosphate-based self-immolative entity 256 for release of two reporter groups.249 | ||
Recently, Rotger has introduced a new linker based upon a squaramide group 261 (Scheme 58).250 When the linker nitrogen proximal to the squaramide is N-methylated, the preferred E,Z-conformation 261 (boxed structure) favours cyclisation through an apparent 7-endo-trig mode. The combination of stability of linker under physiological conditions, with a good rate of cyclisation–elimination for aniline-based reporter groups (alkylamines are significantly slower) makes this a valuable addition to the chemist's arsenal. Use of the linker was exemplified with delivery system 262 which undergoes reduction of the nitro group with nitroreductase in the presence of NADH, followed by 1,6-elimination of 263 to give 264. Cyclisation of 264 occurs smoothly to release the aniline-based nitrogen mustard 265 and bicyclic squaramide 266.
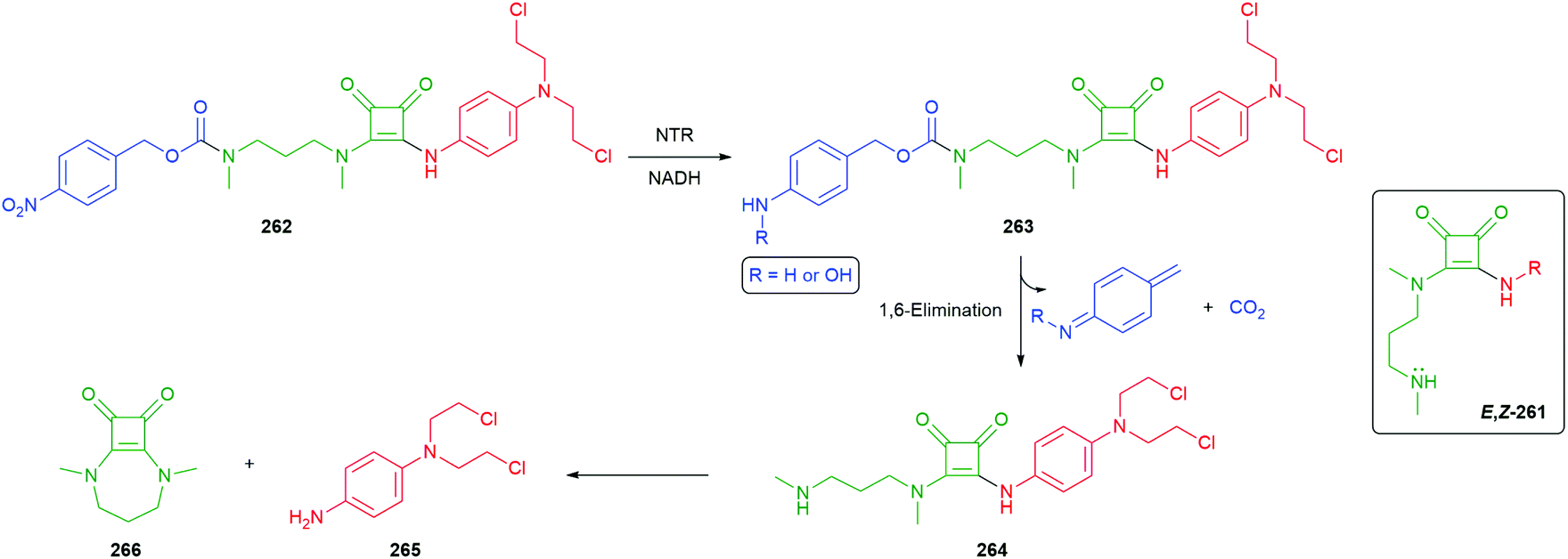 | ||
| Scheme 58 Rotger's novel squaramide self-immolative linker, illustrated with delivery of a nitrogen mustard 265 following triggering by nitroreductase (NTR).250 | ||
3 Amplification of self-immolative systems through dendron-based and related approaches
It was quickly understood that release of reporter groups could be amplified following a single triggering event by either linking self-immolative units together in the appropriate manner (to yield self-immolative polymers, see section 4 below) or alternatively utilising multiple electronic cascade processes through branching units (i.e. self-immolative dendritic systems). To this end, self-immolative dendritic systems are a unique class of molecules defined by the release of multiple reporter groups via a series of electronic elimination processes arising from a single activation event.23,208,251,252 Suitable structural modification of linear self-immolative aromatic linker units to permit degradation via additional pathways enables the creation of branched self-immolative linker systems and ultimately amplified release of multiple reporter groups following the triggering event.In the past decade, the synthetic focus towards self-immolative systems capable of amplified release of reporter units has shifted from the construction of dendritic compounds of high generation number; their syntheses are frequently far too complex and practically challenging, not to mention extremely costly in terms of labour, in relation to the advantage of amplification offered. More pragmatic studies on self-immolative systems offering amplified release have thus focused primarily on the use of first generation dendrons to demonstrate amplification for new trigger or linker groups or indeed application of these systems under realistic environmental conditions.
3.1 Phenol-based AB2, AB3 and AB6 based linkers
Shabat and co-workers described an example of a phenol-based AB2 linker system to control the disassembly rate of self-immolative dendrons.208 Dendrons 267 and 268 contained the methyl and ethylcarboxy-ester substituent on a core benzene building-block (Scheme 59). Upon a single trigger event via photo-irradiation of the ortho-nitrophenyl residue to promote 1,6- and 1,4-elimination, the dendrons release para-nitroaniline from 269/270via diamine cyclisation and fragmentation from the cresol derivative 271. The dendron 268 featuring the electron-withdrawing substituent exhibited a higher disassembly rate when compared to dendron 267 that possessed a para-methyl substituent. It was proposed that control of the rate of self-immolative dendrimer disassembly would be applicable in the field of diagnostic or therapeutic applications when a specific release of reporter rate is needed.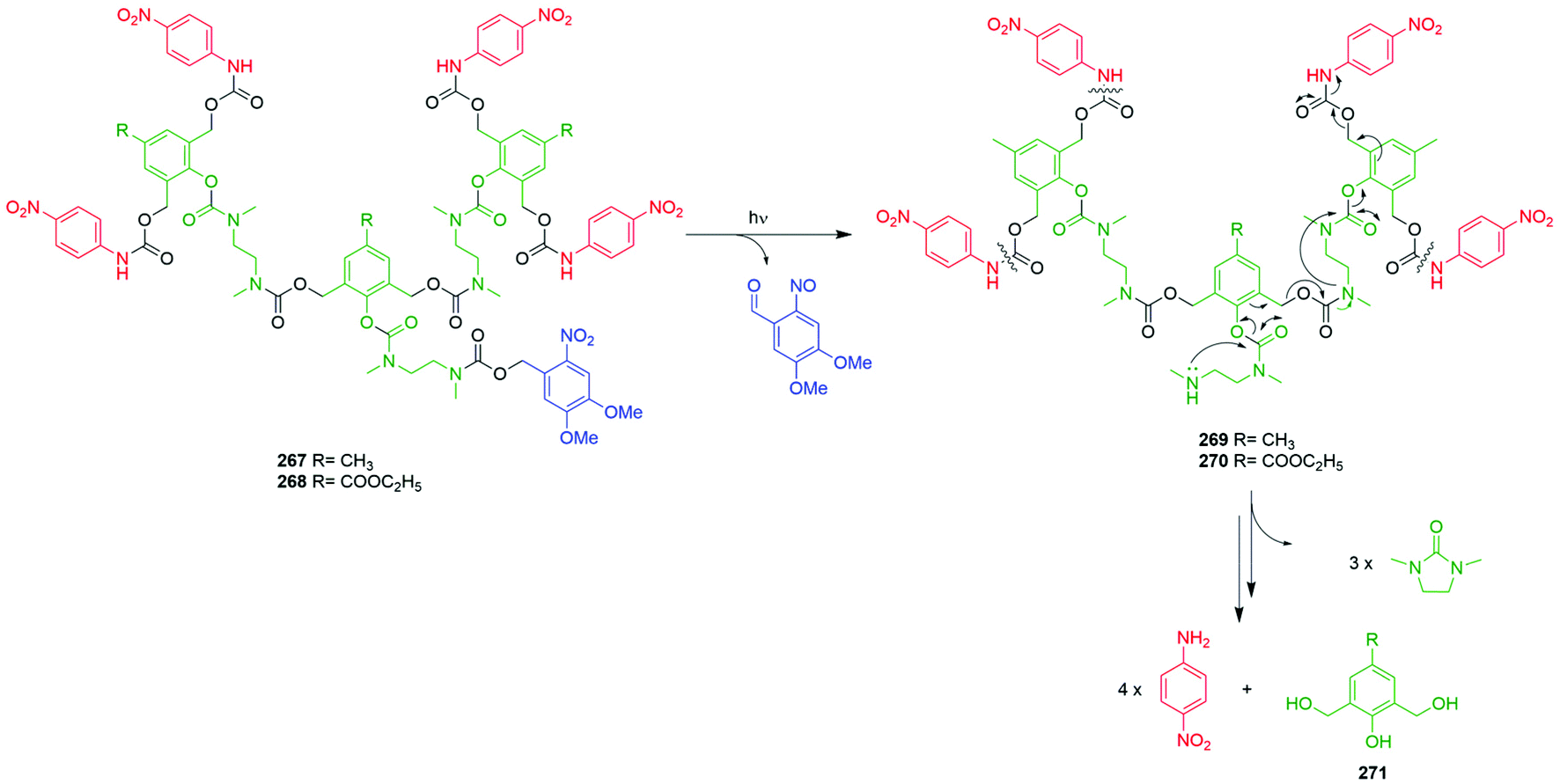 | ||
| Scheme 59 Chemical structure and disassembly of second-generation 2,4-bis(hydroxymethyl)phenol AB2 linker-based self-immolative dendrons.208 | ||
Shabat and co-workers subsequently developed a divergent strategy wherein a phenol-based self-immolative dendritic pro-drug system,198,253 featuring a 2,4,6- tris (hydroxymethyl)phenol branching unit, allowed preparation of a number of derivatives. This approach facilitated the stepwise coupling of doxorubicin (DPX) directly via its primary amine, whereas etoposide (ETP) and camptothecin (CPT) were coupled through the N,N′-dimethylethylenediamine self-immolative cyclisation linker (as exemplified in Scheme 43, 186a). Self-immolative elimination was carried out under physiological conditions in the presence of catalytic antibody 38C2 enzymes198 and facilitated the disassembly via retro-aldol, retro-Michael cleavage of the trigger group and release of three different drugs (i.e. from a single molecular scaffold) through a combination of 1,6-and two 1,4-elimination processes after only a single trigger event. It was, however, observed for self-immolative higher generation dendrons based on AB2 repeat units that the aqueous solubility was compromised in the light of their hydrophobic character, which in turn led to aggregation in this protic medium.254,255 An alternative approach was thus developed to overcome this solubility issue whereby the multiplicity of a low generation self-immolative dendron was increased to permit high substrate loading and also improve the aqueous solubility.256 The self-immolative dendron 272 featured 2,4,6-triformylphenol as the branch point, phenylacetamide as the trigger and six tryptophan reporter groups (Scheme 60). The self-immolative dendron was then incubated in phosphate buffer of saline (PBS, pH 7.4) both in the presence and absence of penicillin G amidase (PGA). PGA cleaved the phenylacetamide from dendron 272 and the resulting phenoxide 273 disassembled to release a total of six equivalents of tryptophan and the hexahydroxyphenol 274. The release of tryptophan was completed within 48 hours in the presence of PGA; whereas, release was not observed in the absence of the enzyme.
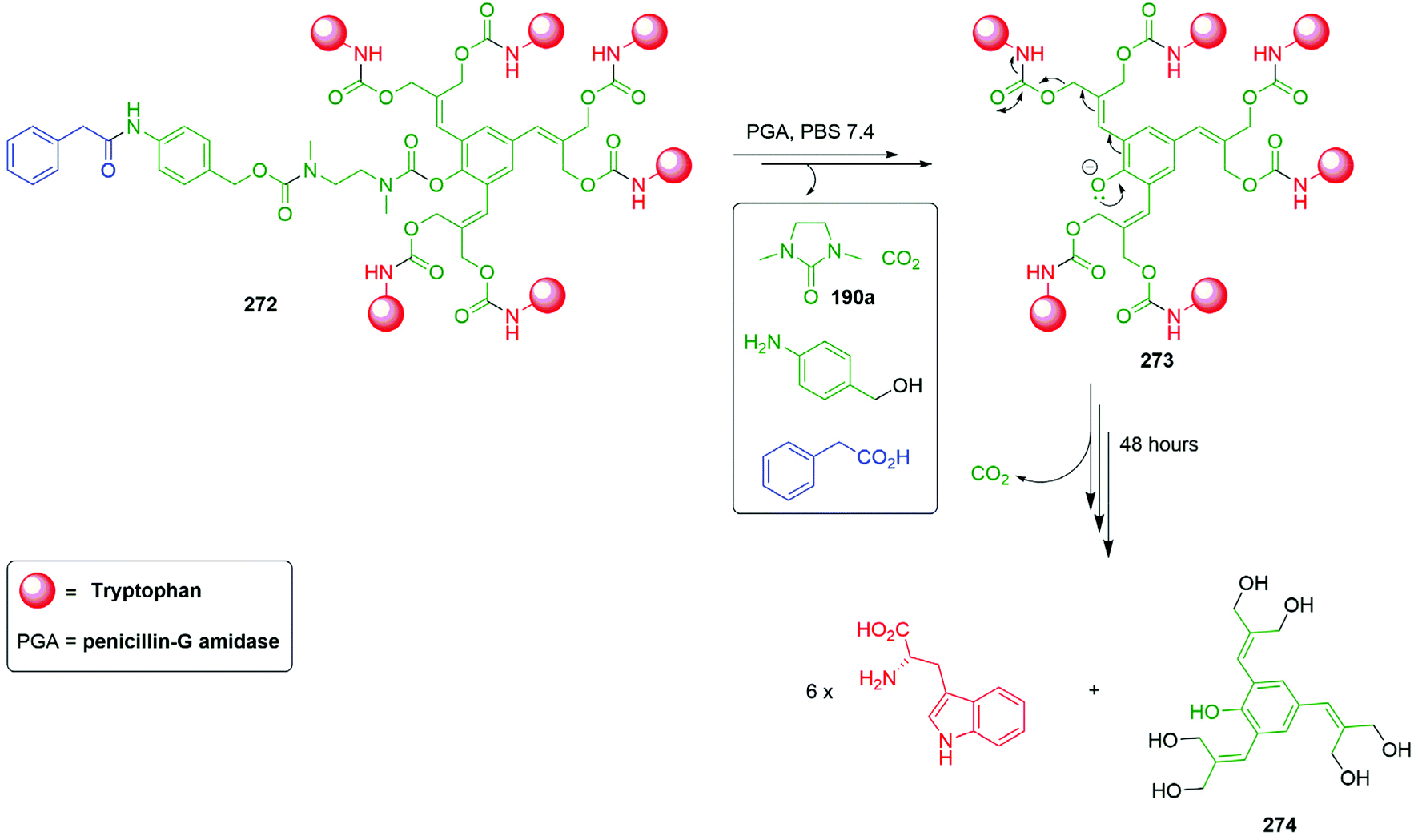 | ||
| Scheme 60 The structure of a self-immolative AB6 dendron 272 with tryptophan units and phenylacetamide group as a trigger.254 | ||
Shabat and co-workers have also utilised a first generation dendritic system where the trigger unit was a phenyl boronic ester (susceptible to the presence of hydrogen peroxide, vide supra) to develop FRET sensors (see 275 and 276 in Fig. 16).257 These dendrons feature fluorophores (a fluorescein system and CY5, respectively) whose visible emission was suppressed by the close proximity of the quencher compounds coupled onto the branched unit. The turn-on FRET sensors shown in Fig. 16 operate by exposure to hydrogen peroxide (in buffered PBS plus DMSO as a co-solvent) to facilitate release of the fluorophores and quencher compounds into solution via two 1,4-elimination processes thus permitting observation of the fluorescence.
 | ||
| Fig. 16 The phenolic-based self-immolative FRET systems 275 and 276 reported by Shabat and co-workers.257 | ||
Akkaya has demonstrated the applicability of an AB2 dendron 277 to the detection of fluoride, utilising Schaap's 1,2-dioxetane-based chemiluminescent reporter group (Scheme 61).258 Related reporter groups have also made a significant impact in a number of areas; in particular, the compounds developed by Shabat (see Fig. 8).118 Treatment of dendron 277 with a source of fluoride ions chemoselectively cleaves the Si–O bond of the trigger group and releases phenoxide 278 that subsequently undergoes successive 1,4-elimination reactions to liberate two equivalents of chemiluminescent reporter group 279 for each ion sensed. The reporter group fragments to 280*, relaxation of which to its ground state occurs with emission in the blue region of the spectrum. The sensor proved effective both in solution and when impregnated into a polymer support (PMMA).258
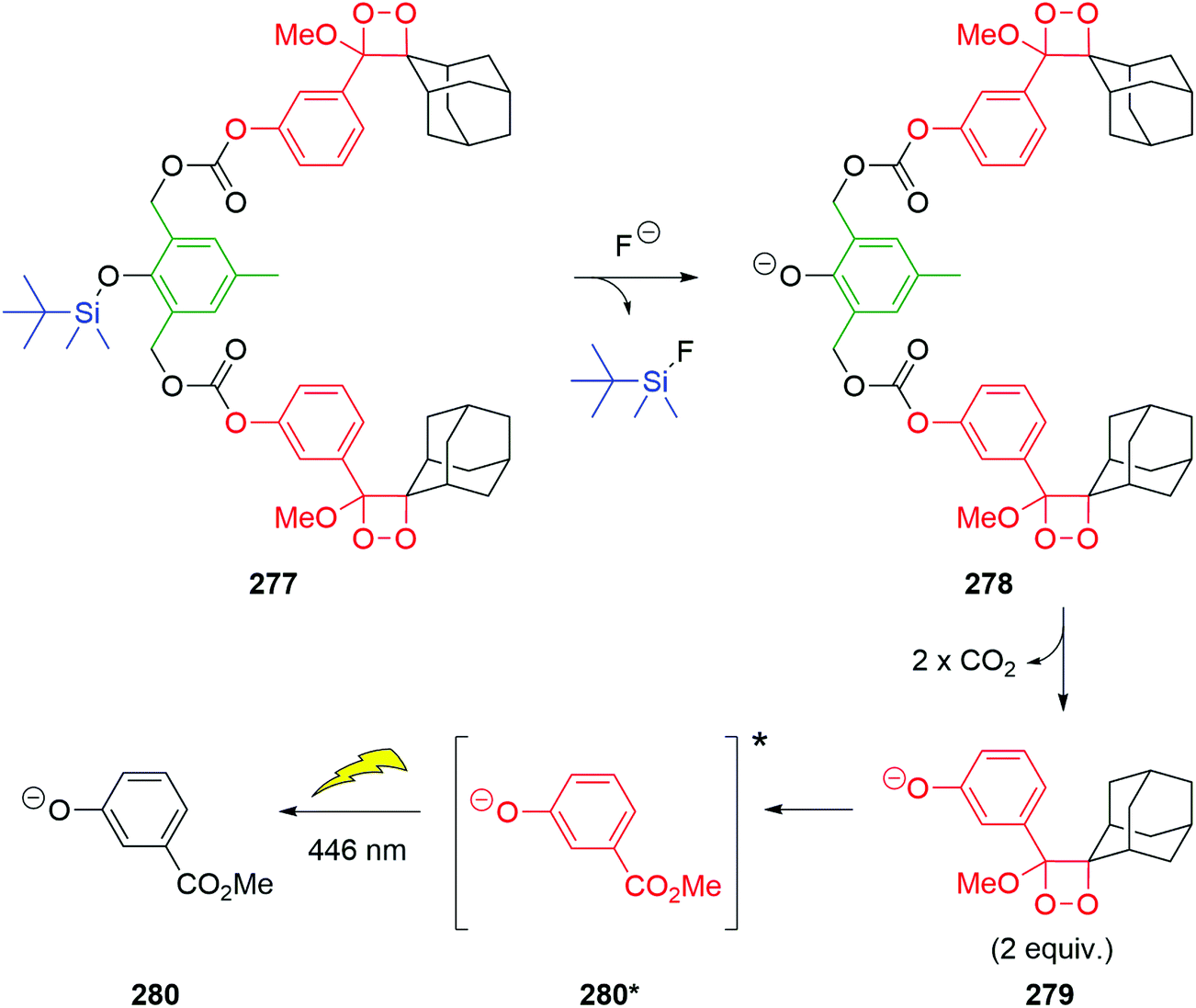 | ||
| Scheme 61 Chemiluminescent sensor for the detection of fluoride using an AB2 linker-based self-immolative dendron 277.258 | ||
Use of two-photon excitation, with near infrared (NIR) light, to trigger a self-immolative delivery system allows good spatial and temporal control of the process and is effective in biological systems. An action cross-section of 1 GM (Goeppert–Mayer) units at 740 nm has been reported for a 4-bromo-7-hydroxycoumarin chromophore. As an alternative to redesigning the trigger chromophore, to increase the action cross-section and hence it's efficiency at triggering an SIE, Almutairi and co-workers have prepared a second-generation (G2) dendritic system 281 that should release four L-glutamic acid units per two-photon excitation (see Fig. 17).209 In practice, some background hydrolysis and side reactions from the irradiation of 281 were observed, but the amount of L-glutamic acid released from the dendron, after 30 minutes exposure to NIR light, was 2.8-fold higher than that from the coumarin conjugate of L-glutamic acid 282. Thus, this work demonstrates the important lesson that while dendrons are effective in this role, it is important to consider potential side reactions.
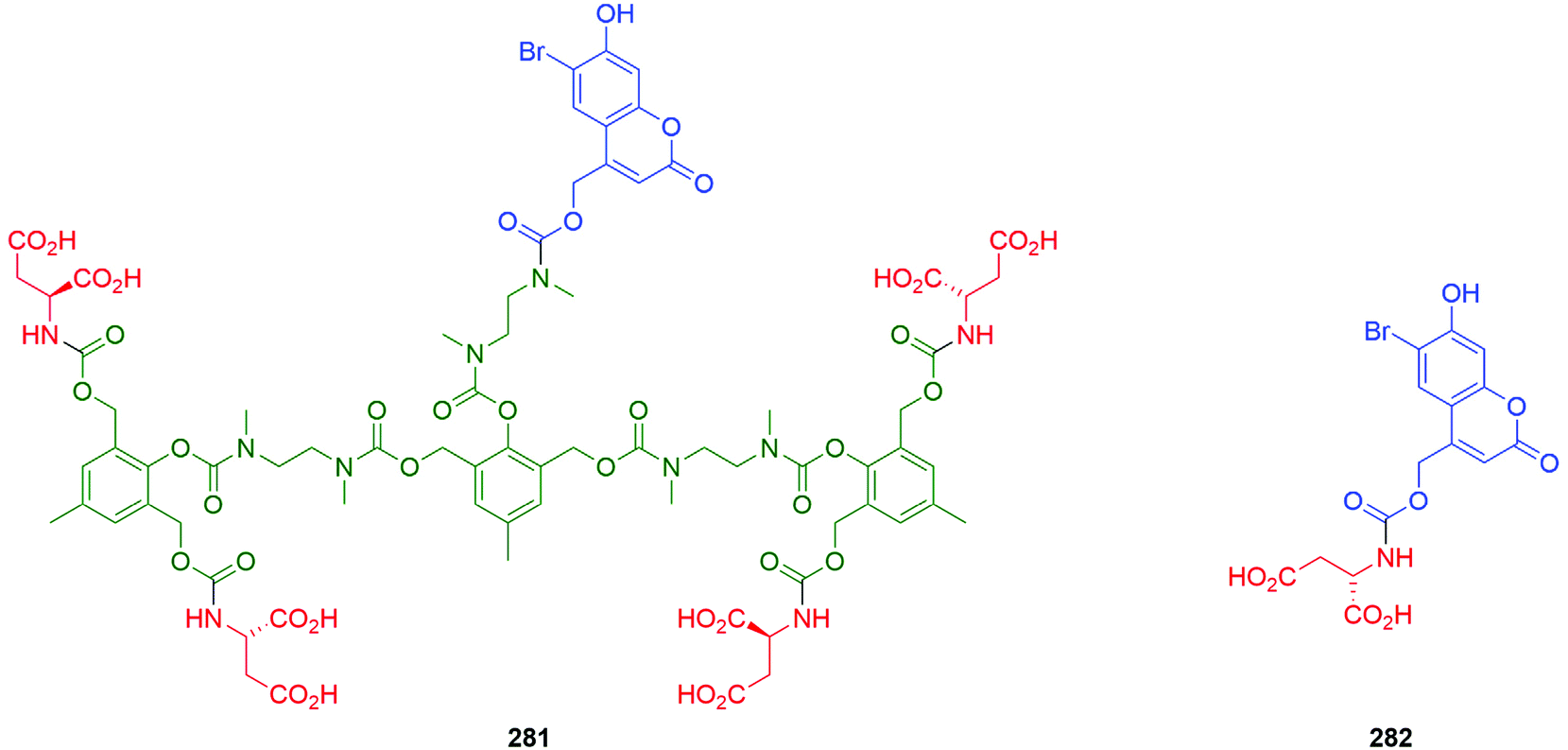 | ||
| Fig. 17 Almutairi's two-photon, NIR triggered G2 dendron 281 for the release of L-glutamic acid.209 | ||
Hennig has described a range of efficiently quenched, NIR fluorescent, activatable (PGA) probes based on the compounds 283 and 284 but varying the fluorescent species, here illustrated with a fluorescein reporter group that forms a non-fluorescent H-dimer in the dendron (Fig. 18).259 The particular advantage of such systems being that once the dye and quencher interaction is optimised, to give the maximum change from the quenched to free state, the trigger component can be varied with little effect on those optical properties. Investigation of the optical properties of these compounds led to a detailed understanding of their behaviour. Interestingly, whether a 2,6 or 2,4-substitution pattern was used in the dendron portion of the molecule had little effect on the relative quantum yields.
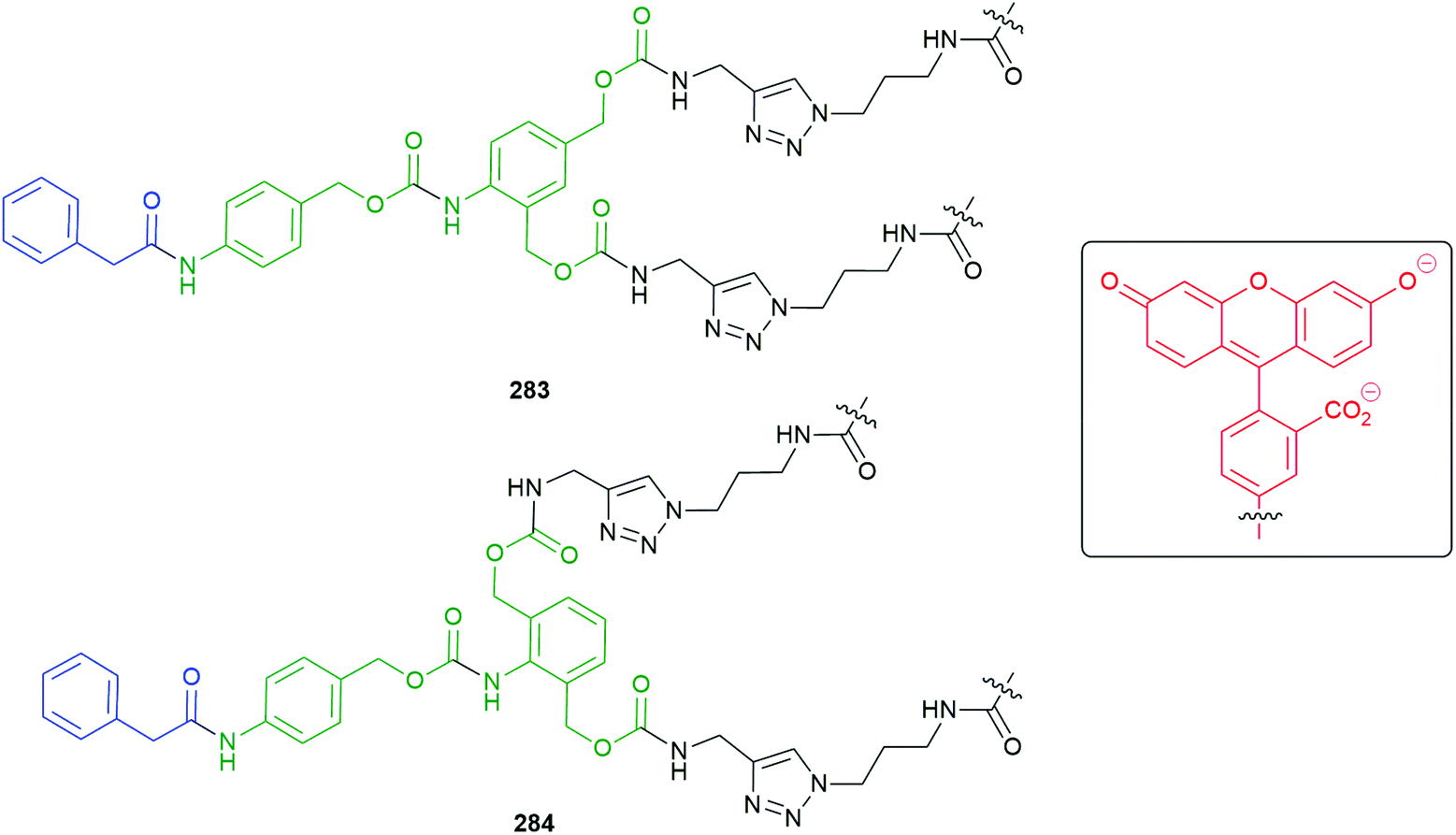 | ||
| Fig. 18 Self-immolative probes 283 and 284 used to detect PGA via a NIR fluorescence response.259 | ||
Gillies and co-workers have developed a route to photo-triggered self-immolative hydrogels260 using a combination of cyclisation linkers and electronic cascade processes. The bis-alkyne dendron 285 shown in Scheme 62 was crosslinked with the four-armed PEG featuring azide end groups via the well-established copper-catalysed ‘click’ cycloaddition to yield a network hydrogel. The dendron component of this network featured an ortho-nitrobenzyl unit end group that could be photochemically triggered to permit liberation of the secondary amine and induce self-immolation via an initial cyclization event followed by electronic cascade (1,4-elimination) to degrade the links of the network, thus disrupting the physical form of the hydrogel as established by studying the compressive moduli.
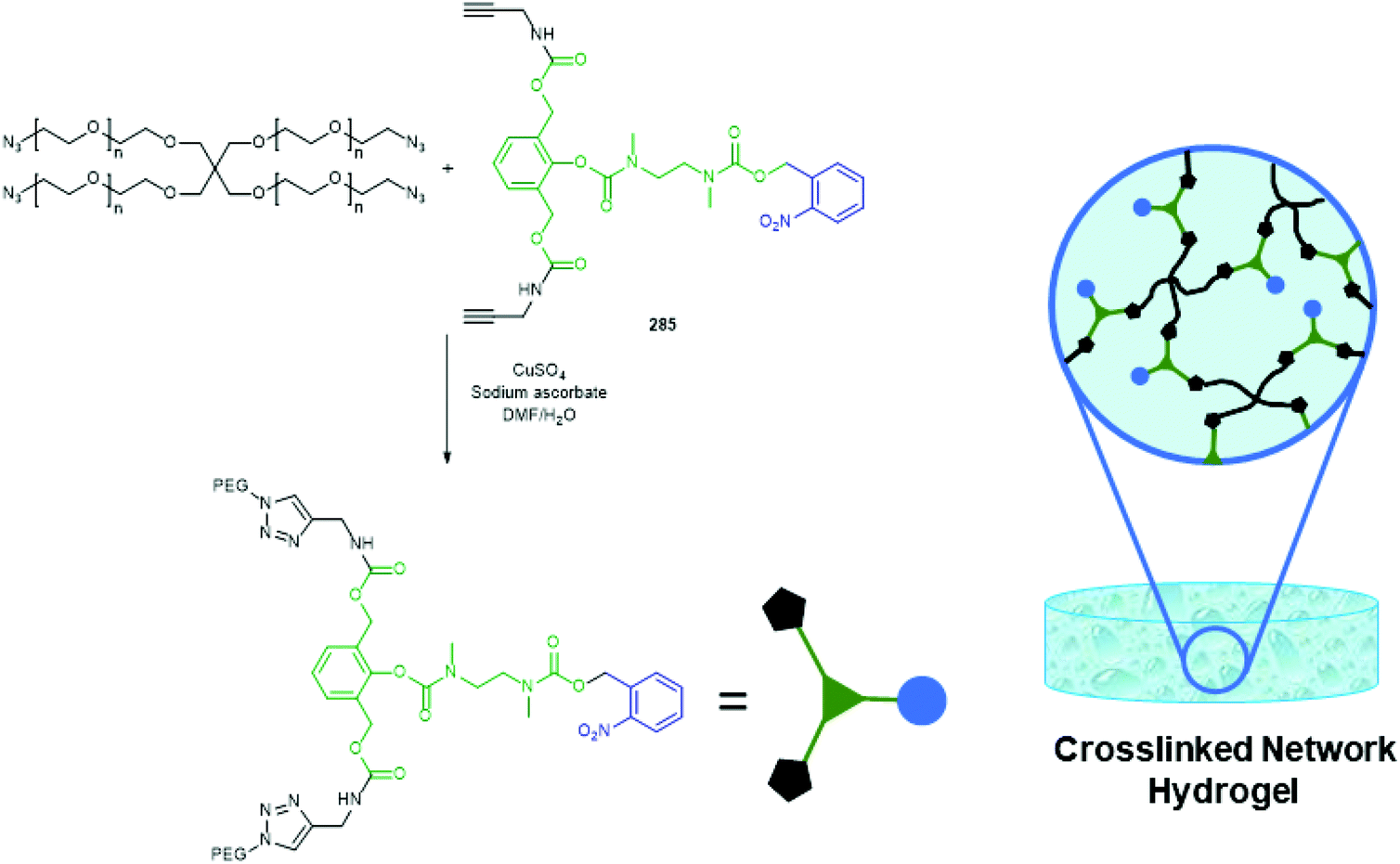 | ||
| Scheme 62 The light-triggered self-immolative hydrogel reported by Gillies and co-workers.260 | ||
An excellent example of how self-immolative electronic cascade processes can be utilised in biotechnology has been reported53 by Fakhari and Rokita in the development of dynamic DNA crosslinking agents to modify biochemical transportation and ultimately disrupt DNA repair as part of a strategy for cancer chemotherapy. A silyl ether protected phenol 286 featuring a DNA intercalator (acridine) unit and two benzyl acetates was synthesised – triggering with a fluoride ion source activated the system to the corresponding phenolate which then could degrade via 1,4-elimination to afford a quinone methide intermediate (Scheme 63). This intermediate was able to reversibly couple to DNA via the nucleophilic nitrogen atoms of adenine or guanine in turn leading to formation of another quinone methide that can then bind to the nitrogen base of an adjacent nucleotide to form a crosslink (either intra or interchain crosslinking is viable). This crosslinking process was shown to be reversible and thus the quinone methide unit can migrate along the DNA duplex in a bipedal fashion.
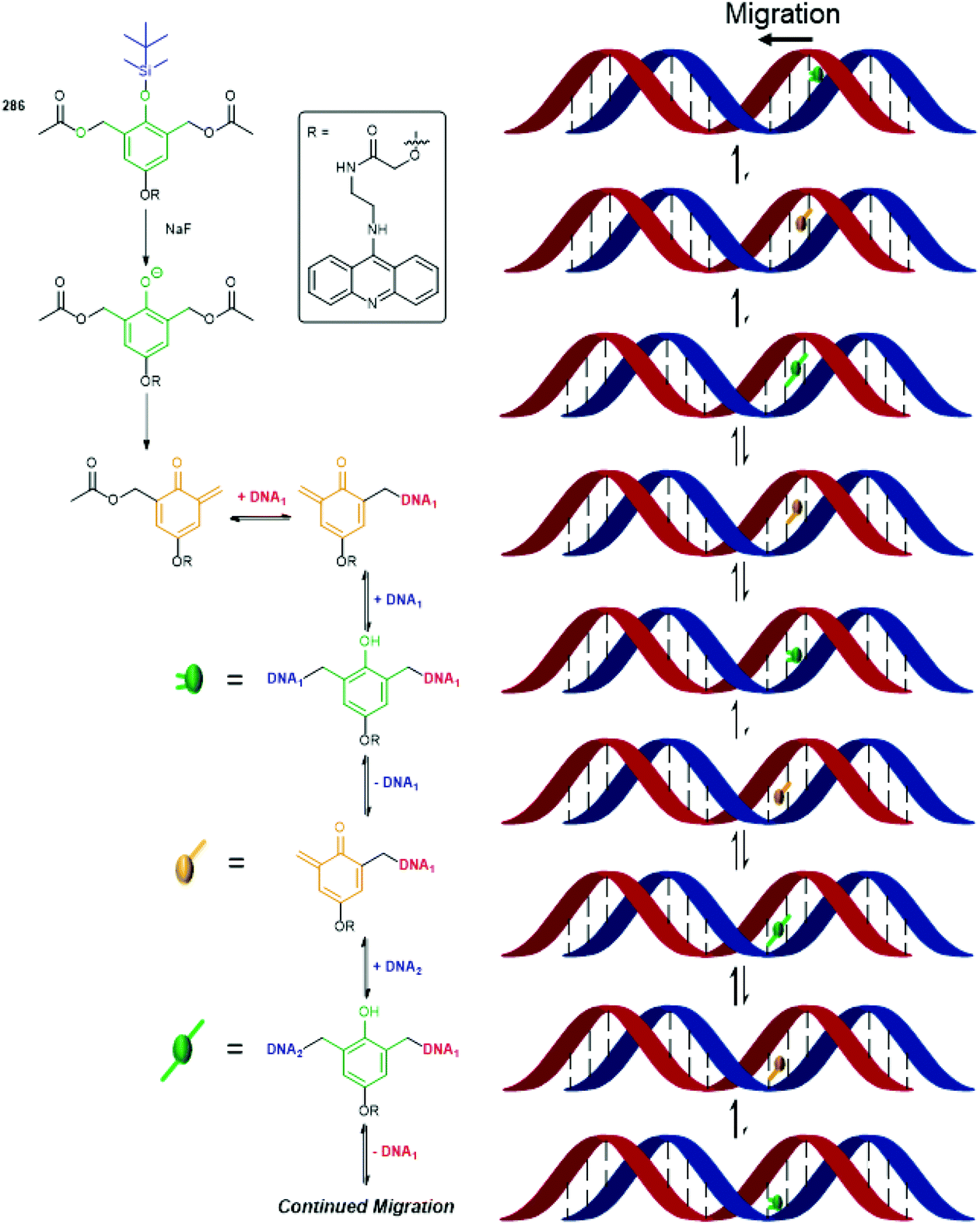 | ||
| Scheme 63 The self-immolative DNA crosslinking system 286 capable of migrating in a bipedal fashion along DNA sequences reported by Fakhari and Rokita.53 | ||
Zhang and co-workers have cleverly deployed an AB3 dendron, conjugated to a range of DNA strands by azide ‘click’ chemistry, to create amphiphiles.261 These amphiphiles assemble into micellar nanostructures in water, whose structure can be varied, that undergo ready endocytosis (within 6 hours). Subsequent UV irradiation (365 nm) triggers release of three equivalents of camptothecin (CPT). The conjugated system affords much lower cell toxicity than the camptothecin itself. For example, a DNA 20-mer 287 was conjugated to an AB3 dendron 288 to afford the amphiphile 289 (Scheme 64).
 | ||
| Scheme 64 A camptothecin bearing AB3 dendron 288 equipped with a UV trigger, conjugated to a strand of DNA 287 to yield amphiphile 289.261 | ||
In 2017, Zeng and Wu described122 a glutathione (GSH)-triggered and self-immolative ABB′ dendritic platform 290 comprising the reporter groups, the anticancer drug camptothecin (CPT) and a two-photon NIR fluorophore (dicyanomethylene-4H-pyran, DCM) for the in situ tracking of drug release and anti-tumour therapy (Scheme 65). GSH is an endogenous tripeptide that is present in cells at the millimolar concentration level and it serves as the major antioxidant in many biological systems. The dendritic drug release system featured 2,4-dinitrobenzenesulfonyl (DNS) as the thiol-responsive trigger group that was cleaved by the action of GSH, presumably via Meisenheimer complex 291, to liberate the corresponding phenol 292 that in turn facilitated self-immolation via a combination of 1,4-elimination processes to deliver CPT and DCM within cellular environments. This prodrug was shown to be effective both in vitro and in vivo and was able to achieve sustained release of CPT to afford efficient inhibition of tumour growth.
 | ||
| Scheme 65 The self-immolative theranostic dendritic drug delivery system 290 reported by Zeng and Wu.122 | ||
In the same manner that the elevated levels of glutathione in some cancer cells was targeted by ABB′ dendron 290 (Scheme 65), a number of malignant cancers e.g. non-small cell lung carcinoma, overexpress DT-diaphorase, sometimes by as much as 50-fold compared to normal cells also can be targeted. Developing their work on a drug delivery system that is both triggered by the enzyme DT-diaphorase and able to measure its level within the cell (Fig. 12), Zeng and Wu have prepared an AB2 dendron 293 (Fig. 19).228 Following reduction of the 1,4-benzoquinone to the corresponding hydroquinone, successive cyclisation of the trimethyl-lock and N,N′-ethylenediamine linkers occurs, followed by 1,4-elimination of phenol moiety and decarboxylation to release the CPT reporter group. The fluorescence that arises from irradiation of camptothecin is quenched by the quinone in 293. However, fragmentation of the dendron not only releases the topoisomerase I inhibitor, but restores camptothecin fluorescence (λmax 446 nm), self-indicating the fragmentation and quantifying DT-diaphorase activity. The release was shown to occur in A549 cells (a DT-Diaphorase overexpressing cell line) but not in L929 cells (a non-overexpressing cell line). Kim and Kim have used a related, theranostic dendron using biotin as a cancer targeting unit an SN-38 as the therapeutic agent.262In vitro and in vivo studies showed inhibition of tumour growth.
 | ||
| Fig. 19 The DT-Diaphorase triggered AB2 dendron 293 for the amplified release of camptothecin reported by Zeng and Wu.228 | ||
3.2 Aniline-based AB2 and AB3 linkers
First and second-generation aniline-based self-immolative dendrons were first introduced in 2003 by De Groot et al.251 A self-immolative second generation dendron featuring three 2-(4-aminobenzylidene)propane-1,3-diol AB2 type self-immolative linkers was able to degrade via two 1,8-elimination processes from a single trigger activation event to liberate four equivalents of paclitaxel (PTX). The self-immolative process for this second generation dendron was triggered by reduction of the nitro focal point under mild acidic conditions (zinc in the presence of acetic acid in MeOH). Subsequent investigations by De Groot et al. assessed the potential of self-immolative AB3 linkers featuring the same trigger group chemistries with the release of 2-phenylethanol from a first-generation dendron 294 with a 2,4,6- tris (hydroxymethyl(aniline) dendritic core unit (Scheme 66).251 In the light of the modest basicity of anilines (pKaH+ of aniline ca. 5) generated post-reduction of the nitro trigger group, a significant percentage was present in the non-protonated form and can, therefore, trigger 1,6-elimination to lead to decarboxylation and liberation of the reporter molecule and aza-quinone methide. Quenching the aza-quinone methide with methanol reformed the electron-donating aniline, which then in turn permitted two sequential 1,4-eliminations to occur with concomitant decarboxylation to release two additional reporter molecules. Overall, a total of three equivalents of 2-phenylethanol were released from a single reduction activation event in the appropriate solvents.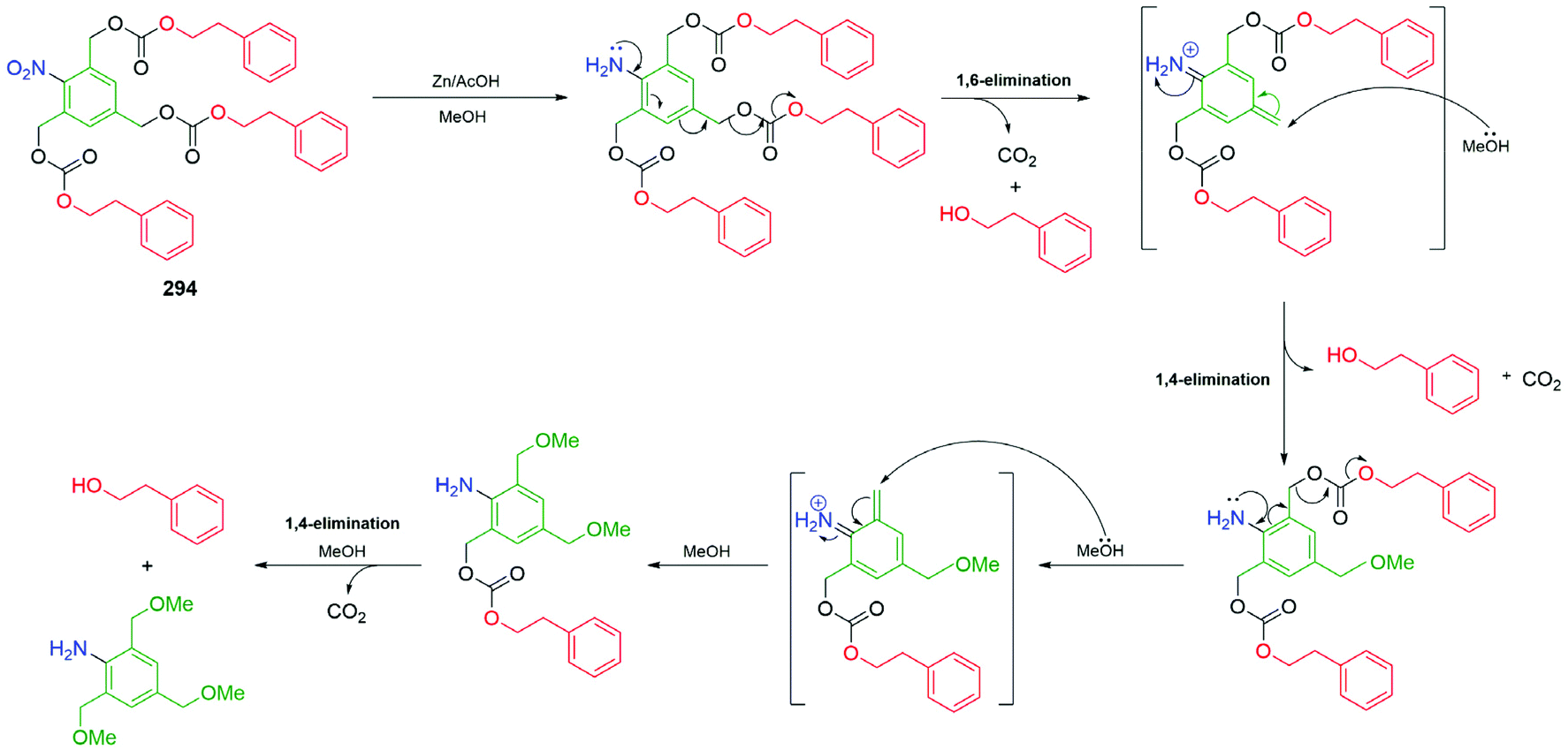 | ||
| Scheme 66 Triple self-immolation elimination of 2-phenylethanol from first generation 2,4,6-tris(hydroxymethyl)aniline based AB3 dendron 294.251 | ||
In 2011, Papot and co-workers demonstrated263 successfully the action of an ABB′ dendritic prodrug system that featured two back-to-back self-immolative linkers (Fig. 20). The first being phenol-based and attached to the glucuronic trigger group; whereas, the second is an aniline-based branched amplifier unit carrying two distinct reporter groups. This first generation dendron (295) featured doxorubicin and the histone deacetylase inhibitor MS-275 as the reporter groups. The phenol-based linker carried an ortho-nitro substituent that will serve to decrease the pKa of the phenol and, as discussed by Hou in the context of their galactosidase-based SIE, it may affect the Michaelis–Menten constant (Km) of the substrate for the enzyme (Scheme 18).136 This linker provides a suitable distance between the enzyme substrate and the two bulky drugs to allow easy recognition of 295 by β-glucuronidase. Enzyme-catalyzed cleavage of 295 led to the generation of a phenoxide intermediate that then induced the release of an aniline via a 1,6-elimination process. Once activated, the aniline amplifier unit first liberated doxorubicin via a 1,4-elimination followed by spontaneous decarboxylation. Reaction with the water in the aqueous environment traps the resultant ortho-azaquinone methide to regenerate the electron-rich aniline thereby permitting the subsequent release of MS-275 via a 1,6-elimination process. Normally, 1,6-eliminations are observed to be faster than their 1,4 counterparts; however, this appears to be significantly influenced by the nature of the leaving group. The authors also note that as azaquinone methides are potential alkylating species (see Scheme 62 for a related example), there is potential for these intermediates to also prove toxic to cancerous cells. The one issue with 295 was effective cell penetration. Additional work on amplification, with delivery of two doxorubicin reporter groups was reported in 2012.264 To enhance the cell specificity and efficiency of the action of drugs in dendritic prodrug systems, Papot and co-workers have cleverly refined their prodrug design to produce 296 that features a folate moiety to target the folate receptors frequently overexpressed in cancerous cell lines and facilitate endocytosis of the prodrug (Fig. 20).265 The structure of the phenoxy linker was modified at the benzylic position to allow the folate to be attached by ‘click’ chemistry. The lysosomal enzyme, β-galactosidase, triggered self-immolation to release two doxorubicin molecules in a highly targeted fashion. A similar, folate directed approach has been reported for delivery of monomethylauristatin E.266 More recently, Papot has extended these studies to give a β-glucuronidase-responsive drug delivery system that contains a maleimide unit that allows in vivo functionalisation of cysteine-34 of plasmatic albumin.267,268 Such albumin conjugates prevent rapid clearance of the drug and concentrate that conjugate in tumour tissues where β-glucuronidase triggers release of the anticancer agent e.g. monomethyl auristatin E or doxorubicin; these drug delivery systems were shown to be effective against, inter alia, pancreatic tumors and murine LLC xenografts. The studies were extended to include trimeric versions of the system.269
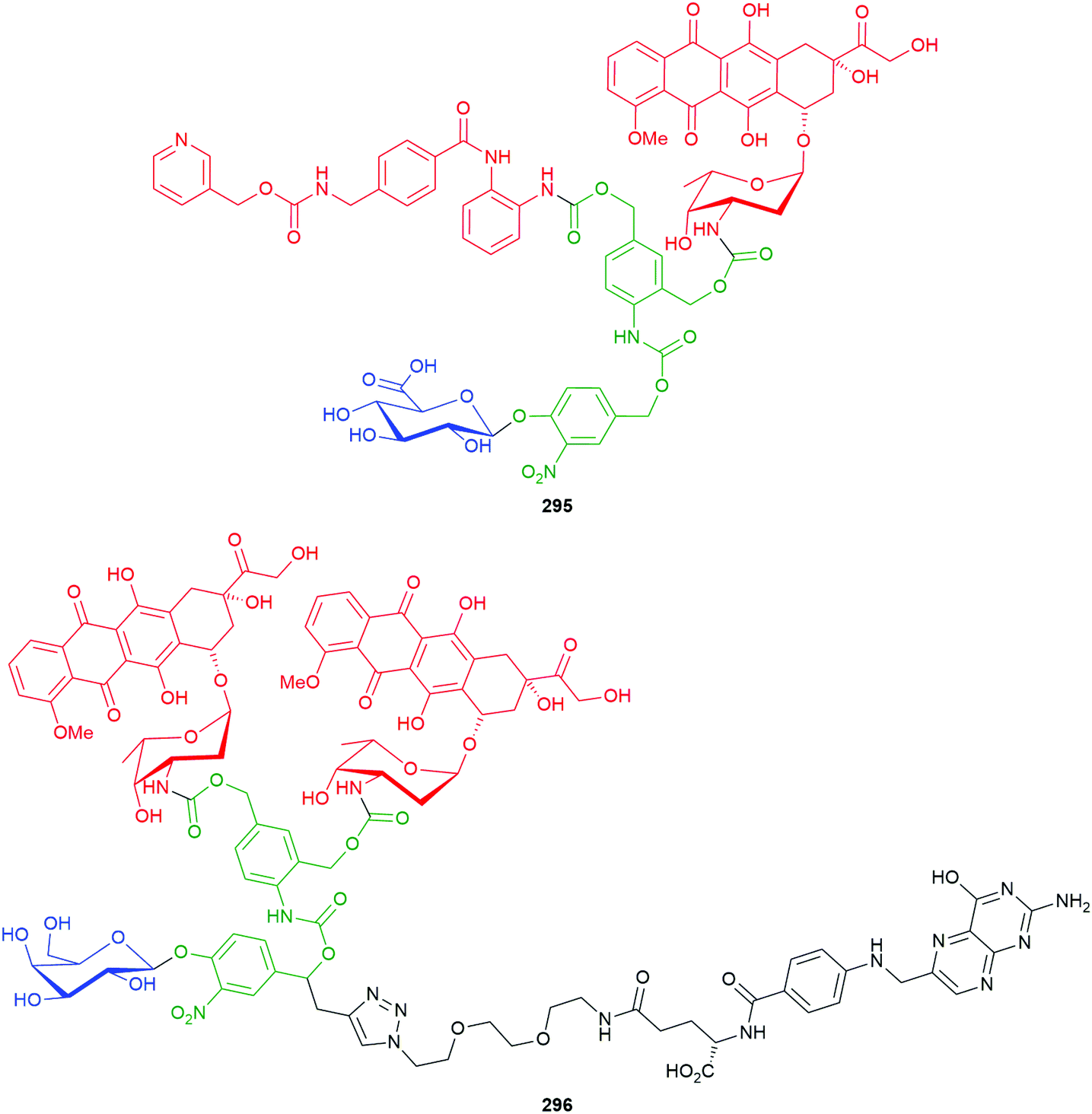 | ||
| Fig. 20 The anti-cancer self-immolative dendritic prodrug systems (295 and 296) developed by the Papot group.263,265 | ||
In the same paper257 in which FRET sensors were reported from first generation self-immolative dendrons bearing a phenyl boronic ester trigger group to permit a turn-on FRET response upon exposure to hydrogen peroxide following two 1,4-elimination processes, Shabat and co-workers also reported an analogous Homo-FRET sensor (see 297 in Fig. 21). Unlike the phenol-based FRET dendritic systems, activation of this aniline-based Homo-FRET probe was triggered by the action of the enzyme PGA on the phenylacetamide group to liberate the Cy5 fluorophores via both 1,4- and 1,6-elimination processes from the branching unit to achieve the desired fluorescence response.
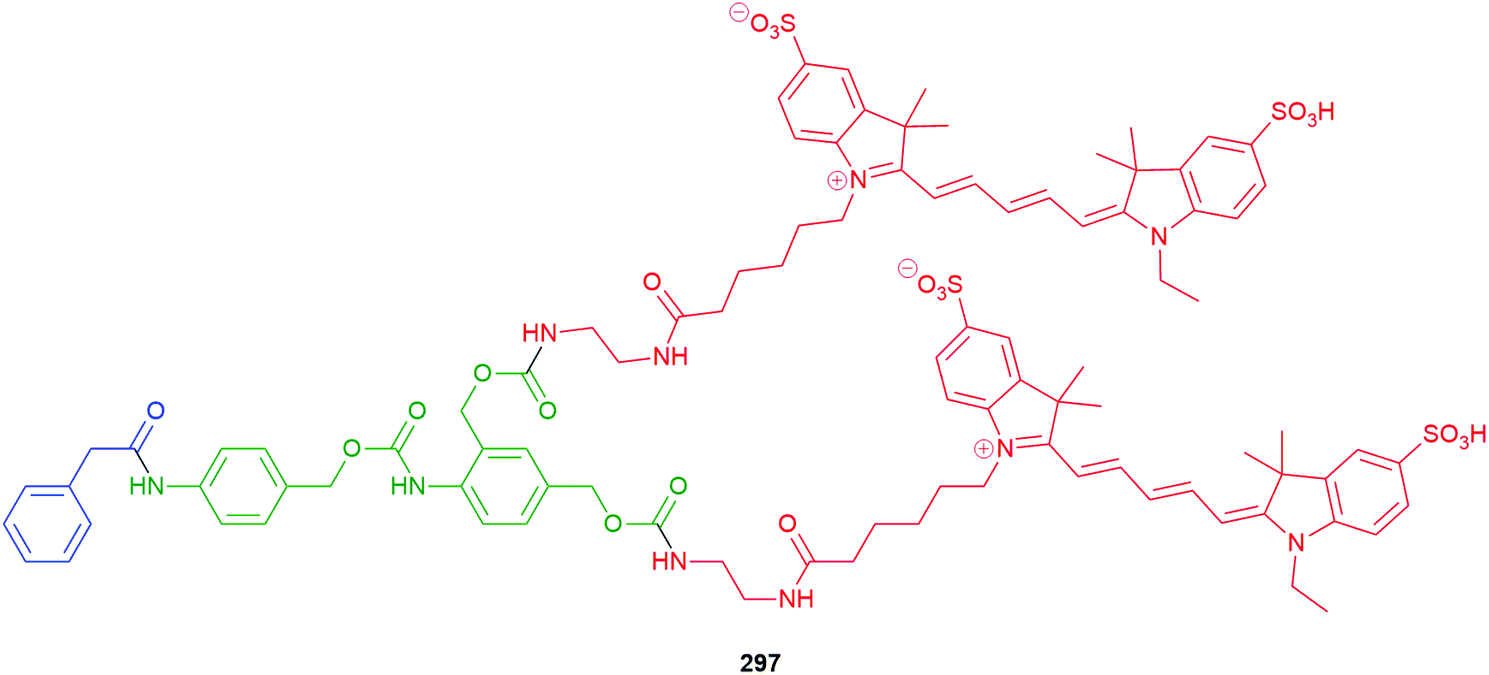 | ||
| Fig. 21 The aniline-based self-immolative Homo-FRET system 297 reported by Shabat and co-workers.257 | ||
3.3 Dendritic chain reactions (DCR) and amplification systems
Although self-immolative dendrons are elegant systems, there are structural and practical limitations (i.e. very labour-intensive syntheses) in terms of achieving significant amplification values for reporter release from dendritic architectures as high generation dendritic species are required. In 2009 Shabat and co-workers reported a solution for this limitation in the form of the dendritic chain reaction (DCR).270 This approach takes inspiration from the polymerase chain reaction (PCR) and Mirkin's example of a PCR-like casade.271,272 Shabat's initial DCR system 298, a first generation dendron which featured an arylboronic acid was triggered in response to H2O2 to provide a phenolic species capable of a 1,6-elimination via a quinone methide and decarboxylation reaction to release the core unit.227 The core unit was able to undergo three further eliminations of the substituents in the ortho- and para-positions to release 4-nitroaniline (yellow) and the two choline molecules. The choline units were then oxidized by choline oxidase (COX) present in solution to produce four molecules of hydrogen peroxide, enabling amplification (Scheme 67).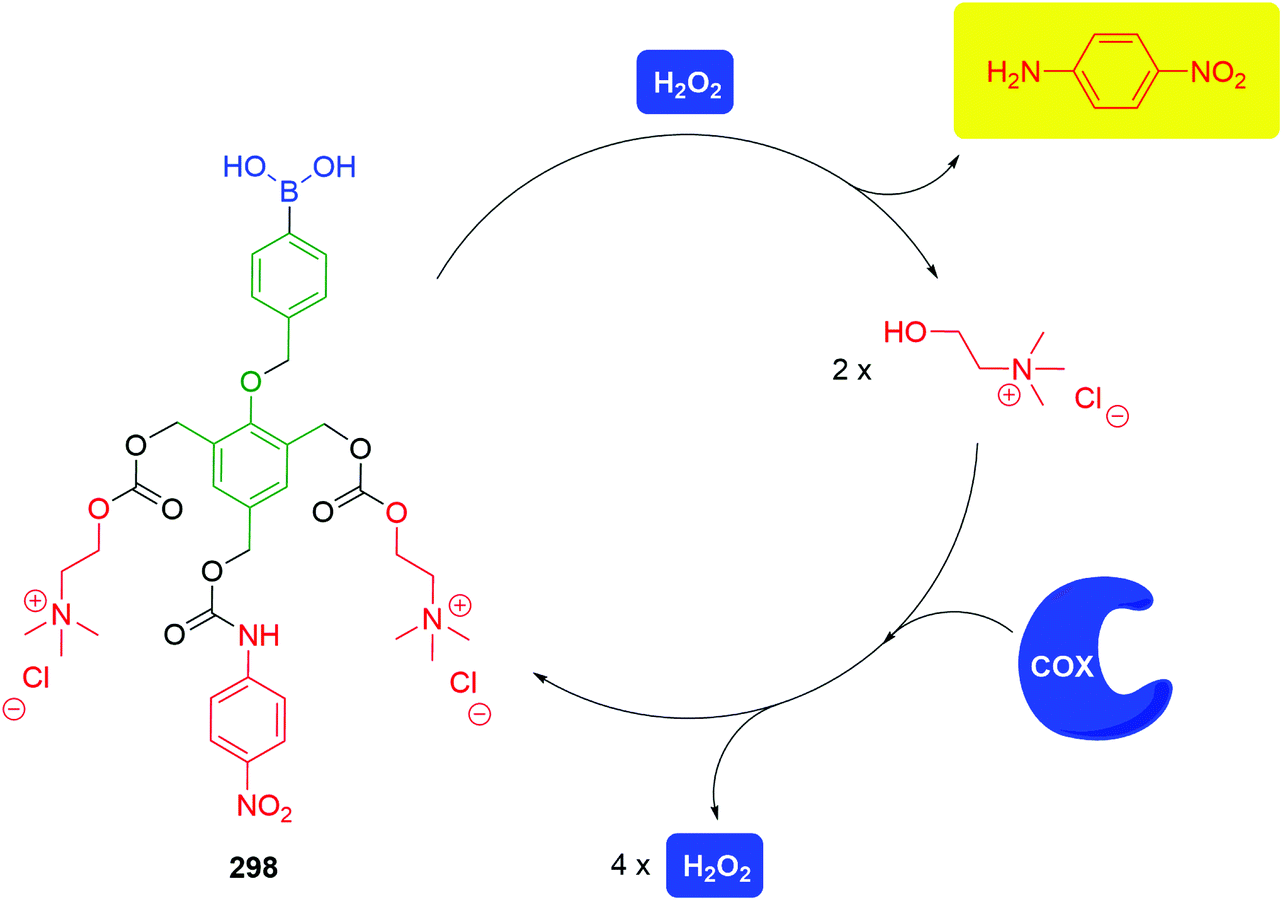 | ||
| Scheme 67 Self-immolative dendritic chain reaction involving the dendron 298 triggered by the oxidation of a phenylborinic acid releasing 4-nitroanline (coloured readout) and two choline molecules that undergo oxidation by COX generating H2O2 for further trigger activation.270 | ||
Developing the concept of a hydrogen peroxide sensitive first-generation dendron 299 as amplifier, Shabat has reported an exponential signal amplification using a dendritic chain reaction. Thus, exposure of 299 to hydrogen peroxide releases three molecules of glucose that upon exposure to glucose oxidase generates three equivalents of hydrogen peroxide (Scheme 68).45 This hydrogen peroxide generator is coupled with a second component 300, sensitive to H2O2, that gives a spectroscopically monitorable signal. During this work, the authors compared the effectiveness of the AB3 dendron 299 with an AB2 and two second-generation dendrons of AB4 and AB6 design, respectively. The higher order dendrons gave the fastest rate of disassembly but it was noted that they are subject to more background hydrolysis; overall, the AB3 dendron 299 represents the best balance of stability and amplification.
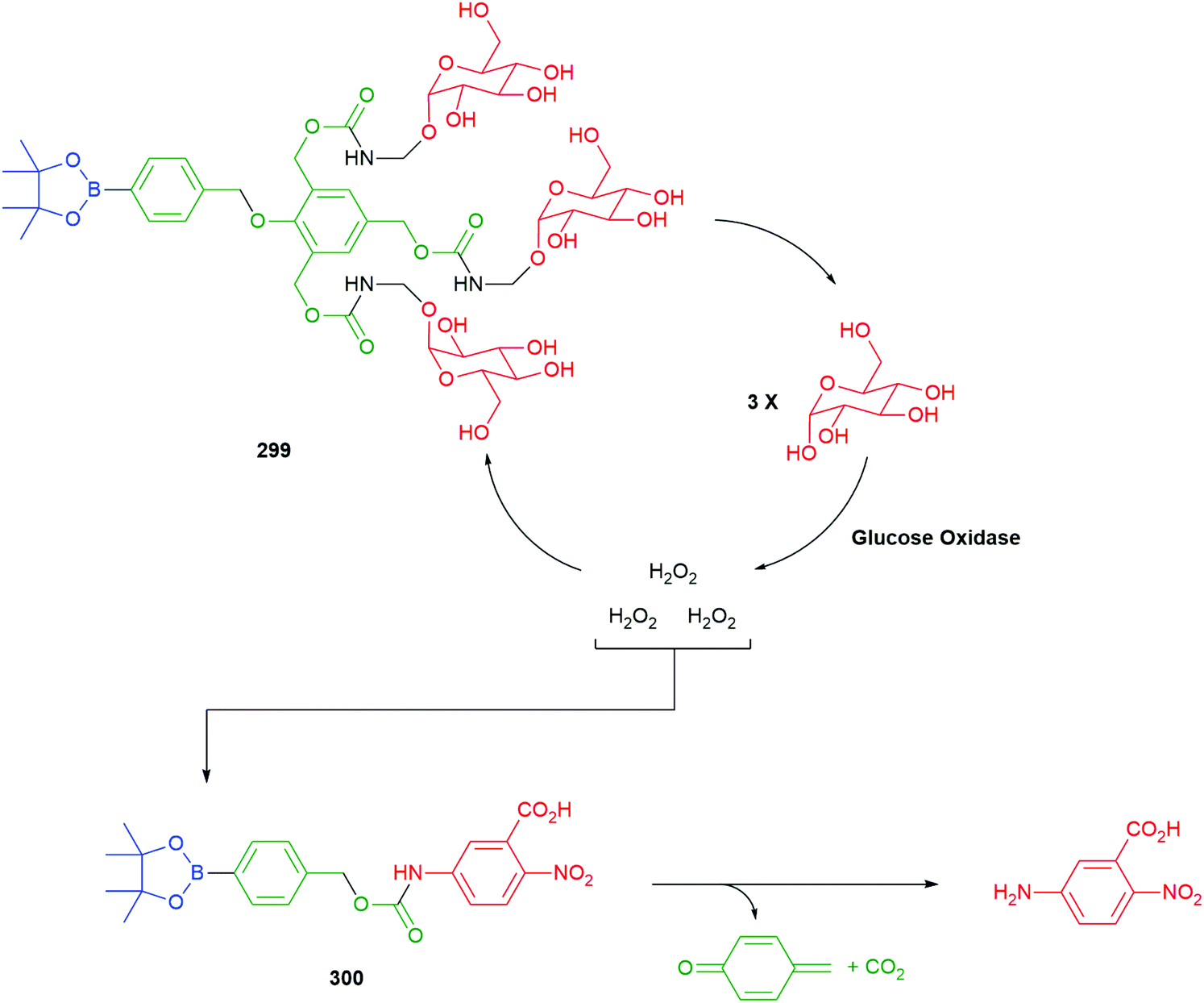 | ||
| Scheme 68 Shabat's self-immolative dendritic chain reaction involving the AB3 dendron 299.45 | ||
In 2015, Hamachi and co-workers described the application of a dendritic chain reaction process within a multicomponent supramolecular hydrogel to permit detection of small-molecule biomarkers by a gel–sol transition, with the naked-eye.273 The hydrogel was composed of a hydrogen peroxide-responsive supramolecular triphenylalanine-based hydrogel (BPmoc-F3) plus an embedded dendritic self-immolative system and corresponding enzymes (sarcosine oxidase (SOx)/urate oxidase (UOx)) to afford a signal amplification system (see Scheme 69). Degradation of both the peptide BPmoc-F3 and self-immolative first generation dendritic system 301 was triggered by hydrogen peroxide, produced by reaction of the biomarker (uric acid) with UOx. In order to effect amplified degradation of the hydrogel network, the multi-component material was designed so that dendritic component would release two sarcosine units upon self-immolation; the sarcosine produced was then consumed by SOx present in the gel matrix, in turn liberating two equivalents of hydrogen peroxide to amplify degradation of BPmoc-F3 and 301, producing the gel–sol transition.
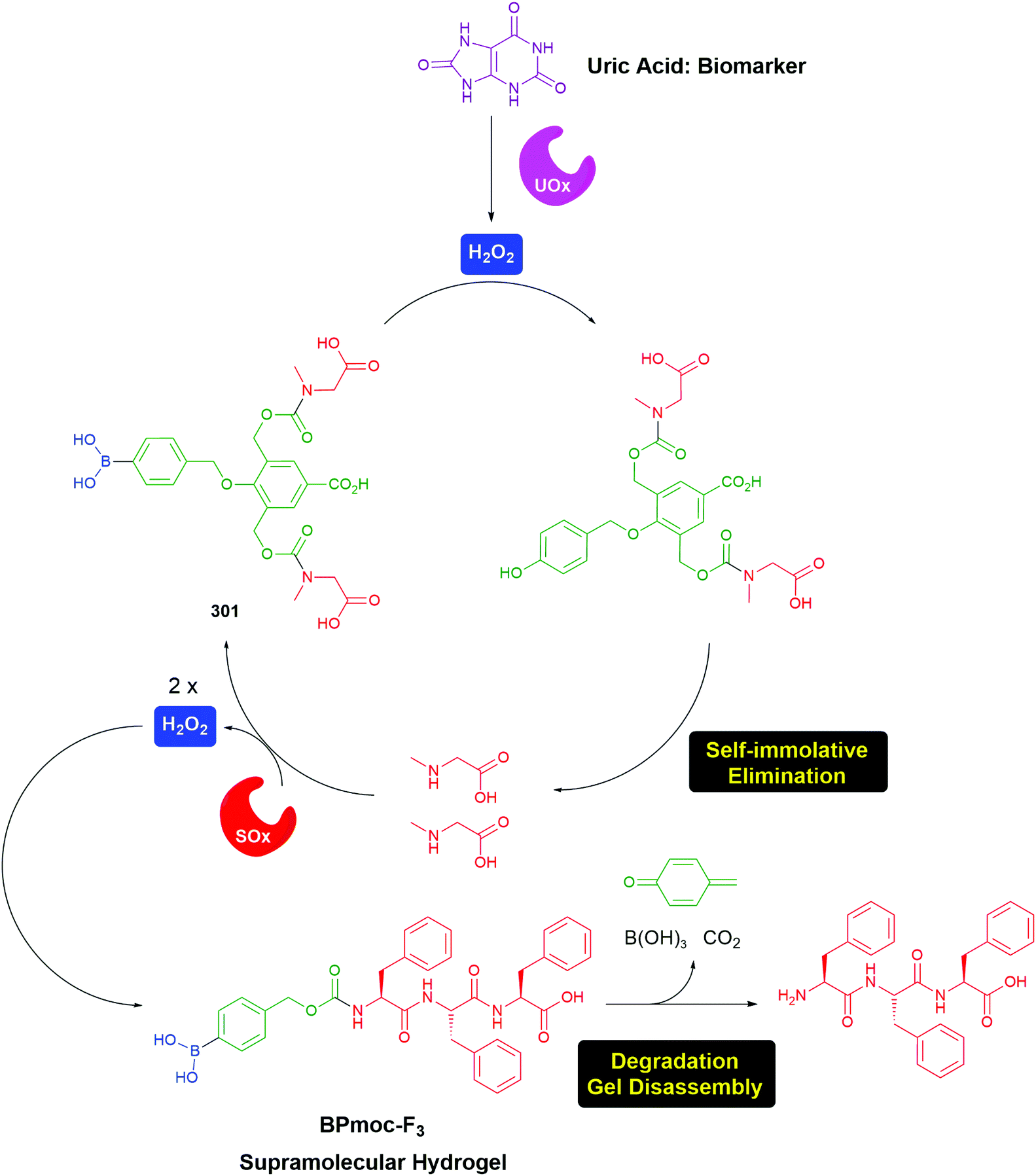 | ||
| Scheme 69 The enhanced degradation of a peptide hydrogel via a dendritic chain reaction from the first generation dendritic self-immolative system 301.273 | ||
Papot and Taran have used their β-glucuronidase sensitive trigger/linker combination to drive controlled release of a payload from a micelle effectively amplifying the analyte signal (Scheme 70).274 An amphiphilic, micelle forming, compound 302 was synthesised around an iminosydnone (coloured brown). The micelles formed from 302 (CMC = 50 μM, 10 ± 1 nm) were shown to carry payloads and to breakdown on exposure to a strained alkyne e.g.303 to give 304 by a ‘click’ reaction with the iminosyndone moiety, cleaving the hydrophilic from the hydrophobic segments. Breakdown of the micelle releases the payload. Use of a self-immolative molecule 305 with a hydrophilic tale slows transport of the strained alkyne into the micelle core; however, upon triggering with β-glucuronidase (a tumour associated enzyme), the released alkyne 303 rapidly penetrates to the hydrophobic core, reacting with concomitant release of the payload e.g. a fluorescent probe. This combination of reagents was found to be effective both in KB cells and subcutaneous KB mouth epidermal carcinoma xenografts in mice.
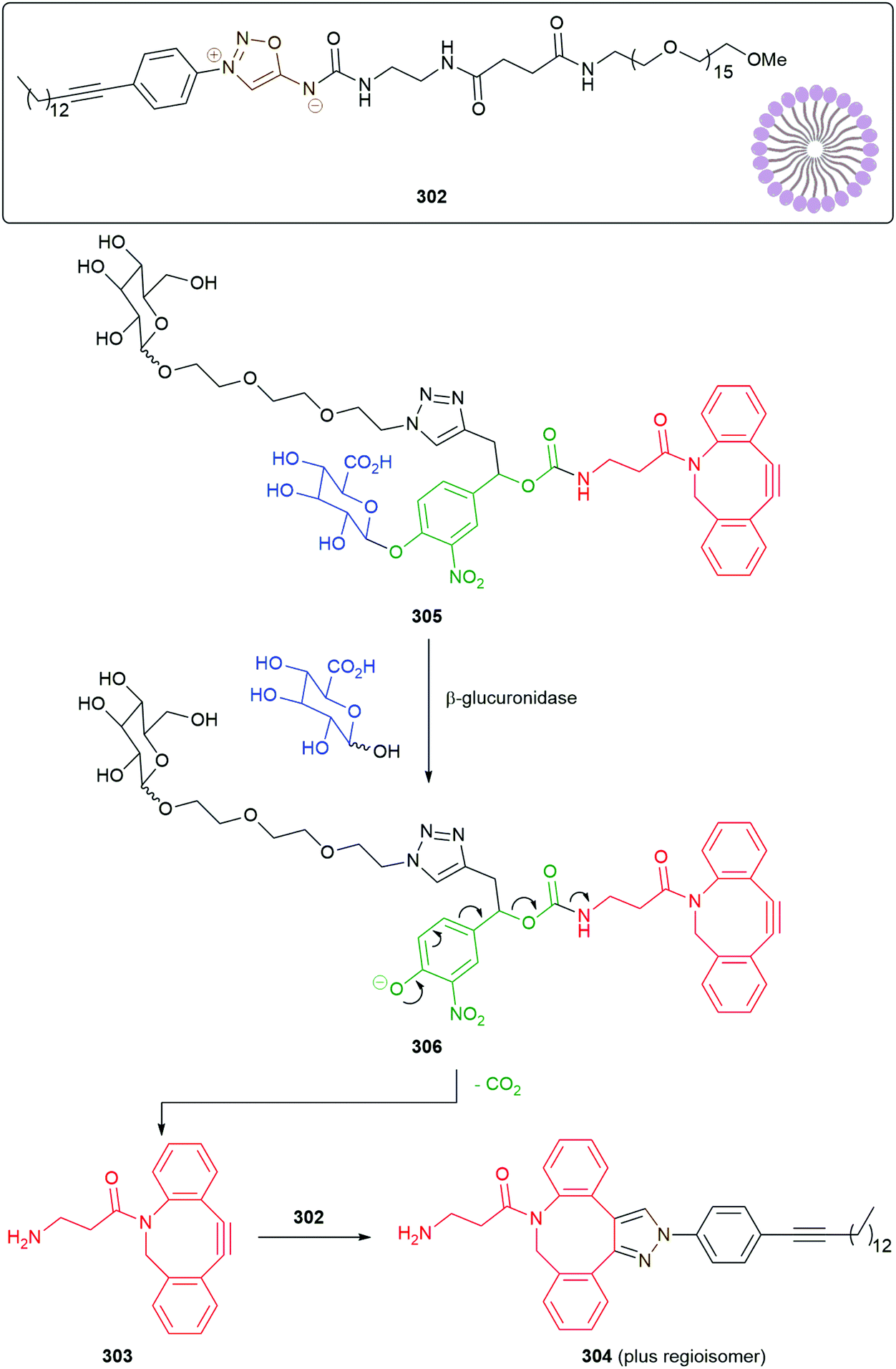 | ||
| Scheme 70 Papot and Taran's self-immolative molecule 305 for biorthogonal ‘click to release’ degradation of a payload bearing micelle.274 | ||
In 2012, Phillips described an autocatalytic signal amplification system that couples a molecule sensitive to a particular analyte (Pd(0)) that produces a chemical signal with another that serves to amplify that initial signal (Scheme 71).275 Thus, 307 detects the analyte with decomposition initiated by a π-allylpalladium species, followed by rapid 1,6-elimination to release a molecule of piperidine. Once formed, the piperidine serves as a base to cleave the Fmoc group from amplifier 308, itself being regenerated in the process, and triggering formation of a second molecule of piperidine. The process then repeats with the total number of piperidine molecules being 2n (n = the number of cycles). As discussed previously, the methoxy group in 308 was found to confer an increase in rate of fragmentation (factor of 2.3) compared to its desmethoxy analogue. Additionally, a dendron-based amplification reagent 309 was also found to be successful in this process, but releases two additional molecules of the base for each complete cycle. Readout occurred spectroscopically, via dibenzofulvene (λmax = 305 nm), or via a pH indicator.
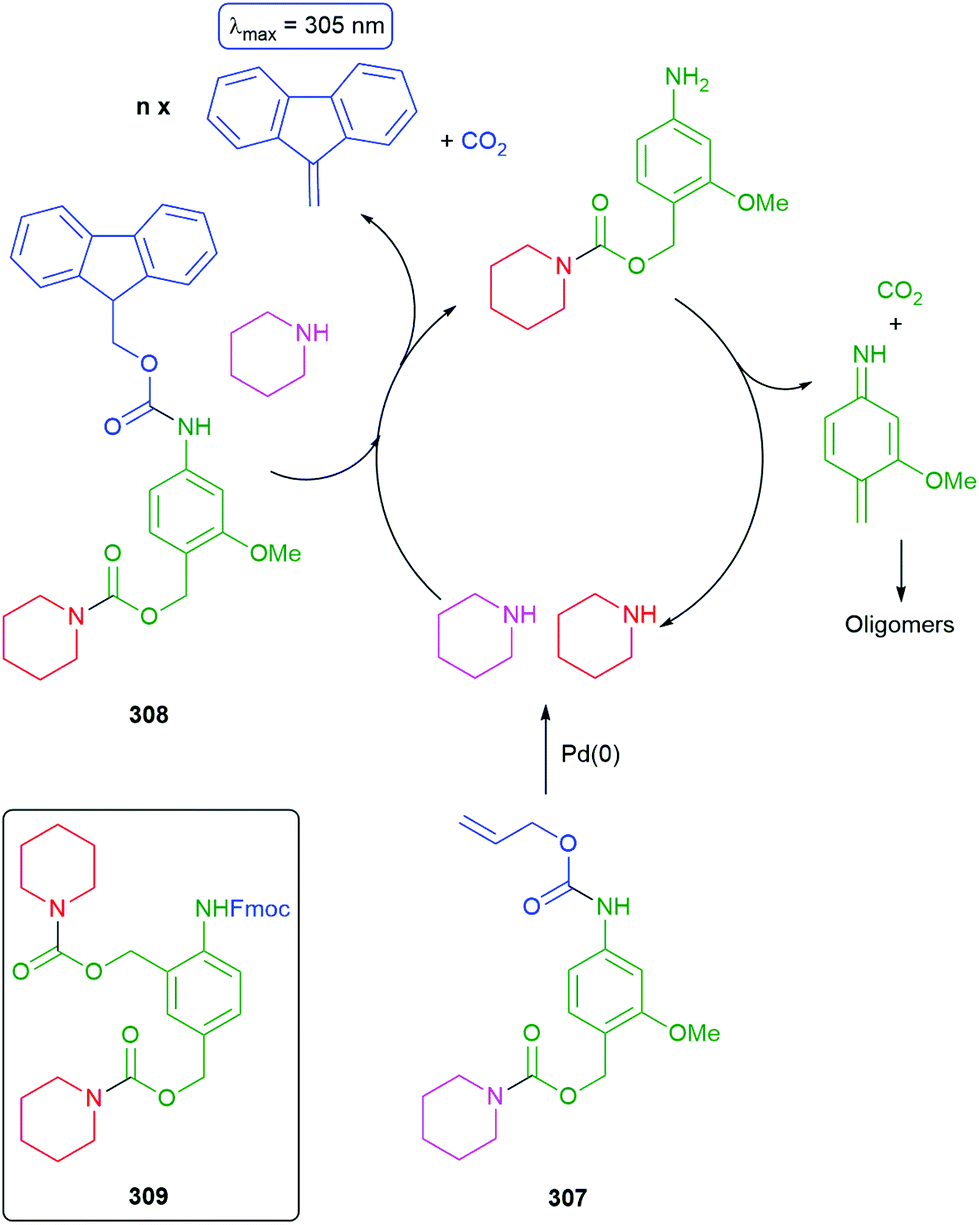 | ||
| Scheme 71 Phillips's autocatalytic amplifier system for the detection of a palladium analyte.275 | ||
In a similar fashion to the palladium detection system outlined in Scheme 71, Phillips outlined275 an amplification system for the detection of β-galactosidase as the analyte, with readout from the assay being colourimetric (2-hydroxy-1,4-benzoquinone).276 Exposure of 310 to β-galactosidase, followed by 1,6-elimination, releases hydroxyquinol 311 that is auto-oxidised in air to give coloured benzoquinone 312 and an equivalent of H2O2 (Scheme 72). Compound 313 serves as a hydrogen peroxide sensitive amplifier.
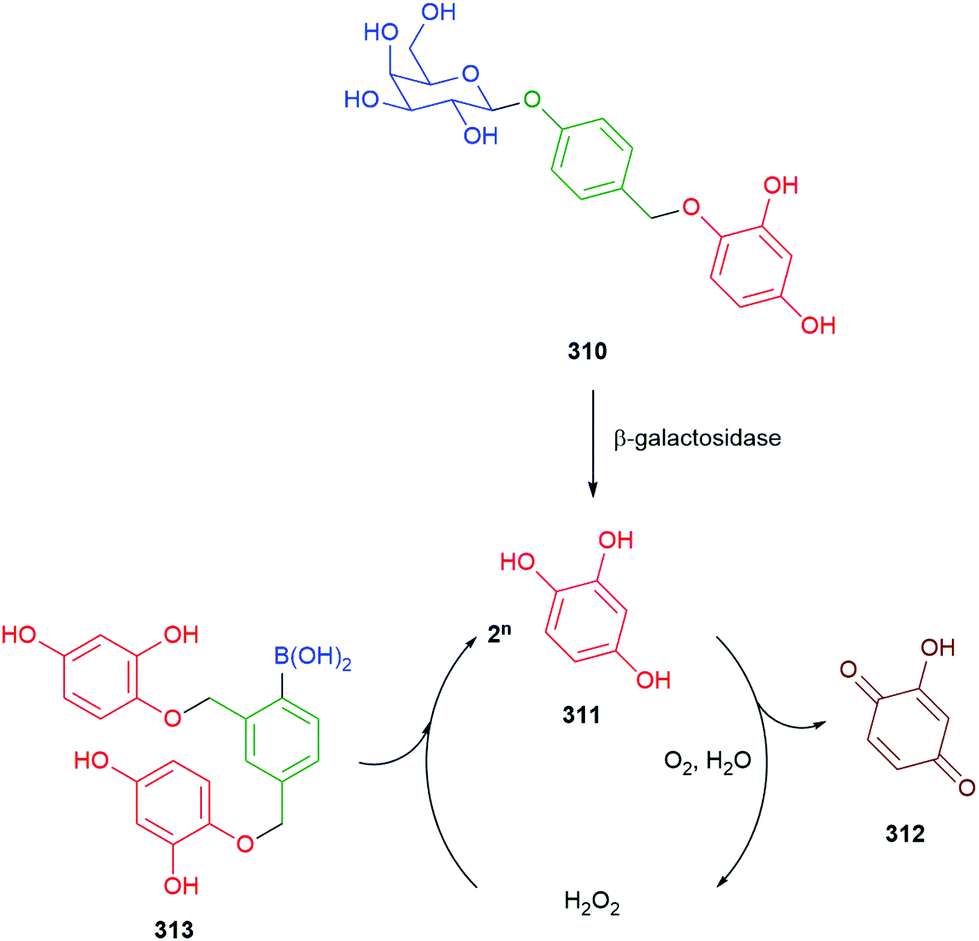 | ||
| Scheme 72 Phillips dendron-based signal amplification system for colourimetric detection of β-galactosidase.276 | ||
Phillips and co-workers have described277 a system for in situ signal amplification that is activated by the presence of the analyte (glucose) and minimises background reactions, an important issue for amplification processes. Thus, in the presence of the analyte (glucose), oxidation takes place by glucose oxidase delivering hydrogen peroxide with the expected lowering of pH. The hydrogen peroxide is used by amplification reagent 314 to mediate oxidation of the C–B bond with subsequent 1,4-elimination of glucose from the so-formed enol/enolate (Scheme 73). As the pH lowers from >6.2 to <4.2 methyl red 315 protonates to afford 316 with a concomitant colour change from yellow to pink, giving a colourimetric readout. In respect of background hydrolysis of the amplification reagent 314, use of an ether bond to connect the linker to the reporter group affords a highly stable molecule that, when highly purified, gives a minimal background reading. A non-amplified assay for penicillin-G-amidase, based upon a self-immolative molecule that releases glucose has also been reported by Mohapatra and Phillips.278
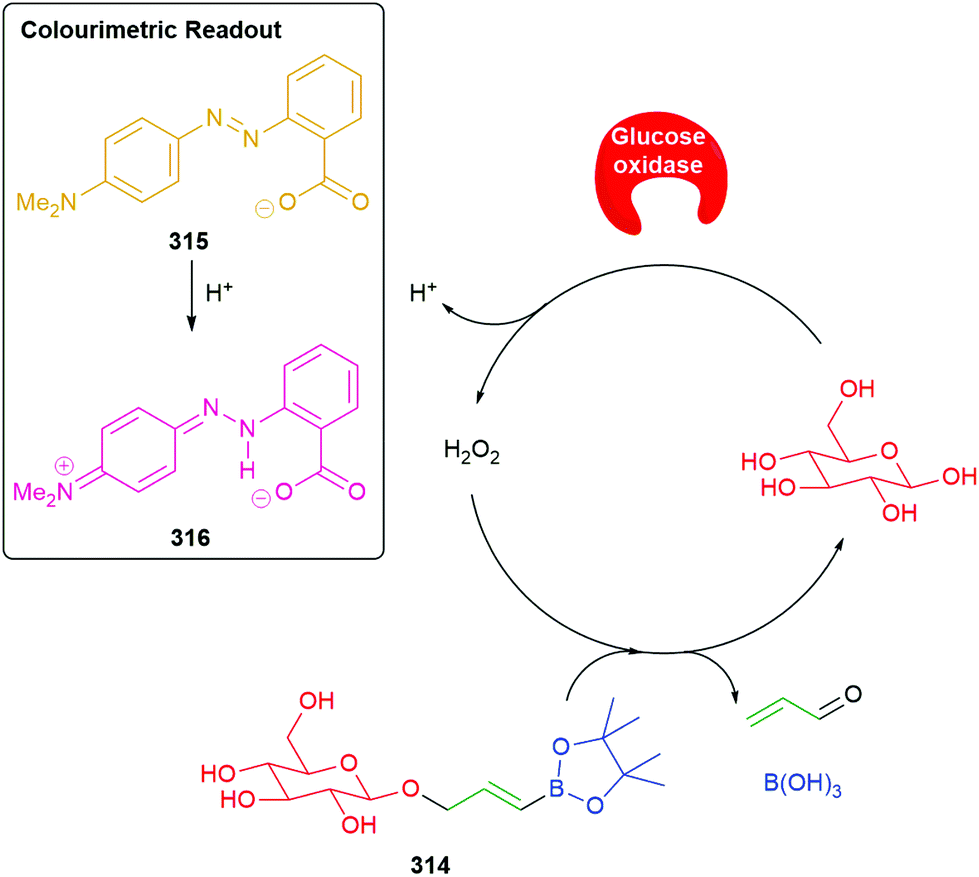 | ||
| Scheme 73 The amplified colourimetric reporter system 314 developed by Phillips and co-workers.277 | ||
Detection and amplification of the analyte fluoride has proved to be of significance in a number of roles e.g. the disclosure and quantification of G series nerve agents (vide infra). Shabat and co-workers have reported279 a dendron-based amplification system 317 that takes advantage of the high chemoselectivity of fluoride for silicon (Scheme 74). Treatment of 317 with as little as 0.004 equivalents fluoride leads to its complete disassembly with an exponential rate of progress. Each disassembly of 317 is accompanied by release of one equivalent of para-nitroaniline, facilitating spectroscopic readout.
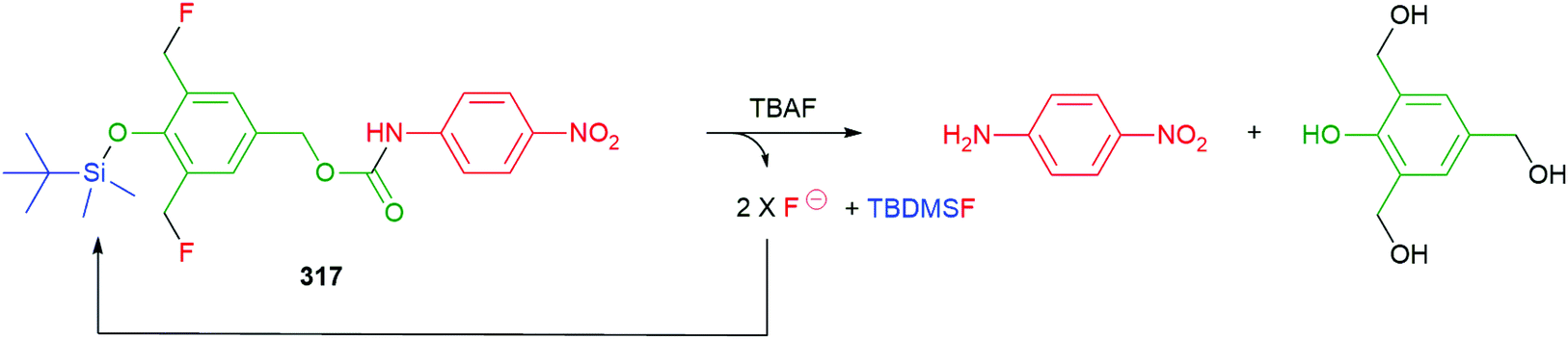 | ||
| Scheme 74 Shabat's fluoride triggered autoinductive amplification system 317.279 | ||
Baker and Phillips have provided a further example of the flexibility of a two-component analytical system discussed in Schemes 71 and 72.280,281 In the case below, the allylic carbamate in 318 again serves as the specifier for the Pd (0); whereas, 319 serves as the amplifier (Scheme 75). Following cleavage of the allylic carbamate, generation of two fluoride ions by 1,6-elimination occurs with concomitant formation of para-aminobenzaldehyde, the latter allowing for colourimetric reporting. In respect of this reporting mechanism, see Scheme 75 for the likely identity of the coloured species. Once the fluoride is released, its high affinity for silicon triggers the amplifier 319. Trapping of the quinone methide from 319 by nucleophiles other than water, to afford 320 and 321, was also noted, again reinforcing this consideration in Katzenellenbogen's seminal paper. Thus, it can be seen that the self-immolative linkers in 318 and 319 serve as both linker and reporter, a feature frequently encountered with self-immolative polymers (vide infra).
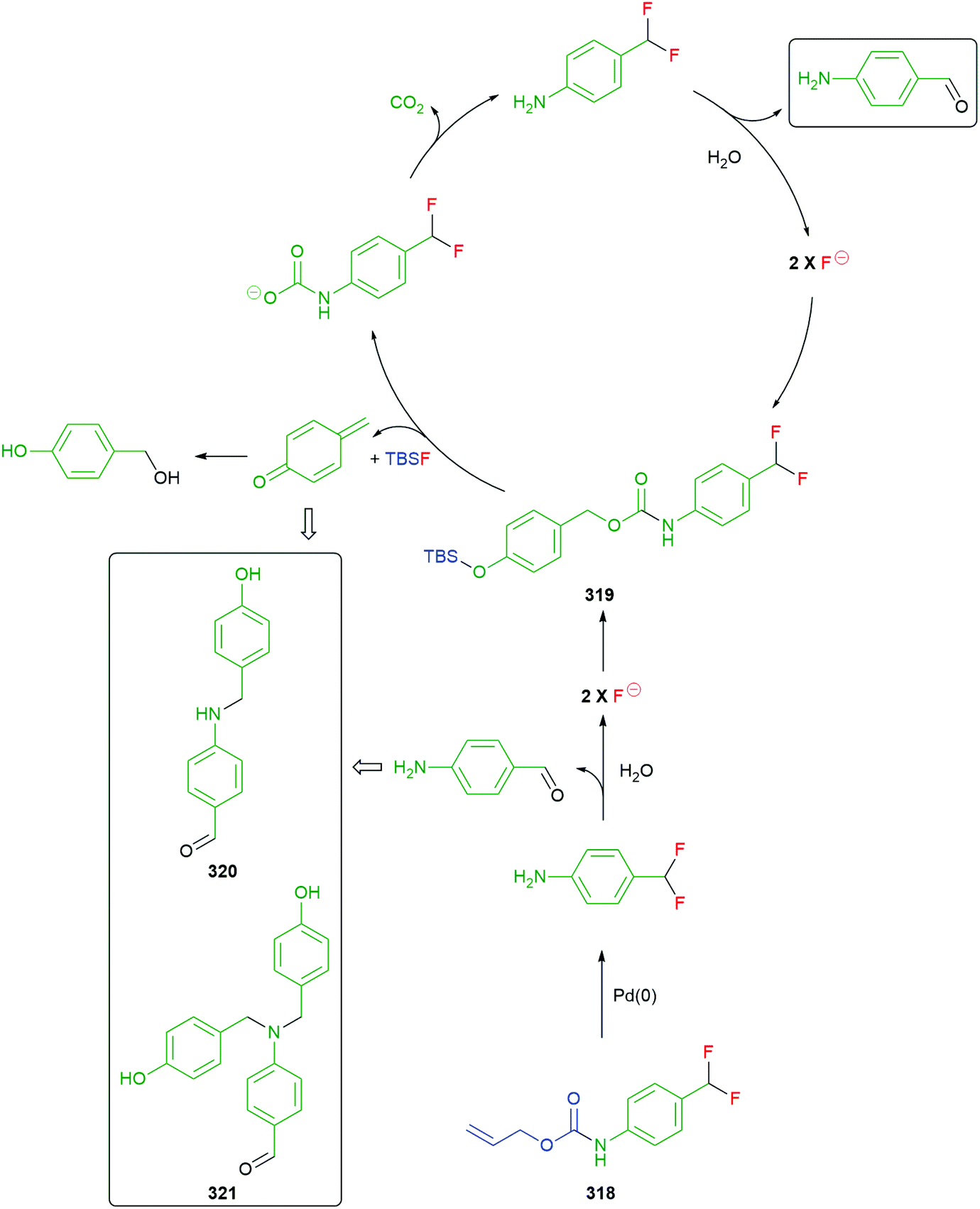 | ||
| Scheme 75 The two-component difluoride specifier/amplifier 318/319 with colourimetric readout reported by Baker and Phillips.280,281 | ||
Huang and co-workers have described282 a self-immolative fluorometric probe 322 that uses autoinductive signal amplification for enhanced sensitivity and combines selectivity for fluoride with generation of a fluorogenic coumarin 323 (λem = 445 nm), for the readout (Scheme 76). As seen above, the affinity of fluoride for silicon drives the selectivity whilst the generation of two fluorides from the disassembly of each molecule of 322via intermediate 324 affords an exponential increase in the signal. Additionally, an alternative fluorometric probe 325, that fluoresces at longer wavelength (λem = 595 nm), was shown to couple with this amplifier and is promising for applications with biological samples. Encouragingly, a limit of detection of 0.5 pM was reported.
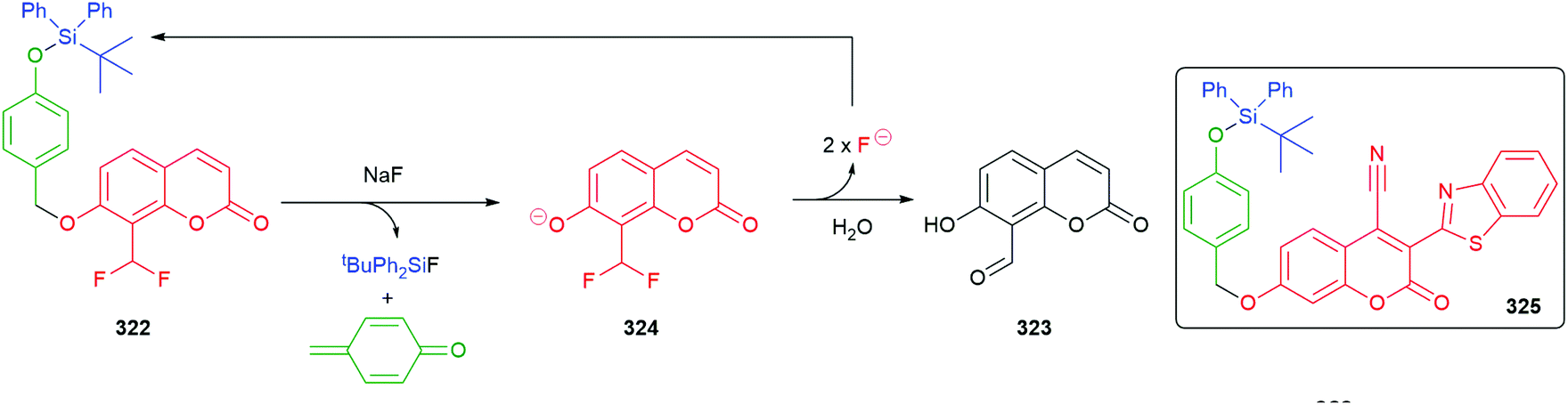 | ||
| Scheme 76 Huang's fluoride triggered autoinductive signal amplification system 322.282 | ||
In an interesting extension of their solution-phase methodology in which detection of an analyte signal with concomitant release of fluoride is coupled to triggering of an amplification cycle, Phillips has demonstrated that within a suitable copolymeric material, a fleeting signal can be converted to a macroscopic response (Scheme 77).283 Exposure of a small section of copolymer 326 to 300 nm light gives cleavage of the ortho-nitrobenzyl moiety with release of a linker–reporter group unit that undergoes both 1,4 and 1,6-elimination to deliver four fluoride ions and copolymer 327. These fluoride ions then trigger an amplification unit on a separate side-chain causing the effect to spread throughout the sample changing its physical properties from hydrophobic in 326 to hydrophilic in 328. The idea has been further developed by Phillips and Sen to give a self-powered microsphere, based on a modified tentagel bead (329), that responds to a short stimulus of 365 nm light by pumping the surrounding fluid toward itself (Scheme 77).284 A similar example of a macroscopic change to a polymer's properties, triggered by a fleeting thiol signal, has also been described.285
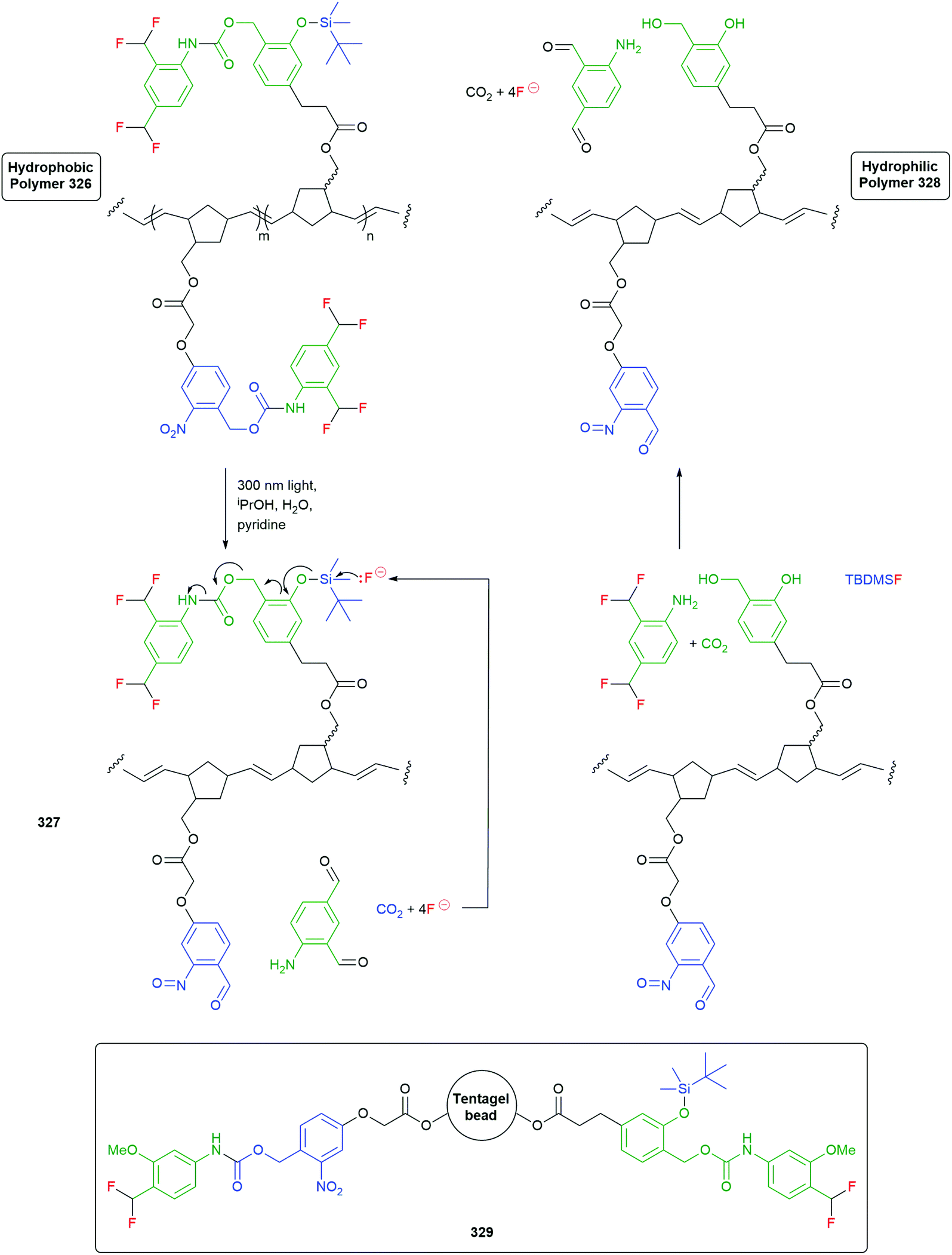 | ||
| Scheme 77 Polymeric species 326 and modified Tentagel beads 329 that undergo macroscopic responses to a fleeting signal.283 | ||
Anslyn and Phillips have applied the methodology set out above to the detection and quantification of G-class nerve agents. Taking the established reactivity of such compounds toward nucleophiles, G-series surrogate 330 was treated with benzaldoxime to give 331 and an initial fluoride signal (Scheme 78).10 Transfer of this sample to a second vessel containing autoinductive amplifier molecule 332, led to cleavage of the Si–O bond to afford phenolate 333 that disassembles by a series of 1,4- and 1,6-eliminations to give six fluoride ions and, initially, para-aminobenzaldehyde. Careful examination of the reaction products revealed that the colourimetric response was attributable to a group of short oligomers 334 (2–10 monomer units, monitored at λ = 425 nm) derived from the first-formed para-aminobenzaldehyde; by inference, this is probably true in other systems that have used this indicator as readout. The fluoride ions produced during amplification can then be used in a third vessel to release fluorophore 91 from G1 dendron 335, allowing readout by fluorescence methods (λmax = 520 nm).
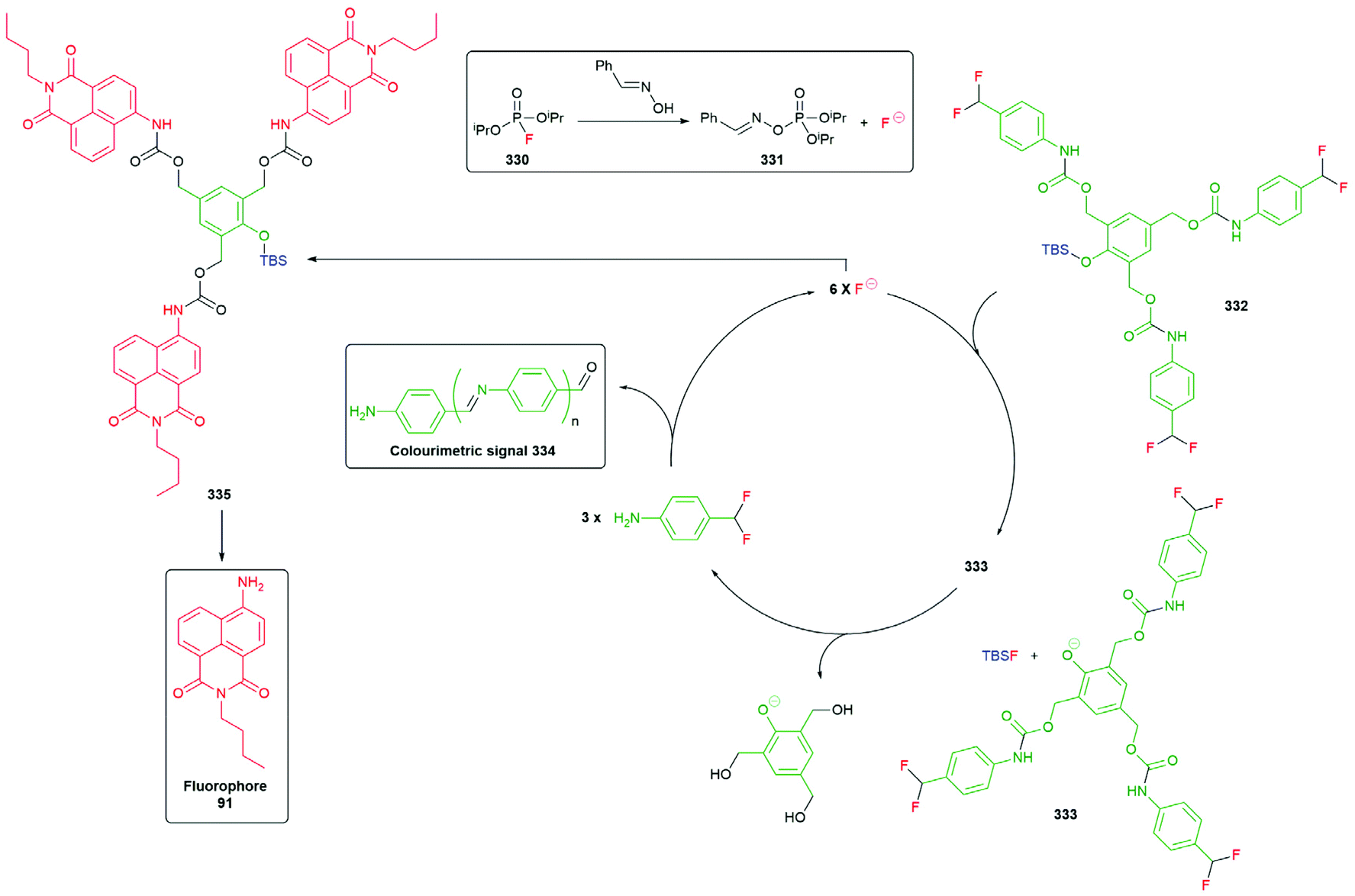 | ||
| Scheme 78 The detection and quantification system for G-series nerve agents reported by Anslyn and Phillips.10 | ||
The high effectiveness of readout from an assay by chemiluminesence has led a number of groups to explore this in the context of signal amplification; for example, in fluoride assays. Below is exemplified three approaches based around the Schaap dioxetane; first, Shabat has synthesised autoinductive molecule 336 wherein the cleavage of the O–Si bond to give phenolate 337 simultaneously promotes chemiluminescence and, by 1,6-elimination, release of a fluoride ion to restart the cycle (Scheme 79).286 Second, Akkaya has developed their dendritic probe 277 (Scheme 61) to accommodate a third arm in the dendron that carries a self-immolative aniline (338).287 This aniline moiety, by 1,6-elimination, releases two fluoride ions, facilitating a dendritic chain reaction. Third, Hisamatsu et al. used a two-component system (Scheme 79) where dendron 339 upon triggering with fluoride releases two fluoride ions, leading to exponential amplification; once amplification is complete, 340 is added inducing its fragmentation with chemiluminescence from the excited state of 341.288 A straightforward system for fluoride amplification that works with particularly simple molecular components was reported by Gabrielli and Mancin in 2017.289
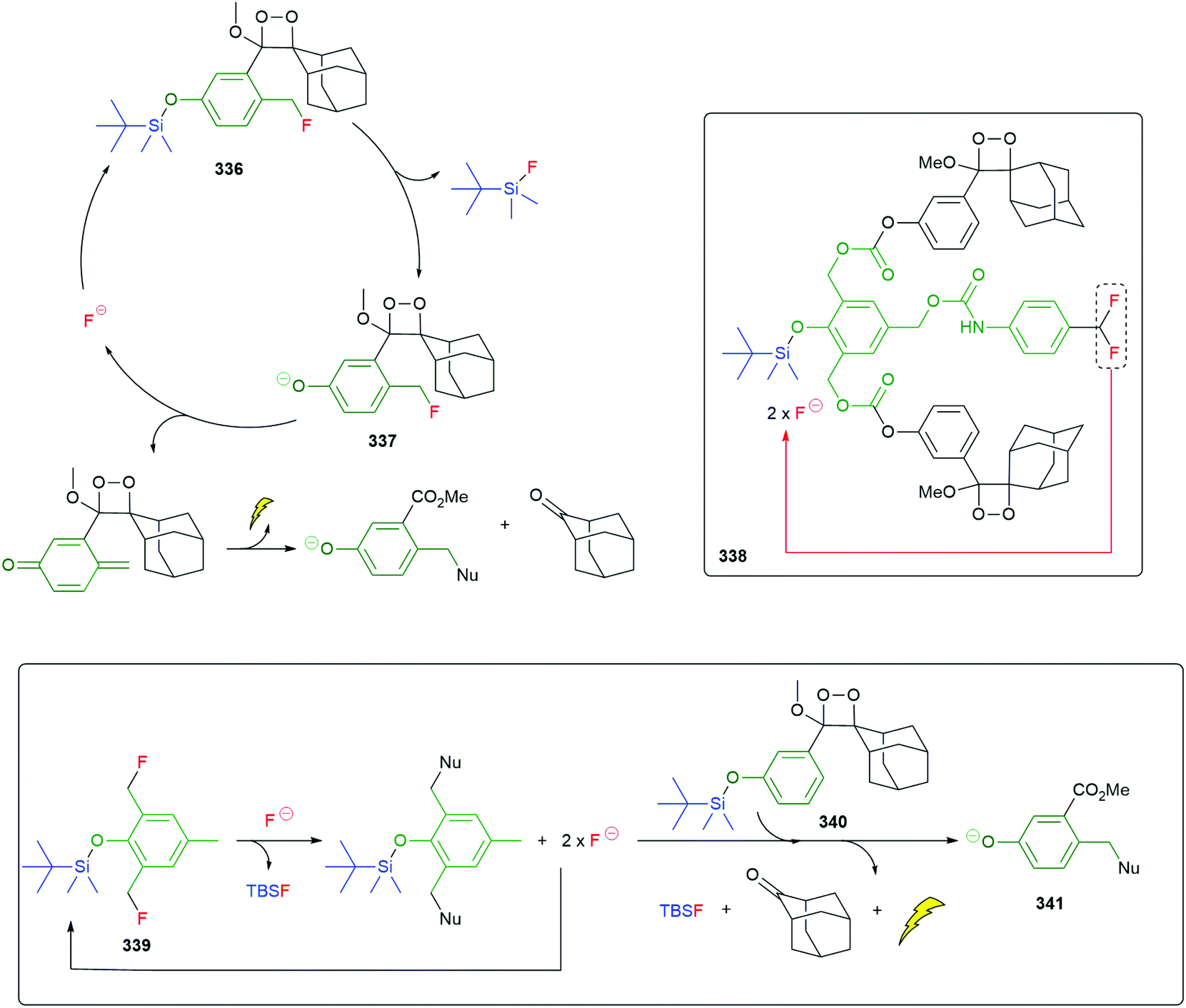 | ||
| Scheme 79 Autoinductive, DCR and two-component fluoride sensors with a chemiluminescent response.286–288 | ||
The fluoride dependent DCR systems noted above, proceed by cleavage of strong C(sp3)–F bonds. This release profile is inherently slow as fluoride is a poor nucleofuge; also, in the case of difluromethyl groups, a high energy fluorinated quinomethide is formed. Anslyn and co-workers overcame this structural issue with the addition of benzoyl fluoride as a latent source of fluoride (Scheme 80).290 Cleavage of the tert-butyldimethylsilyl ether trigger group of the dendron 342 led to the release of three equivalents of 4-amino-1,8-naphthalimide 91, as the reporting fluorophore. The resultant dendritic core, 2,4,6-tris(hydroxymethyl)phenolate, was able to react with benzoyl fluoride (catalysed by 1,5-diazabicyclo[4.3.0]non-5-ene) to release up to four fluoride anions for subsequent triggering events.
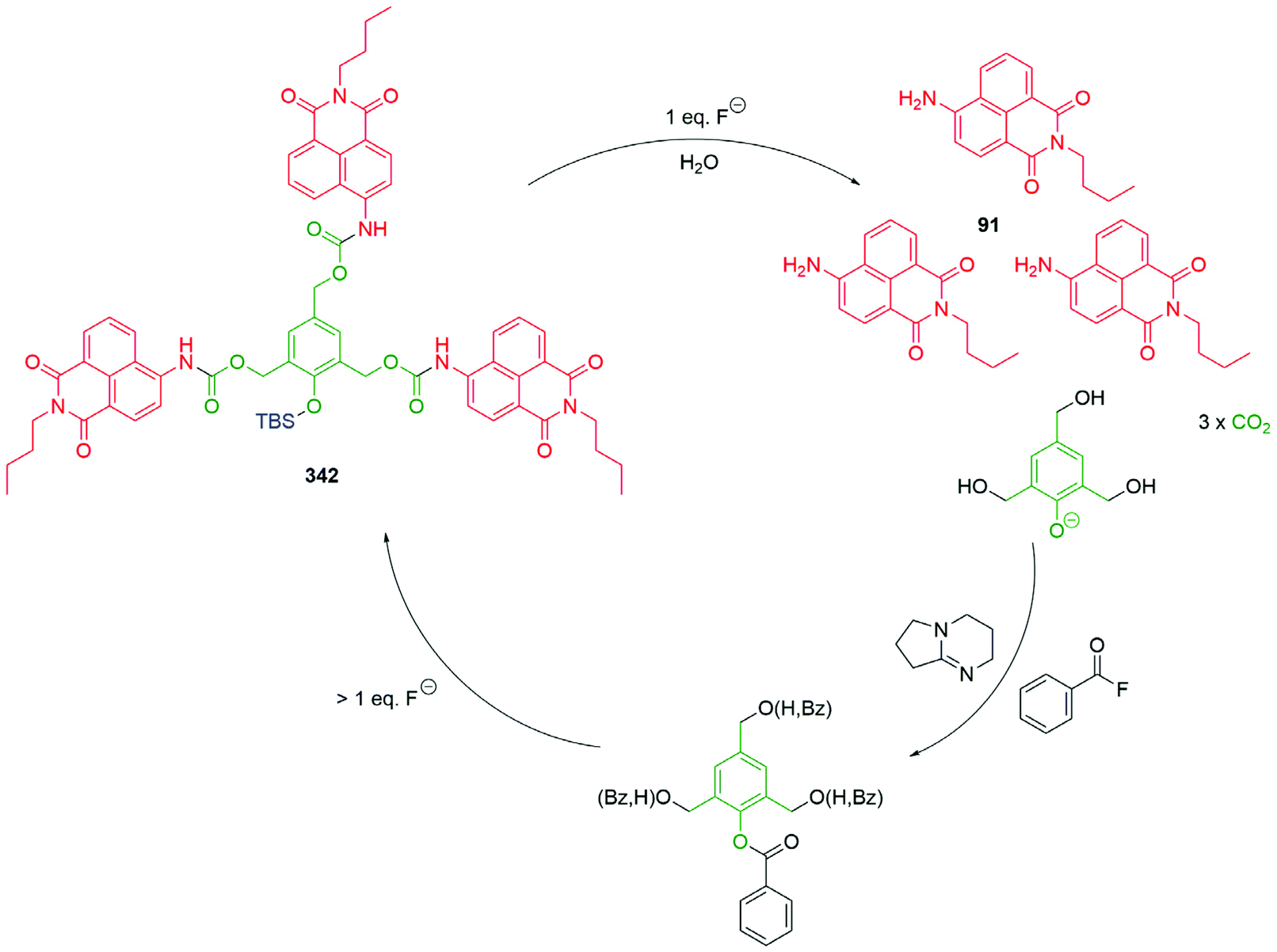 | ||
| Scheme 80 Silyl protected self-immolative dendron 342 capable of DCR with the addition of benzoyl fluoride as a latent source of fluoride to enhance the rate of disassembly, as described by Anslyn and co-workers.290 | ||
Developing their work on an autoinductive cascade for sensing of thiols, to detect V-class nerve agents, Anslyn and Marcotte utilised a cyclisation–elimination, self-immolative linker to release a fluorescent 4-amino-1,8-naphthalimide 91 that can be the subject of ratiometric fluorescence sensing (Scheme 81).291,292 Thus, a thiol is generated from the analyte e.g. VX nerve agent simulant demeton-S-methyl (DSM), and enters an amplification cycle by cleavage of 2-hydroxyethyl disulfide 343 to afford two equivalents of β-mercaptoethanol which then reacts with Meldrum's acid derivative 344 to liberate two equivalents of methylmercaptan for each molecule of β-mercaptoethanol consumed. Once the amplification has occurred, the sample may be analysed by addition of disulfide 345. Upon cleavage of the disulfide to give, for example, 346 cyclisation–elimination occurs to afford 196 with concomitant decarboxylation and release of fluorophore 91. A detailed explanation of a simple-to-apply method of data analysis, based upon chromaticity, using a cell phone was presented.
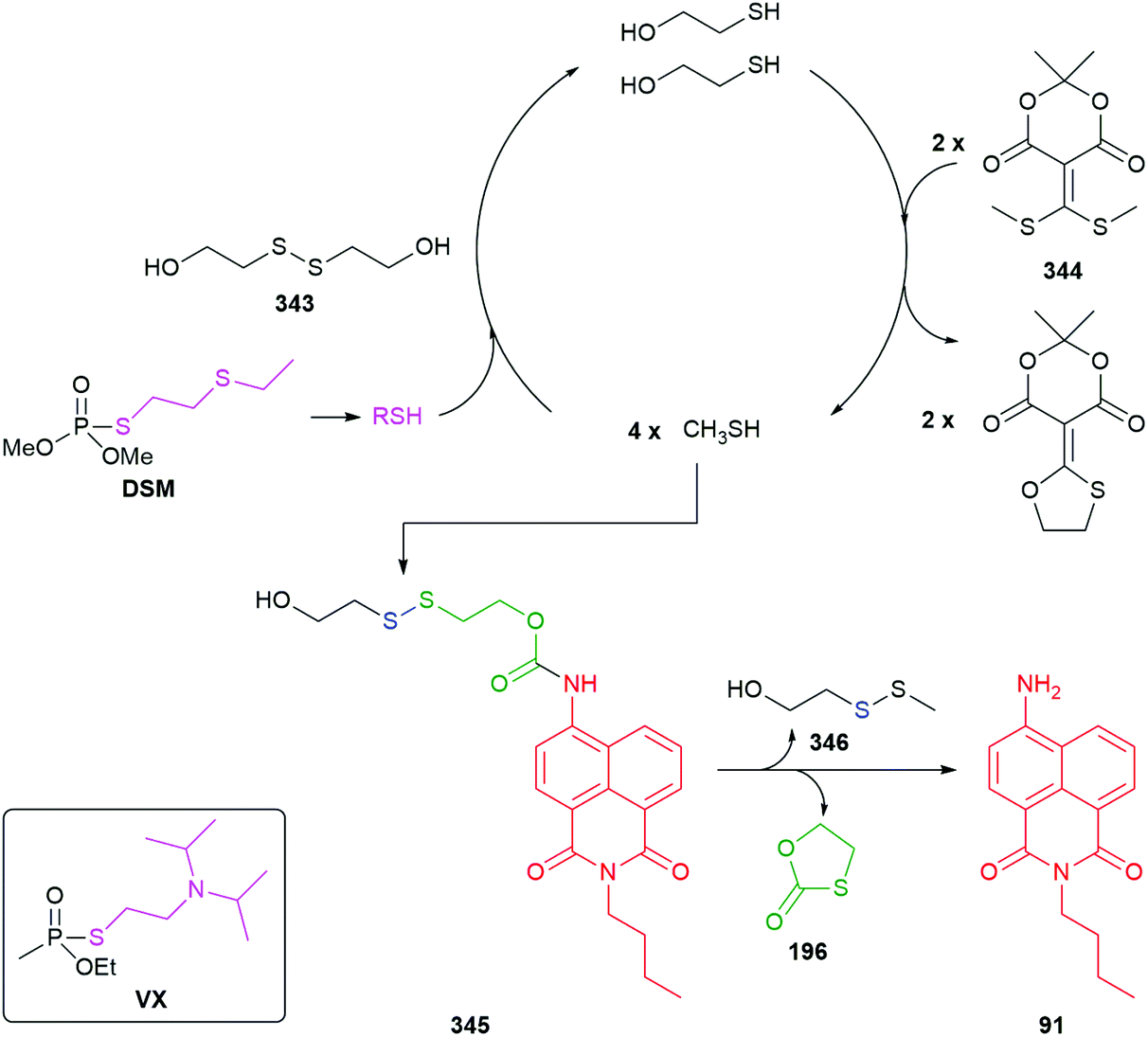 | ||
| Scheme 81 Anslyn and Marcotte's self-propagating chemical cascade for the detection and quantification of V-series nerve agents.291,292 | ||
Recently, Milliron and Anslyn have incorporated Meldrum's acid derivates related to 344 as a linker into hydrogel structures.293 This has allowed their facile, self-propagating decomposition via a ‘declick’ reaction in response to the presence of thiols, with reporting by significant changes to their physical properties and release of dyes. These hydrogels have been applied to the detection of tabun mimics.
4 Self-immolative polymeric systems
4.1 Self-immolative polycarbamates and polycarbonates
Shabat and co-workers reported the first example of a self-immolative linear polymer in 2008.294 The polycarbamate 347 was based on a 4-aminobenzyl alcohol linker that had been used previously in prodrugs,295 self-immolative dendritic systems,296 and oligomers.297 In this system, enzymatic action by BSA in PBS-buffered media promoted cleavage of the butanone trigger group and led to amplified release of fluorogenic aniline-acrylate monomers (Scheme 82). In the context of this self-immolative polymer system, the short oligomers of a 4-aminobenzyl alcohol linker prepared by Scheeren et al.,162 to create extended linkers and activatable anticancer prodrugs plus the studies of Warnecke and Kratz297 on structurally-related materials whereby elimination was triggered by reduction, are both worthy of mention as they laid out key principles in terms of the degradation process for linker units of this type. Trigger activation and cleavage allows for the aniline linker to undergo a head-to-tail disassembly via a series of sequential decarboxylation and 1,6-aza-quinone methide eliminations. Upon the depolymerisation the side product, aza-quinone methide, undergoes rapid conversion to the corresponding 4-aminobenzyl alcohol under the aqueous conditions used. The success of these initial studies on self-immolative polybenzylcarbamates have laid the foundation for extensive exploitation of this system as outlined in the following section. | ||
| Scheme 82 Self-immolation linear polymer 347 capable of 1,6-elimination resulting in head-to-tail depolymersation.294 | ||
The degradable 4-aminobenzyl alcohol linker unit has now been utilised successfully as the scaffold upon which to build complex polycarbamate drug delivery or detection systems whose degradation can be triggered by a diverse range of stimuli and in aqueous media.298–301 For example, Matson and co-workers derivatised this linkage as a polythiocarbamate 348, the trigger was activated by reduction with Na2S forming an aniline, depolymerisation via aza-quinone methides resulted in the 4-aminobenzyl alcohol linker and carbonyl sulfide (COS) which was converted into H2S in the presence of carbonic anhydrase, thus amplifying the release (Scheme 83).110 In contrast to the example shown in Scheme 82, in this case, the linker serves just that function and reporting is done via the COS liberated from the thiocarbamate.
 | ||
| Scheme 83 A self-immolation linear polythiocarbamate 348 capable of 1,6-elimination resulting in amplification of an H2S signal.110 | ||
Systematic developments by the Phillips group on self-immolative oligocarbamates featuring a boronic ester trigger unit have led to the development of elegant microfluidic point of care colorimetric indicator devices for the quantification (at femtomole levels) of active enzymes.158,302,303 Related studies using the same boronic ester trigger group have also led to the successful generation of colorimetric microfluidic multilayer devices capable of assessing the concentration of heavy metal salts (Pb2+ and Hg2+) in water.304 Within discrete layers within the multilayer device are located a DNAzyme, bead-bound glucose oxidase (GOX), glucose, the hydrophobic self-immolative polycarbamate, a food colouring and finally at the external viewing location in the assembly a layer of absorbent white paper. When the metal contaminated water sample is applied to the device, in the case of the Pb2+ metal ions, they activate the DNAzyme which cleaves the GOX from its bound state enabling it to travel to the region of the device where it can react with the store of glucose, in turn generating hydrogen peroxide that then triggers the self-immolation of the polycarbamate. The hydrophobicity of this polymer layer decreases as the material degrades, enabling the aqueous phase to permeate into the food dye and wet the external white paper layer affording a colorimetric response. Redy and Shabat have employed305 an oligocarbamate featuring side chains to yield a modular prodrug system that generates a fluorescent signal through a FRET-activated self-immolative linker.
Following their earlier studies294 wherein the function of linker and reporter are combined (Scheme 82), Gnaim and Shabat have adapted43 the approach to allow chemiluminescent sensing of a range of analytes, according to the nature of the trigger group, 349a (fluoride), 349b (H2O2) and 349c Pd(0) (Fig. 22). The polymeric nature of the sensor offers significant sensitivity advantages over the equivalent small self-immolative molecules (for examples see section 2.1).
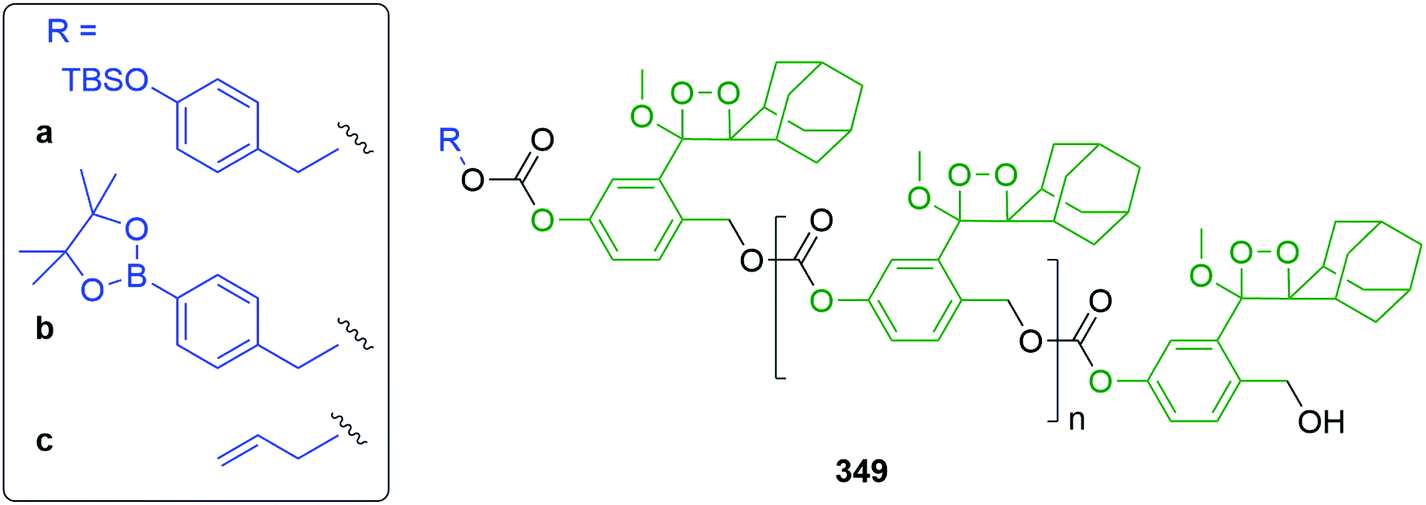 | ||
| Fig. 22 Shabat's chemiluminescent self-immolative polymers 349a–c equipped with various triggers.43 | ||
In 2014, Peterson et al. described306 the site-specific thermal activation of a self-immolative polybenzylcarbamate-PDMA block copolymer 350. In this block copolymer system, the two polymer blocks were joined by a temperature-sensitive oxazine linker unit (Scheme 84). At temperatures ranging between room temperature and 85 °C, the oxazine trigger unit was found to undergo thermal retro-[4π + 2π] reaction followed by hydrolysis of the acyl nitroso moiety to permit depolymerisation of the block polymer in solution. These degradation temperatures are far lower than observed for poly(phthalaldehyde) block copolymers307 (>150 °C, however, the ceiling temperature of self-immolative poly(phthalaldehyde)s can be tuned to easily accessed conditions, see section 4.3). The oxazine unit degrades to the corresponding free amine via the unstable carbamoylnitroso and carbamic acid intermediates to permit depolymerisation of the polybenzylcarbamate block.
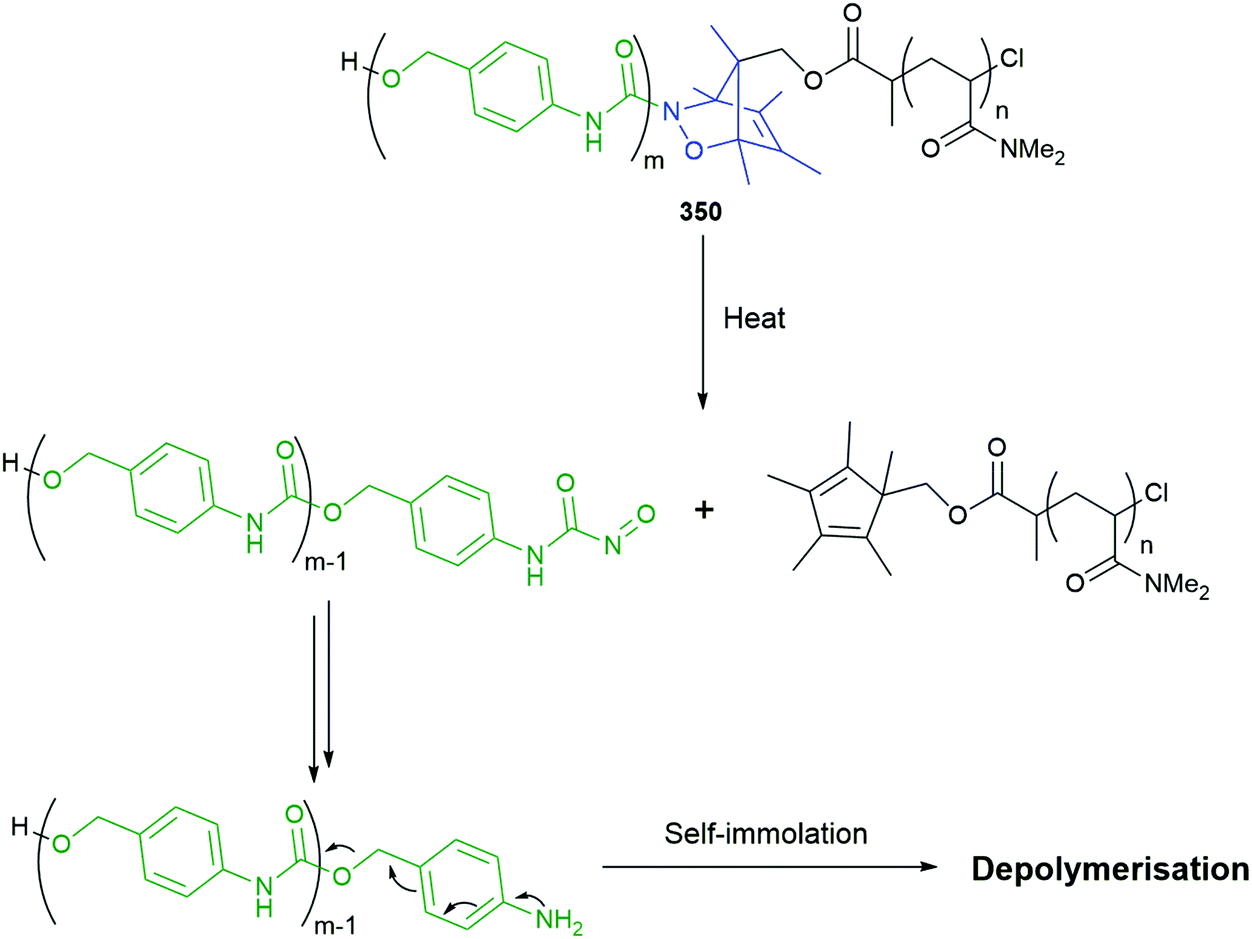 | ||
| Scheme 84 The self-immolative polybenzylcarbamate-PDMA block copolymer 350 reported306 by Peterson et al. featuring a temperature sensitive oxazine linker unit which can be degraded thermally to permit self-immolation of the polybenzylcarbamate block. | ||
Trigger groups sensitive to UV irradiation such as perylene-3-yl units have been coupled to amphiphilic polybenzylcarbamates capable of self-assembling into polymersomes.300 These stable polymersome assemblies can encapsulate photodynamic therapy agents such as eosin Y or other drug payloads. Exposure of the photo-sensitive polymersomes to UV light (420 nm) for a period of thirty minutes leads to self-immolation of the carbamate blocks and results in disintegration of the polymersomes (so-called ‘SIPsomes’) and release of the reactive payload (Scheme 85). Liu et al. have also employed the perylene-3-yl trigger unit successfully in a hyperbranched polycarbamate functionalised with doxorubicin and hydrophilic PEG chains plus cell membrane targeting RGD units to facilitate spatiotemporal intracellular drug delivery in aqueous media.308 Cornelissen and co-workers have reported an analogous photochemically initiated approach for the triggered degradation of polycarbamates and release of monomeric 4-aminobenzyl alcohol subunits from virus-like assemblies using 5-methoxy-2-nitro benzyl alcohol as the photolabile unit.309 In 2020, Kumar et al. reported301 the use of the photolabile 2-nitrobenzyl alcohol trigger system in conjunction with self-immolative amphiphilic polybenzylcarbamates to generate self-assembled vesicles that carry an enzyme payload. Upon photochemical activation, the polybenzylcarbamate structure degrades leading to vesicle disassembly and ultimately payload release. The liberated enzyme promotes the formation of a stable hydrogel and thus triggered destruction of a polymer can be utilised to construct a different polymer-based material. The use of photolabile units in conjunction with degradable polycarbamates based on the 4-aminobenzyl alcohol repeat unit has also been utilised by Chujo et al. to yield highly efficient self-immolative hybrid colorimetric materials.310
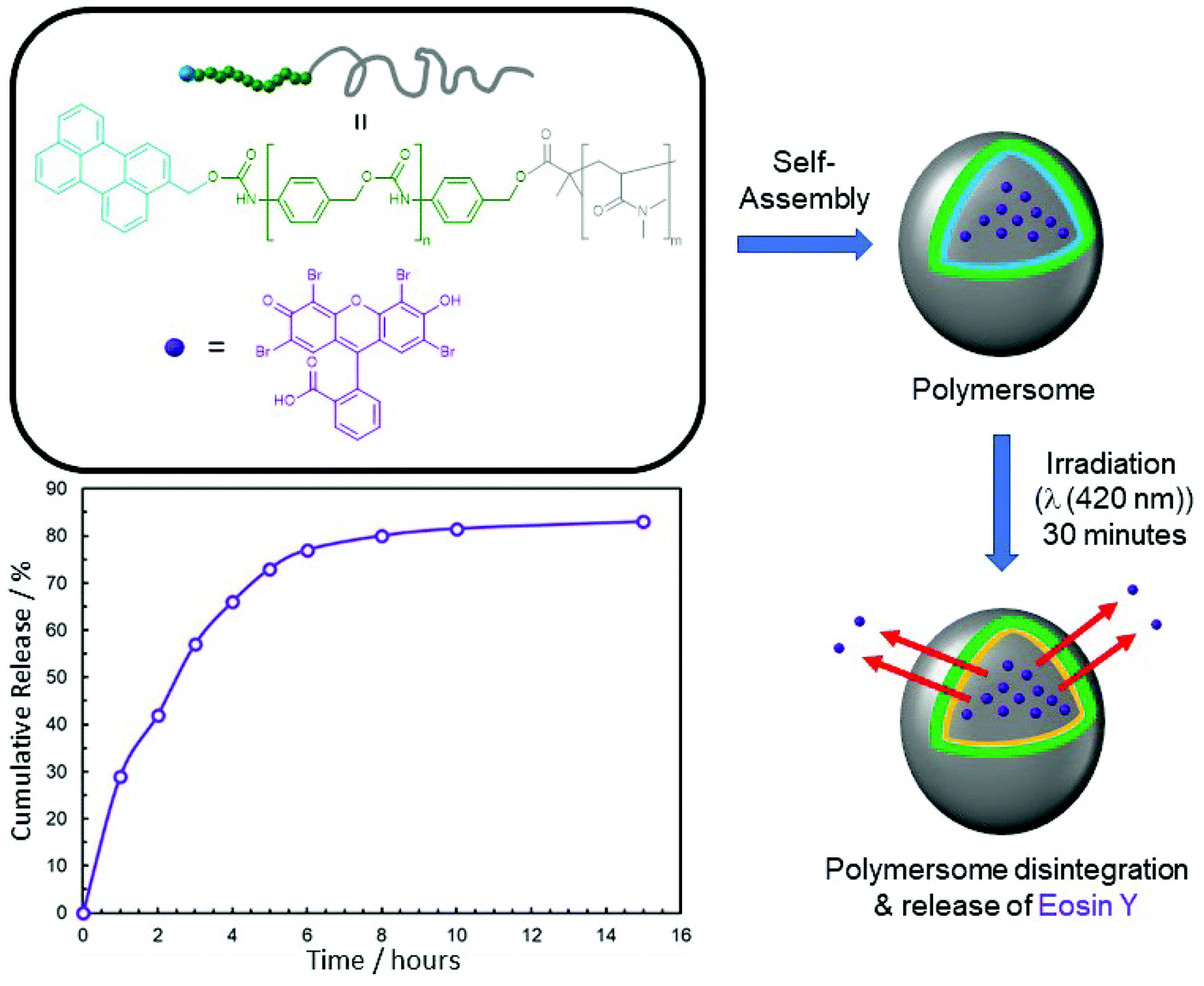 | ||
| Scheme 85 The self-assembly of self-immolative polymersomes (‘SIPsomes’) from amphiphilic block copolymers capable of releasing an encapsulated payload of the photodynamic therapy agent Eosin Y as reported by Liu et al.300 | ||
Liu, Hu and co-workers have recently developed amphiphilic micellar polyurethane nanoparticles that can be degraded in a self-immolative fashion via exposure to only trace quantities of the appropriate stimulus.311 The nanoparticles feature both external and internal ‘built-in’ triggers, activation of the external trigger liberates primary amine species capable of penetrating the micelle to react with and degrade the contents of the micelle in an amplified manner. This approach to highly efficient degradation of micellar systems (as demonstrated in this study using a range of stimuli including enzymes or irradiation) offers an attractive route to the development of nanovectors to treat disease states312 such as cancer via targeted means.
In contrast to the polymer degradation processes outlined above, Zhang et al. reported313 in 2021 an alternative use of the quinone methide elimination cascade process whereby polycarbamates are produced via the controlled ring opening polymerisation314,315 of macrocyclic monomers (Scheme 86). In this innovative study, a nucleophilic amine (such as N-hexylamine) was used to trigger the ring opening of a macrocyclic carbonate-benzylic carbamate 351 that then, in turn, degrades via the quinone methide elimination cascade process to yield a newly formed carbamate featuring a nucleophilic amine terminus that can then propagate chain growth. The lability of the carbonate unit in these carbonate–carbamate macrocycles was enhanced by the presence of an electron-withdrawing nitro group located on the phenyl ring ortho- to the carbonate and control of the polymerisation was improved by the addition of a nucleophilic trapping agent. In these studies, the relatively weak nucleophile, N-methyaniline, was employed to trap the reactive quinone methide generated in the self-immolative ring opening step. This aniline was insufficiently nucleophilic in nature to trigger ring opening and chain growth. Using this strategy, excellent control of the polymerisation was afforded with monomer conversions up to 97% and polydispersity (Đ) values as low as 1.06.
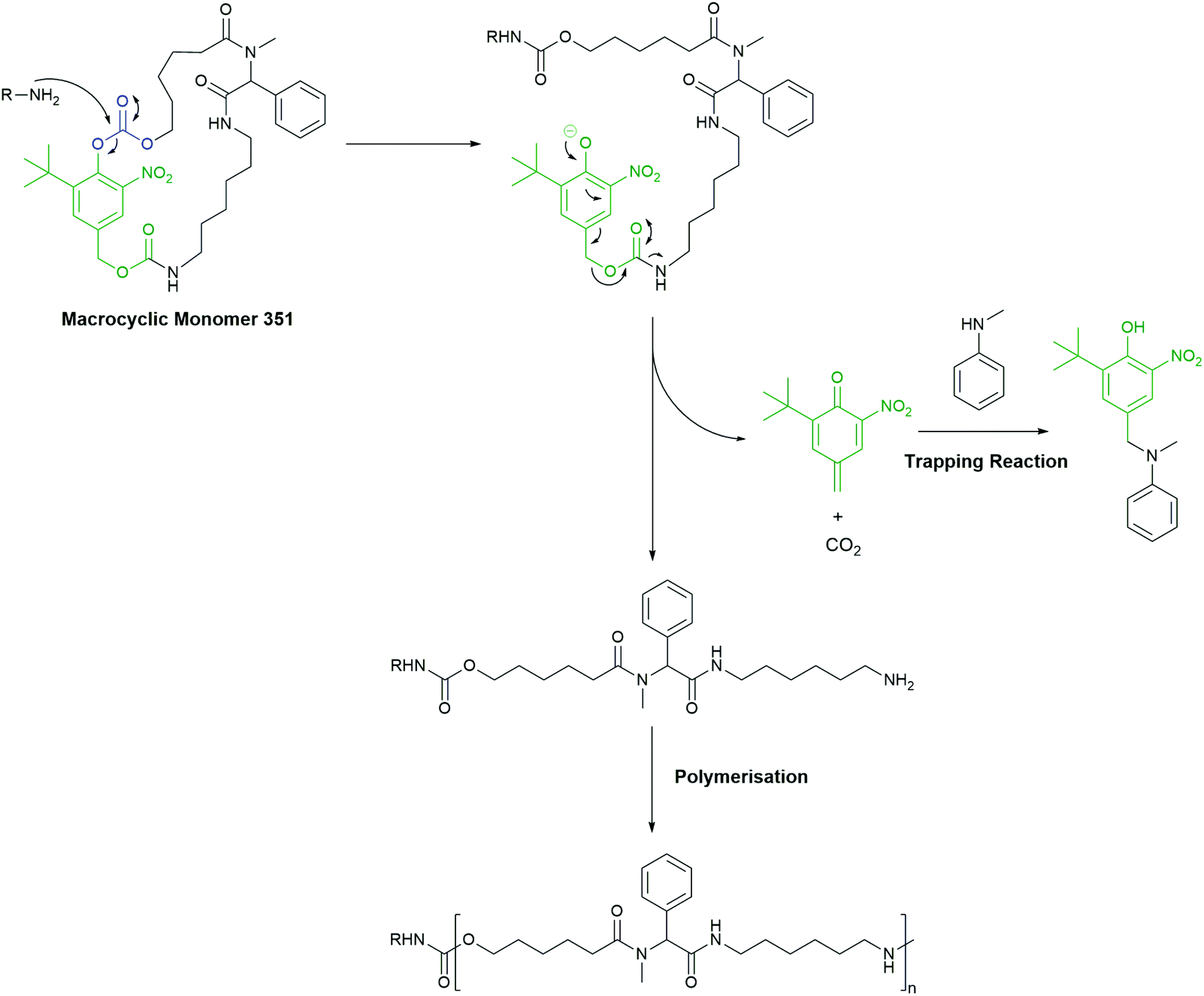 | ||
| Scheme 86 The controlled ring-opening polymerization of a macrocyclic monomer 351 employing the quinone methide elimination cascade reaction.313 | ||
DeWit and Gillies developed the first self-immolative polymer containing alternating elimination and cyclization spacers in 2009.316 This system, 352, was constructed using 4-hydroxybenzyl alcohol as the 1,6-elimination linker and N,N′-dimethylethylenediamine as the cyclization linker (Scheme 87). A self-immolative polycarbamate capped with a Boc protecting group was prepared and subsequent cleavage of the tigger group afforded self-immolative depolymerisation, yielding N,N′-dimethylimidazolidinone, 4-hydroxybenzyl alcohol, and CO2. In an extension of this study, Gillies and co-workers have been able to tune the rates of polymer degradation by replacing the cyclization linker, N,N′-dimethylethylenediamine, with N-methylaminoethanol 353 or 2-mercaptoethanol 354 (Scheme 87).317 In 2013, McBride and Gillies described a detailed study of the effect of the chain length on the kinetics of self-immolative depolymerisation involving alternating elimination and cyclization; a proportional relationship between polymer chain length and the depolymerization time was established.318 Further refinement of this elimination/cyclization approach by Gillies and co-workers involved319 the use of an orange-coloured diazo trigger group to permit a colourimetric indicator for depolymerisation – in an approach related to the report of Hay and co-workers on the effect of leaving group effects in reductively triggered fragmentation of 4-nitrobenzyl carbamates,50 reduction of the azo unit with hydrazine affords the corresponding electron rich hydrazine320 that can then promote self-immolation and depolymerisation to yield a colourless solution.
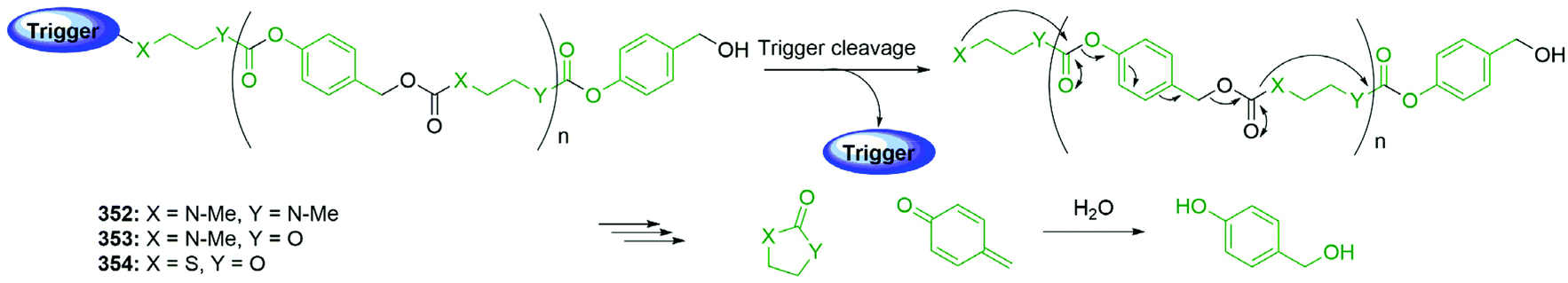 | ||
| Scheme 87 Linear self-immolative polymers 352–354 containing alternating linkers as described by Gillies and co-workers.316,317 | ||
Almutairi and co-workers have developed321 a photochemically driven cyclization/elimination triggering process to initiate the depolymerisation of polycarbamates (Fig. 23).322 In this study, the trigger groups used on the polymer chain ends were either an ortho-nitrobenzyl residue 355 (for cleavage via UV-irradiation) or 4-bromo-7-hydroxycoumarin 356 (sensitive to NIR light) that were coupled to the polycarbamate via a diamine cyclisation linker. Photo-irradiation cleaves the trigger group (via single photon and two photon absorption processes for UV and NIR exposure, respectively) permitting cyclization by the liberated amine end group that in turn promotes depolymerisation. Further work from these authors has studied photochemically triggered depolymerisation of polycaprolactone particles, with payload release.323 Similarly, controlled degradation of poly(lactic-co-glycolic acid)-based and ornithine-based polymers were studied.324,325
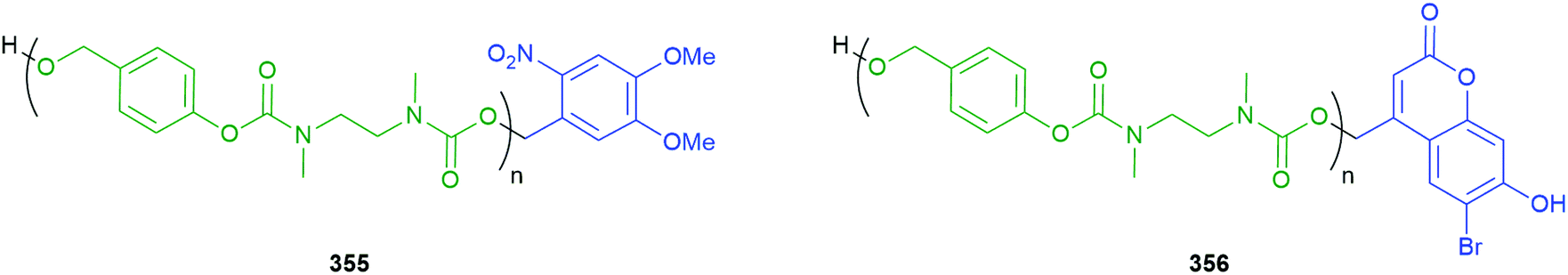 | ||
| Fig. 23 Example polycarbamate structures 355 and 356 reported by Almutairi and co-workers that undergo photochemically induced depolymerisation via a cyclisation/elimination process.322 | ||
UV-triggered self-immolative polycarbamate systems have also been reported by the Cheng research group that successfully demonstrate 2-directional polymer cleavage326 and the ability to exhibit controlled delivery of the anti-cancer drug, 9-aminocamptothecin.2
Zimmerman and co-workers have demonstrated a new type of base-triggered self-amplifying self-immolating polycarbamate based on the Fmoc protecting group 357.327 The polymer was obtained through polycondensation of the Fmoc-derivative, a diol, with hexamethylene diisocyanate and a catalytic amount of dibutyltin dilaurate. Several bases were screened to optimise the degradation of the polymer, hexylamine was selected as the appropriate base with which to initiate the E1cB elimination pathway. Deprotonation of the acidic methine proton of the fluorenylmethyl unit triggers the E1cB elimination and chain scission via a decarboxylation process leading to the production of two shorter polymer chains, one possessing a dormant fluorenyl end group and the other bearing a primary amine that is then capable of triggering further depolymerisation (Scheme 88). Evidence for the autocatalytic nature of this self-immolative depolymerisation process was provided by the sigmoidal shape of the plots of the 1H NMR spectroscopic data vs. time for the degradation of the polycarbamate 357.
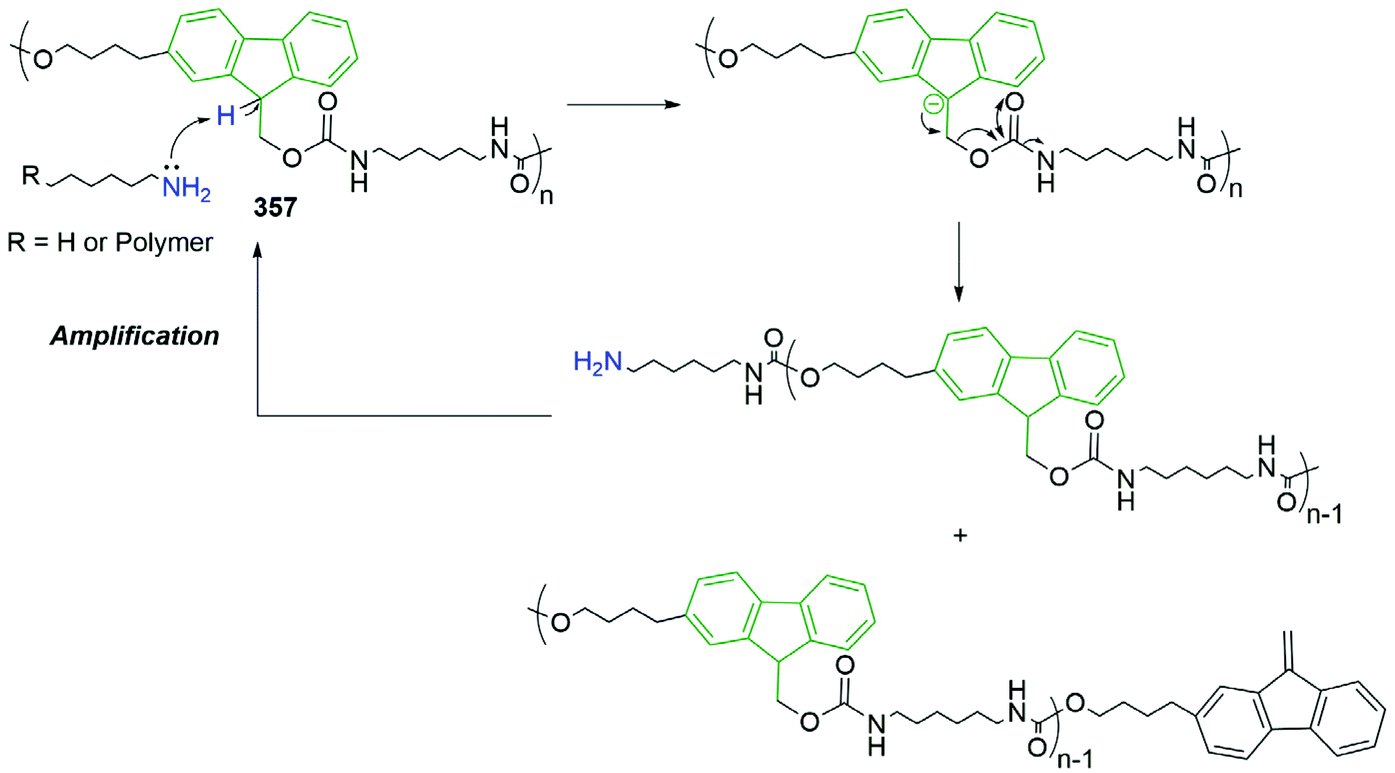 | ||
| Scheme 88 The base-triggered Fmoc-derivative self-immolative polycarbamate 357 as described by Zimmerman and co-workers.327 | ||
Zimmerman and co-workers advanced this further with the addition of the photo-cleavable trigger ortho-nitrobenzyl carbamate.328 Exposure to UV irradiation (365 nm) promoted photolytic cleavage, activating the trigger through a Norrish-type II reaction mechanism, generating a nitrosobenzaldehyde and a primary aliphatic amine. The free amino groups can then act as a trigger to induce further self-immolation of the base sensitive Fmoc carbamate units in the polymer backbone. After irradiation at room temperature for 40 minutes and incubation at room temperature for 24 hours, only low molecular weight species were observed via GPC analysis. Zimmerman and co-workers noted that unlike the Fmoc containing polymer 357,327 the degradation kinetics of the photo-triggered polymer did not show an exponential profile as expected for a self-amplifying system. This was attributed to quenching of the free amine via a Schiff base type reaction with the nitrosobenzaldehyde generated in the photocleavage of the trigger. More recently, Barner-Kowollik and co-workers have reported closely-related studies that involved the non-amplified photolysis of polyurethanes.329
Phillips and co-workers have developed recently a new class of self-immolative polymer, poly(carboxypyrrole)s, that are capable of undergoing complete head-to-tail depolymerisation in the solid state, offering a potential route to selectively depolymerisable adhesive and coating materials (Scheme 89).330 Pyrrole was selected as the core for the repeat unit based upon observations in an earlier study by the Phillips group that reduction in the aromaticity of the central ring (using 1,4-disubstituted benzene, 1,4-disubstituted naphthalene and 9,10-disubstituted phenanthrene as the model substrates) provided an increase in the rate of triggered release of the pendant benzylic group.331 Analogous calculations predicted that pyrrole would be 40% less aromatic than benzene,332,333 hence it was anticipated that the depolymerisation rate would be faster. Polymerisation of bifunctional pyrrole monomers bearing phenyl carbamate at position 1 and a nucleophilic alcohol at position 3 of the heterocycle was achieved by heating at 60 °C using catalytic quantities of 1,8-diazabicycloundec-7-ene (DBU) in the presence of an alcohol functionalised end-capping unit equipped with a stimuli-responsive moiety. Depolymerisation of 358 that featured an aryl boronate trigger group ((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol) via exposure to hydrogen peroxide resulted in rapid head-to-tail depolymerization to the corresponding azafulvene units within 40 minutes (Scheme 89). Within this study, Phillips and co-workers also reported the synthesis of an analogous poly(carboxyindole) 359 to strengthen the hypothesis that lower aromaticity affords more rapid depolymerization (see Table 1 for the resonance energy per π-electron).330,331 Exposure of poly(carboxyindole) 359 to hydrogen peroxide under the same degradation conditions as 358 revealed that poly(carboxyindole) depolymerizes twelve times slower than the poly(carboxypyrrole), thus supporting this hypothesis. Phillips and co-workers further demonstrated that 358 could undergo depolymerisation in the solid state within 9 hours when moulded into a disc with 40 wt% poly(ethylene glycol) (Mn = 400) used as a plasticizer.
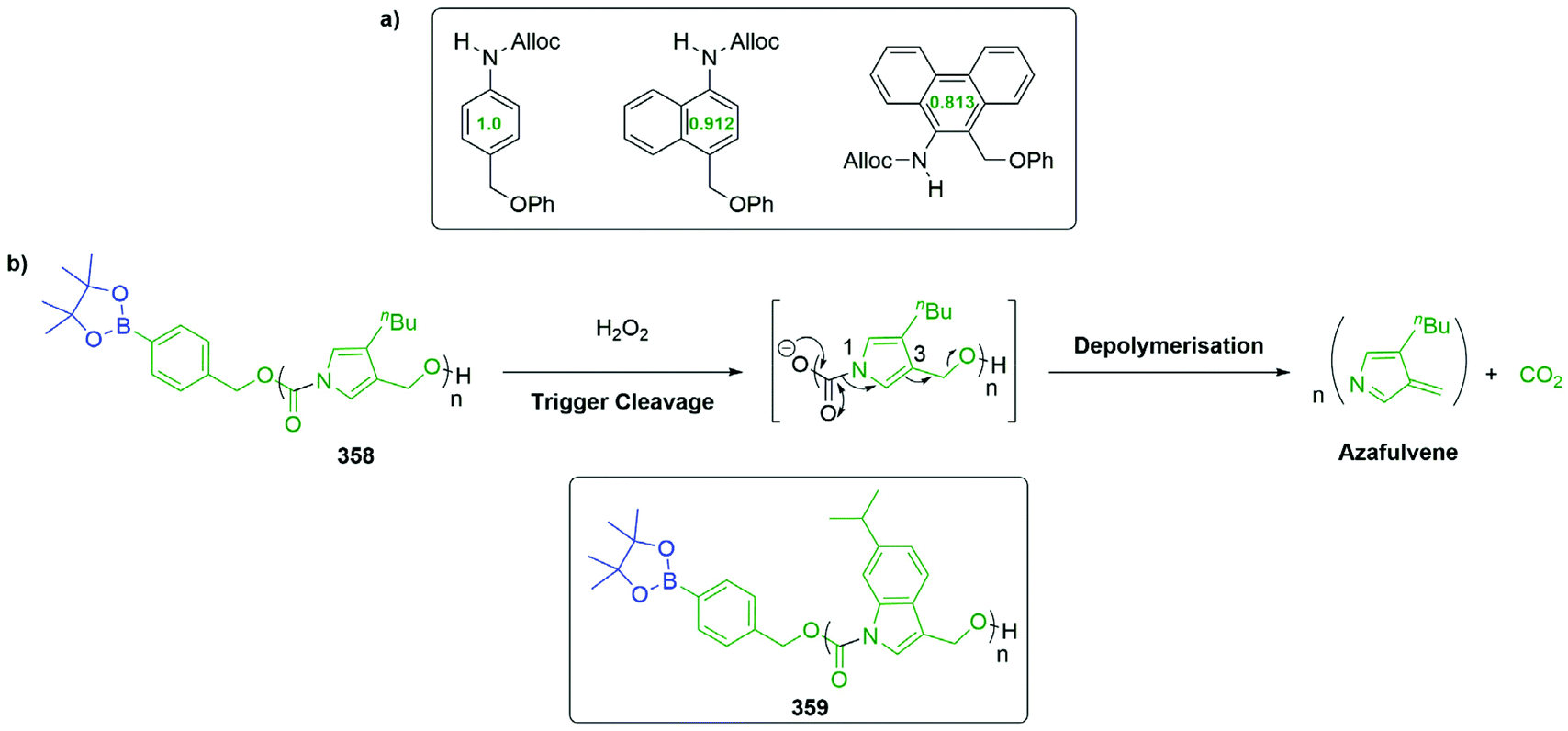 | ||
| Scheme 89 (a) Calculated relative aromaticity values for three different aromatic systems (values shown in green correspond to rings without substituents);331 (b) head-to-tail depolymerisation of poly(carboxypyrrole) 358 as described by Phillips and co-workers plus the analogous poly(carboxyindole) 359 assessed in this study.330 | ||
| Compound | Resonance energy per π-electron (unit = β) |
|---|---|
| Benzene | 0.065 |
| Pyrrole | 0.039 |
| Indole | 0.047 |
Almutairi and co-workers have also developed biocompatible polymeric capsules, prepared from polyesters that contain self-immolative dendron units; these capsules were able to carry cargo molecules, e.g. Nile Red or fluorescein diacetate. The dendron units are triggered by, and degrade in response to biologically relevant levels of H2O2 (produced by cells in oxidative stress).334 Capsules generated from polymers 360 and 361 (Fig. 24) were shown to undergo significant morphological changes on exposure to hydrogen peroxide, with release of Nile Red. Polymer 361 showed release of the cargo molecule fluorescein diacetate. Thus, these self-immolative materials are candidates for delivery of bioactive molecules.
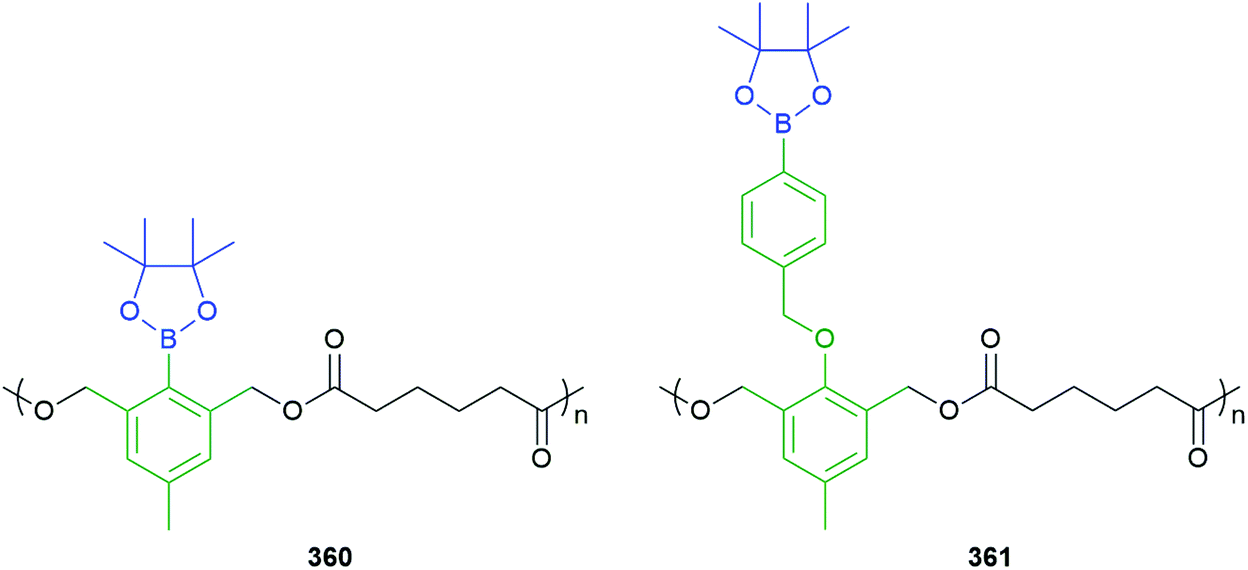 | ||
| Fig. 24 The repeat units used in self-immolative polymer-based capsules by Almutairi and co-workers.334 | ||
In similar style, Du and Li have reported amphiphilic poly(amino ester)s that are degraded in the presence of hydrogen peroxide.51 The copolymers represented by 362 in Scheme 90 had PEG chains attached, primarily at the primary terminal amines with limited attachment at the internal secondary amines. These amphiphilic copolymers self-assemble in aqueous media into nanoparticles and have proved responsive to both pH and hydrogen peroxide. Of general interest, is an extensive study of trapping of the quinone methides formed during self-immolation, with the amines (reporter groups) produced during the fragmentation process. These can give rise to compounds such as 363 and 364.
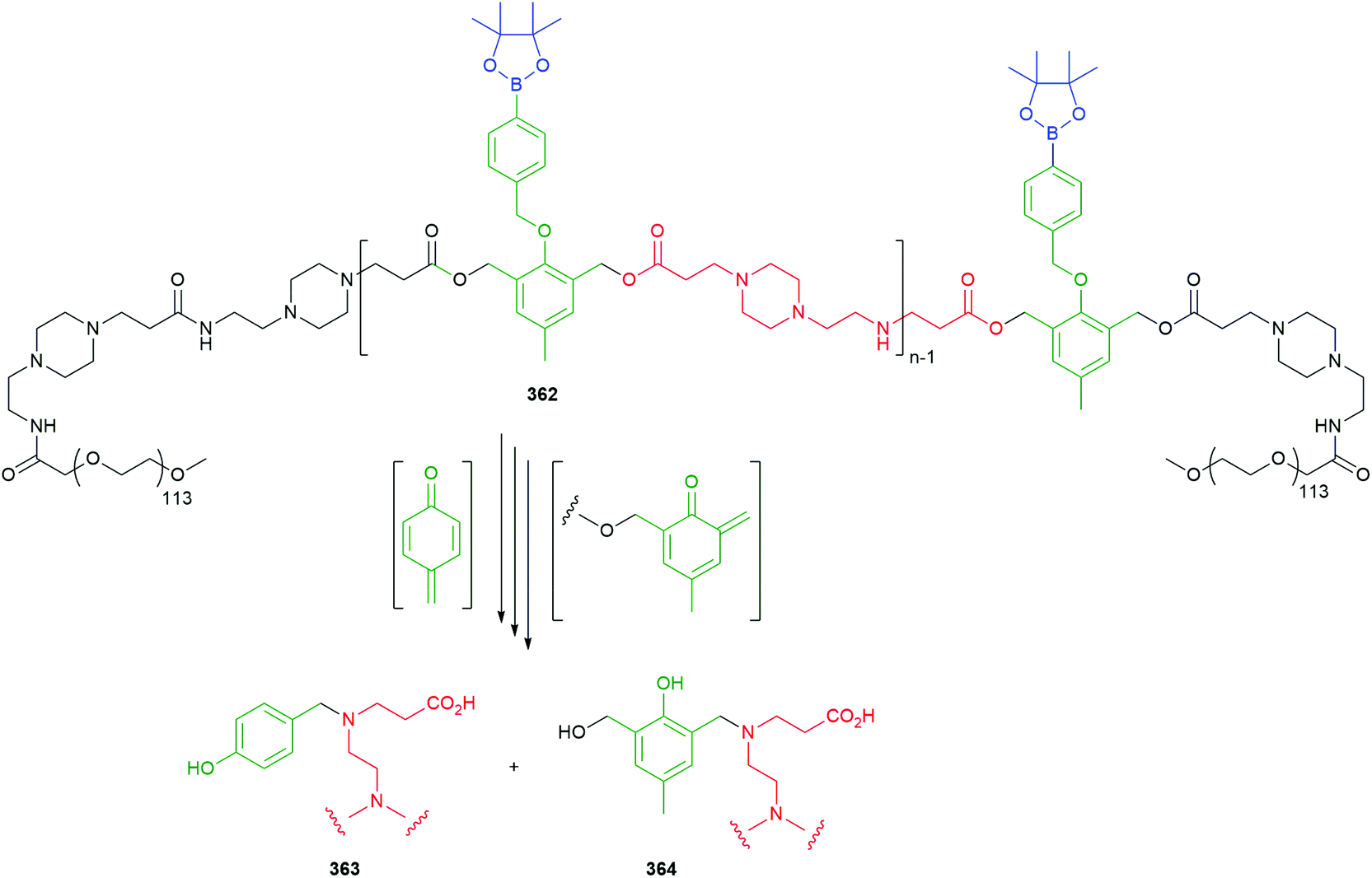 | ||
| Scheme 90 Du and Li's pH/H2O2 sensitive self-immolative, amphiphilic poly(amino ester) 362.51 | ||
Anslyn has introduced a fascinating and important set of sequence defined N-to-O repeating oligocarbamates such as 365 shown in Scheme 91 that have been shown to be readable using a single LC/MS trace as a result of their self-immolative nature.335 Taking inspiration from Li's lactonization depolymerisation, Anslyn et al. established conditions for the controlled self-immolation of the oligocarbamates, initiated by reversible deprotonation of the primary alcohol as trigger (K3PO4, MeOH/H2O, 70 °C) and proceeding via a series of 5-exo-trig cyclisations (Scheme 91).336 Excitingly, the potential in information storage has been addressed in a recent paper.337 In addition to information storage, this system also has potential in areas such catalysis and self-assembly.
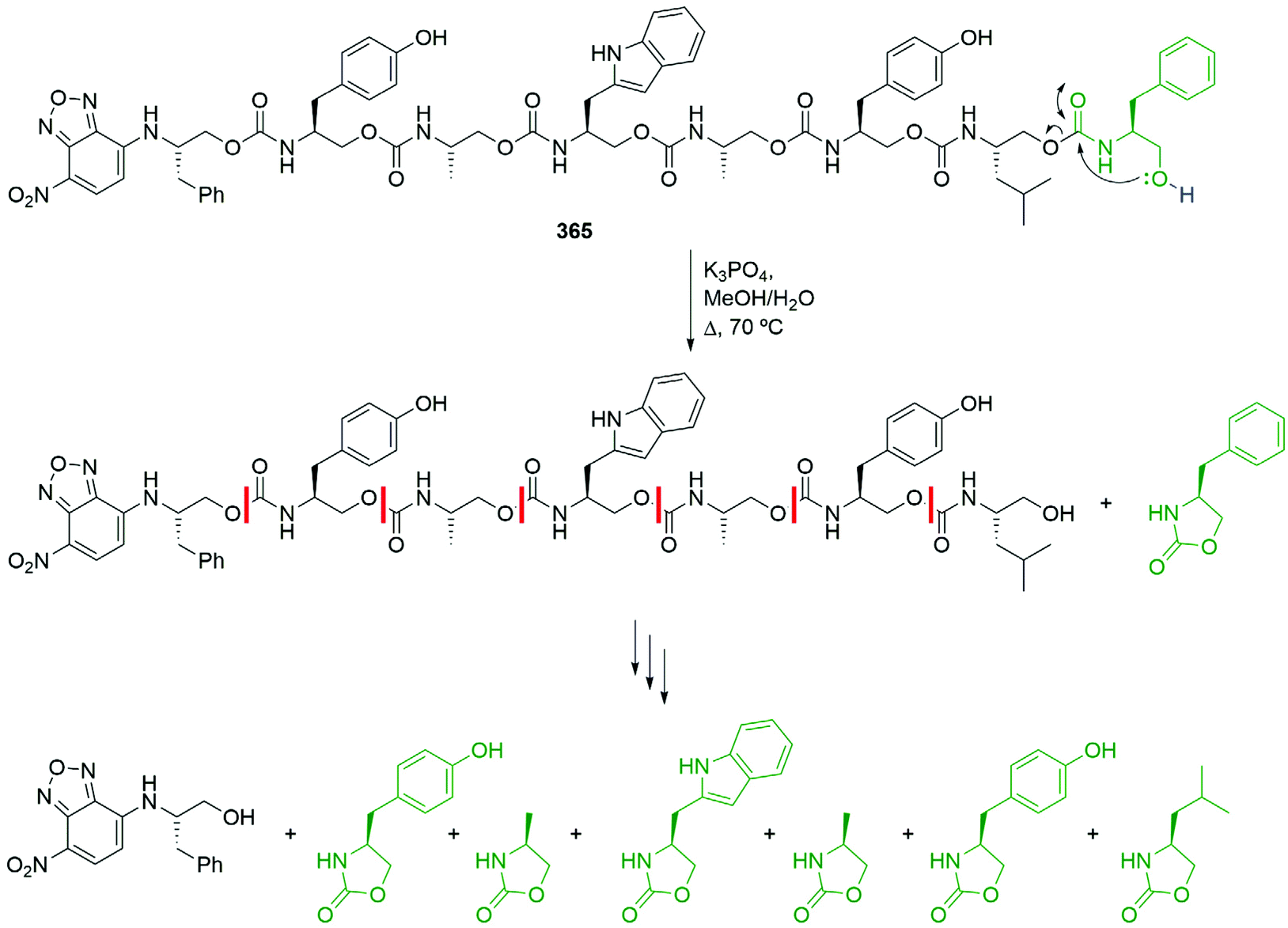 | ||
| Scheme 91 Oligocarbamates information storage systems such as 365 reported by Anslyn and co-workers.335,337 | ||
4.2 Self-immolative poly(benzyl ether)s
In 2003 McGrath and coworkers demonstrated the triggered degradation of branched polybenzyl ethers (PBEs) to permit both linear252 and geometric dendrimer disassembly (so-called ‘dendritic amplification’) via a 1,6-elimination pathway analogous to that observed in the self-immolation of materials featuring 4-aminobenzyl alcohol repeat units (vide supra).338 Subsequent studies of the linear disassembly of dendritic oligomers339 or oligomers involved structures that were based upon vanillin or ortho-vanillin as the cleavage vectors340 in conjunction with either allyl or ortho-nitrobenzyl trigger groups (Fig. 25).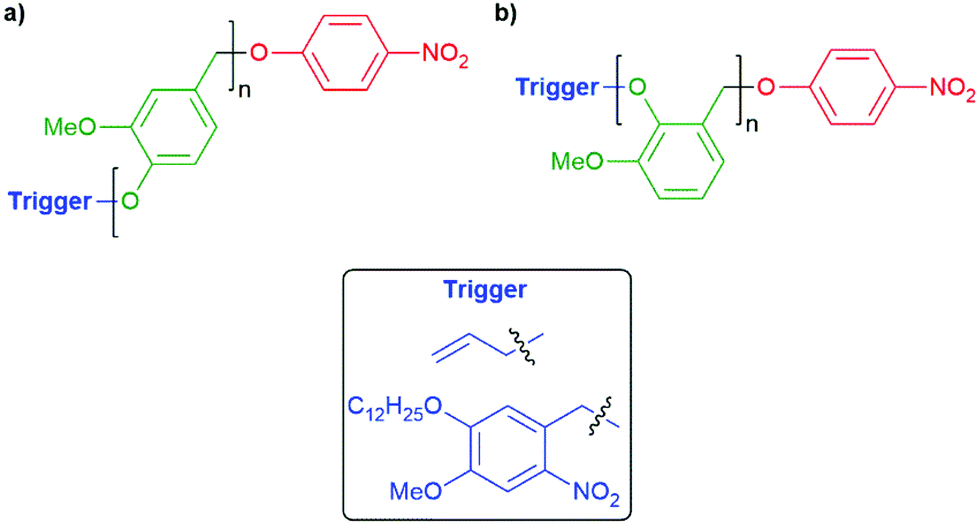 | ||
| Fig. 25 (a) Vanillin and (b) ortho-vanillin-based oligomers developed by McGrath and coworkers that degrade via the 1,6-elimination pathway upon activation of either the allyl or an ortho-nitrobenzyl trigger groups (n = 1–3).340 | ||
In the light of the above developments, in 2013 Phillips and co-workers reported the synthesis and degradation analysis of self-immolative linear PBEs (Scheme 92).341 In this study, PBEs 366 were synthesised via anionic polymerisation of quinone methide monomers and capped with a range of stimulus-responsive (trigger) groups. Cleavage of the triggering group generates a phenolate which is capable of a 1,6-elimination cascade, releasing the quinone methide monomeric units and resulting in rapid depolymerisation within a few hours. The ether linkages of the PBEs offer a higher stability to acid, base, and heat which would cause unwanted degradation of a self-immolative polycarbamate or polycarbonate.
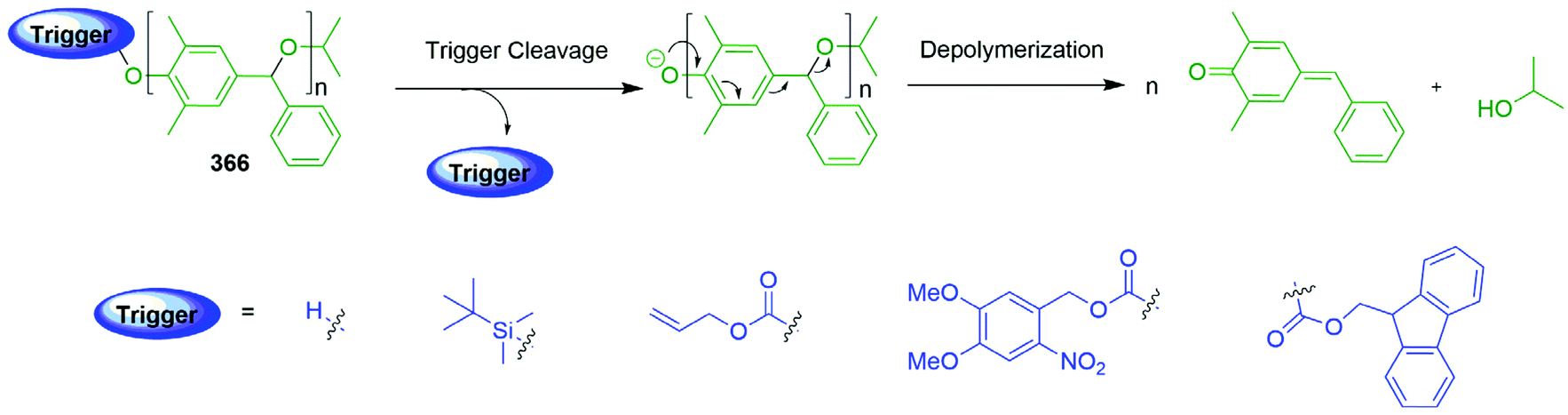 | ||
| Scheme 92 Disassembly pathway of a self-immolative poly(benzyl ether)-based polymer 366 with different triggers.341 | ||
Subsequent evolution of this system led to the development of PBE-based materials (Fig. 26) that incorporated detection units onto each repeating unit and amplified depolymerisation in the solid-state was demonstrated in a colorimetric fashion.47 Treatment of a rigid solid disk of PBE 367 with a solution of TBAF led to cleavage of the silyl ether trigger group and self-immolation of the polymer, with rapid reduction in the size of the polymer disk and release of the purple coloured, deprotonated quinone methide 368 (Fig. 26) into the solvent used (acetonitrile). In contrast, in the case of the analogous PBE where the trigger group was situated only at the polymer chain end, the degradation of cast disks proved to be negligible, even after exposure to TBAF over more than seven days. A triggered degradative response of this type is extremely attractive in applications where dramatic changes in physical form of a polymer is needed (such as debond on demand) or where colorimetric disclosure of the presence a reactive agent on a surface may be needed. Indeed, the Phillips group extended the triggered decay of PBEs featuring silyl ethers using fluoride anion sources to demonstrate successful degradation of a cross-linked prototype adhesive.4
 | ||
| Fig. 26 The self-immolative PBE system 367 capable of revealing a colorimetric response (in the form of the purple coloured deprotonated quinone methide 368) in the solid state.47 | ||
In an approach to demonstrate the applicability of self-immolative processes in polymer recycling, PBEs modified with tri(ethylene glycol) or fluoroalkyl groups to access desirable physical and mechanical properties of plastics were described in 2015 by Phillips and co-workers.342 These modified PBEs featured a phenolic end group and thus were depolymerised in a head-to-tail depolymerisation fashion at ambient temperatures upon exposure to the base, 1,8-diazabicycloundec-7-ene (DBU), and the recovered monomeric materials were of sufficient purity to permit repolymerisation, thus demonstrating end-of-life recycling capabilities.
Successful triggering of the self-immolation of PBEs bearing silyl ethers by fluoride anions is a strategy that has been employed by several other research groups to achieve materials with antimicrobial activity with reduced hemolytic toxicity343,344 and degradable polymer brush architectures.345 Bringing key aspects from these studies and those led by Phillips4,47,342 (vide supra) have enabled Zhang and co-workers to develop so-called ‘side chain-immolative polymers (ScIPs)’ based upon the PBE scaffold.346 The ScIP PBEs in this case bear pendant pyridine disulfide units (Scheme 93), that, in the presence of thiols can undergo thiol-disulfide exchange reactions to prepare ScIP-g-PEG graft polymers and organogel networks. The self-immolative degradation of these ScIP PBE-based materials could, in theory, be triggered via two different approaches – (i) exposure to reductive cleavage conditions for the disulfide linkages in either the grafted side chains or organogel or (ii) treatment with a fluoride ion source to target reaction with the silyl ether chain end of the PBE backbone. On account of the higher density of the disulfide groups present in either of these ScIP PBE-based materials when compared to the chain end silyl ether trigger unit, it was found that treatment using reductive conditions in a basic environment (DTT in combination with DBU) led to rapid depolymerisation both in the solution and solid state. In stark contrast, when exposed to TBAF, a solid pellet of the parent ScIP PBE featuring the pendant pyridine disulfide units did not degrade over the same time frame (ca. 5 hours) demonstrating the advantage to rapid depolymerisation of solid-state materials that possess active side chains to permit self-immolation, in agreement with Phillips.
 | ||
| Scheme 93 The side chain-immolative polybenzylether system (ScIP) reported by Zhang and co-workers.346 | ||
4.3 Self-immolative poly(phthalaldehyde)s
Polyphthalaldehydes (PPAs), first reported in the late 1960s by Aso and Tagami,347,348 are known to degrade under acidic conditions.349,350 They have now, however, been modified351 with stimuli-responsive end-capping units by Seo and Phillips in order to introduce chemoselective self-immolative character to these materials. For example, the PPA 369 shown in Scheme 94a was generated from 1,2-benzenedicarboxaldehyde via anionic polymerisation and the reactive chain end capped with either an allyl or TBDMS group.351 This approach to self-immolative materials was based upon a PPA which had been used successfully in photoresist technologies by Willson and Fréchet352 and further refined for greater sensitivity by Ito and Willson.353,354 In the case of the PPA bearing the TBDMS end capping group 370, exposure to a solution of TBAF resulted in the cleavage of the silyl trigger leading to formation of the hemiacetal and subsequent depolymerisation within 15 minutes. As part of this study, it was demonstrated that the self-immolative degradation of this type of polymer was very effective in the solid state. Patterned films (approximately 0.5 mm thick) were cast where defined regions of the film (a cylinder) were composed of the polyphthalaldehyde 370 and the rest of the film was a made from control polyphthalaldehyde that did not feature an end group sensitive to fluoride ions. When the patterned composite film was exposed to a buffered solution of TBAF for a period of only fifteen minutes and then washed with diethyl ether (to remove the 1,2-benzenedicarboxaldehyde) a cylindrical hole in the plastic film was revealed (Scheme 94b). In a manner similar to PBEs, PPAs are known to undergo rapid depolymerisation because of a significant lowering in ceiling temperature (TC) when the capping group is cleaved (the TC of the hemiacetal is −40 °C). Further studies by the Phillips group led to the development of PPAs with triggering groups located at both ends of the polymer chains in order to optimise the degradation rates.355,356 | ||
| Scheme 94 a) Head-to-tail depolymerisation of PPAs 369 as described by Phillips and co-workers; (b) selective degradation of a cylindrical region of a patterned film featuring a self-immolative PPA 370 reactive to fluoride ion sources such as TBAF.351 | ||
Sen, Phillips and co-workers reported further advances in self-immolative PPAs by reporting self-powered microscale pumps which undergo depolymerisation with a fluoride stimulus.357 The depolymerisation of an insoluble polymer causes the release of highly soluble monomers simulating a concentration gradient capable of moving fluids on a micrometer-scale. It was further demonstrated that the pump could be activated (or turned “on”) with the enzyme β-D-glucuronidase by synthesizing a separate self-immolative spacer capable of releasing fluoride ions upon activation by the enzyme. Phillips and co-workers also developed core–shell microcapsules which resulted in head-to-tail depolymerisation in an amplified response to fluoride.358 The ability to employ PPAs in melt extrusion additive manufacturing processes is limited as a result of the thermal instability of linear PPAs. Phillips and co-workers proposed that polymer degradation may arise from non-specific cleavage of either the end-cap or the polymer backbone, or a combination of the two mechanisms.359,360 To overcome the issue of instability, Phillips and co-workers synthesised polymers with the addition of electron-withdrawing chloride groups para- to the benzylic acetals (poly(4,5-dichlorophthalaldehyde (PCl2PA)); by doing so, the formation of the oxocarbenium ion intermediates would be disfavoured. This hypothesis was proved as variants of PCl2PA featuring different trigger groups (371 and 372) was shown to be suitable for selective laser sintering and selective layers of the resultant 3D image (that were doped with different coloured dyes to aid visualisation of the degradative processes) could be degraded via controlled exposure to the different chemical triggering agents (Scheme 95).359
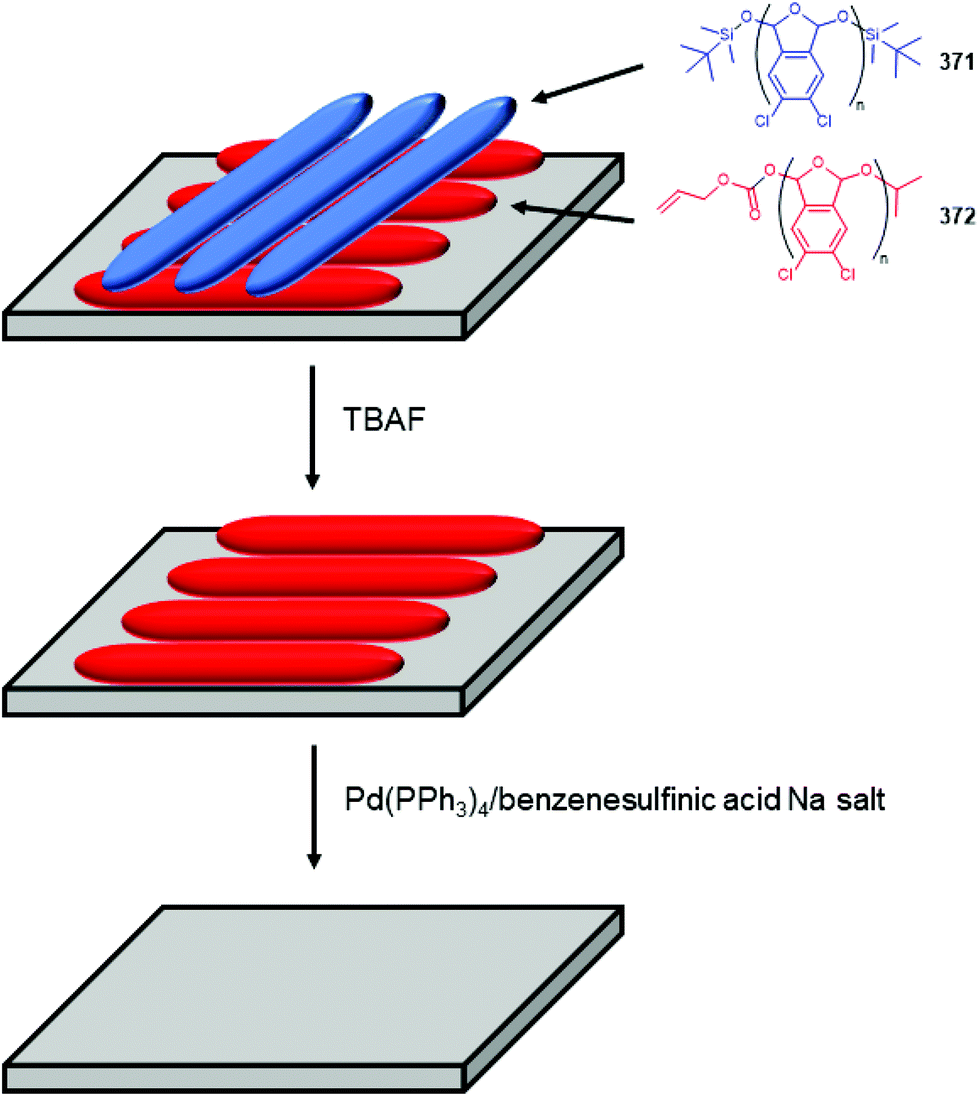 | ||
| Scheme 95 Selective laser-sintered 3D gratings of two PCl2Pas (371 and 372) that feature different triggering groups – exposure of the grating to a solution of TBAF leads to self-immolation and selective degradation of the blue coloured rods of the grating followed by the use of a Pd(0) source and the sodium salt of benzenesulfinic acid to trigger the conversion of the alloc-functionalised PCl2PA into soluble products.359 | ||
Moore and co-workers have reported the synthesis of PPAs, however, these systems did not include a stimuli-responsive end-cap but we include them here for contrast. These polymers could be depolymerised via acetal hydrolysis or mechanical degradation.361,362 In 2013 their studies led to inadvertent synthesis of a cyclic PPA (cPPA) 373via a cationic polymerization using a Lewis acid to generate high-molecular-weight polymer with high purity.363 They highlighted the increased stability of cPPA in comparison to PPA, noting that cPPAs are benchtop stable for several months whereas PPAs require diligent storage or risk untoward depolymerisation. Furthermore, it was demonstrated that the macrocycles could be opened with the addition of a Lewis acid and undergo ring expansion with the addition of more monomer. Interestingly, this phenomenon could be reversed resulted in ring contraction and was attributed to polymerization occurring close to the polymer ceiling temperature, rendering propagation and depropagation dependent on monomer concentration (Scheme 96). This interesting property was further explored by Moore and it was demonstrated that the scrambling of distinct homopolymer mixtures to copolymers under cationic polymerisation conditions occurred.364
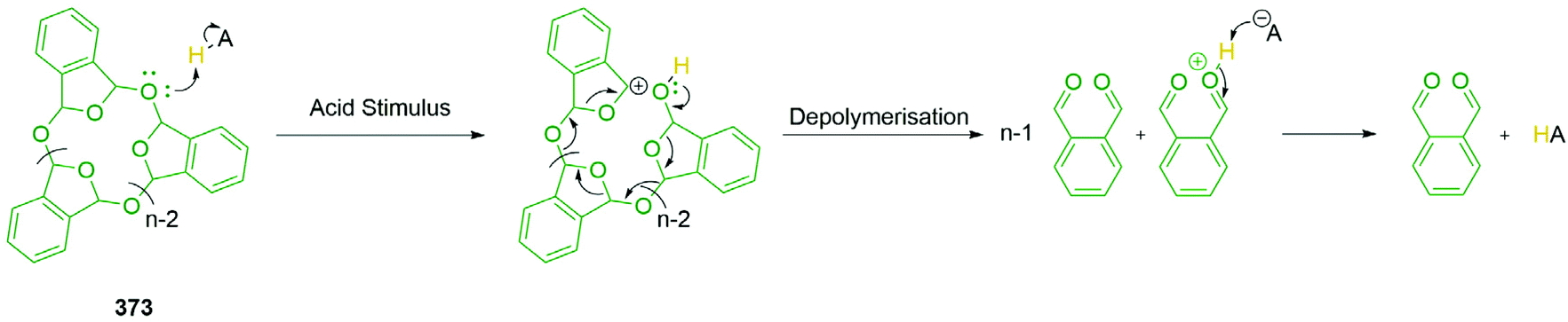 | ||
| Scheme 96 cPPA 373 and self-immolation in the presence of an acidic stimulus as described by Moore and co-workers.363 | ||
4.4 Self-immolative polyglyoxylates
Polyglyoxylates (PGs) are another class of self-immolative polymers that were reported by Gillies and co-workers in 2014.365 In a manner similar to PPHAs, without conjugation of an end-capping group, PGs undergo depolymerisation back to monomeric material via a hemiacetal mechanism. Gillies and co-workers reported the chain-growth polymerization of an ethyl glyoxylate followed by end-capping with the UV trigger, 4,5-dimethoxy-2-nitrobenzyl alcohol to afford poly(ethyl glyoxylate) (PetG) 374. Irradiation with UV light (300–350 nm) resulted in cleavage of the trigger unit that, in turn, led to depolymerisation with 70% degradation to ethyl glyoxylate hydrate after 24 hours in the solution state (Scheme 97). Photo-triggered degradation of 374 in the solid state was further explored with polymer films exposed to UV irradiation, then immersed in pH 7.4 phosphate buffer. Solid state depolymerisation proceeded slower but after 17 days the films of 374 had undergone complete degradation. Gillies and co-workers have demonstrated subsequently that PGs are versatile self-immolative polymers via the incorporation of different end-capping groups that cleave upon exposure to heat, hydrogen peroxide or DTT.162,365,366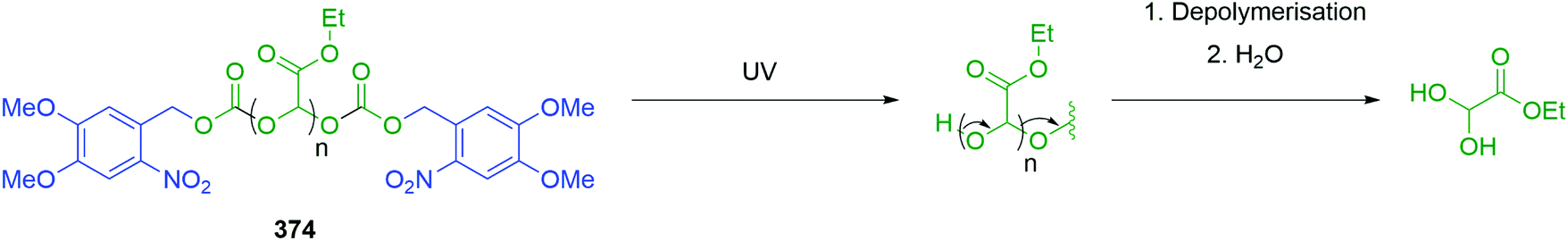 | ||
| Scheme 97 Head-to-tail depolymerisation of PetG 374 upon exposure to UV radiation, as described by Gillies and co-workers.365 | ||
More recently, Gillies and co-workers have reported the preparation and synthesis of self-immolative polyglyoxylamides (PGAms) 375; whilst retaining the desirable stimuli-responsive depolymerisation (Scheme 98), they were able to obtain these materials through monomer polymerisation or post-polymerisation amidation of PetG under mild conditions without degradation of the polymers.367 Furthermore, PGAms exhibit very different properties to PGs with higher glass transition (Tg) values as a result of hydrogen bonding from the amide motif (85 °C for PGAm-Net-MMT 375vs. −10 °C for PetG-MMT) and higher water solubility at ambient temperature. This increased water solubility of PGAms, compared to the predominantly hydrophobic PetG systems, allows for exploration of drug delivery applications. This work has also been reported with an application as kinetic hydrate inhibitors by Gillies and co-workers.368
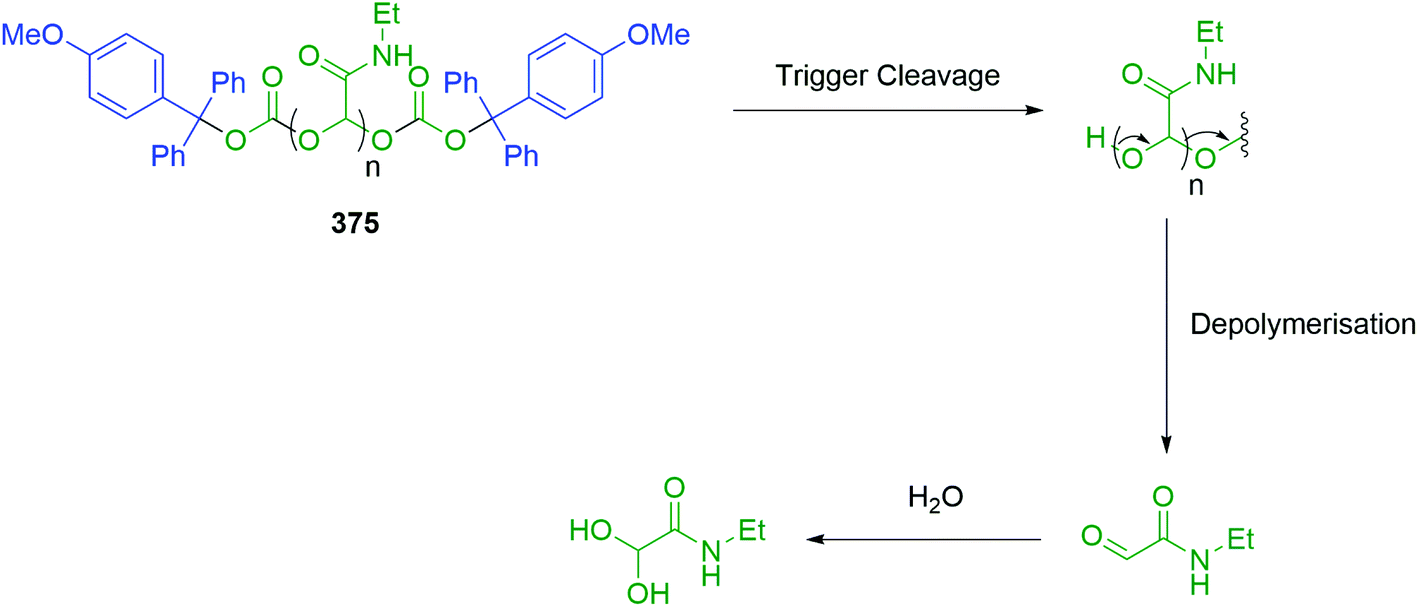 | ||
| Scheme 98 Head-to-tail depolymerisation of PGAm-Net-MMT 375 upon exposure to 0.9 M acetic acid as described by Gillies and co-workers.367 | ||
In another innovative development of self-immolative PG-based systems, Fan and Gillies have conjugated poly(ethyl glyoxylate) (PEtG) with poly(ethylene oxide) (PEO) to generate amphiphilic block copolymers 376–379. As a consequence of their amphiphilic nature, these block copolymers self-assemble to form nanoparticles in aqueous solution.369 The apolar interior of these nanoparticles enables uptake of hydrophobic payloads such as dyes (e.g. Nile Red) or anti-cancer drugs (e.g. doxorubicin or curcumin). Several different self-immolative linker end caps were used to conjugate the two polymers so that each end of PEtG was coupled to a polar PEG unit. The chemistries of the linker end caps permitted triggering via exposure to UV light, thiols, hydrogen peroxide or importantly, combinations of these stimuli (Scheme 99). The trigger event leads to selective degradation of the linker end caps, thus fragmenting the amphiphilic block copolymer and initiating self-immolation of the PEtG block; the net result of this process is disassembly of the block copolymer-based nanoparticles and release of the payload.
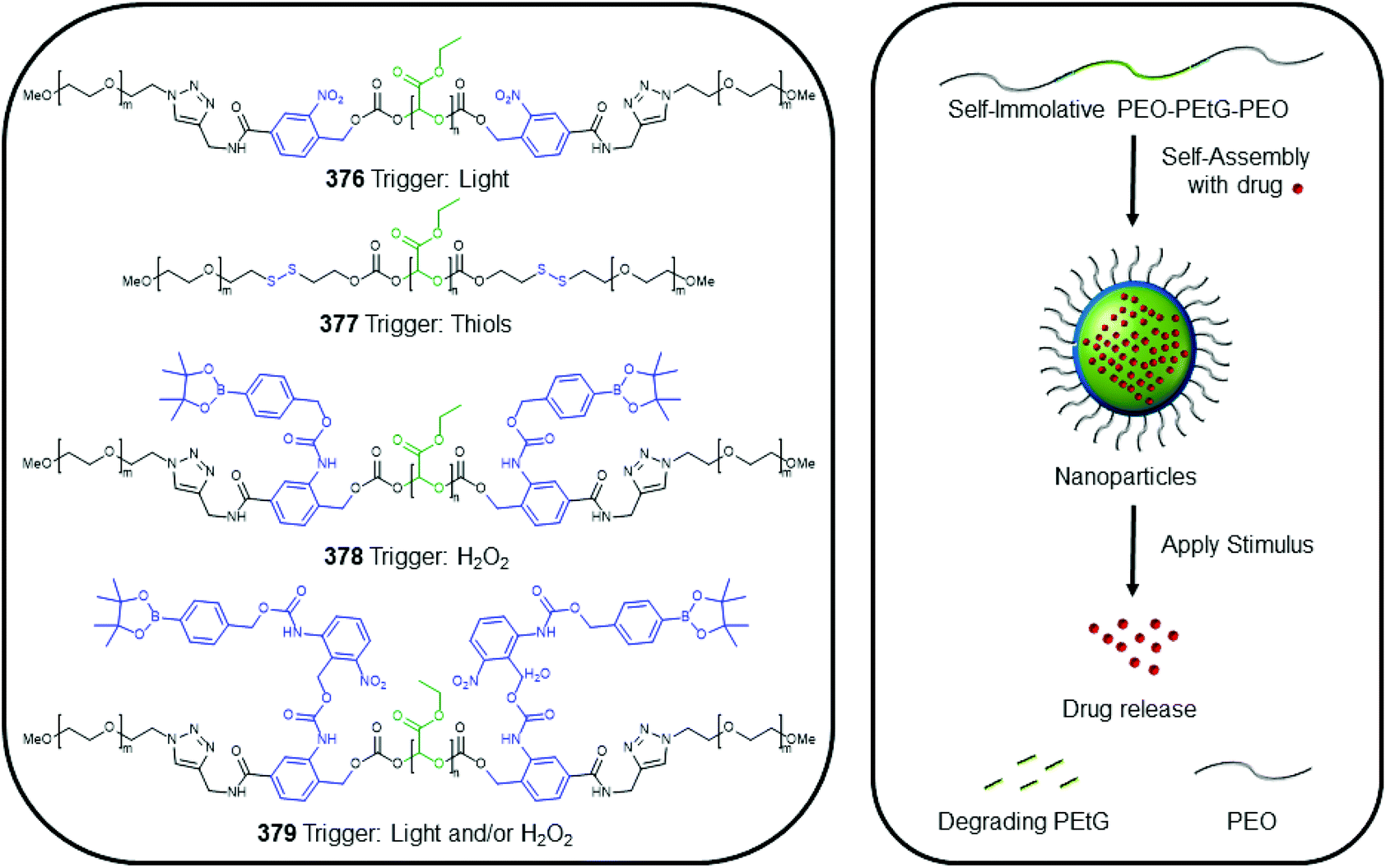 | ||
| Scheme 99 The structures of the PEtG-PEG block copolymers 376–379 that could be triggered via exposure to light, thiols or hydrogen peroxide and a schematic illustrating the self-assembly of nanoparticles plus the mode of payload delivery post application of the key stimulus.369 | ||
In a related approach, Gillies and co-workers have also developed175 thermo-responsive micelles and vesicles utilising a poly(ethyl glyoxylate) block copolymer system 380 equipped with an end cap on the PEtG unit that is susceptible to a retro-[4π + 2π] reaction, liberating a furan. This reaction fragments the hydrophilic and hydrophobic blocks of the copolymer; in addition, furans with this substitution pattern will cleave under the reaction conditions leading to end-to-end depolymerization (Scheme 100). Overall, the PEG-PEtG-PEG architecture with the thermally sensitive cycloadduct at the junction point between the different polymer blocks led to thermo-responsive micelles and vesicles that disassembled upon heating. In addition, stable assemblies could be generated that incorporated iron oxide nanoparticles within the hydrophobic core – exposure to magnetic fields led to localised heating within the thus triggering disassembly.
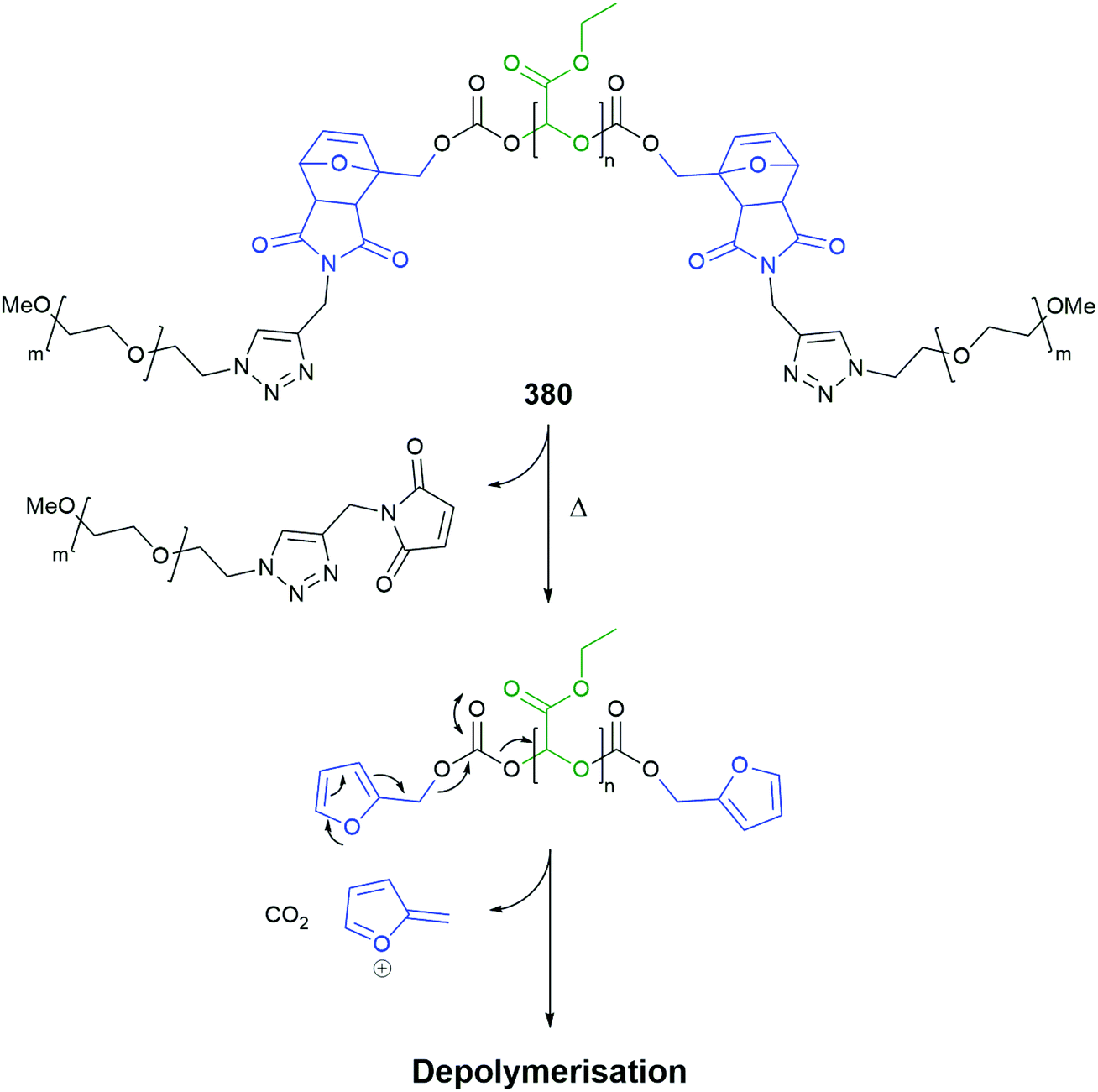 | ||
| Scheme 100 The PEG-PEtG-PEG block copolymer 380 reported by Gillies and co-workers that featured thermally degradable cycloadducts at the polymer block junctions.175 | ||
5 Self-immolative supramolecular systems
Developing their work on β-galactosidase triggered release of anticancer agents (Fig. 20), Papot et al. reported an inventive supramolecular drug delivery system based on a [2]-rotaxane.370 In 381, the taxol prodrug, with its esterase cleavable linker, is mechanically interlocked with a macrocyclic ring (Scheme 101). The macrocyclic ring has an in-built self-immolative linker triggered by β-galactosidase; on exposure to that enzyme, 1,6-elimination of the first, phenol-based linker is initiated to afford 382. Following this, the 2,4-substituted aniline undergoes either 1,6 or 1,4-elimination to fragment the locking ring, exposing the scissile bond of prodrug 383. Hence, this system falls into the category of SIEs that require two-trigger events to liberate the reporter group; with well-chosen triggering events, this has been shown to give improved targeting of the drug.40,217,229 The galactosidase trigger unit was also employed by Leigh, Aucagne and Papot in rotaxane-based peptide delivery systems (e.g. Met-enkephalin) as part of the stopper.371,372 The enhanced solubility of 381, as compared the taxol, owes much to the inclusion of a hydrophilic stopper, attached through a ‘click’ reaction. The stopper features hydrophilic glucose units and was pioneered in earlier work toward a drug delivery system for cyclopamine.373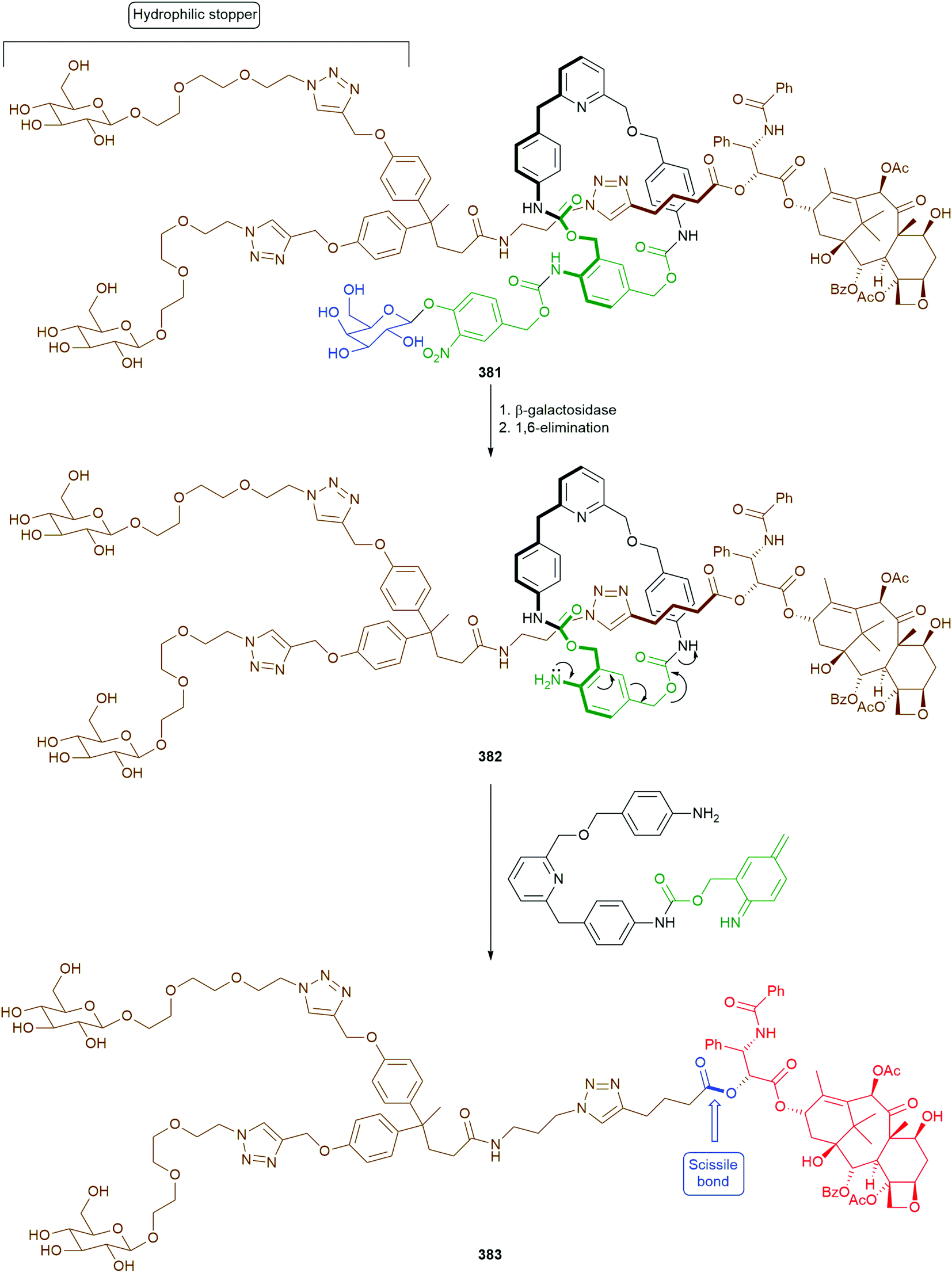 | ||
| Scheme 101 Papot's [2]-rotaxane-based drug delivery system 381 for intracellular delivery of taxol.370 | ||
6 Perspectives and concluding remarks
This update assessment of the field of self-immolative linker systems and their implementation into polymeric materials has revealed a significant degree of progress over the past decade – the number of publications has burgeoned with innovative uses of self-immolative linkers that undergo either an electronic cascade or a cyclisation event (or indeed a combination of both processes) following triggering events. There has been a natural progression from studies that targeted establishment of fundamentals of the self-immolative pathways to the application of these degradable materials. It is notable that self-immolative linkers have now found use within small molecular and macromolecular drug delivery or sensor systems that, with further development, can transition to commercially viable systems. The fate of the by-products of the self-immolative process have also been considered and an overview of their role in the reactive milieu is being gained – this aspect of these degradable systems has particular relevance in the context of the ability to recycle and re-use materials. However, more in-depth studies are needed to comprehensively understand the reactive pathway of these potentially reactive by-products to permit use of self-immolative polymers, especially in biological applications such as drug delivery systems or biosensors. Practical challenges to the commercial uptake of these materials still exist (especially relating to the synthetic complexity of many of the systems reported in this review) but it is not unreasonable to expect that within the next decade we will see the introduction into modern society of self-immolative materials in the form of ‘smart’ medicines, highly sensitive/selective sensor and security devices, or adhesives or coatings that can be debonded on demand to permit recycling of valuable components.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Dstl (contract DSTLX1000116213R) for A. G. G.References
- R. C. Elgersma, R. G. E. Coumans, T. Huijbregts, W. M. P. B. Menge, J. A. F. Joosten, H. J. Spijker, F. M. H. de Groot, M. M. C. van der Lee, R. Ubink, D. J. van den Dobbelsteen, D. F. Egging, W. H. A. Dokter, G. F. M. Verheijden, J. M. Lemmens, C. M. Timmers and P. H. Beusker, Mol. Pharmaceutics, 2015, 12, 1813–1835 CrossRef CAS PubMed.
- Y. Zhang, Q. Yin, L. Yin, L. Ma, L. Tang and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 6435–6439 CrossRef CAS PubMed.
- J. A. Sáez, B. Escuder and J. F. Miravet, Tetrahedron, 2010, 66, 2614–2618 CrossRef.
- H. Kim, H. Mohapatra and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 13063–13067 CrossRef CAS PubMed.
- T. S. Babra, A. Trivedi, C. N. Warriner, N. Bazin, D. Castiglione, C. Sivour, W. Hayes and B. W. Greenland, Polym. Chem., 2017, 8, 7207–7216 RSC.
- T. S. Babra, M. Wood, J. S. Godleman, S. Salimi, C. Warriner, N. Bazin, C. R. Siviour, I. W. Hamley, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 119, 260–271 CrossRef CAS.
- J. Yan, S. Lee, A. Zhang and J. Yoon, Chem. Soc. Rev., 2018, 47, 6900–6916 RSC.
- A. L. Acton, F. Leroux, A. Feula, K. Melia, M. R. Sambrook, W. Hayes and A. T. Russell, Chem. Commun., 2019, 55, 5219–5222 RSC.
- W. Tuo, J. Bouquet, F. Taran and T. Le Gall, Chem. Commun., 2019, 55, 8655–8658 RSC.
- X. Sun, J. F. Reuther, S. T. Phillips and E. V. Anslyn, Chem. – Eur. J., 2017, 23, 3903–3909 CrossRef CAS PubMed.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS PubMed.
- C. A. Blencowe, A. T. Russell, F. Greco, W. Hayes and D. W. Thornthwaite, Polym. Chem., 2011, 2, 773–790 RSC.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS.
- G. I. Peterson, M. B. Larsen and A. J. Boydston, Macromolecules, 2012, 45, 7317–7328 CrossRef CAS.
- A. D. Wong, M. A. DeWit and E. R. Gillies, Adv. Drug Delivery Rev., 2012, 64, 1031–1045 CrossRef CAS PubMed.
- N. Krall, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2013, 52, 1384–1402 CrossRef CAS PubMed.
- S. T. Phillips and A. M. Dilauro, ACS Macro Lett., 2014, 3, 298–304 CrossRef CAS PubMed.
- S. T. Phillips, J. S. Robbins, A. M. Dilauro and M. G. Olah, J. Appl. Polym. Sci., 2014, 131, 40992 CrossRef.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970–2984 CrossRef CAS PubMed.
- R. E. Wang, F. Costanza, Y. Niu, H. Wu, Y. Hu, W. Hang, Y. Sun and J. Cai, J. Controlled Release, 2012, 159, 154–163 CrossRef CAS PubMed.
- A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
- J. A. Kaitz, O. P. Lee and J. S. Moore, MRS Commun., 2015, 5, 191–204 CrossRef CAS.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2016, 116, 1309–1352 CrossRef CAS PubMed.
- S. Goggins and C. G. Frost, Analyst, 2016, 141, 3157–3218 RSC.
- Y. J. Wang, Y. Y. Li, X. Y. Liu, X. L. Lu, X. Cao and B. H. Jiao, Mar. Drugs, 2017, 15, 18 CrossRef PubMed.
- M. Gisbert-Garzarán, M. Manzano and M. Vallet-Regí, Chem. Eng. J., 2018, 340, 24–31 CrossRef.
- X. Sun, D. Shabat, S. T. Phillips and E. V. Anslyn, J. Phys. Org. Chem., 2018, 31, e3827 CrossRef PubMed.
- S. Gnaim, O. Green and D. Shabat, Chem. Commun., 2018, 54, 2073–2085 RSC.
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 6342–6360 CrossRef CAS.
- A. Dal Corso, L. Pignataro, L. Belvisi and C. Gennari, Chem. – Eur. J., 2019, 25, 14740–14757 CrossRef CAS PubMed.
- V. Saluja, A. Mankoo, G. K. Saraogi, M. M. Tambuwala and V. Mishra, J. Drug Delivery Sci. Technol., 2019, 52, 15–26 CrossRef CAS.
- N. Hananya and D. Shabat, ACS Cent. Sci., 2019, 5, 949–959 CrossRef CAS PubMed.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2019, 52, 2806–2817 CrossRef CAS PubMed.
- Q. E. A. Sirianni and E. R. Gillies, Polymer, 2020, 202, 122638 CrossRef CAS.
- Y. Xiao, X. Tan, Z. Li and K. Zhang, J. Mater. Chem. B, 2020, 8, 6697–6709 RSC.
- O. Shelef, S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS PubMed.
- Y. Xue, H. Bai, B. Peng, B. Fang, J. Baell, L. Li, W. Huang and N. H. Voelcker, Chem. Soc. Rev., 2021, 50, 4872–4931 RSC.
- C. M. Geiselhart, H. Mutlu and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2021, 60, 17290–17313 CrossRef CAS PubMed.
- A. Dal Corso, S. Arosio, N. Arrighetti, P. Perego, L. Belvisi, L. Pignataro and C. Gennari, Chem. Commun., 2021, 57, 7778–7781 RSC.
- P. K. Chakravarty, P. L. Carl, M. J. Weber and J. A. Katzenellenbogen, J. Med. Chem., 1983, 26, 638–644 CrossRef CAS PubMed.
- F. M. H. De Groot, L. W. A. Van Berkom and H. W. Scheeren, J. Med. Chem., 1999, 42, 5277–5283 CrossRef CAS PubMed.
- A. Kastrati and C. G. Bochet, J. Org. Chem., 2019, 84, 7776–7785 CrossRef CAS PubMed.
- S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2017, 139, 10002–10008 CrossRef CAS PubMed.
- L. Peles-Strahl, R. Sasson, G. Slor, N. Edelstein-Pardo, A. Dahan and R. J. Amir, Macromolecules, 2019, 52, 3268–3277 CrossRef CAS.
- N. Karton-Lifshin and D. Shabat, New J. Chem., 2012, 36, 386–393 RSC.
- E. M. Lloyd, H. Lopez Hernandez, E. C. Feinberg, M. Yourdkhani, E. K. Zen, E. B. Mejia, N. R. Sottos, J. S. Moore and S. R. White, Chem. Mater., 2019, 31, 398–406 CrossRef CAS.
- K. Yeung, H. Kim, H. Mohapatra and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 5324–5327 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim, T. N. Dell and M. M. Stevens, Chem. Sci., 2020, 11, 3713–3718 RSC.
- K. M. Schmid, L. Jensen and S. T. Phillips, J. Org. Chem., 2012, 77, 4363–4374 CrossRef CAS PubMed.
- B. M. Sykes, M. P. Hay, D. Bohinc-Herceg, N. A. Helsby, C. J. O’Connor and W. A. Denny, J. Chem. Soc., Perkin Trans. 1, 2000, 1601–1608 RSC.
- C. C. Song, R. Ji, F. S. Du and Z. C. Li, Macromolecules, 2013, 46, 8416–8425 CrossRef CAS.
- S. A. Nuñez, K. Yeung, N. S. Fox and S. T. Phillips, J. Org. Chem., 2011, 76, 10099–10113 CrossRef PubMed.
- F. Fakhari and S. E. Rokita, Nat. Commun., 2014, 5, 5591 CrossRef CAS PubMed.
- Y. S. Cho, S. W. Chung, H. R. Kim, T. H. Won, J. U. Choi, I. S. Kim, S. Y. Kim and Y. Byun, J. Controlled Release, 2019, 296, 241–249 CrossRef CAS PubMed.
- S. W. Chung, Y. S. Cho, J. U. Choi, H. R. Kim, T. H. Won, S. Y. Kim and Y. Byun, Biomaterials, 2019, 192, 109–117 CrossRef CAS PubMed.
- Y. Anami, C. M. Yamazaki, W. Xiong, X. Gui, N. Zhang, Z. An and K. Tsuchikama, Nat. Commun., 2018, 9, 2512 CrossRef PubMed.
- B. Q. Wei, J. Gunzner-Toste, H. Yao, T. Wang, J. Wang, Z. Xu, J. Chen, J. Wai, J. Nonomiya, S. P. Tsai, J. Chuh, K. R. Kozak, Y. Liu, S.-F. Yu, J. Lau, G. Li, G. D. Phillips, D. Leipold, A. Kamath, D. Su, K. Xu, C. Eigenbrot, S. Steinbacher, R. Ohri, H. Raab, L. R. Staben, G. Zhao, J. A. Flygare, T. H. Pillow, V. Verma, L. A. Masterson, P. W. Howard and B. Safina, J. Med. Chem., 2018, 61, 989–1000 CrossRef CAS PubMed.
- A. R. M. Dias, L. Bodero, A. Martins, D. Arosio, S. Gazzola, L. Belvisi, L. Pignataro, C. Steinkühler, A. Dal Corso, C. Gennari and U. Piarulli, ChemMedChem, 2019, 14, 938–942 CrossRef PubMed.
- P. D. Senter, W. E. Pearce and R. S. Greenfield, J. Org. Chem., 1990, 55, 2975–2978 CrossRef CAS.
- F. Groot, E. Damen and H. Scheeren, Curr. Med. Chem., 2012, 8, 1093–1122 CrossRef PubMed.
- R. Erez and D. Shabat, Org. Biomol. Chem., 2008, 6, 2669 RSC.
- I. Niculescu-Duvaz, D. Niculescu-Duvaz, F. Friedlos, R. Spooner, J. Martin, R. Marais and C. J. Springer, J. Med. Chem., 1999, 42, 2485–2489 CrossRef CAS PubMed.
- R. Perry, R. J. Amir and D. Shabat, New J. Chem., 2007, 31, 1307 RSC.
- E. Bouvier, S. Thirot, F. Schmidt and C. Monneret, Org. Biomol. Chem., 2003, 1, 3343–3352 RSC.
- J. C. Florent, X. Dong, G. Gaudel, S. Mitaku, C. Monneret, J. P. Gesson, J. C. Jacquesy, M. Mondon, B. Renoux, S. Andrianomenjanahary, S. Michel, M. Koch, F. Tillequin, M. Gerken, J. Czech, R. Straub and K. Bosslet, J. Med. Chem., 1998, 41, 3572–3581 CrossRef CAS PubMed.
- R. Perry-Feigenbaum, P. S. Baran and D. Shabat, Org. Biomol. Chem., 2009, 7, 4825–4828 RSC.
- M. P. Hay, B. M. Sykes, W. A. Denny and C. J. O’Connor, J. Chem. Soc., Perkin Trans. 1, 1999, 2759–2770 RSC.
- K. Matsuo, R. Kamada, K. Mizusawa, H. Imai, Y. Takayama, M. Narazaki, T. Matsuda, Y. Takaoka and I. Hamachi, Chem. – Eur. J, 2013, 19, 12875–12883 CrossRef CAS PubMed.
- J. Zhao, B. Zhang, Q. Mao, K. Ping, P. Zhang, F. Lin, D. Liu, Y. Feng, M. Sun, Y. Zhang, Q. H. Li, T. Zhang, Y. Mou and S. Wang, J. Med. Chem., 2022, 65, 4926–4948 CrossRef CAS PubMed.
- X.-b. Zhao, W. Ha, K. Gao and Y.-p. Shi, Anal. Chem., 2020, 92, 9039–9047 CrossRef CAS PubMed.
- B. E. Toki, C. G. Cerveny, A. F. Wahl and P. D. Senter, J. Org. Chem., 2002, 67, 1866–1872 CrossRef CAS PubMed.
- C. J. M. Stirling, Acc. Chem. Res., 1979, 12, 198–203 CrossRef CAS.
- P. J. Burke, P. D. Senter, D. W. Meyer, J. B. Miyamoto, M. Anderson, B. E. Toki, G. Manikumar, M. C. Wani, D. J. Kroll and S. C. Jeffrey, Bioconjugate Chem., 2009, 20, 1242–1250 CrossRef CAS PubMed.
- Y. L. Leu, C. S. Chen, Y. J. Wu and J. W. Chern, J. Med. Chem., 2008, 51, 1740–1746 CrossRef CAS PubMed.
- G. P. Yan, C. W. Ai, L. Li, R. F. Zong and F. Liu, Chin. Sci. Bull., 2010, 55, 3085–3093 CrossRef CAS.
- Y. L. Leu, S. R. Roffler and J. W. Chern, J. Med. Chem., 1999, 42, 3623–3628 CrossRef CAS PubMed.
- D. Zhang, H. Le, J. D. Cruz-Chuh, S. Bobba, J. Guo, L. Staben, C. Zhang, Y. Ma, K. R. Kozak, G. D. Lewis Phillips, B. S. Vollmar, J. D. Sadowsky, R. Vandlen, B. Wei, D. Su, P. Fan, P. S. Dragovich, S. C. Khojasteh, C. E. C. A. Hop and T. H. Pillow, Bioconjugate Chem., 2018, 29, 267–274 CrossRef CAS PubMed.
- R. Weinstain, P. S. Baran and D. Shabat, Bioconjugate Chem., 2009, 20, 1783–1791 CrossRef CAS PubMed.
- D. A. Rose, J. W. Treacy, Z. J. Yang, J. H. Ko, K. N. Houk and H. D. Maynard, J. Am. Chem. Soc., 2022, 144, 6050–6058 CrossRef CAS PubMed.
- G. C. Van De Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316–21321 CrossRef CAS PubMed.
- D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596–4597 CrossRef CAS PubMed.
- J. L. M. Jourden, K. B. Daniel and S. M. Cohen, Chem. Commun., 2011, 47, 7968–7970 RSC.
- R. A. Mosey and P. E. Floreancig, Org. Biomol. Chem., 2012, 10, 7980–7985 RSC.
- R. A. J. Smith, C. M. Porteous, C. V. Coulter and M. P. Murphy, Eur. J. Biochem., 1999, 263, 709–716 CrossRef CAS PubMed.
- A. Muratovska, R. N. Lightowlers, R. W. Taylor, J. A. Wilce and M. P. Murphy, Adv. Drug Delivery Rev., 2001, 49, 189–198 CrossRef CAS PubMed.
- D. V. Santi, E. L. Schneider and G. W. Ashley, J. Med. Chem., 2014, 57, 2303–2314 CrossRef CAS PubMed.
- R. V. Kolakowski, K. T. Haelsig, K. K. Emmerton, C. I. Leiske, J. B. Miyamoto, J. H. Cochran, R. P. Lyon, P. D. Senter and S. C. Jeffrey, Angew. Chem., Int. Ed., 2016, 55, 7948–7951 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, Z. Li, Z. Yuan, C. L. Organ, R. K. Trivedi, S. Wang, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 11749–11753 CrossRef CAS PubMed.
- J. Kang, S. Xu, M. N. Radford, W. Zhang, S. S. Kelly, J. J. Day and M. Xian, Angew. Chem., Int. Ed., 2018, 57, 5893–5897 CrossRef CAS PubMed.
- R. D. Hanna, Y. Naro, A. Deiters and P. E. Floreancig, J. Am. Chem. Soc., 2016, 138, 13353–13360 CrossRef CAS PubMed.
- Y. Kuang, K. Balakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- W. Chen, Y. Han and X. Peng, Chem. – Eur. J., 2014, 20, 7410–7418 CrossRef CAS PubMed.
- S. Cao, R. Christiansen and X. Peng, Chem. – Eur. J., 2013, 19, 9050–9058 CrossRef CAS PubMed.
- L. R. Staben, S. G. Koenig, S. M. Lehar, R. Vandlen, D. Zhang, J. Chuh, S. F. Yu, C. Ng, J. Guo, Y. Liu, A. Fourie-O'Donohue, M. Go, X. Linghu, N. L. Segraves, T. Wang, J. Chen, B. Wei, G. D. L. Phillips, K. Xu, K. R. Kozak, S. Mariathasan, J. A. Flygare and T. H. Pillow, Nat. Chem., 2016, 8, 1112–1119 CrossRef CAS PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Gonçalves, L. F. Veiros, H. F. Florindo and P. M. P. P. Gois, Angew. Chem., 2017, 129, 9474–9478 CrossRef.
- K. E. Broaders, S. Grandhe and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef CAS PubMed.
- Y. Zhao and M. D. Pluth, Angew. Chem., 2016, 128, 14638–14642 CrossRef PubMed.
- Y. Zhao, H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2017, 139, 16365–16376 CrossRef CAS PubMed.
- V. S. Khodade, B. M. Pharoah, N. Paolocci and J. P. Toscano, J. Am. Chem. Soc., 2020, 142, 4309–4316 CrossRef CAS PubMed.
- P. Chauhan, S. Jos and H. Chakrapani, Org. Lett., 2018, 20, 3766–3770 CrossRef CAS PubMed.
- A. K. Sharma, M. Nair, P. Chauhan, K. Gupta, D. K. Saini and H. Chakrapani, Org. Lett., 2017, 19, 4822–4825 CrossRef CAS PubMed.
- P. Štacko, L. Muchová, L. Vítek and P. Klán, Org. Lett., 2018, 20, 4907–4911 CrossRef PubMed.
- A. T. Dharmaraja, G. Ravikumar and H. Chakrapani, Org. Lett., 2014, 16, 2610–2613 CrossRef CAS PubMed.
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed.
- P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62–65 CrossRef CAS PubMed.
- A. K. Steiger, M. Marcatti, C. Szabo, B. Szczesny and M. D. Pluth, ACS Chem. Biol., 2017, 12, 2117–2123 CrossRef CAS PubMed.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Chem. Commun., 2018, 54, 4951–4954 RSC.
- P. Shukla, V. S. Khodade, M. Sharathchandra, P. Chauhan, S. Mishra, S. Siddaramappa, B. E. Pradeep, A. Singh and H. Chakrapani, Chem. Sci., 2017, 8, 4967–4972 RSC.
- C. R. Powell, J. C. Foster, S. N. Swilley, K. Kaur, S. J. Scannelli, D. Troya and J. B. Matson, Polym. Chem., 2019, 10, 2991–2995 RSC.
- A. K. Steiger, Y. Yang, M. Royzen and M. D. Pluth, Chem. Commun., 2017, 53, 1378–1380 RSC.
- K. A. Pardeshi, G. Ravikumar and H. Chakrapani, Org. Lett., 2018, 20, 4–7 CrossRef CAS PubMed.
- C. R. Powell, K. M. Dillon, Y. Wang, R. J. Carrazzone and J. B. Matson, Angew. Chem., Int. Ed., 2018, 57, 6324–6328 CrossRef CAS PubMed.
- P. Bora, P. Chauhan, K. A. Pardeshi and H. Chakrapani, RSC Adv., 2018, 8, 27359–27374 RSC.
- A. P. Schaap, T. Chen, R. S. Handley, R. DeSilva and B. P. Giri, Tetrahedron Lett., 1987, 28, 1155–1158 CrossRef CAS.
- A. P. Schaap, R. S. Handley and B. P. Giri, Tetrahedron Lett., 1987, 28, 935–938 CrossRef CAS.
- A. P. Schaap, M. D. Sandison and R. S. Handley, Tetrahedron Lett., 1987, 28, 1159–1162 CrossRef CAS.
- O. Green, T. Eilon, N. Hananya, S. Gutkin, C. R. Bauer and D. Shabat, ACS Cent. Sci., 2017, 3, 349–358 CrossRef CAS PubMed.
- T. Eilon-Shaffer, M. Roth-Konforti, A. Eldar-Boock, R. Satchi-Fainaro and D. Shabat, Org. Biomol. Chem., 2018, 16, 1708–1712 RSC.
- S. Gnaim, A. Scomparin, A. Eldar-Boock, C. R. Bauer, R. Satchi-Fainaro and D. Shabat, Chem. Sci., 2019, 10, 2945–2955 RSC.
- S. P. Gholap, C. Yao, O. Green, M. Babjak, P. Jakubec, T. Malatinský, J. Ihssen, L. Wick, U. Spitz and D. Shabat, Bioconjugate Chem., 2021, 32, 991–1000 CrossRef CAS PubMed.
- Z. Wang, H. Wu, P. Liu, F. Zeng and S. Wu, Biomaterials, 2017, 139, 139–150 CrossRef CAS PubMed.
- S. Gnaim, A. Scomparin, S. Das, R. Blau, R. Satchi-Fainaro and D. Shabat, Angew. Chem., Int. Ed., 2018, 57, 9033–9037 CrossRef CAS PubMed.
- M. Roth-Konforti, O. Green, M. Hupfeld, L. Fieseler, N. Heinrich, J. Ihssen, R. Vorberg, L. Wick, U. Spitz and D. Shabat, Angew. Chem., Int. Ed., 2019, 58, 10361–10367 CrossRef CAS PubMed.
- J. A. Richard, L. Jean, A. Romieu, M. Massonneau, P. Noack-Fraissignes and P. Y. Renard, Org. Lett., 2007, 9, 4853–4855 CrossRef CAS PubMed.
- J. A. Richard, L. Jean, C. Schenkels, M. Massonneau, A. Romieu and P. Y. Renard, Org. Biomol. Chem., 2009, 7, 2941–2957 RSC.
- S. Gutkin, S. Gandhesiri, A. Brik and D. Shabat, Bioconjugate Chem., 2021, 32, 2141–2147 CrossRef CAS PubMed.
- J. I. Scott, S. Gutkin, O. Green, E. J. Thompson, T. Kitamura, D. Shabat and M. Vendrell, Angew. Chem., Int. Ed., 2021, 60, 5699–5703 CrossRef CAS PubMed.
- S. Gutkin, O. Green, G. Raviv, D. Shabat and O. Portnoy, Bioconjugate Chem., 2020, 31, 2488–2493 CrossRef CAS PubMed.
- B. M. Babin, G. Fernandez-Cuervo, J. Sheng, O. Green, A. A. Ordonez, M. L. Turner, L. J. Keller, S. K. Jain, D. Shabat and M. Bogyo, ACS Cent. Sci., 2021, 7, 803–814 CrossRef CAS PubMed.
- M. Ponomariov, D. Shabat and O. Green, Bioconjugate Chem., 2021, 32, 2134–2140 CrossRef CAS PubMed.
- O. Shelef, A. C. Sedgwick, S. Pozzi, O. Green, R. Satchi-Fainaro, D. Shabat and J. L. Sessler, Chem. Commun., 2021, 57, 11386–11389 RSC.
- M. J. Lobba, C. Fellmann, A. M. Marmelstein, J. C. Maza, E. N. Kissman, S. A. Robinson, B. T. Staahl, C. Urnes, R. J. Lew, C. S. Mogilevsky, J. A. Doudna and M. B. Francis, ACS Cent. Sci., 2020, 6, 1564–1571 CrossRef CAS PubMed.
- X. Wu, L. Li, W. Shi, Q. Gong and H. Ma, Angew. Chem., Int. Ed., 2016, 55, 14728–14732 CrossRef CAS PubMed.
- H. Li, W. Liu, F. Zhang, X. Zhu, L. Huang and H. Zhang, Anal. Chem., 2018, 90, 855–858 CrossRef CAS PubMed.
- X. Chen, X. Ma, Y. Zhang, G. Gao, J. Liu, X. Zhang, M. Wang and S. Hou, Anal. Chim. Acta, 2018, 1033, 193–198 CrossRef CAS PubMed.
- S. Ye, J. J. Hu and D. Yang, Angew. Chem., Int. Ed., 2018, 57, 10173–10177 CrossRef CAS PubMed.
- S. Ye, N. Hananya, O. Green, H. Chen, A. Q. Zhao, J. Shen, D. Shabat and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 14326–14330 CrossRef CAS PubMed.
- S. Ye, B. Yang, M. Wu, Z. Chen, J. Shen, D. Shabat and D. Yang, CCS Chem., 2021, 3, 2181–2188 CrossRef.
- S. Das, J. Ihssen, L. Wick, U. Spitz and D. Shabat, Chem. – Eur. J., 2020, 26, 3647–3652 CrossRef CAS PubMed.
- K. J. Bruemmer, R. R. Walvoord, T. F. Brewer, G. Burgos-Barragan, N. Wit, L. B. Pontel, K. J. Patel and C. J. Chang, J. Am. Chem. Soc., 2017, 139, 5338–5350 CrossRef CAS PubMed.
- T. F. Brewer, G. Burgos-Barragan, N. Wit, K. J. Patel and C. J. Chang, Chem. Sci., 2017, 8, 4073–4081 RSC.
- W. Liu, C. Truillet, R. R. Flavell, T. F. Brewer, M. J. Evans, D. M. Wilson and C. J. Chang, Chem. Sci., 2016, 7, 5503–5507 RSC.
- K. J. Bruemmer, O. Green, T. A. Su, D. Shabat and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 7508–7512 CrossRef CAS PubMed.
- X. Wu, W. Shi, X. Li and H. Ma, Angew. Chem., Int. Ed., 2017, 56, 15319–15323 CrossRef CAS PubMed.
- K. Gorska and N. Winssinger, Angew. Chem., Int. Ed., 2013, 52, 6820–6843 CrossRef CAS PubMed.
- T. N. Grossmann, A. Strohbach and O. Seitz, ChemBioChem, 2008, 9, 2185–2192 CrossRef CAS PubMed.
- A. P. Silverman and E. T. Kool, Chem. Rev., 2006, 106, 3775–3789 CrossRef CAS PubMed.
- K. Gorska, A. Manicardi, S. Barluenga and N. Winssinger, Chem. Commun., 2011, 47, 4364–4366 RSC.
- A. Shibata, Y. Ito and H. Abe, Chem. Commun., 2013, 49, 270–272 RSC.
- A. Alouane, R. Labruère, T. Le Saux, I. Aujard, S. Dubruille, F. Schmidt and L. Jullien, Chem. – Eur. J., 2013, 19, 11717–11724 CrossRef CAS PubMed.
- K. K. Sadhu and N. Winssinger, Chem. – Eur. J., 2013, 19, 8182–8189 CrossRef CAS PubMed.
- M. Roth and O. Seitz, Chem. – Eur. J., 2021, 27, 14189–14194 CrossRef CAS PubMed.
- R. Labruère, A. Alouane, T. Le Saux, I. Aujard, P. Pelupessy, A. Gautier, S. Dubruille, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2012, 51, 9344–9347 CrossRef PubMed.
- M. Staderini, A. Gambardella, A. Lilienkampf and M. Bradley, Org. Lett., 2018, 20, 3170–3173 CrossRef CAS PubMed.
- C. Zang, H. Wang, T. Li, Y. Zhang, J. Li, M. Shang, J. Du, Z. Xi and C. Zhou, Chem. Sci., 2019, 10, 8973–8980 RSC.
- B. A. Hess and L. J. Schaad, Tetrahedron Lett., 1977, 18, 535–538 CrossRef.
- J. S. Robbins, K. M. Schmid and S. T. Phillips, J. Org. Chem., 2013, 78, 3159–3169 CrossRef CAS PubMed.
- S. Goggins, B. J. Marsh, A. T. Lubben and C. G. Frost, Chem. Sci., 2015, 6, 4978–4985 RSC.
- H. Wang, M. B. Cleary, L. C. Lewis, J. W. Bacon, P. Caravan, H. S. Shafaat and E. M. Gale, Angew. Chem., 2022, 61, e202114019 CAS.
- R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553–555 RSC.
- E. W. P. Damen, T. J. Nevalainen, T. J. M. Van den Bergh, F. M. H. De Groot and H. W. Scheeren, Bioorg. Med. Chem., 2002, 10, 71–77 CrossRef CAS PubMed.
- F. M. H. De Groot, W. J. Loos, R. Koekkoek, L. W. A. Van Berkom, G. F. Busscher, A. E. Seelen, C. Albrecht, P. De Bruijn and H. W. Scheeren, J. Org. Chem., 2001, 66, 8815–8830 CrossRef CAS PubMed.
- P. Bertrand and J. P. Gesson, J. Org. Chem., 2007, 72, 3596–3599 CrossRef CAS PubMed.
- C. A. Blencowe, D. W. Thornthwaite, W. Hayes and A. T. Russell, Org. Biomol. Chem., 2015, 13, 8703–8707 RSC.
- C. A. Blencowe, PhD Thesis, University of Reading, UK, 2012.
- T. N. Forder, P. G. Maschmeyer, H. Zeng and D. A. Roberts, Chem. – Asian J., 2021, 16, 287–291 CrossRef CAS PubMed.
- P. G. Maschmeyer, X. Liang, A. Hung, O. Ahmadzai, A. L. Kenny, Y. C. Luong, T. N. Forder, H. Zeng, E. R. Gillies and D. A. Roberts, Polym. Chem., 2021, 12, 6824–6831 RSC.
- H. Kunz, Chem. Ber., 1976, 109, 2670–2683 CrossRef CAS.
- D. Chantreux, J. P. Gamet, R. Jacquier and J. Verducci, Tetrahedron, 1984, 40, 3087–3094 CrossRef CAS.
- A. G. Gavriel, F. Leroux, G. S. Khurana, V. G. Lewis, A. M. Chippindale, M. R. Sambrook, W. Hayes and A. T. Russell, J. Org. Chem., 2021, 86, 10263–10279 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- K. Neumann, A. Gambardella, A. Lilienkampf and M. Bradley, Chem. Sci., 2018, 9, 7198–7203 RSC.
- B. Fan, J. F. Trant, G. Hemery, O. Sandre and E. R. Gillies, Chem. Commun., 2017, 53, 12068–12071 RSC.
- K. Neumann, S. Jain, A. Gambardella, S. E. Walker, E. Valero, A. Lilienkampf and M. Bradley, ChemBioChem, 2017, 18, 91–95 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 7013–7016 CrossRef CAS PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. Ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- S. Davies, B. L. Oliveira and G. J. L. Bernardes, Org. Biomol. Chem., 2019, 17, 5725–5730 RSC.
- J. M. Mejia Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef CAS PubMed.
- R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. Ten Hoeve, H. M. Janssen, A. H. A. M. van Onzen, P. J. Hudson and M. S. Robillard, Nat. Commun., 2018, 9, 1484 CrossRef PubMed.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- W. S. C. Ngai, S. Yang, X. Zeng, Y. Liu, F. Lin, X. Wang, H. Zhang, X. Fan and P. R. Chen, J. Am. Chem. Soc., 2022, 144, 5411–5417 CrossRef CAS PubMed.
- S. Dadhwal, J. M. Fairhall, S. K. Goswami, S. Hook and A. B. Gamble, Chem. – Asian J., 2019, 14, 1143–1150 CrossRef CAS PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- S. S. Matikonda, J. M. Fairhall, F. Fiedler, S. Sanhajariya, R. A. J. Tucker, S. Hook, A. L. Garden and A. B. Gamble, Bioconjugate Chem., 2018, 29, 324–334 CrossRef CAS PubMed.
- R. van Brakel, R. C. M. Vulders, R. J. Bokdam, H. Grüll and M. S. Robillard, Bioconjugate Chem., 2008, 19, 714–718 CrossRef CAS PubMed.
- A. S. Cohen, E. A. Dubikovskaya, J. S. Rush and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 8563–8565 CrossRef CAS PubMed.
- M. Xu, J. Tu and R. M. Franzini, Chem. Commun., 2017, 53, 6271–6274 RSC.
- M. Xu, R. Galindo-Murillo, T. E. Cheatham, III and R. M. Franzini, Org. Biomol. Chem., 2017, 15, 9855–9865 RSC.
- J. Tu, D. Svatunek, S. Parvez, H. J. Eckvahl, M. Xu, R. T. Peterson, K. N. Houk and R. M. Franzini, Chem. Sci., 2020, 11, 169–179 RSC.
- J. Tu, M. Xu, S. Parvez, R. T. Peterson and R. M. Franzini, J. Am. Chem. Soc., 2018, 140, 8410–8414 CrossRef CAS PubMed.
- H. Stöckmann, A. A. Neves, S. Stairs, K. M. Brindle and F. J. Leeper, Org. Biomol. Chem., 2011, 9, 7303–7305 RSC.
- B. P. Imming, R. Mohr, E. Müller, W. Overheu and G. Seitz, Angew. Chem., Int. Ed. Engl., 1982, 21, 284 CrossRef.
- Z. Shao, W. Liu, H. Tao, F. Liu, R. Zeng, P. A. Champagne, Y. Cao, K. N. Houk and Y. Liang, Chem. Commun., 2018, 54, 14089–14092 RSC.
- K. Haba, M. Popkov, M. Shamis, R. A. Lerner, C. F. Barbas and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 716–720 CrossRef CAS PubMed.
- S. Huvelle, A. Alouane, T. Le Saux, L. Jullien and F. Schmidt, Org. Biomol. Chem., 2017, 15, 3435–3443 RSC.
- Y. Meyer, J. A. Richard, M. Massonneau, P. Y. Renard and A. Romieu, Org. Lett., 2008, 10, 1517–1520 CrossRef CAS PubMed.
- Y. Meyer, J. A. Richard, B. Delest, P. Noack, P. Y. Renard and A. Romieu, Org. Biomol. Chem., 2010, 8, 1777–1780 RSC.
- R. J. Amir and D. Shabat, Chem. Commun., 2004, 4, 1614–1615 RSC.
- I. Artaud and E. Galardon, ChemBioChem, 2014, 15, 2361–2364 CrossRef CAS PubMed.
- J. Tang, Z. Zeng, J. Yan, C. Chen, J. Liu and X. Feng, J. Controlled Release, 2019, 307, 90–97 CrossRef CAS PubMed.
- W. A. Henne, D. D. Doorneweerd, A. R. Hilgenbrink, S. A. Kularatne and P. S. Low, Bioorg. Med. Chem. Lett., 2006, 16, 5350–5355 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- A. R. M. Dias, A. Pina, A. Dean, H.-G. Lerchen, M. Caruso, F. Gasparri, I. Fraietta, S. Troiani, D. Arosio, L. Belvisi, L. Pignataro, A. Dal Corso and C. Gennari, Chem. – Eur. J., 2019, 25, 1696–1700 CrossRef PubMed.
- R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494–4499 CrossRef CAS PubMed.
- N. Fomina, C. L. Mc Fearin and A. Almutairi, Chem. Commun., 2012, 48, 9138–9140 RSC.
- A. Dal Corso, V. Borlandelli, C. Corno, P. Perego, L. Belvisi, L. Pignataro and C. Gennari, Angew. Chem., Int. Ed., 2020, 59, 4176–4181 CrossRef CAS PubMed.
- M. A. Dewit and E. R. Gillies, Org. Biomol. Chem., 2011, 9, 1846–1854 RSC.
- T. Yamamoto, D. R. Caldwell, A. Gandioso and M. J. Schnermann, Photochem. Photobiol., 2019, 95, 951–958 CAS.
- R. R. Nani, A. P. Gorka, T. Nagaya, T. Yamamoto, J. Ivanic, H. Kobayashi and M. J. Schnermann, ACS Cent. Sci., 2017, 3, 329–337 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed.
- R. R. Nani, J. A. Kelley, J. Ivanic and M. J. Schnermann, Chem. Sci., 2015, 6, 6556–6563 RSC.
- A. P. Gorka, R. R. Nani, J. Zhu, S. Mackem and M. J. Schnermann, J. Am. Chem. Soc., 2014, 136, 14153–14159 CrossRef CAS PubMed.
- W. Feng, C. Gao, W. Liu, H. Ren, C. Wang, K. Ge, S. Li, G. Zhou, H. Li, S. Wang, G. Jia, Z. Li and J. Zhang, Chem. Commun., 2016, 52, 9434–9437 RSC.
- N. Singh, A. Gupta, P. Prasad, R. K. Sah, A. Singh, S. Kumar, S. Singh, S. Gupta and P. K. Sasmal, J. Med. Chem., 2021, 64, 17813–17823 CrossRef CAS PubMed.
- N. Gagey, P. Neveu, C. Benbrahim, B. Goetz, I. Aujard, J.-B. Baudin and L. Jullien, J. Am. Chem. Soc., 2007, 129, 9986–9998 CrossRef CAS PubMed.
- S. Chen, X. Zhao, J. Chen, J. Chen, L. Kuznetsova, S. S. Wong and I. Ojima, Bioconjugate Chem., 2010, 21, 979–987 CrossRef CAS PubMed.
- D. Shan, M. G. Nicolaou, R. T. Borchardt and B. Wang, J. Pharm. Sci., 1997, 86, 765–767 CrossRef CAS PubMed.
- P. B. Carvalho and M. A. Avery, J. Med. Chem., 2001, 14, 369–378 Search PubMed.
- O. A. Okoh and P. Klahn, ChemBioChem, 2018, 19, 1668–1694 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef CAS PubMed.
- B. Yu, Y. Zheng, Z. Yuan, S. Li, H. Zhu, L. K. De La Cruz, J. Zhang, K. Ji, S. Wang and B. Wang, J. Am. Chem. Soc., 2018, 140, 30–33 CrossRef CAS PubMed.
- R. Wang, K. Cai, H. Wang, C. Yin and J. Cheng, Chem. Commun., 2018, 54, 4878–4881 RSC.
- P. Liu, J. Xu, D. Yan, P. Zhang, F. Zeng, B. Li and S. Wu, Chem. Commun., 2015, 51, 9567–9570 RSC.
- B. Li, P. Liu, D. Yan, F. Zeng and S. Wu, J. Mater. Chem. B, 2017, 5, 2635–2643 RSC.
- Z. Chen, B. Li, X. Xie, F. Zeng and S. Wu, J. Mater. Chem. B, 2018, 6, 2547–2556 RSC.
- S. Son, M. Won, O. Green, N. Hananya, A. Sharma, Y. Jeon, J. H. Kwak, J. L. Sessler, D. Shabat and J. S. Kim, Angew. Chem., Int. Ed., 2019, 58, 1739–1743 CrossRef CAS PubMed.
- N. Hananya, J. P. Reid, O. Green, M. S. Sigman and D. Shabat, Chem. Sci., 2019, 10, 1380–1385 RSC.
- B. Prasai, W. C. Silvers and R. L. McCarley, Anal. Chem., 2015, 87, 6411–6418 CrossRef CAS PubMed.
- S. U. Hettiarachchi, B. Prasai and R. L. McCarley, J. Am. Chem. Soc., 2014, 136, 7575–7578 CrossRef CAS PubMed.
- C. Ji and M. J. Miller, Bioorg. Med. Chem., 2012, 20, 3828–3836 CrossRef CAS PubMed.
- W. Wang and B. Wang, Chem. Commun., 2017, 53, 10124–10127 RSC.
- J. C. Kern, D. Dooney, R. Zhang, L. Liang, P. E. Brandish, M. Cheng, G. Feng, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, Y. Sun and R. M. Garbaccio, Bioconjugate Chem., 2016, 27, 2081–2088 CrossRef CAS PubMed.
- M. W. Becker, H. H. Chapman, T. Cihlar, E. J. Eisenberg, G. X. He, M. R. Kernan, W. A. Lee, E. J. Prisbe, J. C. Rohloff and M. L. Sparacino, WO2002008241, 2002.
- Y. Wei, G. Qiu, B. Lei, L. Qin, H. Chu, Y. Lu, G. Zhu, Q. Gao, Q. Huang, G. Qian, P. Liao, X. Luo, X. Zhang, C. Zhang, Y. Li, S. Zheng, Y. Yu, P. Tang, J. Ni, P. Yan, Y. Zhou, P. Li, X. Huang, A. Gong and J. Liu, J. Med. Chem., 2017, 60, 8580–8590 CrossRef CAS PubMed.
- C. McGuigan, R. N. Pathirana, N. Mahmood, K. G. Devine and A. J. Hay, Antiviral Res., 1992, 17, 311–321 CrossRef CAS PubMed.
- C. McGuigan, R. N. Pathirana, N. Mahmood and A. J. Hay, Bioorg. Med. Chem. Lett., 1992, 2, 701–704 CrossRef CAS.
- C. McGuigan, R. N. Pathirana, S. S. M. Choi, D. Kinchington and T. J. O'Connor, Antiviral Chem. Chemother., 1993, 4, 97–101 CrossRef CAS.
- D. Cahard, C. McGuigan and J. Balzarini, Mini-Rev. Med. Chem., 2004, 4, 371–381 CrossRef CAS PubMed.
- Y. Mehellou, J. Balzarini and C. McGuigan, ChemMedChem, 2009, 4, 1779–1791 CrossRef CAS PubMed.
- C. J. Choy, J. J. Geruntho, A. L. Davis and C. E. Berkman, Bioconjugate Chem., 2016, 27, 824–830 CrossRef CAS PubMed.
- B. S. Backer, C. J. Choy, A. L. Davis, Z. S. Browne and C. E. Berkman, Tetrahedron Lett., 2020, 61, 151650 CrossRef CAS PubMed.
- C. J. Choy, C. R. Ley, A. L. Davis, B. S. Backer, J. J. Geruntho, B. H. Clowers and C. E. Berkman, Bioconjugate Chem., 2016, 27, 2206–2213 CrossRef CAS PubMed.
- F. P. Olatunji, J. W. Herman, B. N. Kesic, D. Olabode and C. E. Berkman, Tetrahedron Lett., 2020, 61, 152398 CrossRef CAS PubMed.
- F. P. Olatunji, B. N. Kesic, C. J. Choy and C. E. Berkman, Bioorg. Med. Chem. Lett., 2019, 29, 2571–2574 CrossRef CAS PubMed.
- P. Šimon, M. Tichotová, M. García Gallardo, E. Procházková and O. Baszczyňski, Chem. – Eur. J., 2021, 27, 12763–12775 CrossRef PubMed.
- M. Ximenis, A. Sampedro, L. Martínez-Crespo, G. Ramis, F. Orvay, A. Costa and C. Rotger, Chem. Commun., 2021, 57, 2736–2739 RSC.
- F. M. H. De Groot, C. Albrecht, R. Koekkoek, P. H. Beusker and H. W. Scheeren, Angew. Chem., Int. Ed., 2003, 42, 4490–4494 CrossRef CAS PubMed.
- S. Li, M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 10516–10517 CrossRef CAS PubMed.
- M. Shamis, H. N. Lode and D. Shabat, J. Am. Chem. Soc., 2004, 126, 1726–1731 CrossRef CAS PubMed.
- M. Shamis and D. Shabat, Chem. – Eur. J., 2007, 13, 4523–4528 CrossRef CAS PubMed.
- M. Avital-Shmilovici and D. Shabat, Bioorg. Med. Chem. Lett., 2009, 19, 3959–3962 CrossRef CAS PubMed.
- N. Pessah, M. Reznik, M. Shamis, F. Yantiri, H. Xin, K. Bowdish, N. Shomron, G. Ast and D. Shabat, Bioorg. Med. Chem., 2004, 12, 1859–1866 CrossRef CAS PubMed.
- O. Redy, E. Kisin-Finfer, E. Sella and D. Shabat, Org. Biomol. Chem., 2012, 10, 710–715 RSC.
- I. S. Turan and E. U. Akkaya, Org. Lett., 2014, 16, 1680–1683 CrossRef CAS PubMed.
- J. Sloniec, U. Resch-Genger and A. Hennig, J. Phys. Chem. B, 2013, 117, 14336–14344 CrossRef CAS PubMed.
- K. Gill, X. Mei and E. R. Gillies, Chem. Commun., 2021, 57, 11072–11075 RSC.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS PubMed.
- W. S. Shin, J. Han, P. Verwilst, R. Kumar, J. H. Kim and J. S. Kim, Bioconjugate Chem., 2016, 27, 1419–1426 CrossRef CAS PubMed.
- M. Grinda, J. Clarhaut, I. Tranoy-Opalinski, B. Renoux, A. Monvoisin, L. Cronier and S. Papot, ChemMedChem, 2011, 6, 2137–2141 CrossRef CAS PubMed.
- M. Grinda, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski and S. Papot, MedChemComm, 2012, 3, 68–70 RSC.
- M. Grinda, T. Legigan, J. Clarhaut, E. Peraudeau, I. Tranoy-Opalinski, B. Renoux, M. Thomas, F. Guilhot and S. Papot, Org. Biomol. Chem., 2013, 11, 7129–7133 RSC.
- T. Legigan, J. Clarhaut, I. Tranoy-Opalinski, A. Monvoisin, B. Renoux, M. Thomas, A. Le Pape, S. Lerondel and S. Papot, Angew. Chem., Int. Ed., 2012, 51, 11606–11610 CrossRef CAS PubMed.
- B. Renoux, F. Raes, T. Legigan, E. Péraudeau, B. Eddhif, P. Poinot, I. Tranoy-Opalinski, J. Alsarraf, O. Koniev, S. Kolodych, S. Lerondel, A. Le Pape, J. Clarhaut and S. Papot, Chem. Sci., 2017, 8, 3427–3433 RSC.
- T. Legigan, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski, A. Monvoisin, J.-M. Berjeaud, F. Guilhot and S. Papot, J. Med. Chem., 2012, 55, 4516–4520 CrossRef CAS PubMed.
- R. Châtre, J. Lange, E. Péraudeau, P. Poinot, S. Lerondel, A. Le Pape, J. Clarhaut, B. Renoux and S. Papot, J. Controlled Release, 2020, 327, 19–25 CrossRef PubMed.
- E. Sella and D. Shabat, J. Am. Chem. Soc., 2009, 131, 9934–9936 CrossRef CAS PubMed.
- C. M. Niemeyer, M. Adler and R. Wacker, Trends Biotechnol., 2005, 23, 208–216 CrossRef CAS PubMed.
- J. Y. Hyo and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 11590–11591 CrossRef PubMed.
- T. Yoshii, S. Onogi, H. Shigemitsu and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 3360–3365 CrossRef CAS PubMed.
- K. Porte, B. Renoux, E. Péraudeau, J. Clarhaut, B. Eddhif, P. Poinot, E. Gravel, E. Doris, A. Wijkhuisen, D. Audisio, S. Papot and F. Taran, Angew. Chem., Int. Ed., 2019, 58, 6366–6370 CrossRef CAS PubMed.
- H. Mohapatra, K. M. Schmid and S. T. Phillips, Chem. Commun., 2012, 48, 3018–3020 RSC.
- K. Yeung, K. M. Schmid and S. T. Phillips, Chem. Commun., 2013, 49, 394–396 RSC.
- A. D. Brooks, K. Yeung, G. G. Lewis and S. T. Phillips, Anal. Methods, 2015, 7, 7186–7192 RSC.
- H. Mohapatra and S. T. Phillips, Chem. Commun., 2013, 49, 6134–6136 RSC.
- R. Perry-Feigenbaum, E. Sella and D. Shabat, Chem. – Eur. J., 2011, 17, 12123–12128 CrossRef CAS PubMed.
- M. S. Baker and S. T. Phillips, J. Am. Chem. Soc., 2011, 133, 5170–5173 CrossRef CAS PubMed.
- M. S. Baker and S. T. Phillips, Org. Biomol. Chem., 2012, 10, 3595–3599 RSC.
- J. A. Gu, V. Mani and S. T. Huang, Analyst, 2015, 140, 346–352 RSC.
- H. Kim, M. S. Baker and S. T. Phillips, Chem. Sci., 2015, 6, 3388–3392 RSC.
- M. S. Baker, V. Yadav, A. Sen and S. T. Phillips, Angew. Chem., Int. Ed., 2013, 52, 10295–10299 CrossRef CAS PubMed.
- H. Mohapatra, H. Kim and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 12498–12501 CrossRef CAS PubMed.
- S. Gnaim and D. Shabat, Chem. Commun., 2018, 54, 2655–2658 RSC.
- I. S. Turan, O. Seven, S. Ayan and E. U. Akkaya, ACS Omega, 2017, 2, 3291–3295 CrossRef CAS PubMed.
- S. Hisamatsu, S. Suzuki, S. Kohmoto, K. Kishikawa, Y. Yamamoto, R. Motokawa and T. Yaita, Tetrahedron, 2017, 73, 3993–3998 CrossRef CAS.
- L. Gabrielli and F. Mancin, J. Org. Chem., 2016, 81, 10715–10720 CrossRef CAS PubMed.
- X. Sun, S. D. Dahlhauser and E. V. Anslyn, J. Am. Chem. Soc., 2017, 139, 4635–4638 CrossRef CAS PubMed.
- X. Sun and E. V. Anslyn, Angew. Chem., Int. Ed., 2017, 56, 9522–9526 CrossRef CAS PubMed.
- X. Sun, A. A. Boulgakov, L. N. Smith, P. Metola, E. M. Marcotte and E. V. Anslyn, ACS Cent. Sci., 2018, 4, 854–861 CrossRef CAS PubMed.
- D. H. Lee, S. A. Valenzuela, M. N. Dominguez, M. Otsuka, D. J. Milliron and E. V. Anslyn, Cell Rep. Phys. Sci., 2021, 2, 100552 CrossRef CAS PubMed.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 8, 5434–5435 CrossRef PubMed.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS PubMed.
- E. Sella and D. Shabat, Chem. Commun., 2008, 5701 RSC.
- A. Warnecke and F. Kratz, J. Org. Chem., 2008, 73, 1546–1552 CrossRef CAS PubMed.
- M. F. Nichol, K. D. Clark, N. D. Dolinski and J. R. de Alaniz, Polym. Chem., 2019, 10, 4914–4919 RSC.
- A. P. Esser-Kahn, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 10266–10268 CrossRef CAS PubMed.
- G. Liu, X. Wang, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2014, 136, 7492–7497 CrossRef CAS PubMed.
- V. Kumar, J. T. Harris, A. Ribbe, M. Franc, Y. Bae, A. J. McNeil and S. Thayumanavan, ACS Macro Lett., 2020, 9, 377–381 CrossRef CAS.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Macromolecules, 2013, 46, 5177–5183 CrossRef CAS.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Anal. Chem., 2013, 85, 10432–10439 CrossRef CAS PubMed.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Chem. Commun., 2014, 50, 5352–5354 RSC.
- O. Redy and D. Shabat, J. Controlled Release, 2012, 164, 276–282 CrossRef CAS PubMed.
- G. I. Peterson, D. C. Church, N. A. Yakelis and A. J. Boydston, Polymer, 2014, 55, 5980–5985 CrossRef CAS.
- S. Kostler, B. Zechne, B. Trathnigg, H. Fasl, W. Kern and V. Ribitsch, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1499–1509 CrossRef.
- G. Liu, G. Zhang, J. Hu, X. Wang, M. Zhu and S. Liu, J. Am. Chem. Soc., 2015, 137, 11645–11655 CrossRef CAS PubMed.
- M. Brasch, I. K. Voets, M. S. T. Koay and J. L. M. Cornelissen, Faraday Discuss., 2013, 166, 47–57 RSC.
- H. Okada, K. Tanaka, W. Ohashi and Y. Chujo, Bioorg. Med. Chem., 2014, 22, 3435–3440 CrossRef CAS PubMed.
- J. Tan, Z. Deng, C. Song, J. Xu, Y. Zhang, Y. Yu, J. Hu and S. Liu, J. Am. Chem. Soc., 2021, 143, 13738–13748 CrossRef CAS PubMed.
- J. Huang, X. Chen, Y. Jiang, C. Zhang, S. He, H. Wang and K. Pu, Nat. Mater., 2022, 21, 598–607 CrossRef CAS PubMed.
- Y. He, Y. Wu, M. Zhang, Y. Zhang, H. Ding and K. Zhang, Macromolecules, 2021, 54, 5797–5805 CrossRef CAS.
- P. Hodge, Chem. Rev., 2014, 114, 2278–2312 CrossRef CAS PubMed.
- P. Hodge and H. M. Colquhoun, Polym. Adv. Technol., 2005, 16, 84–94 CrossRef CAS.
- M. A. DeWit and E. R. Gillies, J. Am. Chem. Soc., 2009, 131, 18327–18334 CrossRef CAS PubMed.
- E. K. Y. Chen, R. A. McBride and E. R. Gillies, Macromolecules, 2012, 45, 7364–7374 CrossRef CAS.
- R. A. McBride and E. R. Gillies, Macromolecules, 2013, 46, 5157–5166 CrossRef CAS.
- A. D. Wong, T. M. Güngör and E. R. Gillies, ACS Macro Lett., 2014, 3, 1191–1195 CrossRef CAS PubMed.
- H. Mutlu, C. M. Geiselhart and C. Barner-Kowollik, Mater. Horiz., 2018, 5, 162–183 RSC.
- N. Fomina, C. L. McFearin, M. Sermsakdi, J. M. Morachis and A. Almutairi, Macromolecules, 2011, 44, 8590–8597 CrossRef CAS PubMed.
- C. de Gracia Lux, C. L. McFearin, S. Joshi-Barr, J. Sankaranarayanan, N. Fomina and A. Almutairi, ACS Macro Lett., 2012, 1, 922–926 CrossRef CAS PubMed.
- C. de Gracia Lux and A. Almutairi, ACS Macro Lett., 2013, 2, 432–435 CrossRef CAS PubMed.
- J. Olejniczak, M. Chan and A. Almutairi, Macromolecules, 2015, 48, 3166–3172 CrossRef CAS.
- C. de Gracia Lux, J. Olejniczak, N. Fomina, M. L. Viger and A. Almutairi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3783–3790 CrossRef CAS.
- Y. Zhang, L. Ma, X. Deng and J. Cheng, Polym. Chem., 2013, 4, 224–228 RSC.
- Y. Xu, S. Sen, Q. Wu, X. Zhong, R. H. Ewoldt and S. C. Zimmerman, Chem. Sci., 2020, 11, 3326–3331 RSC.
- Y. Xu, E. G. Morado and S. C. Zimmerman, Polym. Chem., 2020, 11, 6215–6220 RSC.
- C. Petit, J. Bachmann, L. Michalek, Y. Catel, E. Blasco, J. P. Blinco, A. N. Unterreiner and C. Barner-Kowollik, Chem. Commun., 2021, 57, 2911–2914 RSC.
- H. Kim, A. D. Brooks, A. M. Dilauro and S. T. Phillips, J. Am. Chem. Soc., 2020, 142, 9447–9452 CrossRef CAS PubMed.
- K. M. Schmid and S. T. Phillips, J. Phys. Org. Chem., 2013, 26, 608–610 CrossRef CAS.
- Z. Zhou and R. G. Parr, J. Am. Chem. Soc., 1989, 111, 7371–7379 CrossRef CAS.
- B. A. Hess, L. J. Schaad and C. W. Holyoke, Tetrahedron, 1972, 28, 3657–3667 CrossRef CAS.
- C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef CAS PubMed.
- S. D. Dahlhauser, P. R. Escamilla, A. N. Vandewalle, J. T. York, R. M. Rapagnani, J. S. Shei, S. A. Glass, J. N. Coronado, S. R. Moor, D. P. Saunders and E. V. Anslyn, J. Am. Chem. Soc., 2020, 142, 2744–2749 CrossRef CAS PubMed.
- L. J. Zhang, X. X. Deng, F. S. Du and Z. C. Li, Macromolecules, 2013, 46, 9554–9562 CrossRef CAS.
- S. D. Dahlhauser, S. R. Moor, M. S. Vera, J. T. York, P. Ngo, A. J. Boley, J. N. Coronado, Z. B. Simpson and E. V. Anslyn, Cell Rep. Phys. Sci., 2021, 2, 100393 CrossRef CAS PubMed.
- M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 15688–15689 CrossRef CAS PubMed.
- A. Ortiz, C. S. Shanahan, D. T. Sisk, S. C. Perera, P. Rao and D. V. McGrath, J. Org. Chem., 2010, 75, 6154–6162 CrossRef CAS PubMed.
- R. M. Kevwitch, C. S. Shanahan and D. V. McGrath, New J. Chem., 2012, 36, 492–505 RSC.
- M. G. Olah, J. S. Robbins, M. S. Baker and S. T. Phillips, Macromolecules, 2013, 46, 5924–5928 CrossRef CAS.
- M. S. Baker, H. Kim, M. G. Olah, G. G. Lewis and S. T. Phillips, Green Chem., 2015, 17, 4541–4545 RSC.
- C. Ergene and E. F. Palermo, Biomacromolecules, 2017, 18, 3400–3409 CrossRef CAS PubMed.
- C. Ergene and E. F. Palermo, J. Mater. Chem. B, 2018, 6, 7217–7229 RSC.
- Y. Xiao, H. Li, B. Zhang, Z. Cheng, Y. Li, X. Tan and K. Zhang, Macromolecules, 2018, 51, 2899–2905 CrossRef CAS PubMed.
- Y. Xiao, Y. Li, B. Zhang, H. Li, Z. Cheng, J. Shi, J. Xiong, Y. Bai and K. Zhang, ACS Macro Lett., 2019, 8, 399–402 CrossRef CAS.
- C. Aso and S. Tagami, Macromolecules, 1969, 2, 414–419 CrossRef CAS.
- C. Aso, S. Tagami and T. Kunitake, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 497–511 CrossRef CAS.
- H. Lopez Hernandez, S. K. Kang, O. P. Lee, S. W. Hwang, J. A. Kaitz, B. Inci, C. W. Park, S. Chung, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2014, 26, 7637–7642 CrossRef PubMed.
- K. M. Lee, O. Phillips, A. Engler, P. A. Kohl and B. P. Rand, ACS Appl. Mater. Interfaces, 2018, 10, 28062–28068 CrossRef CAS PubMed.
- W. Seo and S. T. Phillips, J. Am. Chem. Soc., 2010, 132, 9234–9235 CrossRef CAS PubMed.
- S. A. MacDonald, C. G. Willson and J. M. J. Fréchet, Acc. Chem. Res., 1994, 27, 151–158 CrossRef CAS.
- H. Ito, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3863–3870 CrossRef CAS.
- H. Ito and C. G. Willson, Polym. Eng. Sci., 1983, 23, 1012–1018 CrossRef CAS.
- A. M. Dilauro, J. S. Robbins and S. T. Phillips, Macromolecules, 2013, 46, 2963–2968 CrossRef CAS.
- A. M. Dilauro, H. Zhang, M. S. Baker, F. Wong, A. Sen and S. T. Phillips, Macromolecules, 2013, 46, 7257–7265 CrossRef CAS.
- H. Zhang, K. Yeung, J. S. Robbins, R. A. Pavlick, M. Wu, R. Liu, A. Sen and S. T. Phillips, Angew. Chem., Int. Ed., 2012, 51, 2400–2404 CrossRef CAS PubMed.
- A. M. Dilauro, A. Abbaspourrad, D. A. Weitz and S. T. Phillips, Macromolecules, 2013, 46, 3309–3313 CrossRef CAS.
- A. M. Dilauro, G. G. Lewis and S. T. Phillips, Angew. Chem., Int. Ed., 2015, 54, 6200–6205 CrossRef CAS PubMed.
- A. M. Dilauro and S. T. Phillips, Polym. Chem., 2015, 6, 3252–3258 RSC.
- J. A. Kaitz and J. S. Moore, Macromolecules, 2013, 46, 608–612 CrossRef CAS.
- J. A. Kaitz, C. M. Possanza, Y. Song, C. E. Diesendruck, A. J. H. Spiering, E. W. Meijer and J. S. Moore, Polym. Chem., 2014, 5, 3788–3794 RSC.
- J. A. Kaitz, C. E. Diesendruck and J. S. Moore, J. Am. Chem. Soc., 2013, 135, 12755–12761 CrossRef CAS PubMed.
- J. A. Kaitz, C. E. Diesendruck and J. S. Moore, Macromolecules, 2013, 46, 8121–8128 CrossRef CAS.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed.
- B. Fan, J. F. Trant and E. R. Gillies, Macromolecules, 2016, 49, 9309–9319 CrossRef CAS.
- Q. E. A. Sirianni, A. R. Kenaree and E. R. Gillies, Macromolecules, 2019, 52, 262–270 CrossRef CAS.
- L. H. S. Ree, Q. E. A. Sirianni, E. R. Gillies and M. A. Kelland, Energy Fuels, 2019, 33, 2067–2075 CrossRef CAS.
- B. Fan and E. R. Gillies, Mol. Pharmaceutics, 2017, 14, 2548–2559 CrossRef CAS PubMed.
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS PubMed.
- A. Fernandes, A. Viterisi, V. Aucagne, D. A. Leigh and S. Papot, Chem. Commun., 2012, 48, 2083–2085 RSC.
- B. Renoux, T. Legigan, S. Bensalma, C. Chadéneau, J.-M. Muller and S. Papot, Org. Biomol. Chem., 2011, 9, 8459–8464 RSC.
| This journal is © The Royal Society of Chemistry 2022 |