
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTaking advantage of β-hydroxy amine enhanced reactivity and functionality for the synthesis of dual covalent adaptable networks†
Dimitri
Berne
,
Guilhem
Coste
,
Roberto
Morales-Cerrada
,
Marine
Boursier
,
Julien
Pinaud
 ,
Vincent
Ladmiral
* and
Sylvain
Caillol
*
,
Vincent
Ladmiral
* and
Sylvain
Caillol
*
ICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: vincent.ladmiral@enscm.fr; sylvain.caillol@enscm.fr
First published on 8th June 2022
Abstract
This study demonstrates the advantage of using β-hydroxy amine monomers for the synthesis of CANs. The increased reactivity of β-hydroxy amines towards the aza-Michael addition compared to their alkyl equivalents was highlighted by kinetic analyses coupled with rheological experiments. New catalyst-free covalent adaptable networks (CANs) were thus synthesized by poly aza-Michael addition using either β-hydroxy amine or its non-hydroxy analog. These CANs exhibit dynamic aza-Michael exchange under thermal stimulus. The synergistic effect of exchange reactions was highlighted by stress-relaxation and frequency sweep analyses. CANs were finally reshaped and their chemical and physical properties were compared to the initial ones.
Introduction
Polymers are designed according to the targeted applications and can be classified into three categories: thermoplastics, thermosets and elastomers. Thermoplastics are composed of entangled linear polymer chains, and are reprocessable by thermal treatment and soluble in suitable solvents. In contrast, thermosets are three-dimensional networks composed of polymer chains cross-linked by covalent bonds, giving these materials strong mechanical properties and chemical resistance. However, thermosets are not easily recycled, which is opposed to the current environmental trend of reducing or eliminating waste production.1 In this context, covalent adaptable networks (CANs) allowing the production of “recyclable thermosets” are gaining increasing attention. These polymer materials are three-dimensional networks but include exchangeable bonds giving them reprocessing properties under pH, light or thermal stimuli.2–4The concept of CAN was pioneered decades ago5–7 and revisited recently8 on a cross-linked rubber that incorporated disulfide linkages as reversible covalent bonds enabling the reprocessing of the polymer network. Numerous exchange reactions have been developed to broaden the scope of CANs, as reported in recent reviews.9,10 Among them, Diels–Alder addition,11–14 thiol–ene addition,15,16 triazolinediones (TAD) chemistry,17,18 transesterification,19–21 transamination,22,23 transcarbamoylation,24 transimination,25 and boronate metathesis26 have been the most studied so far. Other CANs have also been based on disulfide exchange,27,28 alkoxyamine bond dissociation,29 transalkylation,30 transacetalation,31 transamidation32 or acid exchange.33,34
Most of these exchange reactions usually require catalysts to be activated under thermal stimulus.35 The presence of these catalysts can potentially cause leaching, ageing and sintering issues.36,37 To avoid the presence of catalyst in CANs, neighbouring group participation (NGP) and internal catalysis strategies (often via electronic effects) have been recently developed.9,38 For instance, transesterification rate has been accelerated by the presence of a hydroxyl group in β-position of the ester function in epoxy Zn-catalyzed vitrimers.2,39 Transesterification was also promoted by the incorporation of carboxylic or sulfonic acids close to the ester function (although this modification can lead to a dissociative mechanism).40,41 Our group also recently highlighted the possibility to use fluorine groups as accelerating electrostatic substituents in transesterification vitrimers.42 Other strategies consist in the incorporation of amino groups in CANs to promote the transesterification of di(o-aminophenylboronic)43 and silyl-ether exchange44 for example. Finally, Du Prez et al. interestingly demonstrated that the presence of β-amino ester allows catalyst-free transesterification via NGP.45
Recently, Dichtel and Elling reported that the use of vitrimers alone would not solve the current industrial recycling challenges and that CANs coupling associative and dissociative mechanisms should be developed to achieve these objectives.46 In this context, some authors have interestingly developed materials using associative and dissociative exchange reactions to decrease both relaxation and reprocessing times. For instance, Zhang et al. developed a CAN combining disulfide metathesis and carboxylate transesterification.47 Thiol chemistry has also been used to develop materials combining associative and dissociative pathways.48,49 Lately, the reversible character of the aza-Michael addition was demonstrated by kinetic experiments on model molecules.45 This new exchange reaction was then synergistically used with transesterification in CANs produced from pentaerythritol triacrylate and commercial diamines.45 This initial work demonstrated the potential of aza-Michael exchange but was in a way structurally limited by the need of hydroxyl group on acrylate monomers. Moreover, the parameters influencing aza-Michael addition have not been explored in this initial study.
Our group recently studied the reactivity of β-hydroxy amines and demonstrated their higher reactivity compared to regular amines. This effect was observed for the addition of amine on both epoxy50 and cyclocarbonate.51 Moreover, β-hydroxy amines can easily be obtained from biobased epoxy monomers. Here, inspired by the work of Du Prez et al.,45 β-hydroxy amines were used to prepare CANs containing β-amino esters as they represent an innovative way to bring hydroxyl group in a transesterification material. First, the reactivity of β-hydroxy amines toward the aza-Michael addition was kinetically evaluated from a molecular and macromolecular point of view, enabling to gain understanding on the activation of this reaction. Then, aza-Michael addition was employed to synthesize new CAN capable of transesterification and exchange via aza-Michael additions. Next, insolubility, reshaping and relaxation properties were studied for this dual aza-Michael/transesterification CAN.
Results and discussion
Kinetic study
Kinetic studies were carried out with model molecules in order to determine the influence of β-hydroxy amine on the aza-Michael addition rate. Two model molecules were chosen: n-butylamine (Scheme 1a) and 1-amino-2-butanol (Scheme 1b). Butylacrylate was selected as Michael acceptor since it exhibits a high reactivity (notably higher than that of methacrylates) in aza-Michael addition among α,β-unsaturated carbonyl-containing compounds.52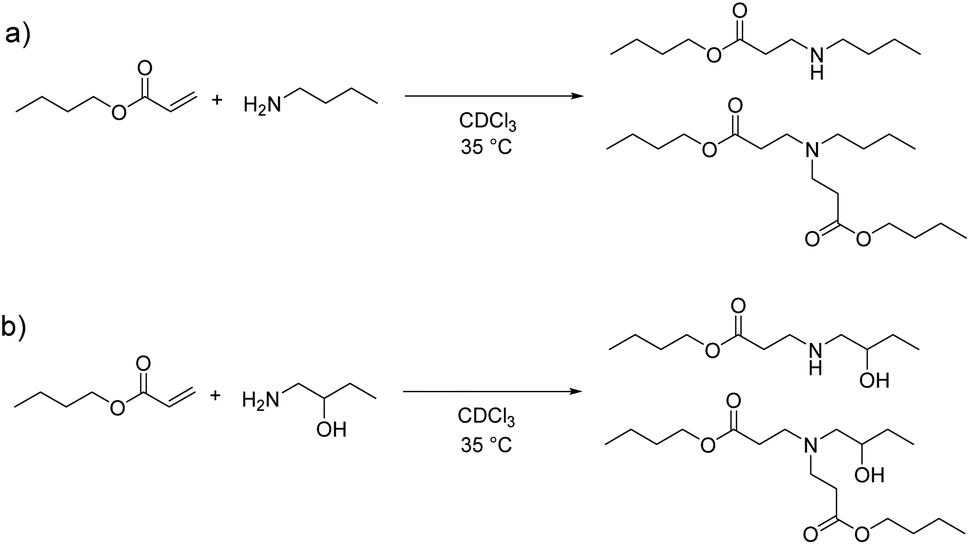 | ||
| Scheme 1 Reaction of butylacrylate with (a) n-butylamine and (b) 1-amino-2-butanol in the presence of 1,3,5-trioxane (internal standard) in CDCl3 at 35 °C. | ||
First, reactions were performed using acrylate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine stoichiometric ratios 2:1 since both primary and secondary amines can react with acrylate double bonds. In addition, to study the influence of an excess of amine protons compare to the acrylate function, a kinetic study was carried out with excess amine (acrylate:amine 1:1). Reagents, equivalents of each reagent and the conversion of the acrylate functions after 15 h of reaction are summarized in Table 1.
:amine stoichiometric ratios 2:1 since both primary and secondary amines can react with acrylate double bonds. In addition, to study the influence of an excess of amine protons compare to the acrylate function, a kinetic study was carried out with excess amine (acrylate:amine 1:1). Reagents, equivalents of each reagent and the conversion of the acrylate functions after 15 h of reaction are summarized in Table 1.
| Entry | n-Butylamine (eq.) | 1-Amino-2-butanol (eq.) | Butylacrylate (eq.) | Ethanol (eq.) | Conversion at t = 15 h (%) |
|---|---|---|---|---|---|
| 1 | 1 | 0 | 2 | 0 | 37 |
| 2 | 0 | 1 | 2 | 0 | 52 |
| 3 | 1 | 0 | 2 | 1 | 55 |
| 4 | 1 | 0 | 1 | 0 | 27 |
| 5 | 0 | 1 | 1 | 0 | 43 |
The disappearance of the double bond of the acrylate at 35 °C was monitored by 1H NMR spectroscopy (e.g. Fig. S1†). Additionally, the 1H NMR peaks assignment of the adducts formed during the aza-Michael reaction of butylacrylate and n-butylamine (entry 4) are exhibited in Fig. 1. As shown in Fig. 2, the conversion of the butylacrylate double bond via aza-Michael addition with n-butylamine (entry 1) progressed slower than with 1-amino-2-butanol (entry 2). After 5 h of reaction, the β-hydroxy amine led to almost 40% conversion of acrylate bonds, whereas the reaction with n-butylamine only reached 20% conversion. The reaction of n-butylamine and butylacrylate was also carried out in the presence of ethanol (2:1:1 butylacrylate:n-butylamine:ethanol, entry 3). At the beginning of the reaction, the β-hydroxy amine reacted faster than n-butylamine in the presence of ethanol. After 7.5 h, the trend was reversed and the n-butylamine/ethanol mixture led to slightly higher conversion than 1-amino-2-butanol. Nevertheless the acrylate reacted faster when hydroxyl groups were present in the reaction medium. These results show that hydroxy groups accelerate the Michael addition of amines on acrylates but whether they are located on the beta carbon of the amines or free in solution (as alcohol) may not be significant. Accordingly, two mechanisms can be accounted for the acceleration of the reaction by the OH group: (i) activation of the carbonyl by H-bonding or (ii) participation of the OH in the proton transfer step similarly to the cyclocarbonate/β-hydroxy amine addition.51 In any case, these results suggest that the use of β-hydroxy amine could allow fast aza-Michael addition in bulk thanks to the catalytic effect of the hydroxyl group.
 | ||
| Fig. 1 1H NMR spectrum of the reaction media corresponding to entry 4 (CDCl3, 400 MHz) at t = 24 h. | ||
 | ||
| Fig. 2 Conversion of n-butylacrylate double bonds versus time in the presence of n-butylamine (entry 1, black square), 1-amino-2-butanol (entry 2, red dot) or n-butylamine with EtOH (entry 3, blue triangle). All these reactions were carried out using one equivalent of acrylate functions and one equivalent of N–H bonds. | ||
Fig. 3 displays the evolution of the acrylate double bond conversion with time during the aza-Michael addition of n-butylamine or 1-amino-2-butanol carried out at different acrylate/amine molar ratios (entries 1, 2, 4 and 5 in Table 1). One primary amine function can react twice with an acrylate. Regardless of the amine, the aza-Michael additions carried out with one equivalent of acrylate functions per one equivalent of N–H bonds (entries 1 and 2) were faster than their counterpart performed using a 1:2 acrylate:N–H bonds ratio from n-butylamine (entries 4 and 5). This expected behavior is explained by the higher concentration of aza-Michael acceptor, which has a strong influence on the kinetics. On the other hand, the kinetic study performed with one equivalent of butylacrylate and two equivalents of N–H bonds of 1-amino-2-butanol (entry 5) remains faster than the one performed with one equivalent of N–H functions of n-butylamine and one equivalent of butylacrylate (entry 1). Thus, stoichiometric amounts of acrylates and N–H bonds (entries 1 and 2) were chosen to carry out the synthesis of the materials presented in this work.
 | ||
| Fig. 3 Conversion vs. time for the aza Michael addition of butyl acrylate and n-butylamine (blue triangle) or 1-amino-2-butanol (inverted red triangle) carried out using 1:1 (full symbols) and 2:1 (open symbols) NH motif:acrylate. | ||
Synthesis of the materials
To demonstrate the potential of β-hydroxy amines for the fast synthesis of CANs, pentaerythritol tetraacrylate (PTA) was reacted with two mono functional amines, one possessing a β-hydroxy group, and one deprived of such hydroxy group (Scheme 2). Butylamine and 1-amino-2-butanol were used in this study because they are commercially available and have relatively simple structures. Moreover, since the two hydrogen atoms of the amine function can perform aza-Michael additions, primary monoamines were used as di-functional Michael donors. Therefore, by combining these primary amines with PTA a three-dimensional network can be obtained. The material syntheses were carried out in bulk at 50 °C overnight, with a ratio of two equivalents of acrylate function for one equivalent of amine function. β-Hydroxy amino networks (BHAN) and alkylamino networks (AN) were respectively prepared from 1-amino-2-butanol and butylamine. PTA possesses ester functions and the use of β-hydroxy amines introduces hydroxy groups in the network structure. Therefore in BHAN, transesterification and aza-Michael exchange reactions may occur. In contrast, in AN (deprived of hydroxy groups) only aza-Michael exchange may take place. | ||
| Scheme 2 Synthesis of AN and BHAN networks. | ||
Model reactions demonstrated that β-hydroxy amines are more reactive than alkylamine in aza-Michael additions. The curing reaction at 50 °C of the two systems was thus monitored by rheometry to confirm this observation on model molecules (Fig. 4). BHAN showed a gel point time of 1.2 h, twice lower than that of AN (2.4 h). As seen on the model compounds, β-hydroxy amines thus also showed higher reactivity than their non hydroxylated analogs during network formation. The same reasons as those developed in the kinetics part can be used to explain the difference of reactivity between β-hydroxyamine and alkylamine. In BHAN, hydroxy groups likely act as internal catalyst for the aza-Michael addition.
 | ||
| Fig. 4 Evolution of G′ and G′′ with curing time for BHAN (blue squaress) and AN (red triangle) formation at a curing temperature of 50 °C. | ||
The networks were cured at 50 °C overnight and then assessed using both chemical and mechanical analyses. Both materials were translucent. However, AN had a slight yellow color compared to BHAN (Fig. 10) likely caused by a slight oxydation of the amine. In order to confirm the network synthesis, swelling tests were carried out in THF (Table 2). The swelling index of AN was slightly higher than that of BHAN. This difference could be explained by a higher network density in BHAN caused by H-bonds interactions or by a lower conversion of the acrylate functions in BHAN. However, no clear difference was observed between BHAN and AN by FTIR analysis and no residual enthalpy peak was observed during DSC analysis for both materials, leading to the conclusion that the swelling index difference is likely caused by the formation of H-bonds in BHAN.53 For both materials, the insoluble ratio were higher than 90% confirming the network formation.
Glass transition temperatures (Tg) were determined by differential scattering calorimetry (DSC), BHAN and AN showed Tg values of 5 °C and of −17 °C respectively. The higher Tg of BHAN is related to the higher number of hydrogen interactions in the material compared to AN, which reduces the mobility of the chains. Dynamic mechanical analyses (Fig. 5) revealed Tα values of 24 °C and −2 °C for BHAN and AN respectively, in agreement with the DSC results. Unsurprisingly, given their structural similarity, BHAN and AN had similar storage modulus values on the glassy and rubbery plateau (Fig. 5).
 | ||
| Fig. 5 Storage modulus (E′) and tan(δ) of BHAN (full blue square and full blue curve) and AN (full red circle and red dashed curve). | ||
TGA analysis showed a 5% weight loss temperature around 260 °C for both materials (Fig. 6 and Table 2). The dissociation of aza-Michael adduct was observed between 260 °C and 380 °C. Indeed, in this temperature range, the adducts obtained after retro-aza Michael could decompose and be removed from the material.45 Therefore, the reprocessing temperature was chosen significantly lower than the decomposition temperature to avoid significant loss of properties during reprocessing.
 | ||
| Fig. 6 TGA of AN (red curves) and BHAN (blue curves) before (full lines) and after (dashed lines) reprocessing. | ||
Dynamic properties
The dynamic properties of the materials were evaluated by stress relaxation experiments. A 5% torsional strain was applied and the relaxation modulus (G(t)) was monitored as a function of time. Relaxation experiments were performed at temperatures ranging from 130 to 190 °C for both BHAN (Fig. 7) and AN materials (Fig. 8). Non-normalized relaxations are respectively provided in Fig. S2 and S3† for BHAN and AN. BHAN is expected to relax faster than AN since it is contains both ester and hydroxy groups whereas only ester functions are present in AN. Hence, both materials may undergo retro-aza Michael/aza Michael exchange but only BHAN can also undergo transesterification. | ||
| Fig. 7 Normalized stress-relaxation curves at temperatures ranging from 130 to 190 °C for BHAN. | ||
 | ||
| Fig. 8 Normalized stress-relaxation curves at temperatures ranging from 130 to 190 °C for AN. | ||
Relaxation times (τ) were shorter for BHAN than for AN For instance at 130 °C, τ = 17.6 h and τ = 29.2 h were obtained for BHAN and AN respectively. Two reasons can be hypothesized to explain these results: first, in AN only aza-Michael exchange reactions can take place while in BHAN transesterification exchanges are also possible due to the presence of hydroxy groups. In addition, the rate of transesterification was shown to be higher than the exchange rate via aza-Michael reactions.45 Secondly, it was also demonstrated48,49 that the use of a combination of exchange reactions entails a synergistic effect for the relaxation of the material and the reshaping temperature required. Finally, it can also be hypothesized that the presence of the hydroxy group favours the retro-aza Michael as H-bond presence could stabilize the ammonium formed after the dissociation step.
BHAN and AN relaxation times followed an Arrhenius behaviour in the studied temperature range (Fig. 9). Flow activation energies of 50.3 and 50.7 kJ mol−1 were determined for BHAN and AN respectively. These values cannot be easily compared due to the number of concomitant effects in the materials. Indeed, Du Prez et al. were also not able to figure out a specific trend for Ea(flow) in their initial studies on β-amino esters. Nevertheless, these Ea(flow) are relatively low compared to others Ea(flow) reported in the literature9 and could be associated to the use of relatively low molar mass monomers which induces a high density of functionalisation in the network and thus a potentially facilitated exchange.
 | ||
| Fig. 9 Fitting of the relaxation times (τ) vs. temperature data by the Arrhenius equation for BHAN (blue squares) and AN (red circle). | ||
It should be noted that Arrhenius behaviour was not expected for these materials as aza-Michael exchange proceeds via a dissociative mechanism. Nevertheless, Montarnal et al. demonstrated that dissociative CANs can show Arrhenius behaviour in a specific temperature range.54 These results may be ascribed to the fact that the retro aza-Michael proceeds much slower than the aza-Michael addition under these experimental conditions. Therefore, the material can relax the applied stress with only a weak influence on the cross-link density.55 This hypothesis is confirmed by the fact that the initial relaxation modulus (G0) is not impacted by the temperature (Fig. S2 and S3†). Frequency sweep data performed at temperature ranging from 120 °C to 180 °C also demonstrate a constant storage modulus, confirming that network connectivity was not dependent of the temperature (Fig. S4 for AN and S5† for BHAN).
Reprocessing properties
These two catalysts-free materials were submitted to reshaping. BHAN and AN were fragmented and compressed for 2 h under 3 tons at 150 °C and 180 °C respectively (Fig. 10). Higher temperatures were required for AN as its relaxation was slower than that of BHAN at a given temperature. The thermo-mechanical properties of the materials observed before and after reshaping were compared (Table 2). No clear visual degradation of the materials was observed after reshaping. Both materials remained translucent although AN acquired a slight yellow tint. Fig. 6 shows the TGA thermograms of the cured and reshaped AN and BHAN samples, from 30 °C to 700 °C. Mass loss profiles were not affected by the reshaping procedures, indicating that materials were not significantly impacted by this procedure. Moreover, the FTIR data confirmed that the chemical compositions of the networks were not affected by the reshaping process (Fig. S6 and S7†). Finally, high insoluble fractions (>84%) were obtained on reprocessed samples even if a slight decrease was observed for both materials (Table 2). These relatively lower values may be attributed to the dissociative nature of the aza-Michael exchange reactions. | ||
| Fig. 10 Reprocessing processes for AN (left) and BHAN (right). | ||
Dynamic mechanical analyses were also performed to test whether the capacity of the network to relax stress was retained after reshaping. Therefore, frequency sweep measurements were performed from 0.1 to 100 rad s−1 at 180 °C for BHAN (Fig. 11) and AN (Fig. S8†). The storage and loss moduli were only slightly affected by the reprocessing, which is coherent with the chemical analyses of the networks. The loss modulus increase at low frequencies is characteristic of the material starting to flow. The conservation of this effect on reshaped samples demonstrated that the capability of the materials to be reprocessed is not lost and that reprocessing cycle could be repeated. The loss modulus increase occurred at lower frequencies for AN compared to BHAN at 180 °C which is in good agreement with the relaxation times determined earlier.
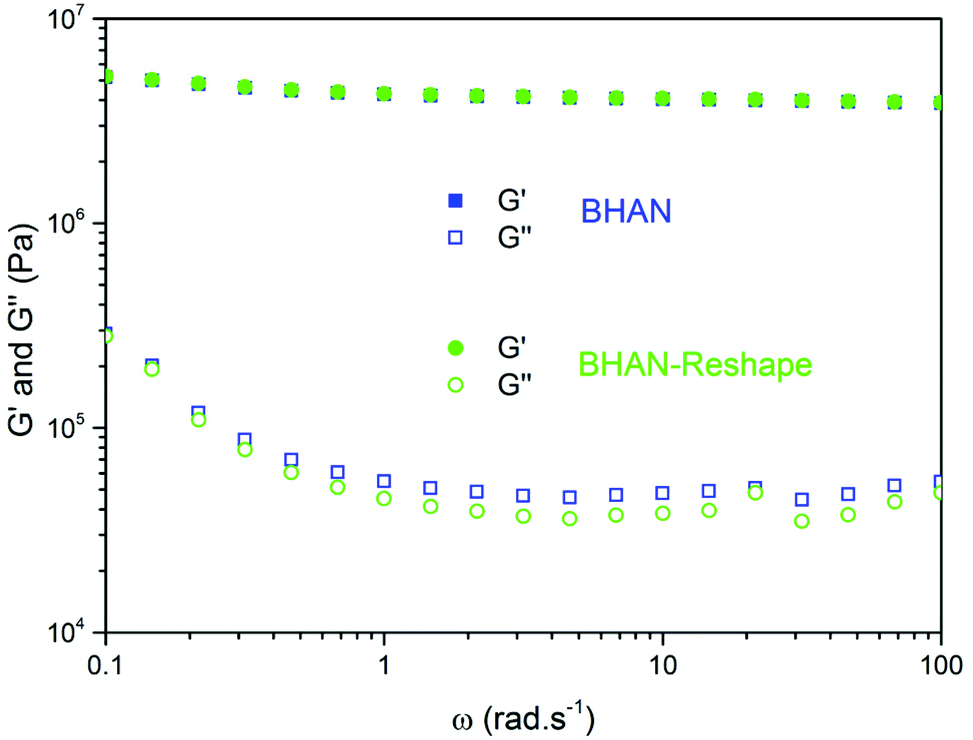 | ||
| Fig. 11 Measurement of G′ (full symbol) and G′′ (open symbol) in frequency sweep at 180 °C of BHAN (blue squares) and BHAN reshape (blue circles). | ||
Experimental
Materials and methods
Pentaerythritol tetraacrylate (purity 99.9%), butyl acrylate (99%), n-butylamine (99%), and 1,3,5-trioxane (99%) were purchased from Sigma-Aldrich (Darmstadt Germany). The 1-amino-2-butanol (99%) was purchased from TCI Europe N.V. (Zwijndrecht, Belgium). Ethanol (96%, VWR Chemicals) was purchased from VWR International S.A.S (Fontenay-sous-Bois, France). CDCl3 (99.5%D) was provided by Eurisotop (Saint-Aubin, France). All the materials were used as received.NMR kinetics
The Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker Avance III 400 MHz spectrometer. The instrumental parameters for recording 1H NMR spectra were as follows: flip angle 30°, acquisition time 4.1 s, pulse delay 1.5 s, number of scans 16, and pulse width 5 μs. NMR spectra were recorded without spinning.As a representative example, for the kinetic study of 2:1 acrylate:amine, 73 mg of n-butylamine (1 eq.), 21 mg of 1,3,5-trioxane as internal standard and 1 mL of CDCl3 were introduced in a vial (solution 1–1 mol L−1 of n-butylamine). 256 mg of butylacrylate (2 eq.) and 1 mL of CDCl3 were introduced in another vial (solution 2–2 mol L−1 of butylacrylate). For the kinetic studies with 1:1 acrylate:amine ratio, 73 mg of n-butylamine (1 eq.), 21 mg of 1,3,5-trioxane as internal standard and 1 mL of CDCl3 were introduced in a vial (solution 1–1 mol L−1 of n-butylamine). 128 mg of butylacrylate (1 eq.) and 1 mL of CDCl3 were introduced in another vial (solution 2–1 mol L−1 of butylacrylate). For the kinetic study carried out in the presence of ethanol, 47 mg of ethanol (1 eq.) were added to solution 2. 300 μL of solution 1 were introduced in an NMR tube. A spectrum was recorded at this time (t = 0), and then 300 μL of solution 2 were added to the NMR tube. The NMR tube was vigorously shaken and introduced in the NMR spectrometer thermostated at 35 °C. The conversion of butylacrylate was monitored by 1H NMR (eqn (1)). The signal of 1,3,5-trioxane (internal standard) was integrated from 4.82 ppm to 5.33 ppm. Then the resonance of the double bond of the butylacrylate was integrated from 5.60 ppm to 6.48 ppm. During the first 6 h, a spectrum was recorded every 15 minutes, and then each hour for 24 h.
Conversion determination
 | (1) |
The same procedure and the was followed for kinetic reaction.
Fourier transform infrared spectroscopy
Infrared spectra were recorded on a Nicolet 210 Fourier Transform Infrared (FTIR) Spectrometer. The characteristic IR absorptions mentioned in the text are reported in cm−1. Materials analyses were recorded using an ATR accessory.Thermogravimetric analyses
Thermogravimetric analyses (TGA) were carried out using a Netzsch TG 209F1 apparatus. Approximately 10 mg of sample were placed in an alumina crucible and heated from room temperature to 600 °C at a heating rate of 20 °C min−1 under nitrogen atmosphere (40 mL min−1). A nitrogen flow was used to protect the apparatus.Differential scanning calorimetry
Differential Scanning Calorimetry (DSC) analyses were carried out using a Netzsch DSC200F3 Maia calorimeter, which was calibrated using indium, n-octadecane and n-octane standards. Nitrogen was used as purge gas. Approximately 10 mg of sample were placed in a perforated aluminum pan and the thermal properties were recorded between −150 °C and 120 °C at 20 °C min−1 to observe the glass transition temperature. The Tg values were measured on the second heating ramp to erase the thermal history of the polymer.Swelling index
Three samples from the same material, of around 40 mg each, were separately immersed in 10 mL of THF for 24 h. The swelling index (SI) was calculated using eqn (2), where m2 is the mass of the swollen material and m1 is the initial mass. The reported swelling indices are average values of the 3 independent samples.Swelling index
 | (2) |
Gel contents
After the SI measurements, the three samples were dried in a ventilated oven at 80 °C for 24 h. The gel content (GC) was calculated using eqn (3), where m3 is the mass of the dried material and m1 is the initial mass. The reported gel contents are the average values of the three samples.Gel contents
 | (3) |
Rheological experiments
Rheology experiments were performed on a Thermo Fisher HAAKE MARS 60 rheometer. The gelation times were measured with a plate-plate geometry with a diameter of 25 mm. The gelation times were analyzed by observing the crossover of the storage modulus (G′) and loss modulus (G′′) during an oscillatory experiment at 1 Hz, 50 °C and 1% of deformation, according to the previously determined linear domain.For stress relaxation experiments, a plate-plate geometry with a diameter of 8 mm was used, a strain of 1% was applied to the material and the relaxation modulus (G(t)) was monitored over time at a constant temperature. Relaxation curves were fitted with the Kohlrausch–Williams–Watts (KWW) model, which is a stretched exponential function (eqn (4)):
Kohlrausch–Williams–Watts (KWW) model equation.
 | (4) |
The dependence of the relaxation times with temperature was fitted with the Arrhenius law (eqn (5)):
Arrhenius equation
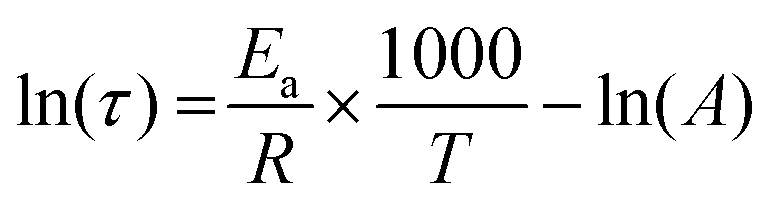 | (5) |
DMA
Dynamic mechanical analyses (DMA) were carried out with a Metravib DMA 25 using Dynatest 6.8 software. The samples were tested in uniaxial tension mode while heating at a rate of 3 °C min−1 at a frequency of 1 Hz and a fixed strain of 10−6 m.Synthesis of the materials
Conclusions
In the course of this study, higher reactivity rate was observed for β-hydroxy amines compared to their alkyl equivalents during the aza-Michael addition with acrylates. Moreover, complementary analyses highlighted the influence of the hydroxy group on this reaction. These kinetic studies were then confirmed by rheological measurements. Catalyst-free CANs, BHAN and AN, were thus synthesized by reacting β-hydroxy amine or alkylamine respectively with TPA, a tetraacrylate, at 50 °C.Both cross-linked materials exhibited aza-Michael exchanges under thermal stimulus. In addition, transesterification exchanges were also possible in the β-hydroxy amine-based networks due to the presence of both ester and hydroxy groups in the material. Stress-relaxation analyses revealed lower relaxation times for the β-hydroxy amine networks due to the combined effects of transesterification and aza-Michael exchanges. Arrhenius behavior was observed for both materials in the temperature range studied. Following the observed trend in dynamic analyses, higher temperature was required to reshape AN (180 °C) compare to BHAN (150 °C) while the reshaping time and pressure applied remained identical. Finally, chemical and mechanical analyses of the materials before and after reshaping were almost identical, confirming that reprocessing was efficient and did not adversely affect the materials.
To conclude, this study highlights the potential of β-hydroxy amine as building blocks for CANs and paves the way for further applications of these compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the French National Research Agency ANR (AFCAN project: ANR-19-CE06-0014).References
- J. P. Pascault and R. J. J. Williams, Handbook of Polymer Synthesis, Characterization, and Processing, John Wiley and Sons, 2013, pp. 519–533 Search PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- P. Chakma, L. H. Rodrigues Possarle, Z. A. Digby, B. Zhang, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2017, 8, 6534–6543 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- A. V. Tobolsky, I. B. Prettyman and J. H. Dillon, Rubber Chem. Technol., 1944, 17, 551–575 CrossRef CAS.
- M. D. Stern and A. V. Tobolsky, Rubber Chem. Technol., 1946, 19, 1178–1192 CrossRef.
- R. C. Osthoff, A. M. Bueche and W. T. Grubb, J. Am. Chem. Soc., 1954, 76, 4659–4663 CrossRef CAS.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- F. Cuminet, S. Caillol, É. Dantras, É. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J.-M. Lehn, Chem. – Eur. J., 2009, 15, 1893–1900 CrossRef CAS PubMed.
- B. Zhang, Z. A. Digby, J. A. Flum, E. M. Foster, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2015, 6, 7368–7372 RSC.
- G. B. Lyon, A. Baranek and C. N. Bowman, Adv. Funct. Mater., 2016, 26, 1477–1485 CrossRef CAS.
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag, U. S. Schubert and M. D. Hager, Macromol. Chem. Phys., 2016, 217, 2541–2550 CrossRef CAS.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- H. A. Houck, K. De Bruycker, S. Billiet, B. Dhanis, H. Goossens, S. Catak, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Chem. Sci., 2017, 8, 3098–3108 RSC.
- F. I. Altuna, U. Casado, I. E. Dell'Erba, L. Luna, C. E. Hoppe and R. J. J. Williams, Polym. Chem., 2020, 11, 1337–1347 RSC.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, RSC Adv., 2016, 6, 88647–88655 RSC.
- M. Hayashi, R. Yano and A. Takasu, Polym. Chem., 2019, 10, 2047 RSC.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5377–5385 RSC.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277–6284 CrossRef CAS.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- M. Liu, J. Zhong, Z. Li, J. Rong, K. Yang, J. Zhou, L. Shen, F. Gao, X. Huang and H. He, Eur. Polym. J., 2020, 124, 109475 CrossRef CAS.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed.
- K. Jin, L. Li and J. M. Torkelson, Adv. Mater., 2016, 28, 6746–6750 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, J. Mater. Chem. A, 2019, 7, 18039–18049 RSC.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- D. Boucher, J. Madsen, L. Yu, Q. Huang, N. Caussé, N. Pébère, V. Ladmiral and C. Negrell, Macromolecules, 2021, 54, 6772–6779 CrossRef CAS.
- D. Boucher, J. Madsen, N. Caussé, N. Pébère, V. Ladmiral and C. Negrell, Reactions, 2020, 1, 89–101 CrossRef.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- J. Wang, S. Chen, T. Lin, J. Ke, T. Chen, X. Wu and C. Lin, RSC Adv., 2020, 10, 39271–39276 RSC.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- H. Zhang, S. Majumdar, R. A. T. M. Van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 12, 272–277 CrossRef PubMed.
- D. Berne, F. Cuminet, S. Lemouzy, C. Joly-Duhamel, R. Poli, S. Caillol, E. Leclerc and V. Ladmiral, Macromolecules, 2022, 55, 1669–1679 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed.
- M. Podgórski, N. Spurgin, S. Mavila and C. N. Bowman, Polym. Chem., 2020, 11, 5365–5376 RSC.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef CAS PubMed.
- A.-S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Green Chem., 2018, 20, 4075–4084 RSC.
- B. Quienne, R. Poli, J. Pinaud and S. Caillol, Green Chem., 2021, 23, 1678–1690 RSC.
- A. Genest, S. Binauld, E. Pouget, F. Ganachaud, E. Fleury and D. Portinha, Polym. Chem., 2017, 8, 624–630 RSC.
- A. S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Eur. Polym. J., 2020, 123, 109460 CrossRef.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- O. Anaya, A. Jourdain, I. Antoniuk, H. Ben Romdhane, D. Montarnal and E. Drockenmuller, Macromolecules, 2021, 54, 3281–3292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, FTIR and frequency sweep analyses. See DOI: https://doi.org/10.1039/d2py00274d |
| This journal is © The Royal Society of Chemistry 2022 |