
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow methacrylated poly(glycerol sebacate) for soft tissue engineering†
Iris Cristina
Becerril-Rodriguez
 and
Frederik
Claeyssens
*
and
Frederik
Claeyssens
*
Kroto Research Institute, Department of Materials Science and Engineering, The University of Sheffield, UK. E-mail: f.claeyssens@sheffield.ac.uk
First published on 23rd May 2022
Abstract
Tissue engineering for soft tissue has made great advances in recent years, though there are still challenges to overcome. The main problem is that autologous tissue implants have not given good results since approximately 60% of tissue is lost or absorbed after implantation. The main strategy to overcome this issue has been the development of biomaterials capable of regenerating damaged tissue and mimicking the host environment. Biopolymers have been widely used for their biocompatibility and hydrophilicity, but they lack structural stability and mechanical properties suitable for the replacement of soft tissue. Synthetic polymers can overcome the drawbacks faced by biopolymers, with synthetic elastomers being of particular interest since they have mechanical properties and elastic moduli close to those of soft tissue. We focused on the physiochemical and biological characterization of poly(glycerol sebacate) methacrylate (PGS-M), and its application in the fabrication of scaffolds for soft tissue through the addition of methacrylate groups to improve its mechanical properties. PGS-M is a relatively new polymer that has not been widely used in soft tissue engineering. Our results confirm that its physicochemical characteristics make it a promising material for tissue engineering to fabricate scaffolds using various techniques like emulsion templating, 3D printing, and soft stereolithography.
Introduction
Soft tissue covers a variety of tissues such as skin, fat, muscle, cartilage, tendon, nerves, and blood vessels, among others. The main function of soft tissues is to provide support and connection for body structures and organs by surrounding them. They are mainly composed of collagen fibres and elastin, giving them an elastic behaviour and characteristic mechanical features (5 KPa–5 MPa Young's modulus).1 However, these values can differ depending on the variability of biological tissue. These tissues are easily damaged by traumatic injuries and thus, replacement and regeneration strategies for damaged tissue have been sought after.2–4 Currently, the main strategy proposed to regenerate soft tissue is autologous implantation. However, its main disadvantage is that implanted tissue is easily absorbed and rapidly loses volume. As a result, only 40 to 60% of cells within soft tissue remain viable after implantation.4There has been considerable effort made to develop a replacement that fulfils all the desired mechanical, biological, and physiological characteristics. The main characteristics sought for tissue replacement are physiochemical and biological attributes that mimic the environment of the target tissue. The matching of mechanical properties represents one of the great challenges in the development of biomaterials for soft tissue engineering, since good stability and biocompatibility are required to allow the regeneration of damaged tissue.5 To date, the creation of soft tissue replacement that approaches the characteristics of a gold standard continues to be a challenge. This replacement should have the following characteristics: (1) ability to mimic the mechanical properties of soft tissue, (2) possess the necessary thickness to be implanted in the patient, (3) show biodegradability, and (4) have an adequate shape that favours and influences cell behaviour, promoting cell migration and proliferation.
Natural polymers, such as collagen, fibrin, and chitosan, have been widely used for the development of soft tissue substitutes due to their biocompatibility and biodegradability, but most lack appropriate mechanical properties and some have reported inflammatory responses.6,7
The use of synthetic polymers has been explored to overcome the drawbacks present in biopolymers. Various polymers have been widely used for the synthesis of scaffolds for soft tissue such as polycaprolactone (PCL), poly(L-lactic acid) (PLLA), and polyethylene glycol (PEG), among others. Synthetic polymers are also advantageous due to their versatility in scaffold fabrication, ranging from heat and UV curing, solvent casting, emulsion templating, and spin coating, among others. Although the problems with mechanical properties have been overcome, the processes involved during their synthesis and manufacturing are complex and time consuming. The hydrophobic nature of some synthetic polymers often requires extra steps such as surface modification to increase cytocompatibility with target cells and improve cell adhesion.8,9 The tuneable mechanical properties, elasticity, and varied techniques for scaffold fabrication make synthetic polymers the most viable option to achieve the gold standard in soft tissue replacement. Thus, synthetic polymers have overcome many drawbacks found in biopolymers.
Polyester polymers such as PGS have tuneable mechanical properties and elastic moduli that can match the mechanical properties of soft tissue.10 PGS in particular has seen increasingly more research interest since its first report by Wang et al. in 2002![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 due to its biocompatibility, biodegradability, inexpensiveness, transparency, and elastomeric nature, with tuneable mechanical properties useful for working with soft tissue.12–14 In addition, PGS has been used in biomedical applications as a retinal graft, vascular tissue, cartilage, cardiac patch, and nerve applications. It has also been used as a biomedical adhesive instead of conventional sutures, and recently as a corneal epithelium replacement.15,16
11 due to its biocompatibility, biodegradability, inexpensiveness, transparency, and elastomeric nature, with tuneable mechanical properties useful for working with soft tissue.12–14 In addition, PGS has been used in biomedical applications as a retinal graft, vascular tissue, cartilage, cardiac patch, and nerve applications. It has also been used as a biomedical adhesive instead of conventional sutures, and recently as a corneal epithelium replacement.15,16
We hereby propose the addition of methacrylate groups in low percentages to PGS for use as a biomaterial for soft tissue engineering based on its physiochemical characterisation. The characterisation confirmed that the addition of methacrylate groups makes it possible to control the mechanical properties, degradation rate, crosslinking density, and elongation through the degree of methacrylation (DM). This tuneability makes PGS-M a promising biomaterial for the development of scaffolds that can more accurately mimic the characteristics of various types of soft tissue (Fig. 1).
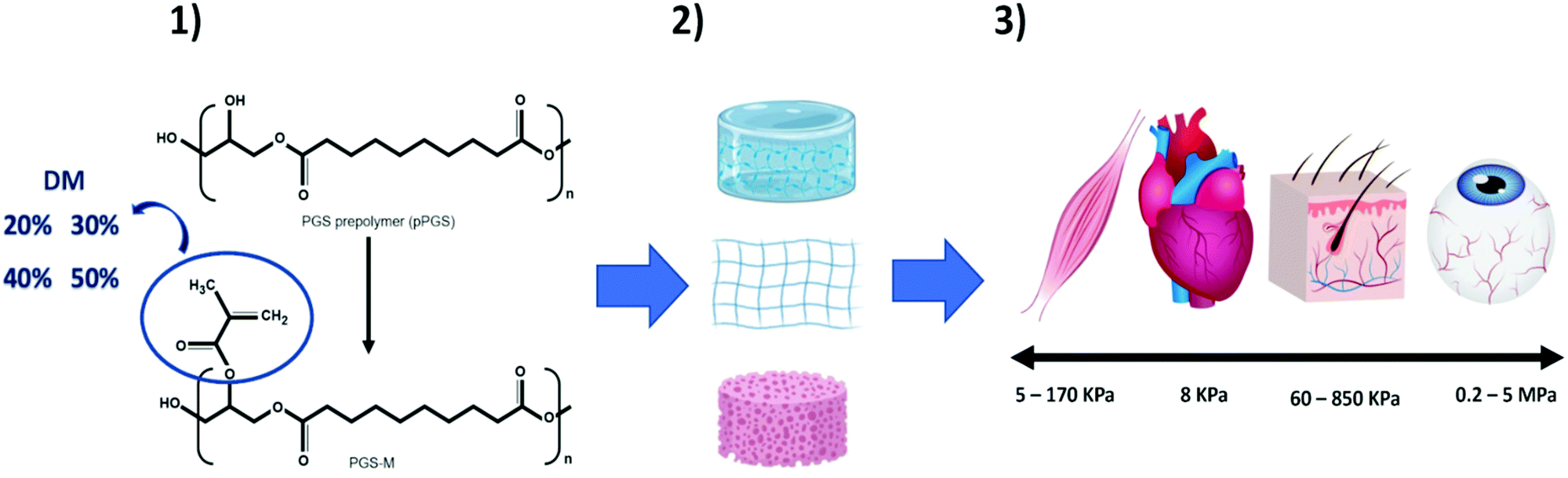 | ||
| Fig. 1 Schematic showing poly(glycerol sebacate) methacrylate (PGS-M) for use in soft tissue engineering. (1) PGS-M degree of methacrylation (20, 30, 40 and 50%) can be modified according to the desirable characteristics of the target tissue. (2) PGS-M has tuneable mechanical properties and can be adapted to various scaffold fabrication techniques. (3) Tailoring DM leads to mechanical properties that mimic native soft tissue. | ||
Materials and methods
Polyglycerol sebacate (PGS) prepolymer (pPGS) standard synthesis
PGS was synthesized through the polycondensation reaction of a 1:1 molar mixture of glycerol (C3H8O3) (Sigma Aldrich) and sebacic acid (C10H18O4) (Sigma Aldrich) added into a two-neck round bottomed flask and reacted at 120 °C under a continuous flow of nitrogen for 24 h. This was heated for an additional 24 h under continuous stirring and vacuum at 120 °C to remove water.
PGS-M synthesis
Methacrylation was carried out as reported previously.17,18 Glycerol has three hydroxyl (OH) groups in its structure. It was assumed that only the two primary hydroxyl groups in glycerol reacted with sebacic acid during the PGS synthesis. Therefore, 3.856 mmol of OH are available for methacrylation.19,20 The methacrylation was carried out by adding methacrylate groups from methacrylate anhydride to the hydroxyl groups of the PGS molecule. PGS was methacrylated with four degrees of methacrylation (DM): 20, 30, 40, and 50%.The methacrylation was carried out using dichloromethane (DCM) (CH2Cl2) (Fisher Scientific, UK) to dissolve the PGS prepolymer in a 1:4 (w/v) ratio. Then, triethylamine ((C2H5)3N) (Sigma Aldrich) was added (equimolar amount) as a neutralizing base for the acidic side products (methacrylic acid). After this, 4-methoxyphenol (MeHQ) (C7H8O2) (Sigma Aldrich) (1 mg g−1 of PGS prepolymer) was added as a photo-polymerisation inhibitor to avoid spontaneous crosslinking. Finally, methacrylic anhydride ([H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)CO]2O) (Sigma Aldrich) was added dropwise at four different concentrations corresponding to the degrees of methacrylation (DM) (20%, 30%, 40% and 50%).
C(CH3)CO]2O) (Sigma Aldrich) was added dropwise at four different concentrations corresponding to the degrees of methacrylation (DM) (20%, 30%, 40% and 50%).
The reaction was performed in the dark (24 h) on ice and allowed to reach room temperature. Extra MeHQ was added at the end of the reaction (0.5 mg g−1 of PGS prepolymer) and the solution was washed four times with 30 mM hydrochloric acid solution (HCl) (Fisher Scientific, UK) at a 1:1 ratio to remove unreacted reagents and impurities. Water was removed from the washing solution using granular calcium chloride (CaCl2) (Fisher Scientific, UK) at 0.4 g−1 of pPGS. The solution was filtered using a 6 μm pore cellulose filter (Whatman – Grade 3, GE Healthcare Life Sciences, UK). Finally, the DCM was removed through rotary evaporation under vacuum in an ice bath until the PGS-M prepolymer was a viscous liquid. To maintain PGS-M stable, the polymer was stored at −8 °C prior to use.
PGS-M samples were analysed before and after crosslinking. The photoinitiator (PI) diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide/2-hydroxy-2-methylpropiophenone (Sigma Aldrich) 1% (w/w) was used to photocure the PGS-M polymer. The mixture was exposed to UV light (200 W, OmniCure Series 2000 curing lamp) for 10 min to photocure. The samples obtained were photocured disks (1 mm thickness and 15 mm diameter). Soxhlet extraction was carried out using methanol (CH3OH) (Sigma Aldrich) as the organic phase, with washing for 24 hours to remove unreacted monomers. Then, PGS-M spin coated coverslips (1 mm thickness) were dried 24 hours under vacuum. Afterwards, the spin coated coverslips were subjected to Soxhlet extraction with dH2O for 24 h to remove both unreacted monomers and the remaining solvent.
Gel permeation chromatography (GPC)
The theoretical number average molecular weight (Mn) and the weight average molecular weight (Mw) were determined through the data obtained from gel permeation chromatography (GPC) (Viscotek GPCmax VE2001 Cirrus with PLgel 3 μm mixed E column). Tetrahydrofuran was used for dissolving samples at 1% w/v. The average molecular weights by number and by weight were calculated using the following equations (eqn (1) and (2)):Average molecular weight by number:
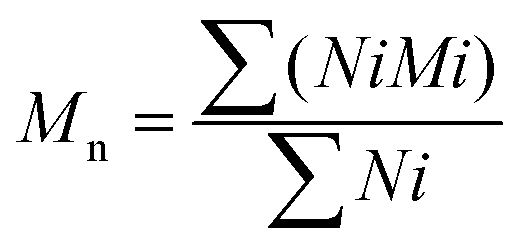 | (1) |
Average molecular weight by weight:
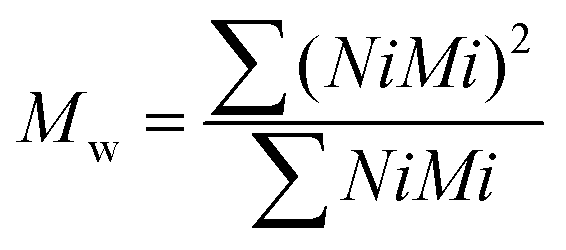 | (2) |
Sol content
Soluble fractions (sol) were determined by washing photocured PGS-M disks in methanol to solubilise the unreacted prepolymer. The disks were dried at 70 °C for 24 h under vacuum (Wi = initial weight) and re-weighed at 24 h (Ws = swollen weight after the first 24 h) intervals, until reaching a constant mass in approximately 3 days (N = 3, n = 3).During weighing, the solvent on the surfaces of the samples was cleaned up, and the samples were placed into sealed vials to reduce solvent evaporation. Controls were subjected to the same drying protocol, but without methanol washing. At the end of the experiment, the samples were dried at 70 °C 24 h under vacuum (Wd = final weight) (eqn (3)).
Sol content:
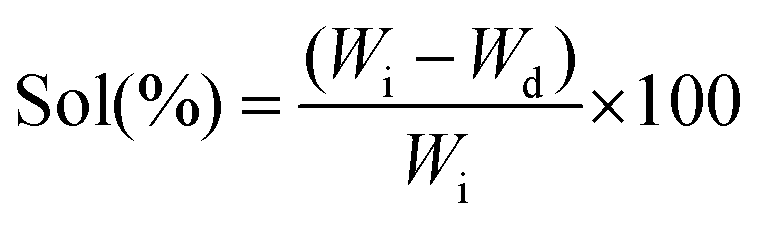 | (3) |
Gel content
Photocured PGS-M disks were dried at 70 °C for 24 h under vacuum (Wi: initial weight) and submerged in methanol at 35 °C for 24 h (N = 3, n = 3). The samples were dried at 70 °C for 24 h under vacuum (Wd: final weight) to determine the degree of crosslinking (eqn (4)).Degree of crosslinking:
 | (4) |
Swelling
Sol free samples were immersed in phosphate buffer saline (PBS), Dulbecco's modified Eagle's media (DMEM), and methanol (Sigma Aldrich) at 35 °C for 24 h. The excess surface liquid was removed, and the swollen samples (Ws) were weighed. The degree of swelling and swelling ratio were calculated from the swollen weight (Ws), final weight (Wd), and initial weight (Wi) using eqn (5) and (6).Degree of swelling:
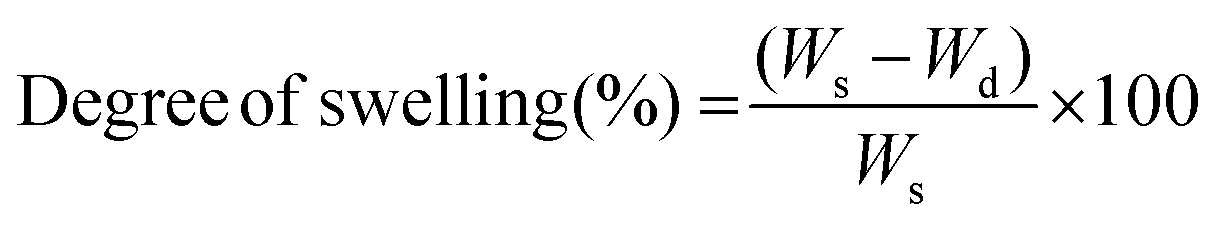 | (5) |
Swelling ratio:
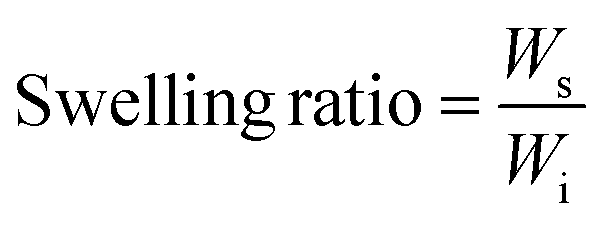 | (6) |
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR)
0.1 ml of PGS prepolymer was analysed using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR), with a Nicolet 380 spectrometer including an ATR device (Golden Gate, 45° single-bounce diamond anvil, Specac). Spectra were obtained between 4000 and 500 cm−1, with a resolution of 4 cm−1. Sol free disks with a 1 mm thickness of photocured PGS-M (DM) 20, 30, 40 and 50%, were washed with methanol (CH3OH) (Sigma Aldrich) 4 consecutive times (24 hours each). After this, the samples were dried in a vacuum oven at 70 °C for 24 h. Afterwards, they were analysed by ATR-FTIR.Nuclear magnetic resonance (NMR)
PGS and PGS-M with different DMs were analysed by nuclear magnetic resonance (NMR) spectroscopy using a Bruker AVIIIHD spectrometer at 500 MHz. The polymer samples were dissolved in 1 ml of deuterated chloroform (CDCl3) at 1% (w/v). The data were analysed using Origin Pro software.Mechanical analysis
Sol free PGS-M samples with different DMs (20, 30, 40 and 50%) were shaped and photocured into tensile test pieces (type 4 dumb-bell, as specified in BS ISO 37:2011) using a silicone mould. Tensile testing was performed using a Zwick Roell system at a crosshead speed of 500 mm min−1, with samples elongated to failure to determine Young's modulus, ultimate tensile strength (UTS), rupture to strain, and maximum elongation.Thermogravimetric analysis (TGA)
Sol free disks were cut into small pieces (∼10 mg). The analysis was done using a Perkin-Elmer Pyris1 TGA with gas purge at 60 mL min−1. The samples were subjected to a heating cycle of 200 °C to 600 °C at a heating rate of 10 °C min−1. The initial degradation temperature (TDI) and peak degradation temperature (TDP) were determined using the first derivative curve to determine the onset of thermal degradation. Thermal stability was determined by calculating the remaining weight at 600 °C. The degradation temperature and temperature range were determined from the first derivative curve (%weight loss/°C).Differential scanning calorimetry (DSC)
Sol free disks were cut into small pieces (∼5 mg). The analysis was done using Perkin-Elmer Pyris1 purged with nitrogen at 30 mL min−1. The samples were heated from −60 °C to 100 °C under N2 at a rate of 10 °C min−1. Thermal properties such as the melting point (Tm), glass transition temperature (Tg), enthalpy of melting (ΔHm) and enthalpy of crystallization (ΔHc) were calculated using the first cooling cycle and second heating cycle. The melting temperature (Tm) and glass transition temperature (Tg) were located at the peak of the process. Recrystallization temperature (Tc) was found at the valley point in the heat capacity curve.In vitro degradation of photocured PGS-M
Sol free disks were weighed and incubated in PBS and media at 37 °C. The samples were analysed at days 3, 7, 10, and 28, and dried at 70 °C overnight under vacuum. The dried samples were weighed and the percentage of weight loss at a specific time point was calculated from the initial (Wi) and final dried weights (Wd) using eqn (7). The surface degradation was analysed using scanning electron microscopy (SEM).Weight loss percentage:
 | (7) |
Scanning electron microscopy (SEM)
Sol free disks were affixed to aluminium stubs, gold coated using a sputter coater (Edwards S150B) and examined by scanning electron microscopy (SEM) using a TESCAN Vega 3 LMU SEM at an accelerating voltage of 20 kV.Contact angle
PGS-M disk hydrophilicity was determined using a goniometer by the sessile drop method. A 3 μL drop of deionized water was placed onto a flat surface of PGS-M with different degrees of methacrylation using a 21-gauge flat needle. Optical images were collected after 10 seconds of contact. The contact angle of water was determined from the optical images with DSA3 software.Surface coating of glass coverslips with PGS-M
Borosilicate glass coverslips (13 mm diameter, no. 2 thickness) (Scientific Laboratory Supplies, UK) were treated with piranha solution for 1 hour and then washed five times with dH2O followed by three washes with methanol. Piranha solution was used as a hydroxylation agent to clean the glass surface and generate additional silanol groups to allow further surface coating.21,22 The piranha solution is made of sulphuric acid (H2SO4) (98%) (Sigma Aldrich) and hydrogen peroxide (H2O2) (30% wt in dH2O) (Sigma Aldrich) (3:1 v/v). The coverslips were immersed in a 10% (w/v) solution of 3-methacryloxypropyltrimethoxysilane in toluene for 24 h in the dark, then washed three times with methanol and dried at room temperature. 3-Methacryloxypropyltrimethoxysilane is a silane coupling agent that promotes the adhesion between the glass surface and PGS-M molecule.23,24
Approximately 50 μl of low methacrylation PGS-M (20, 30, 40 and 50%) and 1% PI (w/w) were deposited at the centre of the coverslip previously treated with piranha solution. The spin coating was carried out at 4000 rpm for 40 seconds (Laurell Technologies WS-400B-6NPP/Lite). The thin layer of the polymer was photocured under UV light for 5 minutes (200 W, OmniCure Series 2000 curing lamp). Unreacted reagents were removed from the coated coverslips by washing them four times in methanol (24 hours each) followed by four washes in dH2O (24 hours each).
Human dermal fibroblast (hDF) culture
hDFs were obtained from primary tissue with informed consent from the NHS (National Health Service) for the donation of waste surgical tissue for research purposes (ethics reference: 15/YH/0177). The fibroblasts were cultured in DMEM supplemented with penicillin/streptomycin (100 IU ml−1), amphotericin (0.625 μg ml−1), L-glutamine (2.5 mM), and foetal calf serum (FCS) (10% (v/v)). Cells were cultured at 37 °C with 5% CO2.Porcine limbal fibroblast (pLF) culture
Limbal explants were isolated from porcine eyes (obtained from “R B Elliott and Son limited”, Chesterfield, United Kingdom). The explants were cultured in DMEM + Glutamax and HAM's 12 media in a 1:1 ratio, supplemented with penicillin/streptomycin (PS) 100 IU ml−1, amphotericin 0.625 μg ml−1, EGF 10 ng ml−1, insulin 5 μg ml−1, and fetal calf serum 10% (v/v). The limbal fibroblasts migrated from the explant after 4 weeks under culture conditions. These fibroblasts were then cultured and expanded until passage 3 (P3). Then, the cells were frozen and stored prior to use. The isolated cells were used in culture with PGS-M.
Cell culture on spin coated PGS-M substrates
PGS-M spin coated coverslips were sterilised in an autoclave at 121 °C for 30 minutes before cell seeding. Cells (hDFs and pLFs) were harvested at a confluence of 90% between passages 3 and 9 with trypsin (0.025%)/EDTA (0.01%), centrifuged at 1000 rpm for 5 minutes (Hettich Zentrifugen Rotofix 32A with a 131 mm rotor radius), re-suspended in fresh media, and counted. The PGS-M spin coated coverslips were placed in a 12-well plate and 50000 cells were seeded in each coverslip. Uncoated glass coverslips seeded with cells were used as the positive control, and unseeded coated coverslips were designated as the negative control. The cells were left to attach for 6 hours. After that time, the coated coverslips were transferred to a new 12 well plate to ensure that only attached cells were included in future analysis. The cells were cultured with 2 ml of media, which were changed every two days. Cell growth was evaluated after incubating the spin coated coverslips at 37.5 °C with 5% CO2 for 7 days.
Resazurin reduction assay
The resazurin working solution was prepared with 1 mM resazurin salt (Scientific Laboratory Supplies) dissolved in dH2O and filtered through a 0.22 μm filter. On days 1, 3, and 7, the growth media were changed prior to resazurin assay and mixed with 10% (v/v) resazurin solution. The samples were incubated at 37 °C with 5% CO2 in the dark, using media with resazurin solution as a blank. After 4 hours in incubation, the samples were taken out and 200 μl of each sample were placed in a 96-well plate in triplicate. The resorufin fluorescence was read at 540 nm excitation and 635 nm emission (BioTek Instruments FLX800). The blank was subtracted from the samples before analysing the data.PicoGreen® DNA quantification assay
The number of cells on PGS-M surfaces was evaluated as follows: on days 1, 3, and 7, the growth media were removed; the samples were washed three times with PBS to remove the remaining media. The samples were incubated in a refrigerator with 500 μl of dH2O at 4 °C for 12 hours. Then, they were subjected to a freeze–thaw regime, passing from −80 °C to 37 °C three times, with time intervals of 30 min freeze and 30 min thaw, with the purpose of lysing the cells and releasing the DNA for quantification. After the final thawing, each solution was removed and placed in a 1 ml microcentrifuge tube. The tubes were vortexed for 15 seconds and centrifuged at 10000 rpm for 5 minutes (Sanyo MSE Micro Centaur MSB010.CX2.5 with a 64 mm rotor). 180 μl were taken from each sample and mixed with 180 μl of stock working solution (0.5% PicoGreen® in TE buffer 10 mM). 180 μl of the stock solution mixed with 180 μl of dH2O was used as a blank. The samples were covered with foil, vortexed for 5 seconds and incubated for 10 minutes at room temperature. 100 μl were taken from each sample and were placed in triplicate in a 96-well plate. The PicoGreen® fluorescence was read at 480 nm excitation and 520 nm emission (BioTek Instruments FLX800); before analysing the data, the blank was subtracted from the samples.
Lactate dehydrogenase (LDH) release assay
The cytotoxicity of PGS-M surfaces was evaluated as follows: on days 1, 3, and 7, 50 μl of the growth media were taken and placed in a 96-well plate in triplicate along with 50 μl of LDH working solution in each well. The samples were incubated at room temperature in the dark for 30 minutes. Afterwards, 50 μl of LDH stop solutions were added to complete the reaction. The absorbance for each well was read at 490 nm excitation and 680 nm emission (BioTek Instruments ELx800). LDH values on days 4 and 7 correspond to the cumulative LDH release of previous days.Statistics
The characterization was carried out with three independent experimental repeats (N = 3) in triplicate per experiment (n = 3). The data were analysed with GraphPad Prism version 7.04 software. The data significance was calculated with one-way ANOVA with Tukey post hoc pairwise multiple comparison analysis for experiments with one independent variable or factor (sample type or condition). Two-way ANOVA (paired samples) with Tukey post hoc pairwise multiple comparison analysis was used for the experiments with two independent variables or factors (sample type or conditions). P ≤ 0.05 was considered statistically significant (*) and P > 0.05 was considered non-significant (ns). Data were presented as means ± SD (standard deviation).Results and discussion
Gel permeation chromatography (GPC)
The average molecular weights (by number and by weight) of the synthesized samples (PGS and PGS-M 20, 30, 40 and 50) were calculated from the data obtained in GPC, using eqn (1) and (2). The results are shown in Table 1.| Sample | Molecular weight (Mw), g mol−1 | Molecular weight (Mn), g mol−1 |
|---|---|---|
| pPGS | 34716.39 |
19488.38 |
| PGS-M 20 | 65547.8 |
59987.37 |
| PGS-M 30 | 48824.39 |
47611.13 |
| PGS-M 40 | 34849.69 |
32126.88 |
| PGS-M 50 | 31235.38 |
20835.17 |
Interestingly, as the DM increases both Mn and Mw decrease. In the PGS-M synthesis section, it was explained that the PGS's OH groups are methacrylated; PGS-M 20 has 20% of its OH groups methacrylated, while PGS-M 50 has 50% of its OH groups methacrylated. Therefore, PGS-M 20 has 80% of its OH groups “free”, which can interact among themselves to form longer polymeric chains that are reflected in larger Mn and Mw. On the other hand, PGS-M 50 has fewer free OH groups to form longer chains, also reflected in lower Mw and Mn compared to those of PGS-M 20.
Sol content
The sol content is the un-crosslinked network in the scaffold. The sol content decreases as the DM increases from 39.95% ± 0.29% to 6.16% ± 0.41% for 20% DM to 50% DM, respectively. This is caused by the higher number of methacrylate groups attached to the PGS molecule, which increased the degree of crosslinking network and the amount of ester bonds within the molecule that make the polymer chains longer. Therefore, unbound chains with low DM result in a higher sol content. The statistical analysis showed that all the samples were significantly different (P < 0.0001) (Fig. 2).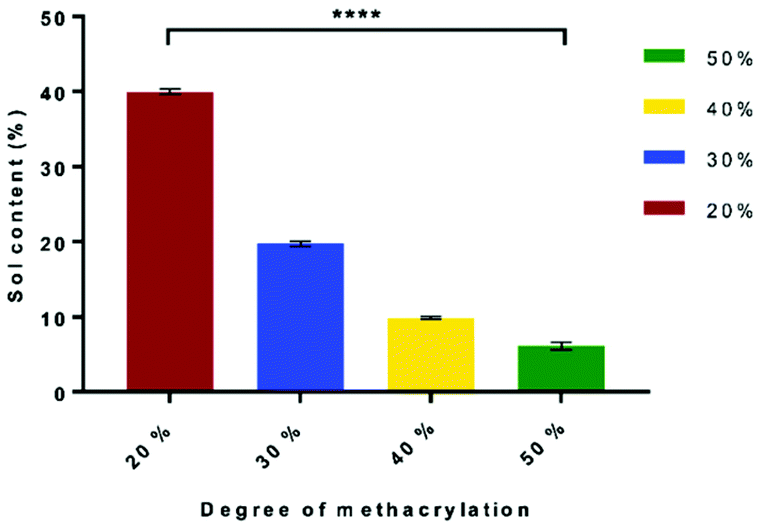 | ||
| Fig. 2 Effects of DM on the sol content percentage. Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by one-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered significant. | ||
Gel content
The percentage of gel content shows the degree of the crosslinked network. The gel fraction increases as the DM rises from 60% ± 0.28% to 93% ± 0.41% for 20% DM to 50% DM, respectively. This is due to the higher number of methacrylate groups attached at the PGS molecule that increase the degree of crosslinking and the amount of ester bonds within the molecule, which make the polymer chains longer. In contrast, unbound chains can be found in PGS-M with a low DM, resulting in a lower gel content. The statistical analysis showed that all the samples were significantly different (p < 0.0001) (Fig. S1†).A range of values have been reported for gel content (60.3% to 93.84%), but these are for thermally crosslinked polymers, not photopolymerised matrices like PGS-M.20,25,26
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR)
The PGS-M molecular structure and the effect of the methacrylation degree were analysed by ATR-FTIR. Peaks related to methacrylate groups appear at 940 cm−1 (C–H bending) and 1640 cm−1 (C stretching).27 These peaks were absent in pPGS and disappeared after polymerisation. The DM was compared with the area under these peaks (Fig. 3, 4, S2† and Table 1).
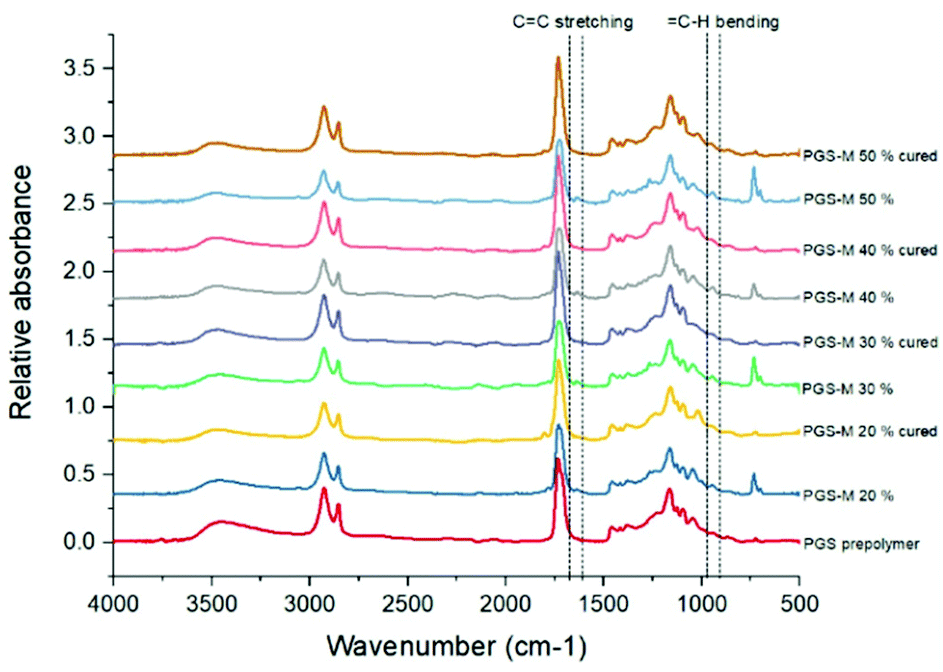 | ||
| Fig. 3 ATR-FTIR spectra of pPGS and PGS-M samples before and after curing. | ||
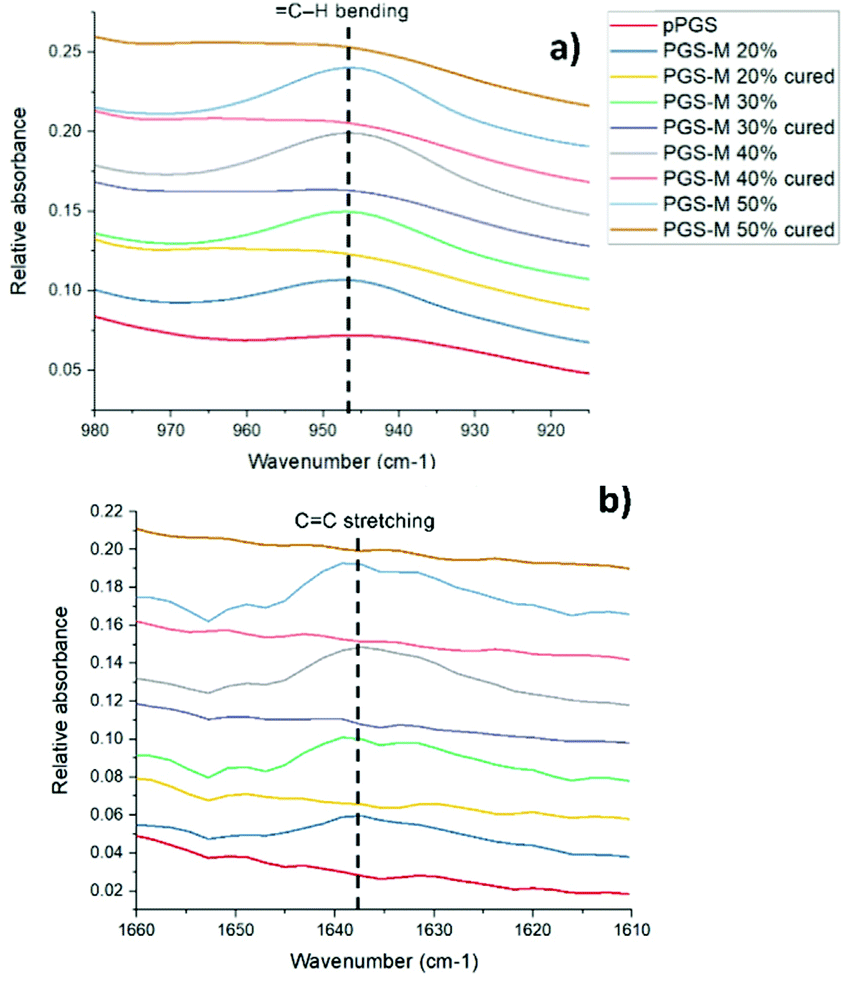 | ||
| Fig. 4 Close up of the ATR-FTIR spectra of pPGS and PGS-M samples before and after curing. (a) Peaks related to methacrylate groups at 940 cm−1 (C–H bending). (b) Peaks related to methacrylate groups at 1640 cm−1 (C stretching). | ||
Fig. 4 shows the peaks related to methacrylate groups with different DM (20%–50%) in PGS-M spectra. These peaks are absent in pPGS and disappear after PGS-M photopolymerization.
The photocuring process was evaluated through the analysis of the ATR-FTIR spectral peaks related to methacrylate groups (Fig. 4 and ESI Fig. S2†).
We can observe that peaks related to the methacrylate groups at 940 and 1640 cm−1 decrease after crosslinking under UV light. The percentage of conversion was calculated through the integration of the peaks and analysis with OriginPro 2017 software (Table 2).
| Degree of methacrylation | Area | Methacrylate conversion after cure (%) | ||
|---|---|---|---|---|
| Wavenumber (cm−1) | ||||
| 1640 | 940 | 1640 | 940 | |
| pPGS | 0.014 | 0.124 | — | — |
| PGS-M 20% | 0.161 | 0.323 | — | — |
| PGS-M 20% cured | 0.018 | 0.146 | 88.82 | 54.8 |
| PGS-M 30% | 0.258 | 0.399 | — | — |
| PGS-M 30% cured | 0.024 | 0.184 | 90.7 | 53.88 |
| PGS-M 40% | 0.310 | 0.497 | — | — |
| PGS-M 40% cured | 0.028 | 0.165 | 90.97 | 66.8 |
| PGS-M 50% | 0.326 | 0.516 | — | — |
| PGS-M 50% cured | 0.014 | 0.165 | 95.7 | 68.02 |
The area of these peaks is well correlated with the DM of the PGS-M samples, as can be observed in Table 1 and Fig. S2.† The data have strong agreement for 940 cm−1y = 0.0128x + 0.3057 and R2 = 0.9691 and for 1640 cm−1y = 0.0054x + 0.0703 and R2 = 0.9039.
Nuclear magnetic resonance (NMR)
pPGS and PGS-M chemical composition was estimated with NMR analysis. It was determined by calculating signal integrals of –COCH2CH2CH2– at 1.2, 1.65, and 2.36 ppm for sebacic acid, –CH2CH– at 3.75, 4.19, and 5.11 ppm for glycerol, and –CH3 and –CH2 at 1.97, 5.3, and 6.17 ppm for the addition of the methacrylate group to the pPGS molecule. CDCl3 was used as a reference at 7.3 ppm. The integrals of the signal for the methacrylate groups (1.97, 5.3 and 6.17 ppm) were calculated with the software OriginPro 2017 and the obtained values correspond to the degree of methacrylation (DM) (Fig. 5).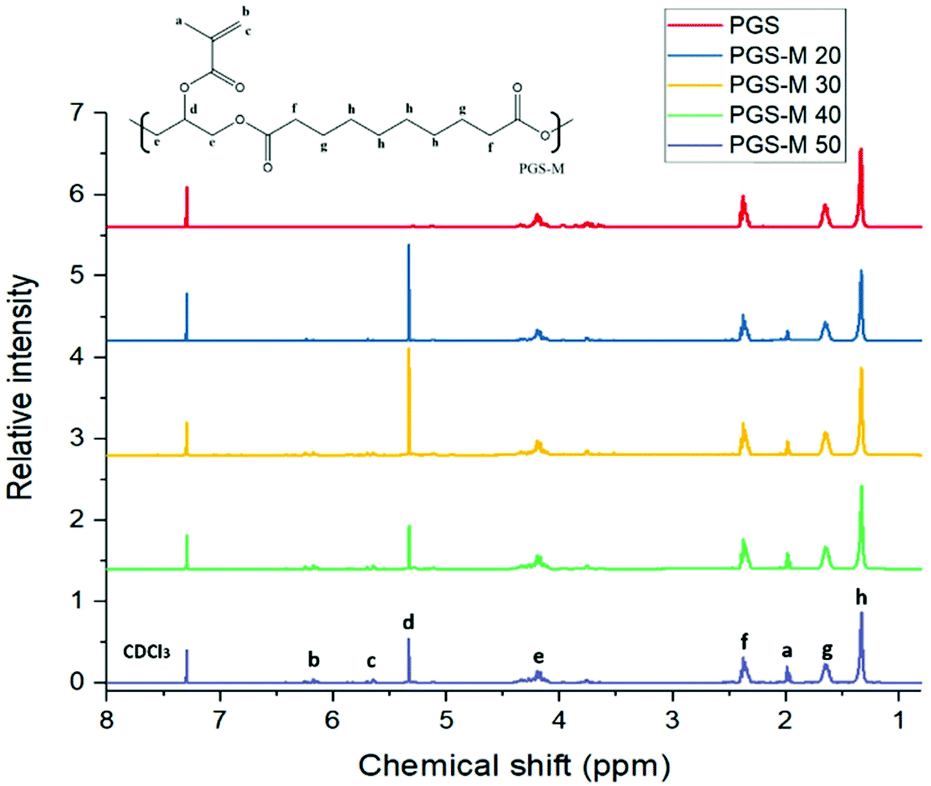 | ||
| Fig. 5 pPGS and PGS-M NMR spectra. PGS-M with different DM (20%, 30%, 40% and 50%). Hydrogen environment peaks of methacrylate groups appear at 1.97, 5.3, and 6.17 ppm (“a”, “b” and “c”). | ||
The obtained DM has a linear relationship and is directly proportional to the molar ratio of methacrylic anhydride per mol of pPGS hydroxyl groups. The results show a correlation of y = 0.923x and an R2 value of 0.9626 (Fig. S3†). From these data, we can conclude that all methacrylate groups added during the reaction are attached in the PGS-M molecule.
NMR analysis suggests that an effective pPGS methacrylation process was carried out, resulting in PGS-M with DMs 20%, 30%, 40% and 50%. This can be confirmed by the appearance of peaks related to methacrylate groups that are absent in pPGS spectra. This result can be compared with the data reported by Pashneh-Tala et al. during the PGS methacrylation (DM: 30%–80%). Even though the methacrylation in that study is higher, the relationship between the DM and the molar ratio of methacrylic anhydride per mol of pPGS hydroxyl groups is consistent.18
Mechanical analysis
Tissue regeneration involves stress and load bearing in the surrounding tissue. Therefore, there should be an equilibrium between the scaffold's mechanical stability and degradation time until tissue regeneration is achieved.28,29 The mechanical properties of scaffolds such as the degree of crosslinking and stiffness affect cell behaviour and biodegradability.30 Ideally, scaffolds for tissue engineering should have the characteristics that mimic the target tissue such as transparency, elasticity, structural and functional requirements of soft tissues.31 They should also support the intraocular pressure, eyelid motion, intraocular pressure and external forces.32 PGS-M mechanical analysis was carried out in order to evaluate each degree of methacrylation to match the mechanical properties in various types of soft tissues and provide appropriate mechanical signals to stimulate the production of new tissue33 (Fig. 6). The scaffold should be strong enough to support deformation, but not too stiff that it stresses the surrounding tissue.34 There is a considerable variation in the soft tissue tensile strength and Young's modulus reported before (strength between 1 and 10 MPa and Young's modulus between 10 kPa and 57 MPa).35 These results depend on different testing mechanisms, tissue anisotropy, and donor variability.34,36,37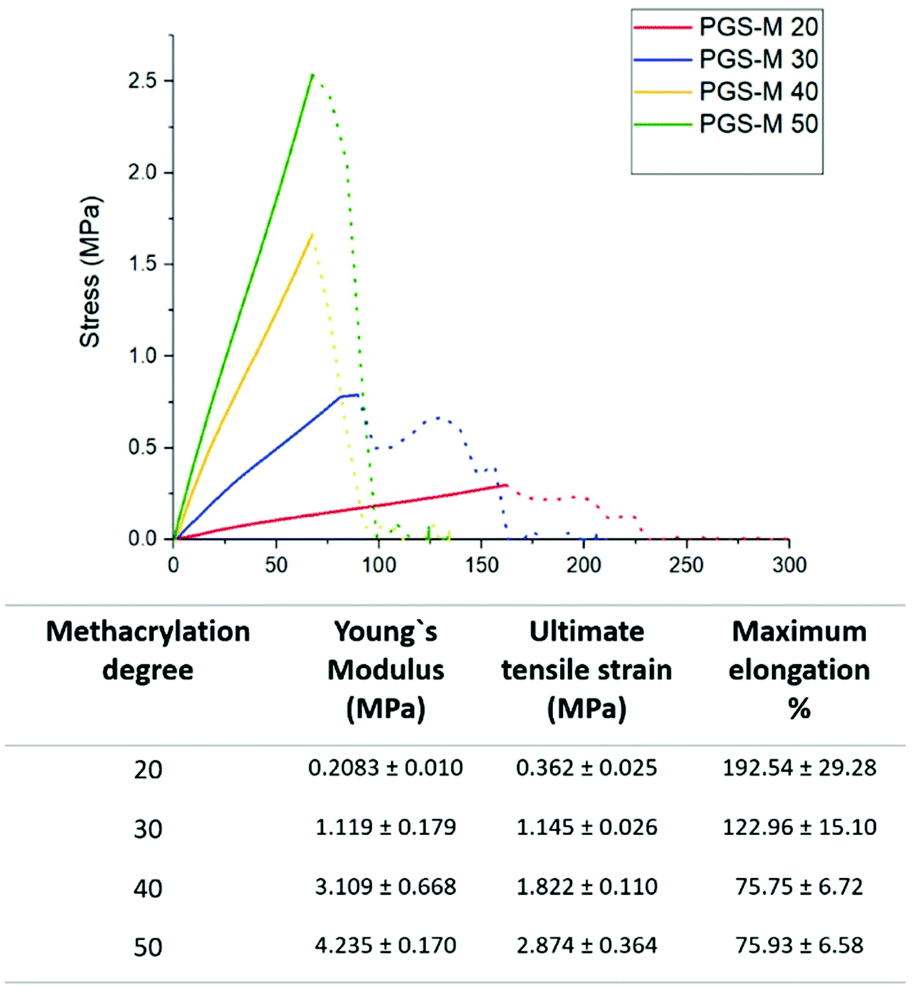 | ||
| Fig. 6 PGS-M mechanical properties in different DMs (20%–50%). The graph values correspond to the mean of load deformation curves (n = 3). | ||
As can be seen in Fig. 8, the Young's modulus and ultimate tensile strain increase along with the DM. However, the maximum elongation decreases as the DM increases. Therefore, PGS-M with a higher DM is stiffer, which coincides with the Young's modulus increment as it is a measurement of stiffness.38 The ultimate tensile strain is the force applied to cause rupture; it makes sense that the values increase with the DM, because the last one is an indication that the polymer matrix is more cross-linked.39 The maximum elongation decreases as a result of the material being stiffer with more covalent bonds in the polymer matrix, making it more ductile.40 From these data we can assume that the methacrylation of PGS-M enhances its mechanical properties compared with PGS (Young's modulus of 0.17 ± 0.018 MPa, tensile strength of 0.264 ± 0.025 MPa and rupture elongation of 292.8 ± 14.1%13).
The data obtained are comparable with other studies for the development of soft tissue scaffolds. Duan et al. obtained dendrimer crosslinked collagen-based scaffolds with a Young's modulus of 1.47 ± 0.1 MPa and an ultimate tensile strength of 1.27 ± 0.17 N.41 Similarly, Bakhshandeh et al. reported similar values for a poly(ε-caprolactone) (PCL) nanofibrous matrix (Young's modulus: 7.5 MPa and ultimate tensile strength: 2.53 ± 0.58 MPa) and polyvinyl alcohol (PVA) hydrogels (Young's modulus: 5.3 MPa and ultimate tensile strength: 0.85 ± 0.55 MPa).42
Thermogravimetry (TGA)
PGS-M thermal stability was determined by TGA analysis (Fig. S4†). Thermal behaviour is an important feature that determines the polymer mechanical stability and degradation.43Fig. 9 shows that PGS-M started to degrade at 436 °C. There is no significant difference in the degradation behaviour for the different DMs. The degradation profile of PGS-M is also comparable with the previously reported PGS profiles by Jiang et al. and Gaharwar et al. with a degradation temperature of 433 °C and 439 °C, respectively.43,44Differential scanning calorimetry analysis (DSC)
PGS-M thermal transitions and crystalline behaviour were determined by DSC analysis (Fig. S5†).Fig. S5† and Table 3 show the glass transition temperature (Tg) for PGS-M with different DMs. The initial and peak thermal decomposition temperatures (TDI and TDP) were obtained from the TGA thermograph. The glass transition temperature (Tg), melting temperature (Tm), crystallization temperature (Tc), enthalpy of melting (ΔHm) and enthalpy of crystallization (ΔHc) were obtained from the DSC thermograph. It was observed that Tg increases as the DM increases, from −37 to −33.95 °C. Melting points (Tm) showed a similar behaviour of increasing as the DM increased, from −28.63 to −21.10 °C. The enthalpy of melting decreases as the DM increases from 3.024 to 2.048 (J g−1). In the cooling cycle the crystallization temperature increases directly proportional to the DM from −22.00 to −14.48 °C. The enthalpy of crystallization decreases as the DM increases from −1.36 to −0.852 (J g−1). Similar results were reported by Singh et al. on PGS-M methacrylation with a DM of 75%, reporting a Tg of −30 °C.17 The low Tg (below 0 °C) suggest that PGS-M is semicrystalline below its melting point and amorphous-elastomeric at physiological temperature. Likewise, similar results were previously reported for PGS.45–48
| Sample | PGS-M 20 | PGS-M 30 | PGS-M 40 | PGS-M 50 |
|---|---|---|---|---|
| TDI (°C) | 350 | 360 | 360 | 360 |
| TDP (°C) | 439.52 | 438.71 | 438.31 | 433.86 |
| T g (°C) | −37 | −36.8 | −34.54 | −33.95 |
| T m1 (°C) | −28.63 | −28.35 | −22.65 | −21.10 |
| ΔHm (J g−1) | 3.024 | 2.183 | 2.182 | 2.048 |
| T c (°C) | −22 | −18.32 | −15.79 | −14.48 |
| ΔHc (J g−1) | 1.36 | 1.052 | 0.936 | 0.852 |
In vitro degradation of photocured PGS-M
Polymeric materials used for tissue engineering should be biocompatible and bioresorbable with controlled degradation rates that match the tissue where they will be implanted.49,50 Current polymers used for soft tissue regeneration have a lack of degradability and stability after implantation. The polymers for soft tissue engineering are expected to be degradable biomaterials that support wound healing processes.51 PGS-M in vitro (PBS) degradation was evaluated through weight loss and SEM analysis (Fig. 7, 8 and S6†).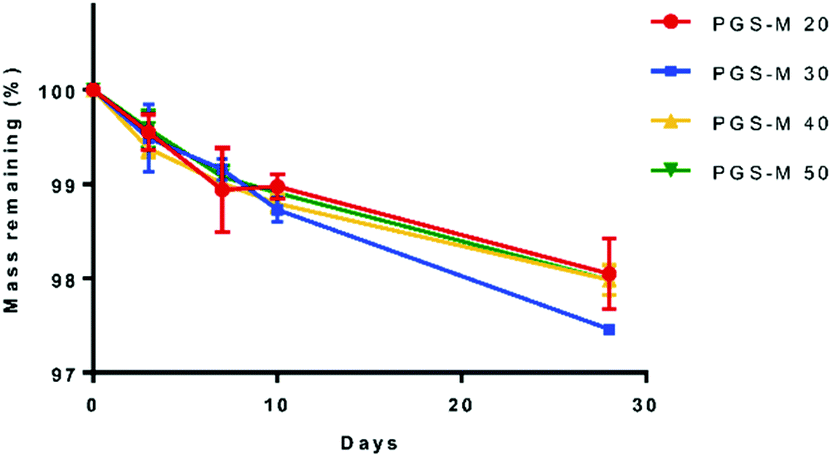 | ||
| Fig. 7 PGS-M weight loss in different DMs (20%–50%). | ||
 | ||
| Fig. 8 Higher magnification images of PGS-M (20–50% DM) degradation analysis in PBS for 0, 3, 7, 10, and 28 days (scale bar = 100 μm). | ||
PGS is a biocompatible polymer but has a fast degradation rate in vivo (around 21 days per mm thickness) and full resorption in 60 days, which limits its biomedical applications.52,53 Comparatively, PGS-M shows a lower degradation rate, remaining stable in vitro for 30 days, and showing a degradation rate of ∼3% throughout this time. This is possibly due to the higher degree of crosslinking in PGS-M compared with that in PGS. The increment in gel content indicated a higher degree of crosslinking and hence a lower degradation rate.54 The gel content of PGS has been reported around 70%13,25,55 while the gel content in PGS-M has been reported in this study as 60 to 93%, for 20% and 50% DM, respectively.
PGS degradation in PBS occurs by hydrolysis, which breaks the ester bonds between glycerol and sebacic acid after an extended period of time.56 Degradation of PGS is caused by surface erosion with a linear loss of mass, which allows the scaffold geometry and mechanical properties to be maintained.53,57 In comparison, PGS-M is an amorphous polymer at physiological temperature. The amorphous region in the polymers are more susceptible to hydrolysis, but by increasing the DM the degradation rate decreases, as reported by Singh et al. and Pashneh-Tala et al.17,18,58Fig. 8 and S6† show the SEM images of PGS-M (20–50% DM) degradation in PBS at 0, 3, 7, 10, and 28 days.
In Fig. 8 and S6,† it is possible to observe that PGS-M has no significant mass loss in PBS. The addition of methacrylate groups increases the degradation time compared with that of PGS, with PGS-M showing degradation by surface erosion. This results in a small reduction of strength and structure. Therefore, the PGS-M scaffold maintains its integrity throughout the degradation process. Similar degradation rates of PGS-M in PBS were reported previously by Singh et al. and Pashneh-Tala et al.17,18
The degradation rate is similar in all DMs because PGS-M is highly crosslinked due to the methacrylate groups, and this study was carried out in a bulk scaffold. The low degradation rate offers certain advantages since the scaffold can maintain its shape without losing integrity when exposed to tensile forces, pressure, motion, and external forces.31,32 However, depending on the biomaterial application, it is necessary to have control over the degradation rate.
As mentioned above, PGS-M bulk scaffolds degrade through surface erosion and have hydrophobic characteristics. In order to control their degradation rate, we propose different strategies that could be studied in future works: (1) decrease the scaffold thickness, (2) add functional groups that increase their hydrophilic nature (peptides, carbonates, esters, etc.), (3) blend the PGS-M with another polymer with more hydrophilic nature to increase water uptake which leads to degradation by hydrolysis, and (4) subject to surface treatment (magnetron sputtering, ion irradiation, plasma polymerization, and plasma treatment).30,59,60
Surface analysis
Human dermal fibroblast (hDF) culture on spin-coated PGS-M surfaces
Fibroblasts from human skin, oral mucosa, periodontal membrane, or embryonic lung have been widely used in studies for the evaluation of materials cytotoxicity.61,62 Fibroblasts are useful in studies of adhesion to surfaces, replication, and cellular integrity due to their characteristics.63 The biocompatibility, cell viability, cell proliferation and cytotoxicity of hDFs on PGS-M surfaces were evaluated with resazurin reduction assay, LDH release assay, PicoGreen® DNA quantification assay, and F-actin staining.hDFs were seeded on PGS-M spin coated substrates with 20, 30, 40, and 50% DMs to evaluate materials cytotoxicity. Cell proliferation and cytotoxicity were evaluated with resazurin reduction assay in spin coated PGS-M 20–50% (Fig. 9, 10 and S8†). PicoGreen® DNA quantification assay was performed only in spin coated PGS-M 40% DM (Fig. 11).
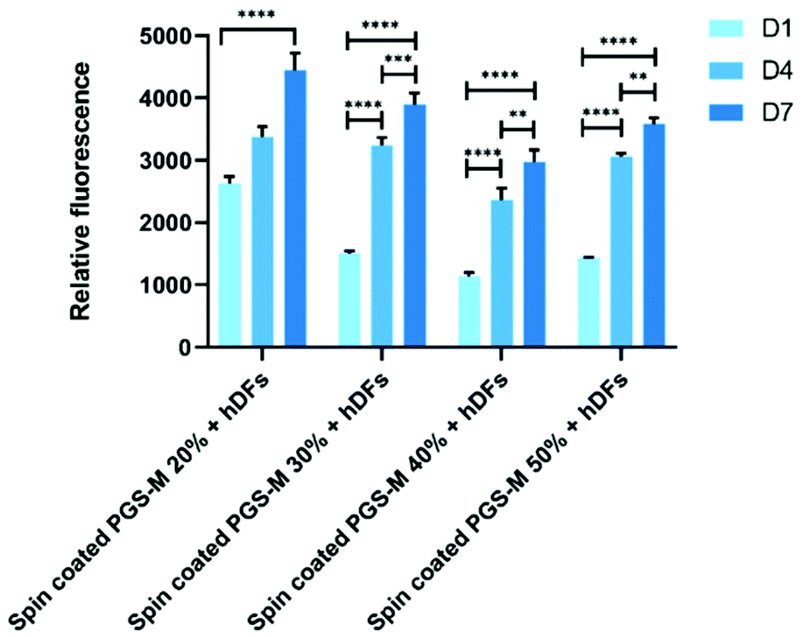 | ||
| Fig. 9 Resazurin reduction assay of hDF culture on spin coated PGS-M (20–50% DM). The assay was carried out at days 1, 4 and 7 (N = 3). Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by two-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered statistically significant (*). | ||
 | ||
| Fig. 10 LDH release assay of hDF culture on spin coated PGS-M (20–50% DM). The assay was carried out at days 1, 4 and 7. Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by two-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered statistically significant (*). | ||
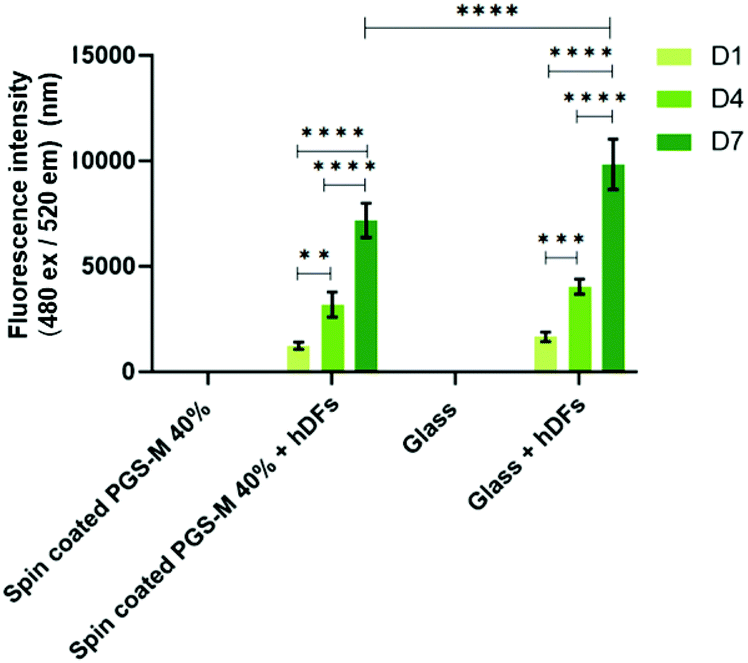 | ||
| Fig. 11 PicoGreen® DNA quantification assay of hDFs cultured on spin coated PGS-M (40% DM). The assay was carried out at days 1, 4 and 7. Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by two-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered statistically significant (*). | ||
In Fig. 9 and S8† it is possible to observe that hDFs show similar behaviour growing on PGS-M spin coated substrates, but cells grow better on lower DM. There is a significant difference with hDFs seeded in each DM and timepoint (P ≤ 0.0001).
The spontaneous LDH release in positive controls (hDF culture on borosilicate glass) was compared with spontaneous LDH release in hDF culture on spin coated PGS-M (Fig. 10 and S9†).
Spontaneous LDH release of hDFs seeded on spin coated PGS-M is lower for 20, 30 and 50% DM compared with the positive control. LDH release is higher in 40% DM, but it is still lower than the control (Fig. S9†).
PicoGreen® DNA quantification assay was carried out only on PGS-M 40% because it was the DM with mechanical properties that showed better match with the soft tissue requirements and with the lower percentage of sol content. Fig. 11 shows that the PicoGreen® DNA quantification assay is well correlated with the data obtained with resazurin reduction assay: hDFs grow on PGS-M spin coated substrates.
The cell morphology was evaluated and compared with the control. The cells were stained with phalloidin-FITC (495 nm excitation and 520 nm emission) (Fig. 12). hDFs grew well on spin coated PGS-M substrates, showing cell adhesion and proliferation. It is possible to observe that the cell morphology on all DMs (20 to 50%) is spindle-shaped since day 1. This suggests that cell behaviour is not affected by the DM and that PGS-M is a promising material for soft tissue engineering.
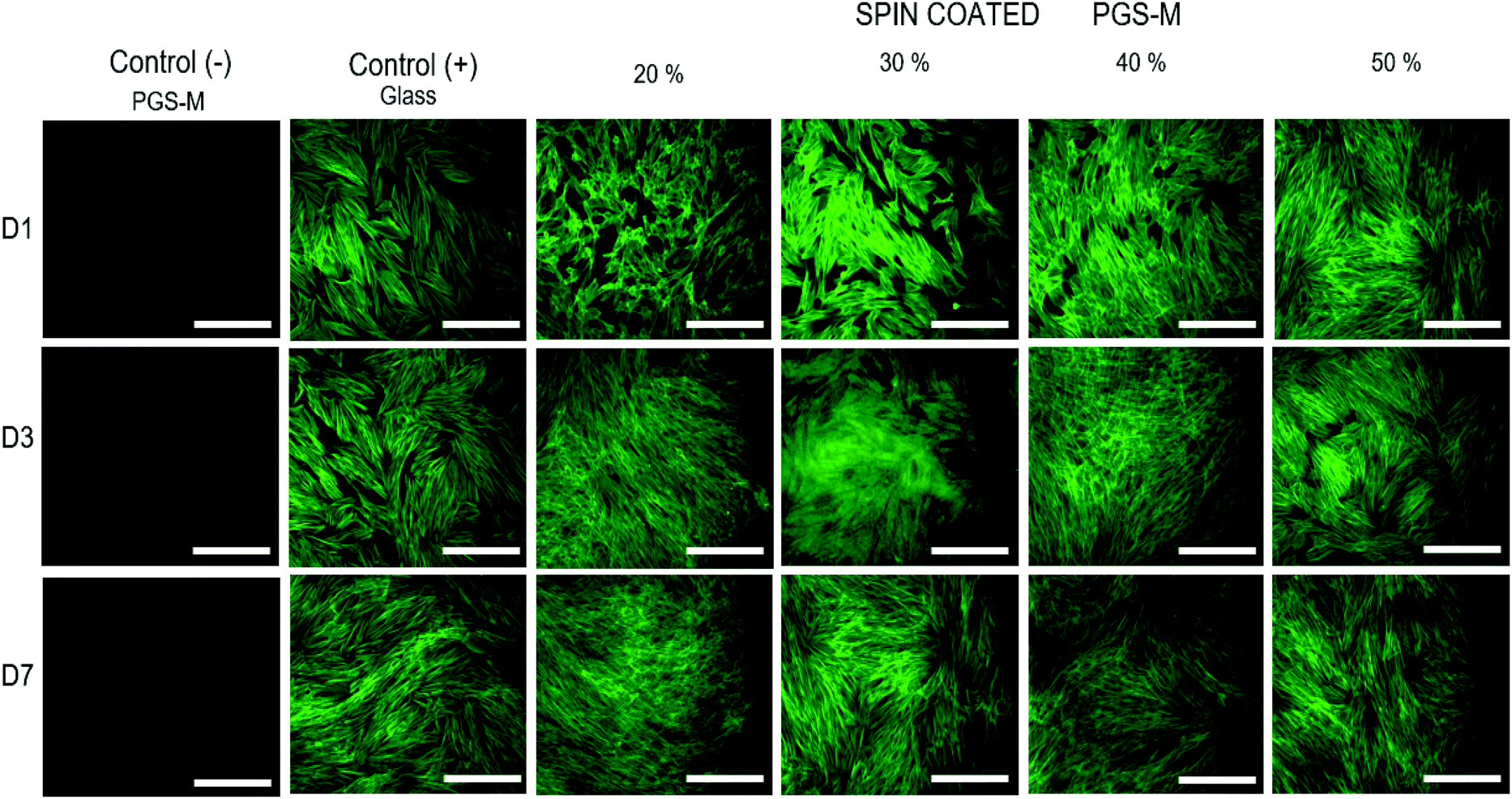 | ||
| Fig. 12 Fluorescence microscopy images of hDFs cultured on spin coated PGS-M (20–50% DM). Positive controls were hDFs cultured on uncoated borosilicate glass. Negative controls were PGS-M spin coated substrates. The images were taken at days 1, 4 and 7. Scale bars are 200 μm. All images were acquired using the same exposure and display settings. | ||
Porcine limbal fibroblast (pLF) culture on spin-coated PGS-M surfaces
pLFs were seeded on PGS-M spin coated substrates with 20, 30, 40, and 50% DMs to evaluate materials cytotoxicity and proliferation with the resazurin reduction and LDH release assays in spin coated PGS-M 20–50% (Fig. 13, 14 and S10†). PicoGreen® DNA quantification assay was used to evaluate spin coated PGS-M 40% DM (Fig. 15).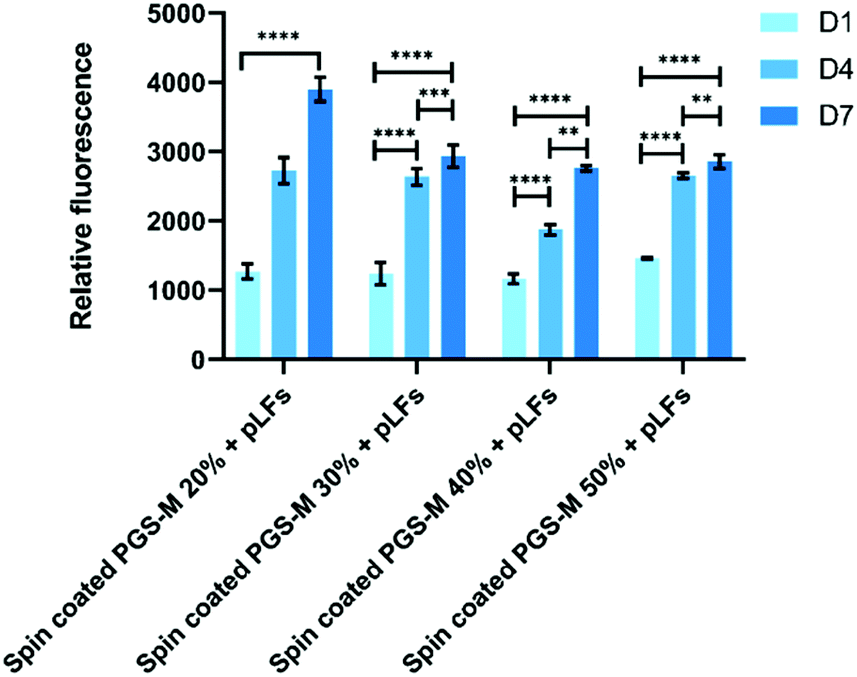 | ||
| Fig. 13 Resazurin reduction assay of pLFs cultured on spin coated PGS-M (20–50% DM). The assay was carried out at days 1, 4, and 7. Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by two-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered statistically significant (*). | ||
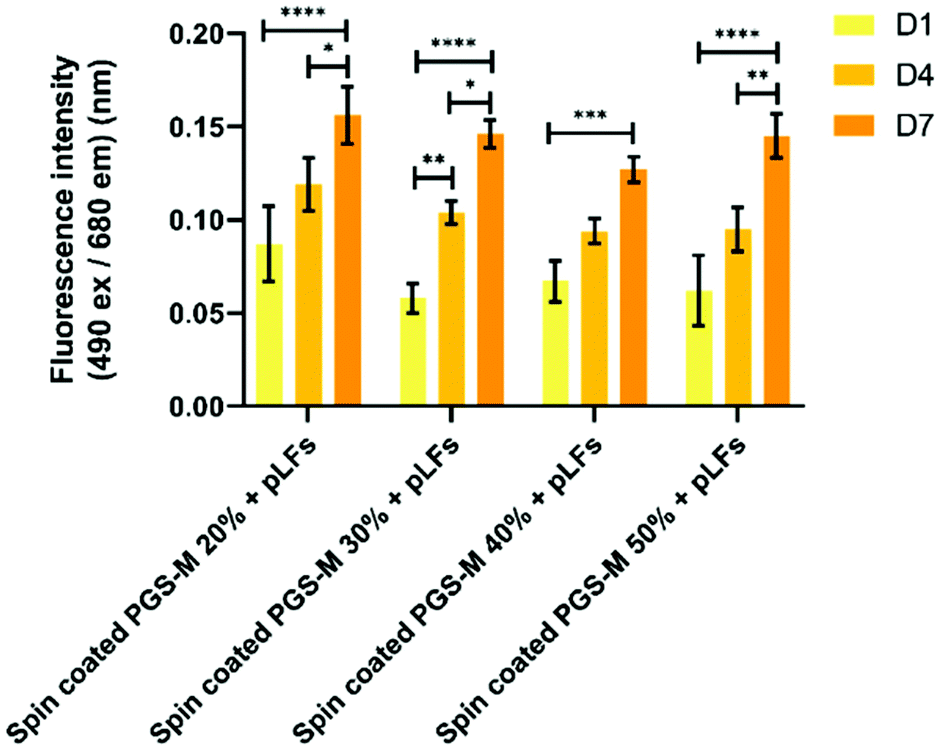 | ||
| Fig. 14 LDH release assay of pLFs cultured on spin coated PGS-M (20–50% DM). The assay was carried out at days 1, 4, and 7 (N = 3). Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by two-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered statistically significant (*). | ||
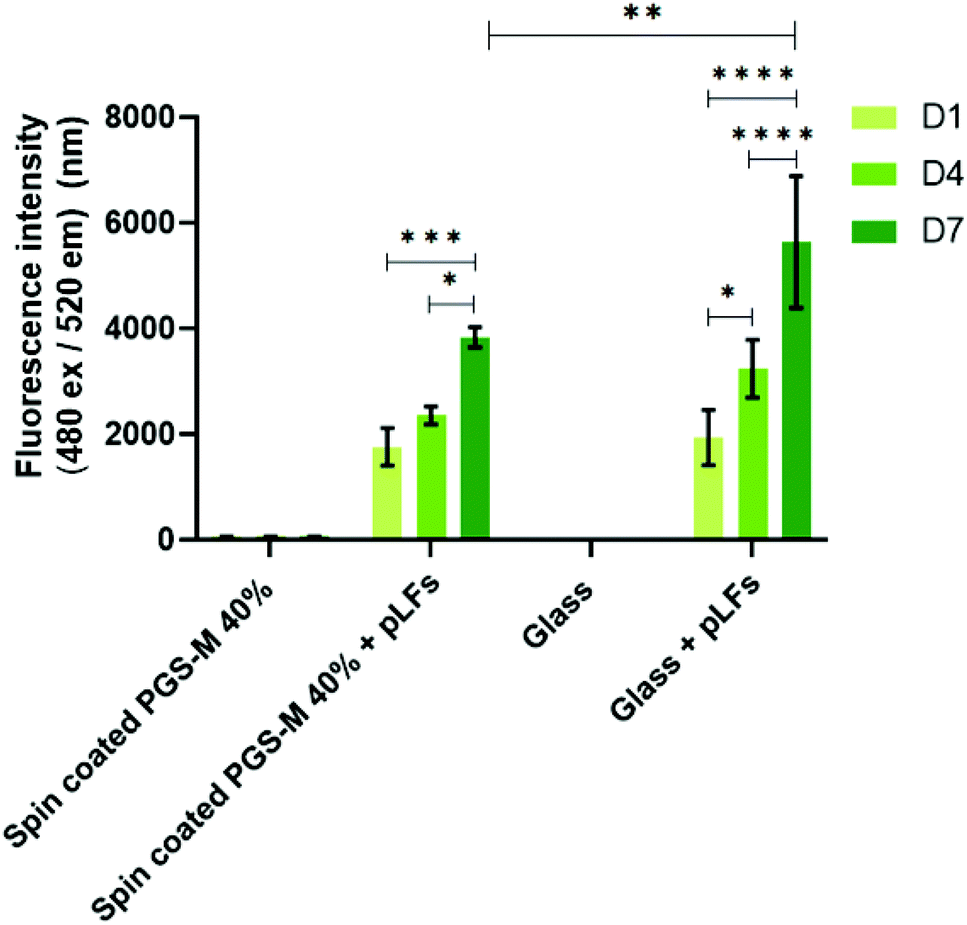 | ||
| Fig. 15 PicoGreen® DNA quantification assay of pLFs cultured on spin coated PGS-M (40% DM). The assay was carried out at days 1, 4, and 7 (N = 3). Samples show means and error bars corresponding to ±SD (N = 3, n = 3), analysed by two-way ANOVA and Tukey's post hoc pairwise comparison. P ≤ 0.05 was considered statistically significant (*). | ||
Fig. 13 shows that pLFs grow on PGS-M spin coated surfaces, with cells seemingly growing better on lower DM. The most likely cause of high proliferation is the low amount of gel fraction in lower DMs: around 60% gel in 20% DM which increases up to 80% gel in 50% DM. There is a significant difference in the cell growth in each time point for all the DMs (P ≤ 0.0001).
The spontaneous LDH release in positive controls (pLFs cultured on borosilicate glass) was compared with spontaneous LDH release in pLF culture on spin coated PGS-M (Fig. 14 and S11†).
Spontaneous LDH release of pLFs seeded on spin coated PGS-M is comparable with the one obtained in the positive control. Based on these results, we can infer that spin coated PGS-M substrates do not cause significant cytotoxicity regardless of DMs.
We decided to utilize the PicoGreen® DNA quantification assay only on PGS-M 40% as it is the DM with mechanical properties that better matched the soft tissue and had a lower percentage of sol content.
In Fig. 15 it is possible to observe that the PicoGreen® DNA quantification assay results corroborate the data obtained with resazurin reduction assay: pLFs grow on PGS-M spin coated substrates. There is no significant difference between pLFs seeded on PGS-M and pLFs seeded on glass at days 1 and 4, but there is a significant difference at day 7 (P ≤ 0.01).
The cell morphology on spin coated PGS-M substrates was also analysed by fluorescence microscopy. Image acquisition was carried out only with phalloidin-FITC (495 nm excitation and 520 nm emission) because PGS-M was found to show fluorescence at 405 nm excitation and 450 nm emission (which overlaps with DAPI absorption and emission) (Fig. 16).
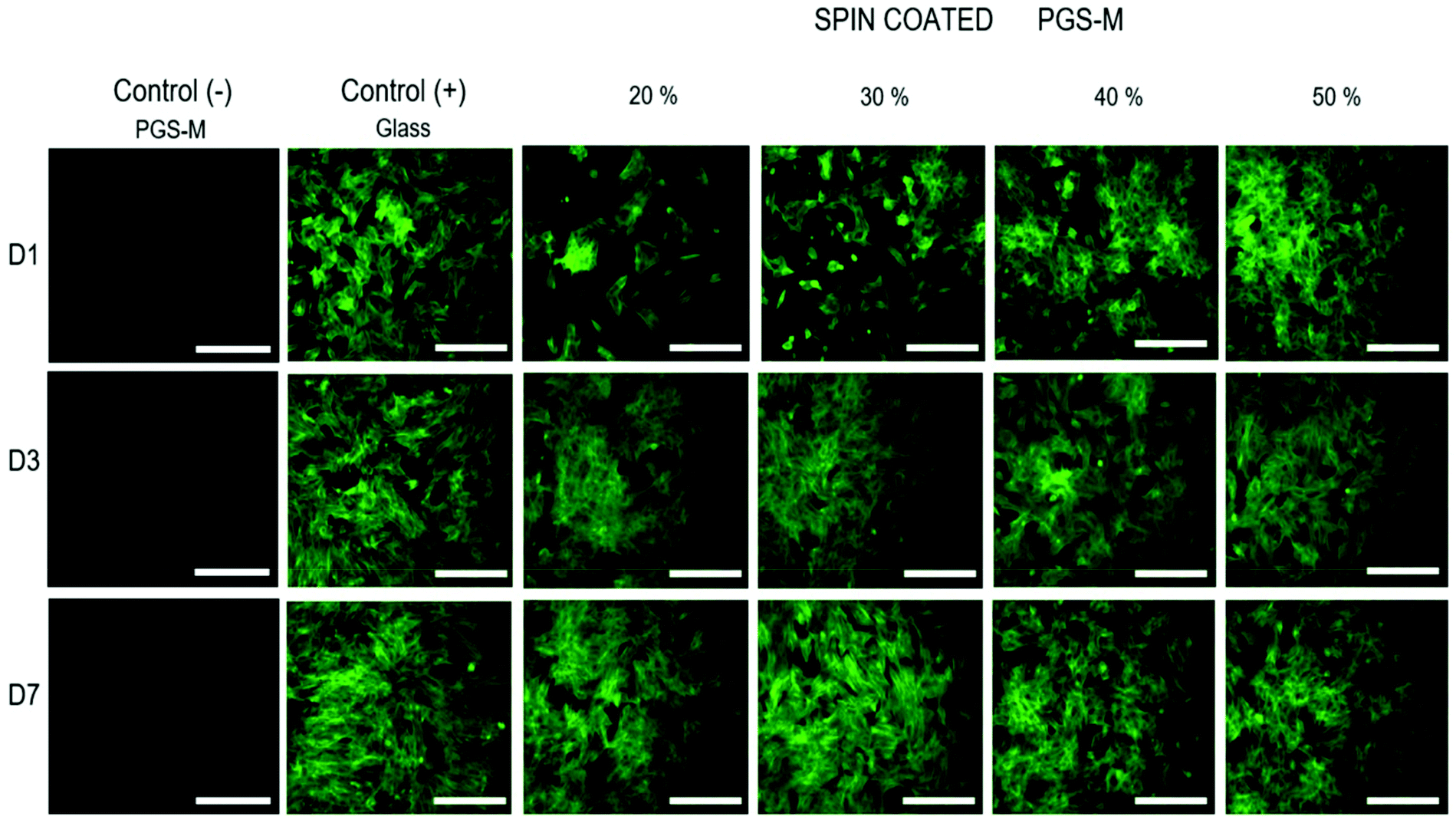 | ||
| Fig. 16 Fluorescence microscopy images of pLFs cultured on spin coated PGS-M (20–50% DM). Positive controls were pLFs cultured on uncoated borosilicate glass. Negative controls were PGS-M spin coated substrates. The images were taken at days 1, 4, and 7. All images were acquired using the same exposure and display settings. Scale bars are 200 μm. | ||
pLFs grew and proliferated on PGS-M spin coated surfaces after 7 days of culture, and the morphology of cells seeded on PGS-M surfaces is comparable with that of pLFs grown on glass. Morphology is an important feature that can tell us about cell behaviour, stability, and stress responses to a variety of conditions.64,65 Spindle-shaped fibroblasts are observed on glass and PGS-M substrates. Interestingly, lower DMs seem to affect pLF growth in D1. This can be caused by the lower stiffness in lower DMs, as was reported previously by Yeung et al., where it was shown that fibroblasts grew better on stiffer substrates. Also, Jones et al. demonstrated that stiffer collagen gels promote cell differentiation using hLECs.66 Cells grown on softer substrates presented a rounded shape like pLFs on PGS-M surfaces in this present work.67 Also, the high amount of sol fraction in lower DMs can affect cell growth as was observed in the resazurin assays. Previous reports show that the cell morphology can be a response of the interaction with a material (mechanical, biochemical and architectural features).68–70
Conclusion
PGS-M with different degrees of methacrylation was successfully synthesized through the standard synthesis for evaluation as a biomaterial for soft tissue engineering. The physicochemical properties of the polymer were analysed using different characterization techniques. Based on the data, all the DMs match the mechanical properties of soft tissue. During chemical characterisation (FTIR and NMR analyses), the addition of methacrylate groups was confirmed, by the analysis of the peaks related to the methacrylate groups. The degradation of PGS-M was only carried out in vitro (PBS): further evaluation is still necessary under physiological conditions that resemble the physiological environment. The low degradation rate of PGS-M has advantages for biomedical applications, such as stable long-term implants that retain their shape, low risk of uncontrollable degradation, and unvarying mechanical properties. The optimization of the PGS-M degradation rate opens a new area of research for using different techniques that allow the synthesis of PGS-M scaffolds with different physical characteristics. In comparison with the bulk material which undergoes surface degradation, structures such as fibres and pores may allow a more uniform degradation both within and throughout the scaffold. In addition, the combination of PGS-M with other polymers with better biodegradability such as PLA, PGA, and PHB, could positively impact its degradation rate, and are certainly an area of interest that can be explored in further work. Hydrophilic evaluation resulted in PGS-M being a more hydrophobic polymer than PGS, indicating that its biocompatibility still needs to be studied to determine suitability as a material for soft tissue engineering. hDFs and pLFs were seeded on PGS-M spin coated substrates and showed positive results on cell growth. The cell morphology was as expected on PGS-M substrates. Cell morphology is an important feature that can tell us about cell behaviour, stability, and stress responses to a variety of conditions. Cells seeded on lower DMs showed a slightly rounded shape with pseudopodial retraction in comparison with cells seeded on higher DMs and glass, which showed a spindle shape. Our hypothesis is that lower stiffness and higher percentages of sol fractions in lower DMs possibly affect cell growth based on resazurin reduction and LDH release assays. In conclusion, physiochemical characterisation of low DM PGS-M showed that it has excellent tuneable physicochemical properties, which are highly advantageous for soft tissue engineering. Further work with PGS-M should carefully select specific tissue by considering all evaluated parameters to best mimic the target tissue and improve cell proliferation.Author contributions
ICBR: conceptualization, data curation (equal), formal analysis, funding acquisition (supporting), investigation (lead), methodology (lead), project administration (lead), resources (equal), supervision (supporting), validation (lead), visualization (lead), writing – original draft (lead), and writing – review and editing (equal). FC: funding acquisition (lead), resources (equal), methodology (equal), supervision (equal), and validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant (CVU-559759) from the Mexican Federal Government through the National Council of Science and Technology and The University of Sheffield.Notes and references
- C. F. Guimarães, L. Gasperini, A. P. Marques and R. L. Reis, The stiffness of living tissues and its implications for tissue engineering, Nat. Rev. Mater., 2020, 5(5), 351–370, DOI:10.1038/s41578-019-0169-1.
- O. Janouskova, Synthetic Polymer Scaffolds for Soft Tissue Engineering, Physiol. Res., 2018, 67 Search PubMed.
- Y. Sang, M. Li, J. Liu, Y. Yao, Z. Ding and L. Wang, et al., Biomimetic Silk Scaffolds with an Amorphous Structure for Soft Tissue Engineering, ACS Appl. Mater. Interfaces, 2018, 10(11), 9290–9300 CrossRef CAS PubMed.
- B. Pei, W. Wang, Y. Fan, X. Wang, F. Watari and X. Li, Fiber-reinforced scaffolds in soft tissueengineering, Regener. Biomater., 2017, 257–268 CrossRef CAS PubMed.
- P. Kakkar, S. Verma, I. Manjubala and B. Madhan, Development of keratin-chitosan-gelatin composite scaffold for soft tissue engineering, Mater. Sci. Eng., C, 2014, 45, 343–347, DOI:10.1016/j.msec.2014.09.021.
- V. Perez-Puyana, M. Jiménez-Rosado, A. Romero and A. Guerrero, Polymer-based scaffolds for soft-tissue engineering, Polymers, 2020, 12(7), 1–21 CrossRef PubMed.
- A. Sionkowska, Biopolymeric nanocomposites for potential biomedical applications, Polym. Int., 2016, 65(10), 1123–1131 CrossRef CAS.
- Z. Ma, Z. Mao and C. Gao, Surface modification and property analysis of biomedical polymers used for tissue engineering, Colloids Surf., B, 2007, 60(2), 137–157 CrossRef CAS PubMed.
- D. Katti, R. Vasita and K. Shanmugam, Improved Biomaterials for Tissue Engineering Applications: Surface Modification of Polymers, Curr. Top. Med. Chem., 2008, 8(4), 341–353 CrossRef PubMed.
- H. Ye, K. Zhang, D. Kai, Z. Li and X. J. Loh, Polyester elastomers for soft tissue engineering, Chem. Soc. Rev., 2018, 47(12), 4545–4580 RSC.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, A tough biodegradable elastomer, Nat. Biotechnol., 2002, 20(6), 602–606 CrossRef CAS PubMed.
- R. Rai, M. Tallawi, A. Grigore and A. R. Boccaccini, Synthesis, properties and biomedical applications of poly(glycerol sebacate) (PGS): A review, Prog. Polym. Sci., 2012, 37(8), 1051–1078, DOI:10.1016/j.progpolymsci.2012.02.001.
- Y. Jia, W. Wang, X. Zhou, W. Nie, L. Chen and C. He, Synthesis and characterization of poly(glycerol sebacate)-based elastomeric copolyesters for tissue engineering applications, Polym. Chem., 2016, 7(14), 2553–2564 RSC . Available from: https://xlink.rsc.org/?DOI=C5PY01993A.
- Y. Li, W. D. Cook, C. Moorhoff, W. C. Huang and Q. Z. Chen, Synthesis, characterization and properties of biocompatible poly(glycerol sebacate) pre-polymer and gel, Polym. Int., 2013, 62(4), 534–547 CrossRef CAS.
- X. J. Loh, A. A. Karim and C. Owh, Poly(glycerol sebacate) biomaterial: synthesis and biomedical applications, J. Mater. Chem. B, 2015, 3(39), 7641–7652 RSC . Available from: https://xlink.rsc.org/?DOI:=C5TB01048A.
- Q. Liu, M. Tian, T. Ding, R. Shi, Y. Feng, L. Zhang, D. Chen and W. Tian, Preparation and Characterization of a Thermoplastic Poly(Glycerol Sebacate) Elastomer by Two-Step Method, Appl. Polym. Sci., 2007, 10, 1412–1419 CrossRef.
- D. Singh, A. J. Harding, E. Albadawi, F. M. Boissonade, J. W. Haycock and F. Claeyssens, Additive manufactured biodegradable poly(glycerol sebacate methacrylate) nerve guidance conduits, Acta Biomater., 2018, 78, 48–63, DOI:10.1016/j.actbio.2018.07.055.
- S. Pashneh-Tala, R. Owen, H. Bahmaee, S. Rekštytė, M. Malinauskas and F. Claeyssens, Synthesis, Characterization and 3D Micro-Structuring via 2-Photon Polymerization of Poly(glycerol sebacate)-Methacrylate–An Elastomeric Degradable Polymer, Front. Phys., 2018, 6, 41 CrossRef . Available from: https://journal.frontiersin.org/article/10.3389/fphy.2018.00041/full.
- C. L. E. Nijst, J. P. Bruggeman, J. M. Karp, L. Ferreira, A. Zumbuehl and C. J. Bettinger, et al., Synthesis and characterization of photocurable elastomers from poly(glycerol-co-sebacate), Biomacromolecules, 2007, 8(10), 3067–3073 CrossRef CAS PubMed.
- J. L. Ifkovits, J. J. Devlin, G. Eng, T. P. Martens, G. Vunjak-Novakovic and J. A. Burdick, Biodegradable fibrous scaffolds with tunable properties formed from photo-cross-linkable poly(glycerol sebacate), ACS Appl. Mater. Interfaces, 2009, 1(9), 1878–1886 CrossRef CAS PubMed.
- N. H. Nordin and Z. Ahmad, Monitoring chemical changes on the surface of borosilicate glass covers during the silanisation process, J. Phys. Sci., 2015, 26(2), 11–22 CAS.
- W. Guo and E. Ruckenstein, Modified glass fiber membrane and its application to membrane affinity chromatography, J. Membr. Sci., 2003, 215(1–2), 141–155 CrossRef CAS.
- F. Monticelli, M. Toledano, R. Osorio and M. Ferrari, Effect of temperature on the silane coupling agents when bonding core resin to quartz fiber posts, Dent. Mater., 2006, 22(11), 1024–1028 CrossRef CAS PubMed.
- P. Chaijareenont, H. Takahashi, N. Nishiyama and M. Arksornnukit, Effects of silane coupling agents and solutions of different polarity on PMMA bonding to alumina, Dent. Mater. J., 2012, 31(4), 610–616 CrossRef CAS PubMed.
- X. Zhang, C. Jia, X. Qiao, T. Liu and K. Sun, Porous poly(glycerol sebacate) (PGS) elastomer scaffolds for skin tissue engineering, Polym. Test., 2016, 54, 118–125, DOI:10.1016/j.polymertesting.2016.07.006.
- A. Patel, A. K. Gaharwar, G. Iviglia, H. Zhang, S. Mukundan and S. M. Mihaila, et al., Highly elastomeric poly(glycerol sebacate)-co-poly(ethylene glycol) amphiphilic block copolymers, Biomaterials, 2013, 34(16), 3970–3983, DOI:10.1016/j.biomaterials.2013.01.045.
- R. W. Silverstein and G. C. Bassler, Spectrometric identification of organic compounds, J. Chem. Educ., 1962, 39(11), 546–553 CrossRef CAS.
- E. Carletti and A. Motta, Scaffolds for Tissue Engineering and 3D Cell Culture, 3D Cell Cult., 2011, 17–39 CAS.
- A. Kopp, T. Derra, M. Müther, L. Jauer, J. H. Schleifenbaum and M. Voshage, et al., Influence of design and postprocessing parameters on the degradation behavior and mechanical properties of additively manufactured magnesium scaffolds, Acta Biomater., 2019, 98, 23–35 CrossRef CAS PubMed.
- H. Zhang, L. Zhou and W. Zhang, Control of scaffold degradation in tissue engineering: A review, Tissue Eng., Part B, 2014, 20(5), 492–502 CrossRef CAS PubMed.
- Z. Chen, J. You, X. Liu, S. Cooper, C. Hodge and G. Sutton, et al., Biomaterials for corneal bioengineering, Biomed. Mater., 2018, 13, 032002 CrossRef PubMed.
- S. Masterton and M. Ahearne, Mechanobiology of the corneal epithelium, Exp. Eye Res., 2018, 177(August), 122–129, DOI:10.1016/j.exer.2018.08.001.
- A. Shah, J. Brugnano, S. Sun, A. Vase and E. Orwin, The development of a tissue-engineered cornea: Biomaterials and culture methods, Pediatr. Res., 2008, 63(5), 535–544 CrossRef PubMed.
- M. Ahearne, J. Fernández-Pérez, S. Masterton, P. W. Madden and P. Bhattacharjee, Designing Scaffolds for Corneal Regeneration, Adv. Funct. Mater., 2020, 1908996 CrossRef CAS.
- C. Q. Zhao, W. G. Liu, Z. Y. Xu, J. G. Li, T. T. Huang and Y. J. Lu, et al., Chitosan ducts fabricated by extrusion-based 3D printing for soft-tissue engineering, Carbohydr. Polym., 2020, 236, 116058 CrossRef CAS PubMed.
- B. J. Blackburn, M. W. Jenkins, A. M. Rollins and W. J. Dupps, A review of structural and biomechanical changes in the cornea in aging, disease, and photochemical crosslinking, Front. Bioeng. Biotechnol., 2019, 7, 66 CrossRef PubMed.
- N. Garcia-Porta, P. Fernandes, A. Queiros, J. Salgado-Borges, M. Parafita-Mato and J. M. González-Méijome, Corneal Biomechanical Properties in Different Ocular Conditions and New Measurement Techniques, ISRN Ophthalmol., 2014, 2014, 1–19 CrossRef PubMed.
- H. Li, Y. S. Choi, M. W. Rutland and R. Atkin, Nanotribology of hydrogels with similar stiffness but different polymer and crosslinker concentrations, J. Colloid Interface Sci., 2020, 563, 347–353, DOI:10.1016/j.jcis.2019.12.045.
- B. Pukánszky, Influence of interface interaction on the ultimate tensile properties of polymer composites, Composites, 1990, 21(3), 255–262 CrossRef.
- D. Palomba, G. E. Vazquez and M. F. Díaz, Prediction of elongation at break for linear polymers, Chemom. Intell. Lab. Syst., 2014, 139, 121–131 CrossRef CAS.
- X. Duan and H. Sheardown, Dendrimer crosslinked collagen as a corneal tissue engineering scaffold: Mechanical properties and corneal epithelial cell interactions, Biomaterials, 2006, 27(26), 4608–4617 CrossRef CAS PubMed.
- H. Bakhshandeh, M. Soleimani, S. S. Hosseini, H. Hashemi, I. Shabani, A. Shafiee, A. H. B. Nejad, M. Erfan, R. Dinarvand and F. Atyabi, Poly (epsilon-caprolactone) nanofibrous ring surrounding a polyvinyl alcohol hydrogel for the development of a biocompatible two-part artificial cornea, Int. J. Nanomed., 2011, 6, 1509–1515 CAS.
- L. Jiang, Y. Jiang, J. Stiadle, X. Wang, L. Wang and Q. Li, Electrospun nanofibrous thermoplastic polyurethane/poly (glycerol sebacate) hybrid scaffolds for vocal fold tissue engineering applications, Mater. Sci. Eng. C Mater. Biol. Appl., 2019, 94, 740–749 CrossRef CAS PubMed.
- P. Kerativitayanan and A. K. Gaharwar, Elastomeric and mechanically stiff nanocomposites from poly(glycerol sebacate) and bioactive nanosilicates, Acta Biomater., 2015, 26, 34–44, DOI:10.1016/j.actbio.2015.08.025.
- W. Cai and L. Liu, Shape-memory effect of poly (glycerol-sebacate) elastomer, Mater. Lett., 2008, 62(14), 2175–2177 CrossRef CAS.
- S. Liang, W. D. Cook and Q. Chen, Physical characterization of poly(glycerol sebacate)/Bioglass ® composites, Polym. Int., 2012, 61(1), 17–22 CrossRef CAS.
- A. K. Gaharwar, A. Patel, A. Dolatshahi-Pirouz, H. Zhang, K. Rangarajan and G. Iviglia, et al., Elastomeric nanocomposite scaffolds made from poly(glycerol sebacate) chemically crosslinked with carbon nanotubes, Biomater. Sci., 2015, 3(1), 46–58 RSC.
- D. Lin, B. Cai, L. Wang, L. Cai, Z. Wang and J. Xie, et al., A viscoelastic PEGylated poly(glycerol sebacate)-based bilayer scaffold for cartilage regeneration in full-thickness osteochondral defect, Biomaterials, 2020, 253(April), 120095, DOI:10.1016/j.biomaterials.2020.120095.
- F. Khan and M. Tanaka, Designing smart biomaterials for tissue engineering, Int. J. Mol. Sci., 2018, 19(1), 1–14 Search PubMed.
- I. O. Smith, X. H. Liu, L. A. Smith and P. X. Ma, Nanostructured polymer scaffolds for tissue engineering and regenerative medicine, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1(2), 226–236 CAS.
- S. L. Liang, X. Y. Yang, X. Y. Fang, W. D. Cook, G. A. Thouas and Q. Z. Chen, In Vitro enzymatic degradation of poly (glycerol sebacate)-based materials, Biomaterials, 2011, 32(33), 8486–8496 CrossRef CAS PubMed.
- I. Pomerantseva, N. Krebs, A. Hart, C. M. Neville, A. Y. Huang and C. A. Sundback, Degradation behavior of poly(glycerol sebacate), J. Biomed. Mater. Res., Part A, 2008, 91(4), 1038–1047 CrossRef PubMed.
- C. A. Sundback, J. Y. Shyu, Y. Wang, W. C. Faquin, R. S. Langer and J. P. Vacanti, et al., Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material, Biomaterials, 2005, 26(27), 5454–5464 CrossRef CAS PubMed.
- C. C. Lau, M. Al Qaysi, N. Owji, M. K. Bayazit, J. Xie and J. C. Knowles, et al., Advanced biocomposites of poly(glycerol sebacate) and β-tricalcium phosphate by in situ microwave synthesis for bioapplication, Mater. Today Adv., 2020, 5, 100023, DOI:10.1016/j.mtadv.2019.100023.
- Q. Liu, M. Tian, R. Shi, L. Zhang, D. Chen and W. Tian, Structure and Properties of Thermoplastic Poly(glycerol sebacate) Elastomers Originating from Prepolymers with Different Molecular Weights, Appl. Polym. Sci., 2007, 104(5), 1131–1137 CrossRef CAS.
- Q. Z. Chen, A. Bismarck, U. Hansen, S. Junaid, M. Q. Tran and S. E. Harding, et al., Characterisation of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue, Biomaterials, 2008, 29(1), 47–57 CrossRef CAS PubMed.
- Y. Wang, Y. M. Kim, R. Langer and C. Truesdell, In vivo degradation characteristics of poly (glycerol sebacate), J. Chem. Phys., 2002, 37(85), 2336 Search PubMed . Available from: https://scitation.aip.org/content/aip/journal/jcp/37/10/10.1063/1.1733007.
- E. S. Yoo and S. S. Im, Effect of crystalline and amorphous structures on biodegradability of poly(tetramethylene succinate), J. Environ. Polym. Degrad., 1999, 7(1), 19–26 CrossRef CAS.
- P. Slepička, S. Trostová, N. S. Kasálková, Z. Kolská, P. Malinský, A. MacKová, L. Bačáková and V. Švorčík, et al., Nanostructuring of polymethylpentene by plasma and heat treatment for improved biocompatibility, Polym. Degrad. Stab., 2012, 97(7), 1075–1082 CrossRef.
- Z. Yang, H. Peng, W. Wang and T. Liu, Crystallization behavior of poly(ε-caprolactone)/layered double hydroxide nanocomposites, J. Appl. Polym. Sci., 2010, 116(5), 2658–2667 CAS.
- R. J. Schmidt, L. Y. Chung, A. M. Andrews and T. D. Turner, Toxicity of L-ascorbic acid to L929 fibroblast cultures: Relevance to biocompatibility testing of materials for use in wound management, J. Biomed. Mater. Res., 1993, 27(4), 521–530 CrossRef CAS PubMed.
- C. Wiegand and U. C. Hipler, Evaluation of biocompatibility and cytotoxicity using keratinocyte and fibroblast cultures, Skin Pharmacol. Physiol., 2009, 22(2), 74–82 CrossRef CAS PubMed.
- A. Pizzoferrato, G. Ciapetti, S. Stea, E. Cenni, C. R. Arciola and D. Granchi, et al., Cell culture methods for testing Biocompatibility, Clin. Mater., 1994, 15(3), 173–190 CrossRef CAS PubMed.
- Y. Tamada and Y. Ikada, Fibroblast growth on polymer surfaces and biosynthesis of collagen, J. Biomed. Mater. Res., 1994, 28(7), 783–789 CrossRef CAS PubMed.
- W. I. Welch and J. P. Suhan, Morphological study of the mammalian stress response: Characterization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance of intranuclear actin filaments in rat fibroblasts after heat-shock treatment, J. Cell Biol., 1985, 101(4), 1198–1211 CrossRef CAS PubMed.
- R. R. Jones, I. W. Hamley and C. J. Connon, Ex vivo expansion of limbal stem cells is affected by substrate properties, Stem Cell Res., 2012, 8(3), 403–409, DOI:10.1016/j.scr.2012.01.001.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz and M. Funaki, et al., Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion, Cell Motil. Cytoskeleton, 2005, 60(1), 24–34 CrossRef PubMed.
- I. E. Palamà, S. D'Amone, V. Arcadio, M. Biasiucci, A. Mezzi and B. Cortese, Cell mechanotactic and cytotoxic response to zinc oxide nanorods depends on substrate stiffness, Toxicol. Res., 2016, 5(6), 1699–1710 CrossRef PubMed.
- C. Dickens and C. Dickens, Nanomaterials for regenerative medicine: Chapter 22 MECHANICAL GUIDANCE OF CELL MIGRATION, in Nanomedicine, 2010. pp. 565–578. DOI:10.2217/nnm.10.146.
- B. Cortese, G. Gigli and M. Riehle, Mechanical gradient cues for guided cell motility and control of cell behavior on uniform substrates, Adv. Funct. Mater., 2009, 19(18), 2961–2968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00212d |
| This journal is © The Royal Society of Chemistry 2022 |