
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free transesterification vitrimers: activation via α-difluoroesters†
Florian
Cuminet
 ab,
Dimitri
Berne
a,
Sébastien
Lemouzy
a,
Éric
Dantras
b,
Christine
Joly-Duhamel
a,
Sylvain
Caillol
a,
Éric
Leclerc
a and
Vincent
Ladmiral
*a
ab,
Dimitri
Berne
a,
Sébastien
Lemouzy
a,
Éric
Dantras
b,
Christine
Joly-Duhamel
a,
Sylvain
Caillol
a,
Éric
Leclerc
a and
Vincent
Ladmiral
*a
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: Vincent.ladmiral@enscm.fr
bCIRIMAT, Université Toulouse 3 Paul Sabatier, Physique des Polymères, 118 Route de Narbonne, 31062 Toulouse, France
First published on 6th April 2022
Abstract
Transesterification vitrimers often require high catalyst loadings to achieve 3D networks reprocessable at moderately high temperature. The addition of an activating group close to the ester bonds allows to synthesize catalyst-free transesterification vitrimers. Here, we unveil the effect of the α-difluoromethylene group as a novel activating group for such materials. Fluorine features exceptional properties, in particular a strong electronegativity enabling CF2 groups to activate the epoxy-acid polymerization, and more interestingly also the transesterification reaction on adjacent esters. Consequently, this fluorinated group affords the easy synthesis of a highly crosslinked reprocessable material that do not require any metallic or organic catalyst. This vitrimer is endowed with advantageous reprocessing abilities and underwent 10 consecutive cycles without loss of mechanical properties. In brief, the vitrimer combines durability, recyclability and is catalyst-free. This discovery is one step further towards recyclable greener polymers.
Introduction
For decades, polymer materials have been classified depending on their macromolecular structure. On the one hand are amorphous thermoplastics, composed of entangled linear chains. In these materials, polymer chains are not covalently bonded, and are able to slide on one another.1 This translates, at the macroscopical scale, in their solubility in suitable solvents and in their ability to be reshaped upon heating. On the other hand are thermosets, composed of a 3D network. Theoretically, a thermoset is a single infinite macromolecule forming a covalently crosslinked network. This permanent structure leads to insolubility in solvents, and prevents recycling by simple thermal methods.1,2 In the pursuit of greener, more sustainable polymers, thermosets remain a challenge for recycling.3 Yet, they are assets in many applications for which thermal and chemical resistances are required. A third class of polymer materials consisting in a covalent crosslinked network in which the covalent bonds can be exchanged by reversible chemical reactions was proposed. They were called CANs, short for Covalent Adaptable Networks. In 2005, Bowman et al.4 synthesized a covalently bonded 3D network alike thermosets, endowed with covalent bonds able to exchange via an associative mechanism upon irradiation with visible light. This concept was further developed by Leibler et al.5 in 2011 with a material based on transesterification activated upon heating. This material was insoluble in common organic solvents even at high temperature, but able to flow and to be reshaped upon heating. Interestingly, this new kind of material exhibited an Arrhenian viscosity decrease with increasing temperature, a feature usually observed for inorganic glasses, or more generally for “strong” glasses in accordance with the work of Angell.6 This analogy inspired the name vitrimers for such materials. Since this discovery, other kinds of exchangeable bonds such as carbonates,7 boronic esters,8,9 disulfides,10,11 silyl ethers,12,13 urethanes,14–18 olefins,19 imines,20–23 vinylogous urethanes,24–26 diketoenamines,27 acylated acetals,28,29 and trialkylsulfonium salts30,31 have been investigated. Despite the success of vitrimers in materials research, they have yet to be implemented in industrial and commercial applications as they are not adapted to the recycling processes existing for thermoplastics.32 Moreover, long reprocessing times at high temperatures can trigger premature degradation of the vitrimer after repeated reshaping processes.33 To accelerate the reshaping process, one solution is the use of catalysts. However, some exchange reactions such as transesterification are so slow that they often require high loadings.5 Yet, the use of catalysts raise concerns about the materials ageing or the risk of leaching,34,35 which is undesirable for application such as food-contact materials for example. The first catalyst-free vitrimer was based on vinylogous urethanes exchange. This reaction is fast enough to afford short reprocessing times without catalyst.25 Since this seminal article, other examples of catalyst-free vitrimers based on fast exchange reactions were proposed, such as hydroxyurethanes,16 hemiacetal esters,28 trialkylsulfonium salts,31 oxime-esters33 and imines.36 Lowering the crosslinking density and increasing the number of exchangeable moieties were proven to enhance the reshaping abilities of catalyst-free materials.37–41 However, since these parameters also influence the materials mechanical properties, this strategy may not be suitable to a wide range of applications. To overcome the risk of catalyst leaching, some vitrimers were also synthesized with a catalyst (amines for instance) embedded in the network.42–44In 2015, Guan et al. proposed to implement in vitrimers the concept of neighboring group participation (NGP), well-known in the field of organic chemistry but new to the field of vitrimers.45 Materials crosslinked using difunctional dioxaborolane and possessing neighboring amino groups relaxed faster than the one deprived of such groups. The potential of internal catalysis or internal activation and neighboring group participation (NGP) to tune CANs has recently been reviewed.46,47 In their review, Van Lijsebetten et al.46 made a clear distinction between NGP for which the neighboring group is involved with a covalent bond at some point of the reaction, and the broader concept of internal catalysis including effects such as inductive effects.
The present work focuses on transesterification vitrimers, as they usually require a catalyst. To address the concerns raised by the use of external catalysts, some functional groups were reported to be efficient for NGP, such as phthalate monoesters48 or benzenesulfonic acid groups.49 Such groups modify the transesterification mechanism and facilitate the exchange. Examples of activation by inductive effect were reported on Meldrum's acids in PDMS50 or in polyimine vitrimers for instance.51 In transesterification vitrimers, malonates were used to activate the exchange reaction.52 Because of its high electronegativity, fluorine has a strong potential to activate bond exchanges. Indeed, the carbonyl group of fluorinated esters is known to be very electrophilic,53 which facilitates hydrolysis and nucleophilic attack.54,55 Fluorine substitution thus appear as a good strategy to activate transesterification reactions. Recently, a CF3 group positionned on the α carbon of esters was demonstrated to have a significant activating effect on transesterification in polyester networks.56 In the present work, fluorine atoms were added one atom closer to the ester bond and α-diflluoro esters are implemented for the first time to design a catalyst-free transesterification vitrimer. A trifunctional α-difluoro carboxylic acid was prepared and used in combination with a commercial diepoxide (butanediol diglycidyl ether, BDGE) to prepare an epoxy-acid network which displayed insolubility in organic solvents, but was able to be reshaped under relatively mild conditions without catalyst.
Results and discussion
α-Difluoro carboxylic acid monomer synthesis
Transesterification vitrimers require dangling hydroxy groups for the exchange reactions to happen. Fortunately, the epoxy opening by a carboxylic acid produces such groups in beta position to the ester. The strategy developed here therefore relies on the use of a trifunctional carboxylic acid monomer, and a commercial diepoxide to obtain a 3D network. To maximize the inductive effect of fluorine on the ester bond, the strongest fluorinated activating group (CF2) should be positioned as close as possible to the exchangeable bond. A trifunctional monomer bearing three α-difluorocarboxylic acids was thus designed and synthesized in two steps from a commercial triphenol (1,1,1-tris(4-hydroxyphenyl)ethane, “TPE”). TPE was reacted with ethyl bromodifluoroacetate at 70 °C for 40 hours to obtain by nucleophilic substitution a mixture of the di- and trisubstituted esters (15 and 60 mol% respectively). After column chromatography, the resulting α-difluoro triester then underwent facile saponification to yield, after appropriate workup, the desired α-difluoro triacid (TPE-TAF) as a white waxy solid with an overall yield of 50% (Scheme 1).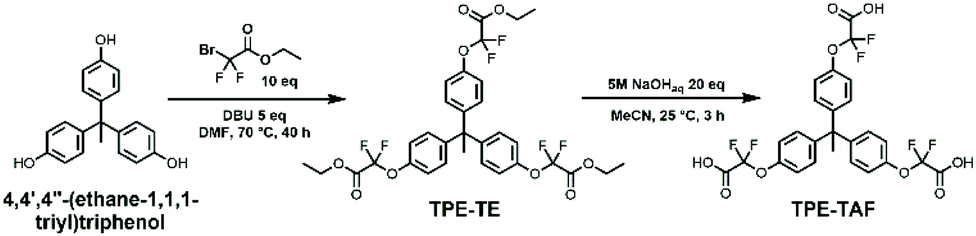 | ||
| Scheme 1 2-Step synthesis of the trifunctional α-difluoro carboxylic acid TPE-TAF. | ||
Polymerization and curing
Butanediol diglycidyl ether (BDGE, ESI Fig. S14, S15 and sections A, B†) was identified as a promising commercial epoxy resin, as its viscosity is very low and because it can dissolve TPE-TAF before gelation happens. In addition, its structure advantageously adds flexibility to the network, may counterbalance the rigidity of the TPE-TAF structure, and might prevent a high Tg which would make the reprocessability of the material more difficult (Scheme 2).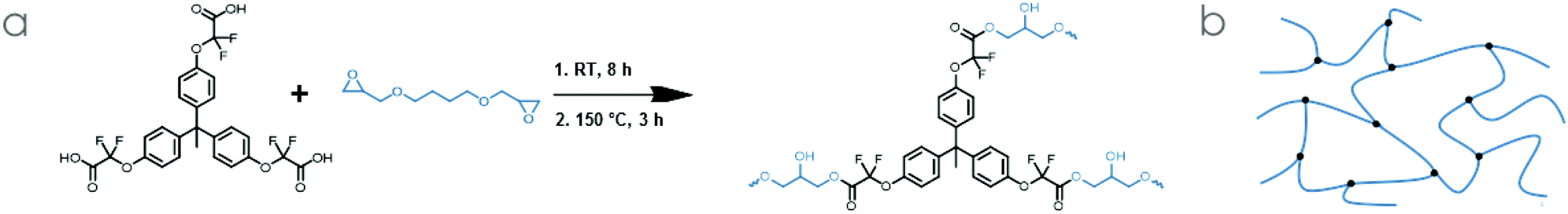 | ||
| Scheme 2 (a) Network synthesis from TPE-TAF and BDGE and (b) schematic representation of the network. | ||
TPE-TAF was dissolved in BDGE at room temperature (ca. 20 °C). The mixing time needed to be reduced to a few minutes only, to avoid the gelation caused by the fast reaction between the α-difluoro acids and the epoxides (even at room temperature) and ensure the homogeneity of the mixture. The mixture was then left at room temperature for at least 8 h, resulting into a brittle gel, which was then cured at 150 °C.
The gel time observed visually was consistent with the gel time determined by rheological analysis (Fig. S17†). At 20 °C, a gel time of 1.3 h was estimated at the crossover of loss and storage moduli, confirming that the epoxy opening by the α-difluoro carboxylic acid readily happens at room temperature. This behavior is highly unusual for epoxy-acid systems, which usually require catalysis and relatively high temperatures (80–120 °C).57 For catalyst-free systems, even higher temperatures (around 200 °C) are required.58 This fast polymerization reaction at low temperature strongly suggests the activating inductive effect of fluorine atoms on the carboxylic acid towards the epoxy-opening reaction.
The polymerization kinetics at room temperature were monitored via FTIR. The disappearance of the epoxy band at 908 cm−1 was clearly observed but the conversion of acid functions at 1758 cm−1 into esters at 1761 cm−1 could not be determined, because the acid and ester bands overlapped too much (Fig. S22†). The epoxy band decreased significantly for the first 6 h of reaction, and then decreased at a much lower rate until the end of the acquisition after 64 h (Fig. 1). This rate change was probably due to the gelation of the mixture which slowed down the reaction between the remaining reactive species.
 | ||
| Fig. 1 Evolution of the epoxy FTIR band at 908 cm−1 of the TPE-TAF/BDGE binary mixture versus time at room temperature (ca. 20 °C). | ||
The material obtained after 4 days at room temperature was left for 3 h in an oven at 150 °C to ensure complete polymerization. Three consecutive DSC ramps were carried out on the resulting material. The thermograms overlaid perfectly, did not show any residual exotherm and revealed a Tg of 47 °C which did not evolve after any of these heating ramps (Fig. S18†). The curing of the material was thus deemed complete.
Solubility tests were performed in acetone, THF, toluene, cyclohexane, DMSO, DCM and acetonitrile (Table S1†). The highest values were found for acetone and THF. In particular, after 24 h under agitation, an insoluble fraction of 94 ± 2% was measured in acetone (the best solvent for BDGE and TPE-TAF), thus proving that the curing process led to the formation of a 3D crosslinked material, as expected.
Vitrimer characterization
TGA under air showed that no significant mass loss was observed up to 200 °C, with a 2% and 5% degradation temperatures Td2% and Td5% of 205 °C and 262 °C respectively (Fig. S20† and Table 1). The Tg of the TPE-TAF/BDGE material was determined to be 47 °C (Fig. S18† and Table 1). TPE-TAF contains three aromatic cycles which bring rigidity to the polymer structure, whereas the linear structure of BDGE adds flexibility. This balance explains the moderate Tg value observed.A few preliminary reshaping trials allowed to set the reshaping temperature value at 100 °C. The material stability to reprocessing cycles was then determined by isothermal TGA experiments performed at 100 °C for 16 h under air, to simulate the oxidative environment during reprocessing (Fig. 2 and Table 1). After 4 h, a mass loss of 4.3% was observed. The value stabilized to 4.4% after 5 h and did not evolve afterwards. This loss might be due to the evaporation of remaining traces of solvents trapped in the TPE-TAF after synthesis.
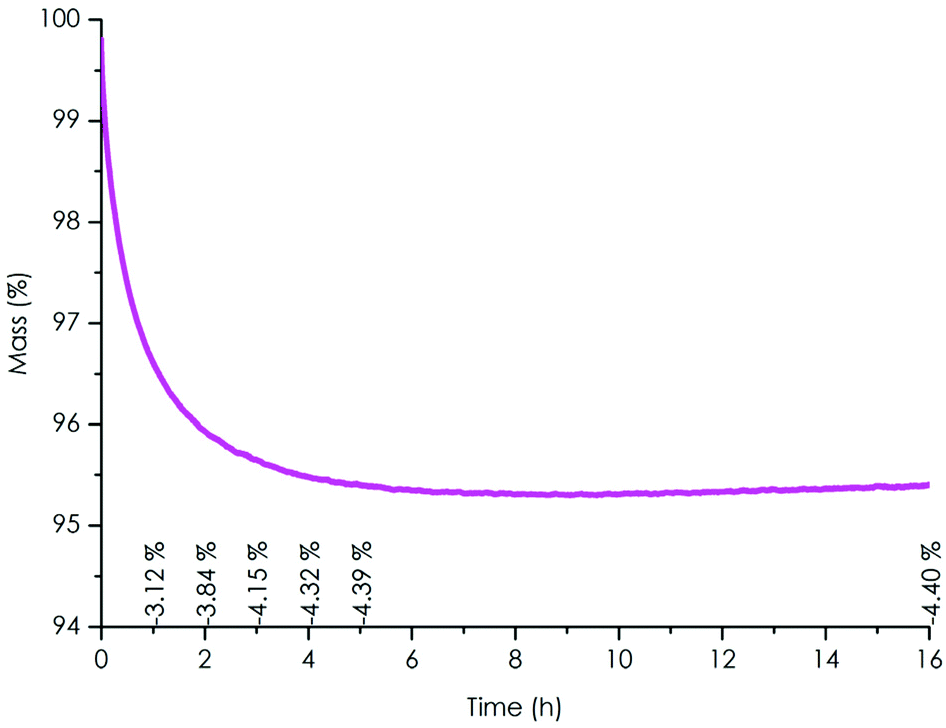 | ||
| Fig. 2 Isothermal TGA thermogram of TPE-TAF/BDGE material under air at 100 °C (% mass loss). | ||
The kinetics of the flow behavior of the material was studied using stress-relaxation experiments. The relaxation modulus was monitored with time between 170 and 210 °C with 10 °C steps (Fig. 3). It is important to state here that analogous non fluorinated polyester epoxy network prepared using 0.1 mol% Zn catalyst by Leibler et al. did not show vitrimer properties.5
 | ||
| Fig. 3 Normalized stress–relaxation curves from 170 to 210 °C with 10 °C steps fitted with the Kohlrausch–Williams–Watts equation (KWW) and τKWW relaxation times reported in the Arrhenius diagram (inset, R2 = 0.98985). | ||
Normalized stress-relaxation experimental curves shown in Fig. 3 reveal that, in the 170 °C–210 °C temperature range, the TPE-TAF/BDGE network relaxed the stress applied. This behavior is expected only if the network is able to reorganise. The fact that this material flowed on the rubbery plateau proves that fluorine atoms activated the transesterification. A contribution from minute amounts of unreacted carboxylic acid function might as well favor the transesterification.60 Furthermore, the relaxation rate depended on the temperature, as expected for a vitrimer.
The usual fitting model for such experiments is the exponential decay (Maxwell model). Nevertheless, this model did not fit well the experimental data obtained in the logarithmic time scale (R2 between 0.934 and 0.989). A Kohlrausch–Williams–Watts “stretched exponential” equation was found to better fit (see Table S2† for the equation and the fitting parameters) the stress relaxation dataset1 (R2 between 0.995 and 0.99975, depending on the temperature). In KWW model, a stretch parameter β is added to the exponential decay. The closer to 1 is β, the closer to the Maxwell model are the data. From 170 °C to 210 °C, the value of β was about 0.56 ± 0.02, indicating that the flow kinetics was associated with a distribution of relaxations rather than with a single relaxation time kinetics.1
The relaxation time values obtained from the KWW fitting equations were plotted in an Arrhenius diagram (Fig. 3 inset) to determine the value of the flow activation energy Ea. This Ea was determined to be 77 kJ mol−1, in good agreement with the 29–163 kJ mol−1 range reported so far for transesterification vitrimers,47 especially for catalyst-free vitrimers activated by neighboring groups for which the activation energy values are ranging from 78 to 94 kJ mol−1.42,59–61
The material relaxation observed in the absence of external catalyst demonstrated the activating effect on the transesterification of the two fluorine atoms located on the α-carbon of the esters, as non-catalyzed epoxy-acid networks usually exhibit no relaxation on a measurable time scale. As previously mentioned, tiny amounts of unreacted carboxylic acid, if present, could also add a contribution on this effect. A slight contribution from the phenoxy group is plausible, but would be much weaker than the contributions of the two fluorine atoms given the relative electronegativities of these elements (χF = 4.2, χO = 3.6). This is well illustrated by the pKas of acetic acid 4.7, glycolic acid 3.8, phenoxyacetic acid 3.2 and difluoroacetic acid 1.2.63,64 This range stresses out the difference in electron-withdrawing ability of two fluorine vs. one oxygen atom. The material behavior in temperature followed an Arrhenius law, as expected for a vitrimer. α-Difluoro esters are thus efficient activated esters for the design of catalyst-free transesterification vitrimers.
The discrepancy between the sluggishness in stress-relaxation experiments and the mild reprocessing conditions observed is striking. This difference can be explained by the pressure applied to the material,65,66 which is an important and often overlooked parameter. The force applied onto the material during reprocessing is 8.8 times the force applied during relaxation experiments. The reprocessing experiments are thus carried out at high pressure value in compression, which explains the difference in the behavior observed.
Reprocessing classical thermosets by compression molding is impossible, in contrast to vitrimers which possess exchangeable bonds. The TPE-TAF/BDGE material was successfully reprocessed using compression-molding further demonstrating its vitrimer character. 1 mm3 pieces of the material were reassembled into a small ribbon after 1.5 h at 100 °C under a 6 ton load. The required reprocessing conditions were relatively mild compared to transesterification vitrimers reported in literature, whether they are catalyzed or not (Fig. 4).5,37,40,44,48,52,62,67–72
 | ||
| Fig. 4 Comparison of TPE-TAF/BDGE reprocessing temperature, pressure and time with various catalyzed and catalyst-free transesterification vitrimers reported so far.5,37,40,44,48,52,62,67–72 | ||
Ten successive reprocessing cycles were successfully performed and each time homogeneous transparent samples were recovered. The color of the material did not significantly change with the successive reshaping cycles (Fig. 5). To quantitatively assess the material evolution with reprocessing, thermomechanical analyses were performed.
 | ||
| Fig. 5 TPE-TAF/BDGE aspect after several reprocessing cycles. | ||
The TPE-TAF/BDGE material evolution with reprocessing was studied using DMA experiments. The values of the storage modulus E′ in the glassy plateau and in the rubbery plateau regions, and the evolution of Tα allow to detect whether the TPE-TAF/BDGE material mechanical properties decrease after several reprocessing cycles. A ramp from −100 to +150 °C was performed after each reprocessing cycle to check whether the mechanical properties were fully recovered.
The pristine material exhibited a glassy plateau storage modulus E′G = 3.7 GPa, a rubbery plateau storage modulus E′R = 18 MPa and a Tα = 39 °C, which is consistent with the values reported in the literature37,70,72–74 for epoxy-acid and epoxy-anhydride vitrimers (Table 2 and Fig. S19†).
Upon the first three reprocessing cycles, the Tα increased from 39 °C to 41, 47 and 49 °C respectively, suggesting that the crosslink density slightly increased after each reshaping step, which is consistent with the increase of the rubbery plateau modulus (Table 2). After this 3rd reshaping process, the Tα value did not change with further reprocessing cycles up to the tenth cycle (Table 3).
| Reprocessing cycle | Pristine | 1 | 2 | 3–10 |
|---|---|---|---|---|
| T α (°C) | 39 | 41 | 47 | 49 |
The storage modulus (E′) value in the glassy plateau region slightly changed from 3.7 to 3.9 GPa for the pristine network and after the tenth reprocessing respectively. Similarly, the value of E′ in the rubbery plateau region increased by a mere 0.6 MPa (Table 2) (from 18.1 to 18.7 MPa for the pristine and 10th reprocessing cycle). These results show that there is no significant evolution of the network after 10 cycles.
Conclusion
In summary, polyester vitrimers were prepared out of a synthesized trifunctional α-difluoroacid and a commercially available difunctional epoxy resin. Thanks to the activation of the acid by the fluorine atoms, the epoxy-acid polymerization reaction happened readily at room temperature, whereas catalysts and high temperatures are usually needed for non-fluorinated systems. The resulting TPE-TAF/BDGE material was insoluble yet able to be reprocessed under mild conditions (100 °C, 1.5 h, 6 t). As expected, α-difluoro ester underwent significantly accelerated transesterification. This activation was such that no external catalyst was needed, in contrast to most other transesterification vitrimers. The flow activation energy Ea was evaluated to be 77 kJ mol−1 which is consistent with the values reported for transesterification vitrimers catalysed by internal amines for instance, but also very close to the value (67–72 kJ mol−1) obtained for α-CF3 activation.56 This polymer proved highly stable over repeated reprocessing cycles, with very little degradation of the mechanical properties observed after 10 cycles. The proof-of-concept based on the high electron withdrawing effect of fluorinated groups and exposed here is very promising to tune vitrimers properties and address the concerns related to the use of external catalysts, such as premature ageing, leaching of the catalyst or limited reprocessing abilities.34,35,75Experimental section
Materials
1,4-Butanediol diglycidyl ether (BDGE, Aldrich, ≥95%, NMR spectra, FTIR spectrum and EEW calculations in ESI Fig. S14, S15† and sections A, B), 1,1,1-Tris(4-hydroxyphenyl)ethane (“TPE”, Sigma-Aldrich, 99%), 1,8-Diazabicyclo(5.4.0)undec-7-ene (DBU, Fluorochem, 98%), ethyl bromodifluoroacetate (Fluorochem, 98%), benzophenone (Avocado Research Chemicals Ltd, 99%), succinic acid (ABCR, 99%) were used as received. Solvents were supplied by VWR Chemicals. Deuterated solvents were supplied by Eurisotop (99.8%).Synthetic procedures
Triester TPE-TE characterizations (NMR spectra in Fig. S1 to S3†): 1H NMR 400MHz CDCl3: δ 7.15–7.10 (m, 6H, aromatic protons, m-OCF2), 7.07–7.02 (m, 6H, aromatic protons, o-OCF2), 4.39 (q, 3J = 7.2 Hz, 6H, OCH2CH3), 2.15 (s, 3H, Ar3C-CH3), 1.36 (t, 3J = 7.1 Hz, 9H, OCH2CH3). 19F NMR 377 MHz, CDCl3: δ −76.41. 13C NMR 101 MHz, CDCl3: δ 159.9 (CO2Et, t, 2JC–F = 41.5 Hz), 148.0 (ipso-ArC-O, t, 3JC–F = 2.0 Hz), 146.5 (ortho-ArC-O, t, 4JC–F = 2.0 Hz), 129.9 (ipso-ArC-C-CH3), 121.2 (ortho-ArC-C-CH3), 114.1 (OCF2, t, 1JC–F = 272.5 Hz), 63.8 (O-CH2CH3), 51.7 (Ar-C-CH3), 30.9 (Ar-C-CH3), 14.0 (O-CH2CH3). Rf (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 5:1) = 0.42. HRMS (ESI+) Calc. for [M + Na]+ 695.1686, found 695.1677.
:ethyl acetate 5:1) = 0.42. HRMS (ESI+) Calc. for [M + Na]+ 695.1686, found 695.1677.
Disubstituted ester characterizations (NMR spectra Fig. S4 to S8†): 1H NMR 400MHz CDCl3: δ 7.14–7.08 (m, 4H, aromatic protons, m-OCF2), 7.07–7.03 (m, 4H aromatic protons, o-OCF2), 6.95–6.89 (m, 2H aromatic protons, m-OH), 6.79–6.72 (m, 2H, aromatic protons, o-OH), 5.06 (s, 1H, OH), 4.39 (q, 3J = 7.1 Hz, 4H, OCH2CH3), 2.12 (s, 3H, Ar3C-CH3), 1.36 (t, 3J = 7.2 Hz, 6H, OCH2CH3). 19F NMR 377 MHz, CDCl3: δ −76.34. 13C NMR 101 MHz, CDCl3: δ 160.0 (CO2Et, t, 2JC–F = 41.2 Hz), 154.1, 147.7 (ipso-ArC-O, t, 3JC–F = 2.0 Hz), 147.2, 140.6, 129.9, 129.9, 121.1 (ortho-ArC-O t, 4JC–F = 0.7 Hz), 115.0, 114.1 (OCF2, t, 1JC–F = 272.4 Hz), 63.8 (O-CH2CH3), 51.4 (Ar-C-CH3), 30.9 (Ar-C-CH3), 14.0 (O-CH2CH3). Rf (petroleum ether:ethyl acetate 5:1) = 0.30.
Trifunctional acid TPE-TAF characterizations (NMR spectra, FTIR spectrum and TGA thermogram in Fig. S9 to S13†): 1H NMR 400MHz d6-acetone: δ 7.89 (br s, 3H, COOH), 7.20 (m, 12H, aromatic protons), 2.22 (s, 3H, C-CH3). 19F NMR 377 MHz, d6-acetone: δ −77.50. 13C NMR 101 MHz, d6-acetone: δ 160.9 (COOH, t, 2JC–F = 40.7 Hz), 148.9 (ipso-ArC-O, t, 3JC–F = 2.0 Hz), 147.6, 130.8, 121.8, 115.3 (OCF2, t, 1JC–F = 270.9 Hz), 52.4 (C-CH3), 30.9 (C-CH3). HRMS (ESI+) Calc. for [M + Na]+ 611.0747, found 611.0750.
Determination of the epoxy equivalent weight (EEW)
The EEW of the BDGE was evaluated by NMR titration using benzophenone as standard in deuterated chloroform (experimental details are given in ESI section A†). This value was confirmed by DSC studies using succinic acid as the curing agent (experimental details are given in ESI section B†). The procedure was described in a previous article.76 Several BDGE/succinic acid ratios were used to make a series of thermosets. The highest Tg was achieved for a 1:1 stoichiometry, from which the EEW was calculated.
“TPE-TAF/BDGE” vitrimer
Typically, 1.156 g (5.90 meq COOH) of TPE-TAF was quickly mixed manually in a 10 mL beaker with 0.682 g (5.93 meq epoxy) of BDGE at room temperature (ca. 20 °C) until the acid was fully dissolved. A clear yellowish viscous liquid was obtained and quickly cast in PTFE molds. The mixture was left at least 8 h at room temperature for gelation. The resulting material (TPE-TAF/BDGE) was then removed from the molds and cured 3 h at 150 °C.Instrumentation
Author contributions
Conceptualization: FC, SC, ED, SL, EL, VL. Funding acquisition: SC, ED, EL, VL. Investigation: FC, SL. Methodology: all authors. Project administration: SC, ED, EL, VL. Supervision: FC, SC, ED, EL, VL. Validation: all authors. Visualization: FC. Writing – original draft: FC. Writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Institut Carnot Chimie Balard CIRIMAT (16CARN000801) and the French National Research Agency ANR (AFCAN project – ANR-19-CE06-0014). The authors would like to thank Lise Reymond for her contribution to TPE-TAF synthesis, as well as Dr Marc Guerre and Dr François Tournilhac for fruitful discussions.Notes and references
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- J.-P. Pascault, H. Sautereau, J. Verdu and R. J. J. Williams, Thermosetting Polymers, Marcel Dekker, Inc., New York, 2002 Search PubMed.
- S. J. Pickering, in Wiley Encyclopedia of Composites, John Wiley & Sons, Inc., Hoboken, New Jersey, 2012 Search PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- C. A. Angell, J. Non-Cryst. Solids, 1991, 131–133, 13–31 CrossRef CAS.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- F. Caffy and R. Nicolaÿ, Polym. Chem., 2019, 10, 3107–3115 RSC.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- Z. Ma, Y. Wang, J. Zhu, J. Yu and Z. Hu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1790–1799 CrossRef CAS.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- X. Wu, X. Yang, R. Yu, X. J. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A, 2018, 6, 10184–10188 RSC.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- N. Zheng, J. Hou, Y. Xu, Z. Fang, W. Zou, Q. Zhao and T. Xie, ACS Macro Lett., 2017, 6, 326–330 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- H. Zhang, D. Wang, W. Liu, P. Li, J. Liu, C. Liu, J. Zhang, N. Zhao and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2011–2018 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, K. Huang, Y. Liu and J. Zhu, Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- H. Zheng, Q. Liu, X. Lei, Y. Chen, B. Zhang and Q. Zhang, J. Mater. Sci., 2019, 54, 2690–2698 CrossRef CAS.
- R. Hajj, A. Duval, S. Dhers and L. Avérous, Macromolecules, 2020, 53, 3796–3805 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CrossRef CAS PubMed.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- D. Boucher, J. Madsen, N. Caussé, N. Pébère, V. Ladmiral and C. Negrell, Reactions, 2020, 1, 89–101 CrossRef.
- D. Boucher, J. Madsen, L. Yu, Q. Huang, N. Caussé, N. Pébère, V. Ladmiral and C. Negrell, Macromolecules, 2021, 54, 6772–6779 CrossRef CAS.
- Z. Tang, Y. Liu, Q. Huang, J. Zhao, B. Guo and L. Zhang, Green Chem., 2018, 20, 5454–5458 RSC.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS.
- W. Alabiso and S. Schlögl, Polymers, 2020, 12, 1660 CrossRef CAS PubMed.
- C. He, S. Shi, D. Wang, B. A. Helms and T. P. Russell, J. Am. Chem. Soc., 2019, 141, 13753–13757 CrossRef CAS PubMed.
- J. Wang, S. Chen, T. Lin, J. Ke, T. Chen, X. Wu and C. Lin, RSC Adv., 2020, 10, 39271–39276 RSC.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- C. Hao, T. Liu, S. Zhang, L. Brown, R. Li, J. Xin, T. Zhong, L. Jiang and J. Zhang, ChemSusChem, 2019, 12, 1049–1058 CrossRef CAS PubMed.
- S. Mu, Y. Zhang, J. Zhou, B. Wang and Z. Wang, ACS Sustainable Chem. Eng., 2020, 8, 5296–5304 CrossRef CAS.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS.
- C. Hao, T. Liu, S. Zhang, W. Liu, Y. Shan and J. Zhang, Macromolecules, 2020, 53, 3110–3118 CrossRef CAS.
- Y. Li, T. Liu, S. Zhang, L. Shao, M. Fei, H. Yu and J. Zhang, Green Chem., 2020, 22, 870–881 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- F. Cuminet, S. Caillol, E. Dantras, E. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- S. Wang, N. Teng, J. Dai, J. Liu, L. Cao, W. Zhao and X. Liu, Polymer, 2020, 210, 123004 CrossRef CAS.
- H. Zhang, S. Majumdar, R. A. T. M. Van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 9, 272–277 CrossRef CAS.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Polym. Chem., 2020, 8, 1–3 Search PubMed.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Chem. Sci., 2021, 12, 293–302 RSC.
- S. Debnath, S. Kaushal and U. Ojha, ACS Appl. Polym. Mater., 2020, 2, 1006–1013 CrossRef CAS.
- P. G. Blake and B. F. Shraydeh, Int. J. Chem. Kinet., 1981, 13, 463–471 CrossRef CAS.
- G. Schmeer and P. Sturm, Phys. Chem. Chem. Phys., 1999, 1, 1025–1030 RSC.
- J.-P. Brégué and D. Bonnet-Delpont, in Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons Inc., Hoboken, 2008 Search PubMed.
- D. Berne, F. Cuminet, S. Lemouzy, C. Joly-Duhamel, R. Poli, S. Caillol, E. Leclerc and V. Ladmiral, Macromolecules, 2022, acs.macromol.1c02538 Search PubMed.
- P.-J. Madec and E. Maréchal, Adv. Polym. Sci., 1985, 71, 153–228 CrossRef CAS.
- L. Shechter, J. Wynstra and R. P. Kurkjy, Ind. Eng. Chem., 1957, 49, 1107–1109 CrossRef CAS.
- M. Hayashi, ACS Appl. Polym. Mater., 2020, 2, 5365–5370 CrossRef CAS.
- A. Adjaoud, A. Trejo-Machin, L. Puchot and P. Verge, Polym. Chem., 2021, 12, 3276–3289 RSC.
- T. Liu, C. Hao, L. Shao, W. Kuang, L. Cosimbescu, K. L. Simmons and J. Zhang, Macromol. Rapid Commun., 2020, 2000458 Search PubMed.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. R. de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS.
- K. U. Goss, Environ. Sci. Technol., 2008, 42, 456–458 CrossRef CAS PubMed.
- N. V. Hayes and G. E. K. Branch, J. Am. Chem. Soc., 1943, 65, 1555–1564 CrossRef CAS.
- A. M. Hubbard, Y. Ren, D. Konkolewicz, A. Sarvestani, C. R. Picu, G. S. Kedziora, A. Roy, V. Varshney and D. Nepal, ACS Appl. Polym. Mater., 2021, 3, 1756–1766 CrossRef CAS.
- H. Fang, W. Ye, Y. Ding and H. H. Winter, Macromolecules, 2020, 53, 4855–4862 CrossRef CAS.
- H. Zhang, C. Cai, W. Liu, D. Li, J. Zhang, N. Zhao and J. Xu, Sci. Rep., 2017, 7, 11833 CrossRef PubMed.
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2017, 50, 6742–6751 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 108248 CrossRef CAS.
- T. Liu, S. Zhang, C. Hao, C. Verdi, W. Liu, H. Liu and J. Zhang, Macromol. Rapid Commun., 2019, 40, 1800889 CrossRef PubMed.
- M. Giebler, C. Sperling, S. Kaiser, I. Duretek and S. Schlögl, Polymers, 2020, 12, 1148 CrossRef CAS PubMed.
- Y. Liu, S. Ma, Q. Li, S. Wang, K. Huang, X. Xu, B. Wang and J. Zhu, Eur. Polym. J., 2020, 135, 109881 CrossRef CAS.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS.
- J. Han, T. Liu, S. Zhang, C. Hao, J. Xin, B. Guo and J. Zhang, Ind. Eng. Chem. Res., 2019, 58, 6466–6475 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- F. Jaillet, M. Desroches, R. Auvergne, B. Boutevin and S. Caillol, Eur. J. Lipid Sci. Technol., 2013, 115, 698–708 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 19F NMR spectra, FTIR spectra, TGA, DSC, DMA thermograms, EEW calculation details, gel time determination, gel content details and fitting parameters for stress relaxation experiments. See DOI: https://doi.org/10.1039/d2py00124a |
| This journal is © The Royal Society of Chemistry 2022 |