
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoresponsive oligo(ethylene glycol) methyl ether methacrylate based copolymers: composition and comonomer effect†
Qian
Li
 ,
Lezhi
Wang
,
Feihong
Chen
,
Anna P.
Constantinou
and
Theoni K.
Georgiou
*
,
Lezhi
Wang
,
Feihong
Chen
,
Anna P.
Constantinou
and
Theoni K.
Georgiou
*
Department of Materials, Imperial College London, Royal School of Mines, Exhibition Road, SW7 2AZ, London, UK. E-mail: t.georgiou@imperial.ac.uk
First published on 5th April 2022
Abstract
Thermoresponsive polymers based on oligo(ethylene glycol) (OEG) methyl ether methacrylate monomers have drawn much attention in recent years. In this investigation, copolymers based on oligo(ethylene glycol) methyl ether methacrylate (OEGMA, 300 g mol−1) and di(ethylene glycol) methyl ether methacrylate (DEGMA) or/and n-butyl methacrylate (n-BuMA) were successfully synthesised via group transfer polymerisation (GTP). The experimental molar mass was around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, while the OEGMA content was varied from 80% w/w to 50% w/w. Three different structures, including diblock bipolymer, diblock terpolymer and statistical copolymers, were synthesised and compared. The thermoresponsive properties of the copolymers were investigated in deionised water and phosphate buffered saline (PBS), and in aqueous mixtures with Pluronic® F127. Interestingly, while the diblock polymers solutions based on OEGMA and DEGMA were not able to form gels upon heating, these polymers were found to lower the critical gelation concentration (CGC) of Pluronic® F127 from 15% w/w to 10% w/w and increase the gelation temperature from room temperature to near body temperature.
000 g mol−1, while the OEGMA content was varied from 80% w/w to 50% w/w. Three different structures, including diblock bipolymer, diblock terpolymer and statistical copolymers, were synthesised and compared. The thermoresponsive properties of the copolymers were investigated in deionised water and phosphate buffered saline (PBS), and in aqueous mixtures with Pluronic® F127. Interestingly, while the diblock polymers solutions based on OEGMA and DEGMA were not able to form gels upon heating, these polymers were found to lower the critical gelation concentration (CGC) of Pluronic® F127 from 15% w/w to 10% w/w and increase the gelation temperature from room temperature to near body temperature.
Introduction
“Smart” materials, the physical properties of which can change in response to external stimuli such as pH, temperature, pressure, etc., have found a wide range of applications in different areas. These include self-healing materials, monitors, sensors, and biomedical devices and healthcare applications.1–8 A special type of “smart” materials are thermogels, which undergo a solution–gel (sol–gel) transition triggered by the change of the temperature and have been extensively investigated for biomedical applications.7–10 Thermogels, also called thermoresponsive gels, with a lower critical solution temperature (LCST) behaviour are preferred for in vivo applications, such as drug delivery,11–15 gene delivery,16–18 tissue engineering,14,19,20 injectable gel,7,14,21–25etc., because the sol–gel transition happens upon increasing the temperature.An intriguing group of thermoresponsive polymers that exhibit LCST behaviour are oligo(ethylene glycol) (OEG) based (meth)acrylate polymers.26–37 These polymers, when compared with other thermoresponsive polymers, present several advantages. For example, there is a wide choice of the OEG-based methacrylate monomers from hydrophobic ones, like mono(ethylene glycol) ethyl ether methacrylate (MEGMA), to hydrophilic ones, like nona(ethylene glycol) methyl ether methacrylate (NEGMA).1,26,29,38–43 Moreover, the thermoresponsive properties of the OEG-based methacrylate polymers can be easily tuned by changing the hydrophobic–hydrophilic balance of the polymer using different comonomers. These polymers are also non-ionic; thus, they will not affect the pH of the internal environment of human body and in fact EG-based polymers were found to be resistant to protein or cell absorption.19,44,45 Crucially, there are also biocompatible and EG based polymers in many FDA approved products.4,7,14,24,25,27,46 Therefore, polymers based on OEG-methacrylate monomers were extensively investigated. Oligo(ethylene glycol) methyl ether methacrylate (OEGMA) based polymers have been successfully synthesised and investigated for contact lenses,44,47 tissue engineering,48–52 drug delivery,22,53–58 and 3-D bioprinting.7,59–64 There were several papers focusing on the OEGMA-co-DEGMA [where DEGMA stands for di(ethylene glycol) methyl ether methacrylate] statistical copolymers.1,65–69 To the best of our knowledge, no study has previously reported the synthesis and investigation of diblock copolymers based on OEGMA and DEGMA. Here, we report the synthesis of (i) OEGMA–DEGMA copolymers, (ii) OEGMA based copolymers where n-butyl methacrylate (n-BuMA), a hydrophobic, non-ionic monomer was used for comparison, and (iii) terpolymers based of these three monomers. Their self-assembly and thermoresponsive properties were investigated. Furthermore, formulations i.e., mixtures of these polymers with Pluronic® F127 were investigated. Pluronic® F127 was chosen because it is the most commonly used thermoresponsive polymer in preclinical and clinical applications of thermoresponsive gels.3
Pluronic®, also known as poloxamer, is a commercially available thermoresponsive gelling system that consists of poly(ethylene glycol)-b-poly(propylene glycol)-b-poly(ethylene glycol) (PEG-PPG-PEG).19,70–72 The gelation temperature (Tgel) of Pluronic® can be influenced by the concentration, the composition of Pluronic (the length of the PEG and PPG), and the additives. The Pluronic® F127 specifically consists of EG at 70 mol% and PG at 30 mol%, with an average molar mass (MM) of 12600 g mol−1. Pluronic® F127 was reported with a critical gelation concentration (CGC) of 15% w/w and a critical gelation temperature (CGT) of 35 °C in deionised water.24,73–76 The Tgel decreases as the concentration of Pluronic® F127 increases. Specifically, at a concentration of 20% w/w the Tgel decreases to room temperature. The high CGC and low Tgel of Pluronic® F127 is limiting its use in injectable applications because it is difficult to handle and inject the formulation at room temperature.
To improve the applicability of Pluronic, studies on trying to tailor the CGC and CGT by adding salts have been reported.77–82 The addition of the salt can change the hydrogen bond formed between the Pluronic and water, and lead to the decrease of the Tgel, which is called “salting-out” effect, or the increase of the Tgel, which is called “salting-in” effect, depending on the chemical nature of the salt. For example, NaCl, KCl, MgSO4 and Na3PO4 cause the “salting-out” effect on the Pluronic, while NaSCN and KI cause the “salting-in” effect.77–81
Other studies focused on adding polymers to Pluronic and investigated how it affects the gelation behaviour of the CGT and CGC.71,73,74,83–85 The addition of PEG homopolymers in Pluronic formulations was reported firstly by Gilbert et al.84 It was reported that the addition of PEG homopolymers increase the gelation temperature of Pluronic®F127. Following this, many studies investigated mixing polymers with Pluronic®F127 and found that the polymeric additions could either increase or lower the Tgel and influence the gelation concentration (Cgel) of Pluronic®F127 depending on the copolymer added. For example, Pragatheeswaran and Chen70 investigated the addition of PEG with different molar masses (200, 600, 1k, 2k, 10k, 20k, and 35k g mol−1) to the 20% w/w Pluronic®F127 solutions, and found that short-chain PEG (MM < 1000 g mol−1) lowers the Tgel, while long-chain PEG (MM > 2000 g mol−1) either increases the Tgel or disrupts the gel formation. In another study by Ricardo et al.,71 it was found that the addition of PEG (MM = 6000, 35000 g mol−1) to 30% w/w Pluronic®F127 solutions, increased the Cgel. The addition of PEG6000 slightly decreases the Tgel, while the addition of PEG35000 increases the Tgel by 10 °C. It was also reported that when adding copolymers to Pluronic solutions,86,87 the hydrophobic content of the copolymer could enhance the sol–gel transition, decrease the Cgel and improve the encapsulation of the hydrophobic drugs.
It would be interesting to study the effect of mixing the PEG-methacrylate copolymers with Pluronic®F127 on the Tgel or/and Cgel. Therefore, in this study it was investigated how the composition, architecture, and the hydrophobic content influences the thermoresponsive behaviour of the copolymer based on OEG-methacrylate monomer(s). Furthermore, it was studied how the copolymer chemistry affects the thermogelling of Pluronic®F127 when these copolymers are used as additives. The target molar mass of the copolymer was 8100 g mol−1, and the OEGMA content was varied from 80% to 50%. The investigated architectures of the copolymers were statistical terpolymer, diblock bipolymer, and diblock terpolymer (AB diblock where one block was a statistical copolymer).
Experimental section
Materials
Fig. 1 shows the chemical structure and the abbreviations of the monomers, and the initiator used in this paper. The monomers: DEGMA (MM = 188.22 g mol−1, 95%), OEGMA (average MM = 300 g mol−1) and n-BuMA (MM = 142.20 g mol−1, 99%) were purchased from Aldrich.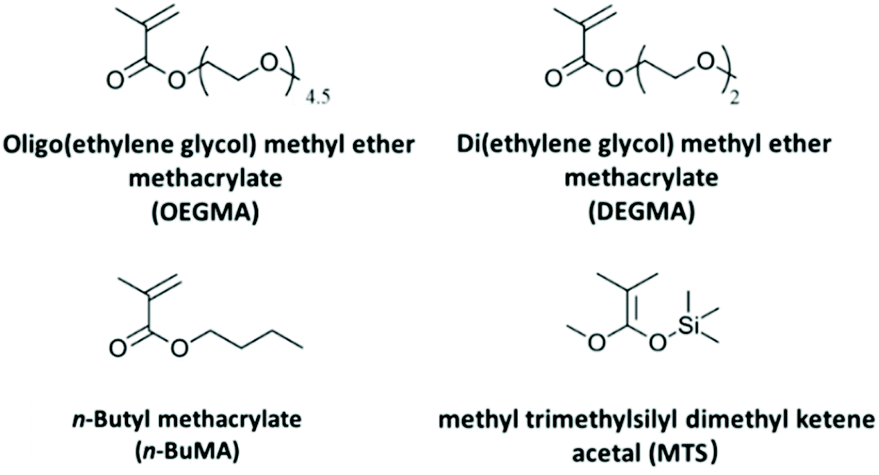 | ||
| Fig. 1 Chemical structures and abbreviations of the monomers and the initiator. | ||
The calcium hydride (CaH2, ≥90%) and the free radical inhibitor 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH), the initiator, methyl trimethylsilyl dimethyl ketene acetal (MTS, 95%), and the polymerisation solvent, tetrahydrofuran (THF, HPLC grade, ≥99.9%) were purchased from Aldrich.
The catalyst, tetrabutylammonium bibenzoate (TBABB), was in-house synthesized, following the procedure reported by Dicker et al.88
For the characterisation, the Proton Nuclear Magnetic Resonance (1H NMR) samples of the precursors and the final products were prepared in deuterated chloroform (chloroform-d, 99.8 atom% D, Aldrich). The solvent in chromatography, tetrahydrofuran (THF, GPC grade), and the visual test solvent, phosphate buffered saline (PBS, solution) were purchased from Fischer Scientific. The final products were precipitated in n-hexane, which was purchased from VWR chemicals.
Group transfer polymerisation (GTP)
All the polymers investigated in this paper were synthesised by group transfer polymerisation (GTP). As shown in Fig. 2, three different architectures were synthesised: diblock bipolymers, diblock terpolymer and statistical terpolymers. | ||
| Fig. 2 Schematic of the architectures of all the copolymers synthesised in this paper. From top to the bottom: OEGMA-b-DEGMA diblock polymer, OEGMA-b-BuMA diblock polymer, OEGMA-b-(DEGMA-co-BuMA) diblock terpolymer and OEGMA-co-DEGMA-co-BuMA statistical polymer. The dark red, blue, and brown represents the DEGMA, BuMA and OEGMA repeated units, respectively. | ||
The other diblock polymers were synthesised in a similar way. The amounts of the TBABB (∼10 mg) and MTS (0.0025 mol, 0.43 g) were kept the same for all the polymers. The concentration of reagents in solution was kept constant at 25% w/w.
The synthesis of the diblock terpolymers, i.e., one of the two blocks of the diblock copolymer is a random copolymer, was similar to the diblock bipolymers reported above.
For example, OEGMA22-b-(DEGMA4-co-BuMA6) was synthesised via sequential GTP similar to the polymers above. The first step, i.e., the homopolymerisation of OEGMA, was the same, but the second step included the copolymerisation of DEGMA and BuMA to form the second statistical block. The simultaneous addition speed of both monomers was kept as consistent as possible in order to allow random distribution of the units in the second block.
The synthesis of the statistical polymer was performed via simultaneous GTP of all three monomers. After the addition of the TBABB and THF, the three monomers, BuMA, DEGMA and OEGMA were added to the reaction flask. Then the polymerisation initiator (MTS) was added to the flask.
In order to collect the polymers after the polymerisation, the products were precipitated in n-hexane and then dried in vacuum oven for a week under room temperature.
Characterisation
000, 50000, and 100000 g mol−1.
The GPC samples were prepared by mixing 10 mg of the precursors/the final products and 1 mL of GPC solvent. The solvent was either pure THF or THF with 5% vol of triethylamine. The samples were filtered through a 0.45 μm PTFE syringe filter.
The measurements were conducted at room temperature (25 °C) and over a temperature range from 20 °C to 80 °C. The samples were allowed 3 min to equilibrate before each measurement. During the temperature ramp mode, the samples were measured with an increment of 5 °C first; more temperature points (at every 2 °C) were obtained near their respective cloud points for better accuracy.
Results and discussion
In this study, polymers based on BuMA, OEGMA and DEGMA were successfully synthesised via GTP. The molar mass was aimed at 8100 g mol−1, which was chosen from the range of the optimal molar mass for the gelation in similar thermogelling systems.89 The OEGMA mass fraction was varied from 80% w/w, 70% w/w, 65% w/w, 60% w/w to 50% w/w. Three different architectures including diblock bipolymer, diblock terpolymer and statistical copolymer, as shown in Fig. 2, were investigated.Molar mass characterisation
As shown in Table 1, the theoretical molar mass and composition was compared to the number average molar mass (Mn) given by GPC, and the composition given by NMR. From the GPC results, we found that the dispersity (Đ) of all the final products were smaller than 1.21, similar to previous reported studies on OEGMA based polymers,26,90–93 indicating a successful synthesis of well-defined polymers. The Mns of the final products were between 9400 g mol−1 and 11800 g mol−1, higher than the theoretical molar mass (8100 g mol−1). This is typical for GTP synthesis as it is highly sensitive to any protic impurities and in this case the OEGMA monomer was not able to be distilled due to its high MM. This can also be attributed to the difference in radius between OEGMA based polymers and the PMMA standards that were used to calibrate the GPC.
| Sample no. | Chemical structurea | Molar mass (g mol−1) | Đ (±0.01) | % w/w OEGMA–BuMA–DEGMA | ||
|---|---|---|---|---|---|---|
| MMtheoreticalb | M n (±250) | Theoretical | 1H NMRd | |||
| a DEGMA: di(ethylene glycol) methyl ether methacrylate, OEGMA: oligo(ethylene glycol) methyl ether methacrylate, BuMA: n-butyl methacrylate. b The theoretical molar mass was calculated as MMtheoretical = MMmonomer × DP + 100 g mol−1, here the MMmonomer was the molar mass of the monomer; the DP was the degree of polymerisation of the corresponding block; the 100 g mol−1 was the MM of the fragment of the MTS initiator remaining on the polymer backbone. c As determined by GPC using poly(methyl methacrylate) PMMA standard samples. d Calculated by the integration of the corresponding characteristic peak determined by the Jeol 400 MHz spectrometer. | ||||||
| 1 | OEGMA22 | 6400 | 8300 | 1.15 | 80-0-20 | 80-0-20 |
| OEGMA22-b-DEGMA9 | 8100 | 10300 |
1.17 | |||
| 2 | OEGMA19 | 5600 | 7800 | 1.10 | 70-0-30 | 74-0-26 |
| OEGMA19-b-DEGMA13 | 8100 | 10100 |
1.18 | |||
| 3 | OEGMA18 | 5200 | 7000 | 1.12 | 65-0-35 | 71-0-29 |
| OEGMA18-b-DEGMA15 | 8100 | 9500 | 1.20 | |||
| 4 | OEGMA16 | 4800 | 6200 | 1.11 | 60-0-40 | 64-0-36 |
| OEGMA16-b-DEGMA17 | 8100 | 10000 |
1.13 | |||
| 5 | OEGMA22 | 6400 | 7600 | 1.15 | 80-20-0 | 79-21-0 |
| OEGMA22-b-BuMA11 | 8100 | 9600 | 1.15 | |||
| 6 | OEGMA19 | 5600 | 7600 | 1.14 | 70-30-0 | 75-25-0 |
| OEGMA19-b-BuMA17 | 8100 | 10200 |
1.14 | |||
| 7 | OEGMA18 | 5200 | 7300 | 1.17 | 65-35-0 | 68-32-0 |
| OEGMA18-b-BuMA20 | 800 | 10700 |
1.19 | |||
| 8 | OEGMA22 | 4800 | 5700 | 1.18 | 60-40-0 | 64-36-0 |
| OEGMA22-b-DEGM31 | 8100 | 10200 |
1.21 | |||
| 9 | OEGMA22 | 6400 | 7600 | 1.15 | 80-10-10 | 86-7-7 |
| OEGMA22-b-(BuMA6-co-DEGMA4) | 800 | 9500 | 1.10 | |||
| 10 | OEGMA19 | 5600 | 6900 | 1.13 | 70-15-15 | 74-11-15 |
| OEGMA19-b-(BuMA9-co-DEGMA6) | 8100 | 9400 | 1.08 | |||
| 11 | OEGMA18 | 5200 | 6700 | 1.15 | 65-17.5-17.5 | 65-15-20 |
| OEGMA18-b-(BuMA10-co-DEGMA8) | 8100 | 9600 | 1.20 | |||
| 12 | OEGMA16 | 4800 | 6400 | 1.15 | 60-20-20 | 55-20-25 |
| OEGMA16-b-(BuMA11-co-DEGMA9) | 8100 | 10900 |
1.20 | |||
| 13 | OEGMA13-co-BuMA14-co-DEGMA11 | 8100 | 11800 |
1.17 | 50-25-25 | 53-21-26 |
The GPC chromatograms of OEGMA19-b-(BuMA9-co-DEGMA6) and its precursor (OEGMA19) are shown in Fig. 3 in dashed and solid lines, respectively. The GPC chromatograms of rest of the polymers can be found in Fig. S1 (in the ESI†). No monomer peaks were found in any of the chromatograms, indicating full conversion of the monomers to the polymers. The right shift of the peak confirms the addition of the DEGMA and BuMA monomers to the growing chain during the GTP. Both peaks are sharp and narrow, which is consistent with the narrow MMD. The experimental composition in Table 1 was calculated by using the integral of the characteristic peaks of the 3 monomers. As shown in the NMR spectrum (Fig. S2†), two peaks appeared at 3.28–3.36 ppm had each singlet assigned to the methoxy proton from OEGMA and DEGMA, respectively. Specifically, the peak at 3.32 ppm corresponds to the three methoxy protons from OEGMA; and the peak at 3.34 ppm corresponds to the three methoxy protons from DEGMA. The singlet at 3.88 ppm corresponds to the two methylene protons next to the ester bond (–OCH2CH2CH2CH3) of BuMA. These 3 peaks were used to calculate the experimental composition of the polymer. The deviation between the theoretical and the experimental composition is satisfactorily small, with the hydrophobicity trend of the polymers being followed experimentally.
 | ||
| Fig. 3 GPC chromatograms: (i) the precursor (OEGMA19) in solid line and (ii) the final product OEGMA19-b-(BuMA9-co-DEGMA6) in dashed line. | ||
Aqueous solution properties
The cloud point is a typical index to evaluate the thermoresponsiveness (if any) of polymers. It was determined as the temperature at which the transmittance at the wavelength of 550 nm of the 1% w/w solution dropped to 50%. The cloud points of the 1% w/w polymer solutions with various solvents given by the UV-VIS are listed in Table 2. The cloud point of a polymer can be influenced by the molar mass,26 composition,32,94–97 structure,98–101 architecture,62,91,102 and solvent.103,104
| Sample no. | Chemical structure | Cloud pointa (±1 °C) | ||
|---|---|---|---|---|
| DI water | PBS | 20% EtOH/80% H2O | ||
| a As determined by using a Cary 3500 Compact Peltier UV–Vis System (Agilent). The samples are 1% w/w DI solution in DI water, PBS, and 20% of EtOH and 80% of water. The measurements were carried out at 550 nm, and the heating rate was controlled at 1 °C min. | ||||
| 1 | OEGMA22-b-DEGMA9 | 58 | 56 | — |
| 2 | OEGMA19-b-DEGMA13 | 54 | 52 | — |
| 3 | OEGMA18-b-DEGMA15 | 53 | 51 | — |
| 4 | OEGMA16-b-DEGMA17 | 51 | 50 | 91 |
| 5 | OEGMA22-b-BuMA11 | 59 | 55 | — |
| 6 | OEGMA19-b-BuMA17 | 57 | 53 | — |
| 7 | OEGMA18-b-BuMA20 | 55 | 51 | — |
| 8 | OEGMA22-b-BuMA31 | 51 | 51 | — |
| 9 | OEGMA22-b-(BuMA6-co-DEGMA4) | 59 | 56 | — |
| 10 | OEGMA19-b-(BuMA9-co-DEGMA6) | 58 | 55 | — |
| 11 | OEGMA18-b-(BuMA10-co-DEGMA8) | 57 | 54 | 88 |
| 12 | OEGMA16-b-(BuMA11-co-DEGMA9) | 56 | 54 | 86 |
| 13 | OEGMA13-co-BuMA14-co-DEGMA11 | 26 | 26 | 25 |
As shown in Fig. 4, the cloud points tested in PBS were 1 °C–3 °C lower than the ones in deionised water which agrees with previous studies.1,30,32,68,105 Due to the ions in the PBS, the polar interactions between PEG moieties and water are sensitive to the interference from these well-hydrated anions in aqueous solutions, the cloud points of the polymer/PBS solutions are consequently lower compared to the polymer/deionised water solutions.
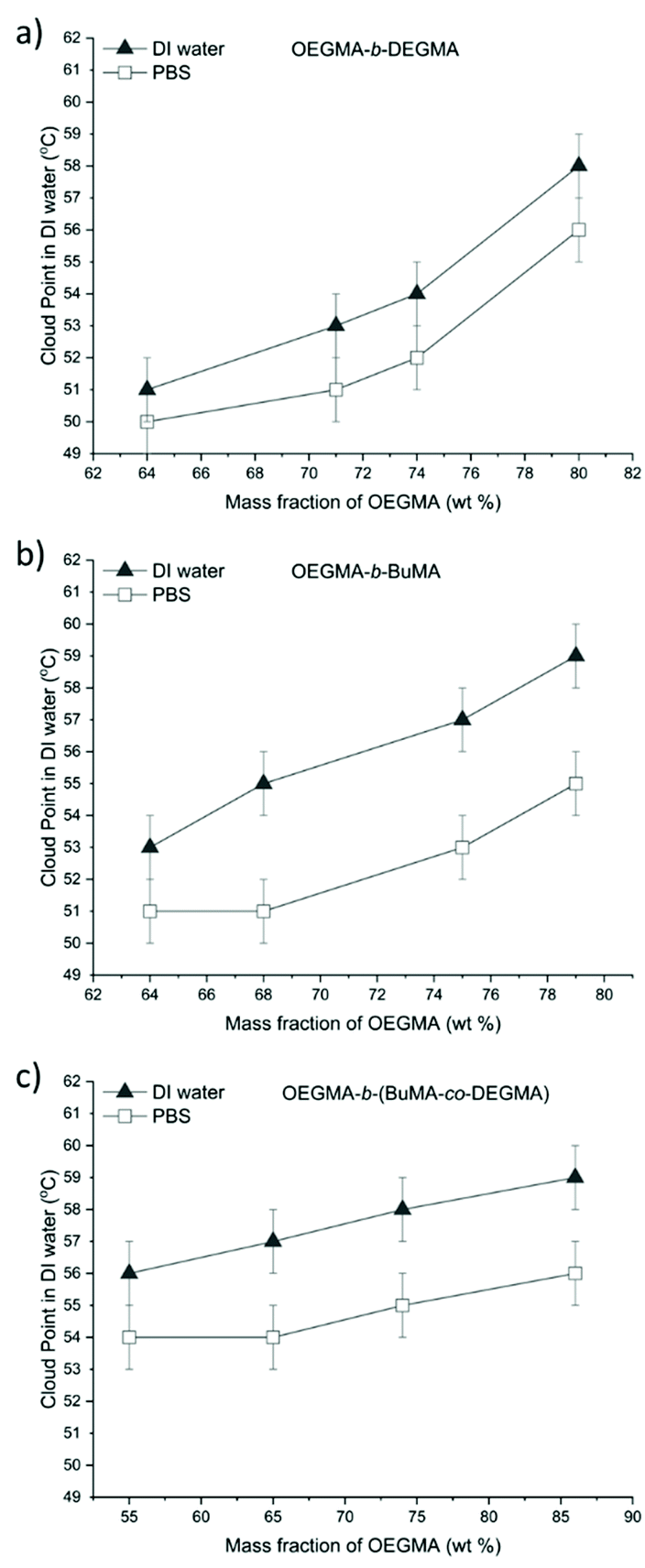 | ||
| Fig. 4 The CPs measured by UV-VIS of 1% w/w DI water and PBS solutions plotted against the OEGMA mass fraction. The black triangles and white squares represent the CP in DI water and PBS, respectively: (a) OEGMA-b-DEGMA; (b) OEGMA-b-BuMA; and (c) OEGMA-b-(BuMA-co-DEGMA). | ||
Furthermore, it can be clearly concluded that increasing the content of the more hydrophilic OEGMA monomer in any of the three different types of copolymers, the CP increases, as it was expected and observed before.1,42,48,106 In Fig. 5, all different copolymers are compared in DI water Fig. 5(a) and PBS, Fig. 5(b). The DEGMA containing diblock polymers at lower OEGMA content always present lower cloud point than the BuMA or BuMA-co-DEGMA containing copolymers.
 | ||
| Fig. 5 The CPs of the copolymers tested in (a) DI water (up) and (b) PBS (down). The black triangles, white squares and the half black stars represent the OEGMA-b-DEGMA, OEGMA-b-BuMA and OEGMA-b-(BuMA-co-DEGMA), respectively. | ||
When comparing the OEGMA-co-BuMA-co-DEGMA (OEGMA mass fraction of 53%) and the diblock terpolymer OEGMA-b-(BuMA-co-DEGMA) (OEGMA mass fraction of 55%), the CP of the statistical polymer (26 °C) was significantly lower than the CP of the diblock terpolymer (56 °C). The CPs of OEGMA-co-DEGMA statistical polymers were reported in many papers.1,65,67,107 According to Ramírez-Jiménez's study, the CPs of OEGMA-co-DEGMA statistical polymers were in the range of 32–56 °C, while in our study, the cloud points of OEGMA-b-DEGMA diblock polymers are in the range of 51–57 °C. Specifically, it was found that the OEGMA-co-DEGMA (with OEGMA mol fraction of 50%) exhibited a CP of 46 °C, while the diblock polymer OEGMA-b-DEGMA (with OEGMA mol fraction of 49%) exhibited a CP of 51 °C. This observation in consistent with other findings according to which the statistical polymers present lower CP than their corresponding block copolymers.62,108 This is because amphiphilic block copolymers can easily self-assemble to form micelles in the solution and stabilise themselves at higher temperatures, while the statistical polymers tend to precipitate due to their inability to form stable micelles. Specifically, statistical copolymers either do not form micelles and exist as unimers or self-assembly is observed if long side groups are present but the self-assembled structures have a different internal structure compared to block copolymers and are not as colloidally stable.
However, when comparing the OEGMA-b-BuMA and OEGMA-b-DEGMA diblock polymers with the diblock terpolymer, OEGMA-b-(BuMA-co-DEGMA) of similar composition, initially the OEGMA-b-(BuMA-co-DEGMA) had lower CP than the two diblock bipolymers, but this trend changes when the OEGMA content is increased above 75 wt%, as seen in Fig. 5. For example, the CP of the OEGMA-b-DEGMA, OEGMA-b-BuMA and OEGMA-b-(BuMA-co-DEGMA) when the OEGMA mass fraction was 64%, the CP was 51 °C, 56 °C and 57 °C, respectively, while above 75 wt% of OEGMA there is no difference between the CPs within experimental error. This could be attributed to the DEGMA block thermoresponsiveness. We believe that if micelles are formed at room temperature, when a permanently hydrophobic block is incorporated into the structure (OEGMA-b-BuMA), then they are colloidally more stable and thus precipitate at higher temperature compared to micelles that are formed upon thermoresponse (OEGMA-b-DEGMA). This was confirmed for the OEGMA-b-DEGMA copolymers by performing DLS at different temperatures.
Specifically, the CPs of PEGMA-b-DEGMA diblock polymers were confirmed by the temperature ramp on DLS. From the result of DLS, we found that the temperature of the micelle formation of PEGMA-b-DEGMA diblock polymers (with PEGMA mass fraction of 74%, 71% and 64%) was 2–4 °C lower than the CP given by the UV-VIS. This is discussed further in the hydrodynamic diameter section that follows.
| 〈dg2〉1/2 = 2 × (2 × 2.20 × (6.75 + DPtotal)/3)1/2 × 0.154 nm | (1) |
| d = (13.5 + DP1 + 2 × DP2) × 0.252 nm. | (2) |
 | ||
| Fig. 6 Schematic representation of left: random coil configuration; right: micelle configuration. With red, yellow, and blue the DEGMA, OEGMA and BuMA are represented, respectively. | ||
| Polymer no. | Theoretical structure | D h (nm) | ||
|---|---|---|---|---|
| Theoretical | Experimental (by intensity, 20 °C ± 0.5) | |||
| Micellea | Random coilb | |||
| a The theoretical diameter was calculated by assuming the polymer formed micelle in the solution based on the equation: d = (13.5 + DP1 + 2 × DP2) × 0.252 nm; here DP1 and DP2 is the degree of polymerisation of the first and second block, calculated based on the result of GPC and NMR, 15 is the converted DP of the ethylene glycol groups on the side chain. b The theoretical diameter was calculated by assuming the polymer formed random coil in the solution based on the equation: 〈dg2〉1/2 = 2 × (2 × 2.20 × (6.75 + DPtotal)/3)1/2 × 0.154 nm; here DPtotal is the total degree of polymerization of both blocks, calculated based on the result of GPC and NMR. | ||||
| P1 | OEGMA22-b-DEGMA9 | 20.2 | 2.5 | 5.6 |
| P2 | OEGMA19-b-DEGMA13 | 19.7 | 2.5 | 5.6 |
| P3 | OEGMA18-b-DEGMA15 | 18.7 | 2.4 | 5.6 |
| P4 | OEGMA16-b-DEGMA17 | 19.1 | 2.5 | 5.4 |
| P5 | OEGMA22-b-BuMA11 | 19.0 | 2.4 | 21.0 |
| P6 | OEGMA19-b-BuMA17 | 19.8 | 2.5 | 32.7 |
| P7 | OEGMA18-b-BuMA20 | 20.4 | 2.5 | 114.0 |
| P8 | OEGMA22-b-BuMA31 | 19.2 | 2.5 | 52.7 |
| P9 | OEGMA22-b-(BuMA6-co-DEGMA4) | 18.9 | 2.4 | 16.5 |
| P10 | OEGMA19-b-(BuMA9-co-DEGMA6) | 18.5 | 2.4 | 19.1 |
| P11 | OEGMA18-b-(BuMA10-co-DEGMA8) | 18.7 | 2.4 | 28.2 |
| P12 | OEGMA16-b-(BuMA11-co-DEGMA9) | 20.3 | 2.6 | 37.8 |
| P13 | OEGMA13-co-BuMA14-co-DEGMA11 | — | 2.9 | 18.2 |
It was found that the hydrodynamic diameters of OEGMA-b-DEGMA diblock polymers were small (5.4–5.6 nm) which is closer to the random coil theoretical calculation and suggests that the polymers are unimers and not self-assembling in a micelle. This was expected as both blocks in the OEGMA-b-DEGMA copolymers are hydrophilic at low temperatures, i.e., below the CP of DEGMA. Once DEGMA becomes hydrophobic at its CP, the diblock polymer is amphiphilic. Thus at 20 °C the OEGMA-b-DEGMA copolymers are considered as hydrophilic polymers, therefore the polymer exists as random coil in the solution, as confirmed by DLS.
The experimental hydrodynamic diameters of OEGMA22-b-BuMA11 and OEGMA19-b-(BuMA9-co-DEGMA6) were close to the theoretical values for micelles, confirming that the polymers are amphiphilic due to the presence of the hydrophobic BuMA and they form micelles. In some cases where the hydrophobic content of BuMA was higher, bigger aggregates were observed by DLS and this was supported visually as the polymer solutions appear slightly turbid at room temperature. The statistical copolymer was also not well soluble, so even though no self-assembly is expected, the diameter by DLS is higher compared to the random coil calculation. This is believed to be due to the poor solubility of the polymer and because some minor aggregation may be observed due to the OEG segments of the OEGMA monomer. Furthermore, since all monomers were polymerised simultaneously and the OEGMA monomer polymerises slower this polymer may have more of a tapered architecture where more OEGMA groups are at the end of the polymer chain. In that case it is expected to self-assemble similar to the OEGMA-b-(BuMA-co-DEGMA) architecture.
The DLS for the OEGMA-b-DEGMA series was also performed at different temperatures. Interestingly, as temperature increases, the increase of the hydrodynamic diameters of the OEGMA-b-DEGMA polymers was observed, as shown in Fig. 7. It was found that for all OEGMA-b-DEGMA polymers, the hydrodynamic diameters increased from a few nanometres to above 1000 nm as the temperature increases.
 | ||
| Fig. 7 Hydrodynamic diameters measured at different temperatures. The black triangles, red stars, blue squares, and the green circles represent the OEGMA-b-DEGMA with OEGMA mass fraction of 80%, 74%, 71% and 64%, respectively. The schematics of the random coils (<10 nm), micelles (10 nm < d < 100 nm), and aggregates (>500 nm) are also illustrated in the figure. | ||
As shown in Fig. 8, the intensity distribution of hydrodynamic diameters of P4: OEGMA16-b-DEGMA17 measured at 25, 48 °C (onset point) and 51 °C (CP) was plotted. From the combination of Fig. 7 and 8, the process of the micelle formation triggered by the temperature of the diblock polymers is self-evident. At room temperature, the hydrodynamic diameter measured at the highest intensity was 5.6 nm, indicating that the polymer chains existed as random coils in the solution. The other peak at around 300 nm was due to the smaller presence of some aggregates. At 48 °C, the hydrodynamic diameter at the highest intensity was 18.2 nm, indicating that the polymers self-assembled into micelles. At the CP of this diblock polymer (51 °C), micelles (21 nm) and aggregates of various sizes (531 nm and >1000 nm) were observed. Similar trends were observed for the other two OEGMA–DEGMA diblock polymers with intermediate OEGMA content. Similar observations on systems that form micelles when increasing the temperature was made by Hoogenboom et al.109–113
 | ||
| Fig. 8 Intensity distribution for OEGMA16-b-DEGMA17, (a) at 25 °C (room temperature, random coils and aggregations coexisted); (b) at 48 °C (onset temperature, micelles formed at this temperature); (c) at 51 °C (CP, micelles, aggregations, and precipitation coexisted). | ||
On the other hand, for OEGMA22-b-DEGMA9, which has the highest OEGMA content (80%), the hydrodynamic diameter was kept at 5.6 nm until the CP was reached. The hydrodynamic diameter suddenly increased to 500 nm at the CP. No intermediate micelle formation was detected during the temperature increase. Ramírez-Jiménez and co-workers1 reported similar observations on OEGMA-co-DEGMA statistical polymers. They also reported the sudden increase of the hydrodynamic diameter at the CP of the polymer. Thus, OEGMA22-b-DEGMA9 behaved differently from other OEGMA-b-DEGMA diblock polymers, like the statistical copolymers, due to its higher OEGMA content. Since the micelle formation is a thermodynamic process, and the stability of the micelle depends on the interaction of the polymer chain inside the hydrophobic core, the hydrophobic content of the polymer chain is crucial. The high OEGMA content of OEGMA22-b-DEGMA9 makes it difficult for this polymer to form stable micelles in the solution, and thus the polymer chains aggregate and collapse immediately when temperature reaches the CP.
For the ones able to form micelles during the heating process, the onset point of the micelle formation (the lowest temperature at which the hydrodynamic diameter starts to increase) was 2–4 °C lower than the cloud point of the diblock polymer, and higher than the cloud point of the DEGMA homopolymers (27–30 °C).26 For example, the OEGMA-b-DEGMA (OEGMA mass fraction of 74%) showed a CP at 54 °C, while the onset temperature was 50 °C. This observation confirms that the micelles self-assembled a few degrees before the CP. While for the OEGMA-b-DEGMA (OEGMA mass fraction of 80%) and the OEGMA-co-DEGMA statistical polymers in Ramírez-Jiménez's study,1 the onset point was very close (0.3 °C lower) to the CP. This difference indicates the formation of the micelles of our polymers. Since the statistical polymers are not able to form micelles, the polymer chains tend to entangle with each other due to the ‘hydrophobic effect’. Therefore, the polymer chain aggregated more easily and precipitated out of solution immediately. However, the diblock polymers reported in this study were able to form micelles to form a more stable system, thus broaden the temperature window for the polymer to stay dissolved in the solution.
Phase diagrams of copolymers in DI water
To investigate the effect of the concentration on the cloud point, the phase diagrams of all the polymers were constructed at the following concentrations: 2%, 5%, 10%, 15%, and 20% w/w. The images shown in Fig. 9, from (a) to (f), demonstrated what is visually observed when there is (a) a transparent solution, (b) a slightly cloudy solution, (c) cloudy solution, (d) a transparent gel, (e) a cloudy gel and (f) precipitation. | ||
| Fig. 9 Visual observations of (a) transparent solution, (b) slightly cloudy solution, (c) cloudy solution, (d) transparent gel, (e) cloudy gel, and (f) precipitation. | ||
The phase diagrams of OEGMA22-b-DEGMA9, OEGMA22-b-BuMA11 and OEGMA22-b-(BuMA11-co-DEGMA4) were plotted based on the observations of visual test as shown in Fig. 10. The polymer solution undergoes visually observable changes including transparent, slightly cloudy, cloudy and precipitation (phase separation) sequentially in response to an increasing temperature, with no gelation observed. The solutions of OEGMA-b-BuMA diblock polymers with OEGMA content of 64% and 68% were slightly cloudy at room temperature due the higher hydrophobicity of the BuMA monomers. From the phase diagrams of all the copolymers, it was found that the phase separation was not significantly affected by the concentration.
 | ||
| Fig. 10 Phase diagrams of OEGMA22-b-DEGMA9, OEGMA22-b-BuMA11 and OEGMA22-b-(BuMA11-co-DEGMA4). The concentration of the solution was varied from 1% w/w to 20% w/w. The transparent solution, slightly cloudy solution, cloudy solution, and precipitation was indicated by square, triangle, circle, and green square respectively. | ||
Mixtures of OEGMA based copolymers with Pluronic F127
Currently the only formulations of thermoresponsive gels that are in clinical trials involve Pluronic F127.3 In these formulations, Pluronic F127 is the main component but often other polymers are present too. Here we wanted to investigate the effect of adding the new synthesised polymers in Pluronic F127 formulations since these are of great interest. Specifically, to investigate how the copolymers influence the gelation behaviour of Pluronic®F127, visual tests were carried out across a range of temperatures and concentrations. In particular, the concentrations were varied in 2 ways: (1) the total concentration of the mixture was kept the same at 25% w/w, while the concentrations of Pluronic®F127 were varied from 5% w/w, 10% w/w, 12.5% w/w, 15% w/w to 20% w/w; (2) the concentration of the copolymer was kept the same at 5% w/w, while the concentrations of Pluronic®F127 were varied from 5% w/w, 10% w/w, 15% w/w and 20% w/w. The gelation temperatures were also confirmed by rheometer. The rheological curves can be found in ESI (Fig. S3, Tables S1 & S2†). One diblock copolymer from each different series OEGMA-b-DEGMA, OEGMA-b-BuMA and OEGMA-b-(BuMA-co-DEGMA) was mixed with Pluronic®F127 and the phase diagrams are shown in Fig. 11. To summarise the main results, the Tgels of the mixtures were plotted against the concentration of Pluronic®F127 and this are shown in Fig. 12. | ||
| Fig. 11 Phase diagram of the mixtures of the copolymers and Pluronic®F127. The concentration of the Pluronic®F127 in the solution was varied from 5% w/w to 20% w/w, with the overall concentration was kept constant at 25% w/w. The transparent solution, cloudy solution, precipitation, stable transparent gel, and stable cloudy gel was indicated by square, circle, green square, blue triangle, and blue circle, respectively. | ||
 | ||
| Fig. 12 Effect of different copolymers on the Tgel of Pluronic®F127. The black triangles, white squares, and black stars represent the OEGMA-b-DEGMA, OEGMA-b-BuMA, and OEGMA-b-(BuMA-co-DEGMA), respectively. | ||
The statistical polymer OEGMA-co-BuMA-co-DEGMA was also mixed with Pluronic®F127, however, the results are not presented in these figures and tables. This is because only the mixtures with (1) 5% w/w OEGMA-co-BuMA-co-DEGMA and 5% w/w Pluronic®F127; and (2) 5% w/w OEGMA-co-BuMA-co-DEGMA and 10% w/w Pluronic®F127, formed homogeneous solutions. The others spontaneously phase separated after standing for about half an hour and did not form homogeneous solutions even at low temperature (4 °C). The visual tests were carried out on the two homogenous mixtures and no gelation was observed.
As reported in previous studies, the minimum gelation concentration Cgel of Pluronic®F127 is 15% w/w with a gelation temperature window from 32 °C to 41 °C.24,84 In our study, we found that the addition of our copolymers to the mixture lowers the Cgel and broadens the temperature window of the stable gel. In the phase diagrams with the same total concentration (25% w/w, shown in Fig. 11), we found that OEGMA-b-BuMA and OEGMA-b-(BuMA-co-DEGMA) exhibited similar influence on the gelation of Pluronic®F127. When increasing the Pluronic®F127 concentration, the first gel was formed at 12.5% w/w Pluronic®F127, with a temperature window from 31 °C to 47 °C. Furthermore, at 20% w/w Pluronic®F127 on its own without an additive will form gels that will collapse when the temperature exceeds 55 °C. When 20% w/w Pluronic®F127 was mixed with 5% w/w of the copolymers of this study on the other hand, the gel formed stays stable at higher temperatures.
The mixture of diblock polymer OEGMA-b-DEGMA and Pluronic®F127 behaved differently from the other diblock polymer mixtures discussed above. It was found that OEGMA-b-DEGMA can lower the Cgel to 10% w/w with a temperature window from 42 °C to 58 °C. In general, the Tgel of OEGMA-b-DEGMA and Pluronic®F127 mixture was a few degrees higher than the other mixtures with similar concentrations. It is worth noting that the gel of the mixture with 10% w/w of OEGMA-b-DEGMA and 15% w/w of Pluronic®F127 was formed at body temperature 37 °C and stayed stable as high as 74 °C. The other mixtures also exhibited Tgels either slightly lower or higher than the body temperatures (34 °C–42 °C), which are potential candidates for injectable gel or drug delivery. The fact that the copolymer chemistry had such an effect on the thermogelling of the copolymer/Pluronic mixture is very interesting. It seems that the OEGMA-b-DEGMA copolymers that are hydrophilic and only form micelles at higher temperatures enhance gelation, decrease the Cgel and shift the Tgel at higher temperatures more than the amphiphilic counterparts. This demonstrates how the mixing with the right copolymer can be used to tailor both the Cgel and Tgel of a thermogelling formulation.
Conclusions
Thermoresponsive polymers based on the BuMA, DEGMA and OEGMA monomers were successfully synthesised via GTP. Three architectures: diblock bipolymer, diblock terpolymer and statistical terpolymer were investigated. The mass fraction of the OEGMA was varied from 50% to 80%. The CPs increased by increasing the OEGMA composition in the copolymer. The architecture also affected the CPs. The statistical copolymer was less water soluble and presented a lower CP than all other copolymers. The copolymers that are hydrophilic at room temperature, thus they do not form micelles (i.e., OEGMA-b-DEGMA), present lower cloud points when compared to the amphiphilic copolymers, which form micelles at room temperature (i.e., OEGMA-b-BuMA and OEGMA-b-(BuMA-co-DEGMA)).Interestingly, the DLS investigation revealed that depending on the OEGMA composition there are some intermediate temperatures, close to the DEGMA CP, where micelles are formed before the polymer phase separates. Furthermore, the synthesised copolymers were used as an additive to control the gelation temperature (Tgel) and concentration (Cgel) of Pluronic®F127. It was demonstrated that by mixing with the appropriate copolymers both Tgel and Cgel can be tailored. The OEGMA-b-DEGMA copolymer was especially promising when mixing with Pluronic®F127, as the Cgel was reduced and the Tgel shifted towards body temperature, so the polymer formulation could be injected at room temperature and be a gel at body temperature. The above observations give directions for further applications in drug delivery, tissue engineering, and sensors.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Ramírez-Jiménez, K. A. Montoya-Villegas, A. Licea-Claverie and M. A. Gónzalez-Ayón, Polymers, 2019 DOI:10.3390/polym11101657.
- Z. Ye, Y. Li, Z. An and P. Wu, Langmuir, 2016, 32, 6691–6700 CrossRef CAS PubMed.
- A. P. Constantinou and T. K. Georgiou, Polym. Int., 2021, 70, 1433–1448 CrossRef CAS.
- S. Bahl, H. Nagar, I. Singh and S. Sehgal, Mater. Today: Proc., 2020, 28, 1302–1306 CAS.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- P. Lai, D. Hong and K. Ku, Nanomedicine, 2014, 10, 553–560 CrossRef CAS PubMed.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2021, 31(8) DOI:10.1002/adfm.202008123.
- K. Zhang, K. Xue and X. J. Loh, Gels, 2021, 7 DOI:10.3390/gels7030077.
- Z. Lei, Q. Wang and P. Wu, Mater. Horiz., 2017, 4, 694–700 RSC.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30(42) DOI:10.1002/adfm.202002759.
- L. Klouda, Eur. J. Pharm. Biopharm., 2015, 97, 338–349 CrossRef CAS PubMed.
- A. Gandhi, A. Paul, S. O. Sen and K. K. Sen, Asian J. Pharm. Sci., 2015, 10, 99–107 CrossRef.
- X. Wang, S. Li, Z. Wan, Z. Quan and Q. Tan, Int. J. Pharm., 2014, 463, 81–88 CrossRef CAS PubMed.
- K. J. Hogan and A. G. Mikos, Polymer, 2020, 211, 123063 CrossRef CAS.
- X. Song, Z. Zhang, J. Zhu, Y. Wen, F. Zhao, L. Lei, N. Phan-Thien, B. C. Khoo and J. Li, Biomacromolecules, 2020, 21, 1516–1527 CrossRef CAS PubMed.
- S. Li and C. M. Schroeder, ACS Macro Lett., 2018, 7, 281–286 CrossRef CAS.
- H. Cheng, J. L. Zhu, Y. X. Sun, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Bioconjugate Chem., 2008, 19, 1368–1374 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- M. Cao, Y. Wang, X. Hu, H. Gong, R. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS PubMed.
- Y. J. Choi, H. G. Yi, S. W. Kim and D. W. Cho, Theranostics, 2017, 7, 3118–3137 CrossRef CAS PubMed.
- Z. Cui, B. H. Lee, C. Pauken and B. L. Vernon, J. Biomed. Mater. Res., Part A, 2011, 98 A, 159–166 CrossRef PubMed.
- M. T. Cidade, D. J. Ramos, J. Santos, H. Carrelo, N. Calero and J. P. Borges, Materials, 2019, 12(7) DOI:10.3390/ma12071083.
- A. Alexander, J. Khan, S. Saraf and S. Saraf, J. Controlled Release, 2013, 172, 715–729 CrossRef CAS PubMed.
- Q. Li, A. P. Constantinou and T. K. Georgiou, J. Polym. Sci., 2021, 59, 230–239 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J. F. Lutz, Macromolecules, 2009, 42, 33–36 CrossRef CAS.
- J.-F. L. Nezha Badi, J. Controlled Release, 2009, 140, 224–229 CrossRef PubMed.
- J. Buller, A. Laschewsky, J. F. Lutz and E. Wischerhoff, Polym. Chem., 2011, 2, 1486–1489 RSC.
- J. F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- J. Lutz, A. Hoth, K. Schade, J. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 CrossRef CAS.
- D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915–920 CrossRef CAS.
- D. Bera, O. Sedlacek, E. Jager, E. Pavlova, M. Vergaelen and R. Hoogenboom, Polym. Chem., 2019, 10, 5116–5123 RSC.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Börner, A. Laschewsky, C. Duschl and J. F. Lutz, Angew. Chem.,– Int. Ed., 2008, 47, 5666–5668 CrossRef CAS PubMed.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- J. F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- J. F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- T. Li, F. Huang, D. Diaz-Dussan, J. Zhao, S. Srinivas, R. Narain, W. Tian and X. Hao, Biomacromolecules, 2020, 21, 1254–1263 CrossRef CAS PubMed.
- I. Lilge, M. Steuber, D. Tranchida, E. Sperotto and H. Schönherr, Macromol. Symp., 2013, 328, 64–72 CrossRef CAS.
- A. E. Dunn, D. J. Dunn, A. Macmillan, R. Whan, T. Stait-Gardner, W. S. Price, M. Lim and C. Boyer, Polym. Chem., 2014, 5, 3311–3315 RSC.
- J. F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- Z. Jin, W. Feng, S. Zhu, H. Sheardown and J. L. Brash, J. Biomed. Mater. Res., Part A, 2010, 95, 1223–1232 CrossRef PubMed.
- V. Stadler, R. Kirmse, M. Beyer, F. Breitling, T. Ludwig and F. R. Bischoff, Langmuir, 2008, 24, 8151–8157 CrossRef CAS PubMed.
- K. Li, L. Yu, X. Liu, C. Chen, Q. Chen and J. Ding, Biomaterials, 2013, 34, 2834–2842 CrossRef CAS PubMed.
- T. Teraya, A. Takahara and T. Kajiyama, Polymer, 1990, 31, 1149–1153 CrossRef CAS.
- H. Xiang, M. Xia, A. Cunningham, W. Chen, B. Sun and M. Zhu, J. Mech. Behav. Biomed. Mater., 2017, 72, 74–81 CrossRef CAS PubMed.
- M. L. Bravi Costantino, T. G. Oberti, A. M. Cortizo and M. S. Cortizo, J. Biomed. Mater. Res., Part A, 2019, 107, 195–203 CrossRef CAS PubMed.
- W. Kim and J. Jung, BMB Rep., 2016, 49, 655–661 CrossRef CAS PubMed.
- K. J. MacKenzie and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 293–300 CrossRef CAS PubMed.
- S. Dey, B. Kellam, M. R. Alexander, C. Alexander and F. R. A. J. Rose, J. Mater. Chem., 2011, 21, 6883–6890 RSC.
- Y. Tian, S. Bian and W. Yang, Polym. Chem., 2016, 7, 1913–1921 RSC.
- M. Zhang, Y. Liu, J. Peng, Y. Liu, F. Liu, W. Ma, L. Ma, C. Y. Yu and H. Wei, Polym. Chem., 2020, 11, 6139–6148 RSC.
- P. Z. Elias, G. W. Liu, H. Wei, M. C. Jensen, P. J. Horner and S. H. Pun, J. Controlled Release, 2015, 208, 76–84 CrossRef CAS PubMed.
- X. Li, Y. Qian, T. Liu, X. Hu, G. Zhang, Y. You and S. Liu, Biomaterials, 2011, 32, 6595–6605 CrossRef CAS PubMed.
- Y. Cheng, C. He, J. Ding, C. Xiao, X. Zhuang and X. Chen, Biomaterials, 2013, 34, 10338–10347 CrossRef CAS PubMed.
- T. Zhou, W. Wu and S. Zhou, Polymer, 2010, 51, 3926–3933 CrossRef CAS.
- F. Khan and S. Ahmad, Biomater. Stem Cells Regener. Med., 2012, 101–122 CAS.
- J. T. Lin, D. C. Cheng, K. T. Chen and H. W. Liu, Polymers, 2019, 11, 1–16 Search PubMed.
- J. Oliveira, V. Correia, H. Castro, P. Martins and S. Lanceros-Mendez, Addit. Manuf., 2018, 21, 269–283 CAS.
- A. P. Constantinou, H. Zhao, C. M. McGilvery, A. E. Porter and T. K. Georgiou, Polymers, 2017, 9, 31, DOI:10.3390/polym9010031.
- Y. Guo, L. Li, F. Li, H. Zhou and Y. Song, Lab Chip, 2015, 15, 1759–1764 RSC.
- Y. Dong, S. Wang, Y. Ke, L. Ding, X. Zeng, S. Magdassi and Y. Long, Adv. Mater. Technol., 2020, 5, 1–19 Search PubMed.
- C. Porsch, S. Hansson, N. Nordgren and E. Malmström, Polym. Chem., 2011, 2, 1114–1123 RSC.
- K. Kobayashi, S. H. Oh, C. K. Yoon and D. H. Gracias, Macromol. Rapid Commun., 2018, 39, 1–7 Search PubMed.
- S. Kessel, S. Schmidt, R. Müller, E. Wischerhoff, A. Laschewsky, J. F. Lutz, K. Uhlig, A. Lankenau, C. Duschl and A. Fery, Langmuir, 2010, 26, 3462–3467 CrossRef CAS PubMed.
- J.-F. L. Nezha Badi, J. Controlled Release, 2009, 40, 3459–3470 Search PubMed.
- J. Bassi da Silva, P. Haddow, M. L. Bruschi and M. T. Cook, J. Mol. Liq., 2021, 117906 Search PubMed.
- A. M. Pragatheeswaran and S. B. Chen, Langmuir, 2013, 29, 9694–9701 CrossRef CAS PubMed.
- N. M. P. S. Ricardo, N. M. P. S. Ricardo, F. de M. L. L. Costa, F. W. A. Bezerra, C. Chaibundit, D. Hermida-Merino, B. W. Greenland, S. Burattini, I. W. Hamley, S. Keith Nixon and S. G. Yeates, J. Colloid Interface Sci., 2012, 368, 336–341 CrossRef CAS PubMed.
- E. Gioffredi, M. Boffito, S. Calzone, S. M. Giannitelli, A. Rainer, M. Trombetta, P. Mozetic and V. Chiono, Procedia CIRP, 2016, 49, 125–132 CrossRef.
- A. L. Kjøniksen, M. T. Calejo, K. Zhu, B. Nyström and S. A. Sande, J. Appl. Polym. Sci., 2014, 131, 1–8 CrossRef.
- A. M. Pragatheeswaran and S. B. Chen, Langmuir, 2013, 29, 9694–9701 CrossRef CAS PubMed.
- C. C. Hopkins and J. R. de Bruyn, J. Rheol., 2019, 63, 191–201 CrossRef CAS.
- M. Bohorquez, C. Koch, T. Trygstad and N. Pandit, J. Colloid Interface Sci., 1999, 216, 34–40 CrossRef CAS PubMed.
- Y. L. Su, H. Z. Liu, J. Wang and J. Y. Chen, Langmuir, 2002, 18, 865–871 CrossRef CAS.
- B. Bharatiya, G. Ghosh, P. Bahadur and J. Mata, J. Dispersion Sci. Technol., 2008, 29, 696–701 CrossRef CAS.
- B. C. Anderson, S. M. Cox, A. V. Ambardekar and S. K. Mallapragada, J. Pharm. Sci., 2002, 91, 180–188 CrossRef CAS PubMed.
- N. Pandit, T. Trygstad, S. Croy, M. Bohorquez and C. Koch, J. Colloid Interface Sci., 2000, 222, 213–220 CrossRef CAS PubMed.
- F. A. E. Silva, R. M. C. Carmo, A. P. M. Fernandes, M. Kholany, J. A. P. Coutinho and S. P. M. Ventura, ACS Sustainable Chem. Eng., 2017, 5, 6409–6419 CrossRef.
- K. C. Shih, Z. Shen, Y. Li, M. Kröger, S. Y. Chang, Y. Liu, M. P. Nieh and H. M. Lai, Soft Matter, 2018, 14, 7653–7663 RSC.
- J. C. Gilbert, J. L. Richardson, M. C. Davies, K. J. Palin and J. Hadgraft, J. Controlled Release, 1987, 5, 113–118 CrossRef CAS.
- J. C. Gilbert, C. Washington, M. C. Davies and J. Hadgraft, Int. J. Pharm., 1987, 40, 93–99 CrossRef CAS.
- L. B. Li and Y. B. Tan, J. Colloid Interface Sci., 2008, 317, 326–331 CrossRef CAS PubMed.
- H. Mao, P. Pan, G. Shan and Y. Bao, J. Phys. Chem. B, 2015, 119, 6471–6480 CrossRef CAS PubMed.
- H. Mao, C. Wang, X. Chang, H. Cao, G. Shan, Y. Bao and P. Pan, Mater. Chem. Front., 2018, 2, 313–322 RSC.
- I. B. Dicker, G. M. Cohen, W. B. Farnham, W. R. Hertler, E. D. Laganis and D. Y. Sogah, Macromolecules, 1990, 23, 4034–4041 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Soft Matter, 2012, 2737–2745 RSC.
- A. P. Constantinou, B. Zhan and T. K. Georgiou, Macromolecules, 2021, 54, 1943–1960 CrossRef CAS.
- A. P. Constantinou, K. Zhang, B. Somuncuoğlu, B. Feng and T. K. Georgiou, Macromolecules, 2021, 54, 6511–6524 CrossRef CAS.
- A. I. Triftaridou, M. Vamvakaki and C. S. Patrickios, Polymer, 2002, 43, 2921–2926 CrossRef CAS.
- A. I. Triftaridou, M. Vamvakaki and C. S. Patrickios, Biomacromolecules, 2007, 8, 1615–1623 CrossRef CAS PubMed.
- N. Chen, X. Xiang, A. Tiwari and P. A. Heiden, J. Colloid Interface Sci., 2013, 391, 60–69 CrossRef CAS PubMed.
- P. De and B. S. Sumerlin, Macromol. Chem. Phys., 2013, 214, 272–279 CrossRef CAS.
- J. A. Jones, N. Novo, K. Flagler, C. D. Pagnucco, S. Carew, C. Cheong, X. Z. Kong, N. A. D. Burke and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6095–6104 CrossRef CAS.
- G. Zhang, J. Lei, L. Wu, C. Guo, J. Fang and R. Bai, Polymer, 2018, 157, 79–86 CrossRef CAS.
- X. Xiang, X. Ding, N. Chen, B. Zhang and P. A. Heiden, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2838–2848 CrossRef CAS.
- Z. Wang, S. Seger and N. V. Tsarevsky, Eur. Polym. J., 2019, 111, 63–68 CrossRef.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- N. Chen, X. Xiang, A. Tiwari and P. A. Heiden, J. Colloid Interface Sci., 2013, 391, 60–69 CrossRef CAS PubMed.
- L. G. Weaver, R. Stockmann, A. Postma and S. H. Thang, RSC Adv., 2016, 6, 90923–90933 RSC.
- P. J. Roth, F. D. Jochum and P. Theato, Soft Matter, 2011, 7, 2484–2492 RSC.
- Z. L. Yao and K. C. Tam, Polymer, 2012, 53, 3446–3453 CrossRef CAS.
- P. Lai, D. Hong and K. Ku, Nanomedicine, 2014, 10, 553–560 CrossRef CAS PubMed.
- C. J. Park, J. W. Heo, J. H. Lee, T. Y. Kim and S. Y. Kim, Polymers, 2020, 12, 284 CrossRef CAS PubMed , https://doi.org/10.3390/polym12020284.
- A. C. Santos, A. F. M. Santos, H. P. Diogo, T. Correia, M. Dionísio and P. S. Farinha, Polymer, 2018, 148, 339–350 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Polym. Chem., 2013, 4, 1893–1902 RSC.
- Q. Zhang, L. Voorhaar, S. K. Filippov, B. F. Yeşil and R. Hoogenboom, J. Phys. Chem. B, 2016, 120, 4635–4643 CrossRef CAS PubMed.
- Q. Zhang, N. Vanparijs, B. Louage, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2014, 5, 1140–1144 RSC.
- L. T. Che, M. Hiorth, R. Hoogenboom and A. L. Kjøniksen, Polymers, 2020, 12, 1–16 Search PubMed.
- O. Sedlacek, K. Lava, B. Verbraeken, S. Kasmi, B. G. De Geest and R. Hoogenboom, J. Am. Chem. Soc., 2019, 141, 9617–9622 CrossRef CAS PubMed.
- L. T. T. Trinh, H. M. L. Lambermont-Thijs, U. S. Schubert, R. Hoogenboom and A. L. Kjøniksen, Macromolecules, 2012, 45, 4337–4345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1py01688a |
| This journal is © The Royal Society of Chemistry 2022 |