
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct routes to functional RAFT agents from substituted N-alkyl maleimides†
Rosa
Catania
 ab,
Ruggero
Foralosso
a,
Lampros
Spanos
a,
Emanuele
Russo
a,
Francesca
Mastrotto
c,
Pratik
Gurnani
a,
Kevin
Butler
d,
Huw
Williams
d,
Snow
Stolnik
*a and
Giuseppe
Mantovani
*a
ab,
Ruggero
Foralosso
a,
Lampros
Spanos
a,
Emanuele
Russo
a,
Francesca
Mastrotto
c,
Pratik
Gurnani
a,
Kevin
Butler
d,
Huw
Williams
d,
Snow
Stolnik
*a and
Giuseppe
Mantovani
*a
aDivision of Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: giuseppe.mantovani@nottingham.ac.uk; snow.stolnik@nottingham.ac.uk
bSchool of Life Sciences, University of Nottingham, Queens Medical Centre, Nottingham NG7 2UH, UK
cDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova 35131, Italy
dSchool of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK
First published on 14th November 2022
Abstract
N-substituted maleimides have become an indispensable tool for the synthesis of bioconjugates and functional materials. Herein, we present three strategies for the incorporation of N-alkyl substituted maleimides into RAFT agents and show that these maleimide-derived CTAs can be used to easily introduce a range of chemical functionality at the β-position of polymer chains, resulting in α,β,ω-functional RAFT polymers. With both functional maleimides and RAFT agents that are increasingly available on the market, the approach presented in this study could facilitate the synthesis of end-functional macromolecules and will complement well the range of existing synthetic routes, including those utilising N-substituted maleimides, to functional polymeric materials.
Introduction
Maleimides are versatile molecular tools utilised by chemists and biologists to engineer functional materials and (bio)conjugates. Due to their unique reactivity, maleimides are often part of ligation strategies to incorporate molecular fragments with specific functionalities – e.g. fluorophores, spin probes, polymer chains and drugs – into (bio)molecules with complementarily reactive residues.1,2 This is typically done through (i) thio-Michael and (ii) Diels–Alder reactions, and (iii) use of N-substituted maleimides as comonomers in radical polymerisations. Due to its high efficiency and, under specific conditions, selectivity, thio-Michael reaction between maleimide and sulfhydryl-containing (bio)molecules is often utilised as the ligation strategy for the synthesis of bioconjugates. For example, 8 of the 11 currently approved antibody drug conjugates (ADCs) used as anticancer therapeutics,3,4 as well as clinically PEGylated Fab’ fragment Certolizumab pegol, targeting tumour necrosis factor alpha (TNF-α) and clinically utilised to treat a range of inflammatory autoimmune diseases,5 are generated through maleimide-cysteine coupling. Moreover, the ability of maleimides to act as electron-deficient dienophiles in Diels–Alder (DA) reactions resulted in their use to produce a variety of functional low molar mass intermediates,6 bioconjugates,7 and polymer-based materials.8In radical polymerisations, N-substituted maleimides possess a rather peculiar reactivity, in that they have a little or no tendency for homopolymerisation, but very rapidly cross-propagate in the presence of electron-rich monomers such as styrenes.9 This unique behaviour has been exploited in reversible-deactivation radical polymerisations (RDRP) to insert monomers at predetermined positions of polymer chains, where N-substituted maleimides are added at definite time points during the polymerisation reaction and, due to their preferential crossover propagation, they are very rapidly incorporated into specific short segments of the growing polymer chains.10 The Lutz group have pioneered this approach and very elegantly utilised N-substituted maleimides to control monomer sequence distribution in polymers synthesised by RDRP, inserting functionalities at precisely defined points of polymer chains.9,11–21 For example, 1-alkyne and activated ester functionalities have been used to generate polymer chains with defined intramolecularly crosslinked regions,13 including well-defined structures such as α-shaped21 and more complex19 macromolecular origamis. Bulky substituents such as functional dendrons15 and, following post-polymerisation modification, poly(ethylene oxide) side chains11 can also be introduced within specific segments of polymer chains. Harrisson and co-workers have then investigated the level of monomer insertion precision that can be obtained using chain polymerization, and estimated that improved control can be attained for longer chains, controlling the relative position of a specific monomer with respect to the total chain length.22
Taking advantage from their very low tendency to homopolymerize, Stayton and co-workers successfully inserted single N-substituted maleimide units at the ω (Z)-terminal chain end of RAFT polymers with very good efficiencies.23 Xu et al. utilised photoinduced electron/energy transfer (PET) to develop a single unit monomer insertion (SUMI) strategy based on RAFT polymerisation, where three different monomers, including N-substituted maleimides, are sequentially added, to generate well-defined trimers with high control over monomer sequence.24 Longer discrete and stereospecific oligomers were subsequently synthesised by Xu and Boyer following a similar approach, under red light irradiation and ZnTPP as the photocatalyst, using and indenes and N-substituted maleimides as electron donor and electron acceptor monomers, respectively.25
RAFT polymerisation is ideally suited for the synthesis of heterotelechelic polymers, that is, polymer chains with different functionalities at their α and ω chain-ends, which are derived from the R and Z groups of the chain-transfer agents (CTAs) chosen for their synthesis.26–28 However, for specific applications an additional functionality may be required at one of the two polymer chain-ends, to give α,β,ω-functional polymer chains. In this work, we addressed this need and describe three synthetic routes where functional N-alkyl maleimides are utilised to insert specific functionalities between the R and Z groups of trithiocarbonate CTAs. Following characterisation of these CTAs, we then show that they can be successfully employed to generate α,β,ω-functional polymers using acrylates and acrylamide model monomers. We believe that our approach complements well the existing routes to functional heterotelechelic (co)polymers and, with the increasing number of N-substituted maleimides that are increasingly becoming available it could become a valuable strategy among the wide toolbox of synthetic strategy to produce functional polymeric materials.
Results and discussion
Design of N-alkyl maleimide-derived chain-transfer agents
To produce the required maleimide-derived chain-transfer agents (Mal-CTAs) the three synthetic strategies illustrated in Fig. 1 were designed. These routes rely on a single unit monomer insertion approach, which involves the addition of a single monomer molecule to a preformed RAFT agent,29 or a bis(thioacyl) disulfide. In Route 1a, oxidative coupling of trithiocarbonate salts (D)30 using K3Fe(CN)6 or I2 afforded the bis(thioacyl) disulfides (A). Reaction of the latter with 1.8 equivalent of AIBN, used here as a representative model azo-initiator, and N-alkyl maleimides (B) at 70 °C in toluene, afforded the desired (Mal-CTAs) (chemical structures shown in Chart 1).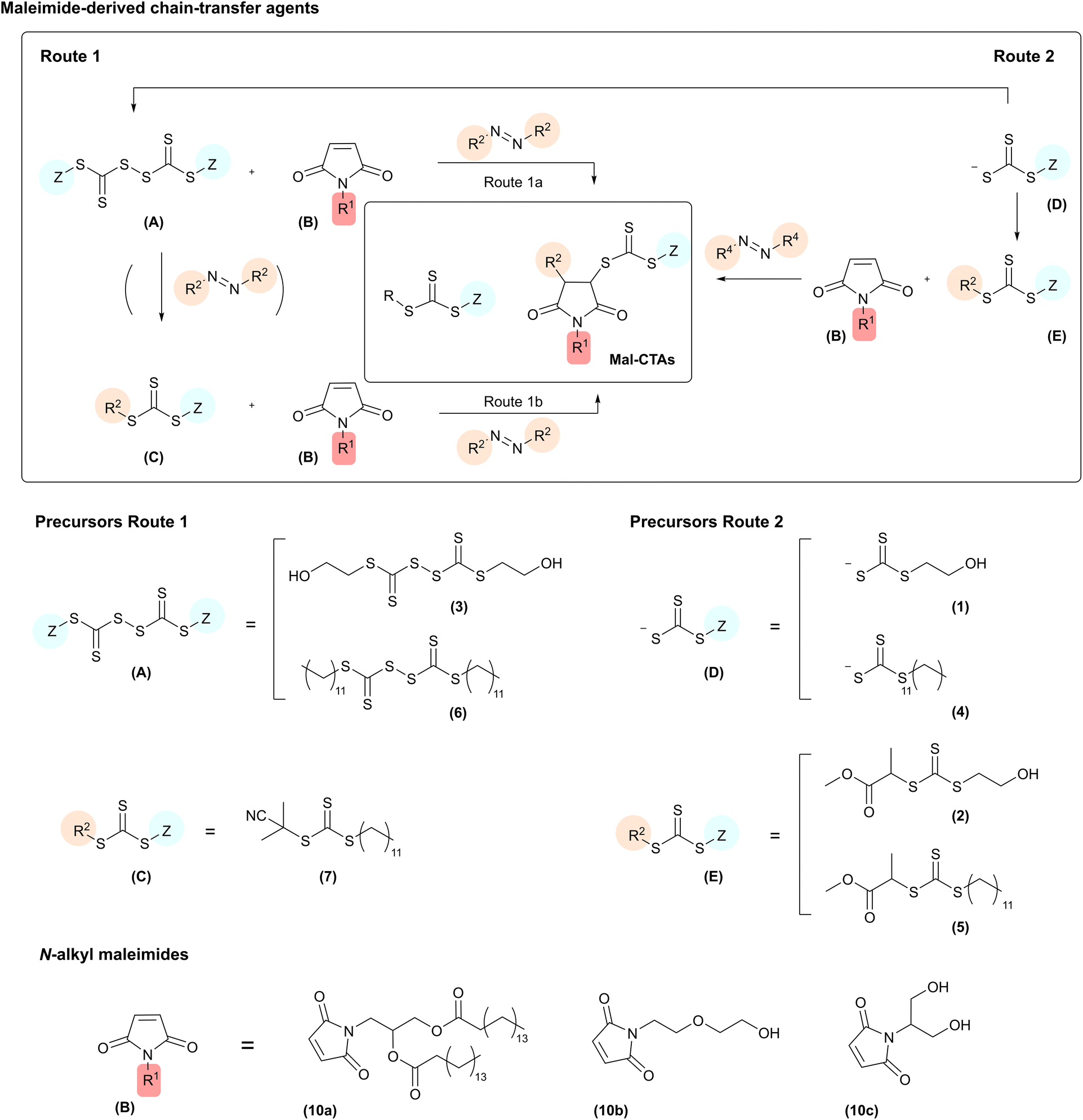 | ||
| Fig. 1 Top. Synthetic routes to N-alkyl-substituted maleimide-derived chain-transfer agents (Mal-CTAs) synthesised in this study. Bottom: precursors utilised to produce the Mal-CTAs. | ||
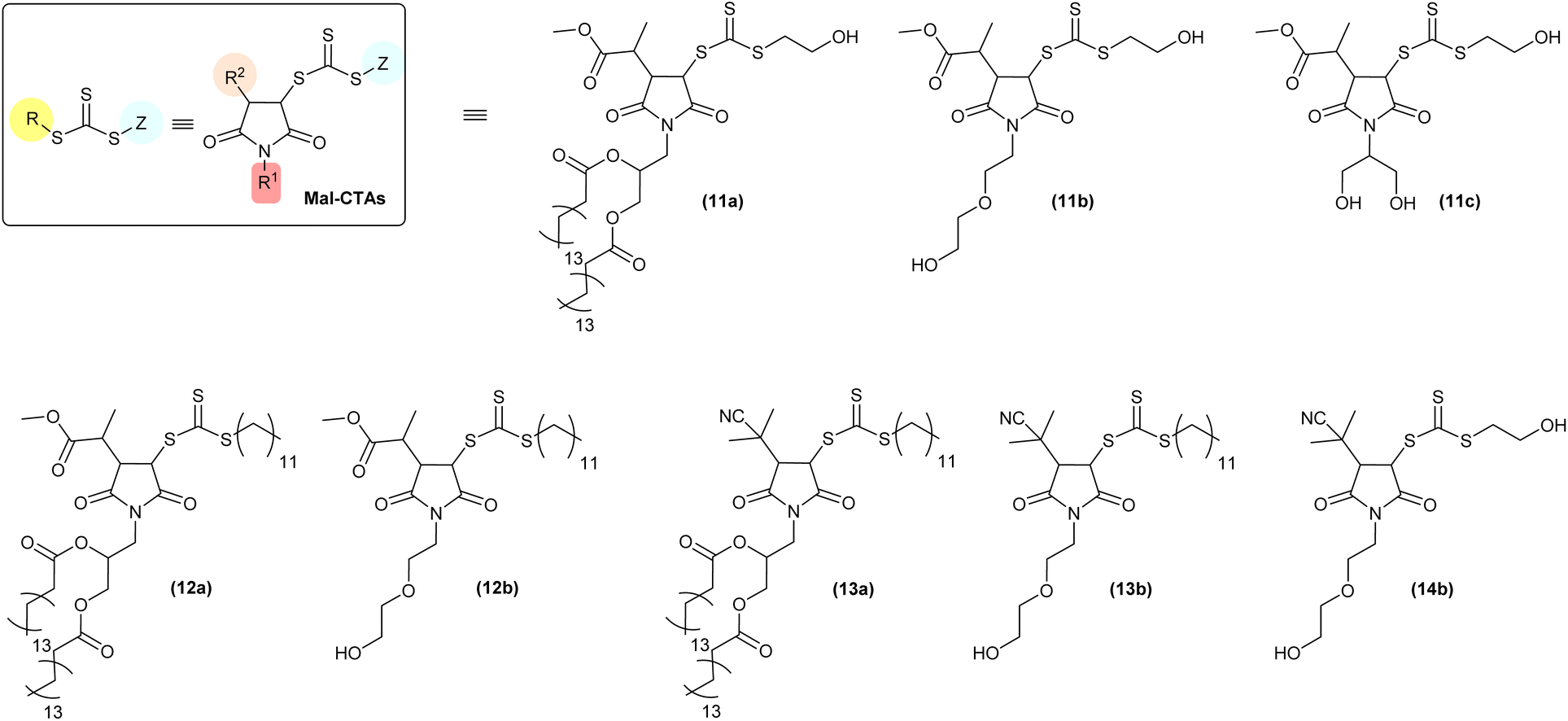 | ||
| Chart 1 Maleimide-derived chain-transfer agents (Mal-CTAs) synthesised in this work. | ||
Route 1b starts from a preformed trithiocarbonate CTA (C), thus it is particularly suited for use with commercially available RAFT agents, e.g. 2-cyano-2-propyl dodecyl trithiocarbonate CTA (7) used in this work. These can be converted into the final N-alkyl maleimide-derived chain-transfer agents Mal-CTAs in one step, by reaction with 0.05 eq. of a radical azo initiator. With an increasing number of trithiocarbonate CTAs, as well as bis(thioacyl) disulfides, that are now becoming available on the market, strategies 1a and 1b provide a direct access to the desired Mal-CTAs from readily accessible precursors. If required, trithiocarbonate CTAs starting materials (C) can also be obtained from bis(thioacyl) disulfides (A) by treatment with a radical azo initiator.29 However, as already discussed, in this case Route 1a allows to bypass this extra step, affording the desired N-alkyl maleimide-derived Mal-CTAs directly from bis(thioacyl) disulfides precursors (A).
Route 2 starts from trithiocarbonate salts (D), prepared from thiols and CS2, again following well-established protocols29 (ESI, Scheme S1†). Reaction of these with an α-halogen propionic ester, in this case methyl 2-bromopropionate, afforded the intermediates with general structure (E), which were then treated with 0.05–0.010 eq. of AIBN to give the desired N-alkyl maleimide-derived Mal-CTAs.
Fig. 1 (bottom) also shows the chemical structure of all intermediates utilised in this work. Hydrophobic (10a) and hydrophilic (10b) and (10c) N-alkyl substituted maleimides were chosen for these proof-of-principle experiments as model maleimide monomers (Fig. 1). (10a) possesses a diglyceride-like structure, and its incorporation at the β position results in polymers with two fatty acid ester moieties at their chain-end. These materials possess a molecular structure largely analogous to that of α-diglyceride polymers described by Hawker and co-workers,31 and to that of phospholipid-terminated polymers utilised as membrane-inserting macromolecules for the modification of artificial lipid bilayers,32 and plasma membranes of selected cells both in vitro33–36 and in vivo.37 Maleimides (10b) and (10c) were chosen as representative example of a hydrophilic maleimide monomer, with free hydroxyl groups available for further chemical modification either prior or after incorporation into the desired functional polymers. Both maleimide derivatives were synthesised by reacting a furan-maleic anhydride Diels–Alder adduct with an appropriate amine in methanol in the presence of Et3N, followed by removal of furan protecting group by retro Diels–Alder reaction in refluxing toluene, to give the maleimide monomers (10a), (10b), (10c) used in this study (ESI, Scheme S2†).
Hydrophilic and hydrophobic bis(thioacyl) disulfides (3) and (6), respectively, were used for Route 1a. It should be noted that although in this study bis(dodecylsulfanylthiocarbonyl) disulfide (6) was synthesised as described in the ESI,† this intermediate is also commercially available, which further simplifies access to the desired Mal-CTAs. The same consideration also applies to 2-cyano-2-propyl dodecyl trithiocarbonate CTA (7), which was chosen as the starting point for Route 1b.
Route 2 started from sodium 2-hydroxyethyl carbonotrithioate (1) and sodium dodecyl carbonotrithioate (4), which following reaction with 2-bromo methylpropionate in acetone at ambient temperature gave the trithiocarbonate CTAs (2) and (5) respectively. Finally, treatment of the latter with 0.05–0.010 eq. of AIBN at 70 °C in the presence of N-alkyl substituted maleimides (10a) and (10b) afforded the required maleimide-derived chain-transfer agents (Mal-CTAs). The chemical structure of all Mal-CTAs synthesised in this work is shown in Chart 1. Overall yields for isolated Mal-CTAs are in the 22–73% range (Tables S1 and S2†), which is in line with previous work by Coote, Boyer, Xu and co-workers, who reported an overall yield of 56% for a single insertion of N-phenylmaleimide (PMI) into n-butyl benzyl trithiocarbonate (BBTC) CTA, using zinc tetraphenylporphyrin (ZnTPP) as the photocatalyst, after 20 h of red-light irradiation (λmax = 635 nm) and subsequent purification by silica chromatography. These authors also reported that further increasing the [maleimide]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA] ratio not only did not improve the reaction yield, but rather led to more by-products due to multiple maleimide monomer insertions.25 For the synthesis of (11a–c) and (12a–b), the complex pattern of signals in the 1H NMR spectra of these RAFT agents do not allow to easily detect potential traces of azo initiator-derived by-products in the crude reaction mixtures, although their presence before the purification steps in principle cannot be ruled out.
:[CTA] ratio not only did not improve the reaction yield, but rather led to more by-products due to multiple maleimide monomer insertions.25 For the synthesis of (11a–c) and (12a–b), the complex pattern of signals in the 1H NMR spectra of these RAFT agents do not allow to easily detect potential traces of azo initiator-derived by-products in the crude reaction mixtures, although their presence before the purification steps in principle cannot be ruled out.
In terms of reaction mechanism and characterisation of the final Mal-CTA, the two sp2-hybridsed CH vinyl carbon atoms of maleimide precursors are prochiral, that is, each can be converted into a chiral centre by addition of a further substituent. 1H NMR analysis of (13b) is discussed here as a representative example, as this Mal-CTAs does not possess any chiral centres in R group of the precursor, or in the N-alkyl substituent R1 of the maleimide monomer, which would result in more complex spectra due to the presence of additional stereoisomers.
In principle, addition of two substituent to maleimide vinyl group could occur in cis (syn)- or trans (anti)-fashions. The presence of a single pattern of signals for –CHSC(S)SZ– (δ = 4.72 ppm, d, JHf–Hg = 6.2 Hz, Hf) and –CHC(CH3)2CN (CHR, δ = 3.22 ppm, d, JHg–Hf = 6.2 Hz, Hg) groups in the 1H NMR spectrum of (13b) indicated that addition of R and Z groups of its CTA precursor (or R group from the azo initiator and Z from bis(thioacyl) disulfides) to the maleimide vinyl group was stereoselective, leading exclusively to a single pair of enantiomers (Fig. 2, top). Although experiments such as single-crystal X-ray diffraction to assign the absolute configuration of the stereocentres of the adducts were not carried out in this study, both reaction mechanism and 1H NMR spectral features of our Mal-CTAs closely resemble those of the trans-(BBTC-PMI) adduct described by Coote, Boyer, Xu and co-workers25 strongly suggest that (13b) exists as a mixture of trans-stereoisomers, namely enantiomers (R,S) and (S,R), at their newly generated saturated 5-membered rings. Moreover, the J coupling constant JHg–Hf = 6.2 Hz observed for the two CH at the newly generated stereogenic centres is in line with those reported in previous studies (J = 6.4–6.5 Hz) for trans-adducts formed by reaction between substituted maleimides and xanthates,38–40 further suggesting a trans-configuration for (13b) and the other Mal-CTAs synthesised in this work.
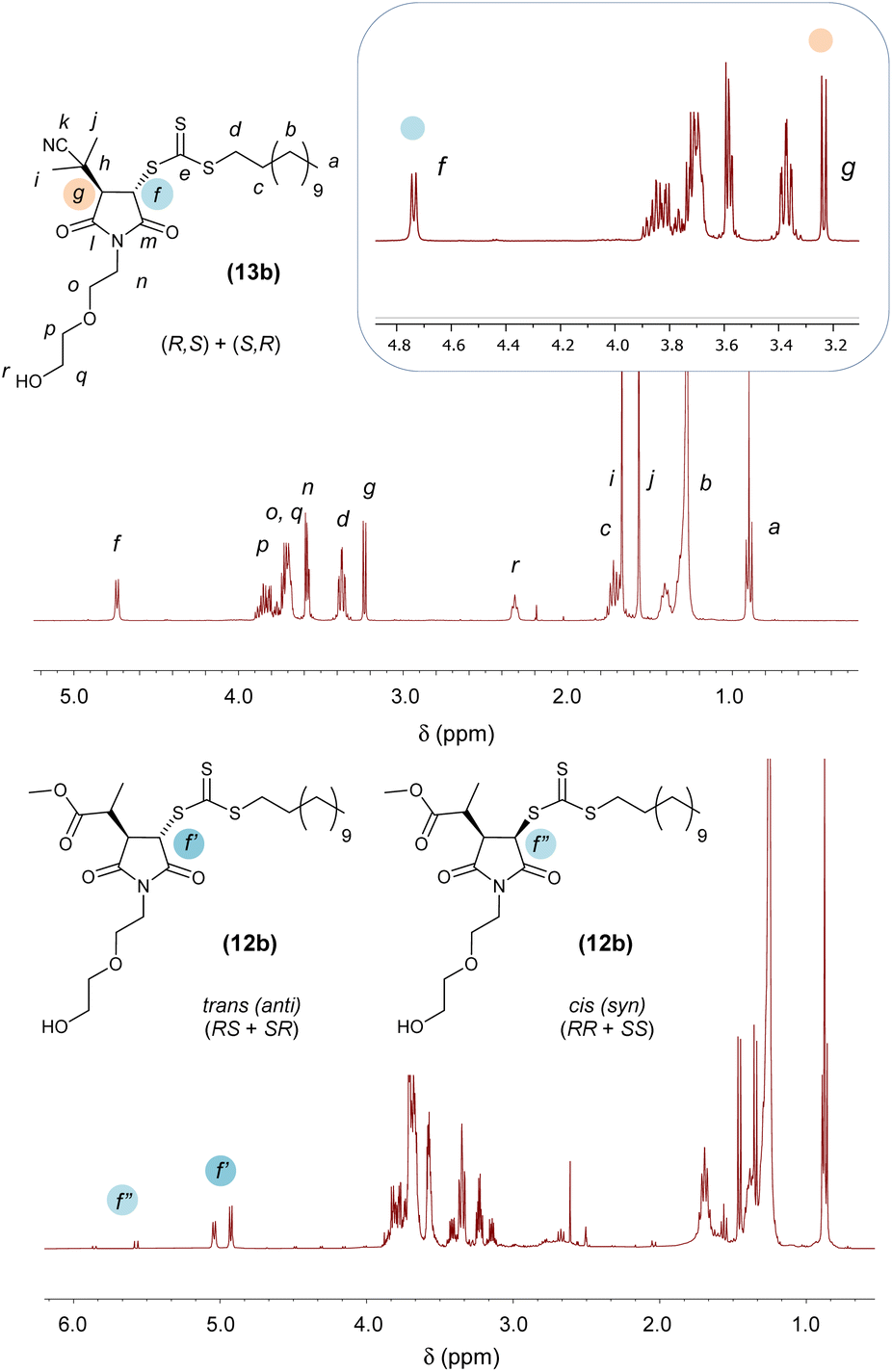 | ||
| Fig. 2 Top: 1H NMR spectrum of racemic trans (anti)-adduct Mal-CTA (13b) in CDCl3. Inset: magnification of the 3.2–4.8 ppm region, highlighting the signals of –CHSC(S)SZ and –CHC(CH3)2CN protons at the stereogenic centres. Bottom: 1H NMR spectrum of adduct Mal-CTA (12b) in CDCl3, showing the presence of minor cis (syn) adduct – anti:sin 92:8. Signal splitting is due to the additional chiral centre in the 2-methylpropionate substituent. | ||
This is consistent with a mechanism by which a 2-(2-methylpropanenitrile) radical is first added to the maleimide ring, providing significant steric hindrance on the face at which this first addition occurred. As a result, addition of the second substituent occurs at the less sterically encumbered opposite face of the 5-membered rings, yielding a trans (anti)-adduct. 1H and 13C NMR spectra of all Mal-CTAs, along with 2D COSY and HSQC spectra are shown in the ESI, Fig. S42–S73.† Conversely when a relatively less hindered 2-methylpropanoate group was added to the maleimide vinyl group the presence of a small amounts of the minor cis (syn) adduct was also observed (Fig. 2, bottom), with signals for the –CHSC(S)SZ proton in their 1H NMR spectrum shifted at lower fields, and with larger J coupling values of around 9 Hz, consistent with a smaller torsion angle (5–10°) for the cis (syn) isomer. NOESY analysis further supported this assignment, by showing that, for the trans (anti), but not cis (syn) isomer, the –CHSC(S)SZ proton is spatially close to the methyl group of the 2-methylpropanoate substituent (Fig. S50 and S58†).
Next, to confirm their suitability to mediate RAFT polymerisations, selected Mal-CTAs were utilised for the polymerisation of (i) n-butyl acrylate (BA) and (ii) N-hydroxyethylacrylamide (HEA), 4-acryloylmorpholine (NAM), and N,N-dimethylacrylamide (DMA) as model acrylate and acrylamide monomers, respectively, initially at a [monomer]0:[Mal-CTA]0 = 50:1 (Fig. 3). Polymerisations of BA were carried out using the relatively hydrophobic Mal-CTAs (11a) and (12a) in DMF, using AIBN as the radical initiator, whereas for HEA, NAM and DMA hydrophilic acrylamide monomers and more hydrophilic CTA (11b) Perrier's ultrafast RAFT conditions,41 with VA-044 azo initiator in water/1,4-dioxane, were chosen (Table S3†). All polymerisations were found to be controlled, with molar mass distributions Đ < 1.20 (Table 1, runs 15–19). Using N,N-dimethylacrylamide (DMA) as a representative acrylamide monomer, polymers with increasing DP – [monomer]0:[Mal-CTA]0 = 50, 100, 200, and 500:1 – were then synthesised.
 | ||
| Fig. 3 Synthesis and SEC characterisation of α,β,ω-functional polymers produced from maleimide-derived CTAs. Reagents and conditions. a. for (Mal-CTA)-(BA)n – [BA]0:[Mal-CTA]0 50:1, DMF, AIBN, 80 °C, 90 min; for (Mal-CTA)-(HEA)n and (Mal-CTA)-(NAM)n – [monomer]0:[Mal-CTA]0 50:1, water:1,4-dioxane, VA-044, 100 °C, 15 min; for (Mal-CTA)-(DMA)n – [DMA]0:[Mal-CTA]0 50, 100, 200, 500:1, DMF water:1,4-dioxane, VA-044, 75 °C, 15 min. HEA = N-hydroxyethylacrylamide, NAM = 4-acryloylmorpholine, DMA = N,N-dimethylacrylamide. Right: SEC traces for the analysis of (Mal-CTA)-(HEA)n, (Mal-CTA)-(NAM)n, and (Mal-CTA)-(DMA)n polymers, in DMF + 0.1% LiBr as the mobile phase. (Mal-CTA)-(BA)n were analysed using CHCl3 as the mobile phase and are shown in the ESI.† | ||
| Code | Polymer | Mal-CTA | Monomer | [M]0:[Mal-CTA]0 |
Conversiona (%) | DPtheob | M n, SEC (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|
|
a Determined by 1H NMR, by comparing the area of the vinyl signals of residual monomer with that of polymer pendant chains.
b Theoretical degree of polymerisation was calculated from monomer conversion and initial [M]0:[Mal-CTA]0.
c Estimated by SEC analysis, using chloroform as the mobile phase and PMMA narrow standards.
d Estimated by SEC analysis, using DMF + 0.1 w/w% LiBr as the mobile phase and PMMA narrow standards.
|
||||||||
| 15 | (11a)-BA48 | (11a) | BA | 50 | 97 | 48 | 7.2c | 1.17c |
| 16 | (12a)-BA34 | (12a) | BA | 50 | 69 | 34 | 7.6c | 1.15c |
| 17 | (11b)-HEA43 | (11b) | HEA | 50 | 87 | 43 | 7.8d | 1.1d |
| 18 | (11b)-NAM49 | (11b) | NAM | 50 | 98 | 49 | 6.1d | 1.17d |
| 19 | (11b)-DMA39 | (11b) | DMA | 50 | 79 | 39 | 5.1d | 1.15d |
| 20 | (11b)-DMA97 | (11b) | DMA | 100 | 97 | 97 | 10.7d | 1.20d |
| 21 | (11b)-DMA184 | (11b) | DMA | 200 | 92 | 184 | 16.9d | 1.17d |
| 22 | (11b)-DMA475 | (11b) | DMA | 500 | 95 | 475 | 35.8d | 1.24d |
A slight shoulder at low molecular masses was observed in the SEC traces of the two shorter polymers (11b)-DMA39 and (11b)-DMA97, potentially suggestive of either a small proportion of early irreversible termination, or slow consumption of the RAFT agent in the pre-equilibrium step. (Mal-CTA)-DMAn materials with higher molar mass (11b)-DMA184 and (11b)-DMA475 had comparably more symmetrical traces, and again good control over molar mass dispersity was achieved (Table 1, runs 19–22).
To further examine the controlled nature of these polymerisations, we periodically sampled a DMA polymerisation, mediated by (13b) at 15, 30, 60, 120 and 360 min. Consistent with conventional RAFT agents, the polymerisation with Mal-CTA (13b) exhibited a linear evolution of molar mass with conversion. SEC chromatograms displayed narrow molar mass distributions for each time point (Đ < 1.20, Fig. S84†). Furthermore, the first order kinetic plot also exhibited the typical linear increase then plateauing due to termination, with a small acceleration between 120 and 360 min likely due to the gel effect at high conversion.
To extend the range of monomer types and Mal-CTAs evaluated, we also conducted a polymerisation using (13b) with methyl methacrylate (MMA) targeting a DP of 125. Interestingly the resulting polymer displayed a broad molar mass distribution (Đ = 2.05) and Mn,SEC (51.0 kg mol−1) significantly higher than the theoretical molar mass (10.5 kg mol−1), suggesting methacrylate polymerisations may not be controlled with this Mal-CTA (Fig. S85 and Table S4†). We theorised this was due to the insertion of the maleimide between the typically stabilising nitrile based reinitiating group and the trithiocarbonate. This would lead to a more stable radical on the MMA initiator derived chains than the maleimide reinitiating group on the Mal-CTA resulting in poor CTA participation in the polymerisation and thus high molar masses with high dispersity. To investigate this, we conducted an identical MMA polymerisation with CTA (7), the precursor to Mal-CTA (13b). In this case, the polymerisation was well controlled, displaying low dispersity (Đ = 1.19) while the theoretical (Mn,th = 10.5 kg mol−1) and experimental (Mn,SEC = 13.2 kg mol−1) were in closer agreement (Fig. S84 and Table S4†). Overall, this highlights a potential limitation for the use of these Mal-CTAs to mediate the polymerisation of methacrylate monomers, while being able to efficiently control acrylate and acrylamide polymerisations.
Conclusions
In summary, in this study we present three general strategies for the synthesis of functional chain-transfer agents starting from N-alkyl substituted maleimides. Typically heterotelechelic polymers can be produced by RAFT polymerisation by using chain-transfer agents with functional R groups and subsequently modifying the thiocarbonylthio group at the ω-chain-end.27,42 By using appropriate N-substituted maleimides and common RAFT agents and precursors, the simple approach we present allows for a facile introduction of an additional residue at the β-position of a polymer chain, resulting in α,β,ω-functional polymers, potentially further expanding the range of applications of RAFT polymers. Moreover, an increasing number of maleimide-containing derivatives are becoming available, largely driven by their application in biology and conjugation chemistry. At the same time, many RAFT agents are also now available on the market. Our strategy can therefore allow for the production of a range of functional chain-transfer agents – incorporating for example fluorophores, ligands, peptide/proteins or drug molecules – in one simple step starting from commercially available maleimide derivatives and RAFT agents. We believe that the strategies presented in this study complements well the existing techniques which utilise N-substituted maleimides for the synthesis of functional polymers and will further expand the palette of tools available to polymer chemists for the design of functional CTAs and resulting polymeric materials.Author contributions
G. M., S. S. and R. C. designed the experiments. R. C. carried out the experiments with the assistance of E. R., F. M., and L. S. for the synthesis of Mal-CTAs, R. F. for the synthesis and characterisation of Mal-CTA-derived polymers, P. G. for the polymerisation reactions, K. B. and H. W. for the NOESY experiments. G. M., S. S. and R. C. wrote, reviewed, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants from Engineering and Physical Sciences Council [grant numbers EP/I01375X/1, EP/N50970X/1, EP/L01646X]. Lorenzo Schiazza and Huitong Li are thanked for their help with NMR and MS analysis.References
- K. Renault, J. W. Fredy, P.-Y. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- C. do Pazo, K. Nawaz and R. M. Webster, Nat. Rev. Drug Discovery, 2021, 20, 583–584 CrossRef CAS PubMed.
- S. Schreiber, P. Rutgeerts, R. N. Fedorak, M. Khaliq-Kareemi, M. A. Kamm, M. Boivin, C. N. Bernstein, M. Staun, O. Ø. Thomsen and A. Innes, Gastroenterology, 2005, 129, 807–818 CrossRef CAS PubMed.
- J.-A. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822–3863 CrossRef CAS PubMed.
- A. H. St Amant, D. Lemen, S. Florinas, S. Mao, C. Fazenbaker, H. Zhong, H. Wu, C. Gao, R. J. Christie and J. Read de Alaniz, Bioconjugate Chem., 2018, 29, 2406–2414 CrossRef CAS PubMed.
- B. Briou, B. Améduri and B. Boutevin, Trends in the Diels–Alder reaction in polymer chemistry, Chem. Soc. Rev., 2021, 50(19), 11055–11097 RSC.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS PubMed.
- M. Ouchi and M. Sawamoto, Polym. J., 2018, 50, 83–94 CrossRef CAS.
- S. Srichan, H. Mutlu, N. Badi and J.-F. Lutz, Angew. Chem., Int. Ed., 2014, 53, 9231–9235 CrossRef CAS PubMed.
- S. Srichan, N. Kayunkid, L. Oswald, B. Lotz and J.-F. Lutz, Macromolecules, 2014, 47, 1570–1577 CrossRef CAS.
- R. K. Roy and J.-F. Lutz, J. Am. Chem. Soc., 2014, 136, 12888–12891 CrossRef CAS PubMed.
- J.-F. Lutz, Acc. Chem. Res., 2013, 46, 2696–2705 CrossRef CAS PubMed.
- N. Baradel, O. Gok, M. Zamfir, A. Sanyal and J.-F. Lutz, Chem. Commun., 2013, 49, 7280–7282 RSC.
- M. Zamfir and J.-F. Lutz, Nat. Commun., 2012, 3, 1138 CrossRef.
- S. Srichan, D. Chan-Seng and J.-F. Lutz, ACS Macro Lett., 2012, 1, 589–592 CrossRef CAS PubMed.
- D. Chan-Seng, M. Zamfir and J.-F. Lutz, Angew. Chem., Int. Ed., 2012, 51, 12254–12257 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS PubMed.
- S. Pfeifer and J.-F. Lutz, Chem. – Eur. J., 2008, 14, 10949–10957 CrossRef CAS PubMed.
- M. Zamfir, P. Theato and J.-F. Lutz, Polym. Chem., 2012, 3, 1796–1802 RSC.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- S. M. Henry, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2009, 20, 1122–1128 CrossRef CAS PubMed.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef CAS PubMed.
- Z. Huang, B. B. Noble, N. Corrigan, Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer and J. Xu, J. Am. Chem. Soc., 2018, 140, 13392–13406 CrossRef CAS PubMed.
- D. Vinciguerra, J. Tran and J. Nicolas, Chem. Commun., 2018, 54, 228–240 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2021, 222, 2000311 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- F. Mastrotto, S. Salmaso, Y. L. Lee, C. Alexander, P. Caliceti and G. Mantovani, Polym. Chem., 2013, 4, 4375–4385 RSC.
- A. Watanabe, J. Niu, D. J. Lunn, J. Lawrence, A. S. Knight, M. Zhang and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1259–1268 CrossRef CAS.
- K. Godula, M. L. Umbel, D. Rabuka, Z. Botyanszki, C. R. Bertozzi and R. Parthasarathy, J. Am. Chem. Soc., 2009, 131, 10263–10268 CrossRef CAS PubMed.
- J. E. Hudak, S. M. Canham and C. R. Bertozzi, Nat. Chem. Biol., 2014, 10, 69–75 CrossRef CAS PubMed.
- E. C. Woods, N. A. Yee, J. Shen and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 15782–15788 CrossRef CAS PubMed.
- J. Mauris, F. Mantelli, A. M. Woodward, Z. Cao, C. R. Bertozzi, N. Panjwani, K. Godula and P. Argüeso, PLoS One, 2013, 8, e72304 CrossRef CAS PubMed.
- Q. Liu, H. Xue, J. Gao, L. Cao, G. Chen and H. Chen, Polym. Chem., 2016, 7, 7287–7294 RSC.
- E. C. Woods, F. Kai, J. M. Barnes, K. Pedram, M. W. Pickup, M. J. Hollander, V. M. Weaver and C. R. Bertozzi, eLife, 2017, 6, e25752 CrossRef PubMed.
- M.-G. Braun and S. Z. Zard, Org. Lett., 2011, 13, 776–779 CrossRef CAS PubMed.
- Q. Huang and S. Z. Zard, Org. Lett., 2017, 19, 3895–3898 CrossRef CAS PubMed.
- J. H. Udding, H. Hiemstra and W. N. Speckamp, J. Org. Chem., 1994, 59, 3721–3725 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation of RAFT agents and relative intermediates, polymerisation reactions. See DOI: https://doi.org/10.1039/d1py01565f |
| This journal is © The Royal Society of Chemistry 2022 |