
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nonionic nontoxic antimicrobial polymers: indole-grafted poly(vinyl alcohol) with pendant alkyl or ether groups†
Xiaoya
Li
 a,
Sedef
İlk
bc,
Yang
Liu
d,
Deepak Bushan
Raina
d,
Deniz
Demircan
a and
Baozhong
Zhang
*a
a,
Sedef
İlk
bc,
Yang
Liu
d,
Deepak Bushan
Raina
d,
Deniz
Demircan
a and
Baozhong
Zhang
*a
aLund University, Centre for Analysis and Synthesis, Department of Chemistry, P. O. Box 124, SE-22100 Lund, Sweden. E-mail: baozhong.zhang@chem.lu.se
bNiğde Ömer Halisdemir University, Faculty of Medicine, Department of Immunology, TR-51240, Niğde, Turkey
cKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Chemistry, Division of Glycoscience, SE-10691 Stockholm, Sweden
dFaculty of Medicine, Department of Clinical Sciences, Orthopedics, Lund University, Lund, Sweden
First published on 24th March 2022
Abstract
A series of new nonionic antimicrobial polymers with a biodegradable polyvinyl alcohol (PVA) backbone grafted with indole units and different hydrophobic alkyl or ether groups were synthesized by facile esterification. The chemical structures and thermal properties of the obtained polymers were characterized by GPC, NMR, FTIR, WAXD, TGA and DSC analyses. All these nonionic polymers showed a significant antibacterial effect similar to gentamicin against 9 food and human pathogenic bacteria according to the disk diffusion assay. The presence of alkyl or ether groups in most cases did not significantly affect the antibacterial effect compared to the polymer with unsubstituted indole units (with N–H moieties). The impacts of the OH conversion and molecular weight of the obtained polymers on their antimicrobial and anti-quorum sensing effects were also preliminarily investigated. Finally, the obtained indole-grafted PVAs were subjected to MTT assay using a mammalian cell line and hemolysis investigations, and the results showed excellent biocompatibility, particularly for those with ether substituents.
Introduction
Antimicrobial polymers (APs) have been under rapid development in the past decade, due to their enhanced antimicrobial activity,1–3 low potential to induce antimicrobial resistance,4–9 and low leaching risk and environmental toxicity compared to small molecular antimicrobials.10–15 Currently most reported APs are ionic, and their ionic interactions with bacterial membranes are essential for their antimicrobial function.16–22 However, for certain applications, ionic polymers may suffer from undesirable water solubility, fouling risk and toxicity.23–28 Therefore, new APs without ions have become an attractive target with high potential.29,30Nonionic polymers do not have ionic interactions with bacteria, so they should be endowed with the ability to interact with bacteria by other grafted nonionic biologically active units, such as glucose, curcumin, astaxanthin, tropolone, aspirin, limonene, indole, isatin, anisole, etc.31–39 These biologically active molecules are known for their ability to interact with bacteria, and they have been adapted by the natural ecosystem and thus tend to be more biodegradable and less eco-toxic.40,41 Some of these biologically active molecules (e.g. limonene, indole, and aspirin) also show anti-quorum sensing effects.37,42,43 Grafting these molecules onto a polymer backbone could enable interactions between the polymers and bacterial membranes leading to an antimicrobial effect.29,44–51 Such nonionic interactions can be complex, including hydrogen bonding, hydrophobic, dipole, and aromatic interactions.10,52–55 However, the general mechanism related to the antimicrobial function of nonionic APs remains to be unravelled.
Indole is a bio-sourced nonionic building block that is ubiquitous in nature and domestic wastes,56,57 and it has received growing attention for the development of bio-based polymers toward various high performance packaging and textile applications.58–61 In the meantime, many indole derivatives are natural antibiotics,31,62,63 and several indole-based polyesters, polyketones and polyurethanes have been reported with antibacterial effects.44,64,65 Indole groups can also have a synergistic antibacterial effect when grafted on polymers together with other antibacterial moieties (e.g. metal ions, ammonium cations, chitosan, and aldehyde).66–69 In addition, indole-based APs can also have desirable miscibility with other matrix polymers so they are potentially suitable antimicrobial additives.30
Despite the promising potential of indole-based nonionic APs, the knowledge regarding the role of indole units and other structural factors in their antimicrobial function remains rather limited. For instance, hydrophobic substituents (e.g. alkyl or ether groups and aromatic units) frequently enhance the antimicrobial effect of ionic APs,70,71 but it remains unclear whether such an effect exists in nonionic APs. Furthermore, the N–H moiety of indole is a hydrogen bond donor, which can form hydrogen bonds with various hydrogen-accepting groups (e.g. ethers or amides in peptidoglycan) in bacterial membranes.72 It remains to be unraveled whether such hydrogen bond interactions are essential for their antimicrobial effects.30 Finally, the effects of molecular weights and the hydrophilic/hydrophobic balance of nonionic APs also remained largely unexplored.
In order to shed some light on the roles of indole and other structural factors such as hydrogen bonding or hydrophobic interactions, we herein report on the synthesis of six nonionic indole-based APs with biodegradable poly(vinyl alcohol) backbones, which contain the N–H unit or different linear or cyclic alkyl or ether groups. The molecular structures, thermal properties, antimicrobial effects and cytotoxicity of the obtained nonionic APs were investigated. The impact of hydrophobic N-substitution as well as the grafting density and molecular weight on the antimicrobial effects of the obtained nonionic APs was preliminarily evaluated.
Experimental
Chemicals and materials
Poly(vinyl alcohol) (Mw 9000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolyzed), indole-3-acetic acid, 1-iodopropane, 1-iodohexane, 2-methoxyethyl-4-methylbenzenesulfonate, 1-bromo-2-cyclohexylethane, phenoxyethyl bromide, 1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide·HCl (EDC–HCl), and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich. N,N-Dimethylformamide (DMF), N,N-dimethylacetamide (DMAc), dimethyl sulfoxide (DMSO), hydrochloric acid (HCl), sodium hydroxide (NaOH), ethanol, acetone, chloroform, ethyl acetate (EtOAc), diethyl ether, potassium carbonate (K2CO3), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3) (ACS, Reag. Ph. Eur.), and anhydrous sodium sulphate (Na2SO4) were purchased from VWR Chemicals. All chemicals were used directly without further purification.
000, 80% hydrolyzed), indole-3-acetic acid, 1-iodopropane, 1-iodohexane, 2-methoxyethyl-4-methylbenzenesulfonate, 1-bromo-2-cyclohexylethane, phenoxyethyl bromide, 1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide·HCl (EDC–HCl), and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich. N,N-Dimethylformamide (DMF), N,N-dimethylacetamide (DMAc), dimethyl sulfoxide (DMSO), hydrochloric acid (HCl), sodium hydroxide (NaOH), ethanol, acetone, chloroform, ethyl acetate (EtOAc), diethyl ether, potassium carbonate (K2CO3), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3) (ACS, Reag. Ph. Eur.), and anhydrous sodium sulphate (Na2SO4) were purchased from VWR Chemicals. All chemicals were used directly without further purification.
Synthesis
N-Propyl-3-indoleacetic acid (2). Yield 90%; brown powder. 1H NMR (400.13 MHz, DMSO-d6) δ, ppm: δ 12.20 (s, 1H), 7.52 (d, 1H), 7.43 (d, 1H), 7.27 (s, 1H), 7.13 (t, 1H), 7.02 (t, 1H), 4.08 (t, 2H), 3.65 (s, 2H), 1.75 (m, 2H), 0.84 (t, 3H). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 173.50, 136.39, 128.07, 127.72, 121.48, 119.36, 118.92, 110.16, 107.45, 47.35, 31.40, 23.67, 11.66. UV (DMF): λmax 288 nm (ε 3.73 × 103 M−1 cm−1). FTIR: ν = 2960, 1709, 1471, 1391, 1231, 1205, 1188, 910, 805, 730 and 654 cm−1. HRMS (ESI+, m/z): exact mass calcd for C13H16NO2+, 218.1181; found, 218.1180.
N-Hexyl-3-indoleacetic acid (3). Yield 94%; brown powder. 1H NMR (400.13 MHz, DMSO-d6) δ, ppm: 1H NMR (400.13 MHz, DMSO-d6) δ, ppm: δ 12.20 (s, 1H), 7.52 (d, 1H), 7.42 (d, 1H), 7.26 (s, 1H), 7.13 (t, 1H), 7.02 (t, 1H), 4.11 (t, 2H), 3.65 (s, 2H), 1.72 (m, 2H), 1.26 (m, 6H), 0.84 (t, 3H). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 173.51, 136.31, 128.03, 127.66, 121.48, 119.35, 118.88, 110.07, 107.45, 45.75, 31.31, 30.33, 26.42, 22.48, 14.32. UV (DMF): λmax 290 nm (ε 3.79 × 103 M−1 cm−1). FTIR: ν = 2929, 1702, 1471, 1369, 1230, 1213, 939, 785, 725 and 645 cm−1. HRMS (ESI+, m/z): exact mass calcd for C16H22NO2+, 260.1651; found, 260.1650.
N-(2-Cyclohexylethyl)-3-indoleacetic acid (4). Yield 75%; brown powder. 1H NMR (400.13 MHz, DMSO-d6) δ, ppm: δ 12.20 (s, 1H), 7.50 (d, 1H), 7.40 (d, 1H), 7.26 (s, 1H), 7.12 (t, 1H), 7.01 (t, 1H), 4.14 (t, 2H), 3.63 (s, 2H), 1.75 (m, 2H), 1.63 (m, 5H), 1.17 (m, 4H), 0.95 (m, 2H). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 173.52, 136.20, 128.07, 127.55, 121.51, 119.38, 118.90, 110.00, 107.52, 43.61, 37.76, 35.10, 33.03, 31.31, 26.52, 26.15. UV (DMF): λmax 292 nm (ε 3.74 × 103 M−1 cm−1). FTIR: ν = 2921, 1696, 1469, 1306, 1138, 1013, 960, 795, 735 and 644 cm−1. HRMS (ESI+, m/z): exact mass calcd for C18H24NO2+, 286.1807; found, 286.1801.
N-Methoxyethyl-3-indoleacetic acid (5). Yield 68%; brown powder. 1H NMR (400.13 MHz, DMSO-d6) δ, ppm: δ 12.20 (s, 1H), 7.50 (d, 1H), 7.45 (d, 1H), 7.25 (s, 1H), 7.13 (t, 1H), 7.02 (t, 1H), 4.29 (t, 2H), 3.64 (m, 4H), 3.22 (t, 3H). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 173.50, 136.54, 128.04, 128.03, 121.53, 119.28, 119.00, 110.23, 107.58, 71.58, 58.52, 45.62, 31.30. UV (DMF): λmax 288 nm (ε 4.15 × 103 M−1 cm−1). FTIR: ν = 2882, 1694, 1469, 1303, 1115, 1011, 947, 833, 736 and 647 cm−1. HRMS (ESI+, m/z): exact mass calcd for C13H16NO3+, 234.1130; found, 234.1126.
N-(2-Phenyloxylethyl)-3-indoleacetic acid (6). Yield 72%; brown powder. 1H NMR (400.13 MHz, DMSO-d6) δ, ppm: δ 12.20 (s, 1H), 7.56–6.86 (m, 10H), 4.54 (t, 2H), 4.27 (t, 2H), 3.65 (s, 2H). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 173.48, 158.54, 136.63, 129.97, 128.10, 128.09, 121.65, 121.29, 119.15, 114.93, 110.35, 107.89, 67.37, 45.35, 31.29. UV (DMF): λmax 288 nm (ε 3.83 × 103 M−1 cm−1). FTIR: ν = 2921, 1702, 1594, 1499, 1464, 1232, 1206, 1054, 903, 782, 753, 734, 688 and 654 cm−1. HRMS (ESI+, m/z): exact mass calcd for C18H18NO3+, 296.1287; found, 296.1285.
PI1 . Yield 45%; brown powder. 1H-NMR (400 MHz, DMSO-d6) δ, ppm: 10.90 (s, 1H, NH), 7.50 (s, 1H, Ar), 7.34 (s, 1H, Ar), 7.22 (s, 1H, Ar), 7.06 (s, 1H, Ar), 6.98 (s, 1H, Ar), 5.24–4.75 (m, CHOOR) 4.75–4.18 (m, OH), 3.96–3.78 (m, CHOH), 3.77–3.57 (m, 2H, ArCH2COOR), 2.04–1.86 (m, CH3COOR), 1.86–1.13 (m, R1CH2R2). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 171.40, 170.19, 136.53, 127.60, 124.42, 121.51, 118.95, 117.82, 107.65, 68.72–63.43, 47.12–44.63, 31.30, 21.24. FTIR: ν = 3411, 2925, 1720, 1458, 1371, 1245 and 742 cm−1.
PI2 . Yield 40%; brown powder. 1H-NMR (400.13 MHz, DMSO-d6) δ, ppm: 7.60–6.79 (m, 5H, Ar), 5.35–4.74 (m, CHOOR) 4.74–4.18 (m, OH), 4.16–3.90 (m, NCH2CH2CH), 3.86–3.75 (m, CHOH), 3.75–3.54 (m, 2H, ArCH2COOR), 2.04–1.84 (m, CH3COOR), 1.84–1.13 (m, NCH2CH2CH3, R1CH2R2), 0.92–0.59 (m, NCH2CH2CH3). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 171.45, 170.34, 136.32, 127.96, 127.68, 121.52, 119.28, 118.95, 110.09, 106.66, 70.63–62.89, 47.33, 46.35–44.57, 31.13, 23.59, 21.17, 11.61. FTIR: ν = 3422, 2929, 1729, 1468, 1370, 1243 and 740 cm−1.
PI3 . Yield 28%; brown powder. 1H-NMR (400.13 MHz, DMSO-d6) δ, ppm: 7.62–6.85 (m, 5H, Ar), 5.32–4.74 (m, CHOOR) 4.74–4.22 (m, OH), 4.18–3.52 (m, NCH2(CH2)4CH3, CHOH, ArCH2COOR), 2.02–1.84 (m, CH3COOR), 1.84–1.02 (m, NCH2(CH2)4CH3, R1CH2R2), 0.88–0.65 (m, CH2CH3). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 171.53, 170.14, 136.28, 127.94, 127.64, 121.54, 119.32, 118.95, 110.04, 106.87, 68.42–63.68, 45.82, 46.93–44.59, 31.32, 30.30, 26.42, 22.52, 21.27, 14.30. FTIR: ν = 3426, 2929, 1729, 1468, 1370, 1240 and 738 cm−1.
PI4 . Yield 35%; brown powder. 1H-NMR (400.13 MHz, DMSO-d6) δ, ppm: 7.84–6.83 (m, 5H, Ar), 5.30–4.75 (m, CHOOR) 4.75–4.17 (m, OH), 4.17–3.54 (m, NCH2CH2(CH2)6, CHOH, ArCH2COOR), 2.08–1.84 (m, CH3COOR), 1.84–0.28 (m, N CH2CH2(CH2)6, R1CH2R2). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 171.45, 170.15, 136.14, 127.98, 127.43, 121.52, 119.31, 118.93, 109.90, 106.83, 71.04–63.36, 47.32–44.47, 43.61, 37.69, 35.04, 32.97, 31.15, 26.47, 26.10, 21.21. FTIR: ν = 3442, 2921, 1732, 1468, 1370, 1243 and 736 cm−1.
PI5 . Yield 23%; brown powder. 1H-NMR (400.13 MHz, DMSO-d6) δ, ppm: 7.58–6.83(m, 5H, Ar), 5.35–4.73 (m, CHOOR), 4.73–4.01 (m, OH, NCH2CH2OCH3), 4.01–3.45 (m, CHOH, ArCH2COOR, NCH2CH2OCH3), 3.27–3.06 (NCH2CH2OCH3), 2.08–1.86 (m, CH3COOR), 1.86–1.18 (m, R1CH2R2). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: 171.61, 170.42, 136.60, 128.05, 127.96, 121.59, 119.26, 119.08, 110.23, 107.08, 71.57, 68.69–63.42, 58.52, 46.53–44.42, 45.67, 31.18, 21.25. FTIR: ν = 3434, 2929, 1729, 1468, 1370, 1243 and 740 cm−1.
PI6 . Yield 30%; brown powder. 1H-NMR (400.13 MHz, DMSO-d6) δ, ppm: 7.63–6.65 (m, 10H, Ar), 5.36–4.75 (m, CHOOR) 4.75–3.98 (m, OH, NCH2CH2O), 3.98–3.54 (m, CHOH, ArCH2COOR), 2.03–1.83 (m, CH3COOR), 1.83–0.98 (m, R1CH2R2). 13C NMR (100.61 MHz, DMSO-d6) δ, ppm: δ, ppm: 171.44, 170.18, 136.61, 129.93, 128.02, 121.70, 121.46, 121.24, 119.23, 114.86, 110.34, 107.37, 67.66–62.91, 67.45, 46.79–44.52, 45.35, 31.15, 21.21. FTIR: ν = 3438, 2925, 1731, 1598, 1494, 1468, 1373, 1241, 742 and 692 cm−1.
Measurements
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DRX400 spectrometer at a proton frequency of 400.13 MHz and a carbon frequency of 100.61 MHz. Fourier transform infrared (FT-IR) spectra were obtained with an attenuated total reflection (ATR) setup using a Bruker Alpha FT-IR spectrometer. Differential scanning calorimetry (DSC) measurements were performed using a TA Instruments DSC Q2000. The samples were studied at a heating rate of 10 °C min−1 under nitrogen with a purge rate of 50 mL min−1. The Tg value was taken as the midpoint of the endothermic step-change observed during the second heating run. Thermogravimetric analysis (TGA) was performed under a nitrogen atmosphere with a Thermogravimetric Analyzer (TA Instrument Q500) at a heating rate 10 °C min−1. Gel permeation chromatography (GPC) was carried out with three Shodex columns in series (KF-805, 2804, and 2802.5) and a refractive index (RI) detector (Viscotek Model 250). All measurements were carried out at room temperature at a concentration of 30 mg mL−1 using chloroform as the eluent, and at an elution rate of 1 mL min−1. Calibration was performed with four polystyrene standard samples (Mn = 650 kg mol−1 from Water Associates, 96 and 30 kg mol−1 from Polymer Laboratories, and 3180 g mol−1 from Agilent Technologies). UV-visible spectra were recorded with an UV-visible spectrometer in the wavelength range from 200 to 600 nm with a resolution of 2 nm, employing quartz cuvettes of 10 mm path length. WXRD (wide angle X-ray diffraction) diffraction patterns were recorded with a Stoe Stadi MP X-ray powder diffractometer in the transmission mode over 2θ ranges 2–60° with Cu Kα radiation. High resolution mass spectrometry (HRMS) was performed by direct infusion on a Water Xevo-G2 QTOF mass spectrometer using electrospray ionization.Antimicrobial bioassay
Anti-quorum sensing (anti-QS)
The quorum sensing inhibitory (anti-QS) activity of three PI4 samples (PI454, PI472, and PI485, 10 μg mL−1 in DMF, w/v) was evaluated by the disk diffusion assay against C. violaceum CV026 according to the literature procedure.30,73,74 The bacteria suspension was sub-cultured in LB broth at 30 °C for 24 h. The Petri plates with LB soft agar containing signal molecule N-hexanoyl-L-homoserine lactone (C6-HSL, 0.25 μg mL−1) were inoculated with the bacteria culture. Afterward, sterile discs (diameter: 6 mm) loaded with samples (20 μL) were placed on the Petri plates and incubated at 30 °C for 24 h. Gentamicin (10 μg per disc) and the solvent (DMF) were used as positive or negative controls, respectively. The anti-QS activity was evaluated by the turbid halo formation around the disc (in contrast to the purple background).Minimum inhibitory concentration (MIC) evaluations
The minimum inhibitory concentration (MIC) of the three PI4 samples (PI454, PI472, and PI485) against two bacteria (Escherichia coli ATCC 8739 and Pseudomonas fluorescens (PCL 1701)) was determined using the method described in the guideline of The Clinical and Laboratory Standards Institute.75 Briefly, the bacterial suspensions were adjusted using 0.5 McFarland's standard (108 CFU mL−1) and the tested polymers were dissolved in DMF. Serial two-fold dilutions of samples’ solution (in a 96-well microtiter plate) in concentrations ranging from 100 μg mL−1 to 0.19 μg mL−1 were used to determine the MIC in the Mueller Hinton broth (MHB). After mixing the samples and MHB, the 96-well microtiter plates were covered with a sterile plate sealer and incubated at 37 °C for 24 h. The only medium was used as the negative control and only bacterial inoculums was used as the positive control. All experiments were performed in triplicate. The MIC value was taken at the lowest concentration of samples where no visible growth is seen in the wells.Leaching evaluations by UV-vis
Suspensions of PI1–6 in PBS buffer (100 μg mL−1) were incubated at 37 °C for 24 h with mild magnetic stirring. The UV-vis spectra of their supernatants at the initial point (PIX-initial) and after 24 h (PIX-24 h) were recorded. The UV-vis spectra of PI1–6 in DMSO (25 μg mL−1) were also recorded as references.MTT assay
MG-63 cells were purchased from American Type Culture Collection (ATCC) (LOT: 70016786). The MG-63 osteoblast-like human cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and 1% streptomycin in a humidified incubator at 37 °C. The medium was replaced every 2 days. Cells were trypsinized and centrifuged at 400g for 4 min to obtain a concentrated cell pellet when the confluence reached 80%. 1 × 104 cells per well were seeded on a 96-well plate and cultured for 24 h before adding the materials. Test compounds (negative control, 1–6, and PI1–6) dissolved in DMSO were then added to the cell culture at a final DMSO concentration of 1% (v/v), which formed suspensions. Fresh culture medium without samples was used as the negative control, and each sample was replicated in four wells. After being cultured for 24 h, the cell culture medium was discarded and the cells were washed with phosphate buffer once. MTT working solution (0.5 mg mL−1) was added to the cells and incubated for 2 h at 37 °C, after which DMSO (200 μL per well) was added to the reaction products for 10 min. The solubilized contents were pipetted and transferred into a clear bottom 96-well plate. Absorbance was determined by spectrophotometry at 600 nm wavelength. In addition, to evaluate the interaction between PI1 and MTT working solution, only the material (i.e.PI1) was subjected to the above procedures under the same conditions without adding the cells.Hemolysis tests
The HaemoScan Biomaterial Haemolytic Assay (HaemoScan, Netherlands) was used to investigate the cytotoxicity of PI1–6 on human erythrocytes according to the manufacturer's protocol.76 The concentrated erythrocytes were included in this kit (LOT: 220307). Briefly, the erythrocyte was prepared by repeatedly rinsing with different wash buffers (dilution buffer I, II and III, provided by the manufacturer) and centrifuged at 400g for 10 minutes. Afterward, 5 mL of dilution buffer III was added to re-suspend the erythrocytes. 0.5 mL of erythrocyte suspension was used to test each sample. PI1–6 samples were first dissolved in DMSO to form 10 mg mL−1 stock solutions. 5 μL of the stock solution was added in 0.5 mL of erythrocyte suspension with a final (suspension) concentration of 100 μg mL−1 for polymers. After 24 h of incubation, the samples were centrifuged at 4500g for 1 min and 20 μL of the supernatant was pipetted into a 96-well plate along with 180 μL of assay buffer. The absorbance was read at a wavelength of 450 nm. Each polymer was tested in triplicate. DMSO (5 μL, 1% v/v) was used as the negative control (0% hemolysis). Lysis buffer was used as the positive control (100% hemolysis). The hemolysis percentage was calculated using the following equation:
Results and discussion
Synthesis of indole-grafted PVAs
Six indole-grafted PVAs (PI1–6) were conveniently synthesized by esterification at room temperature of the PVA backbone using the corresponding carboxylic acids (1–6) as grafting agents (Scheme 1). Indole carboxylic acid (1) is commercially available, while the other five agents (2–6) were synthesized by a straightforward SN2 reaction using 1 and the corresponding electrophilic agents (1-iodopropane, 1-iodohexane, 1-bromo-2-cyclohexylethane, 2-methoxyethyl-4-methylbenzene-sulfonate and phenoxyethyl bromide, Scheme S1, ESI†). The conversion of the OH groups (pOH) on PVA backbones after grafting was evaluated using the integrals of the residual OH signals and the aromatic signals in the 1H-NMR spectra (Fig. S3 for PI1 and Fig. S4 for PI6 as examples, ESI†). As shown in Table 1, pOH for PI1–6 was not identical but has a relatively narrow dispersity range (59–72%) considering the accuracy of the quantification method and intrinsic polydispersity of polymers. To obtain some preliminary insight into the possible impacts of the OH conversion, three PI4 samples with OH conversions of 54%, 72% and 85% were synthesized by using different amounts of grafting agents. Their biological activities were compared in the later session. The resulting crude polymers were purified by precipitation twice to completely remove the unreacted grafting agents and solvents, which causes loss of the products and decreased yields (23–45%). The obtained polymers showed good solubility in polar aprotic solvents (e.g. DMF, DMSO, and DMAc) and low solubility in protic solvents (e.g. H2O and ethanol). In addition, the low water solubility of these polymers was further demonstrated by the low UV-vis absorption of the aqueous phase where the polymers were immersed for 24 h (Fig. S10A, ESI†). The same result also indicated that the purification of the polymers was successful without any noticeable small molecular agent left.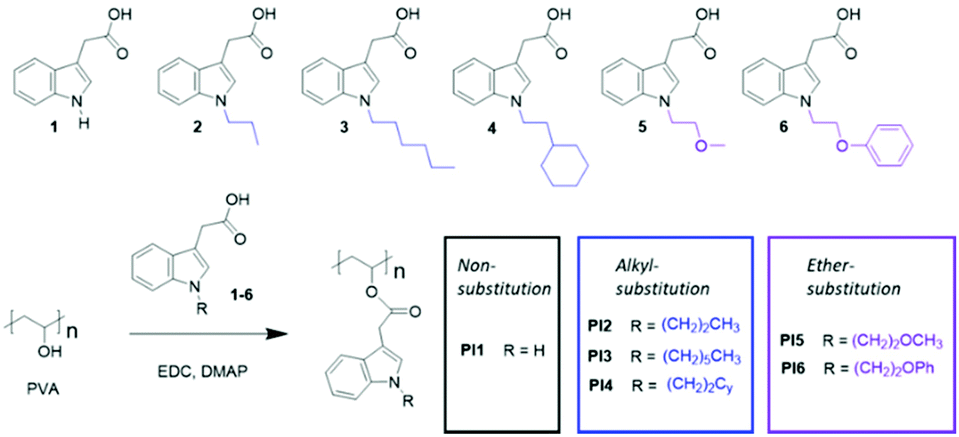 | ||
| Scheme 1 Synthesis of various indole-grafted PVAs (PI1–6), using the corresponding carboxylic acid as grafting agents (1–6). Note that the starting PVA contains 80% OH and 20% acetate (OAc) groups. The OAc groups remained unchanged after the reaction, which were omitted in the chemical structures. | ||
| p OH (%) | M NMR (g mol−1) | M n (g mol−1) | M w (g mol−1) | Đ | T g (°C) | T 95d (°C) | T max (°C) | CY (%) | |
|---|---|---|---|---|---|---|---|---|---|
| PVA | — | — | — | — | 67 | 267 | 312 | 5.4 | |
| PI1 | 59 | 25000 |
— | — | — | 93 | 277 | 322 | 11.0 |
| PI2 | 63 | 31000 |
31300 |
76600 |
2.4 | 61 | 267 | 316 | 4.7 |
| PI3 | 64 | 40000 |
37700 |
67400 |
1.8 | 38 | 264 | 318 | 7.0 |
| PI4 | 72 | 42000 |
38100 |
92000 |
2.4 | 56 | 275 | 320 | 5.8 |
| PI5 | 68 | 34000 |
66100 |
167000 |
2.5 | 53 | 287 | 320 | 9.6 |
| PI6 | 64 | 39000 |
62800 |
108200 |
1.7 | 61 | 277 | 322 | 5.4 |
Molecular and thermal characterization
The chemical structures of the obtained polymers were confirmed by 1H NMR spectroscopy analysis (Fig. 1). The starting polymer PVA contains both OH and OAc substituents, as shown in its 1H NMR spectrum (Fig. 1A). The signal at ∼1.97 ppm (2) is attributed to the Oac protons. The broad signals at 3.97–3.56 ppm (3) and 5.17–4.73 ppm (5) correspond to the backbone CH groups that are adjacent to the OH and OAc groups, respectively. According to the ratio between the integrals of signals 3 and 5, the initial PVA contains ∼78% of OH and ∼22% of OAc groups, which is consistent with the data from the supplier (80 and 20%). The signals for OH protons were observed at 4.73–4.19 ppm (4), containing three discernible signals at 4.69, 4.51, and 4.26 ppm that correspond to mm, mr and rr triads, respectively (Fig. S2, ESI†).77 The backbone CH2 signals were observed at 1.85–1.20 ppm (1).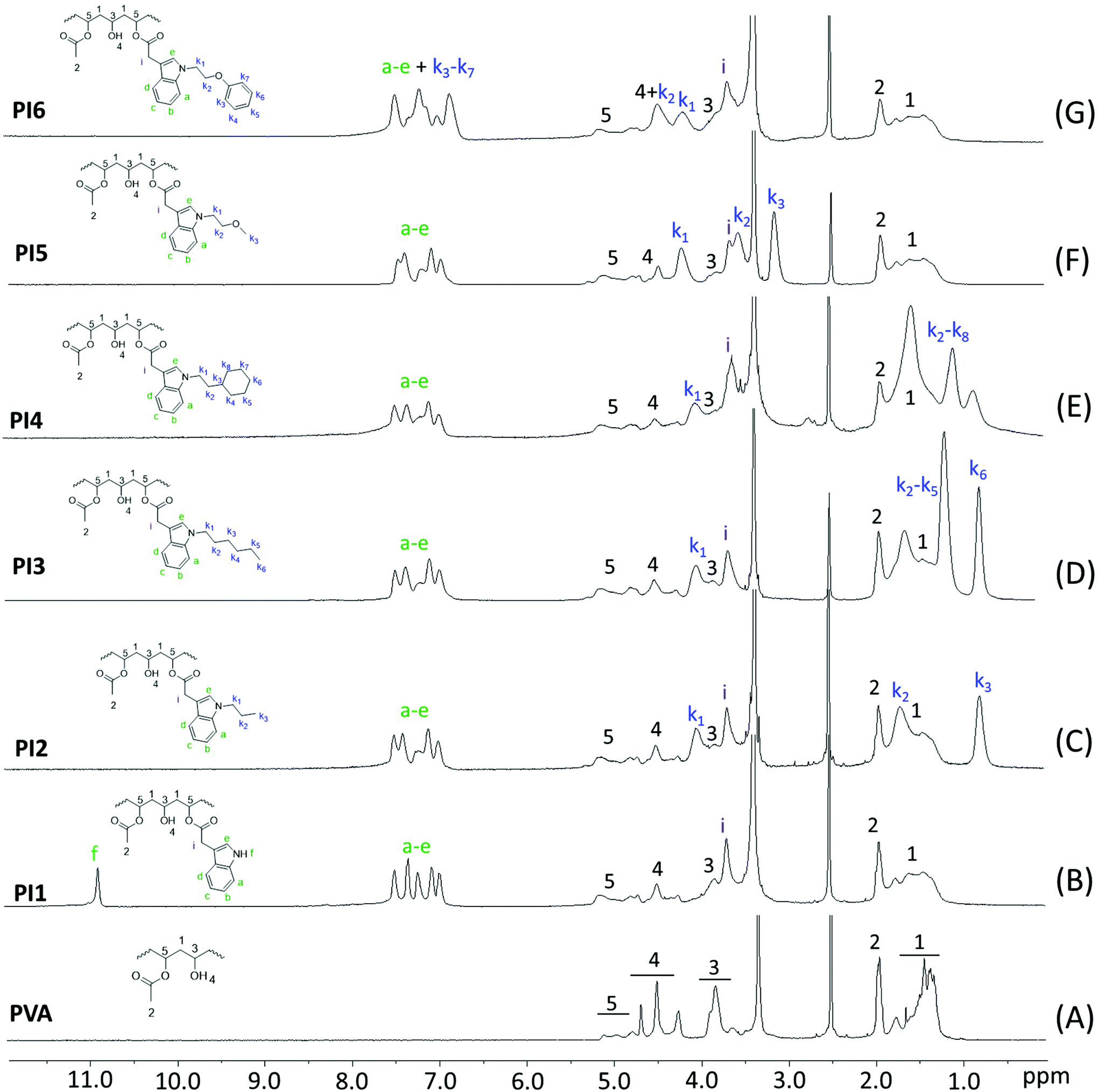 | ||
| Fig. 1 1H NMR spectra of (A) initial PVA and (B–G) indole-based PI1–6, respectively. | ||
The 1H NMR spectra of PI1–6 (Fig. 1B–G) showed significantly reduced intensities of the OH (4) and CHOH (3) signals and increased intensity of the CHOOR signal (5), which confirmed the conversion of OH groups. In addition, new broadened signals appeared in the 1H NMR spectra of PI1–6, including the indole aromatic signals (a–e) and various N-substituted signals (k1, k2, k3, etc.), which further confirmed the occurrence of grafting. These new signals were consistent with the peaks for their corresponding grafting agents 1–6 (Fig. S1, ESI†). In addition, the integrals of the OH, methyl, and aromatic signals were compared and used to calculate the pOH values, as discussed earlier. The molecular weight of these polymers (MNMR) was also calculated using the pOH values. For PI2–4, these values were quite consistent with the Mn values measured by GPC in chloroform. For PI5–6, the calculated MNMR values were lower than the GPC measured Mn values. The differences in the measured values were due to the inherent inaccuracy of the NMR calculations (e.g. signal broadening and overlapping) and GPC measurements (e.g. different behavior of the measured polymers and standard polymers in solution). PI1 was insoluble in chloroform so GPC was not measured.
The chemical structures of PI1–6 were further characterized by 13C NMR spectroscopy (Fig. S7, ESI†). First, all the signals for the grafting agents 1–6 were unambiguously assigned (Fig. S6, ESI†). The signal observed at ∼173.51 ppm (j) corresponded to the carbons of COOH groups. In the spectrum of PVA (Fig. S7A†), the signals for methyl, methylene, tertiary carbon and carbonyl carbons (signal 1, 2, 3 and 4, respectively) were observed at 21.26, 46.92–44.74, 68.79–63.58 and 170.34 ppm, respectively. After grafting, all the characteristic signals for the PVA structures remained discernible (peaks 1–4, Fig. S7B–G, ESI†). In the meantime, a new signal at ∼171.35 ppm (4′) appeared in all the 13C NMR spectra of PI1–6, which corresponded to the carbonyl carbons with indole units grafted. The COOH carbon signal (j) of the grafting agents 1–6 (Fig. S6, ESI†) was not observed in the spectra of the corresponding polymers (PI1 as an example shown in Fig. 2), indicating the complete removal of the grafting agents. All other characteristic signals corresponding to grafting agents 1–6 were observed in the 13C NMR spectra of all the polymers PI1–6 (i.e. aromatic signals a–h, methylene signal i, and aliphatic signals k1–8), which confirmed the successful grafting.
 | ||
| Fig. 2 Zoom-in 13C NMR spectra of (A) initial PVA, (B) resulting PI1 and (C) grafting agent 1 in the carbonyl range (165–176 ppm). | ||
Polymers PI1–6 were further characterized by FTIR spectroscopy (Fig. 3). Before grafting, a broad and strong band centered at 3330 cm−1 related to the O–H stretching vibrations with strong hydrogen bonding was observed in the FTIR spectrum of PVA. The intensity of this band was dramatically reduced after grafting, which confirmed the reaction of OH groups. However, the strong characteristic ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band (centered at ∼1732 cm−1) did not show any noticeable shift after grafting, which was due to the overlapping of the unchanged residual acetate groups of PVA. In the meantime, the C–H stretching band at 3000–2800 cm−1 was also observed for PI1–6, which was due to the overlapping of the C–H stretching of the unchanged PVA backbone and the OAc groups. In addition, a new band appeared at 740 cm−1 in the FTIR spectra of PI1–6, which could be attributed to the out-of-plane bending vibrations of aromatic C–H of the indole ring. Two other new bands at 1589 and 691 cm−1 in the spectra of PI6 corresponded to the vibrations of aromatic CC and C–H of benzene ring, respectively.
O stretching band (centered at ∼1732 cm−1) did not show any noticeable shift after grafting, which was due to the overlapping of the unchanged residual acetate groups of PVA. In the meantime, the C–H stretching band at 3000–2800 cm−1 was also observed for PI1–6, which was due to the overlapping of the C–H stretching of the unchanged PVA backbone and the OAc groups. In addition, a new band appeared at 740 cm−1 in the FTIR spectra of PI1–6, which could be attributed to the out-of-plane bending vibrations of aromatic C–H of the indole ring. Two other new bands at 1589 and 691 cm−1 in the spectra of PI6 corresponded to the vibrations of aromatic CC and C–H of benzene ring, respectively.
 | ||
| Fig. 3 FTIR spectra of initial PVA and indole-based PI1–6. | ||
Next, the thermal properties (e.g. thermal stability and glass transition temperature) were investigated, because these are related to their processing and manufacturing, which to some extent decide their potential application ranges as biomedical materials. The thermal behavior of the obtained PI1–6 was evaluated by DSC analyses. According to the second heating DSC curves (Fig. 4), PI1–6 were all amorphous without any melting endotherm. This was consistent with the results from WAXD measurements (Fig. S8, ESI†), which showed only amorphous halos for PI1–6 while three sharp diffraction peaks (2θ = 19.5, 22.7 and 40.7°, for crystalline planes 〈101〉, 〈200〉 and 〈111〉) for the initial PVA.78 Furthermore, DSC results also revealed the glass transition temperatures (Tg) of PI1–6 in the range of 38–93 °C, depending on the N-substituent on indole. PI1 with unsubstituted indole units showed the highest Tg value (93 °C), which was even higher than that of PVA (67 °C). This enhanced Tg was due to the increased bulkiness around the chains (compared to PVA) and the preserved ability to form hydrogen bonds (by N–H). PI2–6 showed lower Tg values than PVA, due to the loss of hydrogen bonding forming ability as well as the additional flexibility of alkyl and ether units,79,80 despite their increased bulkiness compared to PVA and PI1. Except for PI3 (Tg ∼ 38 °C), other polymers have Tg values ranging from 53–93 °C, which are similar to those of nylon 6/6 (50–60 °C) and poly(vinyl chloride) (65–85 °C). Such Tgs are favourable for plastics toward various commodity applications.
 | ||
| Fig. 4 DSC second heating curves of initial PVA and indole-grafted polymers (PI1–6). | ||
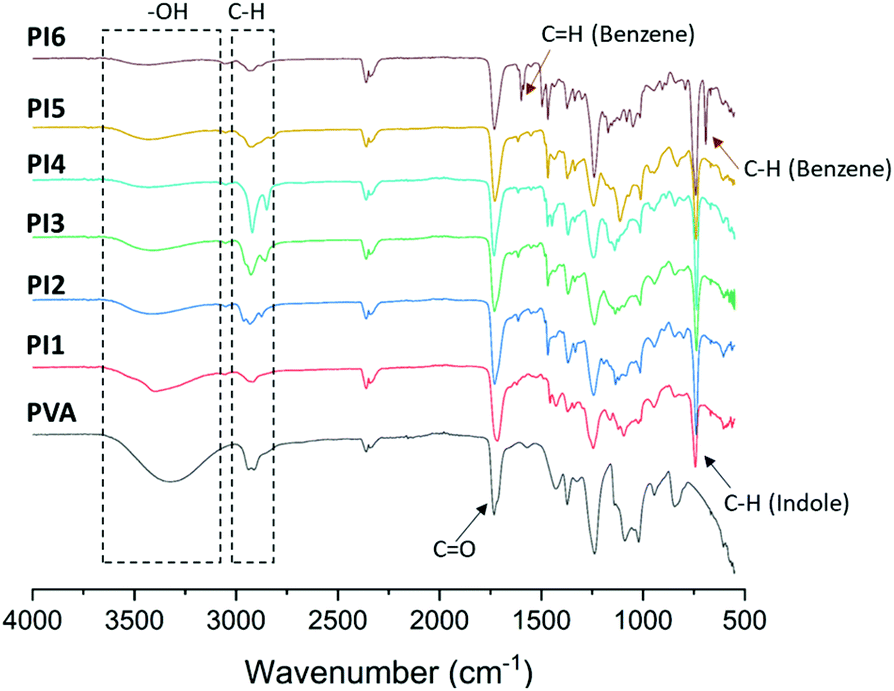
The thermal stability of PI1–6 was evaluated by TGA measurements (Fig. 5 and Table 1). It was observed that PVA presented two decomposition rate maxima (Fig. 5B). According to the literature, the first major decomposition rate maximum at ∼312 °C can be attributed to the side chain decomposition, and the second minor decomposition rate maximum at a higher temperature (∼428 °C) can be related to the main chain decomposition.81,82 For PI1–6, similar thermal decomposition processes with two rate maxima were observed, which was consistent with the presence of their PVA backbones. Compared to their precursor PVA, PI1–6 showed similar or slightly higher T95d (temperature for 5% mass loss) and Tmax values (temperature for the maximal rate of weight loss, Fig. 5B, Table 1), as well as higher residual char yields (CY) of PI1–6, which could be attributed to the presence of aromatic units.29,83
 | ||
| Fig. 5 TGA (A) weight loss and (B) derivative weight loss curves of initial PVA and indole-grafted PVAs (PI1–6). | ||
Antibacterial effects
Antibacterial activities of PI1–6 against six Gram-negative (G−) and three Gram-positive (G+) bacteria were conveniently evaluated according to a disk diffusion method.29 As presented in Fig. S9,†PI1–6 all showed larger inhibition zones than the negative control for all the tested bacteria. By analyzing the significance in differences (reflected by the p values), most tested polymers showed significantly larger zones of inhibition compared to the negative control (p values < 0.05, Table S2, ESI†). The only exception was PI4, for which the inhibition zones against two bacteria Sa and Sm did not show significant difference compared to the negative control (p values ≥ 0.05, Table S2, ESI†). This clearly indicates the broad antibacterial properties of indole-based PVAs, regardless of the N-substitution.Next, the antibacterial efficiency of PI1–6 was compared with that of a commonly used small molecular antibiotic, gentamicin (Fig. 6). As a result, PI1–6 showed generally comparable effects to gentamicin. For most tested bacteria, the zones of inhibition of PI1–6 and gentamicin did not show significant difference (p values ≥ 0.05, Table S3, ESI†). The exceptional cases are shown in Fig. 6 when there was a significant difference in the zones of inhibition of PI1–6 and gentamicin (ΔPIx–G, x = 1–6) reflected by p values < 0.05 (Table S3, ESI†). Furthermore, for the six Gram-negative bacteria all PI1–6 showed either a significantly stronger (ΔPIx–G > 0) or comparable (not shown) antibacterial effect to gentamicin. This indicated the effectiveness of these polymers particularly against Gram-negative bacteria. For the three tested Gram-positive bacteria, the comparison with gentamicin was more complex. For Sm, all polymers showed a comparable antibacterial effect to gentamicin. For Bt, PI3–5 showed a significantly lower (ΔPIx–G < 0) antibacterial effect than gentamicin, while that of the other polymers were comparable to gentamicin. For Sa, PI2 and PI5 showed a significantly better (ΔPIx–G > 0) antibacterial effect than gentamicin, PI1 showed a significantly lower (ΔPIx–G < 0) antibacterial effect than gentamicin, and the rest of the polymers were comparable to gentamicin. As such a preliminary conclusion is that the obtained polymers were in general comparable to gentamicin against Gram-positive bacteria, while they could be more effective against Gram-negative bacteria.
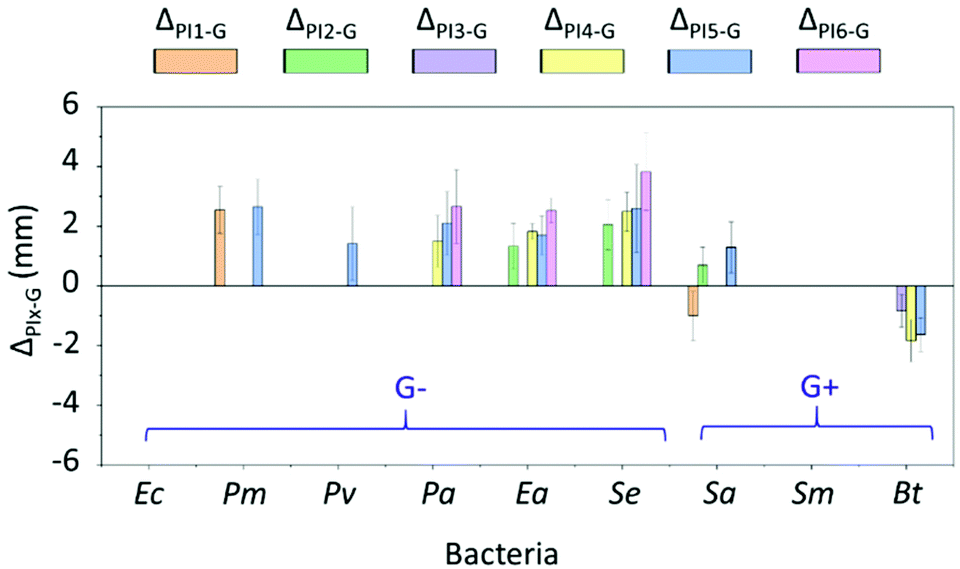 | ||
| Fig. 6 Difference between the inhibition zones of PI1–6 and gentamicin (ΔPIx–G, x = 1–6). Note, only those ΔPIx–G values with significant differences (p values < 0.05, Table S3, ESI†) are shown in the figure. The absence of the data column displayed in the corresponding place means that the inhibition zones of the polymer and gentamicin were comparable without significant difference (p values ≥ 0.05, Table S3, ESI†). For instance, for bacteria Ec and Sm there were no data plotted, which indicated that for these bacteria the effect of the obtained polymers was very similar (no significant difference) to that of gentamicin. | ||
Next, the impact of N-substitution on the antibacterial function of indole-grafted PVAs was investigated. The differences between the zones of inhibition of PI2–6 with various hydrophobic substituents and PI1 with N–H units are illustrated in Fig. 7. In most cases, the inhibition zones of PI2–6 were comparable with that of PI1 without significant differences (p values ≥ 0.05, Table S4, ESI†). This may suggest that neither the indole N–H nor the various hydrophobic substituents were essential in the interactions between the polymers and bacterial membranes. Only on several occasions were the substitution effects observed. For instance, N-substituted ether groups (i.e. in PI5–6) effectively enhanced the antibacterial function against two bacteria (Pv and Sa for PI5, and Se and Sa for PI6). However, N-substituted alkyl groups (i.e. in PI2–4) were less effective. Only N-substituted propyl groups (i.e. in PI2) enhanced the antibacterial function against one bacterium (Sa). The presence of larger linear or cyclic alkyl groups (PI3–4) showed no enhancement of antibacterial effects against most bacteria and even negative impact in the case of bacterium Pm. These observations suggested that ether groups may have a synergistic effect with indole units for the interactions with certain bacterial membranes.84
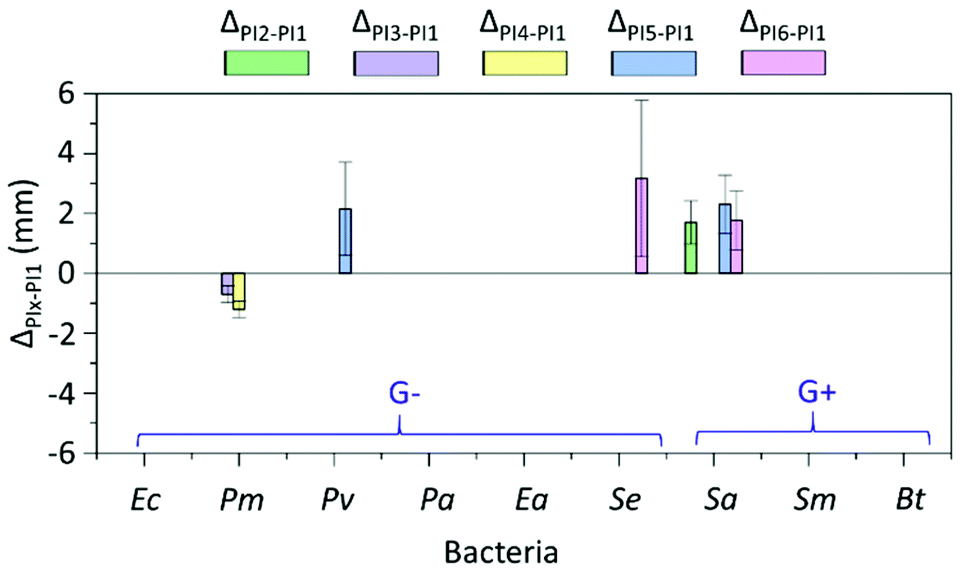 | ||
| Fig. 7 Investigation on the effect of N-substituents. Comparison of the inhibition zones between PI2–6 and PI1 (ΔPIx–PI1, x = 2–6). Note, only those ΔPIx–PI1 values with significant differences (p values < 0.05, Table S4, ESI†) were shown. No data displayed means that the inhibition zones between the hydrophobic substituted PI2–6 and H-substituted (i.e. nonsubstituted) PI1 were comparable without significant difference (p values ≥ 0.05, Table S4, ESI†). | ||
In addition, the possible impact of grafting density (conversion of the OH groups) on the antimicrobial effects for PI4 was investigated. Two PI4 samples with lower or higher grafting density were prepared by using 0.5 or 1.5 equivalents of grafting agent 4. The resulting polymers were characterized by 1H NMR spectroscopy (Fig. S11†), by which their conversion of the OH groups were calculated as 54 and 85%, respectively. Their corresponding molecular weights were also measured by SEC as ∼6 and 32 kDa, respectively. The antimicrobial effects of these two new PI4 samples, namely PI454 and PI485 were compared with that of the PI4 sample synthesized before with 72% OH conversion, namely PI472. As a result (Fig. 8), all the tested PI4 samples presented the antibacterial zone of inhibition against the two tested bacteria. In addition, the largest inhibition zones were observed when PI454 was used against both bacteria. This observed high efficiency of PI454 could be related to the relatively low molecular weight of the sample (thus high molecular mobility), as well as its relatively large number of unreacted OH groups (thus increased water solubility).
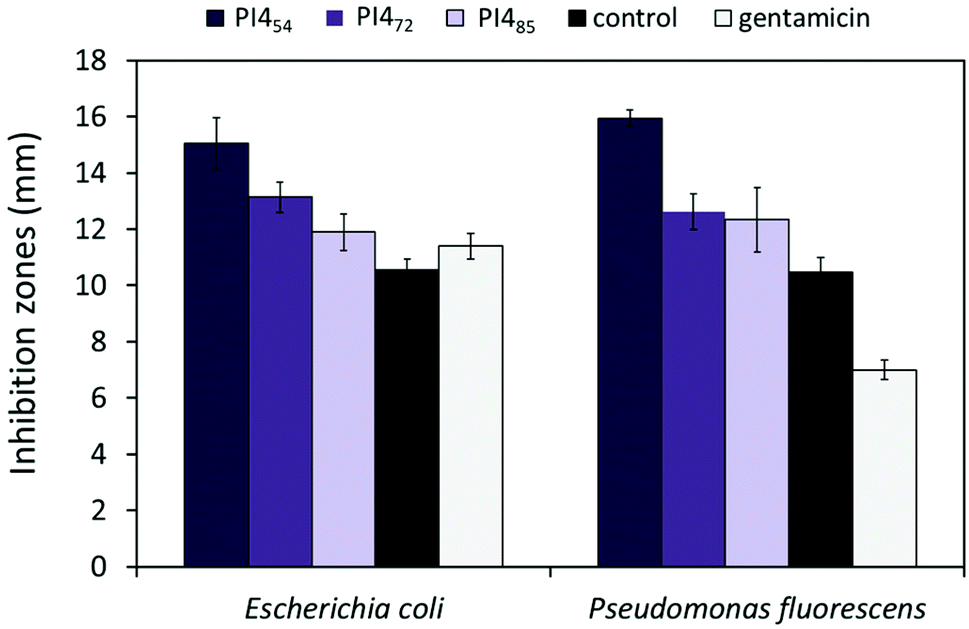 | ||
| Fig. 8 Inhibition zones of PI4 samples with different grafting density (conversion of OH groups). The subscript number after PI4 indicated the OH conversion of the sample. Pure DMF and gentamicin (10 μg per disk) were used as negative and positive controls, respectively. Bacteria: Escherichia coli ATCC 8739 and Pseudomonas fluorescens (PCL 1701). All experiments were performed in triplicate. The error bars stand for standard deviations. | ||
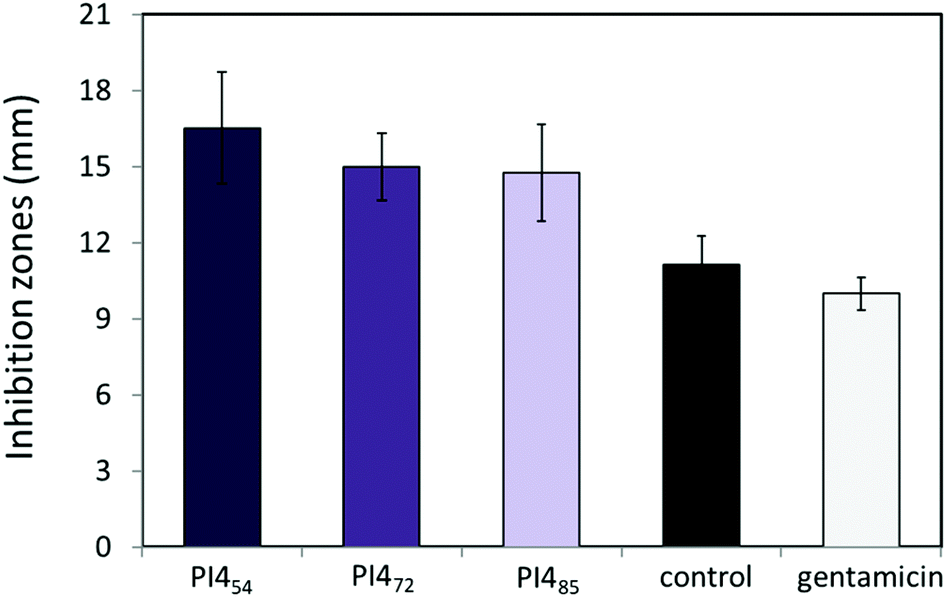
The antibacterial activities of PI454, PI472 and PI485 against two bacteria, Escherichia coli ATCC 8739 and Pseudomonas fluorescens (PCL 1701), were also evaluated by the standard broth dilution method to determine their minimum inhibitory concentration (MIC) values. After 24 h of incubation at 37 °C of the 96-well microtiter plates, turbidity was noticed in the wells containing ≤12.5 μg mL−1 of PI454, ≤12.5 μg mL−1 of PI472, and ≤25 μg mL−1 of PI485, indicating the growth of bacteria. However, with the concentrations of ≥25 μg mL−1 for PI454, ≥25 μg mL−1 for PI472, and ≥50 μg mL−1 for PI485, no turbidity was observed, indicating the inhibition of bacterial growth. These values were then estimated as close to the MIC values for these polymers (Fig. 9). According to this result, PI454 showed the most effective bacterial inhibition among the three PI4 samples, which was consistent with the disk diffusion results discussed earlier. It should be noted that the water solubility of these nonionic polymers was in general low, which may have an impact on their observed MIC values.
 | ||
| Fig. 9 Minimum inhibition concentrations (μg mL−1) of PI454, PI472 and PI485. The only medium was used as the negative control and only bacterial inoculums were used as a positive control. All experiments were performed in triplicate. Bacteria: Escherichia coli ATCC 8739 (dark grey bars) and Pseudomonas fluorescens PCL 1701 (light grey bars). The error bars stand for standard deviations. | ||
Finally, the anti-quorum sensing (anti-QS) effects of the three PI4 samples with different OH conversions were investigated by the zone of pigment inhibition that results in an opaque, halo zone of clearance (Fig. S14, ESI†). As shown in Fig. 10, all the three PI4 samples showed similar anti-QS effects, which were more significant compared to that of gentamicin. Furthermore, it was also noted that the PI4 sample with the lowest grafting density (i.e.PI454) showed a more significant anti-QS effect for the inhibition of the violacein pigment production of C. violaceum. This result preliminarily suggested that these polymers may be potentially interesting targets for further investigations toward materials that resist biofilm formation, which is also regulated by quorum sensing.85
 | ||
| Fig. 10 The anti-QS activity of the three PI4 samples with different OH conversions (PI454, PI472, and PI485) against C. violaceum CV026. Pure DMF and gentamicin (10 μg per disk) were used as negative and positive controls, respectively. All experiments were performed in triplicate. The error bars stand for standard deviations. | ||
Cytotoxicity
The cytotoxicity of PI1–6 to MG-63 osteoblast-like human cells was determined by a standard MTT assay. The results were presented as a relative percentage of the negative control (100% of cell viability). As seen in Fig. 11, less than 30% of reduction of cell viability was observed for all tested polymers during the evaluation period at three different concentrations (10, 100 and 1000 μg mL−1). This indicated that PI1–6 were non-cytotoxic according to the ISO 10993-5 standard.86 Furthermore, it was observed that the cell viability increased gradually with the increasing concentration after treatment with PI1. This suggested that PI1 could enhance cell growth and thus might be potentially suitable for various biomedical applications that require promoting cell proliferation (e.g. wound healing, tissue engineering and drug delivery),87–89 which was consistent with other reported polymers (e.g. chitosan). To address whether the increasing viability by PI1 could be related to its interactions with MTT solution, we have performed an additional experiment for PI1 under the same conditions without adding cells. As shown in Fig. S15 (ESI†), no significant absorbance at 600 nm was observed compared with background at three different concentrations, which demonstrated that PI1 had no significant interaction with the MTT working solution.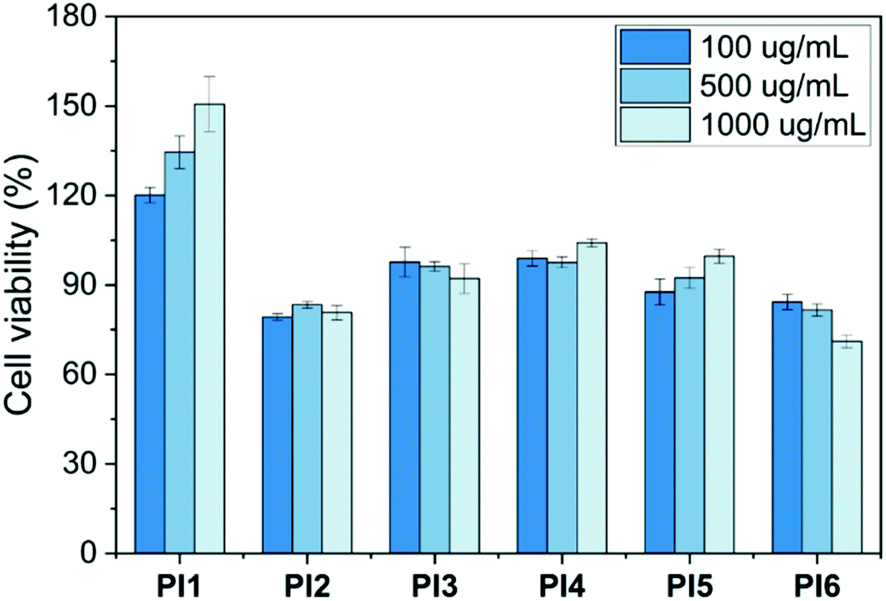 | ||
| Fig. 11 Cytotoxicity of PI1–6 at three concentrations (100, 500 and 1000 μg mL−1). Results were presented as relative percent viability of treated cells compared to that of untreated negative control (100% of cell viability, not shown in the graph). The error bars stand for standard deviations. | ||
Hemotoxicity
The hemolytic activity of the obtained polymers (PI1–6) was evaluated using the HaemoScan Biomaterial Haemolytic Assay (HaemoScan, Netherlands) on human erythrocytes.76 As shown in Fig. 12, the hemolysis rates of PI1, PI5 and PI6 were all negligible (<0.01%) after 24 h of contact with red blood cells (RBCs), indicating that these polymers have good hemocompatibility. For the other three polymers (PI2, PI3 and PI4), various hemolysis rates were observed (3.4%, 6.9% and 2.9%, respectively). This suggested that the substitutions with alky (either linear or cyclic) groups on indole NH could decrease the hemocompatibility of the resulting polymers to some extent, while the substitutions with ether groups have no significant impact on the hemocompatibility, which is consistent with other reported polymers with ether groups.90,91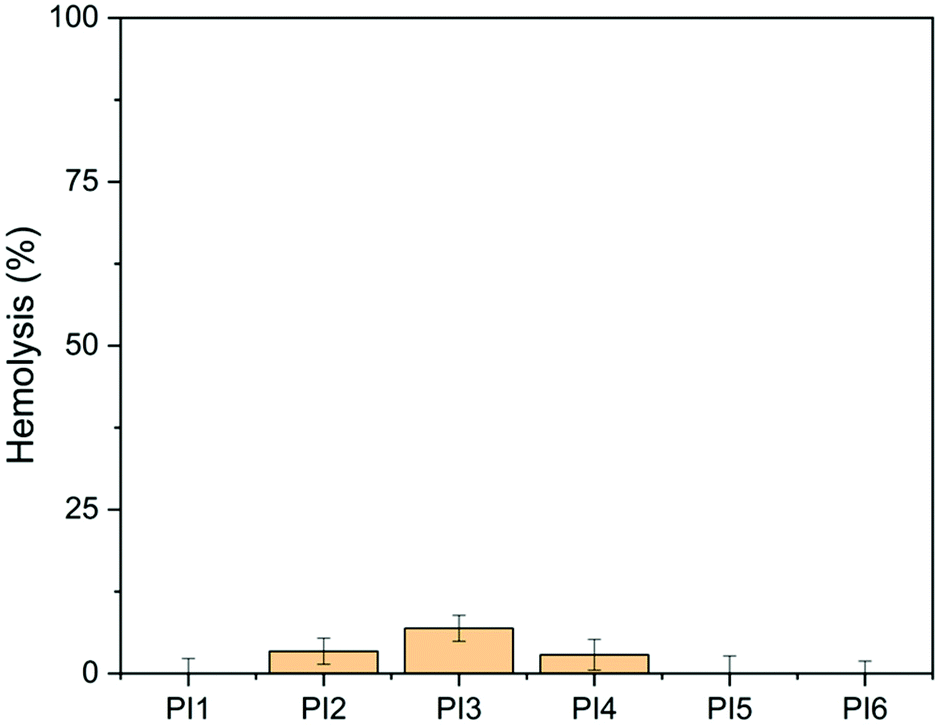 | ||
| Fig. 12 Hemolytic activity of PI1–6 (100 μg mL−1). DMSO (1% v/v) was used as the negative control (0% hemolysis). Lysis buffer was used as the positive control (100% hemolysis). | ||
Conclusions
A new series of nonionic indole-based biodegradable poly(vinyl alcohol)s with different N-substituents were synthesized, which showed tuneable glass transition temperatures (39–93 °C) and desirable thermal stability. According to the disk diffusion assay, all these polymers showed effective antibacterial function against 9 different human pathogens. In particular, these polymers were generally more effective against the 6 tested Gram-negative bacteria, which were comparable or even superior to the effect of gentamicin. For the 3 tested Gram-positive bacteria, the obtained polymers showed a comparable antibacterial effect to gentamicin, while in some cases significant difference was also noticed. Moreover, we discovered that the antimicrobial function of indole-based PVAs generally did not include significant contribution from the N–H unit or various hydrophobic groups. Only in a few cases the effect of N-substitution was observed. This may preliminarily suggest that the antibacterial function of the obtained indole-based PVAs is related to other types of interactions with bacterial membranes (e.g. aromatic interactions, or docking on specific membrane proteins) rather than hydrophobic or hydrogen bonding interactions. In addition, we have noticed that the molecular weight and OH conversion had an impact on the antimicrobial and anti-quorum sensing effects of the obtained indole-based PVAs, which suggested that further investigations on molecular design toward optimized molecular sizes and the hydrophobic/hydrophilic balance could provide more valuable information for the development of such a new class of polymer materials. Finally, these obtained polymers showed excellent biocompatibility, particularly for those with ether substituents, which indicated their potential toward various biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Mistra Foundation (the “STEPS” project, No. 2016/1489), Swedish Research Council for Sustainable Development (Formas, No. 2021-01107), Carl-Trygger Foundation (No. 18:435), Guangzhou Elite Education Program, and Royal Physiographic Society in Lund. We thank María Del Carmen Casado Muñoz at KTH and Laura Quinn at University College Dublin for the help in supplying the strains Escherichia coli ATCC 8739 and Pseudomonas fluorescens (PCL 1701), respectively.Notes and references
- R. Kurapati, M. Vaidyanathan and A. M. Raichur, RSC Adv., 2016, 6, 39852–39860 RSC.
- A. Nagaraja, Y. M. Puttaiahgowda, A. Kulal, A. M. Parambil and T. Varadavenkatesan, Macromol. Res., 2019, 27, 301–309 CrossRef CAS.
- C. Z. Chen and S. L. Cooper, Adv. Mater., 2000, 12, 843–846 CrossRef CAS.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y. Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- A. L. Hook, C. Y. Chang, J. Yang, J. Luckett, A. Cockayne, S. Atkinson, Y. Mei, R. Bayston, D. J. Irvine, R. Langer, D. G. Anderson, P. Williams, M. C. Davies and M. R. Alexander, Nat. Biotechnol., 2012, 30, 868–875 CrossRef CAS PubMed.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 1–14 CrossRef CAS PubMed.
- T. Zhu, Y. Sha, J. Yan, P. Pageni, M. A. Rahman, Y. Yan and C. Tang, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- M. A. Rahman, M. Bam, E. Luat, M. S. Jui, M. S. Ganewatta, T. Shokfai, M. Nagarkatti, A. W. Decho and C. Tang, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- A. Kuroki, A. Kengmo Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock, M. Unnikrishnan and S. Perrier, Biomaterials, 2019, 217, 119249 CrossRef CAS PubMed.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- M. R. E. Santos, A. C. Fonseca, P. V. Mendonça, R. Branco, A. C. Serra, P. V. Morais and J. F. J. Coelho, Materials, 2016, 9, 599–632 CrossRef PubMed.
- Z. Zhu, G. Jeong, S. J. Kim, I. Gadwal, Y. Choe, J. Bang, M. K. Oh, A. Khan and J. Rao, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2391–2396 CrossRef CAS.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. DeGrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46–71 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- D. Raafat and H. Sahl, Microb. Biotechnol., 2009, 2, 186–201 CrossRef CAS PubMed.
- G. Li and J. Shen, J. Appl. Polym. Sci., 2000, 676–684 CrossRef CAS.
- F. Ferrero, M. Periolatto and S. Ferrario, J. Cleaner Prod., 2015, 96, 244–252 CrossRef CAS.
- V. W. L. Ng, J. P. K. Tan, J. Leong, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef CAS.
- N. D. Koromilas, G. C. Lainioti, G. Vasilopoulos, A. Vantarakis and J. K. Kallitsis, Polym. Chem., 2016, 7, 3562–3575 RSC.
- Y. Jiao, L. Niu, S. Ma, J. Li, F. Tay and J. Chen, Prog. Polym. Sci., 2017, 7, 53–90 CrossRef PubMed.
- E.-R. Kenawy and Y. R. Abdel-Fattah, Macromol. Biosci., 2002, 2, 261–266 CrossRef CAS.
- R. Costa, J. L. Pereira, J. Gomes, F. Gonçalves, D. Hunkeler and M. G. Rasteiro, Chemosphere, 2014, 112, 177–184 CrossRef CAS PubMed.
- K. Lewandowska, J. Solution Chem., 2013, 42, 1654–1662 CrossRef CAS PubMed.
- K. Izutsu and K. Shigeo, Phys. Chem. Chem. Phys., 2000, 2, 123–127 RSC.
- Y. T. Hung, L. A. McLandsborough, J. M. Goddard and L. J. Bastarrachea, LWT–Food Sci. Technol., 2018, 97, 546–554 CrossRef CAS.
- L. J. Bastarrachea and J. M. Goddard, Appl. Surf. Sci., 2016, 378, 479–488 CrossRef CAS.
- L. J. Bastarrachea, A. Denis-Rohr and J. M. Goddard, Annu. Rev. Food Sci. Technol., 2015, 6, 97–118 CrossRef CAS PubMed.
- C. R. Arza, S. Ilk, D. Demircan and B. Zhang, Green Chem., 2018, 20, 1238–1249 RSC.
- X. Li, S. Ilk, J. A. Linares-Pasten, Y. Liu, D. B. Raina, D. Demircan and B. Zhang, Biomacromolecules, 2021, 22, 2256–2271 CrossRef CAS PubMed.
- M. G. Ciulla and K. Kumar, Tetrahedron Lett., 2018, 59, 3223–3233 CrossRef CAS.
- H. Guo, Eur. J. Med. Chem., 2019, 164, 678–688 CrossRef CAS PubMed.
- M. Raccach, J. Food Saf., 1984, 6, 141–170 CrossRef CAS.
- S. Y. Teow, K. Liew, S. A. Ali, A. S. B. Khoo and S. C. Peh, J. Trop. Med., 2016, 2016, 1–4 CrossRef PubMed.
- K. Shanmugapriya, H. Kim, P. S. Saravana, B. S. Chun and H. W. Kang, Colloids Surf., B, 2018, 172, 170–179 CrossRef CAS PubMed.
- K. Nakano, T. Chigira, T. Miyafusa, S. Nagatoishi, J. M. M. Caaveiro and K. Tsumoto, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- S. A. El-Mowafy, K. H. Abd El Galil, S. M. El-Messery and M. I. Shaaban, Microb. Pathog., 2014, 74, 25–32 CrossRef CAS PubMed.
- A. C. Justino de Araújo, P. R. Freitas, C. Rodrigues dos Santos Barbosa, D. F. Muniz, J. E. Rocha, A. C. Albuquerque da Silva, C. Datiane de Morais Oliveira-Tintino, J. Ribeiro-Filho, L. Everson da Silva, C. Confortin, W. do Amaral, C. Deschamps, J. M. Barbosa-Filho, N. T. Ramos de Lima, S. R. Tintino and H. D. Melo Coutinho, Food Chem. Toxicol., 2020, 136, 111023 CrossRef PubMed.
- L. Martin, P. Gurnani, J. Zhang, M. Hartlieb, N. R. Cameron, A. M. Eissa and S. Perrier, Biomacromolecules, 2019, 20, 1297–1307 CrossRef CAS PubMed.
- Q. Ma, Y. Qu, X. Zhang, Z. Liu, H. Li, Z. Zhang, J. Wang, W. Shen and J. Zhou, Sci. Rep., 2016, 5, 17674 CrossRef PubMed.
- Q. Ma, X. Zhang and Y. Qu, Front. Microbiol., 2018, 9, 2625 CrossRef PubMed.
- R. Wang, P. Vega, Y. Xu, C. Y. Chen and J. Irudayaraj, J. Biomed. Mater. Res., Part A, 2018, 106, 1979–1986 CrossRef CAS PubMed.
- J. Kim and W. Park, Microbiology, 2013, 159, 2616–2625 CrossRef CAS PubMed.
- S. Karpagam and S. Guhanathan, Prog. Org. Coat., 2014, 77, 1901–1910 CrossRef CAS.
- M. Boopathy, R. Selvam, S. JohnSanthoshkumar and K. Subramanian, Polym. Adv. Technol., 2017, 28, 717–727 CrossRef CAS.
- R. J. Cornell and L. G. Donaruma, J. Med. Chem., 1965, 8, 388–390 CrossRef CAS PubMed.
- L. Erdmann and K. E. Uhrich, Biomaterials, 2000, 21, 1941–1946 CrossRef CAS PubMed.
- R. Jabara, N. Chronds and K. Robinson, Catheter. Cardiovasc. Interv., 2008, 72, 186–194 CrossRef PubMed.
- N. Shpaisman, L. Sheihet, J. Bushman, J. Winters and J. Kohn, Biomacromolecules, 2012, 13, 2279–2286 CrossRef CAS PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- S. Weintraub, T. Shpigel, L. G. Harris, R. Schuster, E. C. Lewis and D. Y. Lewitus, Polym. Chem., 2017, 8, 4182–4189 RSC.
- M. B. Patel, S. A. Patel, A. Ray and R. M. Patel, J. Appl. Polym. Sci., 2002, 89, 895–900 CrossRef.
- D. Demircan and B. Zhang, Carbohydr. Polym., 2017, 157, 1913–1921 CrossRef CAS PubMed.
- T. Nonaka, Y. Uemura, K. Ohse, K. Jyono and S. Kurihara, J. Appl. Polym. Sci., 1997, 66, 1621–1630 CrossRef CAS.
- L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler-Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. Degrado and E. Gazit, Nat. Commun., 2017, 8, 1–10 CrossRef CAS PubMed.
- B. Zhang, Q. Yu, G. Yan, H. Zhu, X. Y. Xu and L. Zhu, Sci. Rep., 2018, 8, 1–11 Search PubMed.
- J. Zhang, Y. Chen, Y. Liao, Q. Wang and J. Yu, Chemosphere, 2022, 286, 131551 CrossRef CAS PubMed.
- C. R. Arza, P. Wang, J. Linares-Pastén and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2314–2323 CrossRef CAS.
- P. Wang, J. A. Linares-Pastén and B. Zhang, Biomacromolecules, 2020, 21, 1078–1090 CrossRef CAS PubMed.
- P. Wang, C. R. Arza and B. Zhang, Polym. Chem., 2018, 9, 4706–4710 RSC.
- P. Wang and B. Zhang, RSC Adv., 2021, 11, 16480–16489 RSC.
- P. Singh, P. Verma, B. Yadav and S. S. Komath, Bioorg. Med. Chem. Lett., 2011, 21, 3367–3372 CrossRef CAS PubMed.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed.
- S. Sathiyaraj, A. Shanavas, K. A. Kumar, A. Sathiyaseelan, J. Senthilselvan, P. T. Kalaichelvan and A. S. Nasar, Eur. Polym. J., 2017, 95, 216–231 CrossRef CAS.
- M. Q. Du, Y. Z. Peng, Y. C. Ma, L. Yang, Y. L. Zhou, F. K. Zeng, X. K. Wang, M. L. Song and G. J. Chang, Chin. J. Polym. Sci., 2020, 38, 187–194 CrossRef CAS.
- C. Ni, K. Feng, X. Li, H. Zhao and L. Yu, Prog. Org. Coat., 2020, 148, 105824 CrossRef CAS.
- M. A. Hassan, A. M. Omer, E. Abbas, W. M. A. Baset and T. M. Tamer, Sci. Rep., 2018, 8, 1–14 CAS.
- K. E. S. Locock, T. D. Michl, N. Stevens, J. D. Hayball, K. Vasilev, A. Postma, H. J. Griesser, L. Meagher and M. Haeussler, ACS Macro Lett., 2014, 3, 319–323 CrossRef CAS.
- A. Srivastava, P. Singh, R. Kumar, S. K. Verma and R. N. Kharwar, Polym. Int., 2013, 62, 210–218 CrossRef CAS.
- R. Namivandi-Zangeneh, R. J. Kwan, T. K. Nguyen, J. Yeow, F. L. Byrne, S. H. Oehlers, E. H. H. Wong and C. Boyer, Polym. Chem., 2018, 9, 1735–1744 RSC.
- V. Sambhy, B. R. Peterson and A. Sen, Angew. Chem., 2008, 120, 1270–1274 CrossRef.
- F. Wang, H. Zhou, O. P. Olademehin, S. J. Kim and P. Tao, ACS Omega, 2018, 3, 37–45 CrossRef CAS PubMed.
- S. Ilk, N. Sağlam, M. Özgen and F. Korkusuz, Int. J. Biol. Macromol., 2017, 94, 653–662 CrossRef CAS PubMed.
- D. Demircan, S. Ilk and B. Zhang, Biomacromolecules, 2017, 18, 3439–3446 CrossRef CAS PubMed.
- CLSI, Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Second Informational Supplement, 2022 Search PubMed.
- K. V. Nemani, K. L. Moodie, J. B. Brennick, A. Su and B. Gimi, Mater. Sci. Eng., C, 2013, 33, 4453–4459 CrossRef CAS PubMed.
- T. Congdon, P. Shaw and M. I. Gibson, Polym. Chem., 2015, 6, 4749–4757 RSC.
- L. Jiang, T. Yang, L. Peng and Y. Dan, RSC Adv., 2015, 5, 86598–86605 RSC.
- P. I. Tarraco, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 925–934 Search PubMed.
- C. Gaina, O. Ursache, V. Gaina and D. Ionita, Polym.-Plast. Technol. Eng., 2012, 51, 65–70 CrossRef CAS.
- H. Awada and C. Daneault, Appl. Sci., 2015, 5, 840–850 CrossRef CAS.
- L. T. Sin, W. A. W. A. Rahman, A. R. Rahmat and M. Mokhtar, Carbohydr. Polym., 2011, 83, 303–305 CrossRef CAS.
- X. L. Wang, T. Fu, D. M. Guo, J. N. Wu, X. L. Wang, L. Chen and Y. Z. Wang, Polym. Chem., 2016, 7, 1584–1592 RSC.
- K. Fukushima, K. Kishi, K. Saito, K. Takakuwa, S. Hakozaki and S. Yano, Biomater. Sci., 2019, 7, 2288–2296 RSC.
- V. K. Singh, A. Mishra and B. Jha, Front. Cell. Infect. Microbiol., 2017, 7, 1–16 CAS.
- I. Standard 11266, 61010-1 © Iec2001, 2014, 2014, 13.
- W. Y. Chuang, T. H. Young, C. H. Yao and W. Y. Chiu, Biomaterials, 1999, 20, 1479–1487 CrossRef CAS PubMed.
- B. Duan, X. Yuan, Y. Zhu, Y. Zhang, X. Li, Y. Zhang and K. Yao, Eur. Polym. J., 2006, 42, 2013–2022 CrossRef CAS.
- D. Aggarwal and H. W. T. Matthew, Acta Biomater., 2009, 5, 1575–1581 CrossRef CAS PubMed.
- K. Fukushima, Y. Inoue, Y. Haga, T. Ota, K. Honda, C. Sato and M. Tanaka, Biomacromolecules, 2017, 18, 3834–3843 CrossRef CAS PubMed.
- X. Li, X. Wang, S. Subramaniyan, Y. Liu, J. Rao and B. Zhang, Biomacromolecules, 2022, 23, 150–162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis scheme of grafting agents 2–6; analytical data of grafting agents 1–6 (1H NMR and 13C NMR spectra); zoomed-in 1H NMR spectra of PVA; calculations of OH conversion of PI1–6; analytical data of PI1–6 (solubility data, 13C NMR, GPC and UV-vis spectra); WAXD spectra of PVA and PI1–6; inhibition zones of PI1–6; p values of the antimicrobial results; analytical data of PI454, PI472 and PI485 (1H NMR spectra and GPC spectra); plates of disk diffusion tests and anti-QS tests of PI454, PI472 and PI485; UV absorbance at 600 nm of PI1 in MTT assay. See DOI: https://doi.org/10.1039/d1py01504d |
| This journal is © The Royal Society of Chemistry 2022 |