
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlended vinylogous urethane/urea vitrimers derived from aromatic alcohols†
Philipp
Haida
a,
Gloria
Signorato
a and
Volker
Abetz
 *ab
*ab
aInstitute of Physical Chemistry, Universität Hamburg, Grindelallee 117, 20146 Hamburg, Germany
bInstitute of Membrane Research, Helmholtz-Zentrum Hereon, Max-Planck-Straße 1, 21502 Geesthacht, Germany. E-mail: volker.abetz@hereon.de
First published on 1st January 2022
Abstract
Vitrimers belong to the class of covalent adaptable networks and are cross-linked polymers, which undergo dynamic, associative exchange reactions under thermal treatment, making these networks permanent as well as dynamic. In this work a feasible synthesis route for the acetoacetylation of aromatic alcohols, which expands the selection of acetoacetate monomers for the synthesis of vitrimers, is introduced. Bisphenol-A, resorcin, 2,7-naphthalenediol and 1,1,1-tris(4-hydroxyphenyl)ethane are chosen as examples for commercially relevant di- and trifunctional alcohols used for countless applications, e.g. epoxy resins, phenolic resins and polyester-networks, which are in general not reprocessable. In contrast, aromatic alcohols provide the basis for the prepared vitrimers, representing a reprocessable alternative to the established materials. Model studies are conducted to enlighten the undergoing condensation, substitution and transamination reactions in the emerging vinylogous urethane/urea mixtures, investigating solvent and catalyst effects and determining activation energies. Utilizing the findings from the model studies for the preparation of vitrimers, 16 elastomeric and thermosetting blended poly(vinylogous urethane/urea) networks are prepared by bulk/solvent polymerization, showing short stress-relaxation times of up to 0.7 s at 130 °C and activation energies of ca. 45–150 kJ mol−1 with a broad range of material properties. Moreover, the materials show remarkable reprocessing, reshaping, shape-memory and self-healing properties.
Introduction
The outstanding properties of thermosets with respect to their mechanical stability and chemical resistance make this class of polymers attractive for various applications. However, thermosets cannot be reprocessed or recycled due to their permanent cross-linked networks.1–3 For this reason, a great deal of focus has been placed on the field of chemically and physically robust, but still reprocessable polymer networks over the last few decades. The use of reversible covalent bonds within polymer networks enables a dynamic reorganization of cross-linking points and leads to a macroscopic flow behavior of the material without a permanent change in the physical or chemical properties. Polymers with covalently exchangeable bonds are assigned to the class of covalent adaptable networks (CANs) and classified according to the underlying associative, dissociative, or concerted exchange mechanism.4–10 Considering CANs based on an associative exchange mechanism, the number of cross-linking points always remains constant during the rearrangement process, providing permanent and dynamic polymer networks at the same time.11–13In 2011, Leibler and co-workers established a novel class of CANs by adding a suitable transesterification catalyst to polyester-based networks. The thermally activated, catalytic transesterification reaction based on an associative exchange mechanism resulted in permanent polyester/polyol networks that showed an astonishing viscosity decrease upon heating, while maintaining a cross-linked polymer network. Hence, Leibler invented the name “vitrimers” for this novel subcategory of thermosetting polymers.14–16 Since this initial finding, various dynamic covalent exchange mechanisms, e.g. transcarbonation,17 transalkylation,18 transcarbamoylation,19,20 thiol–disulfide exchange,21 trans-N-alkylation,22 transamination23–25 and transimination reactions26 have been investigated and applied with innumerable molecules, oligomers or modified polymers.11,22,27–31 In 2014, Du Prez, Leibler and co-workers examined vinylogous amide-type enamine-one species as a suitable covalent dynamic bond for vitrimers, which are synthesized by the condensation reaction of primary amines and acetoacetates.24 This bond can be regarded as a vinylogous urethane, stabilized by the vinylic bond and the conjugation through the ester unit. In contrast to conventional urethane bonds, the electrophilicity of vinylogous urethane bonds is highly increased, thus enabling thermally induced, dynamic exchange reactions with primary amines.32,33
In this work, a synthetic route for the acetoacetylation of aromatic alcohols as suitable raw materials for vinylogous urethane vitrimers was investigated using 2,2,6-trimethyl-1,3-dioxin-4-one (TMDO) as a reactant. In particular, the acetoacetylation of aromatic alcohols and the use of the prepared aromatic acetoacetates have not been described in the literature for vitrimers which increases the number of possible reactants multiple times. For example, phenol, bisphenol-A, resorcin, 2,7-naphthalenediol and 1,1,1-tris(4-hydroxyphenyl)ethane were chosen as commercially accessible and broadly established mono-, di-, and trifunctional reactants. Nowadays, these aromatic alcohols are used for countless applications, e.g. epoxy resins,34–36 phenolic resins (resorcinol),37–40 polyesters,41–46 dyes (fluorescein and eosin),47,48 medical diagnostics49 and other usages,50 which make them promising candidates for the use of recyclable and reprocessable polymer networks. Moreover, we introduce 2,7-naphthalenediol and 1,1,1-tris(4-hydroxyphenyl)ethane as less toxic alternatives to the widely established toxic and carcinogenic bisphenol-A and resorcin-based polymers, which are increasingly being avoided and replaced by industry.51–56
The condensation and substitution reactions of the prepared aromatic acetoacetates with aliphatic primary amines lead to the formation of blended vinylogous urethane and vinylogous urea compounds, which both undergo thermally induced exchange reactions, thus representing the required dynamic covalent bonds for vitrimers. Model studies are conducted to enlighten the undergoing condensation, substitution and exchange reactions in the emerging vinylogous urethane/urea mixtures, investigate the solvent or catalyst effects and compare the activation energies. Utilizing the findings from the model studies for the preparation of vitrimers, 16 elastomeric and thermosetting blended poly(vinylogous urethane/urea) networks have been prepared and characterized in terms of their material properties and remarkable reshaping, reprocessing, shape-memory and self-healing properties.
Results and discussion
Synthesis of aromatic acetoacetates
According to the literature, the established synthetic route for the acetoacetylation of various aliphatic alcohols was carried out with tert-butylacetoacetate (t-buacac).24,25,57 However, another synthesis route using TMDO is described for monofunctional aliphatic and aromatic alcohols (basic organic chemistry) in the literature and turned out to proceed in a much more efficient way for aromatic alcohols, thus enabling aromatic acetoacetates to be used as suitable monomers for this type of vitrimer.58–61 In both synthetic routes the thermal dissociation of the reactant into a highly reactive, electrophilic acetylketene is the crucial step for acetoacetylation with the corresponding nucleophile (Fig. S1, ESI†). TMDO undergoes a retro-Diels Alder reaction at elevated temperatures (>110 °C), thus releasing the reactive acetylketene and acetone, with acetone being removed by distillation. In comparison, the established acetoacetylation with t-buacac proceeds by dissociation into the same reactive acetylketene and tert-butanol. Due to the delocalized electrons, aromatic alcohols show significantly lower nucleophilicity in comparison to aliphatic alcohols. Experiments showed that the acetoacetylation of aromatic alcohols with t-buacac proceeds slowly with low conversions and emerging byproducts after a long time of stirring at high temperatures, while the synthesis could not be significantly improved by changing the solvent, temperature, reaction time, and reactant concentration, or even using a catalyst. Nevertheless, the desired di- and trifunctional acetoacetate products may be isolated by complicated and time-consuming purification steps. In contrast, the use of TMDO instead of t-buacac enables a fast and selective acetoacetylation at 135 °C in xylene after 45 min. Obviously, in the dissociation step, the backward reaction of the byproducts or the removal of acetone proceeds more efficiently. The synthetic route using TMDO works for aliphatic alcohols as well but does not offer any major advantages compared to the established synthesis route using t-buacac. Since the reaction times are significantly increased for aromatic alcohols, the acetoacetylation using TMDO represents an effective synthesis route, which allows easy access to aromatic acetoacetates (Fig. S1, ESI†).Herein, the aromatic alcohols phenol, bisphenol-A, resorcin, 2,7-naphthalenediol and 1,1,1-tris(4-hydroxyphenyl)ethane were used as raw materials for the mono-, di-, and trifunctional monomers phenyl-3-oxobutanoate (PH), 1,3-phenylene-bis(3-oxobutanoate) (RE), naphthalene-2,7-diyl-bis(3-oxobutanoate) (NDO), propane-2,2-diylbis(4,1-phenylene)-bis(3-oxobutanoate) (BPA) and ethane-1,1,1-triyltris(benzene-4,1-diyl)-tris(3-oxobutanoate) (THPE) (Fig. 1). The synthesized acetoacetate monomers were characterized by Attenuated-total-reflection-Fourier-transform infrared (ATR-FT-IR) spectroscopy (Fig. S2, ESI†), 1H/13C nuclear magnetic resonance (NMR) spectroscopy (Fig. S3, ESI†) and mass spectrometry (MS) (Fig. S4, ESI†).
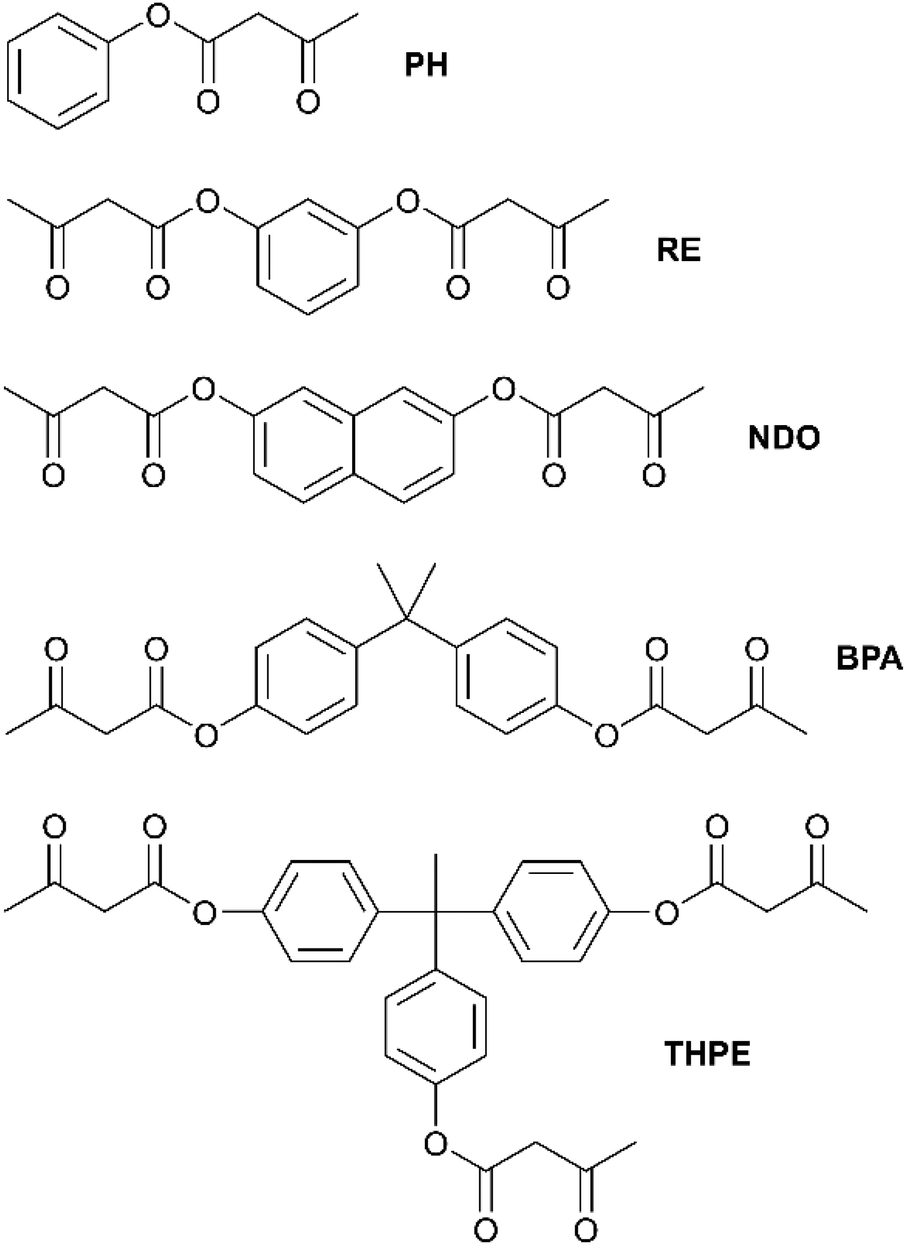 | ||
| Fig. 1 Representation of the synthesized acetoacetate monomers phenyl-3-oxobutanoate (PH), 1,3-phenylene bis(3-oxobutanoate) (RE), naphthalene-2,7-diyl bis(3-oxobutanoate) (NDO), propane-2,2-diylbis(4,1-phenylene) bis(3-oxobutanoate) (BPA) and ethane-1,1,1-triyltris(benzene-4,1-diyl) tris(3-oxobutanoate) (THPE). | ||
Model studies – condensation, substitution and exchange reactions
In order to investigate the characteristic condensation, substitution and exchange reactions, model studies with monofunctional reactants have been carried out. In the following text and illustrations, the vinylogous urethane (VUT) and vinylogous urea (VUA) compounds are abbreviated with the respective substituent on each side of the dynamic covalent bonds (Phe = phenyl, Hex = hexyl, Bz = benzyl, and AcAc = acetoacetate, e.g.: Phe-VUT-Hex). Aliphatic acetoacetates undergo a selective condensation reaction with primary amines to give a vinylogous urethane.24,62 However, aromatic acetoacetates show a different reaction behavior due to their variant electron configuration. For the model study, phenylacetoacetate (PH) was mixed with an excess of hexylamine (2 equivalents) in chloroform at room temperature to give the vinylogous urethane phenyl-3-(hexylamino)but-2-enoate (Phe-VUT-Hex) (Fig. 2). Characterization by 1H/13C NMR spectroscopy, ATR-FT-IR spectroscopy and MS showed the formation of the vinylogous urethane with the characteristic chemical shifts (Fig. S5, ESI†), the typical conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester band (1663 cm−1) and CC band (1606 cm−1) (Fig. S6, ESI†) and the molecular ion peak m/z = 262.181 (M+ + 1) (Fig. S7, ESI†). In addition, the spectra showed the temporary formation of the acetoacetamide Hex-VUA-AcAc, since the ester group of the aromatic acetoacetates is sufficiently electrophilic to undergo a substitution reaction with the nucleophilic hexylamine. Subsequently, the acetoacetamide undergoes a fast condensation reaction with another hexylamine, giving the vinylogous urea compound Hex-VUA-Hex together with Phe-VUT-Hex (1H NMR, MS, and ATR-FT-IR, Fig. S8–S10, ESI†). This is proven by individual reference measurements of the pure vinylogous urea compound Hex-VUA-Hex (1H/13C NMR, MS, and ATR-FT-IR, Fig. S11–14, ESI†). In this way two dynamic covalent bonds were synthesized simultaneously with an approximate proportion of 37% vinylogous urethane and 63% vinylogous urea products, respectively.
O ester band (1663 cm−1) and CC band (1606 cm−1) (Fig. S6, ESI†) and the molecular ion peak m/z = 262.181 (M+ + 1) (Fig. S7, ESI†). In addition, the spectra showed the temporary formation of the acetoacetamide Hex-VUA-AcAc, since the ester group of the aromatic acetoacetates is sufficiently electrophilic to undergo a substitution reaction with the nucleophilic hexylamine. Subsequently, the acetoacetamide undergoes a fast condensation reaction with another hexylamine, giving the vinylogous urea compound Hex-VUA-Hex together with Phe-VUT-Hex (1H NMR, MS, and ATR-FT-IR, Fig. S8–S10, ESI†). This is proven by individual reference measurements of the pure vinylogous urea compound Hex-VUA-Hex (1H/13C NMR, MS, and ATR-FT-IR, Fig. S11–14, ESI†). In this way two dynamic covalent bonds were synthesized simultaneously with an approximate proportion of 37% vinylogous urethane and 63% vinylogous urea products, respectively.
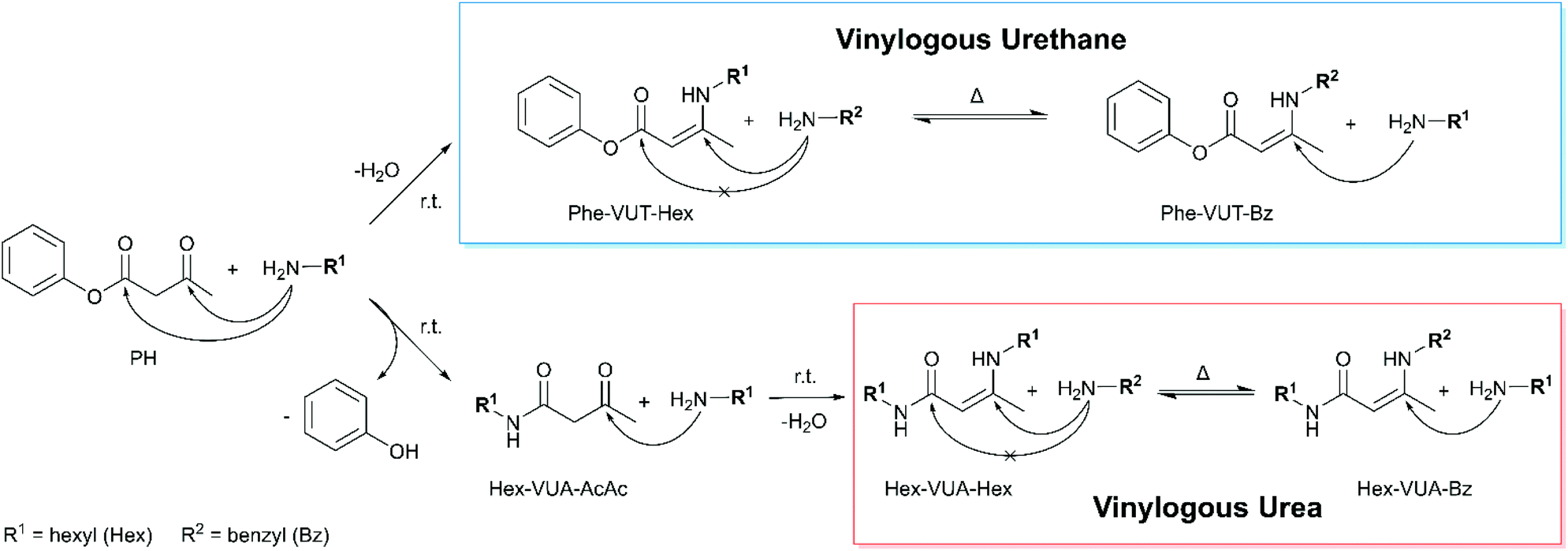 | ||
| Fig. 2 Schematic representation of the condensation and substitution reactions of aromatic acetoacetates and aliphatic primary amines. The nucleophilic attack can either occur at the keto group, producing a vinylogous urethane, or at the ester unit, producing an acetoacetamide and vinylogous urea. Both types represent dynamic covalent bonds, which undergo fast exchange reactions with free primary amines at elevated temperatures in the absence of a catalyst. For the model studies phenylacetoacetate, hexylamine and benzylamine have been used. | ||
Due to the significantly lower electrophilicity of the conjugated CO ester bond of the vinylogous urethane, this bond is resistant to further substitution reactions with free hexylamine, while the CC bond enables dynamic covalent exchange reactions. Thus, mixtures of vinylogous urethane/urea compounds and pendant free amines could be obtained. In general, the nucleophilicity of the amines highly depends on solvation, steric hindrance or coordinating effects and can be adjusted by solvents, catalysts or other additives.63–65 For example, the aromatic amine aniline undergoes a more selective condensation reaction, producing mainly the vinylogous urethane compound Phe-VUT-Phe (67%)(Fig. S15, ESI†), proved by reference measurements (1H/MS and ATR-FT-IR, Fig. S16–21, ESI†).
Moreover, experiments showed that the ratio of the emerging vinylogous urethane and vinylogous urea products can be adjusted by the solvent (dimethyl sulfoxide (DMSO), benzene, and chloroform) (1H NMR, Fig. S22, ESI†) and/or acetic acid as a catalyst (Fig. 3) (1H NMR, Fig. S23, ESI†). Available protons protonate the keto group of the acetoacetate and increase the electrophilicity, which shifts the ratio to the vinylogous urethane products and allows a selective conversion by using a sufficient amount of acetic acid. Furthermore, the conversion rates are highly increased by the addition of some acetic acid, while a higher amount of acetic acid slowed down the reaction and finally totally inhibited it due to the protonation of all nucleophilic amines (Fig. S24, ESI†). In comparison, the condensation reaction of the aliphatic methylacetoacetate and hexylamine selectively yields the vinylogous urethane compound in all considered solvents and acetic acid as a catalyst.62 In addition, the characteristic exchange reactions of the vinylogous urethane and vinylogous urea compounds with pendant free amines were investigated and compared in terms of their activation energies (Fig. 4). For this, an excess of four equivalents of a different amine-type were added to the vinylogous urethane/vinylogous urea mixture to ensure a pseudo first order decay during the beginning of the exchange reaction (eqn (S1), ESI†). The vinylogous urethane exchange was monitored at 70, 90 and 110 °C, while the vinylogous urea exchange was monitored at 12, 22 and 32 °C (schematic overview of the used compounds, Fig. S25, ESI†). By plotting the rate constant (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k) against the reciprocal temperature (1/K), the activation energies can be calculated from the slope of the linear fit. 1H NMR spectra show the exchange of benzyl- and hexylamine during the exchange reaction. Regarding the vinylogous urethanes the spectra show a shift of the characteristic vinylic proton at 4.68 ppm (Phe-VUT-Bz) to 4.58 ppm (Phe-VUT-Hex) (Fig. S26, ESI†). The vinylogous urea compounds show a shift of the characteristic NH protons at 8.93 ppm (Hex-VUA-Hex) and 9.39 ppm (Hex-VUA-Bz) after the exchange reaction (Fig. S27, ESI†). ATR-FT-IR spectra did not show significant shifts of the characteristic CO ester, CO amide or CC bands after the exchange reaction (Fig. S28, ESI†). An activation energy of 64.7 ± 8.1 kJ mol−1 was calculated for the aromatic vinylogous urethane exchange, while 52.2 ± 4.8 kJ mol−1 was calculated for the vinylogous urea exchange (Fig. 4). In addition, the activation energy for the exchange of aliphatic vinylogous urethanes synthesized by methylacetoacetate and hexylamine was determined and a similar activation energy of 64.5 ± 10.3 kJ mol−1 was obtained (Fig. S29, ESI†). The results were supported by comparing the expected exchange products with individual reference compounds (1H/13C NMR, MS, and ATR-FT-IR, Fig. S30–S32, ESI†). Furthermore, the calculated values are in good agreement with the values reported in the literature and point out that aromatic and aliphatic vinylogous urethanes show similar activation energies.24,25
k) against the reciprocal temperature (1/K), the activation energies can be calculated from the slope of the linear fit. 1H NMR spectra show the exchange of benzyl- and hexylamine during the exchange reaction. Regarding the vinylogous urethanes the spectra show a shift of the characteristic vinylic proton at 4.68 ppm (Phe-VUT-Bz) to 4.58 ppm (Phe-VUT-Hex) (Fig. S26, ESI†). The vinylogous urea compounds show a shift of the characteristic NH protons at 8.93 ppm (Hex-VUA-Hex) and 9.39 ppm (Hex-VUA-Bz) after the exchange reaction (Fig. S27, ESI†). ATR-FT-IR spectra did not show significant shifts of the characteristic CO ester, CO amide or CC bands after the exchange reaction (Fig. S28, ESI†). An activation energy of 64.7 ± 8.1 kJ mol−1 was calculated for the aromatic vinylogous urethane exchange, while 52.2 ± 4.8 kJ mol−1 was calculated for the vinylogous urea exchange (Fig. 4). In addition, the activation energy for the exchange of aliphatic vinylogous urethanes synthesized by methylacetoacetate and hexylamine was determined and a similar activation energy of 64.5 ± 10.3 kJ mol−1 was obtained (Fig. S29, ESI†). The results were supported by comparing the expected exchange products with individual reference compounds (1H/13C NMR, MS, and ATR-FT-IR, Fig. S30–S32, ESI†). Furthermore, the calculated values are in good agreement with the values reported in the literature and point out that aromatic and aliphatic vinylogous urethanes show similar activation energies.24,25
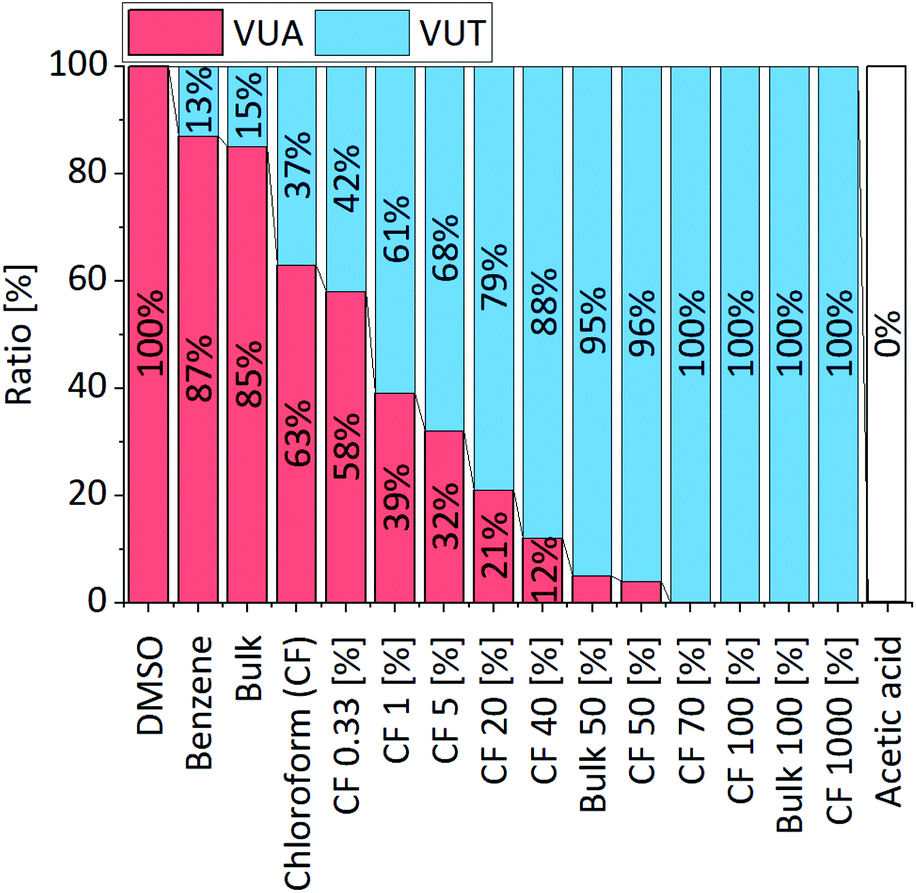 | ||
| Fig. 3 Ratio of the vinylogous urethane and vinylogous urea compounds resulting from the condensation and substitution reactions of phenylacetoacetate and hexylamine in different solvents/bulk and with different amounts of acetic acid. The ratio can be shifted to a selective formation of either the vinylogous urethane or the vinylogous urea compound. | ||
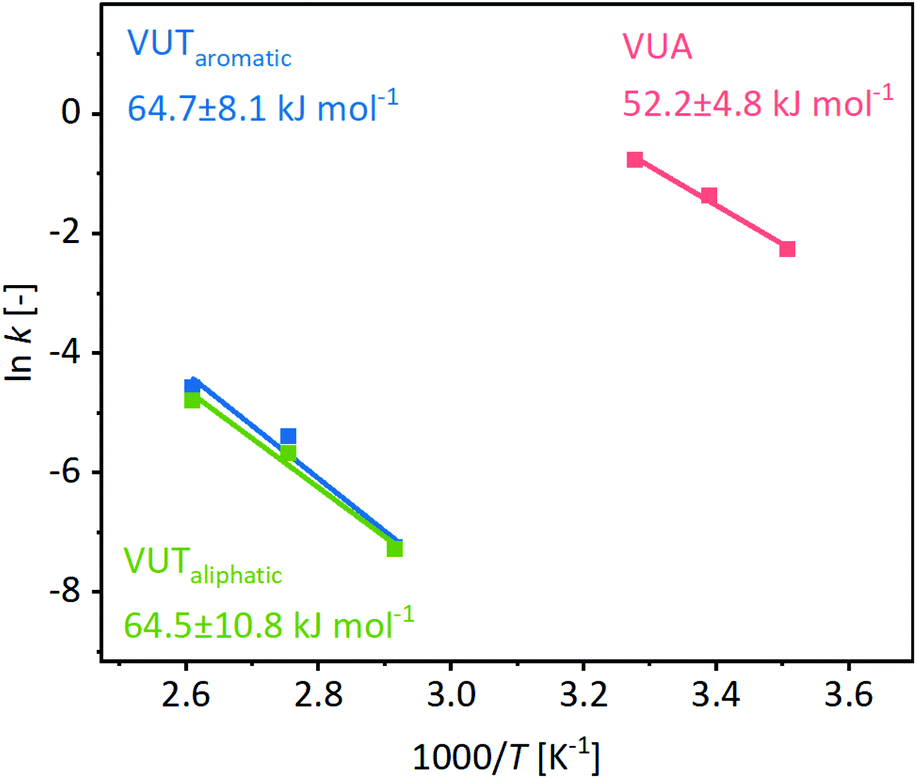 | ||
| Fig. 4 Calculation of the activation energies from the slope of the linear fit by plotting lnk against the reciprocal temperature. The activation energies of aromatic and aliphatic vinylogous urethanes show comparable values of 64.5 ± 10.3 kJ mol−1 and 64.7 ± 8.1 kJ mol−1. The vinylogous urea compounds show an activation energy of 52.2 ± 4.8 kJ mol−1. | ||
Blended poly(vinylogous urethane/urea) networks
In this work four different di- and trifunctional acetoacetate monomers, used for the condensation/substitution reactions with trifunctional primary amines, are introduced. Jeffamine-T403 (JA) and tris(2-aminoethyl)amine (TREN) have been used as trifunctional cross-linking agents (Fig. 5). TREN has already been established as a suitable cross-linker for vinylogous urethane vitrimers and other vitrimer-types.24,57,66,67 Furthermore, JA is a commercial, trifunctional amine based on poly(propylene oxide), which is used as a cross-linker in several industrial applications, especially epoxy resins.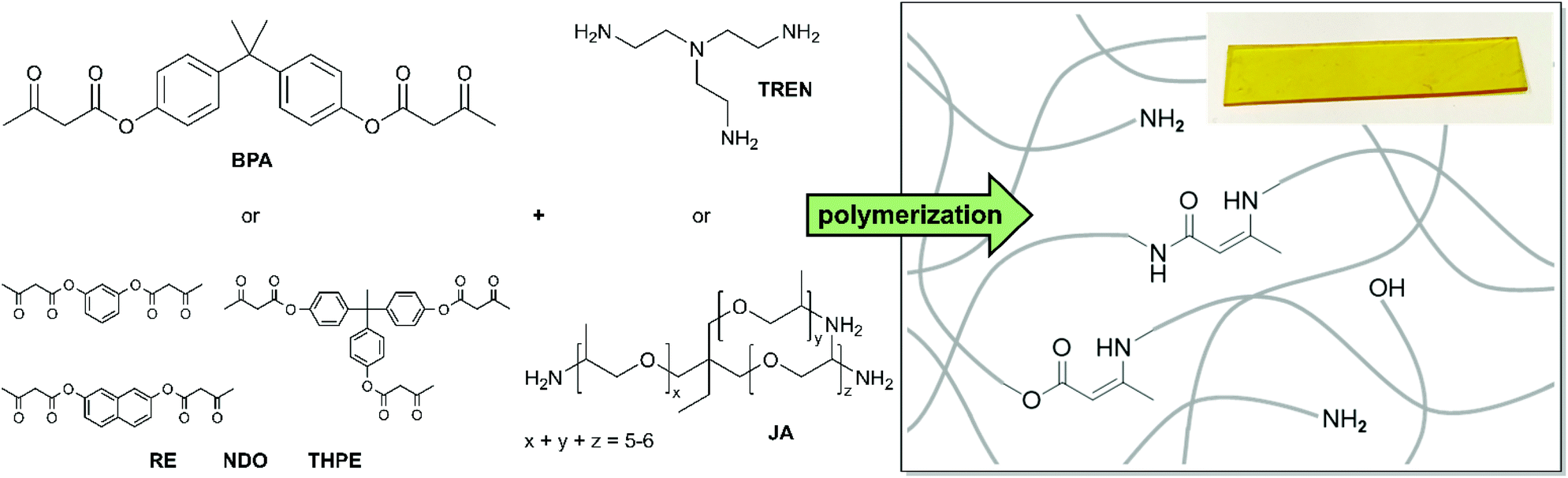 | ||
| Fig. 5 Representation of the used di- and trifunctional acetoacetate and amine monomers and the resulting network structure, containing vinylogous urethane bonds, vinylogous urea bonds, and free aliphatic amine- and aromatic hydroxy groups. | ||
Moreover, JA has been of interest to scientific basic research in terms of epoxy chemistry, composite materials, and other applications, serving as an alternative as a less toxic cross-linker than the highly toxic TREN.68–73 The monomers were mixed at 80 °C in a bulk reaction (with and without acetic acid) until an opaque, homogeneous mass was obtained. A solvent-based polymerization was carried out in chloroform. Herein, the solvent was slowly evaporated at room temperature until gelation of the vitrimers occurred. Next, the vitrimers were cured in vacuo for 20 h at 100 °C to complete the curing process and to remove excess water/acetic acid from the condensation reaction. Afterwards, the vitrimers were post-cured at 150 °C for 1 h and hot-pressed at 150 °C to obtain 1 mm thick films. The stoichiometry of the functional groups and the degree of cross-linking are important factors in the prepared poly(vinylogous urethane/urea) networks. The availability of free primary amine groups accelerates the reorganization process due to exchange reactions within the network. Nevertheless, the network topology is affected by synthesizing a material with an excess of amines, which implies working under non-stoichiometric conditions. Therefore, a compromise between the rearrangement rate and resilient network integrity must be made. In addition, the ratio of the emerging vinylogous urethane/urea compounds determines the required amount of amines (R) and can be estimated from the results of the model study. Furthermore, the choice of the monomer backbone is a decisive factor, since it determines the spatial arrangement of the polymer network and thus has a strong influence on the exchange rate and mechanical properties.24,27 As discussed in the model study, blended poly(vinylogous urethane/urea) networks were synthesized simultaneously, containing two dynamic covalent bond types at the same time (Fig. 5). Because of that, acetoacetate to amine ratios (R) with an excess of amines (R = 0.5, 0.7, and 0.9) were set to ensure sufficient amine groups for the condensation, substitution, and exchange reactions depending on the polymerization conditions (with or without acetic acid). By performing that, 16 vitrimers were polymerized, providing new materials with a broad range of properties. The used monomers, compositions and the measurement results of differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), dynamic mechanical analysis (DMA), and tensile tests as well as the calculated activation energies (Ea) are summarized in Table 1.
| Vitrimer | T g [°C] | T 95% [°C] | τ 130 °C [s] | E a [kJ mol−1] | G′20 °C [GPa] | G′150 °C [MPa] | E t ∅ r.t. [MPa] | σ m∅ r.t. [MPa] | ε m∅ r.t. [%] |
|---|---|---|---|---|---|---|---|---|---|
| The table shows the respective compositions of the vitrimers with the used acetoacetate monomers (RE, NDO, BPA or THPE), the used amine monomers (JA or TREN), the acetoacetate to amine ratio R and H+ for the use of acetic acid (template: VUH+-acetoacetateamine, R). Poly(vinylogous urethane/urea) networks are abbreviated with VU. The glass transition temperatures (Tg on-set), thermal degradation temperatures at 5% mass loss under an ambient atmosphere (T95%), stress–relaxation times at 130 °C (τ130 °C), activation energies (Ea), storage moduli at 20 °C (G′, ω = 10 rad s−1, y = 0.1%), rubbery plateaus at 150 °C (G′150 °C), average Young's moduli (Et ∅), maximum tensile strengths (σm∅) and strains (εm∅) at room temperature are shown. | |||||||||
| Bulk | |||||||||
| VU-REJA, 0.5 | 27 | 244 | 79 | 43.8 ± 2.6 | 0.70 | 2.00 | 290 | 14.6 | 7.4 |
| VU-REJA, 0.7 | 11 | 237 | 343 | 47.3 ± 2.5 | 0.10 | 0.43 | 5.0 | 5.60 | 340 |
| VUH+-REJA, 0.9 | 32 | 222 | 686 | 55.5 ± 2.6 | 0.33 | 0.64 | 260 | 10.1 | 9.6 |
| VU-NDOJA, 0.5 | 39 | 262 | 44 | 48.9 ± 2.4 | 0.42 | 1.70 | 1270 | 52.9 | 5.4 |
| VU-NDOJA, 0.7 | 30 | 241 | 65 | 54.8 ± 5.9 | 0.77 | 0.80 | 350 | 31.1 | 6.5 |
| VUH+-NDOJA, 0.9 | 43 | 253 | 36 | 79.6 ± 3.8 | 0.78 | 0.70 | 1150 | 50.0 | 5.4 |
| VU-THPEJA, 0.5 | 45 | 290 | 133 | 46.4 ± 1.4 | 0.81 | 1.40 | 870 | 62.8 | 6.7 |
| VU-THPEJA, 0.7 | 42 | 267 | 136 | 44.6 ± 2.8 | 0.33 | 0.42 | 1240 | 30.9 | 2.5 |
| VUH+-THPEJA, 0.9 | 68 | 279 | 3193 | 149.7 ± 41.3 | 0.74 | 0.82 | 1540 | 20.3 | 1.8 |
| VU-BPAJA, 0.5 | 33 | 260 | 58 | 52.2 ± 6.7 | 0.67 | 2.05 | 850 | 35.0 | 4.9 |
| VU-BPAJA, 0.7 | 20 | 250 | 133 | 43.9 ± 10.6 | 0.47 | 0.25 | 320 | 12.5 | 5.9 |
| VUH+-BPAJA, 0.9 | 48 | 258 | 275 | 97.9 ± 2.6 | 0.78 | 0.12 | 1160 | 5.20 | 0.5 |
| VU-BPATREN, 0.5 | 68 | 244 | 0.7 | 50.2 ± 2.2 | 0.78 | 1.04 | 1100 | 33.6 | 2.7 |
| VU-BPATREN, 0.7 | 73 | 242 | 1.2 | 59.9 ± 2.4 | 0.67 | 1.57 | 1450 | 12.7 | 1.2 |
| Solvent-based | |||||||||
| CFVUH+-BPAJA, 0.7 | 45 | 266 | 355 | 90.2 ± 6.9 | 0.63 | 0.32 | 920 | 7.8 | 1.3 |
| CFVUH+-BPAJA, 0.9 | 57 | 269 | 814 | 149.5 ± 8.1 | 0.54 | 0.59 | 1170 | 53.0 | 4.6 |
ATR-FT-IR spectra show the disappearance of the characteristic CO ester and CO ketone bands of the acetoacetate monomers after the polymerization (Fig. 6) (Fig. S33, ESI†). The vitrimers show the characteristic CC band (1610 cm−1) and CO ester band (1664 cm−1) of the vinylogous urethane bonds as well as the CC band (1593 cm−1) and CO amide band (1616 cm−1) of the vinylogous urea bonds, proving the formation of the blended poly(vinylogous urethane/urea) networks (all spectra, Fig. S34, ESI†). Nevertheless, the spectra show several overlapping bands, which lead back to the aromatic CC stretching bands of the conjugated phenylic rings (1587–1605 cm−1) and free primary amine groups (1591 cm−1) in the same area. The reference spectra of the individual model compounds support these findings by measuring similar bands (Fig. S6–S12, ESI†). Furthermore, a qualitative shift regarding the vinylogous urethane/urea bond ratios could be observed, using acetic acid as an additional catalyst and/or carrying out a controlled polymerization in chloroform. As shown in the model study, this shifts the ratio towards the vinylogous urethane bonds. IR spectra support this finding by measuring the increased CO ester band (1664 cm−1) (Fig. 6). Nevertheless, it is not possible to determine exact ratios from the spectra.
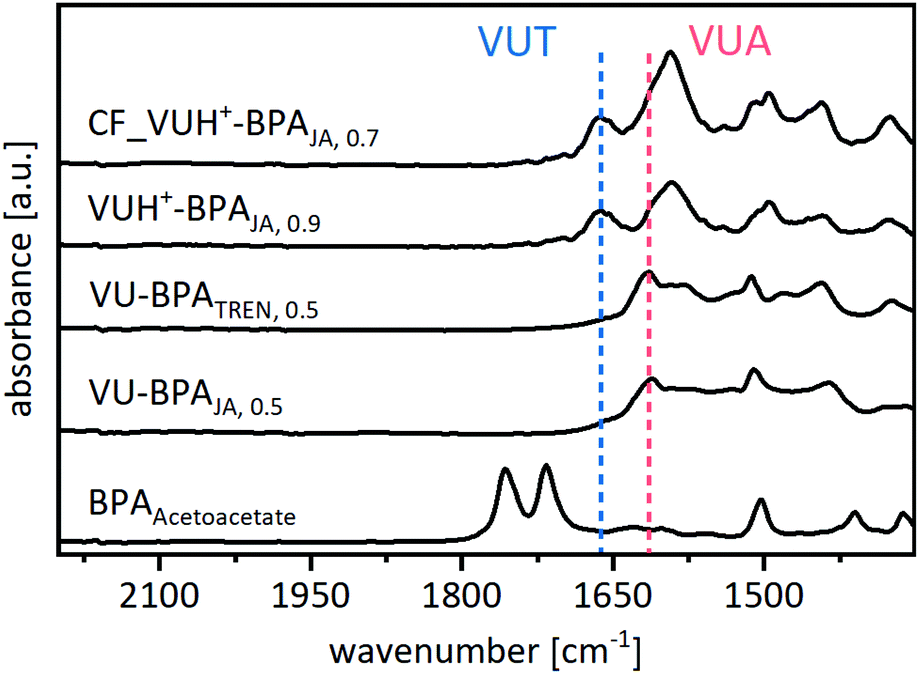 | ||
| Fig. 6 Exemplary ATR-FT-IR spectra of the vitrimers cured with BPA and JA/TREN by different synthetic routes. The blue line marks the characteristic CO ester band of the vinylogous urethane bond (1664 cm−1), while the red line marks the characteristic CO amide band of the vinylogous urea bond (1623 cm−1). The vitrimers polymerized with additional acetic acid show a significantly higher intensity of the ester band (blue), while the vitrimers show a higher intensity of the amide band (red). | ||
Thermogravimetric analysis of the materials showed thermal degradation temperatures of 222–290 °C (5% mass loss) at ambient atmosphere (Fig. S35, ESI†). Relating the results to the values of vinylogous urethane vitrimers described in the literature (230–280 °C), the prepared blended vitrimers showed similar degradation temperatures.27 The glass transition temperatures of the vitrimers have differed significantly by using various acetoacetate to amine ratios, cross-linking agents or acetoacetate backbones. With this, elastomeric and thermosetting vitrimers with Tg values between 11 and 73 °C could be prepared. In particular, the use of JA enabled low Tg, partially elastomeric vitrimers, while the use of TREN led exclusively to thermosetting vitrimers. By using additional acetic acid (ratio shifted to vinylogous urethane bonds) the materials show higher glass transition temperatures (Fig. S36, ESI†). Temperature-dependent DMA measurements showed maximum storage moduli (G′) of 0.10–0.81 GPA at 20 °C and rubbery plateaus of 0.12–2.05 MPA at 150 °C, measured within the linear viscoelastic regime, so that the storage modulus G′ and the loss modulus G′′ were independent of strain (ω = 10 rad s−1 and y = 0.1%) (Fig. S37, ESI†). In addition, the materials displayed stress-relaxation times of 0.7–3193 s at 130 °C, measured by a torsional deformation of 1% (Fig. S38, ESI†).
Moreover, the cross-linking agents JA and TREN, polymerized with BPA (R = 0.5), were compared in terms of the material properties. Interestingly, VU-BPATREN, 0.5 shows significantly shorter stress-relaxation times of 0.7 s at 130 °C compared to VU-BPAJA, 0.5 with 58 s at 130 °C, even if VU-BPATREN, 0.5 showed a significantly higher Tg (JA: 33 °C and TREN: 68 °C) (Fig. 7). Reasons for this might be the closer spatial arrangement of the functional groups regarding the network with the small molecule TREN in comparison to the bulkier molecule JA as well as a higher nucleophilicity of the terminal primary amine groups of TREN in comparison to the sterically hindered isopropyl-terminated primary amine groups of JA. The activation energies were calculated by using Maxwell's model for viscoelastic fluids and an Arrhenius law (eqn (S2), ESI†). Therefore, stress-relaxation curves at different temperatures (110–150 °C) were measured and normalized (Fig. S38, ESI†).
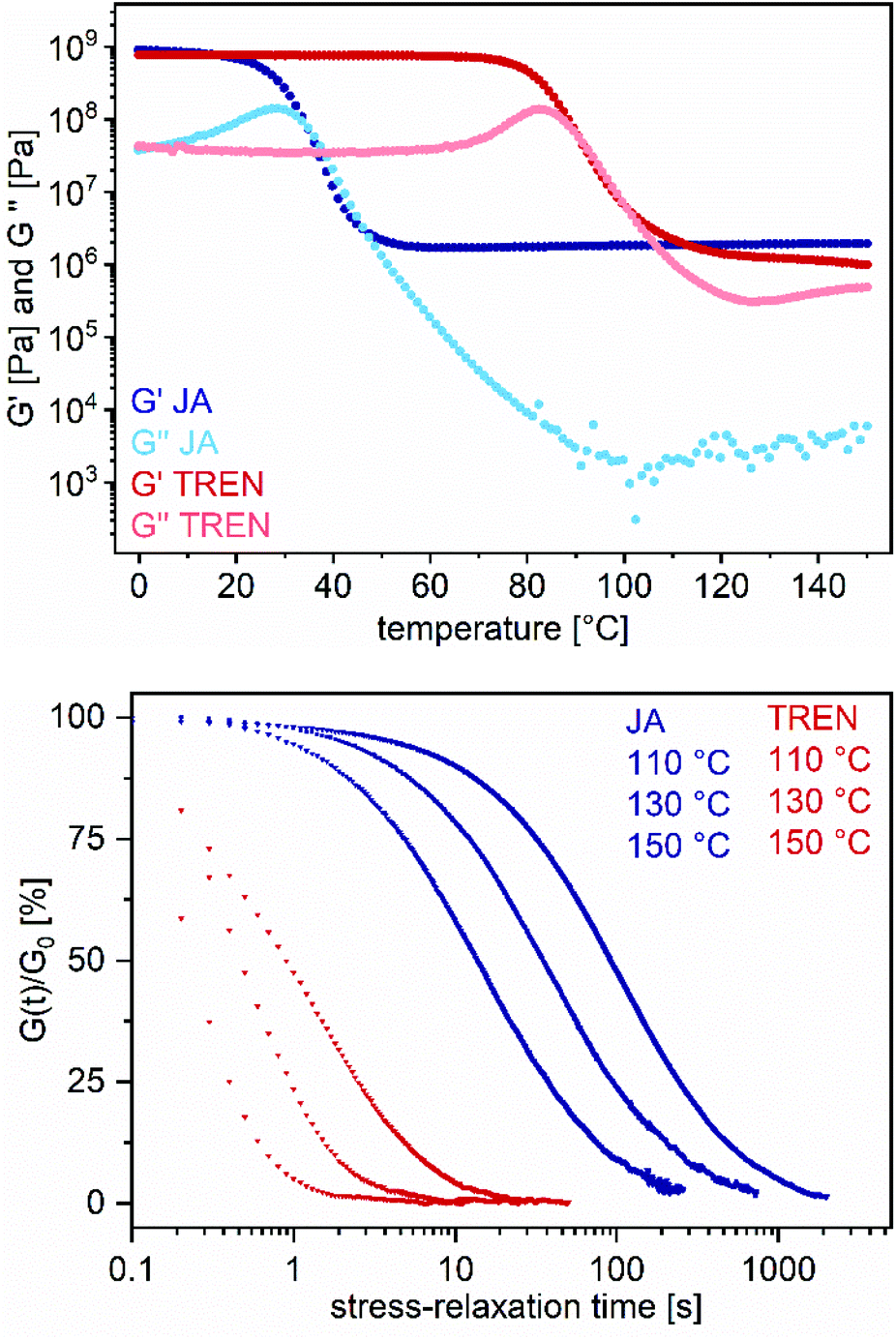 | ||
| Fig. 7 Exemplary temperature-dependent DMA and stress-relaxation measurements in comparison with the vitrimers VU-BPATREN, 0.5 and VU-BPAJA, 0.5. Temperature-dependent DMA measurements show cross-linked networks with the corresponding glass transition temperatures and the characteristic rubbery plateaus (top). Stress-relaxation measurements at 110, 130 and 150 °C show short stress-relaxation times (TREN notably shorter than JA) with an Arrhenius dependence (bottom). | ||
Next, the logarithm of the stress-relaxation time at 37% of the normalized stress-relaxation was plotted against 1000/T to calculate the activation energy from the slope. Even if the stress-relaxation times and Tg values differ significantly, the calculated activation energies of the vitrimers BPATREN, 0.5 and VU-BPAJA, 0.5 showed comparable values, which were 50.2 kJ mol−1 and 52.2 kJ mol−1, respectively (Fig. 8). The different acetoacetate monomers RE, NDO, BPA and THPE were equally cured with JA and compared in terms of the stress relaxation times and calculated activation energies (Ea). The activation energies were calculated in the range of 43.8–54.8 kJ mol−1, while higher activation energies of 55.5–149.7 kJ mol−1 were determined when acetic acid was used during the synthesis. This is reasonable because the catalyst shifts the ratio of vinylogous urethane and vinylogous urea bonds towards the side of the vinylogous urethanes, which show higher activation energies (Fig. 8). Comparing these results with activation energies reported for pure vinylogous urethane vitrimers (68–149 kJ mol−1) and pure vinylogous urea vitrimers (49–53 kJ mol−1), the calculated activation energies in this work reflect the results in good accordance.25,27,57
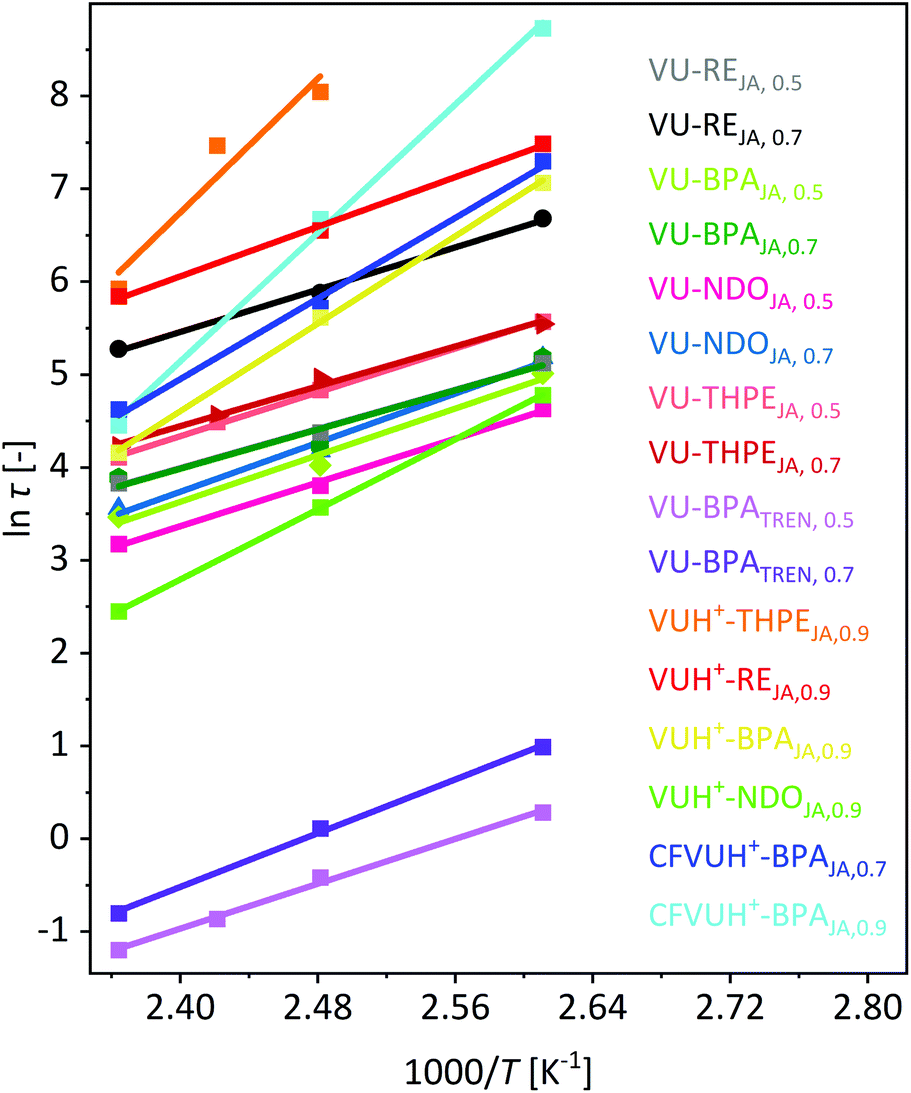 | ||
| Fig. 8 Plot of lnτ against 1000/T to calculate the activation energies from the slope of the linear fit. The stress relaxation times τ (37%) were determined by different stress-relaxation experiments in the temperature range of 110–150 °C. The calculated activation energies of all 16 prepared vitrimers were calculated as summarized in Table 1. | ||
For further investigations of the mechanical properties of the materials, several stress-strain measurements have been carried out at room temperature for each vitrimer and showed significantly different resilience with averaged tensile strengths (σm∅) of 5.2–62.8 MPa, elongation in tensile strengths (εm∅) of 0.5–340% and Young's moduli of 5–1540 MPa (Table 1) (Fig. S39, ESI†). For comparison, one representative curve of each vitrimer is shown in Fig. 9. As expected, the elastomeric vitrimer VU-RE0.7 shows the highest elongation of 340%, with a low tensile strength of 5.6 MPa and a Young's modulus of 5 MPA. The vitrimer with the highest tensile strength turned out to be VU-THPE0.5 with a εm∅ of 6.7%, a σm∅ of 62.8 MPa and a Young's modulus of 870 MPA. The highest Young's modulus was measured for VUH+-THPEJA, 0.9 with a value of 1540 MPa.
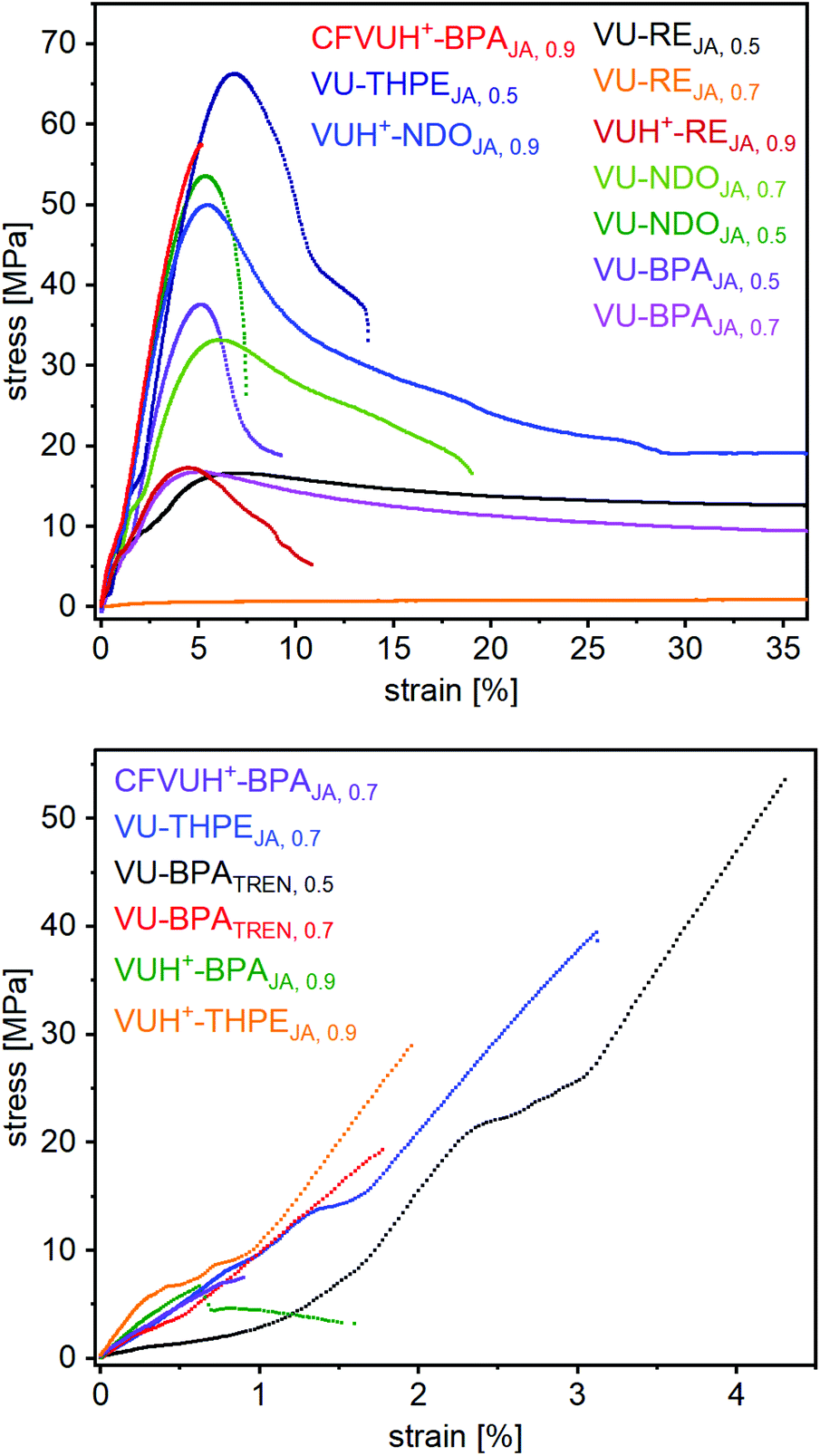 | ||
| Fig. 9 Stress–strain curves of the prepared vitrimers. For comparison, one representative curve is shown for each vitrimer. The elastomeric vitrimer VU-RE0.7 shows the stress–strain curve of an elastomer, with a high elongation at break of 340% strain. The most resilient vitrimer turned out to be VU-THPE0.5 with a strain at break of 6.7%, a tensile strength of 62.8 MPa and a Young's modulus of 866 MPA. The highest Young's modulus was measured for VUH+-THPEJA, 0.9 with 1540 MPa. | ||
Reprocessing and reshaping properties
Experiments were carried out to determine the reshaping, reprocessing and self-healing properties of the vitrimers. Two exemplary cycles of successive grinding and compression molding of the elastomeric vitrimer VU-RE0.7 demonstrated a good reprocessability of the material, proven by repetitively measured ATR-FT-IR spectra and tensile tests. The reprocessing cycles produced always homogeneous, transparent, and bubble-free specimens (Fig. 10). Besides, the ATR-FT-IR spectra are rather similar (if not the same), while the stress–strain measurements show comparable results with slightly decreasing stress and strain at break after the first cycle (Fig. S40, ESI†).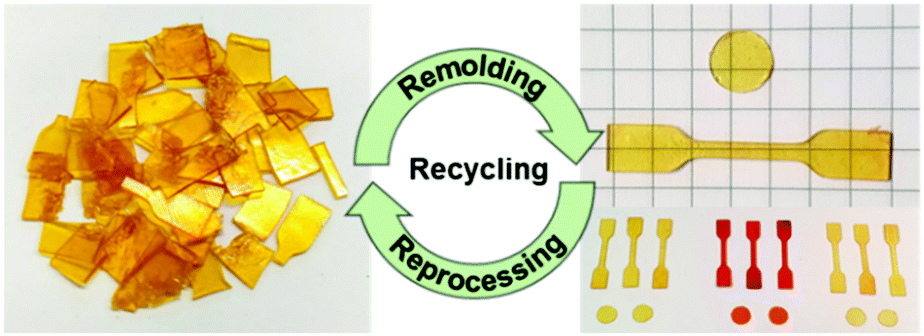 | ||
| Fig. 10 Reprocessing cycles of the vitrimers by consecutive grinding and compression molding (150 °C, 300 s), generating homogeneous, transparent, and bubble-free films (specimen). | ||
Furthermore, the materials present reshaping and shape-memory properties using different temperature protocols (Fig. 11). Polymers with shape-memory properties are smart materials that are capable of memorizing their temporary shape and recovering the permanent shape due to entropic elasticity under an external stimulus,74,75 while traditionally, the refinement of permanent shapes is limited by molding and processing methods.76 However, the investigation of dynamic bonds with polymer networks provides an elegant solution to overcome this limitation. Vitrimers can be reshaped and reprocessed in the solid state due to the thermally induced associative exchange mechanism in the network. Thus, well defined shapes can be easily transformed into new defined shapes without the intermediate step of a mold.77 The presented materials show both thermo-adaptive shape-memory and solid state plasticity, depending on the applied temperature and duration. Heating up above the respective Tg for a short time (80 °C, 5 s) and twisting the material led to topology freezing with subsequent shape-memory when heating the material again above the Tg. Moreover, heating up at 150 °C for 5 min and bending the material, the thermal-induced exchange reactions enable a reorientation of the cross-linking points, leading to a new defined and permanent shape of the material. In addition, self-healing properties were observed by cutting the tensile test specimen (1 mm) in half in the middle (0.5 mm) and heating them for 30 minutes at 150 °C. Afterwards, the cut and non-cut specimens were compared using tensile tests and they show self-healing by almost full recovery and comparable results (Fig. S41, ESI†).
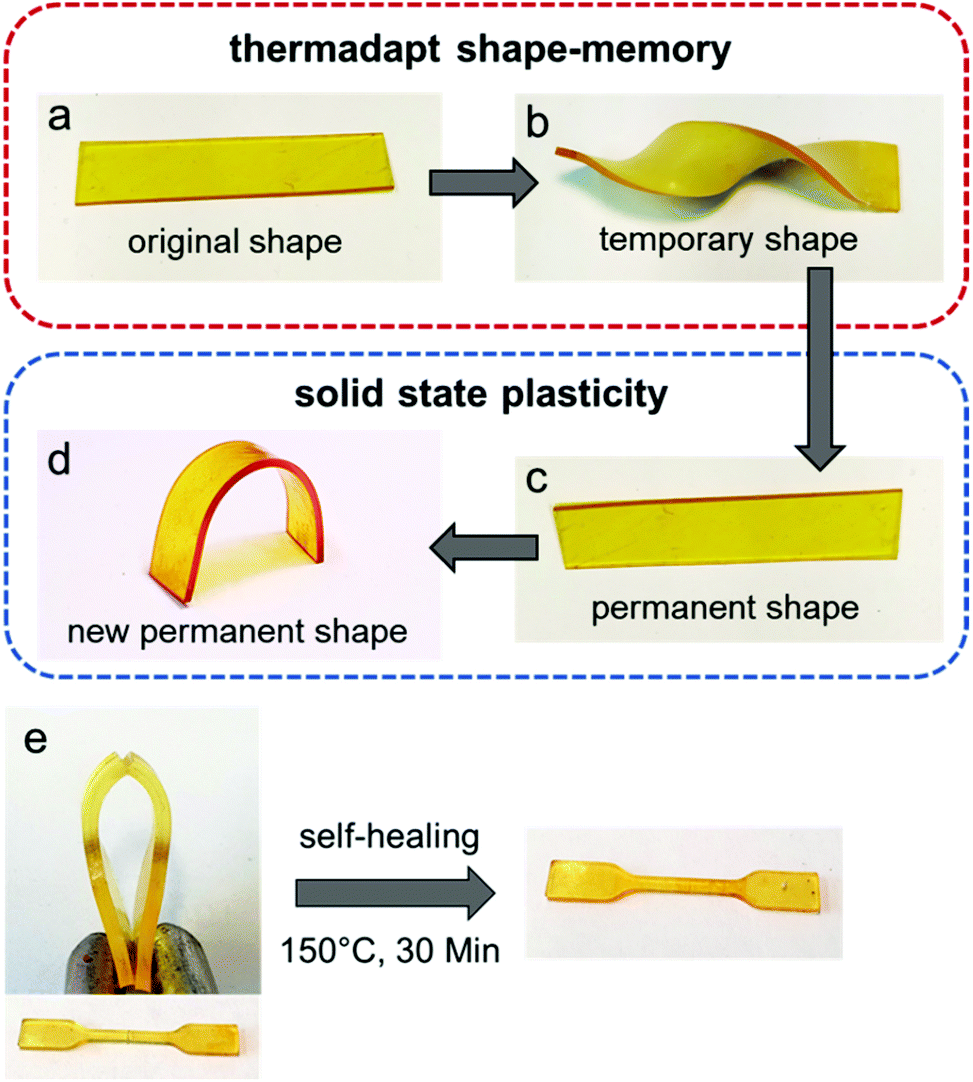 | ||
| Fig. 11 Exemplary thermoadapt shape-memory properties (VU-REJA, 0.5) were investigated by twisting the original strip (a) at 80 °C for 5 s, freezing the topology below the Tg at room temperature (b) and letting it relax back to the original shape at 80 °C for 30 s (c). Solid state plasticity properties were explored by bending the permanent strip (c) at 150 °C for 300 s and reshaping the strip into a new shape (d). The specimens for tensile tests (1 mm) were cut in half in the middle (0.5 mm) and heated for 30 minutes at 150 °C. The cut and non-cut specimens were compared using tensile tests and they show self-healing by almost full recovery (e). | ||
Experimental
Materials
Acetic acid and chloroform were purchased from VWR chemicals. Bisphenol-A (97%), 2,7-naphthalenediol (98%), and hexylamine (99%) were purchased from Sigma-Aldrich. 2,2,6-Trimethyl-1,3-dioxin-4-one (95%), benzylamine (99%), aniline (98%), tert-butylacetoacetate (98%) and 1,1,1-tris(4-hydroxyphenyl)ethane (98%) were obtained from TCI. Tris(2-aminoethyl)amine (97%) was purchased from Alfa Aesar. Phenol (99.5%) was purchased from MERCK. Xylene (99%, isomeric mixture) and resorcin (98%) were purchased from Grüssing. CDCl3, benzene-d6 and DMSO-d6 were purchased from Deutero GmbH. Jeffamine T-403 was purchased from Huntsman.Instrumentation
Nuclear magnetic resonance ( 1 H NMR, 13C NMR) spectroscopy: the spectra (broadband decoupled, 13C NMR (DEPTQ135)) were recorded on a Bruker AVANCEII system (Bruker BioSpin GmbH, Karlsruhe, Germany) at 300 MHz/75 MHz with chloroform-d1 (CDCl3) as a solvent and tetramethylsilane (TMS) as an internal standard. Sample concentrations were between 20 and 40 mg mL−1 and measurements were recorded at 298 K. Data processing was carried out using MestReNova (Version 9.0.1, Mestrelab Research S.L., Santiago de Compostela, Spain). Mass spectrometry (MS): electrospray ionization (ESI) was measured using an Agilent 6224 ESI-TOF device coupled with an Agilent HPLC 1200 Series and direct injection (110–3200 m/z) system. Electron ionization (EI-DIP) was measured using a Thermo ISQ LT EI device coupled with a Thermo Trace 1300 and direct injection (40–800 m/z) system. Data processing was carried out using MestReNova (Version 9.0.1, Mestrelab Research S.L., Santiago de Compostela, Spain). Attenuated total reflection-Fourier transform Infrared (ATR-FT-IR) spectroscopy: ATR-FT-IR spectra were recorded on a Bruker FT-IR Vertex 70 spectrometer (Bruker, Ettlingen, Germany) with a diamond ATR-probe. The measurement software was Opus 8.1. All samples were measured in the range of 3600–600 cm−1 with a resolution of 4 cm−1 and 32 scans. Thermogravimetric analysis (TGA): for thermogravimetric analysis, a TGA 209 F1 Iris system (NETZSCH-Gerätebau GmbH, Selb, Germany) was used to determine the mass loss during heat treatment. A temperature range of 25–600 °C with a heating rate of 10 K min−1 was chosen. About 5–10 mg of the polymer was weighed into a ceramic crucible. A normal atmosphere (with oxygen) was used with a flow rate of 20 mL min−1. Data processing was performed using Proteus analysis (NETZSCH-Gerätebau GmbH, Selb, Germany). Differential scanning calorimetry (DSC): to determine the glass transition temperature (Tg) of the vitrimers, a DSC 204 F1 Phoenix differential scanning calorimeter (NETZSCH-Gerätebau GmbH, Selb, Germany) was used. 5–10 mg of the polymer was weighed into an aluminum crucible. The measurements were performed at one bar under a nitrogen atmosphere (flow rate of 20 mL min−1) in the temperature range between −50 and 150 °C. The heating and cooling rates were set to 10 K min−1 and the thermal properties were analyzed using the DSC data of the second and third heating curves. Data processing was performed by Proteus analysis (NETZSCH-Gerätebau GmbH, Selb, Germany). Dynamic-mechanical analysis (DMA): for oscillatory shear experiments, a rotational rheometer (MCR 502, Anton Paar GmbH, Graz, Austria) with an 8 mm plate-plate geometry and a heat chamber with a nitrogen atmosphere was used. The temperature was controlled using a Peltier plate and by a constant nitrogen flow into the chamber. The gap between the lower and the upper plates was usually set to 1 mm. Amplitude sweeps between 0.01% and 1% shear strain (y) at a constant angular frequency (ω) of 10 rad per s were carried out at 110 °C before each measurement. This should ensure that the chosen strain amplitude y0 was within the linear viscoelastic regime, so that the storage modulus G′ and the loss modulus G′′ were independent of strain. The temperature-dependent measurements were carried out at 10 rad s−1 and a deformation of 0.1%. The temperature was first increased from 110 to 150 °C at a rate of 2 K min−1, and then, after a waiting period of 5 min, it was decreased from 150 to 0 °C. Usually, one measuring point per 1 K was recorded. The stress relaxation experiments were carried out with a deformation of 1% and the relaxation modulus was recorded as a function of time. The comparative measurements were carried out in temperature ranges of 110–150 °C. Data processing was performed using the RheoCompass software (version 1.19.266, Anton Paar GmbH, Graz, Austria). The test specimens for the dynamic-mechanical analysis and the relaxation tests were pushed out with an 8 mm punch. The vitrimer films were previously placed in a drying chamber at 100 °C for a few seconds in order to heat them above the glass transition temperature to prevent splintering during processing. Tensile tests: tensile tests were carried out at room temperature on a universal testing machine type Z020 (Zwick Roell) using a 5 kN load cell. The measurements were carried out in accordance with the test standard DIN EN ISO 527-1. The initial force was 0.1 MPa and the clamping length was 13.24 mm. The modulus of elasticity was determined at a speed of 1 mm min−1 and the rest of the test was carried out at 10 mm min−1. The test specimens for the tensile test were punched out with a cutting press type ZCP020 (Zwick Roell). The attachment was used for test specimens of type 5B (according to DIN EN ISO 527-2). The vitrimer films were placed in a drying cabinet at 100 °C for a few minutes in order to heat them above the glass transition temperature, to prevent splintering during processingSynthetic procedures
O), 165.83 (q, COO), 150.49 (q, Ph), 129.63 (t, Ph), 126.33 (t, Ph), 121.15 (t, Ph), 50.09 (s, CH2), 30.37 (p, CH3). δH (300 MHz, DMSO-d6, 298 K): 7.47–7.39 (2H, m, Ph), 7.31–7.24 (1H, m, Ph), 7.16–7.10 (2H, m, Ph), 3.89 (2H, s, CH2), 2.27 (3H, s, CH3). δC (75 MHz, DMSO-d6, 298 K): 201.46 (q, CO), 166.17 (q, COO), 150.28 (q, Ph), 129.58 (t, Ph), 126.02 (t, Ph), 121.65 (t, Ph), 49.47 (s, CH2), 30.19 (p, CH3). δH (300 MHz, benzene-d6, 298 K): 7.11–6.97 (4H, m, Ph), 6.92–6.85 (1H, m, Ph), 3.00 (2H, s, CH2), 1.62 (3H, s, CH3). δC (75 MHz, benzene-d6, 298 K): 198.77 (q, CO), 165.52 (q, COO), 151.23 (q, Ph), 129.62 (t, Ph), 126.08 (t, Ph), 121.89 (t, Ph), 49.55 (s, CH2), 29.43 (p, CH3). EI-DIP: m/z 179.152 (M+ + 1, 2.73%) 178.113 (M+, 24.72), 94.128 (90.19), 43.024 (100). Calculated for C10H10O3 (M+) 178.063, found 178.113.
O), 165.38 (q, COO), 150.90 (q, Ph–O), 130.05 (t, Ph), 119.42 (t, Ph), 115.38 (t, Ph), 49.46 (s, CH2), 30.41 (p, CH3). EI-DIP: m/z 278.146 (M+, 0.37%), 194.118 (17.92), 110.066 (100) 43.014 (31.53). Calculated for C14H14O6 (M+) 278.079, found 278.146.
O), 165.86 (q, COO), 148.89 (q, Ph–O), 134.25 (q, Ph), 129.86 (q, Ph), 129.63 (t, Ph), 121.08 (t, Ph), 118.53 (t, Ph), 50.13 (s, CH2), 30.47 (p, CH3). EI-DIP: m/z 328.064 (M+, 0.03%), 160.087 (100.00), 131.086 (26.20), 43.002 (15.06). Calculated for C18H16O6 (M+) 328.094, found 328.064.
O), 165.87 (q, COO), 148.37 (q, Ph), 148.28 (q, Ph–O), 127.96 (t, Ph), 120.86 (t, Ph), 50.06 (s, CH2), 42.57 (q, C–CH3), 30.95 (p, CH3), 30.31 (p, OC–CH3). EI-DIP: m/z 396.210 (M+, 0.05%), 228.162 (21.73), 213.154 (100.00), 119.081 (18.09), 43.020 (12.17). Calculated for C23H24O6 (M+) 396.157, found 396.210.
O), 165.81 (q, COO), 148.77 (q, Ph–O), 146.48 (q, Ph–C), 129.86 (t, Ph), 120.93 (t, Ph), 51.80 (q, C–CH3) 50.15 (s, CH2), 30.95 (p, C–CH3), 30.40 (p, OC–CH3). EI-DIP: m/z 558.271 (0.01%) 306.146 (9.05), 291.136 (100.00), 197.105 (10.57) 43.038 (13.32). Calculated for C32H30O9 (M+) 558.189, found 558.271.
O), 158.61 (q, C–CH3), 84.32 (t, CH), 43.04 (s, CH2), 31.07 (s, CH2), 30.73 (s, CH2), 30.31 (s, CH2), 26.83 (s, CH2), 26.75 (s, CH2), 22.72 (s, CH2), 22.66 (s, CH2), 19.53 (p, CH3). 14.15 (p, CH3). δH (300 MHz, DMSO-d6. 298 K): 8.92 (1H, t, NH), 6.89 (1H, t, NH), 4.28 (1H, s, CH), 3.08 (2H, dd, CH2), 2.97 (2H, dd, CH2), 1.77 (3H, s, CH3), 1.51–1.05 (16H, m, CH2), 0.86 (3H, t, CH3), 0.85 (3H, t, CH3). δC (75 MHz, DMSO, 298 K): 170.56 (q, CO), 156.94 (q, C–CH3), 85.90 (t, CH), 42.33 (s, CH2), 38.24 (s, CH2), 31.57 (s, CH2), 31.48 (s, CH2), 30.78 (s, CH2), 30.17 (s, CH2), 26.69 (s, CH2), 26.49 (s, CH2), 22.58 (s, CH2), 22.54 (s, CH2), 19.36 (p, CH3), 14.38 (p, CH3), 14.35 (p, CH3). δH (300 MHz, benzene-d6, 298 K): 9.68 (1H, s, NH), 4.63 (1H, t, NH), 4.25 (1H, s, CH), 3.26 (2H, d, CH2), 2.77 (2H, dd, CH2), 1.61 (3H, s, CH3), 1.42–1.02 (16H, m, CH2, 0.84 (3H, t, CH3), 0.83 (3H, t, CH3). δC (75 MHz, benzene-d6, 298 K): 171.04 (q, CO), 157.40 (q, C–CH3), 85.78 (t, CH), 42.90 (s, CH2), 32.02 (s, CH2), 31.89 (s, CH2), 31.08 (s, CH2), 30.81 (s, CH2), 27.09 (s, CH2), 26.89 (s, CH2), 23.03 (s, CH2), 22.91 (s, CH2), 19.24 (p, CH3). 14.29 (p, CH3), 14.26 (p, CH3). ESI: m/z 270.262 (M+ + 2, 16.07%), 269.259 (M+ + 1, 100.00). Calculated for C16H32N2O (M) 268.251.
O), 157.38 (q, C–CH3), 139.71 (q, Ph), 138.91 (q, Ph), 129.08 (t, Ph), 129.00 (t, Ph), 124.56 (t, Ph), 124.18 (t, Ph), 123.37 (t, Ph), 120.11 (t, Ph), 88.66 (t, CH), 20.57 (p, CH3). EI-DIP: m/z 252.174 (M+, 13.25%), 160.134 (100.00), 132.109 (15.36), 118.105 (11.17). Calculated for C16H16N2O6 (M+) 252.126, found 252.174. HexylamineδH (300 MHz, CDCl3, 25 °C): 2.62 (2H, t, N–CH2), 1.43–1.32 (2H, m, CH2), 1.32–1.17 (6H, m, CH2), 1.11 (2H, s, NH2), 0.83 (3H, t, CH3). δC (75 MHz, CDCl3): 42.35 (s, CH2), 33.93 (s, CH2), 31.78 (s, CH2), 26.63 (s, CH2), 22.70 (s, CH2), 14.08 (s, CH3). BenzylamineδH (300 MHz, CDCl3, 25 °C): 7.31–7.15 (5H, m, Ph), 3.80 (2H, s, CH2), 1.37 (2H, s, NH2). δC (75 MHz, CDCl3): 143.42 (q, Ph), 128.56 (t, Ph), 127.08 (t, Ph), 126.79 (t, Ph), 46.56 (s, CH2). AnilinδH (300 MHz, CDCl3, 25 °C): 7.06 (2H, t, Ph), 6.66 (1H, t, Ph), 6.57 (2H, d, Ph), 3.52 (2H, s, NH2). δC (75 MHz, CDCl3): 146.47 (q, Ph), 129.35 (t, Ph), 118.59 (t, Ph), 115.17 (t, Ph).
C), 151.54 (q, Ph–O), 129.28 (t, Ph), 124.96 (t, Ph), 122.32 (t, Ph), 80.82 (t, CH), 43.38 (s, CH2), 31.59 (s, CH2), 31.32 (s, CH2), 26.62 (s, CH2), 22.49 (s, CH2), 31.6 (p, C–CH3) 14.11 (p, CH2–CH3). δH (300 MHz, DMSO-d6, 298 K): 8.52 (1H, s, NH), 7.19–7.06 (2 H, m, Ph), 6.79–6.66 (3H, m, Ph), 4.57 (s, 1H; CH), 3.23 (2H, m, CH2) 1.77 (3H, s, CH3), 1.57–1.40 (2H, m, CH2), 1.35–1.14 (6H, m, CH2), 0.90–0.78 (3H, t, CH3). δH (300 MHz, benzene-d6, 298 K): 8.90 (1H, s, NH), 4.90 (1H, s, CH), 2.54 (2H, dd, CH2) 1.41 (3H, s, CH3), 1.33–1.03 (8H, m, CH2), 0.89–0.78 (6H, t, CH3). ESI: m/z 263.184 (M+ + 2, 17.15%), 262.181 (M+ + 1, 100.00). Calculated for C16H23NO2 (M) 261.172.
C), 151.15 (q, Ph–O), 137.92 (q, Ph), 129.10 (t, Ph), 128.54 (t, Ph), 126.94 (t, Ph), 125.54 (t, Ph), 121.64 (t, Ph), 81.95 (t, CH), 46.86 (s, CH2), 19.34 (p, CH3). ESI-MS: m/z 269.138 (M+ + 2, 17.16%), 268.36 (M+ + 1, 100.00). Calculated for C17H17NO2 (M) 267.126.
C), 151.21 (q, Ph–O), 129.45 (t, Ph), 129.10 (t, Ph), 124.74 (t, Ph), 122.24 (t, Ph), 118.72 (t, Ph), 84.86 (t, CH), 20.74 (p, CH3). ESI: m/z 255.119 (M+ + 2, 4.41%), 254.116 (M+ + 1, 24.44). Calculated for C16H15NO2 (M) 253.111.
Model study – exchange reaction (Ea calculation)
VUTaromatic: 4 equivalents of hexylamine were added to a mixture of Phe-VUT-Bz (1 M) and stirred for 4 hours at 70, 90 and 110 °C in xylene, resulting in the formation of the transamination product Phe-VUT-Hex. VUTaliphatic: the reaction was carried out under the same conditions for Me-VUT-Bz (aliphatic vinylogous urethane). VUA: 4 equivalents of benzylamine were added to a mixture of Hex-VUA-Hex (0.0015 M) and stirred for 30 minutes at 12, 22 and 32 °C in benzene, resulting in the formation of the transamination product Hex-VUA-Bz.Vitrimer synthesis
Bulk: All vitrimers were prepared by the following exemplary synthesis route: propane-2,2-diylbis(4,1-phenylene)bis(3-oxobutanoate) (BPA) (1.38 g, 3.48 mmol) was provided in a rolled rim glass and heated up in an oil bath at 80 °C. When the vitrimer was cured with additional acetic acid, the acid was added to the acetoacetate monomer before the amine. Jeffamine-T403 (2.05 g, 6.96 mmol) was added under constant stirring. As soon as a homogeneous, opaque and cross-linked mass was obtained, the vitrimer-gel was filled onto a Teflon sheet and cured for 20 h at 100 °C in vacuo (oil pump). Afterwards, the vitrimer was post-cured for 1 h at 150 °C in vacuo, in order to ensure full conversion and the removal of the remaining reaction water. Next, the samples were pressed for 5–10 min at 150 °C into a 1 mm thick film. Solvent-based: the acetoacetate monomer was dissolved in chloroform (0.3 M) and acetic acid was added to the solution (70 mol%). Afterwards, Jeffamine-T403 was added to the solution and stirred until gelation. The gel was filled onto a Teflon sheet and cured under the same conditions as those used for the bulk reaction.Recycling/reprocessing
The vitrimers were reprocessed by consecutive grinding and remolding, cutting the films and specimens into small pieces and pressing them at 150 °C for 5–10 min into 1 mm films. In each cycle, ATR-FT-IR and tensile test measurements were carried out repeatedly.Shape-memory, solid state plasticity and self-healing experiments
Exemplary thermoadapt shape-memory properties were investigated by twisting the original film at 80 °C for 5 s, freezing the topology by cooling down to room temperature (22 °C) (topology freezing) and letting it relax back to the original shape at 80 °C for 30 s. Solid state plasticity was explored by bending the film at 150 °C for 300 s, reshaping it into a new shape due to the rearrangement of the cross-linking points. The self-healing experiment is exemplarily described for VU-REJA, 0.5. The DIN 5B specimens for tensile tests (1 mm) were cut in half in the middle of the specimen (0.5 mm) and heated for 30 minutes at 150 °C. Thecut and non-cut specimens were compared using tensile tests.Conclusions
A synthetic route for an easily feasible acetoacetylation of aromatic alcohols using 2,2,6-trimethyl-4H-1,3-dioxin-4-one instead of the established tert-butylacetoacetate is introduced and proceeds significantly more efficient by simple heating for 45 minutes at 135 °C. With this, the widely used and commercially relevant alcohols such as bisphenol-A, resorcin, 2,7-naphthalenediol, and 1,1,1-tris(4-hydroxyphenyl)ethane are introduced as raw materials for vinylogous urethane vitrimers by easy acetoacetylation and curing with the commercially available poly(propylene oxide)-based trifunctional amine Jeffamine T-403 (JA) and tris(2-aminoethyl)amine (TREN). Model studies with monofunctional compounds show that aromatic acetoacetates undergo fast condensation- and substitution reactions with primary amines, leading to mixtures of vinylogous urethane and vinylogous urea bonds, both representing dynamic covalent bonds for thermally induced transamination reactions. Furthermore, this ratio can be adjusted by the used solvent (DMSO, benzene, and chloroform) and/or acetic acid as a catalyst, where the ratio can be completely shifted either to vinylogous urethane or vinylogous urea bonds. By transferring the findings from the model study to the preparation of vitrimers, 16 different elastomeric and thermosetting blended poly(vinylogous urethane/urea) networks were synthesized, showing glass transition temperatures of 11–73 °C and thermal degradation temperatures of 222–290 °C at ambient atmosphere. Dynamic mechanical analysis shows short stress-relaxation times of up to 0.7 s at 130 °C, with calculated activation energies of 43.8–149.7 kJ mol−1, storage moduli of 0.10–0.81 GPa at 20 °C and rubbery plateaus of 0.12–2.05 MPa at 150 °C. In addition, tensile tests showed maximum tensile strengths of 5.2–62.8 MPa with elongations of 0.5–340% (at break) and Young's moduli of 5–1540 MPa. Moreover, the prepared smart materials exhibit remarkable reprocessing, reshaping, shape-memory and self-healing properties, by undergoing two consecutive cycles of grinding and remolding, showing thermoadapt shape-memory and solid state plasticity. Notably, this work contributes detailed model- and material studies for the use of aromatic alcohols as an important class of raw materials for several applications and introduces reprocessable alternatives to the established materials, which offer potential for further investigation of highly resilient, but reprocessable cross-linked polymers.Author contributions
P. H. and V. A.: conceptualization; P. H. and G. S.: methodology, investigation, data curation, formal analysis, and validation; V. A.: supervision and resources; P. H.: writing – the original draft; and P. H. and V. A.: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the German Research Foundation (DFG) via SFB986 “M3”, project A2.Notes and references
- J. P. Pascault, H. Sautereau, J. Verdu and R. J. Williams, Thermosetting polymers, CRC press, Boca Raton, 2002 Search PubMed.
- A. D. McNaught and A. Wilkinson, Compendium of Chemical Terminology, Blackwell Science Publications, Oxford, 1997 Search PubMed.
- H. Dodiuk and S. H. Goodman, Handbook of Thermoset Plastics, Elsevier, Amsterdam, 2013 Search PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- H. Yang, K. Yu, X. Mu, X. Shi, Y. Wei, Y. Guo and H. J. Qi, Soft Matter, 2015, 11, 6305–6317 RSC.
- C. Bowman, F. Du Prez and J. Kalow, Polym. Chem., 2020, 11, 5295–5296 RSC.
- A. Khan, N. Ahmed and M. Rabnawaz, Polymers, 2020, 12, 2027 CrossRef CAS PubMed.
- M. Thiessen and V. Abetz, Polymers, 2021, 13, 1189 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- Y. Spiesschaert, M. Guerre, L. Imbernon, J. M. Winne and F. Du Prez, Polymer, 2019, 172, 239–246 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5377–5385 RSC.
- T. Wright, T. Tomkovic, S. G. Hatzikiriakos and M. O. Wolf, Macromolecules, 2018, 52, 36–42 CrossRef.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps III and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 142, 283–289 CrossRef PubMed.
- B. Krishnakumar, R. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, Chem. Eng. J., 2020, 385, 123820 CrossRef CAS.
- C. Ye, V. S. D. Voet, R. Folkersma and K. Loos, Adv. Mater., 2021, 33, 2008460 CrossRef CAS PubMed.
- R. J. Friary, V. Seidl, J. H. Schwerdt, T.-M. Chan, M. P. Cohen, E. R. Conklin, T. Duelfer, D. Hou, M. Nafissi and R. L. Runkle, Tetrahedron, 1993, 49, 7179–7192 CrossRef CAS.
- K. Ostrowska, M. Ciechanowicz-Rutkowska, T. Pilati and G. Zżuchowski, Monatsh. Chem., 1999, 130, 555–562 CAS.
- M. Irfan, Chemistry and Technology of Thermosetting Polymers in Construction Applications, Springer Science & Business Media, Luxembourg, 1998 Search PubMed.
- Epoxy resins: Chemistry and Technology, ed. M. Clayton, Routledge, London, 2018 Search PubMed.
- H. Q. Pham and M. J. Marks, Epoxy Resins, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, Weinheim, 7th edn, 2005 Search PubMed.
- K. W. Schmiedel and D. Decker, Resorcinol, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, Weinheim, 7th edn, 2011 Search PubMed.
- R. B. Durairaj, Chemistry, Technology and Applications, Springer Science & Business Media, Luxembourg, 2005 Search PubMed.
- A. Gardziella, L. A. Pilato and A. Knop, Phenolic resins: Chemistry, Applications, Standardization, Safety and Ecology, Springer Science & Business Media, Luxembourg, 2013 Search PubMed.
- M. Asim, N. Saba, M. Jawaid, M. Nasir, M. Pervaiz and O. Y. Alothman, Curr. Anal. Chem., 2018, 14, 185–197 CrossRef CAS.
- M. Miyasaka, T. Takazoe, H. Kudo and T. Nishikubo, Polym. J., 2010, 42, 852–859 CrossRef CAS.
- H. Domininghaus, P. Eyerer, P. Elsner and T. Hirth, Die Kunststoffe und ihre Eigenschaften, Springer-Verlag, Berlin, 2005 Search PubMed.
- M. Weber and P. Charoensirisomboon, Macromol. Symp., 2003, 199(1), 243–252 CrossRef CAS.
- I. Sluijs, J. Heijl, E. Sluyts and F. Bruynseels, US Pat., US20170369642A1, 2016; Covestro Deutschland AG Search PubMed.
- C. E. Scott, M. E. Stewart, D. S. Wilmoth, J. C. Morris and J. R. Bradley, US Pat., US5942585A, 1997; Eastman Chemical Co Search PubMed.
- W. Kaiser, Kunststoffchemie für Ingenieure, HanserMünchen, 2006 Search PubMed.
- H. Shindy, Chem. Int, 2016, 2, 41–47 Search PubMed.
- H. P. Latscha and H. A. Klein, Organische Chemie: Chemie—Basiswissen II, Springer, Berlin, 1990, pp. 559–567 Search PubMed.
- R. L. Stamper, M. F. Lieberman and M. V. Drake, Becker-Shaffer's Diagnosis and Therapy of the Glaucomas, Elsevier Health Sciences, Amsterdam, 2009 Search PubMed.
- J. M. Dennis, R. J. Mondschein, J. D. Wolfgang, M. Hegde, R. Odle and T. E. Long, ACS Appl. Polym. Mater., 2020, 2, 958–965 CrossRef CAS.
- A. M. Nelson and T. E. Long, Polym. Int., 2012, 61, 1485–1491 CrossRef CAS.
- R. K. Shahni, M. Mabin, Z. Wang, M. Shaik, A. Ugrinov and Q. R. Chu, Polym. Chem., 2020, 11, 6081–6090 RSC.
- T. Wang, J. P. Matinlinna, J. He, K. E. Ahmed and M. F. Burrow, Mater. Sci. Eng., C, 2020, 117, 111309 CrossRef CAS PubMed.
- D. Schmidt, US Pat., US9139690B2, 2012; UMass Lowell Search PubMed.
- N. Jalal, A. R. Surendranath, J. L. Pathak, S. Yu and C. Y. Chung, Toxicol. Rep., 2018, 5, 76–84 CrossRef CAS PubMed.
- C. Erler and J. Novak, J. Pediatr. Nurs., 2010, 25, 400–407 CrossRef PubMed.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- F. J. Zawacki and M. T. Crimmins, Tetrahedron Lett., 1996, 37, 6499–6502 CrossRef CAS.
- Y.-J. Li, H.-Y. Hung, Y.-W. Liu, P.-J. Lin and H.-J. Huang, Tetrahedron, 2011, 67, 927–935 CrossRef CAS.
- V. Sridharan, M. Ruiz and J. C. Menéndez, Synthesis, 2010, 1053–1057 CAS.
- R. J. Clemens and J. A. Hyatt, J. Org. Chem., 1985, 50, 2431–2435 CrossRef CAS.
- P. Haida and V. Abetz, Macromol. Rapid Commun., 2020, 41, 2000273 CrossRef CAS PubMed.
- D. Xin and K. Burgess, Org. Lett., 2014, 16, 2108–2110 CrossRef CAS PubMed.
- W. A. Henderson and C. J. Schultz, J. Org. Chem., 1962, 27, 4643–4646 CrossRef CAS.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Eur. J. Org. Chem., 2009, 6379–6385 CrossRef CAS.
- P. Haida and V. Abetz, Macromol. Rapid Commun., 2020, 41, 2000273 CrossRef CAS PubMed.
- X. Kuang, G. Liu, X. Dong, X. Liu, J. Xu and D. Wang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2094–2103 CrossRef CAS.
- L. Liu and H. D. Wagner, Compos. Sci. Technol., 2005, 65, 1861–1868 CrossRef CAS.
- L.-Q. Liu and H. D. Wagner, Compos. Interfaces, 2007, 14, 285–297 CrossRef CAS.
- H. Lu and G. Tan, Mech. Time-Depend. Mater., 2001, 5(2), 119–129 CrossRef CAS.
- W. Chen and B. Zhou, Mech. Time-Depend. Mater., 1998, 2, 103–111 CrossRef CAS.
- R. J. Morgan, F. M. Kong and C. M. Walkup, Polymer, 1984, 25, 375–386 CrossRef CAS.
- R. J. Morgan and C. M. Walkup, J. Appl. Polym. Sci., 1987, 34, 37–46 CrossRef CAS.
- L. Bai and J. Zheng, Compos. Sci. Technol., 2020, 190, 108062 CrossRef CAS.
- Z. Tang, Y. Liu, B. Guo and L. Zhang, Macromolecules, 2017, 50, 7584–7592 CrossRef CAS.
- N. Zheng, J. Hou, Y. Xu, Z. Fang, W. Zou, Q. Zhao and T. Xie, ACS Macro Lett., 2017, 6, 326–330 CrossRef CAS.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01237a |
| This journal is © The Royal Society of Chemistry 2022 |