
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of Myxoprincomide, a secondary metabolite from Myxococcus xanthus†
Michael
Kohr
a,
Christine
Walt
b,
Jan
Dastbaz
b,
Rolf
Müller
 b and
Uli
Kazmaier
*ab
b and
Uli
Kazmaier
*ab
aOrganic Chemistry, Saarland University, D-66123 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
bDepartment Microbial Natural Products, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), and Department of Pharmacy, Saarland University, D-66123 Saarbrücken, Germany
First published on 18th November 2022
Abstract
Myxoprincomide, a secondary metabolite of the myxobacterium Myxococcus xanthus DK 1622, is synthesised for the first time. The central, unusual α-ketoamide is generated at the end of the synthesis to avoid side reactions during the synthesis of this rather reactive subunit. Nevertheless, the synthetic natural product is obtained as an isomeric mixture. Detailed analytical investigations show that the identical isomeric mixture is found in the isolated natural product.
Myxobacteria, Gram-negative δ-proteobacteria, widespread in nature, are highly efficient producers of natural products.1Myxococcus xanthus DK 1622, the microbe of the year 2020, was the first myxobacterial strain whose complete genome sequence was published in 2006.2 With 9.8 million base pairs, this extremely large bacterial genome contains at least 18 biosynthetic gene clusters encoding for polyketides, nonribosomal peptides or hybrids of both.3 Still, interestingly, only five compound classes can be isolated and characterised. This indicates that there are yet unassigned pathways, where no natural products could be related with so far.4 Modern genome mining techniques, combining targeted mutagenesis, liquid chromatography coupled with HRMS, and statistical data analysis identified Myxoprincomide as a new secondary metabolite from Myxococcus xanthus DK 1622.5
The linear nonapeptide contains some unusual building blocks (red in Fig. 1), such as β-hydroxylated valine, β-lysine and an α-keto-β-amino acid derived from valine. The tiny, isolated amounts allow for the structure elucidation of a new natural product, but only a few investigations of the biological activities of this compound have been conducted.6
 | ||
| Fig. 1 Myxoprincomide. | ||
Because of our interest in the total synthesis of natural products, we began working on the synthesis of Myxoprincomide to verify the proposed structure and obtain enough material in hand for further biological studies. The most remarkable building block is the central α-keto-β-amino acid, whose biosynthesis has yet to be explained. Similar building blocks are found in a series of mainly cyclic peptides such as jamaicensamide A,7 orbiculamide A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and the keramamides.9 Furthermore, the α-ketoamino acid is presumably the most critical subunit in the molecule. In addition, the adjacent β-stereogenic centre should be somewhat acidic and sensitive to epimerisation. Similarly, the α-keto functionality should be relatively reactive, undergoing additions such as hydrate formation.10 Therefore, we decided to generate the α-keto-functionality near the end of the synthesis by oxidation of the corresponding α-hydroxy acid.
8 and the keramamides.9 Furthermore, the α-ketoamino acid is presumably the most critical subunit in the molecule. In addition, the adjacent β-stereogenic centre should be somewhat acidic and sensitive to epimerisation. Similarly, the α-keto functionality should be relatively reactive, undergoing additions such as hydrate formation.10 Therefore, we decided to generate the α-keto-functionality near the end of the synthesis by oxidation of the corresponding α-hydroxy acid.
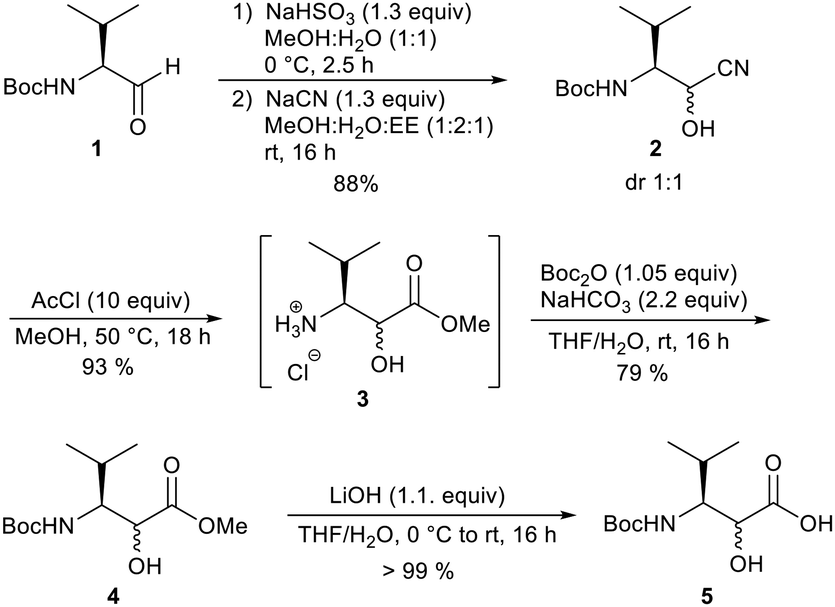
In our work, we obtained the desired N-Boc-protected derivative from Boc-valinal 1 by adding NaCN to provide cyanhydrine 2 as a 1:1 diastereomeric mixture (Scheme 1). The α-hydroxy nitrile 2 was converted into the α-hydroxy ester 3 using acetylchloride in MeOH. Under these conditions, the Boc-protecting group was removed but reintroduced in the next step. Finally, the saponification of the methyl ester provided the free carboxylic acid 5, which could be used in the peptide synthesis. The other unusual amino acids were prepared according to procedures in the literature or purchased.11
 | ||
| Scheme 1 Synthesis of α-hydroxy-β-amino acid 5. | ||
The synthesis of the linear peptide chain, as shown in Scheme 2, started with a prolongation of L-ornithine 612via an Arndt–Eistert homologation to generate the required β-lysine. The activation of 6 as a mixed anhydride and reaction with diazomethane provided diazoketone 7, with only traces of ornithine methyl ester as a side product. This issue was not concerning since the diazoketone could be easily purified by flash chromatography. The diazoketone 7 could directly be reacted with alanine benzyl ester in the presence of silver benzoate to the desired dipeptide 8 in overall high yield. Next, the coupling with the following two protected amino acids proceeded under standard conditions. Tyrosine was used with an allyl-protecting group on the phenolic OH group to avoid side reactions in the final oxidation step. The allyl group could easily be removed later using CpRu(MeCN)3PF6 as a catalyst.13 The subsequent coupling with α-hydroxy acid 5 proceeded without problems, and no O-protecting group was required. The final peptide couplings were performed using TBTU, which provided the best yields.
 | ||
| Scheme 2 Synthesis of myxoprincomide. | ||
The oxidation of the α-hydroxy acid subunit in 15 was performed using Dess Martin periodinane. The reaction had to be carried out in DMSO because of the insolubility of 15 in dichloromethane, which is generally used in Dess Martin oxidations.14 Next, the O-allyl protecting group was removed from the tyrosine using the above-mentioned ruthenium complex. Finally, the benzyl-protecting groups were removed by catalytic hydrogenation. Of note, we had to carry out the reaction in a mixture of chloroform, ethyl acetate and methanol. Without chloroform, only uncomplete conversion was observed, as well as a partial reduction of the α-keto group. Under the optimised conditions, myxoprincomide could be obtained in high yield, sufficient for biological studies (Scheme 2).
The characterisation and identification of the natural product were not as trivial as expected. The NMR spectra showed several sets of signals which could be partly explained by the existence of rotamers since some sets disappeared at a higher temperature. Nevertheless, the synthesised product contained two ‘side products’, also confirmed by UPLC. The purification of myxoprincomide by preparative HPLC and reanalysis indicated that the three products are evidently in equilibrium and converting into each other. A detailed analysis of the NMR spectra of the isolated natural product clearly showed that the same side products could also be found there.
The minor signals can be likely explained by an epimerisation of the α-keto amide or the formation of a hydrate at the keto functionality, but a clear identification was not possible. Therefore, we also performed MS measurements, which gave identical fragmentation patterns for the isolated and synthesised natural product (see ESI†).
Although most 1H NMR signals fit almost perfectly, some signals showed differences, especially for protons close to the amino group of the β-lysine incorporated. This difference might be caused by a different pH or the water content of the NMR solvent. This phenomenon is common with such a highly polar natural product containing basic side chains.
With the synthetic myxoprincomide in hand, we carried out many bioactivity assays, investigating the IC50 values against several bacteria, fungi and cancer cell lines. Unfortunately, no significant activity was observed in any of these assays. Therefore, the ecological function of this secondary metabolite of Myxococcus xanthus remains elusive.
Conclusions
We developed the first total synthesis of the myxobacterial natural product myxoprincomide with the longest linear sequence of 19 steps and in 16% overall yield. The unusual α-ketoamide as the central part of the molecule was generated at the end of the peptide synthesis to avoid side reactions at this subunit. The natural product itself was obtained as a mixture of isomers which equilibrate. Detailed investigations indicate that the same isomers are also found in the isolated natural product. Unfortunately, no significant biological activity of myxoprincomide could be observed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Saarland University and the DFG (Bruker Neo 500 – 447298507) is gratefully acknowledged. We also thank Dr Susanne Kirsch-Dahmen for her biological studies.References
- (a) H. Reichenbach, J. Ind. Microbiol. Biotechnol., 2001, 27, 149–156 CrossRef CAS PubMed; (b) R. O. Garcia, D. Krug and R. Müller, Methods Enzymol., 2009, 458, 59–91 CAS; (c) K. J. Weissman and R. Müller, Nat. Prod. Rep., 2010, 27, 1276–1295 RSC.
- B. S. Goldman, W. C. Nierman, D. Kaiser, S. C. Slater, A. S. Durkin, J. Eisen, C. M. Ronning, W. B. Barbazuk, M. Blanchard, C. Field, C. Halling and G. Hinkle, et al. , Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15200–15205 CrossRef CAS PubMed.
- D. E. Whitworth, Myxobacteria: Multicellularity and Differentiation, ASM Press, Chicago, 2007 Search PubMed.
- (a) H. B. Bode and R. Müller, Angew. Chem., 2005, 117, 6988–7007 ( Angew. Chem., Int. Ed. , 2005 , 44 , 6828–6846 ) CrossRef; (b) H. B. Bode, P. Meiser, T. Klefisch, N. S. Cortina, D. Krug, A. Göhring, G. Schwär, T. Mahmud, Y. A. Elnakady and R. Müller, ChemBioChem, 2007, 8, 2139–2144 CrossRef CAS PubMed; (c) S. C. Wenzel, P. Meiser, T. Binz, T. Mahmud and R. Müller, Angew. Chem., 2006, 118, 2354–2360 ( Angew. Chem., Int. Ed. , 2006 , 45 , 2296–2301 ) CrossRef; (d) V. Simunovic, J. Zapp, S. Rachid, D. Krug, P. Meiser and R. Müller, ChemBioChem, 2006, 7, 1206–1220 CrossRef CAS PubMed; (e) P. Meiser, K. J. Weissman, H. B. Bode, D. Krug, J. S. Dickschat, A. Sandmann and R. Müller, Chem. Biol., 2008, 15, 771–781 CrossRef CAS PubMed.
- N. S. Cortina, D. Krug, A. Plaza, E. Revermann and R. Müller, Angew. Chem., 2012, 124, 836–841 ( Angew. Chem., Int. Ed. , 2012 , 51 , 811–816 ) CrossRef.
- N. S. Cortina, Pathways to products: exploring the biosynthetic depth of Myxococcus xanthus by comprehensive secondary metabolite profiling, dissertation, Saarbrücken, 2013 Search PubMed.
- M. T. Jamison and T. F. Molinski, J. Nat. Prod., 2016, 79, 2243–2249 CrossRef CAS PubMed.
- N. Fusetani, T. Sugawara and S. Matsunaga, J. Am. Chem. Soc., 1991, 113, 7811–7812 CrossRef CAS.
- (a) F. Itagaki, H. Shigemori, M. Ishibashi, T. Nakamura, T. Sasaki and J. Kobayashi, J. Org. Chem, 1992, 57, 5540–5542 CrossRef CAS; (b) H. Uemoto, Y. Yahiro, H. Shigemori, M. Tsuda, T. Takao, Y. Shimonishi and J. Kobayashi, Keramamides K and L, Tetrahedron, 1998, 54, 6719–6724 CrossRef CAS.
- (a) M. Robello, E. Barresi, E. Baglini, S. Salerno, S. Taliani and F. Da Settimo, J. Med. Chem., 2021, 64, 3508–3545 CrossRef CAS PubMed; (b) S. F. Brady, J. T. Sisko, K. J. Stauffer, C. D. Colton, H. Qiu, S. D. Lewis, A. S. Ng, J. A. Shafer, M. J. Bogusky, D. F. Veber and R. F. Nutt, Bioorg. Med. Chem., 1995, 3, 1063–1078 CrossRef CAS PubMed; (c) G. G. Xu and F. A. Etzkorn, Org. Lett., 2010, 12, 696–699 CrossRef CAS PubMed.
- Y. Luo, G. Evindar, D. Fishlock and G. A. Lajoie, Tetrahedron Lett., 2001, 42, 3807–3809 CrossRef CAS.
- G. Pattenden and T. Thompson, Chem. Commun., 2001, 717–718 RSC.
- S. Tanaka, H. Saburi, Y. Ishibashi and M. Kitamura, Org. Lett., 2004, 6, 1873–1875 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, copies of 1H and 13C NMR spectra, HPLC chromatograms and MS2 analysis. See DOI: https://doi.org/10.1039/d2ob02021a |
| This journal is © The Royal Society of Chemistry 2022 |