
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal cations catalyze 16O/18O exchange of catechol motifs with H218O†
Roelant
Hilgers
 a,
Judith
Bijlsma
a,
Luana
Malacaria
b,
Jean-Paul
Vincken
a,
Emilia
Furia
b and
Wouter J. C.
de Bruijn
*a
a,
Judith
Bijlsma
a,
Luana
Malacaria
b,
Jean-Paul
Vincken
a,
Emilia
Furia
b and
Wouter J. C.
de Bruijn
*a
aLaboratory of Food Chemistry, Wageningen University & Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: wouter.debruijn@wur.nl
bDipartimento di Chimica e Tecnologie Chimiche, Via P. Bucci, Cubo 12/D, Università della Calabria, I-87030 Arcavacata di Rende (CS), Italy
First published on 14th November 2022
Abstract
Catechol motifs are ubiquitous in nature, as part of plant, animal and microbial metabolites, and are known to form complexes with various metal cations. Here, we report for the first time that complexation with transition metal cations, especially Fe(III), results in rapid 16O/18O exchange of the catecholic hydroxyl groups with H218O. We discuss the implications of this finding for mechanistic studies using H218O and potential relevance for production of 18O-labeled catechol derivatives.
Isotopic labeling studies are widely used in many fields of chemistry, typically in combination with mass spectrometric detection. For example, 18O2 and H218O are often used to investigate chemical, enzymatic and microbial oxidation and degradation reactions.1–4 In addition, when compounds of interest are available in a stable isotopically labeled form, they can be used for isotope dilution mass spectrometry,5–7 determination of kinetic isotope effects,8–10 mass spectrometric monitoring of reaction kinetics,11,12 and even to follow the metabolic fate of biologically active compounds in human or animal studies.13,14 In recent (to be published) work, we employed H218O to investigate degradation reactions of several catechol derivatives in the presence of metal cations, and observed unexpected rapid 16O/18O exchange between the H218O and the aromatic hydroxyl groups under mild conditions (37 °C, neutral or slightly acidic pH). 16O/18O exchange of catecholic hydroxyl groups with H218O has been previously reported, yet only under harsh catalyst-free conditions (3 M HCl, 150 °C, 20 days).15 To the best of our knowledge, rapid exchange under mild conditions has not been previously described. We investigated this metal-induced 16O/18O exchange in further detail, as it may open up a new and simple approach to produce stable 18O-labeled catechol derivatives. Additionally, this communication serves as a warning for researchers who intend to study the reactions of catechol derivatives in the presence of metal cations using H218O.
The starting point of our work was the observation that 3,4-dihydroxybenzoic acid (3,4-DHBA, Fig. 1) showed rapid 16O/18O exchange of two oxygen atoms in the presence of various metal cations. These 18O labels were exclusively inserted on the aromatic hydroxyl groups, as evidenced by the neutral loss of 44 Da (i.e., unlabeled CO2) in both unlabeled and 18O labeled catechols (Fig. 2).
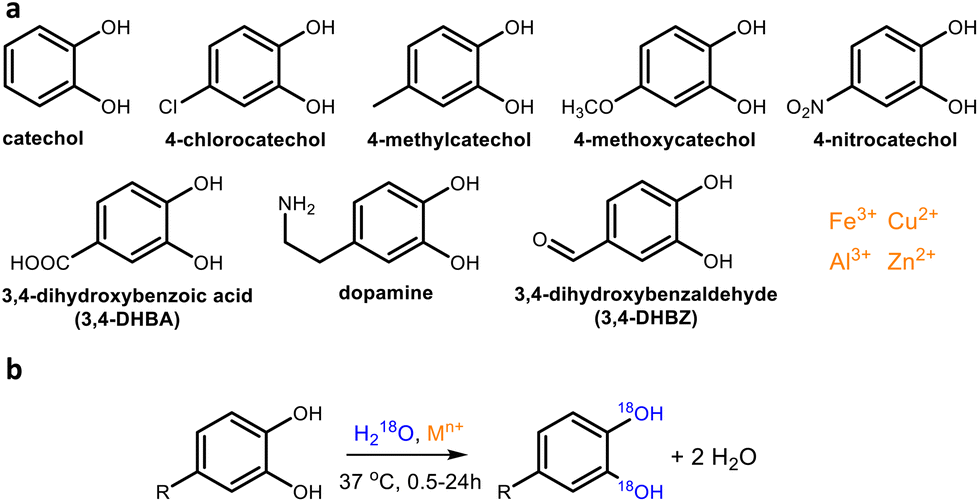 | ||
| Fig. 1 Catechol derivatives and metal cations used for the 18O labeling study (a) and schematic representation of the observed 18O labeling (b). Unless stated otherwise, cations and catechols were used in equimolar concentrations. The R-group in figure (b) represents any of the substituents shown in figure (a). | ||
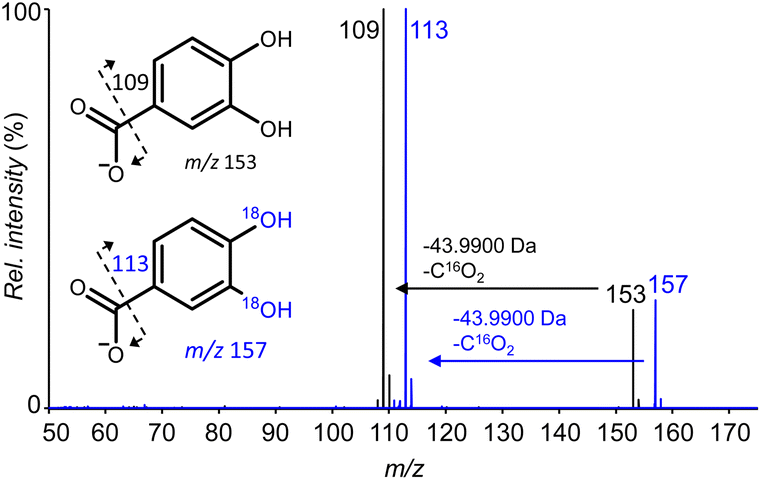 | ||
| Fig. 2 Overlayed Higher-energy Collisional Dissociation (HCD) fragmentation patterns of unlabeled (black) and doubly 18O-labeled (blue) 3,4-DHBA obtained using high resolution ESI-MS in negative ionization mode. The spectrum of doubly labeled 3,4-DHBA was obtained after incubation with FeCl3 (equimolar, 1 h, 37 °C). Fragmentation patterns of other (un)labeled catechol derivatives can be found in Fig. S1.† | ||
Many metal cations are known to form catecholato complexes in the presence of catechol and its derivatives.16 Seemingly, this complexation somehow catalyzes the 16O/18O exchange with H218O. To investigate this phenomenon in greater detail, we first screened the ability of four metal cations to catalyze this 16O/18O exchange. Hereto, catechol and 3,4-DHBA were incubated with the chloride salts of Fe(III), Al(III), Cu(II), and Zn(II) in H218O. We selected these metal cations because they are all able to form catecholato complexes,17,18 but are fundamentally different in terms of redox activity. Whereas Fe(III) and Cu(II) have been described to oxidize catechol derivatives,19 Al(III) and Zn(II) are redox inactive.20 Samples of the incubation mixture were taken at 2 and 24 h, and analyzed using high resolution RP-UHPLC-PDA-ESI-MS to determine the percentage of single and double 18O labeling. As complexation of catechol motifs to metal cations has been reported to be pH-dependent,21 these screening experiments were performed at both pH 7 and pH 3.
For both catechol and 3,4-DHBA, incubation with FeCl3 resulted in extensive labeling at pH 3 and 7, already after 2 hours (Fig. 3). ZnCl2 at pH 7 and CuCl2 at both pH 3 and 7 were found to catalyze 16O/18O exchange in the case of catechol, but no effect was observed in the case of 3,4-DBHA. The higher labeling yield at pH 7 as compared to pH 3 is in line with the expected increased complexation of catechol at elevated pH (Fig. S2†).22 Despite its ability to form catecholato complexes,23 Al(III) did not catalyze 18O labeling (Fig. 3 and Fig. S2†). The observed 16O/18O exchange in the presence of Fe(III) and Cu(II) could be the result of valence tautomerism within the catecholato complexes. Such tautomerism has, indeed, been described for several complexes of transition metal cations with partly filled 3d orbitals.24–26 A H218O molecule coordinated to the metal or present in the bulk could attack the activated keto-tautomer of the catechol and thereby replace the original 16O hydroxyl group (Fig. 4). Such a mechanism seems, furthermore, fully in line with the lack of 16O/18O exchange in the presence of the redox-inactive Al(III). The proposed mechanism, however, cannot explain the observation of 16O/18O exchange in the presence of Zn(II). Since Zn(II) has a completely filled 3d orbital, valence tautomerism, forming Zn(I), is expected to be highly unfavorable. In addition, when catechol incubations were performed with Mn(II), rapid 16O/18O exchange was observed at pH 7 (Fig. S3†). As we expect valence tautomerism between Mn(II) and Mn(I) to be highly unlikely, this finding also challenges valence tautomerism as the only mechanism. To explore other potential mechanisms, we investigated whether molecular oxygen could, somehow, promote 16O/18O exchange, by repeating the incubations of catechol with FeCl3, CuCl2, and ZnCl2 after N2 purging of the H218O solvent and headspace. In none of the incubations, a decrease in 18O labeling was observed after N2 purging (Fig. S4†), excluding a potential role of O2 in the 16O/18O exchange. We therefore propose that the exchange in the presence of Fe(III) and Cu(II) occurs via the mechanism depicted in Fig. 4, whereas the exact mechanism of the Zn(II)-catalyzed exchange remains to be elucidated.
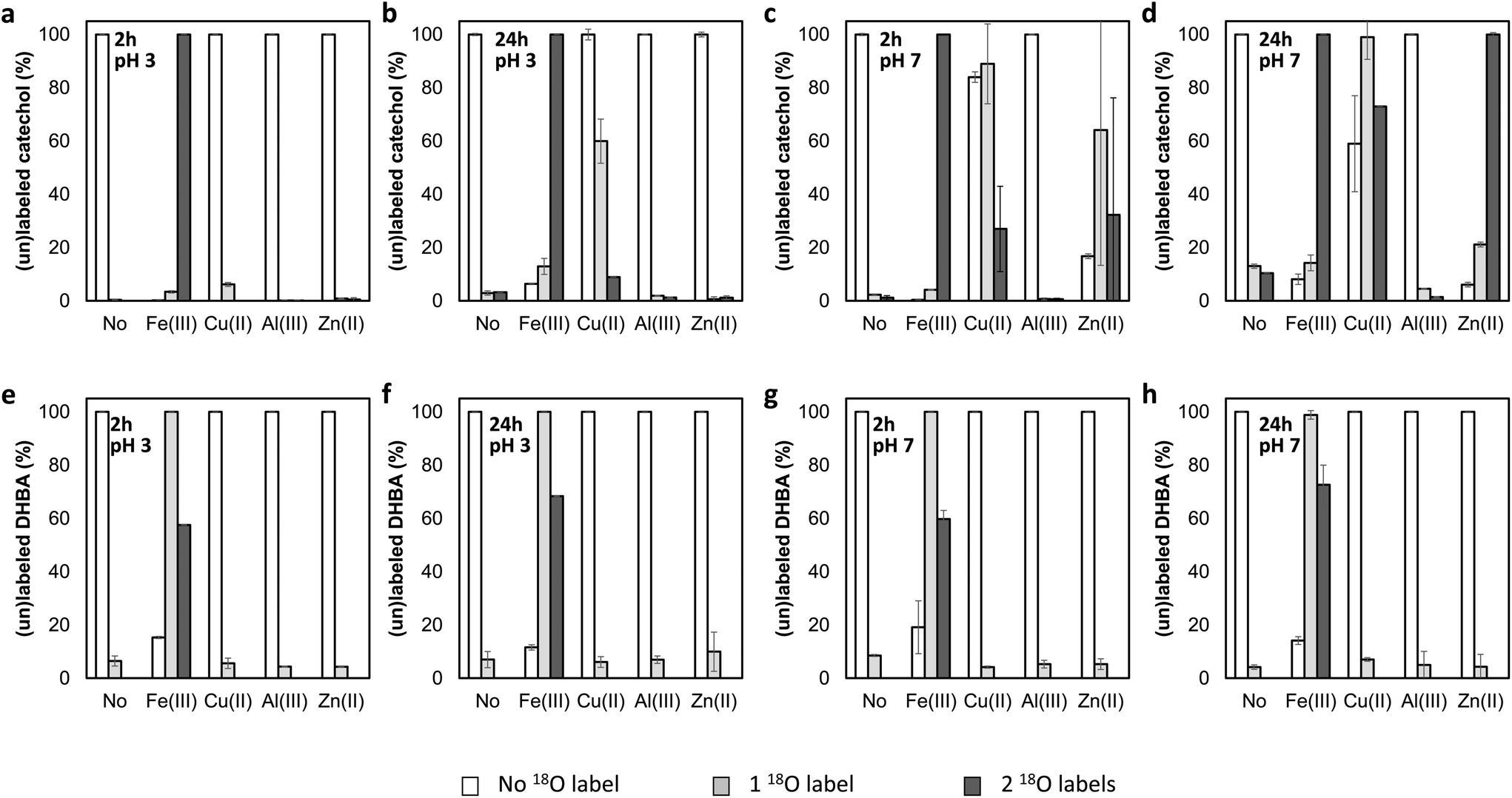 | ||
| Fig. 3 Percentages of unlabeled, singly labeled, and doubly labeled catechol (a–d) and 3,4-DHBA (e–h) after equimolar incubations (1 mM) with FeCl3, CuCl2, AlCl3, or ZnCl2 at 37 °C for 2 or 24 h, as determined by RP-UHPLC-PDA-ESI-MS. Data are presented as average and standard deviation of two separate incubations. | ||
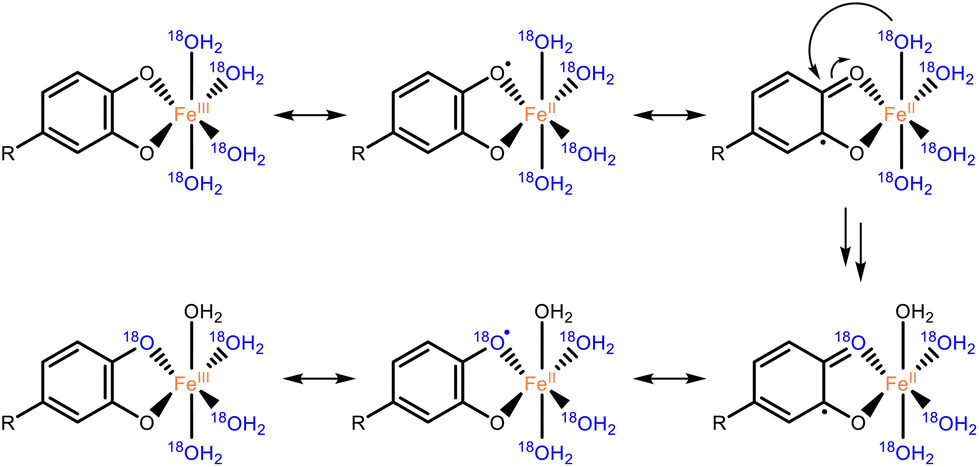 | ||
| Fig. 4 Proposed mechanism for the Fe(III) or Cu(II) catalyzed 16O/18O exchange of catechol motifs, here depicted with Fe(III) as an example. The H218O attacking the catecholato ligand may also originate from the bulk. Examples of valence tautomerism as a mechanism underlying catalysis in Fe and Cu–catecholato complexes have been reported in earlier research.25,26 | ||
As Fe(III) showed the highest extent of 18O labeling in all cases, we continued our research using only FeCl3. No notable differences were observed between the labeling percentages at pH 3 and pH 7 in FeCl3 incubations. Because of the relatively higher solubility of Fe(III) at acidic conditions,27 further experiments were performed at pH 3.
Since the initial screening showed limited differences between the timepoints tested, and the 16O/18O exchange of catechol seemed already complete within 2 h, we proceeded to follow the exchange kinetics by using time-resolved ESI-MS. Hereto, catechol, 3,4-DHBA, 4-chlorocatechol, 4-methylcatechol, and dopamine solutions in H218O (pH 3) were mixed with equimolar amounts of FeCl3 in H218O (pH 3), and infused for ∼60 min into an ion trap MS. The kinetics were found to be strongly dependent on the substitution of the aromatic ring. Catechol, 4-chlorocatechol, and 3,4-DHBA were found to undergo near-to-complete 18O labeling within 30 min (Fig. 5a, b and d), whereas 4-methylcatechol and dopamine were labeled considerably slower and reached around 60% labelling within the duration of the experiment (Fig. 5c and e).
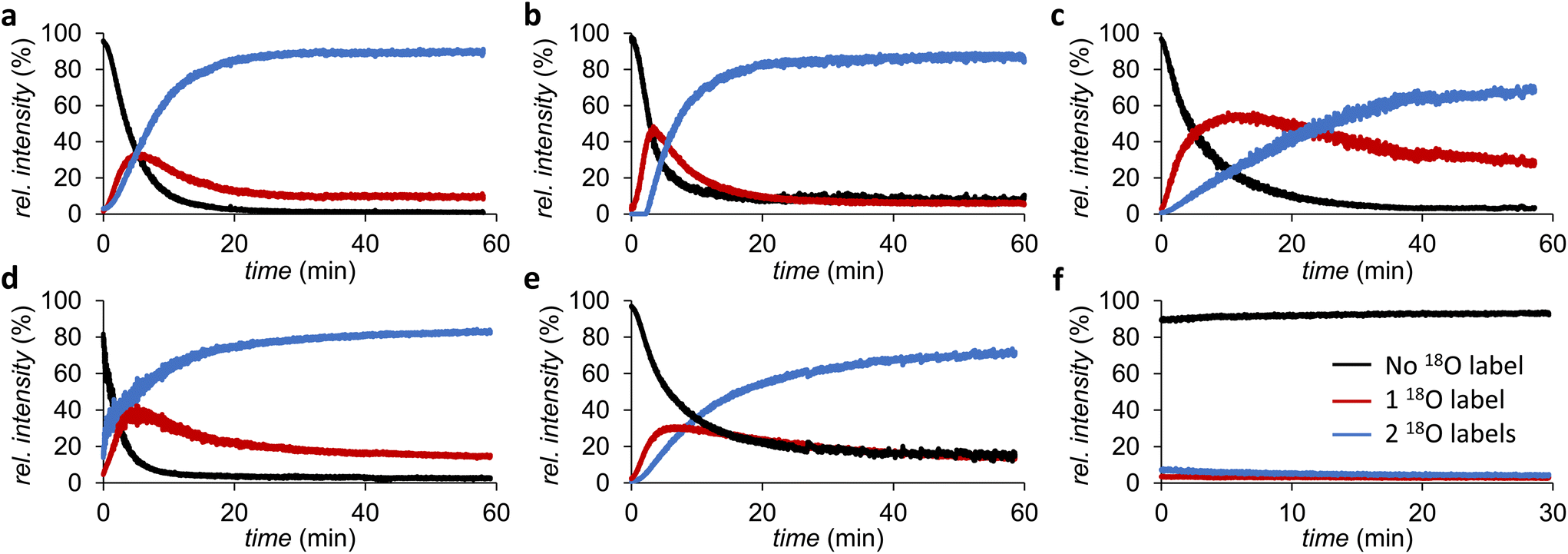 | ||
| Fig. 5 Kinetics of FeCl3 catalyzed 18O labeling of (a) catechol, (b) 4-chlorocatechol, (c) 4-methylcatechol, (d) 3,4-DHBA, and (e) dopamine. Relative intensities of unlabeled, singly 18O-labeled, and doubly 18O-labeled catechols are shown in black, red, and blue, respectively. In absence of FeCl3, no labeling was observed as indicated in (f) for 4-chlorocatechol. Note: the relative intensities shown are not corrected for the fact that 97% 18O-labeled water was used as solvent. | ||
No labeling was observed in H218O in absence of FeCl3, confirming that 16O/18O exchange with H218O only occurs in the presence of a suitable catalyst (Fig. 5f). The latter also indicates that the isotopic labeling will be stable after removal of FeCl3 from the product. When phenol was used instead of the catechols, no labeling was observed after 24 h, which underscores the importance of the catechol motif for the isotope exchange (data not shown).
Although the above described 16O/18O exchange may provide an interesting new approach for production of stable 18O-labeled catechols, Fe(III) has been reported to induce oxidative degradation of various catechols.28 Therefore, we simultaneously determined the labeling yield and recovery of all eight catechol derivatives, shown in Fig. 1, at various time points, using high resolution RP-UHPLC-PDA-ESI-MS. As can be observed from Table 1, complete (i.e., 100% doubly labeled compound) or near-to-complete labeling (i.e., >90% doubly labeled compound) was obtained for catechol, 4-chlorocatechol, 4-methylcatechol, 3,4-DHBA and dopamine with high (i.e., >69%) to very high (i.e., >95%) recoveries. In the case of catechol and 4-chlorocatechol, complete labeling and very high recoveries were obtained for incubations at 0.5 and 1 h. Prolonged incubations (24 h) decreased the recoveries, presumably due to oxidative degradation. For 4-methylcatechol, 3,4-DHBA and dopamine, incubations of 4 h were required to achieve >90% doubly 18O labeled products. The double labeling yields of 3,4-DBHA and dopamine stagnated around 95%, and addition of extra iron or incubating at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 catechol:Fe ratio did not substantially increase the labeling yield, but mainly resulted in a decreased recovery. Significantly lower labeling yields were obtained for 4-nitrocatechol and 3,4-dihydroxybenzaldehyde (3,4-DHBZ), even though both compounds possess an electron withdrawing substituent. No general trend could be observed on the effect of electron withdrawing and electron donating substituents on the labeling yield. The reason for the poor yields of the 16O/18O exchange of 4-nitrocatechol and 3,4-DHBZ remains to be investigated. For 4-methoxycatechol, 66% 18O labeling was observed after 24 h, along with a remarkably low recovery. Various new peaks corresponding to compounds of decreased molecular weight were observed in the RP-UHPLC-PDA-ESI-MS chromatograms, suggesting extensive oxidative degradation of 4-methoxycatechol in the presence of FeCl3 (Fig. S5i†).
:3 catechol:Fe ratio did not substantially increase the labeling yield, but mainly resulted in a decreased recovery. Significantly lower labeling yields were obtained for 4-nitrocatechol and 3,4-dihydroxybenzaldehyde (3,4-DHBZ), even though both compounds possess an electron withdrawing substituent. No general trend could be observed on the effect of electron withdrawing and electron donating substituents on the labeling yield. The reason for the poor yields of the 16O/18O exchange of 4-nitrocatechol and 3,4-DHBZ remains to be investigated. For 4-methoxycatechol, 66% 18O labeling was observed after 24 h, along with a remarkably low recovery. Various new peaks corresponding to compounds of decreased molecular weight were observed in the RP-UHPLC-PDA-ESI-MS chromatograms, suggesting extensive oxidative degradation of 4-methoxycatechol in the presence of FeCl3 (Fig. S5i†).
| Compound | Time (h) | Doubly labeled (%) | Recovery (%) |
|---|---|---|---|
|
a These incubations were performed at a catechol:FeCl3 ratio of 1:3.
|
|||
| Catechol | 0.5 | 100 ± 0.1 | 101 ± 1 |
| Catechol | 1 | 100 ± 0.1 | 102 ± 5 |
| Catechol | 24 | 100 ± 1 | 88 ± 5 |
| 4-Chlorocatechol | 0.5 | 100 ± 1 | 95 ± 4 |
| 4-Chlorocatechol | 1 | 100 ± 0.5 | 91 ± 1 |
| 4-Chlorocatechol | 24 | 100 ± 0.1 | 70 ± 3 |
| 4-Methylcatechol | 1 | 84 ± 1 | 69 ± 1 |
| 4-Methylcatechol | 4 | 100 ± 0.2 | 79 ± 2 |
| 4-Methylcatechol | 24 | 100 ± 0.1 | 70 ± 1 |
| 3,4-DHBA | 1 | 91 ± 10 | 86 ± 13 |
| 3,4-DHBA | 4 | 94 ± 8 | 88 ± 15 |
| 3,4-DHBAa | 4 | 98 ± 2 | 49 ± 3 |
| 3,4-DHBA | 24 | 95 ± 6 | 80 ± 0.3 |
| Dopamine | 1 | 92 | 98 |
| Dopamine | 4 | 96 ± 1 | 92 ± 11 |
| Dopaminea | 4 | 97 ± 2 | 59 ± 3 |
| Dopamine | 24 | 95 ± 2 | 72 ± 13 |
| 4-Nitrocatechol | 24 | 0.2 ± 0.04 | 65 ± 13 |
| 4-Nitrocatechola | 24 | 0.7 ± 0.03 | 60 ± 4 |
| 3,4-DHBZ | 24 | 9 ± 1 | 87 ± 9 |
| 3,4-DHBZa | 24 | 24 ± 9 | 59 ± 6 |
| 4-Methoxycatechol | 24 | 66 | 2 |
| 4-Methoxycatechola | 24 | N.D. | 0 |
Results in Table 1 show that FeCl3 is an excellent catalyst for the 18O labeling of various catechols. Although isotopically labeled compounds are mainly used in mass spectrometric studies, requiring low concentrations, the above described experiments (at 0.1 mM) would only allow synthesis of ∼10–15 μg of labeled compound per 1 mL of solvent. Therefore, we investigated whether the 18O labeling of catechol could be upscaled to concentrations that are more meaningful for lab-scale synthesis. We note that at increased concentrations the catecholato-complexes partly precipitated, but could be completely resolubilized in ethanol after incubation. RP-UHPLC-PDA-ESI-MS analysis of the resolubilized samples showed that using 10 and 50-fold higher concentrations (1 and 5 mM) still resulted in complete and near-to complete labeling, respectively, within 0.5 h (Table 2). Further upscaling to 50 mM, however, showed a drop in the labeling yield. Presumably this is caused by the poor water solubility of the catechol–iron mixtures at increased concentrations. We are currently investigating approaches to circumvent this. Nonetheless, even partially 18O-labeled compounds can be highly valuable as mechanistic probes and quantification standards.
| Concentration (mM) | Time (h) | Doubly labeled (%) | Recovery (%) |
|---|---|---|---|
| 0.1 | 0.5 | 100 ± 1 | 101 ± 1 |
| 1 | 0.5 | 100 ± 0.8 | 101 ± 8 |
| 5 | 0.5 | 95 ± 2 | 90 ± 15 |
| 50 | 4 | 8 | 100 |
| 50 | 24 | 16 | 85 |
In summary, we showed that Fe(III), Cu(II), Zn(II) and Mn(II) cations can catalyze 16O/18O exchange of catechol motifs with H218O. This may provide a new route for facile production of 18O labeled catechol derivatives. At the same time, our findings indicate that extra caution is required when drawing conclusions from studies on the reactions of catechol derivatives that employ H218O labeling in the presence of transition metals.
Conflicts of interest
There are no conflicts to declare.References
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 Search PubMed.
- K. Norrman, S. A. Gevorgyan and F. C. Krebs, ACS Appl. Mater. Interfaces, 2009, 1, 102–112 Search PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. Eijsink, Science, 2010, 330, 219–222 Search PubMed.
- M. D. Hilton and W. J. Cain, Appl. Environ. Microbiol., 1990, 56, 623–627 Search PubMed.
- C. G. Arsene, R. d. Ohlendorf, W. Burkitt, C. Pritchard, A. Henrion, D. M. Bunk and B. Güttler, Anal. Chem., 2008, 80, 4154–4160 Search PubMed.
- N. Tretyakova, M. Goggin, D. Sangaraju and G. Janis, Chem. Res. Toxicol., 2012, 25, 2007–2035 Search PubMed.
- J. Villanueva, M. Carrascal and J. Abian, J. Proteomics, 2014, 96, 184–199 Search PubMed.
- P. Liuni, E. Olkhov-Mitsel, A. Orellana and D. J. Wilson, Anal. Chem., 2013, 85, 3758–3764 Search PubMed.
- D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9358 Search PubMed.
- R. Hilgers, A. Van Dam, H. Zuilhof, J.-P. Vincken and M. A. Kabel, ACS Catal., 2020, 10, 8650–8659 Search PubMed.
- L. Jasikova, M. Anania, S. Hybelbauerova and J. Roithova, J. Am. Chem. Soc., 2015, 137, 13647–13657 Search PubMed.
- R. Hilgers, S. Y. Teng, A. Bris, A. Pereverzev, P. White, J. Jansen and J. Roithová, Angew. Chem., Int. Ed., 2022, 61, e202205720 Search PubMed.
- M. D. Neinast, C. Jang, S. Hui, D. S. Murashige, Q. Chu, R. J. Morscher, X. Li, L. Zhan, E. White and T. G. Anthony, Cell Metab., 2019, 29, 417–429 Search PubMed.
- B. Kluger, C. Bueschl, N. Neumann, R. Stückler, M. Doppler, A. W. Chassy, A. L. Waterhouse, J. Rechthaler, N. Kampleitner and G. G. Thallinger, Anal. Chem., 2014, 86, 11533–11537 Search PubMed.
- K. L. Clay and R. C. Murphy, Biomed. Mass Spectrom., 1980, 7, 345–348 Search PubMed.
- Z. Xu, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- L. Malacaria, C. La Torre, E. Furia, A. Fazio, M. C. Caroleo, E. Cione, L. Gallelli, T. Marino and P. Plastina, J. Mol. Liq., 2022, 345, 117895 Search PubMed.
- A. Primikyri, G. Mazzone, C. Lekka, A. G. Tzakos, N. Russo and I. P. Gerothanassis, J. Phys. Chem. B, 2015, 119, 83–95 Search PubMed.
- E. Nkhili, M. Loonis, S. Mihai, H. El Hajji and O. Dangles, Food Funct., 2014, 5, 1186–1202 Search PubMed.
- M. E. Bodini, G. Copia, R. Robinson and D. T. Sawyer, Inorg. Chem., 1983, 22, 126–129 Search PubMed.
- J. Bijlsma, W. J. de Bruijn, J. A. Hageman, P. Goos, K. P. Velikov and J.-P. Vincken, Sci. Rep., 2020, 10, 1–11 Search PubMed.
- M. Elhabiri, C. Carrër, F. Marmolle and H. Traboulsi, Inorg. Chim. Acta, 2007, 360, 353–359 Search PubMed.
- L. Malacaria, G. A. Corrente, A. Beneduci, E. Furia, T. Marino and G. Mazzone, Molecules, 2021, 26, 2603 Search PubMed.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 Search PubMed.
- N. Shaikh, S. Goswami, A. Panja, X.-Y. Wang, S. Gao, R. J. Butcher and P. Banerjee, Inorg. Chem., 2004, 43, 5908–5918 Search PubMed.
- G. Speier, Z. Tyeklar, P. Toth, E. Speier, S. Tisza, A. Rockenbauer, A. Whalen, N. Alkire and C. Pierpont, Inorg. Chem., 2001, 40, 5653–5659 Search PubMed.
- P. N. Johnson and A. Amirtharajah, J. – Am. Water Works Assoc., 1983, 75, 232–239 Search PubMed.
- J. Bijlsma, W. J. de Bruijn, K. P. Velikov and J.-P. Vincken, Food Chem., 2022, 370, 131292 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01884e |
| This journal is © The Royal Society of Chemistry 2022 |