
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of a tetracyclic tripeptide mimetic frozen in a polyproline type II (PP2) helix conformation†
Marco T.
Klein
a,
Bernhard M.
Krause
 a,
Jörg-Martin
Neudörfl
a,
Ronald
Kühne
b and
Hans-Günther
Schmalz
*a
a,
Jörg-Martin
Neudörfl
a,
Ronald
Kühne
b and
Hans-Günther
Schmalz
*a
aUniversity of Cologne, Department of Chemistry, Greinstrasse 4, 50939 Köln, Germany. E-mail: schmalz@uni-koeln.de
bLeibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Strasse 10, 13125 Berlin, Germany
First published on 14th November 2022
Abstract
A synthesis of the new tetracyclic scaffold ProM-19, which represents a XPP tripeptide unit frozen in a PPII helix conformation, was developed. As a key building block, N-Boc-protected ethyl (1S,3S,4R)-2-azabicyclo[2.2.1]hept-5-ene-2-carboxylate was prepared through a diastereoselective aza-Diels–Alder reaction and subsequent hydrogenolytic removal of the chiral N-1-phenylethyl substituent under temporary protection of the double bond through dihydroxylation and reconstitution by Corey–Winter olefination. The target compound Boc-[ProM-19]-OMe was then prepared via subsequent peptide coupling and Ru-catalyzed ring-closing metathesis steps employing (S)-N-Boc-allylgylcine and cis-5-vinyl-proline methyl ester as additional building blocks. In addition, Ac-[2-Cl-Phe]-[Pro]-[ProM-19]-OMe was prepared by solution phase peptide synthesis as a potential ligand for the ena-VASP EVH1 domain.
Introduction
The search for synthetic small-molecule molecules which are able to selectively inhibit or modulate relevant protein–protein interactions represents an important task in biomedical research.1 In this context, we are interested in molecules capable of replacing natural binding partners of protein domains specialized in the recognition of so-called proline-rich motifs (PRMs).2 Such specific interactions are involved in a variety of relevant cellular processes, including tyrosine kinase receptor signaling,3 endocytosis,4 cytoskeletal restructuring,5 transcription,6 and splicing.7 As a distinctive feature, PRMs adopt a left-handed polyproline type II (PP2) helix secondary structure upon binding to their target domains. This helix type, in addition to the absence of hydrogen bonding, is characterized by a helical pitch of about 9 Å (three residues per turn) with ψ- and ϕ-angles of about 145° and −75°, respectively (Fig. 1).8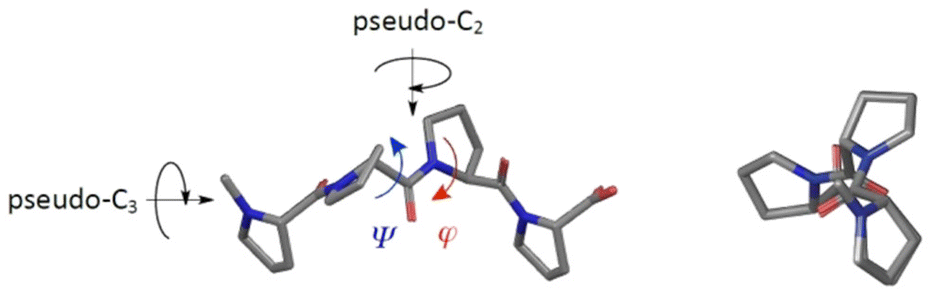 | ||
| Fig. 1 Section of an idealized polyproline type II (PP2) helix in different perspectives. | ||
Following the concept of conformational preorganization, we have previously synthesized tricyclic diproline mimetics, such as ProM-19 and ProM-2,10 (as Pro-Pro equivalents rigidified in a PP2 helix conformation) by introduction of a Z-vinylidene bridge between the two pyrrolidine rings (Fig. 2).
 | ||
| Fig. 2 Top: Structures of ProM-1 and ProM-2. Bottom: Modeling studies show that exchange of the central diproline unit of (Pro)4 (white) by the rigidified units ProM-1 (left) or ProM-2 (right) does not lead to a distortion of the idealized PP2 helix conformation. | ||
We also demonstrated the value of these scaffolds by successfully developing small molecule ligands for the EVH-1 domain.11 For instance, the compound Ac-[2-Cl-Phe]-[ProM-2]-[ProM-1]-OMe (Fig. 3) was shown to selectively bind to the Ena/VASP EVH1 domain with nanomolar affinity and thereby to impair invasion and extravasation of breast cancer cells.12
 | ||
| Fig. 3 Top: Structure of the established EVH-1 ligand (left) and the targeted analog containing the tetracyclic tripeptide mimicking unit ProM-19 (right). Bottom: Simulated fit of the latter ligand (shown in green) on the EVH-1 surface in comparison to Ac-[2-Cl-Phe]-[Pro]-[Pro]-[ProM-1]-OMe (shown in white). Red dots indicate water molecules. | ||
Having successfully synthesized and applied tricyclic diproline mimetics such as ProM-1 and ProM-2, we now asked ourselves whether it would be possible to further expand our toolbox of geometrically defined scaffolds by synthesizing the tetracyclic system ProM-19 (shown in black in Fig. 3) which was designed as an N-terminally extended analog of ProM-1 representing a tripeptide mimetic rigidified in a PPII helix conformation. However, due the structural complexity of this molecule, its synthesis represented a non-trivial task.
We here describe the stereocontrolled synthesis of the new scaffold ProM-19 (in form of the protected derivative 1) and of the novel ligand Ac-[2-Cl-Phe]-[Pro]-[ProM-19]-OMe derived thereof, which according to docking simulations would also perfectly fit to the surface of the Ena/Vasp EVH1 domain in the canonical fashion (Fig. 3).12
Results and discussion
Our initial strategy for the synthesis of the required tetracyclic unit 1 (Boc-[ProM-19]-OMe) is shown in Scheme 1. We intended to apply a domino ring-closing/ring-opening metathesis (ring rearrangement metathesis)13 in the key step employing the precursor 2, which in turn could possibly be assembled from the building blocks 3, 4 and 5 through amide bond formation.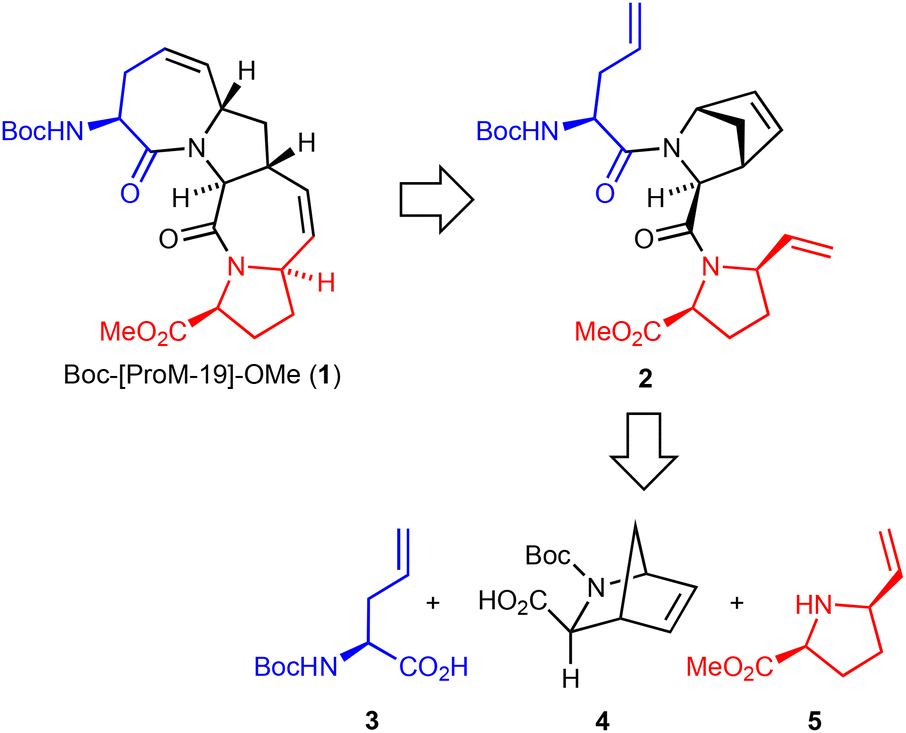 | ||
| Scheme 1 Retrosynthetic analysis. | ||
While the protected cis-5-vinyl-proline 5 was available in our laboratory14 and the allyl-glycine derivative 3 could be prepared in enantiomerically pure form via known methods,15 we considered a Diels–Alder approach as a most attractive approach to construct the 2-aza-bicyclo[2.21]heptane ring system of building block 4.16
The synthesis of 4 commenced with an asymmetric aza-Diels–Alder reaction according to Waldmann16a (Scheme 2). Best results were obtained when the imine 6, prepared in quantitative yield from ethyl glyoxylate and (R)-1-phenylethylamine (Na2CO3, toluene, r.t., 1.5. h), was reacted with cyclopentadiene in the presence of trifluoroacetic acid and catalytic amounts of water.16b Under optimized conditions, the reaction proceeded smoothly even on a 10 g scale to give the desired exo-product 7a in 64% isolated yield after chromatographic purification, besides 7% of the endo-diastereomer 7b.
 | ||
| Scheme 2 Synthesis of the 2-aza-bicyclo[2.2.1]heptane derivative 7a by hetero-Diels–Alder reaction. | ||
Since direct hydrogenolytic cleavage of the chiral N-substituent was not feasible in the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, we decided to temporarily protect this bond by dihydroxylation.17 Thus, 7a was treated with K3Fe(CN)6 and K2CO3 in the presence of 0.025 mol% of K2OsO2 to give the exo-diol 8 as a single diastereomer in 95% yield (Scheme 3). At this stage, hydrogenolysis of the benzylic C–N bond with Pd/C in MeOH was achieved in good yield, and it was found to be advantageous to pass a hydrogen stream directly through the reaction mixture. After filtration of the product solution through a pad of Celite and removal of the solvent, the crude amine was dissolved in dichloromethane and treated directly with Boc2O, after which the N-Boc-protected diol 9 was obtained in 87% yield (2 steps). This compound showed a remarkable tendency to form massive crystals from CH2Cl2 with an edge length of up to 10 mm. Crystallographic analysis confirmed its relative and absolute configuration (Fig. 4).
C double bond, we decided to temporarily protect this bond by dihydroxylation.17 Thus, 7a was treated with K3Fe(CN)6 and K2CO3 in the presence of 0.025 mol% of K2OsO2 to give the exo-diol 8 as a single diastereomer in 95% yield (Scheme 3). At this stage, hydrogenolysis of the benzylic C–N bond with Pd/C in MeOH was achieved in good yield, and it was found to be advantageous to pass a hydrogen stream directly through the reaction mixture. After filtration of the product solution through a pad of Celite and removal of the solvent, the crude amine was dissolved in dichloromethane and treated directly with Boc2O, after which the N-Boc-protected diol 9 was obtained in 87% yield (2 steps). This compound showed a remarkable tendency to form massive crystals from CH2Cl2 with an edge length of up to 10 mm. Crystallographic analysis confirmed its relative and absolute configuration (Fig. 4).
 | ||
| Scheme 3 Synthesis of the 3,5-divinylproline derivative 13 as a synthetic equivalent of the aza-bicyclic building block 4. | ||
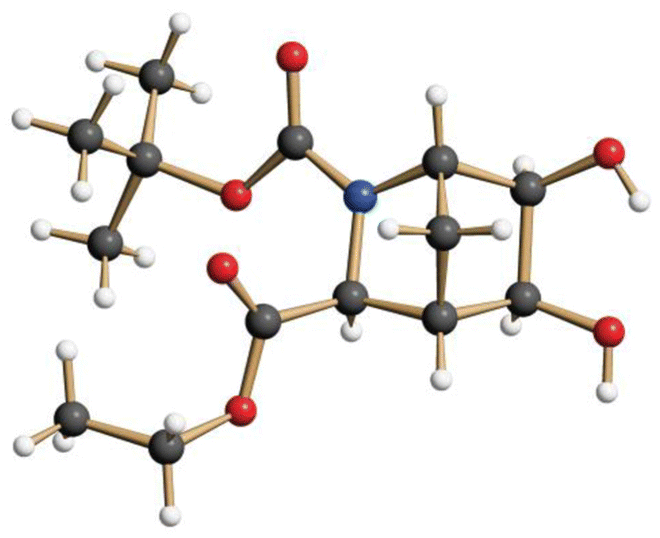 | ||
| Fig. 4 Structure of diol 9 in the crystalline state. | ||
Having successfully accomplished the exchange of the chiral N-phenylethyl substituent by a Boc protecting group, we employed a Corey–Winter reaction18 to reconstitute the double bond. For this purpose, the diol 9 was first reacted with thiophosgene and DMAP, and the resulting cyclic thiocarbonate 10 was subsequently heated with trimethyl phosphite to afford the expected olefin 11 in 72% yield over two steps. When attempting to saponify the ester function of 11, this compound surprisingly proved to be highly sensitive towards aqueous alkali, and we were unable to prepare the targeted aza-bicyclic building block 4 with a free carboxylic acid function. For this reason, we converted 9 into the 3,5-divinylproline derivative 12 by Ru-catalyzed ring-opening metathesis in the presence of ethylene.19 And unlike 11, the ester function in 12 (obtained in 70% yield) could now be readily hydrolyzed with aqueous LiOH, yielding the acid 13 in quantitative yield, which in terms of the planned strategy (Scheme 1) represents a synthetic equivalent of 4. Noteworthy, an attempt to achieve the conversion of 9 to 12 through periodate cleavage of the diol17b and subsequent Wittig olefination gave rise to a mixture of diastereomers, obviously due to enolization/epimerization at the stage of the dialdehyde.
With the ring-opened acid building block 13 (instead of 4) in our hands, we continued the synthesis (Scheme 4) by reacting 13 with the known 5-vinylproline ester 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 under proven peptide coupling conditions (PyBOP, DIPEA).20 Ring closing metathesis of the resulting trivinyldiproline derivative 14 then proved surprisingly difficult. However, repeated addition of small amounts of the Grubbs II catalyst (20 mol% in total) to a dilute solution of 14 in dichloromethane over a period of 48 hours succeeded in achieving a satisfactory conversion, and the tricyclic product 15 was obtained in 58% yield.
14 under proven peptide coupling conditions (PyBOP, DIPEA).20 Ring closing metathesis of the resulting trivinyldiproline derivative 14 then proved surprisingly difficult. However, repeated addition of small amounts of the Grubbs II catalyst (20 mol% in total) to a dilute solution of 14 in dichloromethane over a period of 48 hours succeeded in achieving a satisfactory conversion, and the tricyclic product 15 was obtained in 58% yield.
 | ||
| Scheme 4 Synthesis of the designed tetracyclic tripeptide mimetic Boc-[ProM-19]-OMe (1) and its further transformation to the potential EVH-1 ligand 20. | ||
The tricycle 15, which formally represents a vinyl-ProM-1 derivative, was then treated with TMSOTf to cleave off the Boc protecting group and the resulting amine was directly coupled to (S)-N-Boc-allylglycine (3) in the presence of HATU, DIPEA.21 The subsequent cyclization of 15 through ring-closing metathesis then proceeded smoothly in the presence of the Grubbs II catalyst in hexafluorobenzene to afford the targeted ProM-19 derivative 1 in 61% overall yield from 15 (3 steps).
The final conversion of 1 into ligand 20 (Ac-[2-Cl-Phe]-[Pro]-[ProM-19]-OMe) commenced with the removal of the Boc group (TMSOTf) and HATU-mediated coupling with N-Boc-proline. After renewed removal of the Boc protecting group, the 2-chlorophenylalanine unit was attached employing the Fmoc-protected pentafluorophenyl ester 18 as a reagent.22 Noteworthy, the corresponding N-acetylated reagent could not be employed due to epimerization of the stereocenter – probably via an azlactone intermediate.23 However, the exchange of the Fmoc against an N-acetyl group was smoothly accomplished at the very end of the sequence by reacting the coupling product 19 subsequently with piperidine and acetic anhydride. This way, the devised the potential EVH-1 ligand 20 was obtained in satisfying overall yield as shown in Scheme 4. The biological investigation of 20 is not yet finished and the results will be reported separately in the context of a broader study.
Conclusions
In conclusion, we have elaborated an efficient stereoselective synthesis of the novel tetracyclic scaffold ProM-19 (in form of the protected derivative 1) which was designed as a conformationally defined XPP tripeptide mimetic locked in a polyproline type 2 (PPII) helix conformation. We also demonstrated the applicability of 1 in solution phase peptide synthesis. In the course of the work, a practical asymmetric synthesis of the bridged bicyclic chiral building block 11 was elaborated. Further studies are now required to explore whether ProM-19-based ligands offer advantages over those derived from previously developed PPII helix-inducing diproline equivalents,9,10,14,24,25 due to the higher degree of structural preorganization within the tetracyclic core structure of ProM-19.Experimental
(R,E)-Ethyl 2-((1-phenylethyl)imino)acetate (6)
To a stirred solution of (R)-1-phenylethylamine (7.4 g, 61.1 mmol, 1.0 eq.) in 100 mL of dry toluene were added 31.2 g of anhydrous Na2SO4. Then, a solution of 12.5 g (61.2 mmol, 1.95 eq.) of ethyl glyoxylate in 15 mL of toluene was added dropwise and the mixture was stirred for 1.5 h at room temp. The solid was filtered off and the residue was washed with toluene. The organic phase was concentrated under reduced pressure and the residue dried in vacuum (oil pump). The product 6 (12.54 g, 61.1 mmol, 99%) was obtained as a slightly yellowish oil, which was used directly in the subsequent aza-Diels–Alder reaction. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.73 (d, J = 1.0 Hz, 1H, H-2), 7.39–7.08 (m, 5H, ar-H), 4.61 (q, J = 6.7 Hz, 1H, Ph-CH), 4.33 (q, J = 7.1 Hz, 2H, OCH2), 1.62 (d, J = 6.7 Hz, 3H, CH3), 1.34 (t, J = 7.1 Hz, 3H, CH3).(1S,3S,4R)-Ethyl 2-((R)-1-phenylethyl)-2-azabicyclo[2.2.1]-hept-5-ene-3-carboxylate (7a) and its C-3-epimer (7b)
A solution of 12.54 g of the imine 6 (61.10 mmol, 1.00 eq.) in 100 mL of DMF was cooled to 0 °C before 5.0 mL of TFA (64.9 mmol, 1.05 eq.), 13.0 mL of freshly distilled cyclopentadiene (157 mmol, 2.60 eq.) and 33 μL of water (1.83 mmol, 0.03 eq.) were added. The mixture was then stirred under light exclusion for 24 h at room temperature. Then, 50 mL of a sat. aqueous NaHCO3 solution were added and the pH adjusted to 8 by addition of solid Na2CO3 before the mixture was extracted with 4 × 100 mL of MTBE. The combined organic phases were dried with MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica (EtOAc/CyHex = 1:8) to afford 10.68 g (39.35 mmol, 64%) of 7a (39.35 mmol, 64%) and 1.22 g of 7b (4.48 mmol, 7%), both as yellowish oils. Data for7a: TLC: Rf = 0.21 (CyHex/EtOAc = 10:1). 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.36–7.09 (m, 5H, H-Ar), 6.46–6.34 (m, 1H, H-5), 6.26 (dd, J = 5.6, 1.6 Hz, 1H, H-6), 4.29 (s, 1H, H-1), 3.80 (m, 2H, OCH2), 3.03 (q, J = 6.5 Hz, 1H, H-8), 2.89 (s, 1H, H-4), 2.20 (s, 1H, H-3), 2.13 (d, J = 8.3 Hz, 1H, H-7), 1.40 (d, J = 6.6 Hz, 4H, H-7, CH3), 0.94 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 174.3 (CO), 144.9 (ar-C), 136.4 (C-5), 132.9 (C-6), 128.0 (ar-C), 127.9 (ar-C), 127.0 (ar-C), 65.0 (C-3), 63.9 (benz-C), 62.6 (C-1), 60.2 (OCH2), 49.1 (C-4), 45.3 (C-7), 22.5 (CH3), 14.3; 14.0 (CH3). IR (FT-ATR):ṽ (cm−1): 2976 (m), 1743 (s), 1723 (m), 1454 (m), 1376 (m), 1193 (m), 1163 (s), 1108 (m), 1058 (m), 1034 (m), 701 (s). GC-MS:m/z = 271 [M]+ (1), 204 (9), 176 (21), 160 (16), 131 (15), 105 (100), 91 (11), 77 (16), 51 (5). [α]20 (c = 0.63, CHCl3): −328.8° (365 nm), −178.7° (436 nm), −94.7° (546 nm), −81.8° (579 nm), −78.3° (589 nm). Data for7b: TLC:Rf = 0.39 (CyHex/EtOAc = 10:1). 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.49–7.08 (m, 5H, ar-H), 6.40 (ddd, J = 5.6, 3.1, 1.2 Hz, 1H, H-5), 6.03 (dd, J = 5.6, 2.0 Hz, 1H, H-6), 4.24 (q, J = 7.1 Hz, 2H, OCH2), 3.53 (td, J = 3.1, 1.4 Hz, 1H, H-1), 3.13–3.07 (m, 1H, H-4), 3.03 (q, J = 6.5 Hz, 1H, benz-H), 2.46 (s, 1H, H-3), 1.92 (dt, J = 8.2, 1.7 Hz, 1H, H-7), 1.31 (t, J = 7.1 Hz, 3H, CH3), 1.23 (ψd, J = 6.4 Hz, 4H, CH3 + H-7). 13C NMR (75 MHz, CDCl3): δ (ppm) = 174.6 (CO), 144.9 (ar-C), 135.8 (d, C-5), 133.6 (d, C-6), 128.2 (ar-H); 127.4 (ar-H), 126.9 (ar-H), 64.2 (C-3), 63.4 (d, benz-C), 63.3 (C-1), 60.5 (OCH2), 49.5 (C-4), 45.6 (C-7), 23.7 (CH3), 14.2 (CH3). IR (FT-ATR):ṽ (cm−1): 2977 (m), 1779 (w), 1747 (s), 1723 (m), 1454 (m), 1191 (m), 1172 (s), 1164 (s), 1109 (m), 1053 (m), 1028 (m), 702 (s). GC-MS:m/z = 271 [M]+ (1), 204 (10), 176 (20), 160 (16), 131 (14), 105 (100), 91 (11), 77 (15), 51 (5). [α]20 (c = 0.29, CHCl3) = 406.3° (365 nm), 230.8° (436 nm), 126.0° (546 nm), 109.0° (579 nm), 124.3° (589 nm).
(1S,3S,4S,5S,6R) Ethyl 5,6-dihydroxy-2-((R)-1-phenylethyl)-2-aza-bicyclo[2.2.1]heptane-3-carboxylate (8)
To a solution of 5.5 g of 7a (20.3 mmol, 1.0 eq.) in 120 mL of tBuOH/H2O (1:1) were added 8.4 g of K2CO3 (60.8 mmol, 3.0 eq.), 20.0 g of K3Fe(CN)6 (60.8 mmol, 3.0 eq.), and 37 mg of K2OsO2 × 2H2O (0.005 mmol). The mixture was stirred at room temp. until TLC control indicated full conversion. After addition of water (150 mL) the mixture was extracted with MTBE (3 × 150 mL) and the combined organic layers were washed with sat. aqueous NaCl solution. After removing the solvent under reduced pressure, the residue (yellowish oil) was purified by flash column chromatography on silica gel (EtOAc/CyHex = 1.5:1) and dried in an oil pump vacuum to give 5.88 g of diol 8 (19.3 mmol, 95%) as a colorless oil. TLC: Rf = 0.23 (CyHex/EtOAc = 3:2). 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.35–7.12 (m, 5H, ar-H), 4.31 (d, J = 5.7 Hz, 1H, H-6), 3.85 (d, J = 5.7 Hz, 1H, H-5), 3.76–3.62 (m, 2H, OCH2), 3.60–3.54 (m, 2H, H-1, H-8), 2.50 (s, 1H, H-3), 2.25 (s, 1H, H-1), 1.96 (d, J = 10.7 Hz, 1H, H-7), 1.80 (d, J = 10.6 Hz, 1H, H-7), 1.45 (d, J = 6.5 Hz, 3H, benz-CH3), 0.93 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 173.5 (CO), 143.9 (ar-C), 128.1 (ar-C); 127.9 (ar-C); 127.4 (ar-C), 73.3 (C-5), 67.3 (C-6), 65.6 (C-3), 61.7 (C-1), 60.4 (OCH2), 60.2 (benz-C), 48.9 (C-4), 29.6 (C-7), 22.3 (benz-CH3), 13.9 (CH3). IR (FT-ATR): ṽ (cm−1): 3410 (br), 1739 (s), 1199 (s), 1178 (s), 1144 (m), 1082 (s), 1058 (m), 1031 (s). GC-MS:m/z = 375 [M]+ (17), 290 (13), 244 (74), 232 (54), 214 (4), 184 (22), 172 (37), 140 (36), 105 (100), 79 (15), 68 (34). [α]20 (c = 0.63, CHCl3) = 16.2° (365 nm), 11.8° (436 nm), 7.8° (546 nm), 7.4° (579 nm), 7.3° (589 nm).
2-(tert-Butyl) 3-ethyl (1S,3S,4S,5S,6R)-5,6-dihydroxy-2-aza-bi-cyclo[2.2.1]heptane-2,3-dicarboxylate (9)
To a solution of diol 8 (7.5 g, 24.56 mmol, 1.0 eq.) in 75 mL of dry methanol was added 750 mg of Pd/C (10% Pd) under an atmosphere of argon. Then, the flask was flushed with hydrogen and a slow stream of hydrogen was constantly passed through the stirred suspension using a steel cannula and a balloon. After TLC control indicated complete conversion (1–5 d) the mixture was filtered through a pad of Celite and the solvent was removed under reduced pressure. The crude product was dissolved in 75 mL of CH2Cl2 and 8.1 g of Boc2O (36.8 mmol, 1.5 eq.) were added. The mixture was stirred for 24 h at room temp. Then, all volatiles were removed under reduced pressure and the residue was purified by flash column chromatography on silica (CH2Cl2/MeOH = 30:1) to give 6.4 g of 9 (21.2 mmol, 87%) as a colorless crystalline solid. TLC: Rf = 0.30 (CH2Cl2/MeOH = 20:1). 1H NMR (600 MHz, CDCl3, mixture of rotamers): δ (ppm) = 4.34 (d, J = 4.7 Hz, 0.7H, OHrot1), 4.23–4.15 (m, 2H, OCH2), 4.13 (s, 0.7H, H-1rot1), 4.05 (s, 0.3H, H-1rot2), 4.00–3.98 (m, 0.3H, H-5rot2), 3.95 (t, J = 5.1 Hz, 0.7H, H-6rot1), 3.92 (t, J = 4.9 Hz, 0.7H, H-5rot1), 3.87–3.86 (m, 0.3H, H-6rot2), 3.70 (s, 0.3H, H-3rot2), 3.64 (d, J = 4.7 Hz, 0.7H, -OHrot1), 3.60 (s, 0.7H, H-3rot1), 3.27 (d, J = 4.7 Hz, 0.3H, OHrot2), 3.21 (d, J = 5.1 Hz, 0.3H, OHrot2), 2.57–2.55 (m, 1H, H-4), 1.86–1.79 (m, 1H, H-7), 1.82 (br, 1H, H-7), 1.46 (s, 2.70H, tert-Burot2), 1.38 (s, 6.3H, tert-Burot1), 1.28 and 1.26 (2 × t, J = 7.1, 3H, Me). 13C NMR (151 MHz, CDCl3): δ (ppm) = 170.4/170.3 (COester), 153.8/153.5 (COBoc), 80.9/80.6 (O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) Me3), 73.0/72.6 (C-5), 72.2/71.0 (C-6), 61.3/61.2 (OCH2), 60.2/59.7 (C-3), 60.2/59.2 (C-1), 48.1/47.6 (C-4), 28.7/28.4 (C-7), 28.2/28.0 (C(H3)3), 14.3/14.1 (CH3). IR (FT-ATR): ṽ (cm−1): 3397 (br), 1749 (m), 1701 (s), 1677 (s), 1404 (s), 1368 (m), 1160 (s). GC-MS: m/z = 301 [M]+ (2), 245 (3), 228 (7), 200 (16), 184 (10), 172 (14), 165 (4), 154 (4), 140 (100), 128 (20), 110 (18), 96 (6), 80 (10), 68 (67), 57 (70), 41 (36). [α]20 (c = 0.57, CHCl3) = −165.5° (365 nm), −102.5° (436 nm), −59.1° (546 nm), −51.5° (579 nm), −55.5° (589 nm). M.p.: 147.5–148.5 °C (from CH2Cl2/MeOH).
Me3), 73.0/72.6 (C-5), 72.2/71.0 (C-6), 61.3/61.2 (OCH2), 60.2/59.7 (C-3), 60.2/59.2 (C-1), 48.1/47.6 (C-4), 28.7/28.4 (C-7), 28.2/28.0 (C(H3)3), 14.3/14.1 (CH3). IR (FT-ATR): ṽ (cm−1): 3397 (br), 1749 (m), 1701 (s), 1677 (s), 1404 (s), 1368 (m), 1160 (s). GC-MS: m/z = 301 [M]+ (2), 245 (3), 228 (7), 200 (16), 184 (10), 172 (14), 165 (4), 154 (4), 140 (100), 128 (20), 110 (18), 96 (6), 80 (10), 68 (67), 57 (70), 41 (36). [α]20 (c = 0.57, CHCl3) = −165.5° (365 nm), −102.5° (436 nm), −59.1° (546 nm), −51.5° (579 nm), −55.5° (589 nm). M.p.: 147.5–148.5 °C (from CH2Cl2/MeOH).
2-(tert-Butyl) 3-ethyl (1S,3S,4S,5S,6R)-5,6-thiooxodioxolo-2-aza-bicyclo[2.2.1]heptane-2,3-dicarboxylate (10)
A solution of diol 9 (6.40 g, 21.2 mmol, 1.0 eq.) and 6.23 g of DMAP (50.98 mmol, 2.4 eq.) in 106 mL of dry CH2Cl2 was cooled to 0 °C before 1.95 mL of thiophosgene (25.5 mmol, 1.2 eq.) were added dropwise and the mixture was stirred for 18 h at room temp. For workup, the mixture was diluted with 50 mL of CH2Cl2 and washed with 1 N HCl aqueous solution (2 × 50 mL). The aqueous layer was back-extracted with 50 mL CH2Cl2. The combined organic solutions were dried with MgSO4 and the solvent was removed under reduced pressure. The crude product obtained was purified by flash column chromatography on silica (CH2Cl2/MeOH = 100:1) to afford 5.75 g of thiocarbonate 10 (16.8 mmol, 79%) as a yellowish solid. TLC: Rf = 0.29 (CyHex/EtOAc = 3:1). 1H NMR (600 MHz, CDCl3, mixture of rotamers): δ (ppm) = 5.02–4.99 (m, 1H, H-5), 4.94 (m, H-6rot1), 4.84 (m, 0.5H, H-6rot2), 4.62 (s, 0.5H, H-1rot1), 4.5 (s, 0.48H, H-1rot2), 4.28–4.15 (m, 2H, OCH2), 3.75 (s, 0.5H, H-3rot2), 3.66 (s, 0.5H, H-3rot1), 3.06–3.02 (m, 1H, H-4), 2.19 (m, 1H, H-7), 1.72 (dd, J = 12.0, 5.7 Hz, 1H, H-7), 1.47 (s, 4.5H, tert-Bu), 1.40 (s, 4.5H, tert-Bu), 1.33–1.25 (m, 3H, CH3). 13C NMR (151 MHz, CDCl3): δ (ppm) = 191.0/190.8 (CS), 169.0/168.7 (COester), 152.9/152.3 (COBoc), 85.2/85.1 (C-6), 83.4/83.3 (C-5), 81.9/81.7 (Me3), 62.0/61.9 (OCH2), 58.1/57.7 (d, C-3), 57.9/56.7 (C-1), 46.2/45.4 (C-4), 29.7/27.4 (C-7), 28.2/28.1 (C(H3)3), 14.2/14.1 (CH3). IR (FT-ATR): ṽ (cm−1): 1748 (m), 1703 (s), 1395 (s), 1367 (m), 1346 (m), 1296 (s), 1161 (s), 1119 (m); GC-MS: same as for compound 11 due to rapid thermal fragmentation. [α]20 (c = 0.505, CHCl3) = −192.9° (365 nm), −105.1° (436 nm), −59.4° (546 nm), −51.7° (579 nm), −55.4° (589 nm). M.p.: 71.0–72.0 °C (from CH2Cl2/MeOH).
2-(tert-Butyl) 3-ethyl (1S,3S,4R)-2-azabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (11)
A solution of 5.70 g (16.6 mmol, 1.0 eq.) of thiocarbonate 10 in 30 mL of P(OMe)3 was refluxed under argon (at 125 °C oil bath temp.) for 2 d. After cooling to room temp. the flask was connected to a cold trap (cooled with liquid N2) and an oil pump vacuum was applied until all excess P(OMe)3 had condensed into the external cold trap (to allow proper disposal). The remaining crude product was purified by flash column chromatography on silica gel (CyHex:EtOAc, 7:1) to give 4.04 g of olefin 11 (15.11 mmol, 91%) as a clear oil. TLC: Rf = 0.53 (CyHex/EtOAc, 3:1). 1H NMR (500 MHz, CDCl3; mixture of rotamers): δ (ppm) = 6.47 (br, 0.6H, H-5rot1), 6.37–6.35 (m, 1.5H, H-5rot2/H-6), 4.77 (s, 0.6H, H-1rot1), 4.64 (s, 0.5H, H-1rot2), 4.19 (m, 2H, H-12), 3.46 (s, 0.5H, H-3rot2), 3.37 (s, 0.6H, H-3rot1), 3.25 (s, 1H, H-4), 1.96 (dt, J = 8.6, 1.8 Hz, 1H, H-7), 1.45–1.43 (m, 1H, H-7), 1.41–1.36 (m, 9H, H-10), 1.26 (t, J = 7.2 Hz, 3H, H-13). 13C NMR (125 MHz, CDCl3, mixture of rotamers): δ (ppm) = 171.6 (s, COester), 155.7/154.6 (q, COBoc), 137.1 (d, C-5); 136.8/136.2 (d, C-6), 79.8 (s, Me3), 61.8/60.3 (d, C-1), 61.1/61.0 (t, O-CH2), 59.0 (d, C-3), 48.9/48.2 (d, C-4), 45.4/45.1 (t, C-7), 28.3 (q, C(H3)3), 14.2/14.1 (q, CH3). IR (FT-ATR): ṽ (cm−1): 1748 (m), 1697 (s), 1388 (s), 1365 (s), 1159 (s), 1122 (s). GC-MS: m/z = 267 [M]+ (7), 211 (6), 194 (4), 167 (10), 151 (21), 138 (82), 102 (18), 94 (100), 67 (15), 57 (59), 41 (16). [α]20 (c = 0.96, CHCl3) = −781.0° (365 nm), −461.7° (436 nm), −257.3° (546 nm), −223.6° (579 nm), −217.2° (589 nm).
1-(tert-Butyl) 2-ethyl (2S,3R,5S)-3,5-divinyl-pyrrolidin-1,2-dicarboxylate (12)
A solution of 800 mg of olefin 11 (2.99 mmol, 1.00 eq.) in 250 mL of dry CH2Cl2 was prepared under argon and ethene was passed through the solution (from a balloon as reservoir). After 5 min, 127 mg of Grubbs II catalyst (0.15 mmol, 0.05 eq.) were added and a constant small flow of ethene was passed through the solution for 4 h. The solution was then concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (CyHex:EtOAc, 5:1) to give 12 (613 mg, 2.08 mmol, 70%) as a clear oil. TLC: Rf = 0.34 (CyHex/EtOAc, 5:1). 1H NMR (500 MHz, CDCl3; mixture of rotamers): δ (ppm) = 5.85–5.76 (m, 1.6H, H-12, H-14rot1), 5.70 (ddd, J = 17.4, 10.2, 7.4 Hz, 0.4H, H-14rot2), 5.16–4.96 (m, 4H, H-13, H-15), 4.41 (q, J = 7.0 Hz, 0.6H, H-5rot1), 4.31 (q, J = 7.1 Hz, 0.4H, H-5rot2), 4.23–4.07 (m, 2.4H, H-2rot1, H-10), 4.04 (d, J = 5.2 Hz, 0.6H, H-2rot2), 2.78 (p, J = 6.8 Hz, 1H, H-3), 2.36–2.26 (m, 1H, H-4), 1.66–1.59 (m, 1H, H-4), 1.36 (m, 9H, H-8), 1.22 (dt, J = 11.1, 7.1 Hz, 3H, H-11). 13C NMR (125 MHz, CDCl3): δ (ppm) = 172.3; 171.9 (s, COester), 154.3; 153.4 (s, COBoc), 140.17 139.4 (d, –CH at C-5), 138.1 (d, –CH at C-3), 116.0/115.9 (t, CH2), 114.5/114.2 (t, CH2), 80.1; 80.0 (s, Me3), 65.5/65.1 (d, C-2), 60.8 (t, O-CH2), 60.7/60.6 (d, C-5), 46.7/45.7 (d, C-3), 38.2/37.6 (t, C-4), 28.1/28.1 (q, C(H3)3), 14.2/14.1 (q, CH3). IR (FT-ATR:ṽ (cm−1): 1744 (s), 1694 (s), 1365 (s), 1254 (m), 1184 (s). GC-MS: m/z = 295 [M]+ (1), 239 (3), 222 (17), 194 (58), 166 (98), 148 (2), 122 (100), 105 (8), 94 (8), 77 (8), 67 (16), 57 (74), 41 (39). [α]20 (c = 0.39, CHCl3) = 48.9° (365 nm), 21.0° (436 nm), 8.1° (546 nm), 6.1° (579 nm), 4.6° (589 nm).
(2S,3R,5S)-1-(tert-Butoxycarbonyl)-3,5-divinyl-pyrrolidin-2-carboxylic acid (13)
To a solution of 613 mg of ethyl ester 12 (2.08 mmol, 1.0 eq.) in 20 mL of THF/MeOH (3:1) were added 4 mL of a 2.5 N LiOH solution (10 mmol, 10.0 eq.) and the mixture was stirred for 36 h at room temperature. Then, 10 mL of CH2Cl2 was added and the mixture was concentrated under reduced pressure. The aqueous phase was brought to pH = 1 with aqueous 1 N HCl solution and extracted with CH2Cl2 (4 × 25 mL). The combined organic layers were dried over MgSO4 and all volatiles were removed under reduced pressure to yield 556 mg of carboxylic acid 13 (2.08 mmol, 100%) as a colorless highly viscous oil. TLC: Rf = 0.51 (CyHex/EtOAc/HOAc, 100:100:5). 1H NMR (600 MHz, CDCl3; mixture of rotamers): δ (ppm) = 9.96 (br, 1H, COOH), 5.86 (ddd, J = 17.4, 10.3, 7.6 Hz, 1H, H-10), 5.86–5.80 (m, 0.6 H, H-10rot2), 5.75 (ddd, J = 17.6, 10.2, 7.4 Hz, 0.4H, H-10rot1), 5.22–4.99 (m, 4H, H-11, H-13), 4.45 (q, J = 7.1 Hz, 0.6H, H-5rot2), 4.34 (q, J = 7.2 Hz, 0.4H, H-5rot1), 4.23 (d, J = 4.7 Hz, 0.4H, H-2rot1), 4.11 (d, J = 5.3 Hz, 0.6H, H-2rot2), 2.99–2.88 (m, 1H, H-3), 2.41 (dt, J = 14.1, 7.7 Hz, 0.4H, H-4rot1), 2.35 (dt, J = 13.8, 7.4 Hz, 0.6H, H-4rot2), 1.79–1.64 (m, 1H, H-4), 1.41; 1.40 (2 × s, 9H, tBu). 13C NMR (151 MHz, CDCl3): δ (ppm) = 178.6; 177.0 (s, COacid), 155.0; 153.4 (s, COBoc), 139.9; 139.1 (d, CH), 138.0; 137.8 (d, CH), 116.5; 116.3 (t, CH2), 114.9; 114.6 (t, CH2), 80.8 (s, Me3), 65.5; 65.0 (d, C-2), 60.9 (d, C-5), 46.7; 45.4 (d, C-3), 38.3; 37.8 (t, C-4), 28.2 (q, C(H3)3). IR (FT-ATR):ṽ (cm−1): 3054 (br), 2979 (m), 1745 (m), 1712 (s), 1693 (s), 1645 (m), 1392 (s), 1367 (s), 1307 (m), 1254 (m), 1164 (s). HRMS (ESI): calcd for [M + Na]+ 290.1363; found 290.1363. [α]20 (c = 0.895, CHCl3) = +33.9° (365 nm), +14.8° (436 nm), +5.7° (546 nm), +4.5° (579 nm), +3.9° (589 nm).
tert-Butyl (2S,3R,5S)-2-((2S,5R)-2-(methoxycarbonyl)-5-vinyl-pyrrolidin-1-carbonyl)-3,5-divinylpyrrolidin-1-carboxylate (14)
In an inert Schlenk flask, 1.85 g of acid 13 (6.92 mmol, 1.0 eq.) was dissolved in 25 mL of dry acetonitrile and 1.07 g of amine 5 (6.92 mmol, 1.0 eq.) was added. Subsequently, 4.68 g of PyBOP (9.00 mmol, 1.3 eq.) and 3.54 mL of DIPEA (20.8 mmol, 3.0 eq.) were added successively. The reaction mixture was stirred for 18 h at room temp. before dilution with 50 mL of water and extraction three times with 80 mL of MTBE each. The combined organic phases were dried over MgSO4 and freed from solvent under reduced pressure. The crude product obtained was subsequently purified by flash column chromatography on silica gel (CyHex:EtOAc, 2:3). 1.68 g of dipeptide 14 (4.16 mmol, 95%) was obtained in the form of a yellowish oil. TLC: Rf = 0.36 (CyHex/EtOAc, 2:3). 1H NMR (600 MHz, CDCl3; mixture of rotamers): δ (ppm) = 5.95–5.88 (m, 1H), 5.87–5.75 (m, 1H,), 5.82–5.74 (m, 1H), 5.45 (m, 1H), 5.13 (m, 1H), 5.02 (m, 1H), 5.02 (m, 1H), 4.97 (m, 2H), 4.90–4.84 (t, 0.65H), 4.57–4.55 (m, 0.7H), 4.52–4.47 (m, 1H), 4.41–4.38 (m, 0.65H), 4.34 (m, 0.65H), 4.30 (m), 3.74; 3.72 (2 × s, 3H), 2.85–2.79 (m, 1H), 2.73–2.64 (m, 1H), 2.27–2.11 (m, 2H), 2.01–1.80 (m, 2H), 1.64–1.60 (m, 1H), 1.39; 1.37 (2 × s, 9H). 13C NMR (125 MHz, CDCl3): δ (ppm) = 172.8/172.6; 172.5/172.2, 154.9/153.9, 141.0/140.5, 139.9/139.7, 138.6/138.2, 117.2/116.7, 115.1, 114.1/113.8, 79.8, 63.5/63.2, 61.2/61.0, 60.8, 60.7, 60.0/59.8, 52.2/52.0 46.7/45.7, 37.5/36.2, 32.9/32.6, 28.5/28.3/28.2, 26.9/26.9. For the assignment of NMR signals, see the ESI.†IR (FT-ATR): ṽ (cm−1): 2978 (m), 1750 (s), 1709 (s), 1686 (s), 1657 (s), 1422 (s), 1389 (s), 1366 (s), 1201 (s), 1172 (s). GC-MS: m/z = 404 [M]+ (1), 331 (5), 303 (85), 277 (1), 236 (7), 222 (7), 194 (4), 166 (87), 156 (12), 154 (22), 122 (100), 96 (23), 79 (18), 67 (18), 57 (56), 41 (43). [α]20 (c = 0.51, CHCl3) = 42.2° (365 nm), 21.6° (436 nm), 10.0° (546 nm), 8.3° (579 nm), 6.8° (589 nm). For the assignments of NMR signals, see the ESI.†
1-(tert-Butyl) 8-methyl (2S,3aR,5aR,8S,10aS)-10-oxo-2-vinyl-3,3a,5a,6,7,8,10,10a-octahydrodipyrrolo[1,2-a:3′,2′-e]azepin-1,8(2H)-dicarboxylate (15)
Under an atmosphere of argon, 532 mg of dipeptide 14 (1.32 mmol, 1.0 eq.) were dissolved in 250 mL of absolute CH2Cl2 (distilled from P4O10 and filtered through a pad of basic alumina (activity level 1). Then 56 mg (5 mol%) of Grubbs II catalyst was added and the mixture was heated to 40 °C for 48 h. During this time, additional batches of Grubbs II catalyst (typically 22 mg, 2 mol%) were added every 2 h until a total amount of 20 mol% was reached. For work-up, the reaction solution was concentrated under reduced pressure and the crude product purified by flash column chromatography on silica gel (CyHex:EtOAc, 1:3) to yield 288 mg of 15 (0.77 mmol, 58%) as a grayish solid. TLC: Rf = 0.22 (CyHex/EtOAc, 1:1). 1H NMR (500 MHz, CDCl3; mixture of rotamers): δ (ppm) 5.85–5.74 (m, 1.25H), 5.66 (ddd, J = 17.0, 10.1, 8.3 Hz, 0.75H), 5.55 (ddd, J = 11.2, 2.9, 1.6 Hz, 1H), 5.26–4.97 (m, 2H), 4.80 (dd, J = 7.8, 2.8 Hz, 0.75H), 4.73–4.62 (m, 1.25H), 4.50 (dd, J = 25.5, 10.6 Hz, 1H), 4.32 (ddd, J = 10.5, 8.1, 5.6 Hz, 0.25H), 4.25 (ddd, J = 10.3, 8.3, 5.7 Hz, 0.75H), 3.69 (s, 0.75H), 3.68 (s, 2.25H), 2.99–2.85 (m, 1H), 2.39–2.25 (m, 1H), 2.28–2.17 (m, 1H), 2.11–1.97 (m, 2H), 1.96–1.80 (m, 1H), 1.59–1.46 (m, 1H), 1.41 (s, 6H), 1.38 (s, 2H). 13C NMR (100 MHz, CDCl3; mixture of rotamers): δ (ppm) = 172.5; 172.3, 169.7/169.1, 154.2/153.0, 140.0/138.8, 129.6/129.3, 128.9/128.6, 115.1/114.7, 80.0/79.6, 63.2/62.8, 61.8/61.5, 59.5/59.4, 57.1, 52.3/52.1, 39.7/39.5, 39.2/39.1, 33.1/33.0, 28.3/28.1, 27.2/27.0. IR (FT-ATR): ṽ (cm−1): 1701 (s), 1658 (s), 1427 (m), 1401 (m), 1384 (m), 1364 (m), 1319 (m), (s), 1167 (s). GC-MS: m/z = 376 [M]+ (1), 320 (38), 303 (34), 276 (100), 261 (24), 247 (34), 207 (20), 189 (27), 175 (12), 134 (20), 120 (27), 108 (19), 94 (22), 80 (19), 57 (42), 41 (73). HRMS(ESI): calcd for [M + H]+ 377.2071; found 377.2077, calcd for [M + Na]+ 399.1890; found 399.1890. [α]20 (c = 0.65, MeOH) = −448.3° (436 nm), −266.6° (546 nm), −234.0° (579 nm), −224.8° (589 nm). M.p.: 166–167 °C (CH2Cl2). For the assignments of NMR signals, see the ESI.†
1-(tert-Butyl)-8-methyl-(2S,3aR,5aR,8S,10aS)-10-oxo-2-vinyl-3,3a,5a,6,7,8,10,10a-octahydrodipyrrolo[1,2-a:3′,2′-e]azepin-1,8(2H)-dicarboxylate (16)
A solution of 390 mg of tricycle 15 (1.04 mmol, 1.0 eq.) in 7 mL of dry CH2Cl2 was cooled to 0 °C before 1.6 mL of TFA (20.7 mmol, 20 eq.) were added dropwise. The mixture was stirred at room temp. for 1.5 h before all volatiles were removed under oil pump vacuum. The residue obtained was taken up twice in 3 mL of CH2Cl2 followed by solvent removal in vacuum. Finally, the residue was dissolved in 4 mL of CH2Cl2 and little solid Na2CO3 was added to neutralize any remaining acid. In a second flask, 336 mg of Boc-L-allylglycine-OH (3) (1.56 mmol, 1.5 eq.) were dissolved in 5 mL of dry acetonitrile before HATU (692 mg, 1.82 mmol, 1.75 eq.) and DIPEA (1.55 mmol, 1.5 eq.) were added. To this mixture was then transferred at room temp. by means of a syringe needle the above-prepared CH2Cl2 solution of the deprotected amine (rinsing the Na2CO3 residue with 2 mL of CH2Cl2). After addition of 265 μL of DIPEA (1.55 mmol, 1.5 eq.) and 10 mg of solid Na2CO3 the mixture was stirred for 18 h at room temp. before it was filtered through a short pad of Celite and rinsed with 50 mL of CH2Cl2/MeOH (30:1). Then, the solvents were removed under reduced pressure and the crude product was purified by flash column chromatography on silica gel (CH2Cl2/MeOH, 30:1) to yield 338 mg of 16 (0.71 mmol, 69%) as a yellow foam. TLC: Rf = 0.53 (EtOAc). 1H NMR (500 MHz, CDCl3, data for main rotamer): δ (ppm) = 5.97–5.84 (m, 1.4H), 5.79 (dt, J = 11.2, 2.2 Hz, 1H), 5.75–5.64 (m, 0.6H), 5.63–5.54 (m, 1H), 5.43–5.22 (m, 2H), 5.18–5.04 (m, 2H), 4.93–4.89 (m, 1H), 4.78–4.76 (m, 1H), 4.74–4.70 (m, 2H), 4.42–4.37 (m, 1H), 3.74–3.67 (m, 3H), 2.99–2.91 (m, 1H), 2.55–2.50 (m, 1H), 2.41–2.31 (m, 3H), 2.11–2.00 (m, 2H), 1.91–1.80 (m, 1H), 1.70–1.65 (m, 1H). 13C NMR (125 MHz, CDCl3, data for main rotamer): δ (ppm) = 172.3, 172.1, 168.4, 155.5, 137.9, 133.5, 130.0, 128.1, 118.2, 79.0, 63.9, 62.9, 59.3, 57.2, 52.3, 51.7, 40.9, 38.1, 37.1, 32.8, 28.4, 27.2. IR (FT-ATR): ṽ (cm−1): 3335 (br), 3977 (w), 1705 (m), 1673 (m), 1645 (m), 1501 (w), 1434 (m), 1164 (s). HRMS(ESI): calcd for [M + H]+ 474.2598; found 474.2601, calcd for [M + Na]+ 496.2418; found 496.2418. [α]20 (c = 0.68, CHCl3) = −241.1° (436 nm), −138.0° (546 nm), −120.8° (579 nm), −115.8° (589 nm). For the assignments of NMR signals, see the ESI.†
Boc-[ProM-19]-OMe (1)
Under an atmosphere of argon, a solution of 338 mg of 16 (0.71 mmol, 1.0 eq.) in 50 mL of dry hexafluorobenzene was heated to 45 °C before 10 mol% of Grubbs II catalyst (dissolved in hexafluorobenzene) was slowly added over 2 h. After stirring for another 2 h the solvent was removed under reduced pressure and the residue purified by flash column chromatography on silica gel (CH2Cl2/MeOH, 30:1). The product was purified once again by flash column chromatography (CH2Cl2/MeOH, 25:1). To remove traces of Ru, the obtained gray foam was dissolved in 5 mL of a mixture of CH2Cl2/MeOH, 20:1 and stirred with Quadrasil AP for 1 h. After filtration and rinsing with 20 mL of CH2Cl2/MeOH (20:1) the combined organic solutions were concentrated under reduced pressure and the product was dried in oil pump vacuum to give 277 mg of pure 1 (0.61 mmol, 88%) as a still slightly grayish foam. TLC: Rf = 0.21 (CH2Cl2/MeOH, 20:1). 1H NMR (500 MHz, CDCl3): δ (ppm) = 5.86–5.79 (m, 2H), 5.70–5.65 (m, 1H), 5.61–5.56 (m, 1H), 4.78–4.67 (m, 3H), 4.64–4.59 (m, 1H), 4.34–4.29 (m, 1H), 2.99–2.90 (m, 1H), 2.73 (br, 1H), 2.61–2.54 (1H), 2.42 (dt, J = 11.6, 5.7 Hz, 1H), 2.36–2.29 (m, 1H), 2.17–2.00 (m, 2H), 1.95–1.80 (m, 1H), 1.72–1.63 (m, 1H), 1.45 (s, 10H). 13C-NMR (125 MHz, CDCl3): δ (ppm) = 172.5, 170.8, 168.7, 155.4, 130.0, 128.8, 128.0, 79.7, 64.6, 59.4, 57.5, 57.3, 52.3, 40.1, 37.5, 33.0, 29.2, 28.3, 27.0. IR (FT-ATR):ṽ (cm−1): 3410 (br), 1651 (s), 1433 (s), 1168 (s). HRMS(ESI): calcd for [M + Na]+ 446.2286; found 446.2289, calcd for [M + Na]+ 468.2105; found 468.2104. [α]20 (c = 0.5, CHCl3) = −217.1° (436 nm), −128.9° (546 nm), −113.6° (579 nm), −109.5° (589 nm). For the assignments of NMR signals, see the ESI.†
Peptide 17
Under an atmosphere of argon, a solution of 50 mg of 1 (112 μmol, 1.0 eq.) in 5.0 mL of dry CH2Cl2 was cooled to 0 °C before 20 μL of TMSOTf (112 μmol, 1.0 eq.) were added. Stirring was continued at 0 °C until TLC control indicated complete cleavage of the Boc protecting group. Then, 1 mL of sat. aqueous NaHCO3 was added and the mixture was extracted four times with 5 mL of CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure to a volume of ca. 2 mL. In a separate flask, 36 mg of N-Boc-proline (168 μmol, 1.5 eq.) were dissolved under argon in 3 mL of CH2Cl2 before 64 mg of HATU (168 μmol, 1.5 eq.) and 48 μL of DIPEA (280 μmol, 2.5 eq.) were added and stirring was continued for 1 h at room temp. To the resulting solution of the active ester was then dropwise added the amine prepared above and the mixture was stirred for 18 h at room temp. Then, the solvent was removed under reduced pressure and the residue was purified by flash column chromatography (CH2Cl2/MeOH, 20:1) to give 51 mg of the peptide 17 (94 μmol, 84%) as a yellowish foam. (Note: According to 1H NMR, the product was contaminated with some (≤5%) tetramethylurea which could not be completely separated off even after multiple chromatography.) TLC: Rf = 0.26 (CH2Cl2/MeOH, 20:1). 1H NMR (500 MHz, CDCl3, mixture of rotamers): δ (ppm) = 7.67 (d, J = 7.6 Hz, 1H), 6.04–5.55 (m, 4H), 4.82–4.15 (m, 6H), 3.69 (s, 3H), 3.54–3.22 (m, 2H), 2.98–2.90 (m, 1H), 2.71–1.61 (m, 12H), 1.42 (s, 9H). IR (FT-ATR): ṽ (cm−1): 3484 (br), 3413 (br), 3322 (br), 1678 (s), 1666 (s), 1645 (s), 1513 (s), 1433 (m), 1392 (s), 1365 (m), 1198 (m), 1163 (s). HRMS(ESI): calcd for [M + H]+ 543.2813; found: 543.2816, calcd for [M + Na]+ 565.2633; found 565.2626. [α]20 (c = 0.575, CHCl3) = −278.5° (436 nm), −161.2° (546 nm), −141.2° (579 nm), −136.0° (589 nm). For the assignments of NMR signals, see the ESI.†
Peptide 19
Under an atmosphere of argon, 50 mg of peptide 17 (92 μmol, 1.0 eq.) were dissolved in 2.5 mL of dry CH2Cl2. After cooling the solution to 0 °C, 17 μL of TMSOTf (92 μmol, 1.0 eq.) were added and stirring was continued to 0 °C until complete conversion was detected by TLC. Then, 1 mL of sat. aqueous NaHCO3 was added and the mixture was extracted four times with 5 mL of to CH2Cl2. The combined organic phases were dried over MgSO4 and concentrated under reduced pressure. To the amine residue was then added a solution of 135 mg of Fmoc-L-2-Cl-Phe-OPfp (18) (230 μmol, 2.5 eq.) in 5 mL of dry CH2Cl2 dropwise and the mixture was stirred at room temp. for 18 h. Then, the solvent was removed under reduced pressure and the crude product purified by flash column chromatography on silica (CH2Cl2/MeOH, 20:1) to yield 47 mg of 19 (55 μmol, 60%) as a colorless foam. TLC: Rf = 0.26 (CH2Cl2/MeOH, 20:1). 1H NMR (500 MHz, CDCl3, mixture of rotamers): δ (ppm) = 7.74–7.72 (m, 2H), 7.53–7.11 (m, 8.3H), 4.89 (td, J = 9.1, 5.3 Hz, 0.7H), 4.82–4.49 (m, 6.3H), 5.90–5.67 (m, 3.7H), 5.61–5.54 (m, 1H), 4.89 (td, J = 9.1, 5.3 Hz, 0.7H), 4.75–4.50 (m, 6.3H), 4.26 (dd, J = 10.5, 7.2 Hz, 1H), 4.15 (dd, J = 10.5, 7.4 Hz, 1H), 4.11–4.03 (m, 1H), 3.68 (s, 3H), 3.71–3.44 (m, 0.3H), 3.53–3.44 (m, 0.7H), 3.31 (dd, J = 13.3, 6.7 Hz, 0.3H), 3.20 (dd, J = 13.8, 5.3 Hz, 0.7H),3.08 (dd, J = 13.1, 7.7 Hz, 0.3H), 2.99 (dd, J = 13.8, 9.3 Hz, 0.7H), 2.96 (br, 1H), 2.67–2.55 (m, 2H), 2.47 (dt, J = 11.6, 5.7 Hz, 1H), 2.41–1.81 (m, 8H),1.69 (q, J = 12.2 Hz, 1H). 13C NMR (125 MHz, CDCl3, mixture of rotamers): δ (ppm) = 172.4, 171.2, 170.5, 170.1, 168.4, 155.6, 143.8/143.7, 141.1, 134.4/133.9, 131.9, 129.9/129.6/129.5/128.6/127.9, 127.6, 126.9, 126.8, 125.1, 125.0, 119.9, 66.9, 64.5, 60.0, 59.3, 57.3, 57.0, 53.6, 52.2, 51.9, 47.4, 47.0, 40.1, 37.4, 37.0, 32.9, 29.1, 27.5, 27.0, 25.0. IR (FT-ATR):ṽ (cm−1): 3292 (br), 1643 (s), 1515 (m), 711 (s). HRMS(ESI): calcd for [M + H]+ 846.3264; found: 846.3278, calcd for [M + Na]+ 868.3083; found 868.3092. [α]20 (c = 0.51, CHCl3) = −356.4° (365 nm), −213.3° (436 nm) −121.6° (546 nm), −106.2° (579 nm), −102.0 (589 nm). For the assignments of NMR signals, see the ESI.†
Ac-[2-Cl-Phe]-[Pro]-[ProM-19]-OMe (20)
Under an atmosphere of argon, 45 mg of peptide 19 (53.2 μmol, 1.0 eq.) were dissolved in 3.0 mL of dry acetonitrile before 0.21 mL of piperidine (2.12 mmol, 40 eq.) were added. After stirring the mixture for 1 h at room temperature the solvent was removed under reduced pressure. The residue was dissolved three times in 3.00 mL of dry CH2Cl2 and re-concentrated under reduced pressure. The resulting crude amine was taken up in 3 mL of dry CH2Cl2 and 0.20 mL of Ac2O (2.12 mmol, 40 eq.) were added. The mixture was stirred for 4 h at room temp. before the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (CH2Cl2/MeOH, 20:1 → 10:1) to give 25 mg of the target ligand 20 (37.5 μmol, 71%) as a colorless foam. TLC: Rf = 0.70 (CH2Cl2/MeOH, 10:1). 1H NMR (500 MHz, CDCl3, mixture of rotamers): δ (ppm) = 7.62 (d, J = 9.6 Hz, 0.3H), 7.43 (d, J = 7.2 Hz, 0.7 H), 7.37–7.32 (m, 0.7H), 7.23–7.15 (m, 2.6H), 7.15–7.08 (m, 0.7H), 6.98 (d, J = 8.5 Hz, 0.3H), 6.36 (d, J = 8.5 Hz, 0.7H), 5.90–5.79 (m, 2H), 5.78–5.68 (m, 1H), 5.65–5.55 (m, 1H), 5.09 (td, J = 8.7, 5.5 Hz, 0.7H), 5.01 (td, J = 9.7, 5.0 Hz, 0.3H), 4.80–4.67 (m, 4H), 4.61 (td, J = 7.2, 3.3 Hz, 0.7H), 4.56–4.48 (m, 1H), 4.28 (d, J = 8.1 Hz, 0.3H), 3.72 (q, J = 8.3 Hz, 0.7H), 3.67 (s, 2.1H), 3.59 (s, 0.9H), 3.56 (m, 0.6H), 3.50–3.40 (m, 1H), 3.15 (dd, J = 13.9, 5.5 Hz, 0.7H), 3.05–2.91 (m, 1.7H), 2.80 (dd, J = 13.6, 9.7 Hz, 0.3H), 2.70–2.52 (m, 2H), 2.48 (m, 1H), 2.41–2.27 (m, 2H), 2.25–1.80 (m, 8.1H), 1.77–1.63 (m, 1.9H). 13C NMR (125 MHz, CDCl3, mixture of rotamers): δ (ppm) = 172.4/171.8, 171.3/171.3, 170.5/170.4, 171.1/170.1, 170.1/169.5, 169.1/168.4, 135.4/134.0, 134.4/134.4, 132.0/131.7, 130.0/129.8, 129.5/129.5/129.3/128.6/128.1/127.9/127.8/127.5/126.8/126.2, 64.7/64.5, 60.7/60.1, 59.6/59.3, 57.7/57.4/57.1, 53.7/52.7, 52.4/52.2, 51.0/50.2, 47.5, 40.3/40.1, 37.4/37.3, 36.4/35.9, 32.9/32.8, 29.7/29.1, 27.5, 27.0, 25.0, 23.0, 22.2. IR (FT-ATR): ṽ (cm−1): 3298 (br), 1434 (s). HRMS(ESI): calcd for [M + H]+ 666.2689; found 666.2692, calcd for [M + Na]+ 688.2508; found 688.2508. [α]20 (c = 0.7, CHCl3) = −493.1° (436 nm), −280.2° (546 nm), −244.1° (579 nm), −234.6° (589 nm). For the assignments of NMR signals, see the ESI.†
Author contributions
Chemical syntheses and analyses were conducted by M. T. K. and B. M. K. X-ray crystallographic analyses were contributed by J.-M. N. The conceptualization of the project was performed by R. K. and H.-G. S. Based on a draft by M. T. K., the manuscript was written and polished mainly by H.-G. S. and B. M. K. All authors discussed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Federal Ministry of Education and Research (Project 16GW0187 “EnVision”). We gratefully acknowledge basic support by the University of Cologne.References
- (a) B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed; (b) O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Chem. Rev., 2008, 108, 1225–1244 CrossRef CAS PubMed; (c) D. Gonzalez-Ruiz and H. Gohlke, Curr. Med. Chem., 2006, 13, 2607–2625 CrossRef CAS PubMed; (d) Y. Pommier and J. Cherfils, Trends Pharmacol. Sci., 2005, 26, 138–145 CrossRef CAS PubMed.
- (a) L. Ball, R. Kühne, J. Schneider-Mergener and H. Oschkinat, Angew. Chem., Int. Ed., 2005, 44, 2852–2869 CrossRef CAS PubMed; (b) A. Zarrinpar, R. P. Bhattacharyya and W. A. Lim, Sci. Signal., 2003, 2003(179) DOI:10.1126/stke.2003.179.re8.
- (a) L. Ball, R. Kühne, B. Hoffmann, A. Häfner, P. Schmieder, R. Volkmer-Engert, M. Hof, M. Wahl, J. Schneider-Mergener, U. Walter, H. Oschkinat and T. Jarchau, EMBO J., 2000, 19, 4903–4914 CrossRef CAS PubMed; (b) R. Aasland, C. Abrams, C. Ampe, L. Ball, M. Bedford, G. Cesareni, M. Gimona, J. Hurley, T. Jarchau, V. Lehto, M. Lemmon, R. Linding, B. Mayer, M. Nagai, M. Sudol, U. Walter and S. Winder, FEBS Lett., 2002, 513, 141–144 CrossRef CAS PubMed; (c) L. Ball, T. Jarchau, H. Oschkinat and U. Walter, FEBS Lett., 2002, 513, 45–52 CrossRef CAS PubMed.
- I. Geisler and J. Chmielewski, Bioorg. Med. Chem. Lett., 2007, 17, 2765–2768 CrossRef CAS PubMed.
- (a) M. Dustin, M. Olszowy, A. Holdorf, J. Bromley, N. Desai, P. Widder, F. Rosenberger, P. van der Merwe, P. Allen and A. Shaw, Cell, 1998, 94, 667–677 CrossRef CAS PubMed; (b) V. Laurent, T. Loisel, B. Harbeck, A. Wehman, L. Gröbe, B. Jockusch, J. Wehland, F. Gertler and M. Carlier, J. Cell Biol., 1999, 144, 1245–1258 CrossRef CAS PubMed.
- A. Goldstrohm, T. Albrecht, C. Suné, M. Bedford and M. Garcia-Blanco, Mol. Cell Biol., 2001, 21, 7617–7628 CrossRef CAS PubMed.
- (a) B. Laggerbauer, S. Liu, E. Makarov, H.-P. Vornlocher, O. Makarova, D. Ingelfinger, T. Achsel and R. Lührmann, RNA, 2005, 11, 598–608 CrossRef CAS PubMed; (b) M. Kofler, M. Schuemann, C. Merz, D. Kosslick, A. Schlundt, A. Tannert, M. Schaefer, R. Luhrmann, E. Krause and C. Freund, Mol. Cell. Proteomics, 2009, 8, 2461–2473 CrossRef CAS PubMed.
- (a) C. M. Deber, F. A. Bovey, J. P. Carver and E. R. Blout, J. Am. Chem. Soc., 1970, 92, 6191–6198 CrossRef CAS PubMed; (b) N. Helbecque and M. H. Loucheux-Lefebvre, Int. J. Pept. Protein Res., 2009, 19, 94–101 CrossRef PubMed; (c) H. Okabayashi, T. Isemura and S. Sakakibara, Biopolymers, 1968, 6, 323–330 CrossRef CAS PubMed; (d) R. K. Dukor and T. A. Kiederling, Biopolymers, 1991, 31, 1747–1761 CrossRef CAS PubMed; (e) R. K. Dukor, T. A. Kiederling and V. Gut, Int. J. Pept. Protein Res., 2009, 38, 198–203 CrossRef PubMed; (f) P. M. Cowan and S. McGavin, Nature, 1955, 176, 501–503 CrossRef CAS.
- (a) J. Zaminer, C. Brockmann, P. Huy, R. Opitz, C. Reuter, M. Beyermann, C. Freund, M. Müller, H. Oschkinat, R. Kühne and H.-G. Schmalz, Angew. Chem., Int. Ed., 2010, 49, 7111–7115 CrossRef CAS PubMed; (b) C. Reuter, P. Huy, J. M. Neudörfl, R. Kühne and H.-G. Schmalz, Chem. – Eur. J., 2011, 17, 12037–12044 CrossRef CAS PubMed.
- (a) C. Reuter, R. Opitz, A. Soicke, S. Dohmen, M. Barone, S. Chiha, M. T. Klein, J. M. Neudörfl, R. Kühne and H.-G. Schmalz, Chem. – Eur. J., 2015, 21, 8464–8470 CrossRef CAS PubMed; (b) A. Maaßen, J. M. Gebauer, E. T. Abraham, I. Grimm, J.-M. Neudörfl, R. Kühne, I. Neundorf, U. Baumann and H.-G. Schmalz, Angew. Chem., Int. Ed., 2020, 59, 5747–5755 CrossRef PubMed.
- R. Opitz, M. Müller, C. Reuter, M. Barone, A. Soicke, Y. Roske, K. Piotukh, P. Huy, M. Beerbaum, B. Wiesner, M. Beyermann, P. Schmieder, C. Freund, R. Volkmer, H. Oschkinat, H.-G. Schmalz and R. Kühne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5011–5016 CrossRef CAS PubMed.
- M. Barone, M. Müller, S. Chiha, J. Ren, D. Albat, A. Soicke, S. Dohmen, M. Klein, J. Bruns, M. van Dinther, R. Opitz, P. Lindemann, M. Beerbaum, K. Motzny, Y. Roske, P. Schmieder, R. Volkmer, M. Nazaré, U. Heinemann, H. Oschkinat, P. ten Dijke, H.-G. Schmalz and R. Kühne, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29684–29690 CrossRef CAS PubMed.
- For reviews, see: (a) N. Holub and S. Blechert, Chem. – Asian J., 2007, 2, 1064–1082 CrossRef CAS PubMed; (b) S. Kotha, M. Meshram, P. Khedkar, S. Banerjee and D. Deodhar, Beilstein J. Org. Chem., 2015, 11, 1833–1864 CrossRef CAS PubMed.
- A. Soicke, C. Reuter, M. Winter, J.-M. Neudörfl, N. Schlörer, R. Kühne and H.-G. Schmalz, Eur. J. Org. Chem., 2014, 6467–6480 CrossRef CAS.
- The allyl-glycine derivative 3 (≥ 95% ee) was prepared through chymotrypsin-catalyzed kinetic resolution according to. (a) B. Schricker, K. Thirring and H. Berner, Biorganic Med. Chem. Lett., 1992, 2, 387–390 CrossRef CAS. The required racemic intermediate was synthesised starting from N-benzhydrylamine and ethyl glyoxylate according to. (b) M. Lautens, E. Tayama and D. Nguyen, Org. Lett., 2004, 6, 345–347 CrossRef CAS PubMed; (c) D. J. Hyett, M. Didonè, T. J. A. Milcent, Q. B. Broxterman and B. Kaptein, Tetrahedron Lett., 2006, 47, 7771–7774 CrossRef CAS.
- (a) H. Waldmann and M. Braun, Liebigs Ann. Chem., 1991, 1045–1048 CrossRef CAS; (b) P. D. Bailey, G. R. Brown, P. Korber, A. Reed and R. D. Wilson, Tetrahedron Asymmetry, 1991, 2, 1263–1282 CrossRef CAS.
- (a) W. Maison, D. C. Grohs and A. H. G. P. Prenzel, Eur. J. Org. Chem., 2004, 1527–1543 CrossRef CAS; (b) N. Deppermann, A. H. G. P. Prenzel, A. Beitat and W. Maison, J. Org. Chem., 2009, 74, 4267–4271 CrossRef CAS PubMed.
- (a) E. J. Corey, F. A. Carey and R. Winter, J. Am. Chem. Soc., 1965, 87, 934–935 CrossRef CAS; (b) E. J. Corey and P. B. Hopkins, Tetrahedron Lett., 1982, 23, 1979–1982 CrossRef CAS; (c) E. J. Corey and R. A. E. Winter, J. Am. Chem. Soc., 1963, 85, 2677–2678 CrossRef CAS.
- O. Arjona, M. J. Cabas, J. Nieto-Rubio and A. Querejeta, Heterocycles, 2006, 68, 2079–2086 CrossRef CAS.
- J. Coste, D. Le-Nguyen and B. Castro, Tetrahedron Lett., 1990, 31, 205–208 CrossRef CAS.
- (a) L. A. Carpino, A. El-Fahama and F. Albericio, Tetrahedron Lett., 1994, 35, 2279–2282 CrossRef CAS; (b) Z. J. Kamiński, B. Kolesińska, J. Kolesińska, G. Sabatino, M. Chelli, P. Rovero, M. Błaszczyk, M. L. Główka and A. M. Papini, J. Am. Chem. Soc., 2005, 127(48), 16912–16920 CrossRef PubMed.
- This reagent was prepared according to: Z. Dai, G. Ye, C. U. Pittman and T. Li, Chirality, 2012, 24, 329–338 CrossRef CAS PubMed.
- P. P. de Castro, G. M. F. Batista, H. F. dos Santos and G. W. Amarante, ACS Omega, 2018, 3, 3507–3512 CrossRef CAS PubMed.
- S. Dohmen, M. Reiher, D. Albat, S. Akyol, M. Barone, J.-M. Neudörfl, R. Kühne and H.-G. Schmalz, Chem. Eur. J., 2020, 26, 3049–3053 CrossRef CAS PubMed.
- (a) J.-B. Garsi, P. M. Aguiar and S. Hanessian, J. Org. Chem., 2021, 86, 16834–16847 CrossRef CAS PubMed; (b) J. D. Vasta, A. Choudhary, K. H. Jensen, N. A. McGrath and R. T. Raines, Biochemistry, 2017, 56, 219–227 CrossRef CAS PubMed; (c) N. G. Bandur, K. Harms and U. Koert, Synlett, 2005, 5, 773–776 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and X-ray crystallographic details. CCDC 2211404. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01857h |
| This journal is © The Royal Society of Chemistry 2022 |