
New strategies for the synthesis of 1- and 2-azetines and their applications as value-added building blocks
Michael R.
Gatazka†
 ,
Elvis C.
McFee†
,
Cody H.
Ng†
,
Emily R.
Wearing†
and
Corinna S.
Schindler
*
,
Elvis C.
McFee†
,
Cody H.
Ng†
,
Emily R.
Wearing†
and
Corinna S.
Schindler
*
Willard Henry Dow Laboratory, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: corinnas@umich.edu
First published on 10th November 2022
Abstract
Four-membered nitrogen-containing heterocycles are highly desirable functional groups with synthetic and biological applications. Unsaturated four-membered N-heterocycles, 1- and 2-azetines, are historically underexplored, but have recently been gaining interest due to the development of new synthetic methods to access these compounds, and to their potential as reactive intermediates. This review covers both the synthesis and applications of azetines, with a focus on synthetic methods to access azetines developed since 2018, and a comprehensive review of the reactivity and applications of azetines as starting materials or intermediates to access both other heterocycles and complex products.
Introduction
Four-membered nitrogen-containing heterocycles (Fig. 1A) have recently gained interest and popularity for their potential as underexplored biologically active compounds. While azetidines (1) represent fully saturated four-membered N-heterocycles, azetines (2, 3), also known as dihydroazetes, are their corresponding counterparts with one unit of unsaturation. Specifically, 1-azetines (2) contain an imine functional group with a double bond between the nitrogen atom and the neighbouring carbon atom. In comparison, 2-azetines (3) incorporate a carbon–carbon double bond in the heterocycle. Much of the focus in the area of the synthesis and reactivity of four-membered N-heterocycles has been on azetidines (1), which offer a variety of advantages compared to larger ring heterocycles including three dimensionality, increased metabolic stability,1–3 and potential as bioisosteres for other N-heterocycles such as pyridines.4–6 Although access to azetidines and azetines has historically been challenging, interest in functionalized azetidines has rapidly heightened due to recent synthetic advances.1,2 While development of methods to access azetines has been more limited, the field has seen a recent rise in popularity with the development of five new synthetic methods since 2018.7–12 Azetidines and azetines share desirable physical properties including high ring-strain and the potential to serve as bioisosteres; however, these four-membered heterocycles have important differences in their inherent reactivity, making azetines highly desirable synthetic targets.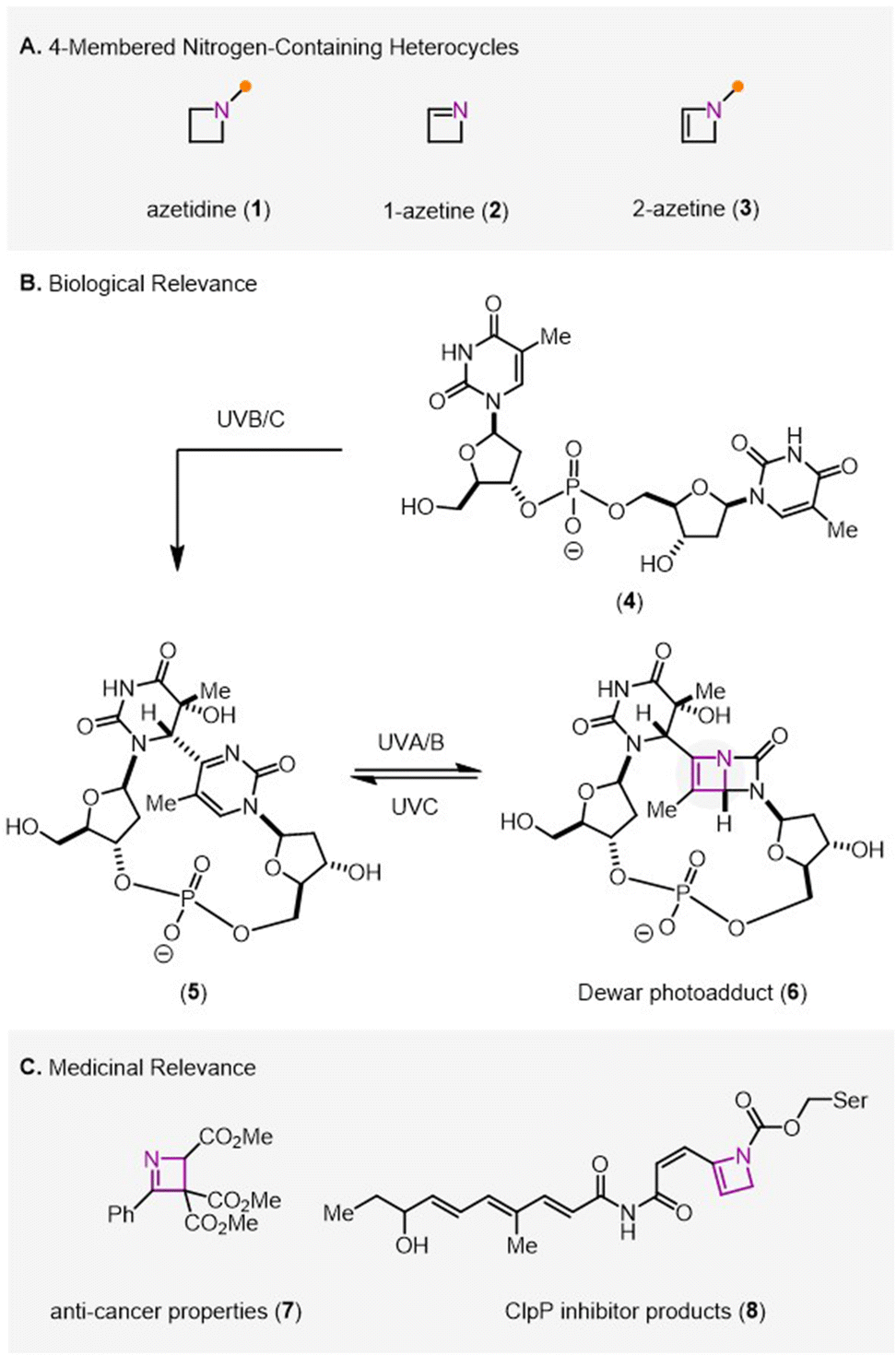 | ||
| Fig. 1 (A) Structure of four-membered N-heterocycles, azetidines and azetines. (B) The biological relevance of 2-azetines is demonstrated by their prevalence in the DNA photodegradation process. (C) Limited medicinal relevance has been demonstrated in studies of synthetic azetines (7) and through the isolation of metabolites from ClpP inhibition (8). | ||
In comparison to azetidines (1), incorporating exclusively sp3-hybridized carbons, the azetine scaffold (2, 3) includes two sp2-hybridized centers, which are responsible for their distinct reactivity. A key challenge in the development of efficient methods to synthesize azetines (2, 3) is their unsaturated nature, resulting in elevated strain compared to azetidines (1). Importantly, azetines (2, 3) are prone to undergo competing thermal fragmentation through electrocyclic ring-opening resulting in the formation of aza-dienes,13–17 which has hampered the development of general synthetic strategies for their construction. Compared to azetidines, 1-azetines (2) possess an imine functionality, which can engage in additional reactivity and modulates its basicity compared to azetidines’ inherently more basic amine functionality.18
Despite challenges in synthetic access, azetines have been shown to be biologically relevant, which holds great promise for future pharmaceutical and biological applications. The photodegradation of DNA is known to proceed through the formation of Dewar isomers (6), which form upon isomerization of (6-4)-pyrimidinone photoproducts (5) arising from UV light-promoted cycloaddition and rearrangement of 4.19–22 The synthetic utility of Dewar photoadducts is reviewed elsewhere.23,24 Additionally, 2-azetine 8 has been found to be the product of the reaction between a recently discovered inhibitor for bacterial caseinolytic protease (ClpP) and a serine residue in the binding pocket. This product is formed when the inhibitor and the serine form a covalent bond, leading to the formation of the azetine 8 and inhibition of the ClpP enzyme.25 Furthermore, azetine-containing compounds hold potential as cancer therapeutics. Specifically, 1-azetine 7 was shown to induce apoptosis in and have high cytotoxicity against THP-1 (human monocytic leukemia) cells.18 Additionally, other azetines similar to 7 were shown to vary greatly in their activity, demonstrating the importance of being able to synthesize a wide range of substituted azetines for further testing.18 As such, the development of more general and efficient methods for the formation of 1- and 2-azetines is expected to lead to important future discoveries in the area of azetine biological activity.
Access to 1- and 2-azetines has historically been limited to three main approaches including [2+2]-cycloadditions (Fig. 2A), elimination reactions (Fig. 2B), and ring expansions (Fig. 2C). General challenges that plague strategies to synthesize azetines include competing aza-diene formation via electrocyclic ring-opening, and facile imine hydrolysis in the case of 1-azetines (2). Consequently, established synthetic methods tend to be limited in terms of scope or generality.
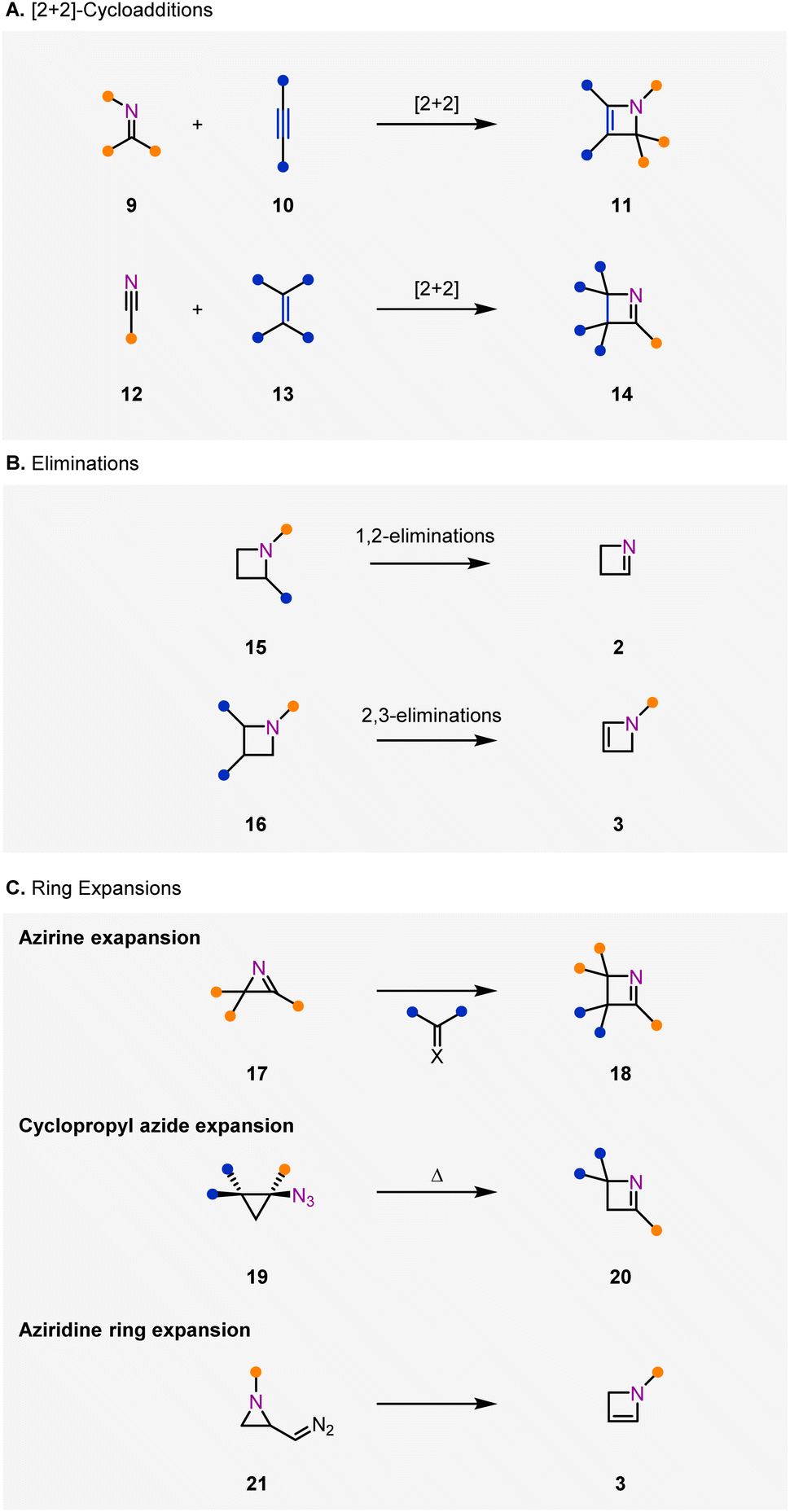 | ||
| Fig. 2 General approaches to synthesize azetines generally fall into three categories. (A) [2+2]-Cycloaddition strategies can yield either 1- or 2-azetines depending on the choice of cycloaddition partners. (B) Elimination of a leaving group on an azetidine compound can yield either 1- or 2-azetines. (C) Ring expansions of three-membered rings can form 1-azetines from azirines or cyclopropyl azides, and 2-azetines from diazoaziridines. | ||
Methods to access azetines through [2+2]-cycloadditions (Fig. 2A) fall into two categories, [2+2]-cycloadditions of imines (9) and alkynes (10) to access 2-azetines (11)26–28 and [2+2]-cycloadditions of nitriles (12) and alkenes (13) to access 1-azetines (14).29–31 Cycloadditions relying on imines (9) and alkynes (10) have been shown to proceed either under UV light irradiation26 or metal-mediated conditions.27 In comparison, cycloadditions between nitriles (12) and alkenes (13) have been promoted exclusively with UV light and are limited to aryl substituted nitrile groups.29–31
Access to azetines via elimination from an azetidine starting material or intermediate has also been demonstrated for both 1-azetines (2)14,32–39 and 2-azetines (3)40–46 (Fig. 2B). Access to 1-azetines (2) can be mediated by elimination of a leaving group on the azetidine nitrogen atom,14,32–34 or by alkylation of a carbonyl, thiocarbonyl, or imine in the azetidine's 2-position, followed by tautomerization to the 1-azetine product.35–38 2-Azetines (3) are similarly accessible by elimination of a leaving group from an azetidine (16),40,45 which can occur in situ from an azetidine intermediate41–43 or by alpha-deprotonation of a 3-azetidinone followed by trapping with an electrophile.44,46
Ring expansions of azirines (17),18,23,47–51 cyclopropanes (19),34,52,53 or aziridines (21)17,54 to form azetines capture the inherent reactivity of three-membered rings to enable access to the strained azetine products (Fig. 2C). In the presence of a carbene, azirine 17 is converted to 1-azetine 18 through a net [3+1]-cycloaddition, often with the aid of a metal catalyst.18,23,47–51 In an alternative approach, cyclopropyl azides (19) can be converted to 1-azetines (20) upon heating.34,52,53 Finally, a ring expansion of a diazo-aziridine (21), formed in situ from aziridination of an α,β-unsaturated diazo-compound, under copper-catalyzed conditions results in a variety of 2-azetines (3).54 Aziridines can also be converted to azetines under basic conditions.17 Notably, [3+1]-cycloaddition reactions resulting in azetines are not limited to three-membered rings. It has been shown that [3+1]-cycloadditions between 5-membered oxazaphospholes and isocyanates can form 1-azetine products, yet these examples are limited.55,56
Other less general methods to access azetines include photochemical ring contraction of 1,4-benzoxazepines yielding polycyclic 1-azetines.57 Azetine products can also be accessed with high diastereoselectivity with the use of chiral auxiliaries in a sequential condensation–cyclization approach.58–60
Although access to and applications of azetine compounds has historically been more limited than those of their saturated azetidine counterparts, evolving synthetic methodology has enabled their further exploration as synthetic intermediates and valuable building blocks. This review aims to provide a comprehensive overview of new synthetic protocols giving rise to 1- and 2-azetines, and to highlight applications of these heterocycles as versatile synthetic intermediates. Specifically, strategies resulting in the formation of azetidines as well as 3-, 5-, 6-membered and larger heterocycles upon conversion of azetine intermediates have been developed and are discussed in this review together with their utilization in complex molecule synthesis.
Azetines have been the topic of some recent reviews,23,24,61–64 though review of this topic including recent methods to access azetines since 2018, and a comprehensive review of the reactivity and applications of azetine products has yet to be published. Previous reviews have primarily focused on providing overviews of established methods to access 1- and 2-azetines, or applications of azetines in a focused area, such as azetidine formation or ring expansions.23,24,61–64 This review will include new synthetic methods to access azetines since 2018, and will comprehensively cover the applications and reactivity of azetines as starting materials and intermediates to access other desirable products.
Synthetic advances since 2018
Methods for the syntheses of 1- and 2-azetines have been the subject of previous reviews.24,61–64Fig. 3 provides an overview of new synthetic methods developed since 2018 that include advances in cycloaddition, ring-expansion, and elimination strategies.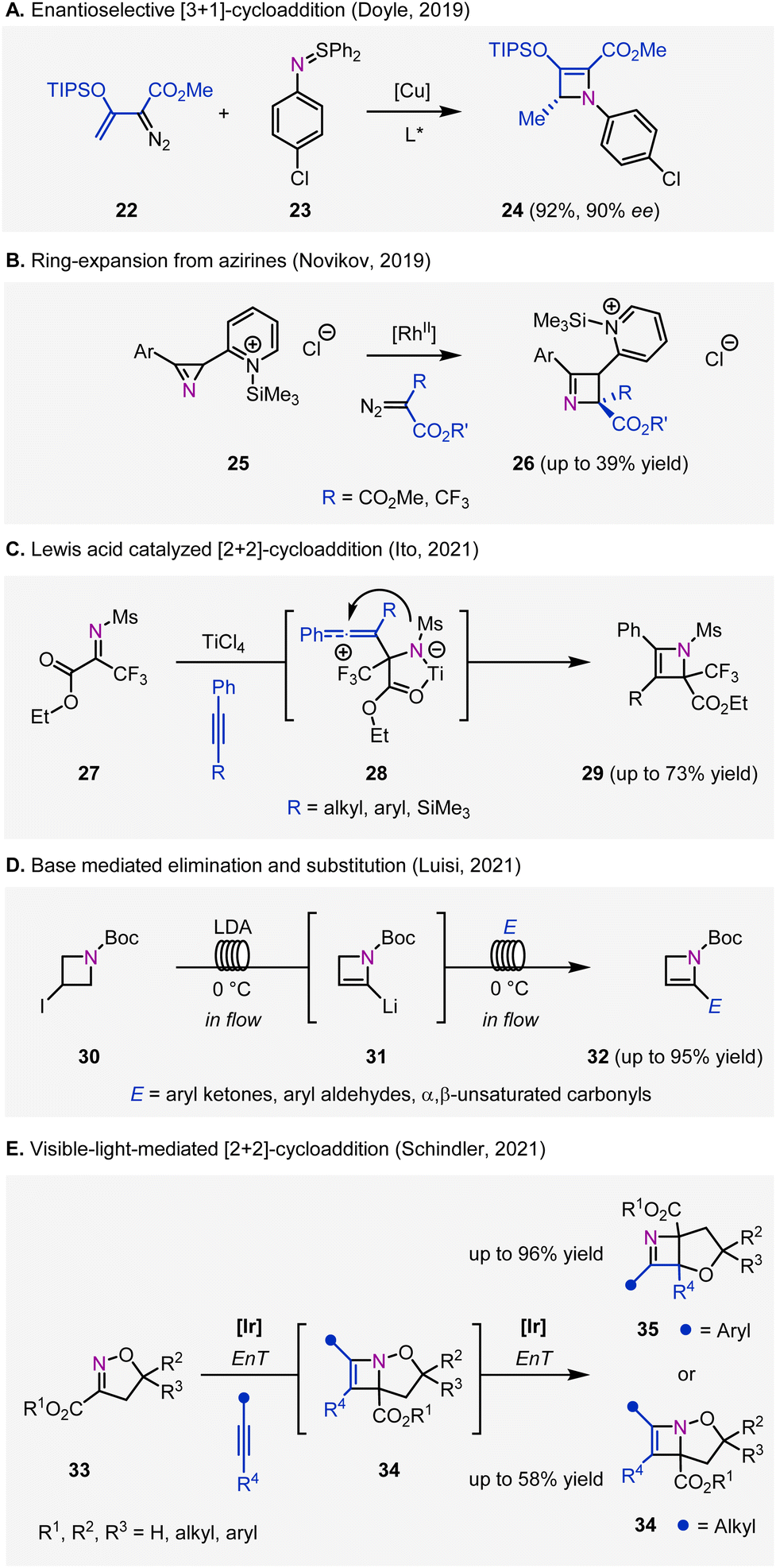 | ||
| Fig. 3 Recent advances in synthetic methods to access azetines. (A) Copper-catalyzed enantioselective [3+1]-cycloaddition to access tetrasubstituted 2-azetines. (B) Ring-expansion of aziridines via rhodium carbenoids to access 1-azetines. (C) Lewis acid catalyzed [2+2]-cycloaddition to access 2-azetines. (D) Flow synthesis of 2-substituted 2-azetines via formation of α-lithiated-2-azetines. (E) Divergent synthesis of 1- and 2-azetines via a [2+2]-photocycloaddition. | ||
In the area of [3+1]-cycloadditions, in 2019, Doyle and co-workers reported a new strategy for the enantioselective synthesis of 2-azetines (24) upon [3+1]-cycloadditions between imido-sulfur ylides (23) and enoldiazoacetates (22) under copper catalysis in the presence of a chiral sabox ligand. The methods achieved high enantiomeric excess of up to 95% (Fig. 3A).7,12 The reports also showed further utility of the azetine products, namely in accessing azetidines by reduction7 and amino acid derivatives upon ring-opening.12
In the area of ring-expansions, Novikov and co-workers’ 2019 report showcased the synthesis of substituted 3-(2′-pyridyl)-1-azetines (26) via ring-expansion of azirines (25) with rhodium carbenes (Fig. 3B).8 To accomplish the transformation it was necessary to incorporate a silyl protecting group on the pyridine nitrogen to limit side reactivity, which was installed and removed in a sequential one-pot reaction.
In 2021, Ito and co-workers reported a Lewis acid catalyzed stepwise [2+2]-cycloaddition to generate 2-azetines (29) from substituted iminopyruvates (27) and aryl alkynes (Fig. 3C).9 The authors proposed that intermediate 28, in which the titanium is chelated by the iminopyruvate, is crucial in preventing side reactivity between the ester and the electrophilic carbon of the alkyne.
Expanding on the area of eliminations to synthesize azetines, Luisi and co-workers’ 2021 report demonstrated a flow-synthesis protocol capable of facilitating an elimination and incorporating an electrophile to synthesize 2-azetines (32). Specifically, in the presence of lithium diisopropylamide (LDA), the N-Boc-3-iodo-azetidines (30) undergo an elimination to form a 2-azetine that is then further deprotonated to generate α-lithiated-2-azetines (31). Downstream in the flow reactor, this intermediate is reacted with an electrophile to yield 2-azetines (32) bearing additional substitution at the 2-position (Fig. 3D). The scope of electrophiles included aldehydes, ketones, imines, and silyl chlorides.10
A divergent synthesis of 1- and 2-azetines enabled by visible-light-mediated triplet energy transfer (EnT) photocatalysis was reported by Schindler and co-workers in 2021 (Fig. 3E).11 This method utilized a [2+2]-photocycloaddition to access these products either directly (2-azetine 34) or from a cycloaddition–rearrangement sequence (1-azetine 35). Therein, 2-azetines formed upon initial [2+2]-cycloaddition between cyclic oximes (33) and aryl alkynes can undergo a consecutive sensitization and subsequent rearrangement to 1-azetines (35). In the absence of aryl substituents on the alkyne component, 2-azetine products (34) are isolable as the initial [2+2]-cycloaddition products between aliphatic alkynes and isoxazolines 33.
Azetines as intermediates
Synthesis of 4-membered heterocycles
1- and 2-azetines contain reactive π-bonds due, in part, to the strained nature of the four-membered ring. The imine moiety in 1-azetines and the alkene subunit in 2-azetines promote imine or alkene addition reactions, which allows access to a wide range of substituted and/or polycyclic azetidines (Fig. 4). There are eight types of addition reactions, not including cycloadditions, that 1- and 2-azetines undergo to produce azetidines. These classes of addition reactions are radical addition to 2-azetines (Fig. 4A), acid-facilitated addition to both 1- and 2-azetines (Fig. 4B), metal-catalyzed addition to 2-azetines (Fig. 4C), β-lactam formation from 1-azetines (Fig. 4D), hydride reduction of 1-azetines (Fig. 4E), base-facilitated addition to 1- and 2-azetines (Fig. 4F), metal-catalyzed hydrogenation of 2-azetines (Fig. 4G), and N-methylation/sulfide formation of 1-azetines (Fig. 4H).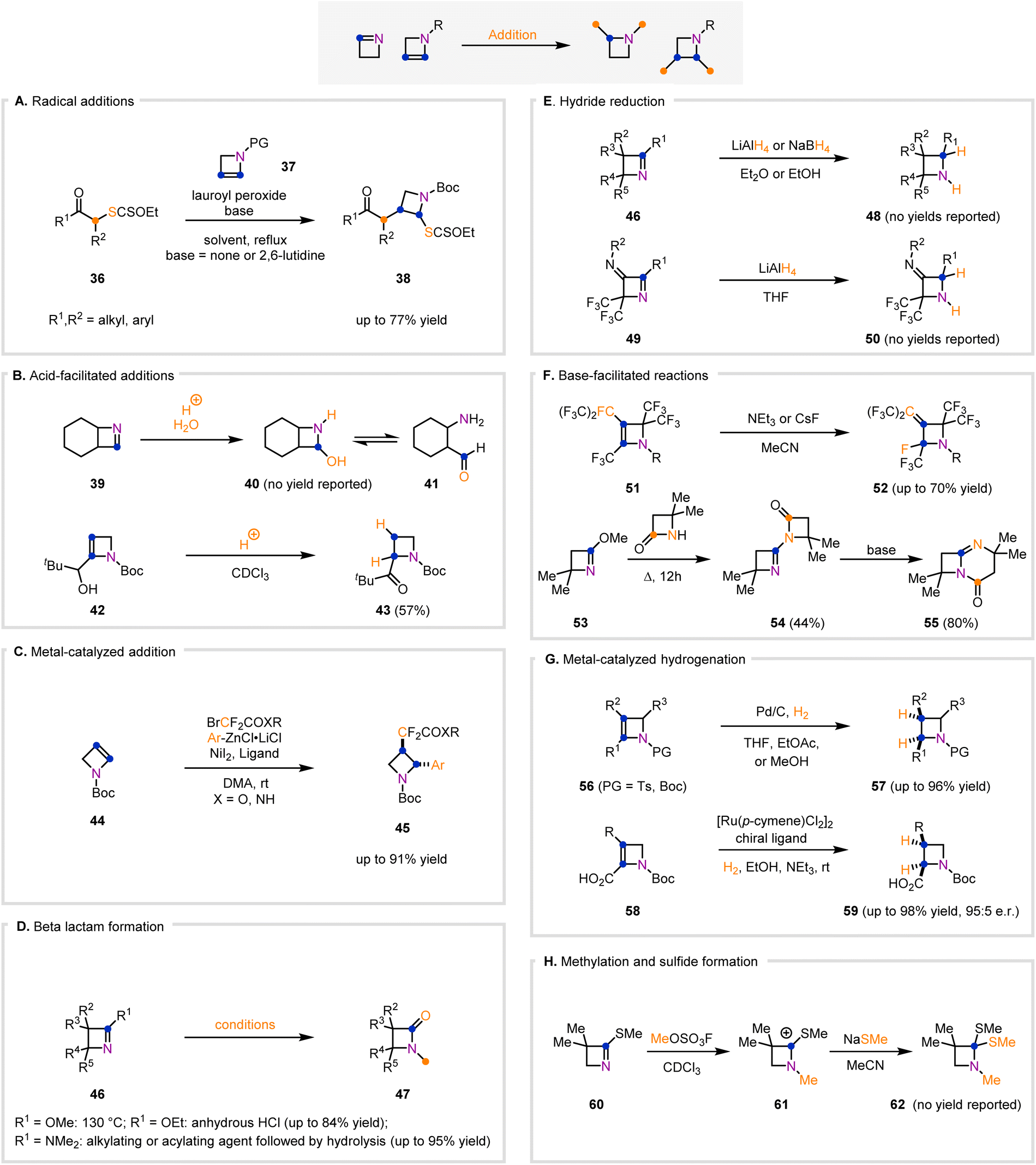 | ||
| Fig. 4 General classes of addition reactions of 1- and 2-azetines that lead to functionalized four-membered nitrogen-containing heterocycles. (A) Addition reactions of 2-azetines through a radical mechanism. (B) Addition reactions of 1- and 2-azetines under acidic conditions. (C) Addition reactions of 2-azetines catalyzed by transition metals. (D) Addition reactions of 1-azetines that lead to the formation of β-lactam rings. (E) Addition reactions of 1-azetines involving hydride reagents. (F) Addition reactions of 1-azetines under basic conditions. (G) Addition reactions of 2-azetines under metal-catalyzed hydrogenation conditions. (H) Addition reaction of a 1-azetine that formed a N-methylated and C-thiomethylated product. | ||
Zard and co-workers developed methods relying on xanthates as radical precursors to facilitate addition across the olefinic π-bond of 2-azetines to access functionalized azetidines (Fig. 4A).65–67 They initially disclosed two examples in which 37 (PG = Bz) in the presence of catalytic lauroyl peroxide and mono-substituted xanthates (36) afforded disubstituted azetidines (38) in up to 50% yield.65 For both substrates, the trans isomer was preferred over the cis isomer, although both isomers were formed.65 This work was furthered by employing similar reaction conditions with a Boc-protected 2-azetine (37) and xanthates (36) to produce azetidines (38) regioselectively.66,67 In these examples, the authors added 2,6-lutidine to their initially disclosed reaction conditions to suppress lauric acid-induced decomposition of the 2-azetine substrates (37).66,67 Incorporation of the xanthate group into the final product was advantageous as it allowed conversion of the resultant azetidines (38) into complex pyrroles, or could be reductively removed to afford monosubstituted azetidines bearing complex carbon skeletons including a steroid scaffold.66,67
The π-systems of 1- and 2-azetines are labile under acidic conditions, and the propensity of the azetine π-system to react with acids can be harnessed to access functionalized azetidine scaffolds (Fig. 4B). Wulfman and Steinheimer reported that the formation of 39 (Fig. 4B), a 1-azetine, via thermolysis of a cyclic azide and upon purification on silica gel provided a mixture of azetidine 40 and its ring-opened isomer 41 in equilibrium.68 Presumably, the acidity of silica gel and nucleophilicity of water account for the formation of the observed products. Hodgson and co-workers also recognized the lability of azetines to mildly acidic conditions.46 They observed an acid-driven isomerization from a 2-azetine (42, Fig. 4B) to form azetidine 43 when 42 was dissolved in deuterated chloroform, which is known to be slightly acidic.46 Another notable example is from Petrov and Marshall who showed that fluorinated tricyclic 1-azetines can react with mCPBA at the imine bond to produce oxaziridine products.69 Although the acid-driven reactivity of 1- and 2-azetines has not been extensively explored, there is great potential for accessing functionalized azetidines using this class of reactivity.
Transition-metal-catalyzed additions across π-systems have been extensively explored and have similarly been employed to control the reactivity of 2-azetines (Fig. 4C). In this area, Zhang and co-workers developed a nickel-catalyzed aryldifluoroalkylation across the alkene of 2-azetines (44) that affords trans-disubstituted azetidines (45).70 This metal-catalyzed transformation of 2-azetines (44) proceeded with a diastereoselectivity greater than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and produced azetidines with ester or amide functionality for subsequent modifications such as amide couplings.70 Mykhailiuk and co-workers also published a single example of a transition-metal-catalyzed addition of a carbene across the alkene of a 2-azetine (44).71 They employed a CuCl catalyst and passed a stream of CF3CHN2 gas into the reaction vessel, leading to the formation of a 2-trifluoromethylcyclopropyl ring fused to a Boc-protected azetidine.71
:1, and produced azetidines with ester or amide functionality for subsequent modifications such as amide couplings.70 Mykhailiuk and co-workers also published a single example of a transition-metal-catalyzed addition of a carbene across the alkene of a 2-azetine (44).71 They employed a CuCl catalyst and passed a stream of CF3CHN2 gas into the reaction vessel, leading to the formation of a 2-trifluoromethylcyclopropyl ring fused to a Boc-protected azetidine.71
Functionalized β-lactams are a well-established motif that have demonstrated medicinal properties.72–75 1-Azetines have been identified by researchers as precursors to functionalized β-lactam rings under a variety of reaction conditions (Fig. 4D). The first example of a 1-azetine being converted to a β-lactam was disclosed by Atkinson and co-workers,49 in which two different 1-azetines (46, R1 = Cl or OMe, R2 = Ph, R3 = Cl, R4 = Me, R5 = H) were converted to the same N-hydro β-lactam (47) upon exposure to 10% hydrochloric acid.49 Likewise, Aue and Thomas were able to synthesize a N-methyl β-lactam (47, R2 = R3 = Me, R4 = R5 = H) from a 1-azetine (46, R1 = OMe) via a thermally induced Chapman rearrangement.76 Ghosez and co-workers developed a general protocol that allows conversion of a 1-azetine (46, R1 = NMe2, R2 = R3 = Me, R4 = Ph, R5 = H) to β-lactams with an N-electrophile bond (47) in the presence of an electrophile such as methyl iodide or acrylonitrile following basic hydrolysis.37 Pifferi and co-workers synthesized a series of 1-azetines (46, R1 = OEt, R2 = alkyl or aryl, R3 = alkyl or aryl, R4 = R5 = H) from beta lactams (47).36 The authors observed that anhydrous hydrochloric acid in ethereal solvents reverted 1-azetines (46) to the corresponding β-lactams (47) from which they were formed.36 The four protocols described (vide supra) allow access to highly functionalized β-lactams from 1-azetines and proceed through different mechanisms, which highlights the potential of 1-azetines as useful building blocks to access β-lactam rings.
Hydride-driven reduction of imines is a well-established class of reactivity that has also been demonstrated to readily convert 1-azetines to azetidines (Fig. 4E). The first example of a hydride reduction of a 1-azetine to form azetidine products was presented by Pifferi and co-workers, who employed LiAlH4 to reduce three different 1-azetines (46, Fig. 4E) to the corresponding azetidine (48) scaffolds.36 Interestingly, the R1 substituent (R1 = OMe) was cleaved in the course of this transformation and replaced with a hydrogen atom.36 The Hassner, Denis, and Khlebnikov groups also demonstrated that various 1-azetine (46) scaffolds could be reduced by LiAlH4 in ethereal solvents.14,50,52 Notably, Wulfman and Steinheimer were able to reduce a [4.2.0]-fused cyclic 1-azetine (46) with the less reactive NaBH4 to afford the corresponding [4.2.0]-fused-cyclic azetidine (48).68 Finally, Burger and co-workers disclosed an intriguing example of a LiAlH4 reduction of 49 (Fig. 4E) to 50, which left the exocyclic imine motif intact.77 Hydride reduction of 1-azetines offers a viable strategy to access the valuable azetidine scaffold in a straightforward manner.
1- and 2-azetines are also reactive under basic conditions, which often leads to rearrangements (Fig. 4F). Del'tsova and co-workers identified that highly fluorinated 2-azetines (51, Fig. 4F) undergo a rearrangement to afford a fluorinated azetidine product (52) with an exocyclic π-bond under basic conditions.78 Petrov and colleagues similarly identified that fluorinated tricyclic 1-azetines react with CF3Si(CH3)3 at the imine bond in the presence of cesium fluoride to produce further fluorinated azetidine products.79 The importance of fluorinated compounds in medicinal chemistry is extensively documented,80–86 which highlights the potential applications of these base-driven reactions. In 1970, Bormann developed a method to couple 1-azetine 53 (Fig. 4F) to a β-lactam moiety, giving adduct 54.87 Under basic conditions, adduct 54 cyclized to produce [4.2.0]-cycloadduct 55, which contains an azetidine structural unit.87 Similar to the acid-driven reactivity of 1- and 2-azetines, basic reaction conditions of azetines have not been extensively explored. However, the research to date highlights the potential to generate useful four-membered nitrogen-containing scaffolds using base-mediated reactions of azetines.
Metal-catalyzed hydrogenation of olefins is a well-established transformation, and 2-azetines can be easily converted to the corresponding azetidine by this approach (Fig. 4G). The first example of a 2-azetine being converted to the corresponding azetidine was presented by Barluenga, López, and co-workers in 2012.54 The authors exposed N-tosyl 2-azetines (56, R1 = CO2Et, R2 = H or Me, R3 = H, Fig. 4G) to hydrogen gas in the presence of palladium on carbon (Pd/C) to produce the desired cis-disubstituted azetidines (57).54 Both Hodgson and co-workers and Didier and colleagues utilized a similar strategy to hydrogenate various N-Boc-2-azetines (56) giving the respective mono- or disubstituted-azetidine frameworks (57).44,46 Notably, Doyle and co-workers have synthesized chiral 2-azetines (56) bearing a stereocenter at the carbon substituted with R3.7 The authors highlighted that Pd/C-catalyzed hydrogenation of chiral 2-azetines (56) allowed access to chiral azetidines (57), preserving the high enantiomeric excess observed in their initially synthesized 2-azetines (56).7 Similarly, Didier and co-workers expanded upon their earlier work with 2-azetines and employed a ruthenium catalyst and chiral ligand to convert achiral 2-azetines (58, Fig. 4G) to chiral azetidines (59) in high yields with enantiomeric ratios up to 95:5.44,88–90 2-Azetines bear a highly strained olefin, which makes them primed to be reduced under metal-catalyzed hydrogenation to the corresponding azetidine scaffolds. As demonstrated (vide supra), this allows facile access to a wide range of functionalized four-membered nitrogen heterocycles.
The cyclic imine contained within 1-azetines provide a unique reactivity profile, which is centered on the nucleophilicity of the imine nitrogen and the electrophilicity of the imine carbon. In 1981, de Boer and co-workers harnessed this reactivity to convert 60 to a highly functionalized azetidine (62, Fig. 4H).53 The authors methylated the imine nitrogen of 60 using methyl fluorosulfonate to result in the resonance stabilized carbenium ion 61 that was subsequently trapped with sodium methanethiolate to give azetidine 62.53 Azetidines have been identified as useful isosteres of other N-heterocycles,4–6 which makes accessing highly functionalized azetidines such as 62 of interest.
1- and 2-azetines contain π-systems that prime them to react via cycloadditions, which afford valuable fused-polycyclic azetidines. Specifically, azetines have been demonstrated to undergo four types of cycloaddition reactions. These classes of reactivity are [3+2]-cycloadditions (Fig. 5A), cycloadditions with ketenes and isocyanates (Fig. 5B), [4+2]-cycloadditions (Fig. 5C), and cycloadditions that lead to dimerization (Fig. 5D).
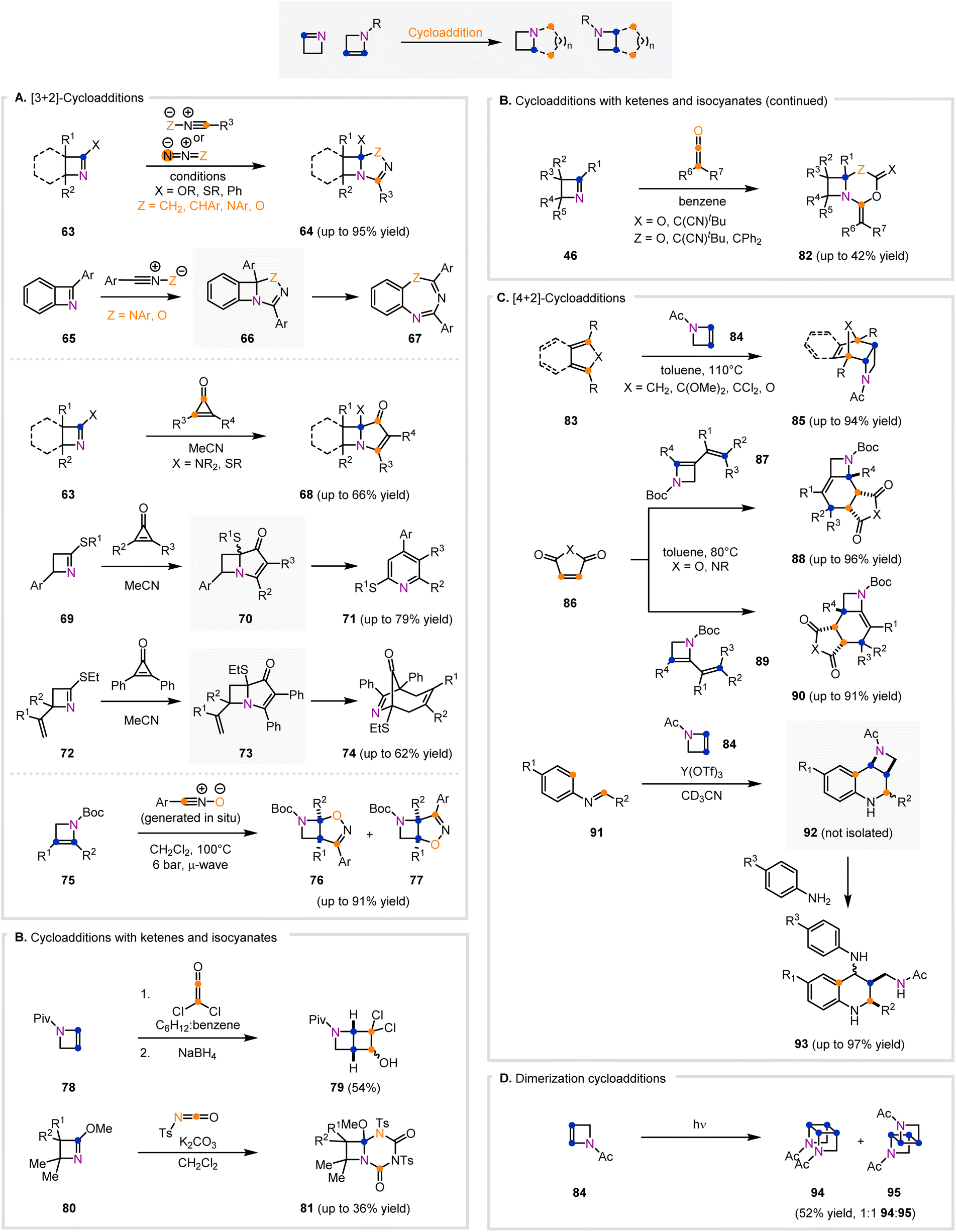 | ||
| Fig. 5 General classes of cycloaddition reactions involving 1- and 2-azetines that lead to polycyclic azetidine adducts. (A) [3+2]-Cycloaddition reactions of 1- and 2-azetines using dipolar reagents and 1,3-dipole equivalents. (B) Cycloaddition reactions of 1- and 2-azetines with ketene and isocyanate reactants. (C) [4+2]-Cycloaddition reactions of 2-azetines acting as both the diene and dienophile. (D) [2+2]-Cycloaddition reaction that affords head-to-head azetidine dimers. | ||
[3+2]-Cycloaddition reactions of 1- and 2-azetines are attractive because they rapidly build complexity and form fused heterocycles containing a four- and five-membered ring, both of which can be challenging to synthesize (Fig. 5A). [3+2]-Cycloadditions involving azetines were first disclosed by Rees, Storr, and colleagues in 1975.91 1-Azetines (63, X = Ph) upon exposure to nitrile oxides (Z = O) cyclized to give polycyclic products (64) that are labile to mild acidic conditions and heat.91 Storr's group expanded upon this work in 1978 when they reacted 2-phenylbenzazete (63, X = Ph) with diazomethane to produce cycloadduct 64 (X = Ph, Z = CH2, orange dot = N).92 Similar to their initial 1975 work, this cycloadduct was also sensitive to acidic and thermal conditions.92 This lability of the azetidine products proved advantageous. It allowed the researchers to access 1,3,5-oxadiazepine and 1,3,5-triazepine products (67) from azetine 65 through the semi-transient intermediate azetidine 66.91,92 Further exploration of [3+2]-cycloadditions was published in 1990 by Smalley and Luheshi.93,94 Upon reaction of 1-azetines (63, X = OR or SR) with nitrile oxides (Z = O), nitrile ylides (Z = CHAr), and nitrilimines (Z = NAr), generated in situ from hydroxyimidoyl, imidoyl, or hydrazonyl halides, highly functionalized bicyclic or tricyclic scaffolds were produced (64).93,94 Polycyclic azetidines (64) resulting from nitrile oxides were synthesized in up to 95% yield, and azetidines formed from nitrile ylides were isolated in up to 68% yield.94 Similarly, fused azetidines (64) formed from nitrilimines were isolated with a maximum of 89% yield.93 This same group expanded their work in 1993 with a more detailed report of their initial findings.95 Hemming and co-workers expanded upon this methodology with the synthesis of bicyclic azetidines (64) from the [3+2]-cycloaddition of 1-azetines (63, X = SR) and hydroxyimidoyl chlorides.96 This reaction proceeded through the in situ generation of nitrile oxides (Z = O) from hydroxyimidoyl chlorides to afford bicyclic-azetidine products (64) in up to 72% yield as single diastereomers.96 Azetidines 64 were further converted to functionalized oxadiazoles upon heating in toluene.96
Cyclopropenones are also highly useful reagents for [3+2]-cycloadditions. Unlike other 1,3-dipoles that commonly contain at least one heteroatom, cyclopropenones provide a source of three contiguous carbon atoms (Fig. 5A). As such, cyclopropenone reagents allow access to fused polycyclic structures (68) with a higher carbon content than nitrile oxides and similar 1,3-dipolar reagents. The first example of a cycloaddition between cyclopropenones and 1-azetines was disclosed by Heimgartner and co-workers in 1983.97 A series of 1-azetines (63, X = NR2) were reacted with difunctionalized cyclopropenones to afford [3.2.0]-cycloadducts containing a fused azetidine ring (68). Smalley and co-workers extended these initial studies in 1992 with the synthesis of fused-bicyclic or fused-tricyclic azetidines (68) from highly functionalized 1-azetines (63, X = SR) and diphenylcyclopropenone.98 In 2006, Hemming and co-workers also presented a single example of a 1-azetine (63, X = SEt) reacting with diphenylcyclopropenone to afford a similar azetidine adduct (68) to the azetidines disclosed by Smalley and colleagues.98,99 Notably, similar 1-azetines (72) underwent [3+2]-cycloaddition to form 73 as a transient intermediate that ultimately formed azabicyclo[4.2.1]nonene products (74).99 Hemming and co-workers expanded upon their 2006 work in 2012 when they presented a [3+2]-cycloaddition reaction of 1-azetines (69) with difunctionalized cyclopropenones to access [3.2.0]-fused azetidines (70).100 These adducts (70) formed from 1-azetines (69) were subsequently converted to highly functionalized pyridines (71) upon heating in toluene, which are useful building blocks.100
Didier and co-workers were also able to leverage a [3+2]-cycloaddition strategy that used 2-azetines (75, Fig. 5A) and nitrile oxides in a telescoped sequence to access fused-isoxazoline-azetidine scaffolds (76 and 77).89 2-Azetines and hydroxyimidoyl chlorides were reacted under microwave irradiation to afford fused azetidines (76 and 77) in up to 83% yield (up to 91% yield final step) over four or five steps. Interestingly, the 2-azetine precursors were generated by a three to four step telescoped sequence from a substituted-Boc-protected azetidine, and the nitrile oxides were generated in situ. Monosubstituted 2-azetines gave single products (76), but disubstituted 2-azetines gave a mixture of regioisomers (76 and 77) with a maximum regioselectivity of 16:1 (the identity of the major regioisomer is substrate dependent).89
Another useful class of reactivity to access fused-azetidine scaffolds from 1- and 2-azetines involves cycloaddition with ketenes and isocyanates (Fig. 5B). Both ketenes and isocyanates are highly reactive species, and cycloadditions involving these compounds with 1- and 2-azetines lead to fused [2.2.0]-bicyclic compounds (79) and [4.2.0]-bicyclic skeletons (81, 82) containing an azetidine. Correia and co-workers published the first example of a [2+2]-cycloaddition between 2-azetine 78 (Fig. 5B) and dichloroketene to afford a [2.2.0]-fused azetidine product (79).101 The initial adduct formed bears a ketone instead of the hydroxyl substituent in 79, but was non-isolable and thus was reduced with NaBH4, giving rise to azetidine alcohol 79.101 The first example of a ketene reacting with a 1-azetine was disclosed in 1973 by Hassner and co-workers, who showed a single example of the reaction of a 1-azetine (46, R1 = Ph, R2 = R3 = Cl, R4 = Me, R5 = H, Fig. 5B) with diphenylketene to give cycloadduct 82 (X = O, Z = CPh2).102 The reactivity of ketenes with 1-azetines was expanded by Aue and Thomas in 1975 when they presented additional examples of a 1-azetine (46, R1 = OMe, R2 = R3 = H or Me, R4 = R5 = Me) reacting with a ketene (R6 = CN, R7 = tBu) to afford cycloadducts 82 (X = O or C(CN)tBu, Z = O or C(CN)tBu).103 Aue and Thomas in the same year published a similar report that used 1-azetines (80, R1 = H or Me, R2 = H or Me, Fig. 5B) and tosylated isocyanates to form cycloadducts 81.104 Ketenes and isocyanates are well established highly reactive species, and their application in reacting with 1- and 2-azetines allows access to complex polycyclic azetidines.
One of the most powerful classes of cycloaddition reactions is [4+2]-cycloadditions, and the olefinic π-bond in 2-azetines allows these strained heterocycles to participate in this class of reactivity (Fig. 5C). The first example of a [4+2]-cycloaddition involving 2-azetines was published by Dave and co-workers, who presented a Diels–Alder reaction of an N-acetyl-2-azetine (84, Fig. 5C) dienophile with various dienes (83) to afford complex cycloadducts (85).105 This Diels–Alder reaction proceeded in a straightforward manner, only requiring heat, giving the endo products selectively in up to 94% yield.105 Didier and co-workers employed an alternate approach utilizing functionalized 2-azetines with a pendent vinyl group (87 or 89, Fig. 5C) as the diene with a succinic anhydride or succinimide dienophile (86).90 The [4+2]-cycloaddition proceeded with good yields and high diastereoselectivity, which favored an endo transition state, to give tricyclic azetidines (88 or 90).90 Notably, the aforementioned two examples demonstrate that 2-azetines can act as both the diene or dienophile component in a Diels–Alder cycloaddition reaction depending on the substitution of the 2-azetine.90,105 Stevenson and co-workers utilized a Y(OTf)3-catalyzed [4+2]-cycloaddition between N-acetyl-2-azetine 84 (Fig. 5C) and various aromatic imines (91) to access azetidines fused to tetrahydroquinolines (92).106,107 These cycloadducts (92) proved unstable upon standing overnight, so they were trapped with aromatic amines to selectively break open the azetidine ring providing tetrahydroquinoline products (93).106,107 These examples highlight the value of [4+2]-cycloaddition reactions resulting in the rapid formation of molecular complexity in the form of polycyclic azetidine scaffolds.
Olefin-containing compounds can often be induced to dimerize or polymerize under thermal or photochemical conditions. 2-Azetines consist of a cyclic enamine that enables a photochemical dimerization process (Fig. 5D). Dave and co-workers disclosed the [2+2]-cycloaddition of N-acetyl-2-azetine 84 to an approximately 1:1 ratio of the two possible head-to-head dimers 94 and 95 in a 52% combined yield.108 This dimerization reaction was initiated by irradiation from a 500 Watt high pressure mercury lamp, and no head-to-tail dimerized products were observed.108
Synthesis of 3-, 5-, 6-membered and larger heterocycles
In addition to being used as intermediates to access azetidines, azetines can also be used to form 3-, 5-, 6-membered and larger heterocycles. The ring-contraction of isolated azetines for the synthesis of azirines has rarely been reported in the literature. Only a single example was reported by Regitz and co-workers in 1988, who observed that upon heating or irradiation of a dihydrotriazole (96), azirine 97 formed (Fig. 6A).109 This was proposed to occur via a zwitterionic intermediate.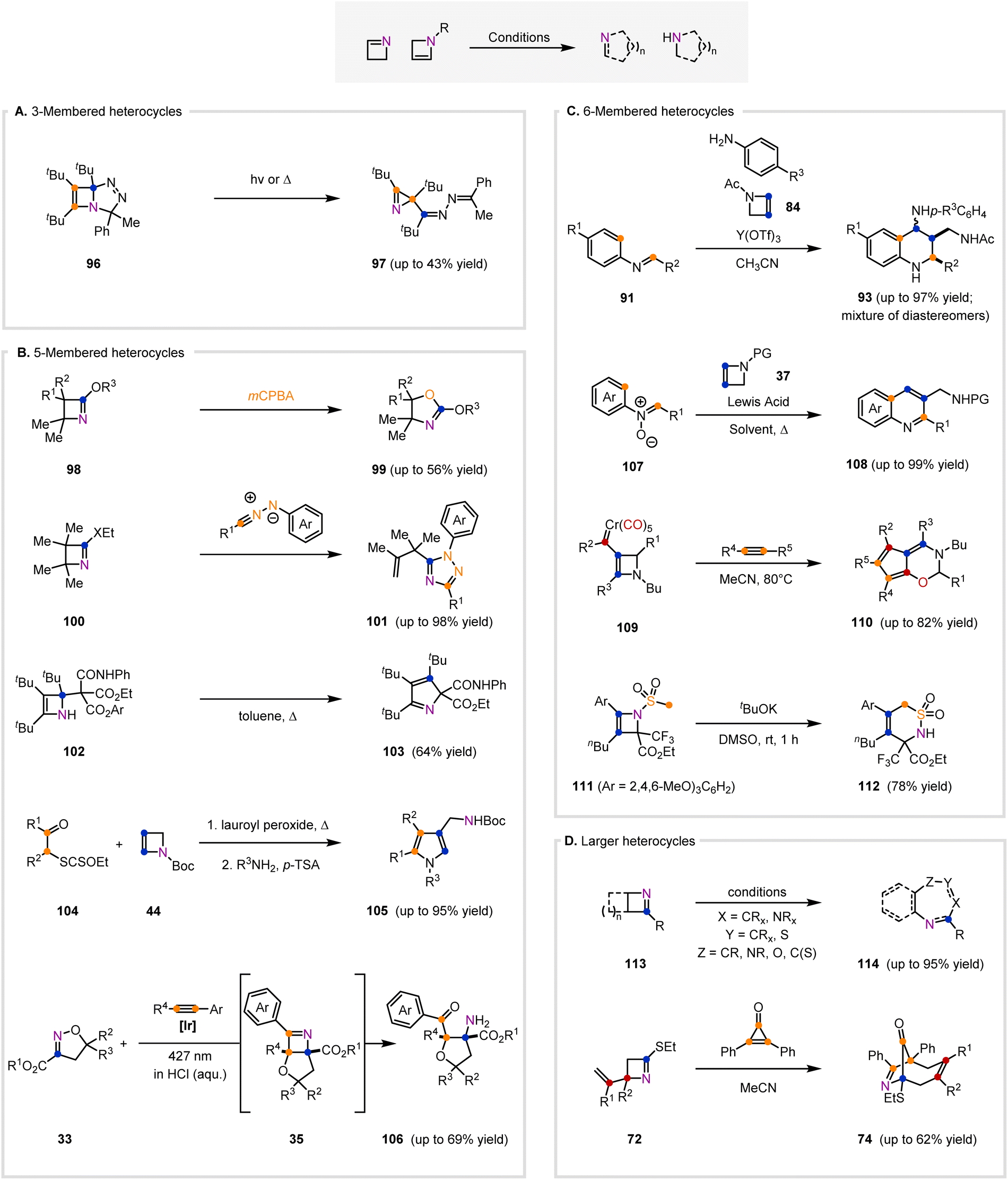 | ||
| Fig. 6 Methods that convert 1- and 2-azetines to 3-, 5-, 6-, and larger heterocycles. (A) Method that converts a fused, bicyclic 2-azetine to 3-membered nitrogen-containing heterocycles in the presence of light or heat. (B) Methods that transform 1- and 2-azetines to 5-membered nitrogen-containing heterocycles. (C) Methods that transform 2-azetines to 6-membered nitrogen-containing heterocycles. (D) Methods that convert 1-azetines to larger nitrogen-containing heterocycles. | ||
Conversely, the ring-expansion of isolated azetines has been more widely studied, mostly to result in 5- and 6-membered heterocycles. In 1973, Aue and co-workers reported one of the first examples of a 5-membered heterocycle that was derived directly from an isolated azetine.110 The initial report described that oxidation of tetra-substituted azetine 98 (R1 = R2 = Me) provided an oxazoline (99) in 56% yield (Fig. 6B). A subsequent report discussed the effect of R1 and R2 substitution on azetine 98 on ring-expansion.111 Oxidation of tri-substituted 98 (R1 = Me, R2 = H) resulted in a product mixture of 99 and a nitroso ester that is proposed to form via a bicyclic oxazirane intermediate. The authors proposed that 99 forms via a Baeyer–Villiger oxidation and is favored when R1 ≠ H or R2 ≠ H due to the increased migratory aptitude of alkyl substituents.
Smalley and co-workers reported that 2-ethoxy- and 2-ethylthio-1-azetines (100) and arylnitrilimines underwent a 1,3-dipolar cycloaddition to afford 1,2,4-triazoles (101), rather than the expected fused bicyclic triazole that would arise from a direct [3+2]-cycloaddition (Fig. 6B).93 The absence of –XEt (X = O, S) in 101 suggests that after the cycloaddition, –XEt is expelled to result in a resonance-stabilized triazolium intermediate. This intermediate can undergo ring-opening and subsequent aromatization to the observed product 101. Nitrilimines incorporating a nitrophenyl substituent resulted in only the non-rearranged bicyclic triazole, likely due to the decreased stability of the proposed triazolium intermediate.
Azetines have also been used to access various pyrrole scaffolds. In 1997, Regitz and co-workers reported that azetine 102 underwent thermolysis in toluene to the 2H-pyrrole 103 (Fig. 6B).112 Zard's group reported a two-step approach towards 4-(aminomethyl)pyrroles (105), which occurs via an isolable azetidine intermediate (38, Fig. 4A) that is observed from the radical addition of 2-azetine 44 (Fig. 6B) to xanthates (104).66,67 Upon addition of excess ammonia or amine to the azetidine intermediate (38, Fig. 4A), the xanthate group on 38 undergoes aminolysis, which induces fragmentation of the azetidine ring and allows for condensation of the amine to furnish products 105. Both 2,4-disubstituted and 2,3,4-trisubstituted pyrroles were obtained in moderate to high yields, 64–95% (from the isolated azetidine intermediate 38, Fig. 4A) and 58–68% (from the xanthate 104 and 2-azetine 44, Fig. 6B), respectively.
Recently, Schindler and co-workers reported a one-pot photochemical method enabled by triplet energy transfer to access highly-functionalized tetrahydrofurans 106 (Fig. 6B).11 Irradiation of 2-isoxazolines (33) and aryl alkynes in a mixture of acetonitrile and 0.1 M HCl furnished tetrahydrofuran products 106 in up to 69% yield. Product 106 forms from in situ hydrolysis of an azetine intermediate (35) that forms via an initial [2+2]-cycloaddition and subsequent rearrangement. Both terminal alkynes (R4 = H) and internal alkynes (R4 = CO2Me, Me) were tolerated. For terminal alkynes, both electron-donating and electron-withdrawing groups were also compatible with the reaction conditions. However, substrates in which R2 and R3 were larger led to lower reactivity.
In an extension of an aza Diels–Alder reaction developed using 2-azetines, Stevenson and co-workers demonstrated a one-pot method to access tetrahydroquinolines from N-acetyl-2-azetine 84 and N-aryl imines (91) in the presence of yttrium(III) triflate as a Lewis acid catalyst. Following the aza Diels–Alder cyclization to generate intermediate tetrahydroquinolines bearing fused azetidines (92, Fig. 5C), the aniline is able to facilitate ring-opening of the azetidines to ultimately generate substituted tetrahydroquinolines with a large preference for the less sterically congested diastereomer (93, Fig. 6C).106
Stevenson and co-workers, while screening conditions to convert their tetrahydroquinolines (93), found that upon heating the substituted tetrahydroquinolines (93) in the presence of yttrium(III) triflate, they were able to form quinolines in good yield. The authors subsequently showed that quinolines (108) could be formed from the corresponding N-acetyl-2-azetine (84) and N-aryl imines (91) in a sequential one-pot reaction sequence.107
Yan and co-workers showed that similar quinolines (108) could be formed from N-aryl-nitrones (107, X = O−) and N-Boc-2-azetines (37) under catalytic silver(I) triflate. In this case, the reaction was proposed to proceed through a [3+2]-cycloaddition to form an intermediate isoxazolidine before subsequent rearrangements.113
In addition to N-containing 6-membered heterocycles, O-containing oxazines can be obtained. As demonstrated by Barluenga and co-workers, 1,3-oxazines (110) were formed through the reaction of stable 2-azetine chromium carbenes (109) and alkynes.27 The proposed mechanism begins with the insertion of the alkyne into the carbene–metal bond, followed by CO insertion to ultimately form a metal–ketene intermediate. Upon an intramolecular nucleophilic attack of the ketene motif by the 2-azetine, a spirocyclic 1-azetinium intermediate is formed. This 1-azetinium intermediate can then undergo a ring-expansion and rearrange to form the product 1,3-oxazine (110).
The synthesis of sulfur-containing heterocycles was demonstrated by Ito and Hara in 2021. Their method shows the formation of 6-membered cyclic sulfonamide 112 upon treating N-mesylated 2-azetine 111 with three equivalents of potassium tert-butoxide.9 The authors hypothesized the reaction proceeded via a [2,3]-rearrangement following deprotonation of the mesyl group.
Azetines (113, Fig. 6D) have also been used as building blocks to construct seven-membered rings such as diazepines, triazepines, oxadiazepines, oxazepines, and a thiazepine (114).91,92,114–117 In 1975, Rees, Storr, and coworkers reported the conversion of 2-phenylbenzazete (113, R = Ph) to four examples of a 1,3,5-oxadiazepine product (114, X = N, Y = CPh, Z = O).91 Storr and colleagues continued this work in 1978 when they published the reaction of 2-phenylbenzazete (113, R = Ph) with nitrile imines to give 1,3,5-triazepine products (114, X = N, Y = CPh, Z = NPh).92 This work was later expanded to allow for access to benzodiazepines (114, X = NMe, Y = CAr, Z = CPh) from 2-phenylbenzazete (113, R = Ph) and munchones. This product likely forms from the thermodynamically favorable decomposition of the initial cycloadduct.114 It is of interest to note that the carbon (indicated by the blue circle) bearing the R group can ultimately end up at the Y or Z position opposed to the indicated position in the final product depending on the operative mechanism. The Tsuchiya group also explored the formation of seven-membered heterocycles from azetines.115,116 In 1986 and 1987, they disclosed the formation of 1,4-oxazepines (114, X = CR, Y = CR, Z = O) from aza-tricycloheptenes (113).115,116 Another interesting example from Schaumann and co-workers demonstrates formation of a thiazepine (114, X = CMe2, Y = S, Z = C(S)) from a bicyclic 2-azetine-thiazolidine precursor opposed to a 1-azetine in the presence of cesium fluoride.117
The formation of larger polycyclic ring systems was shown by Hemming and co-workers in their 2006 report. Therein, the authors demonstrated the formation of azabicyclo[4.2.1]nonene products (74, Fig. 6D) from 1-azetines (72) with cyclopropenones. These reactions were proposed to proceed through a Michael-type addition of the 1-azetine (72) into the cyclopropenone, and subsequent ring expansion of the cyclopropenone followed by an aza-Cope [3,3]-rearrangement.99
Synthesis of complex molecules enabled by azetines
Historically, the use of azetines to synthesize more complex molecules has favored addition and cycloaddition methods, as discussed above. However, the use of these compounds to access other complex molecules shows the potential for the use of azetines to access a diverse range of new products (Fig. 7).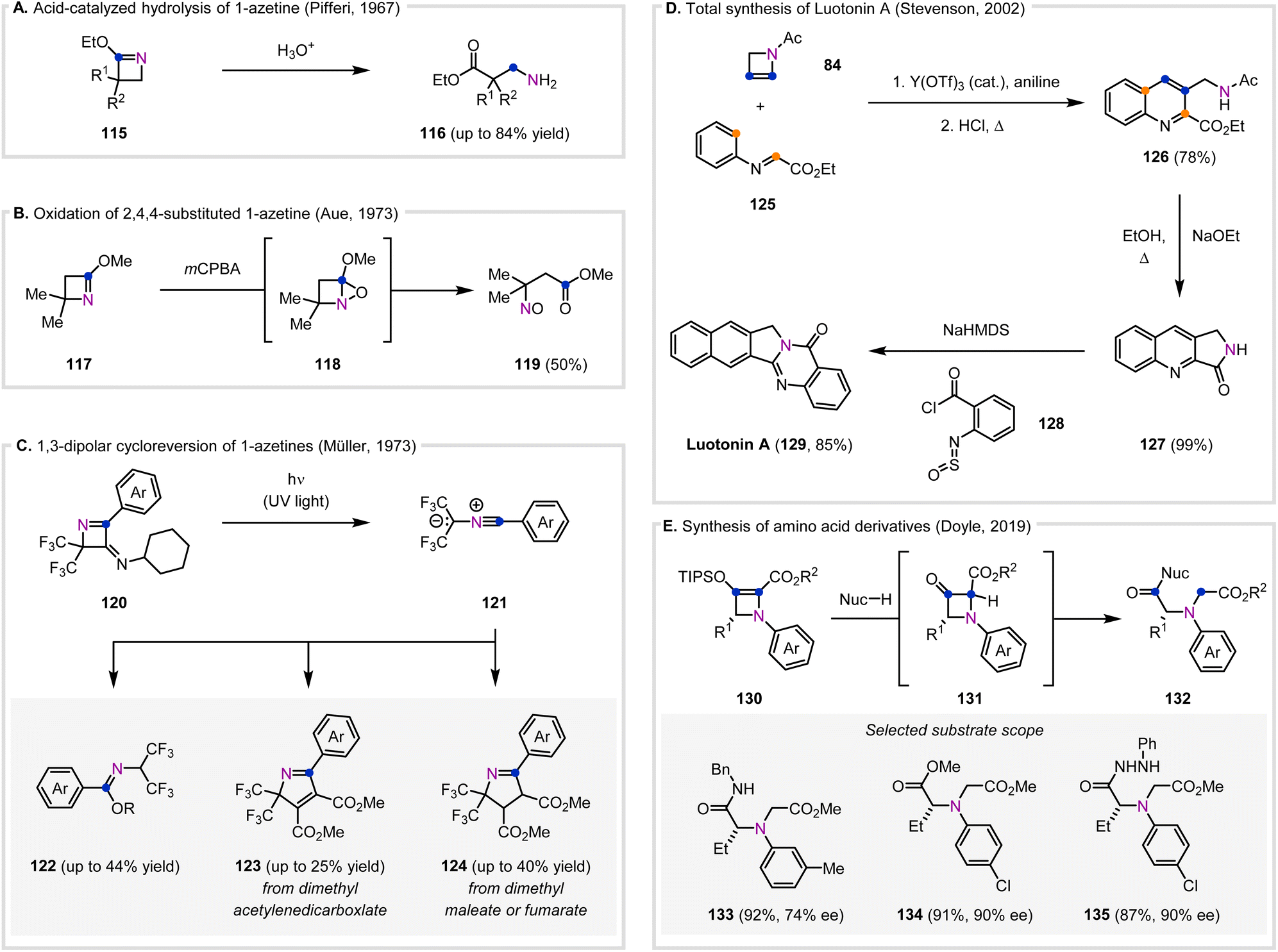 | ||
| Fig. 7 Synthesis of complex molecules enabled by azetines. (A) First known report of acid-catalyzed hydrolysis of 1-azetines. (B) Oxidation of 2,4,4-substituted 1-azetines to acyclic nitroso esters. (C) Photochemical[3+1]-cycloreversion of 1-azetines to nitrile ylide intermediates. (D) A concise, four-step synthesis of Luotonin A, derived from N-acetyl-2-azetine. (E) Synthesis of chiral α-amino acid derivatives from chiral 2-azetines. | ||
While imine hydrolysis of 1-azetines can be detrimental to their stability, this reaction can also be employed to form value-added products. The first known report of 1-azetine hydrolysis was described by Pifferi and co-workers in 1967 (Fig. 7A), who observed that dilute HCl facilitated hydrolysis of azetines 115 to afford ethyl α,α-disubstituted β-aminopropionates 116 (Fig. 7A).36 Additionally, when R1 = R2 = Ph, 3,3-diphenylazetidin-2-one was formed as a minor by-product.
In addition to hydrolysis, the imine moiety of 1-azetines has also been documented to be sensitive to oxidation (Fig. 7B). Aue and co-workers reported that upon exposure to mCPBA, 2-methoxy-4,4-dimethylazetine (117) oxidized to form the non-isolable 1-aza-5-oxabicyclo[2.1.0]pentane 118 that further oxidized to afford the acyclic nitroso ester 119 (Fig. 7B).110
In contrast to the formation of a reactive cyclic intermediate such as 118 (Fig. 7B) from a 1-azetine, Müller and co-workers diversified an acyclic nitrile ylide intermediate (121) that originated from 1-azetine 120 (Fig. 7C) to form a variety of products (122–124).118 Irradiation of 1-azetines (120) in benzene with UV light resulted in the reversible [3+1]-cycloelimination of isocyanide to afford nitrile ylides 121. The nitrile ylides (121) were trapped by alcohols, dimethyl acetylenedicarboxylate, or dimethyl maleate or fumarate to provide benzimidic ester (122), 2H-pyrrole (123), or 1-pyrroline (124) products respectively. The use of acrylic esters as the dipolarophile resulted in a mixture of regioisomers. Importantly, the diversity of both cyclic and acyclic products shows that azetines can be utilized to form desirable products that are not heterocycles, examples of which are much more limited than methods resulting in new heterocyclic products.
There are only two known examples of azetines being used as precursors in natural product synthesis. In 1991, Jung and coworker reported a three-step synthesis of (±)-δ-coniceine, a 1-azabicyclo[4.3.0]nonane, derived from a 1-acyl-2-azetine.119 Initial electrocyclic ring-opening of the 1-acyl-2-azetine allowed for an intramolecular Diels–Alder reaction with a terminal olefin moiety on the acyl-chain. Subsequent hydrogenation afforded (±)-δ-coniceine. In 2000, Stevenson and co-worker reported a concise, four-step synthesis of the biologically active alkaloid Luotonin A (129), in which N-acetyl-2-azetine (84) was used as an initial precursor (Fig. 7D).120 An initial Diels–Alder reaction of 84 and 125, followed by an elimination–aromatization sequence under acidic conditions, afforded the 2,3-disubstituted quinoline 126 in 78% yield. The authors attempted to oxidize 126 with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ), but instead recovered 126 in 80% yield. Alternatively, cyclization of 126 using NaOEt provided lactam 127 in 99% yield. Final addition of 2-sulfinylaminobenzoyl chloride (128) under basic conditions afforded Luotonin A (129). While a yield for 129 was not reported by Stevenson and co-worker,120 they cited a previous report in which this final transformation using 128 was reported to occur in 85% yield.121
More recently, Doyle and co-workers described the synthesis of amino acid derivatives (132) using chiral donor–acceptor azetines (Fig. 7E).12 Upon treatment with a nucleophile, 2-azetine-2-carboxylates 130 underwent strain-induced ring-opening via 3-azetidinone carboxylate intermediates (131, Fig. 7E). Amine nucleophiles with electron-withdrawing substituents on the N-aryl group provided moderate to high yields and high enantiopurities (63–96% yield, up to >99% ee), while electron-donating substituents resulted in diminished enantiopurity (e.g.133). Other nucleophiles such as alcohols, hydrazines, hydroxylamine, and ammonia were also compatible (e.g.134–135) in good yields and enantiopurities. Mechanistic studies suggested that two equivalences of the nucleophile are required, with the first equivalent desilylating the TIPS group and the second equivalent adding into the carbonyl of 131. In contrast to the β-amino acid derivatives obtained via hydrolysis of 1-azetines (Fig. 7A), this method allows for modular access to chiral, highly functionalized α-amino acid derivatives from chiral 2-azetines.
Azetines as proposed intermediates
Many discussions of azetines in early literature propose them as non-isolable intermediates towards various complex structures. A common reaction pathway for azetines is electrocyclic ring-opening to the corresponding azabutadiene, as shown by the reversible reaction between 2-azabutadiene 141 and 1-azetine 142 (Fig. 8B).122 In 1972, Cantrell reported that benzonitrile (136) underwent a photochemical [2+2]-cycloaddition with excess 2,3-dimethyl-2-butene or 1,1-dimethoxy-2,2-dimethylethylene (137) to afford 139 in 71% and 45% yield, respectively (Fig. 8A).123 This was postulated to occur via a [2+2]-cycloaddition of 136 in the singlet excited state to form the 1-azetine intermediate (138), which subsequently underwent electrocyclic ring-opening. While 138 was not detected or isolated in the initial report, its isolation was later reported.30 The electrocyclic ring-opening of 1-azetines was also proposed as a reversible process in the pyrolysis of 2H-azirines 140 by Bergman and co-workers in 1974 (Fig. 8B).122 The authors proposed that 140 undergoes two sequential hydrogen atom transfers to first form a carbene intermediate and then 141, which is in equilibrium with 142. Subsequently, 142 undergoes a retro-[2+2]-cycloaddition to 143 and 144.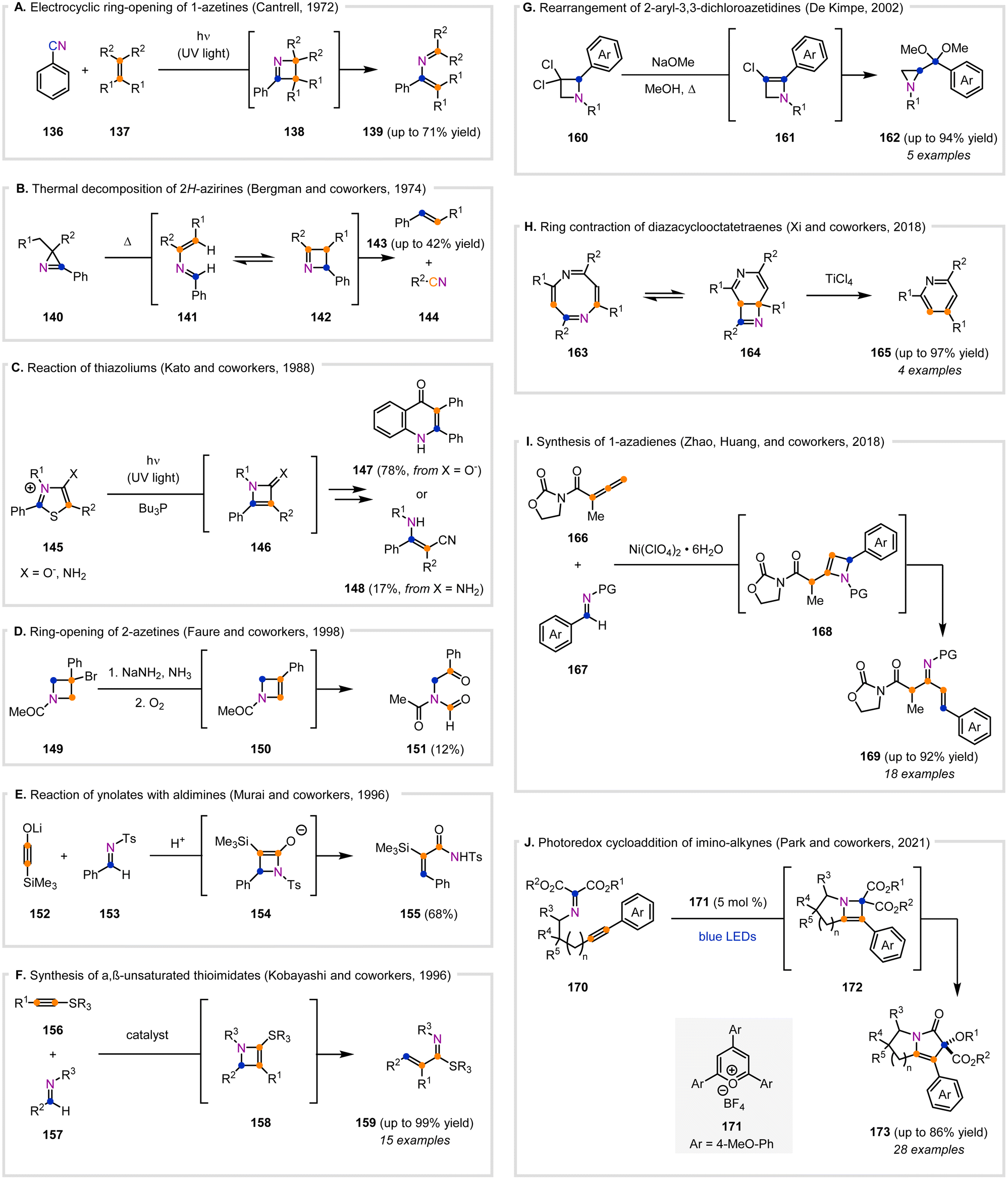 | ||
| Fig. 8 Azetines as proposed intermediates. (A) Electrocyclic ring-opening of a proposed 1-azetine intermediate. (B) Thermal decomposition of 2H-azirines, which is proposed to occur via a 1-azetine intermediate in equilibrium with a 2-azabutadiene. (C) Photochemical reaction of thiazoliums via a 2-azetine intermediate. (D) Ring-opening of proposed 2-azetine intermediates. (E) [2+2]-Cycloaddition of silylynolates and aldimines. (F) [2+2]-Cycloaddition of alkynyl sulfides and imines. (G) Rearrangement of 2-aryl-3,3-dichloroazetidines to azridines via a 2-azetine intermediate. (H) Ring contraction of diazacyclooctatetraenes to pyridines via a 1-azetine intermediate. (I) Formation of azabutadienes from allenyl imides and aldimines via a stepwise [2+2]-cycloaddition. (J) Intramolecular photoredox cycloaddition of imino-alkynes. | ||
1-Azetines have been invoked as intermediates in the photochemical reactions of thiazolium compounds. Kato and co-workers observed that thiazoliums 145 underwent desulfurization using UV light and tributylphosphine (Fig. 8C).124 They postulated that 145 initially isomerizes to a bicyclic intermediate and subsequently desulfurizes to form an azetone (X = O) or imino 2-azetine (X = N) (146) intermediate, which then undergoes ring-opening to the corresponding ketene or ketenimine, respectively. While the ring-closed products (147) were observed when X = O−, acyclic enaminonitriles (148) were observed when X = NH2.
Additionally, azetine intermediates have been proposed to result upon elimination reactions of intermediate azetidines. Faure and co-workers discussed that elimination of HBr from 149 resulted in 151, rather than the expected 2-azetine product (Fig. 8D).125 Instead, the 2-azetine is proposed to form transiently as intermediate 150, which then reacts with atmospheric oxygen to form an oxetane species that undergoes ring-opening to 151. A polymer byproduct arising from 150 was also observed, demonstrating the reactivity of this intermediate.
2-Azetines are commonly invoked as intermediates in the reaction of alkyne derivatives with aldimines. Murai and co-workers disclosed the formation of 155 from the silyl-substituted lithium ynolate 152 and aldimine 153 under acidic conditions (Fig. 8E).126 This reaction proceeds through a 2-azetine intermediate 154 that forms via a [2+2]-cycloaddition. Similarly, in 1996, Kobayashi and co-workers observed the formation of 159 from alkynyl sulfides 156 and imines 157 (Fig. 8F).127 An initial [2+2]-cycloaddition between 156 and 157, catalyzed by Lewis acids including Sc(OTf)3, Yb(OTf)3, or BF3·OEt2 (10–100 mol%), resulted in the formation of the non-isolable intermediate 158. This intermediate (158) subsequently fragmented via cycloreversion to 159. Substituted alkynyl sulfides (156, R1 = Me, Bu) gave moderate to high yields (40–100%), while terminal alkynyl sulfides (156) yielded only 8% in a single example. An intramolecular example provided a cyclohexene thioimidate product in 46% yield. Analogous methods for the synthesis of α,β-unsaturated selenylimidates128 and α,β-unsaturated amidines16,129 have also since been reported.
In contrast to the formation of acyclic products as mentioned above (Fig. 8A–F), azetines have also been proposed as intermediates in the synthesis of aziridines from azetidines (Fig. 8G). De Kimpe and co-workers observed that in the presence of NaOMe, 3,3-dichloroazetidines (160) form aziridines (162) in refluxing methanol (Fig. 8G).130 Elimination of HCl from 160 results in the formation of 161, after which addition of methanol allows for a bicyclic aziridinium intermediate to form that ring-contracts to 162 in high yields of up to 94%. The authors also reported that treatment of 160 with NaH in DMSO and subsequent hydrolysis afforded 2-acylaziridines in modest yields (46–59%). In a later report, the authors disclosed that under similar basic conditions, 2,4-diaryl-3,3-dichloroazetidines afforded benzimidoyl-substituted alkynes.131 This is proposed to proceed through elimination of HCl to a 3-chloro-2-azetine intermediate, which undergoes electrocyclic ring-opening. The acyclic intermediate then undergoes elimination of HCl to furnish the alkyne products.
The reversibility of the electrocyclic ring-opening of azetines has also been exploited for the synthesis of pyridine derivatives. Xi and co-workers reported the formation of 2,4,6-trisubstituted pyridines (165) from 2,4,6,8-tetrasubstituted 1,5-diazacyclooctatetraenes (163, Fig. 8H). This is proposed to proceed through electrocyclic ring-closing of 163 to 164 that then coordinates to TiCl4 through the azetine nitrogen atom to induce a retro-[2+2]-cycloaddition to 165.132 Similar scaffolds of 1H-triazepines were also previously reported by Regitz and co-workers to undergo ring-closing to 1H-pyrazoles via a bicyclic 1-azetine intermediate.112
The mechanism for the reaction between alkyne derivatives and aldimines (Fig. 8E and F) can generally be rationalized by a concerted [2+2]-cycloaddition. In contrast, Zhao, Huang, and co-workers disclosed a method for the synthesis of 1-azadienes (169) through a stepwise [2+2]-cycloaddition between allenyl imides 166 and aldimines 167 (Fig. 8I).133 Mediated by a nickel-based Lewis acid, 166 and 167 underwent a stepwise [2+2]-cycloaddition and two sequential proton shifts to afford a 2-azetine intermediate (168). 2-Azetine 168 subsequently undergoes a conrotatory electrocyclic ring-opening to the trans product 169. Computational studies suggested that the presence of the 2-oxazolidinone group in 166 allows for the Lewis acid to bridge the oxygen atoms of the two carbonyls and facilitate proton transfer to 168.
Finally, Park and co-workers described a photochemical method for the synthesis of fused nitrogen-heterocycles (173) from imino-alkynes (170) using photocatalyst 171 and blue LEDs (Fig. 8J).134 Based on computational studies, it is suggested that the imino-alkyne 170 undergoes an intramolecular [2+2]-cycloaddition to 172, which then undergoes ring-opening and a second oxidation to rearrange to 173.
Conclusions
In summary, this review provides an overview of new methods to access 1- and 2-azetines and the importance of these compounds with respect to both their biological relevance and their utility as reactive intermediates, which allows access to a diverse array of value-added products. While methods to synthesize azetines have historically been limited, recent years have seen the development of innovative new approaches that overcome the challenges of azetine synthesis to access a range of both 1- and 2-azetine products. It is expected that this recent interest will promote further exploration of the properties and uses of these interesting heterocycles for biological and synthetic applications, which will in turn encourage the development of further synthetic approaches. In this area, the development of methods to synthesize azetines using simple or commercially available starting materials and reagents remains a challenge in the development of general methods to access these compounds easily.The comprehensive review of developed synthetic methods to convert 1- and 2-azetines to a variety of complex products, including 3- to 6-membered heterocycles, demonstrates the versatility of these functional groups to enable the synthesis of diverse products. However, to our knowledge, azetines have only been used as intermediates in two total syntheses, demonstrating this as an area of future growth and application of these compounds.
With the potential for development in both the area of synthesis and applications of 1- and 2-azetines, along with the largely unexplored potential biological activity of these compounds, we consider the field of azetine synthesis and applications to be rich with future opportunities that will surely see many exciting developments in the near future.
Author contributions
M.R.G., E.C.M., C.H.N., and E.R.W. contributed equally. E.R.W. and C.S.S. conceptualized the initial structure and topics for this review. All authors contributed to all further research, drafts, and the writing and editing of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Alfred P. Sloan Foundation, the David and Lucile Packard Foundation, the Camille and Henry Dreyfus Foundation, the National Science Foundation (NSF CHE-1654223) and NIH (R01-GM141340) for funding. E. R. W. thanks the National Science Foundation for a predoctoral fellowship.References
- H. Mughal and M. Szostak, Org. Biomol. Chem., 2021, 19, 3274–3286 RSC.
- A. D. Richardson, M. R. Becker and C. S. Schindler, Chem. Sci., 2020, 11, 7553–7561 RSC.
- D. J. St. Jean and C. Fotsch, J. Med. Chem., 2012, 55, 6002–6020 CrossRef CAS.
- N. A. Meanwell, Top. Med. Chem., 2015, 9, 283–382 CAS.
- M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC.
- Y.-Z. Shu, B. M. Johnson and T. J. Yang, AAPS J., 2008, 10, 178–192 CrossRef CAS PubMed.
- K. O. Marichev, K. Wang, K. Dong, N. Greco, L. A. Massey, Y. Deng, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2019, 58, 16188–16192 CrossRef CAS PubMed.
- A. N. Koronatov, N. V. Rostovskii, A. F. Khlebnikov and M. S. Novikov, Chem. Heterocycl. Compd., 2019, 55, 1185–1189 CrossRef CAS.
- S. Hara and S. Ito, Asian J. Org. Chem., 2021, 10, 788–792 CrossRef CAS.
- M. Colella, P. Musci, D. Cannillo, M. Spennacchio, A. Aramini, L. Degennaro and R. Luisi, J. Org. Chem., 2021, 86, 13943–13954 CrossRef CAS PubMed.
- E. R. Wearing, D. E. Blackmun, M. R. Becker and C. S. Schindler, J. Am. Chem. Soc., 2021, 143, 16235–16242 CrossRef CAS.
- K. O. Marichev, K. Dong, L. A. Massey, Y. Deng, L. De Angelis, K. Wang, H. Arman and M. P. Doyle, Nat. Commun., 2019, 10, 5328 CrossRef PubMed.
- L. A. Paquette, M. J. Wyvratt and G. R. Allen, J. Am. Chem. Soc., 1970, 92, 1763–1765 CrossRef CAS.
- J. C. Guillemin, J. M. Denis and A. Lablache-Combier, J. Am. Chem. Soc., 1981, 103, 468–469 CrossRef CAS.
- S. M. Bachrach and M. Liu, J. Org. Chem., 1992, 57, 209–215 CrossRef CAS.
- N. Shindoh, K. Kitaura, Y. Takemoto and K. Takasu, J. Am. Chem. Soc., 2011, 133, 8470–8473 CrossRef CAS PubMed.
- A. Hassner, J. O. Currie, A. S. Steinfeld and R. F. Atkinson, J. Am. Chem. Soc., 1973, 95, 2982–2987 CrossRef CAS.
- I. A. Smetanin, M. S. Novikov, A. V. Agafonova, N. V. Rostovskii, A. F. Khlebnikov, I. V. Kudryavtsev, M. A. Terpilowski, M. K. Serebriakova, A. S. Trulioff and N. V. Goncharov, Org. Biomol. Chem., 2016, 14, 4479–4487 RSC.
- J. S. Taylor and M. P. Cohrs, J. Am. Chem. Soc., 1987, 109, 2834–2835 CrossRef CAS.
- T. Douki and E. Sage, Photochem. Photobiol. Sci., 2016, 15, 24–30 CrossRef CAS PubMed.
- T. Douki, S. Rebelo-Moreira, N. Hamon and P.-A. Bayle, Org. Lett., 2015, 17, 246–249 CrossRef CAS PubMed.
- K. Haiser, B. P. Fingerhut, K. Heil, A. Glas, T. T. Herzog, B. M. Pilles, W. J. Schreier, W. Zinth, R. de Vivie-Riedle and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 408–411 CrossRef CAS PubMed.
- A. F. Khlebnikov and M. S. Novikov, Top. Heterocycl. Chem., 2016, 41, 143–232 CAS.
- R. Lagoutte, Q. Lefebvre, C. Salome and T. Fessard, in Comprehensive Heterocyclic Chemistry IV, ed. D. StC. Black, J. Cossy, C. V. Stevens and G. Evano, Elsevier, Amsterdam, 4th edn, 2022, vol. 2, ch. 2.03, pp. 159–211 Search PubMed.
- E. J. Culp, D. Sychantha, C. Hobson, A. C. Pawlowski, G. Prehna and G. D. Wright, Nat. Microbiol., 2022, 7, 451–462 CrossRef CAS PubMed.
- F. Meigong, Z. Yalin and F. Ping, Acta Chim. Sin., 1985, 43, 778 Search PubMed.
- J. Barluenga, A. Gómez, J. Santamaría and M. Tomás, Angew. Chem., Int. Ed., 2010, 49, 1306–1308 CrossRef CAS PubMed.
- D. P. Del'tsova and N. P. Gambaryan, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1978, 27, 760–763 CrossRef.
- N.-C. C. Yang, B. Kim, W. Chiang and T. Hamada, J. Chem. Soc., Chem. Commun., 1976, 729–730 RSC.
- T. S. Cantrell, J. Org. Chem., 1977, 42, 4238–4245 CrossRef CAS.
- A. Albini, E. Fasani and F. Giavarini, J. Org. Chem., 1988, 53, 5601–5607 CrossRef CAS.
- J.-C. Guillemin, J.-M. Denis, M.-C. Lasne and J.-L. Ripoll, Tetrahedron, 1988, 44, 4447–4455 CrossRef CAS.
- R. G. Kostyanovskii, I. M. Gella and Kh. Khafizov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1971, 20, 819–819 CrossRef.
- H. Bock and R. Dammel, Chem. Ber., 1987, 120, 1971–1985 CrossRef CAS.
- D. Bormann, Justus Liebigs Ann. Chem., 1969, 725, 124–129 CrossRef CAS.
- G. Pifferi, P. Consonni, G. Pelizza and E. Testa, J. Heterocycl. Chem., 1967, 4, 619–624 CrossRef CAS.
- J. Marchand-Brynaert, M. Moya-Portuguez, D. Lesuisse and L. Ghosez, J. Chem. Soc., Chem. Commun., 1980, 173–174 RSC.
- F. Jamshaid, M. Khan and K. Hemming, Molbank, 2015, 2015, M860 CrossRef.
- T. H. Koch, R. H. Higgins and H. F. Schuster, Tetrahedron Lett., 1977, 18, 431–434 CrossRef.
- A. P. Marchand, D. Rajagopal, S. G. Bott and T. G. Archibald, J. Org. Chem., 1994, 59, 1608–1612 CrossRef CAS.
- I. Yavari and M. Nematpour, Synlett, 2012, 2215–2218 CrossRef CAS.
- I. Yavari and M. Nematpour, Synlett, 2013, 1420–1422 CrossRef CAS.
- F. Effenberger and R. Maier, Angew. Chem., Int. Ed. Engl., 1966, 5, 416–417 CrossRef CAS.
- A. N. Baumann, M. Eisold, A. Music, G. Haas, Y. M. Kiw and D. Didier, Org. Lett., 2017, 19, 5681–5684 CrossRef CAS PubMed.
- K. R. Henery-Logan and J. V. Rodricks, J. Am. Chem. Soc., 1963, 85, 3524–3525 CrossRef CAS.
- D. M. Hodgson, C. I. Pearson and M. Kazmi, Org. Lett., 2014, 16, 856–859 CrossRef CAS PubMed.
- M. S. Novikov, I. A. Smetanin, A. F. Khlebnikov, N. V. Rostovskii and D. S. Yufit, Tetrahedron Lett., 2012, 53, 5777–5780 CrossRef CAS.
- M. S. Novikov, A. F. Khlebnikov, N. V. Rostovskii, S. Tcyrulnikov, A. A. Suhanova, K. V. Zavyalov and D. S. Yufit, J. Org. Chem., 2015, 80, 18–29 CrossRef CAS PubMed.
- A. Hassner, J. O. Currie, Jr., A. S. Steinfeld and R. F. Atkinson, Angew. Chem., Int. Ed. Engl., 1970, 9, 731–732 CrossRef CAS.
- A. F. Khlebnikov, M. S. Novikov and A. A. Amer, Tetrahedron Lett., 2004, 45, 6003–6006 CrossRef CAS.
- A. F. Khlebnikov, M. S. Novikov, A. A. Amer, R. R. Kostikov, J. Magull and D. Vidovic, Russ. J. Org. Chem., 2006, 42, 515–526 CrossRef CAS.
- A. Hassner and A. B. Levy, J. Am. Chem. Soc., 1971, 93, 2051–2053 CrossRef CAS.
- R. Jorritsma, H. Steinberg and Th. J. de Boer, Recl. Trav. Chim. Pays-Bas, 1981, 100, 307–312 CrossRef CAS.
- J. Barluenga, L. Riesgo, G. Lonzi, M. Tomás and L. A. López, Chem. – Eur. J., 2012, 18, 9221–9224 CrossRef CAS PubMed.
- A. Gieren, K. Burger and W. Thenn, Z. Naturforsch., B: J. Chem. Sci., 1974, 29, 399–402 CrossRef CAS.
- K. Burger and J. Fehn, Angew. Chem., Int. Ed. Engl., 1972, 11, 47–48 CrossRef CAS.
- H. Hofmann and H. Fischer, Liebigs Ann. Chem., 1990, 1990, 917–921 CrossRef.
- N. B. Ambhaikar, J. P. Snyder and D. C. Liotta, J. Am. Chem. Soc., 2003, 125, 3690–3691 CrossRef CAS PubMed.
- D. C. Liotta, N. B. Ambhaikar and M. Herold, Heterocycles, 2004, 62, 217 CrossRef.
- C. J. MacNevin, R. L. Moore and D. C. Liotta, J. Org. Chem., 2008, 73, 1264–1269 CrossRef CAS PubMed.
- D. Didier, A. N. Baumann and M. Eisold, Tetrahedron Lett., 2018, 59, 3975–3987 CrossRef CAS.
- T. W. Reidl and L. L. Anderson, Asian J. Org. Chem., 2019, 8, 931–945 CrossRef CAS.
- D. Didier and F. Reiners, Chem. Rec., 2021, 21, 1144–1160 CrossRef CAS PubMed.
- M. Andresini, L. Degennaro and R. Luisi, in Comprehensive Heterocyclic Chemistry IV, ed. D. StC. Black, J. Cossy, C. V. Stevens and G. Evano, Elsevier, Amsterdam, 4th edn, 2022, vol. 2, ch. 2.01, pp. 1–115 Search PubMed.
- N. Legrand, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 2000, 41, 9815–9818 CrossRef CAS.
- S. Han and S. Z. Zard, Org. Lett., 2014, 16, 1992–1995 CrossRef CAS PubMed.
- S. Han and S. Z. Zard, Tetrahedron, 2015, 71, 3680–3689 CrossRef CAS.
- D. S. Wulfman and T. R. Steinheimer, Tetrahedron Lett., 1972, 13, 3933–3936 CrossRef.
- V. Petrov and W. Marshall, J. Fluor. Chem., 2018, 207, 1–6 CrossRef CAS.
- C. Xu, R. Cheng, Y. Luo, M. Wang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18741–18747 CrossRef CAS PubMed.
- O. S. Artamonov, E. Y. Slobodyanyuk, D. M. Volochnyuk, I. V. Komarov, A. A. Tolmachev and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 3592–3598 CrossRef CAS.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef PubMed.
- A. L. Demain and R. P. Elander, Antonie Van Leeuwenhoek, 1999, 75, 5–19 CrossRef CAS PubMed.
- L. M. Lima, B. N. M. da Silva, G. Barbosa and E. J. Barreiro, Eur. J. Med. Chem., 2020, 208, 112829 CrossRef CAS PubMed.
- C. A. Townsend, Curr. Opin. Chem. Biol., 2016, 35, 97–108 CrossRef CAS PubMed.
- D. H. Aue and D. Thomas, J. Org. Chem., 1975, 40, 1349–1351 CrossRef CAS.
- K. Burger, J. Fehn and E. Müller, Chem. Ber., 1973, 106, 1–7 CrossRef CAS.
- D. P. Del'tsova, L. L. Gervits and A. A. Kadyrov, J. Fluor. Chem., 1996, 79, 97–102 CrossRef.
- V. A. Petrov, F. Davidson and W. Marshall, J. Fluor. Chem., 2004, 125, 1621–1628 CrossRef CAS.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- C. Jäckel, M. Salwiczek and B. Koksch, Angew. Chem., Int. Ed., 2006, 45, 4198–4203 CrossRef PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- D. Bormann, Chem. Ber., 1970, 103, 1797–1804 CrossRef CAS.
- F. Reiners, E. Joseph, B. Nißl and D. Didier, Org. Lett., 2020, 22, 8533–8537 CrossRef CAS PubMed.
- A. N. Baumann, F. Reiners, T. Juli and D. Didier, Org. Lett., 2018, 20, 6736–6740 CrossRef CAS PubMed.
- A. Music, A. N. Baumann, M. Eisold and D. Didier, J. Org. Chem., 2018, 83, 783–792 CrossRef CAS PubMed.
- C. W. Rees, R. Somanathan, R. C. Storr and A. D. Woolhouse, J. Chem. Soc., Chem. Commun., 1975, 740–741 RSC.
- P. W. Manley, R. Somanathan, D. L. R. Reeves and R. C. Storr, J. Chem. Soc., Chem. Commun., 1978, 396–397 RSC.
- A.-B. N. Luheshi, R. K. Smalley, P. D. Kennewell and R. Westwood, Tetrahedron Lett., 1990, 31, 127–130 CrossRef CAS.
- A.-B. N. Luheshi, R. K. Smalley, P. D. Kennewell and R. Westwood, Tetrahedron Lett., 1990, 31, 123–126 CrossRef CAS.
- K. Hemming, A.-B. N. Luheshi, A. D. Redhouse, R. K. Smalley, J. R. Thompson, P. D. Kennewell and R. Westwood, Tetrahedron, 1993, 49, 4383–4408 CrossRef CAS.
- K. Hemming, M. N. Khan, P. A. O'Gorman and A. Pitard, Tetrahedron, 2013, 69, 1279–1284 CrossRef CAS.
- F. Stierli, R. Prewo, J. H. Bieri and H. Heimgartner, Helv. Chim. Acta, 1983, 66, 1366–1375 CrossRef CAS.
- K. Hemming, A. D. Redhouse, R. K. Smalley, J. Robin Thompson, P. D. Kennewell and R. Westwood, Tetrahedron Lett., 1992, 33, 2231–2234 CrossRef CAS.
- K. Hemming, P. A. O'Gorman and M. I. Page, Tetrahedron Lett., 2006, 47, 425–428 CrossRef CAS.
- K. Hemming, M. N. Khan, V. V. R. Kondakal, A. Pitard, M. I. Qamar and C. R. Rice, Org. Lett., 2012, 14, 126–129 CrossRef CAS PubMed.
- A. C. B. Burtoloso and C. R. D. Correia, Tetrahedron Lett., 2006, 47, 6377–6380 CrossRef CAS.
- A. Hassner, M. J. Haddadin and A. B. Levy, Tetrahedron Lett., 1973, 14, 1015–1018 CrossRef.
- D. H. Aue and D. Thomas, J. Org. Chem., 1975, 40, 2552–2554 CrossRef CAS.
- D. H. Aue and D. Thomas, J. Org. Chem., 1975, 40, 2356–2359 CrossRef CAS.
- P. R. Dave, R. Duddu, R. Surapaneni and R. Gilardi, Tetrahedron Lett., 1999, 40, 443–446 CrossRef CAS.
- P. J. Stevenson, M. Nieuwenhuyzen and D. Osborne, Chem. Commun., 2002, 444–445 RSC.
- P. J. Stevenson, M. Nieuwenhuyzen and D. Osborne, Arkivoc, 2006, 2007, 129–144 Search PubMed.
- P. R. Dave, R. Duddu, J. Li, R. Surapaneni and R. Gilardi, Tetrahedron Lett., 1998, 39, 5481–5484 CrossRef CAS.
- U. J. Vogelbacher, M. Ledermann, T. Schach, G. Michels, U. Hees and M. Regitz, Angew. Chem., Int. Ed. Engl., 1988, 27, 272–274 CrossRef.
- D. Thomas and D. H. Aue, Tetrahedron Lett., 1973, 14, 1807–1810 CrossRef.
- D. H. Aue and D. Thomas, J. Org. Chem., 1974, 39, 3855–3862 CrossRef CAS.
- P. Bach, U. Bergstraesser, S. Leininger and M. Regitz, Bull. Soc. Chim. Fr., 1997, 134, 927–936 CAS.
- H. Yan, X. Li, C. Wang and B. Wan, Org. Chem. Front., 2017, 4, 1833–1838 RSC.
- P. W. Manley, C. W. Rees and R. C. Storr, J. Chem. Soc., Chem. Commun., 1983, 1007 RSC.
- J. Kurita, K. Iwata and T. Tsuchiya, J. Chem. Soc., Chem. Commun., 1986, 1188 RSC.
- J. Kurita, K. Iwata and T. Tsuchiya, Chem. Pharm. Bull., 1987, 35, 3166–3174 CrossRef CAS.
- E. Schaumann, J. Nieschalk, R. Isecke, C. Spanka, H. Mrotzek and W.-R. Förster, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 120, 349–350 CrossRef.
- K. Burger, W. Thenn and E. Müller, Angew. Chem., Int. Ed. Engl., 1973, 12, 155–155 CrossRef.
- M. E. Jung and Y. M. Choi, J. Org. Chem., 1991, 56, 6729–6730 CrossRef CAS.
- D. Osborne and P. J. Stevenson, Tetrahedron Lett., 2002, 43, 5469–5470 CrossRef CAS.
- H. Wang and A. Ganesan, Tetrahedron Lett., 1998, 39, 9097–9098 CrossRef CAS.
- L. A. Wendling and R. G. Bergman, J. Am. Chem. Soc., 1974, 96, 308–309 CrossRef CAS.
- T. S. Cantrell, J. Am. Chem. Soc., 1972, 94, 5929–5931 CrossRef CAS.
- H. Kato, K. Wakao, A. Yamada and Y. Mutoh, J. Chem. Soc., Perkin Trans. 1, 1988, 189 RSC.
- R. Bartnik, R. Faure and K. Gebicki, J. Chem. Crystallogr., 1998, 28, 119–123 CrossRef CAS.
- H. Kai, K. Iwamoto, N. Chatani and S. Murai, J. Am. Chem. Soc., 1996, 118, 7634–7635 CrossRef CAS.
- H. Ishitani, S. Nagayama and S. Kobayashi, J. Org. Chem., 1996, 61, 1902–1903 CrossRef CAS.
- Y. Ma and C. Qian, Tetrahedron Lett., 2000, 41, 945–947 CrossRef CAS.
- N. Shindoh, Y. Takemoto and K. Takasu, Chem. – Eur. J., 2009, 15, 7026–7030 CrossRef CAS PubMed.
- Y. Dejaegher, S. Mangelinckx and N. De Kimpe, J. Org. Chem., 2002, 67, 2075–2081 CrossRef CAS PubMed.
- S. Mangelinckx, V. V. Speybroeck, P. Vansteenkiste, M. Waroquier and N. D. Kimpe, J. Org. Chem., 2008, 73, 5481–5488 CrossRef CAS PubMed.
- Z. Huang, W.-X. Zhang and Z. Xi, Org. Lett., 2018, 20, 485–488 CrossRef CAS PubMed.
- S. Pang, X. Yang, Z.-H. Cao, Y.-L. Zhang, Y. Zhao and Y.-Y. Huang, ACS Catal., 2018, 8, 5193–5199 CrossRef CAS.
- H. Oh, B. Ryou, J. Park, M. Kim, J.-H. Choi and C.-M. Park, ACS Catal., 2021, 11, 13670–13679 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |