
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of fluorinated carbocyclic pyrimidine nucleoside analogues†
Caecilie M. M.
Benckendorff
ab,
Valentyna D.
Slyusarchuk
b,
Ningwu
Huang
c,
Marcelo A.
Lima
 a,
Mark
Smith
c and
Gavin J.
Miller
*ab
a,
Mark
Smith
c and
Gavin J.
Miller
*ab
aCentre for Glycosciences, Keele University, Keele, Staffordshire ST5 5BG, UK. E-mail: g.j.miller@keele.ac.uk
bLennard-Jones Laboratory, School of Chemical and Physical Sciences, Keele University, Keele, Staffordshire ST5 5BG, UK
cRiboscience LLC, 428 Oakmead Pkwy, Sunnyvale, CA 94085, USA
First published on 14th November 2022
Abstract
Analogues of the canonical nucleosides have a longstanding presence and proven capability within medicinal chemistry and drug discovery research. The synthesis reported herein successfully replaces furanose oxygen with CF2 and CHF in pyrimidine nucleosides, granting access to an alternative pharmacophore space. Key diastereoselective conjugate addition and fluorination methodologies are developed from chiral pool materials, establishing a robust gram-scale synthesis of 6′-(R)-monofluoro- and 6′-gem-difluorouridines. Vital intermediate stereochemistries are confirmed using X-ray crystallography and NMR analysis, providing an indicative conformational preference for these fluorinated carbanucleosides. Utilising these 6′-fluorocarbauridine scaffolds enables synthesis of related cytidine, ProTide and 2′-deoxy analogues alongside a preliminary exploration of their biological capabilities in cancer cell viability assays. This synthetic blueprint offers potential to explore fluorocarbanucleoside scaffolds, indicatively towards triphosphate analogues and as building blocks for oligonucleotide synthesis.
Introduction
Nucleoside analogues possess a privileged and accomplished history within therapeutic intervention strategies, most notably in the fight against viruses and cancer.1,2 They are also present within synthetic sequences of DNA and RNA, providing a cornerstone to nucleic acid therapies.3 Since its foundation in the 1950s, the field of nucleoside analogue chemistry has continually evolved, delivering generations of landscape defining therapeutics; notably this includes recent frontline treatments against COVID-19.4,5 As a result of these successes, the requirement for synthetic capability, both chemical and enzymatic, to evolve new pharmacophore space and manufacture these materials efficiently is ever growing.6–8A commonly encountered structural change is modification of the ribose ring (Fig. 1a); examples here include cytarabine, remdesivir and gemcitabine. Within this subtext of ribose ring modification, replacement of furanose oxygen with other heteroatoms or functional groups is often explored (Fig. 1b). Examples here include forodosine, a 4′-azanucleoside, and 4′-thionucleosides.9,10 The templating of additional ribose ring modifications, ontop of furanose oxygen replacement, enables further divergence to explore new modalities.11,12
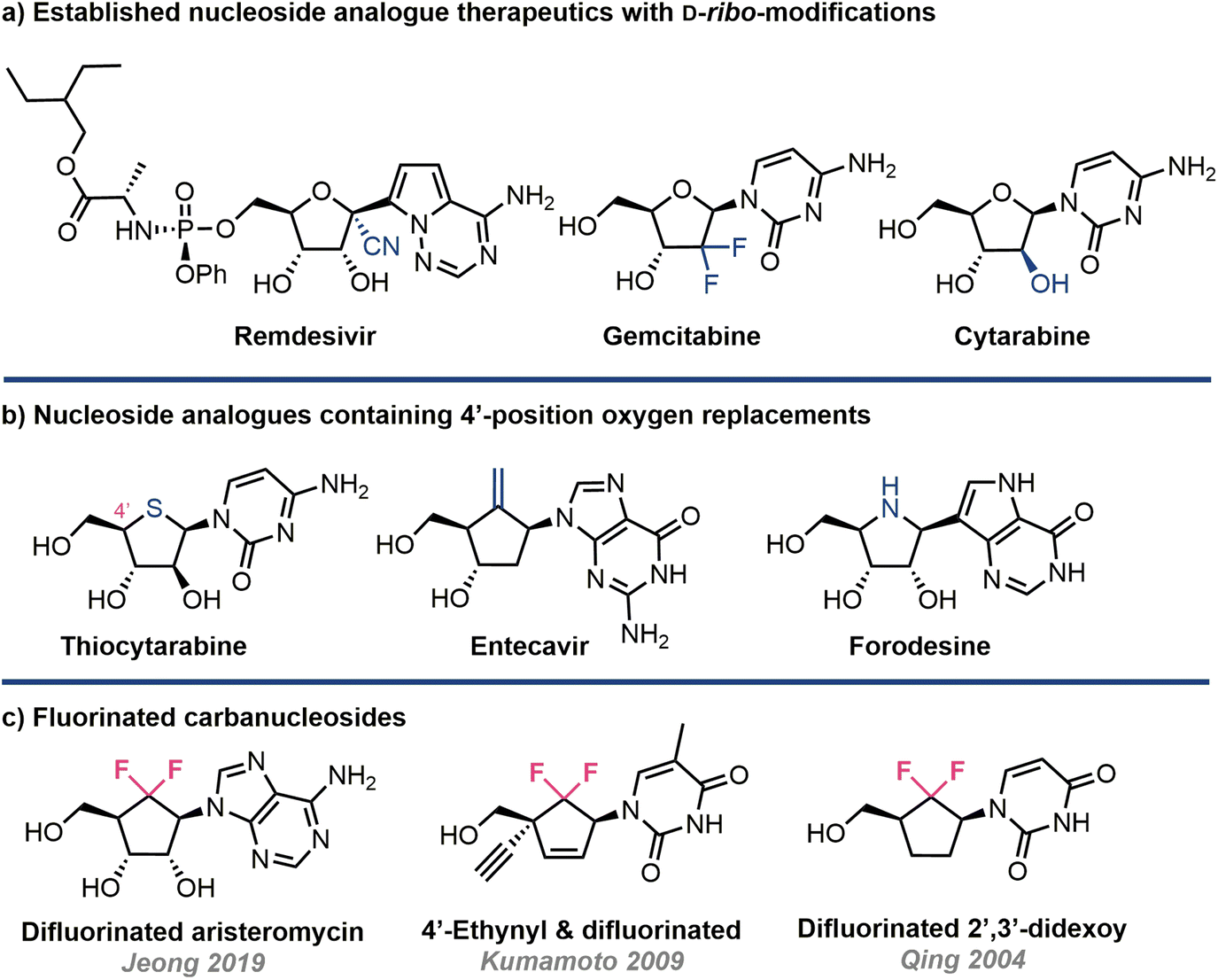 | ||
| Fig. 1 (a) Examples of nucleoside analogue drugs with furanose ring modifications shown in blue (b) replacement of furanose oxygen with other heteroatoms and carbon, in blue (c) key recent examples of the bioisosteric O → CF2 replacement for nucleoside analogues. | ||
A third and perhaps more extensively interrogated class of ethereal replacement harnesses carbon;13 carbanucleosides (a cyclopentane core with oxygen replaced by CH2) exist naturally as highly cytotoxic agents aristeromycin and neplanocin A. Carbocyclic nucleosides are resistant to enzymatic degradation, as the hemi-aminal linkage targeted by nucleoside phosphorylases is absent, and adopt alternative ring conformations compared to classical (C3′ or C2′ endo) systems.14,15 The carbocylic structural motif has successfully underpinned development of the therapeutics abacavir and entecavir and has recently been included within synthetic RNA oligonucleotides.16
In addition to using CH2 to replace the furanose ring oxygen, there are a limited number of reports that concomitantly build fluorine into this carbocylic modification, effectively introducing CF2 (Fig. 1c). For example, 6′6′-difluorinated purines have been developed to target RNA virus activity.17 This suggested bioisosteric replacement (and the related CHF), originally proposed by Blackburn to replace ethereal oxygen in pyrophosphates,18 remains underexplored as a pharmacophore.19–21 Synthetic methods to expedite accessing it,22 and further analogues therefrom, are therefore required to sustain the evolution of new generations of biorelevant nucleoside analogues.17,23–25
Established benefits from introducing fluorine to nucleoside analogues are evidenced by the drugs sofosbuvir (2′-deoxy-2′-C-methyl-2′-fluoro) and gemcitabine (2′,2′-difluoro-2′-deoxycytidine), alongside the biological capabilities of including 2′-fluorinated monomer components within oligonucleotides for medicinal chemistry.23,26 Notwithstanding this, the structural effect of replacing furanose oxygen with CF2, both in free nucleosides and nucleotides, and related DNA/RNA sequences is an area primed for exploration.27–29
In line with the achievements highlighted in Fig. 1c, we present herein our synthesis of a series of pyrimidine (uridine and cytidine) carbocyclic nucleosides, modified with β-mono- and di-fluorination at the 6′-position (Fig. 2). In addition, we access their ProTide prodrug forms and demonstrate the first examples of diversification to 2′-deoxygentated materials.
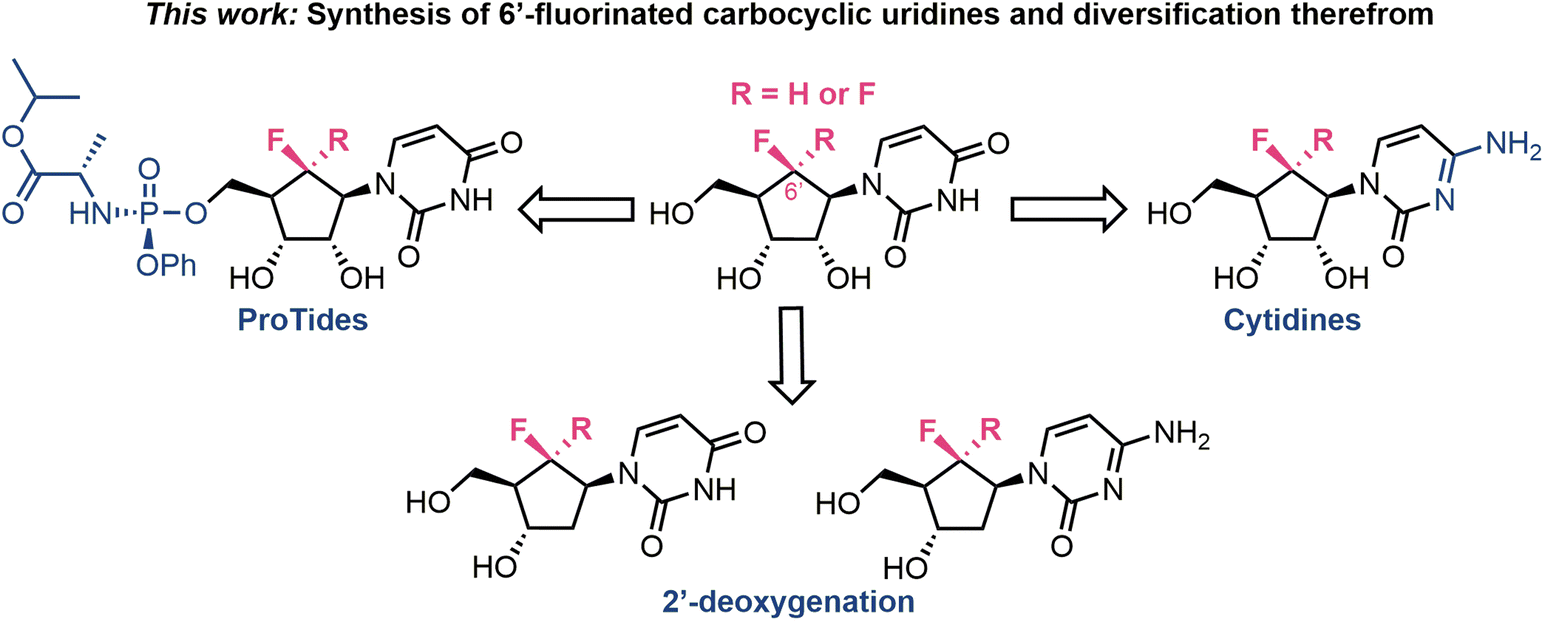 | ||
| Fig. 2 Overview of synthetic targets: access to 6′-mono- and di-fluorinated carbocylic uridines as a point to diversify analogues series to ProTide, cytidine and 2′-deoxygenated forms. | ||
Results and discussion
To access our fluorinated carbocylic nucleoside analogue series, we first required building blocks suitable for nucleobase installation and further diversification. Previous syntheses of bioisosteric CF2 replacement involved starting from commercially available fragments, where the gem-difluorination is already in place,25 alongside methods that add fluorine to the scaffold using electrophilic fluorination.24 Jeong and colleagues have reported syntheses of fluorinated nucleoside analogues, where the core nucleoside is accessed from enone 1, most commonly effecting conjugate addition of a Gilman reagent, to diastereoselectively install a tBu ether protected C5′-hydroxymethyl unit, followed by electrophilic fluorination of an appropriate silyl enol ether.17,30,31Our synthesis began similarly, using commercial isopropylidine protected cyclopentenone 1, where we envisaged conjugate addition of a vinyl group (Scheme 1), effectively masking the C5′-primary alcohol until a later oxidative cleavage step. To achieve this, we adopted a report by Schneller accessing aristeromycin analogues, that used a combination of vinyl magnesium bromide, TMSCl, HMPA, and CuBr·Me2S. However, in our hands we were unable to consistently replicate the yield of 80% reported for this transformation.32 Gratifyingly, when we modified the reagent combination to just vinyl magnesium bromide and CuI in THF, the successful isolation of 2 in 78% yield was enabled. This reaction proved reliable on scales up to 20 g and only one diastereoisomer of 2 was isolated, following chromatography. NMR data matched those reported by Schneller, indicating top face addition of the organocuprate, with the bottom face blocked by the 2,3-di-O-isopropylidine.
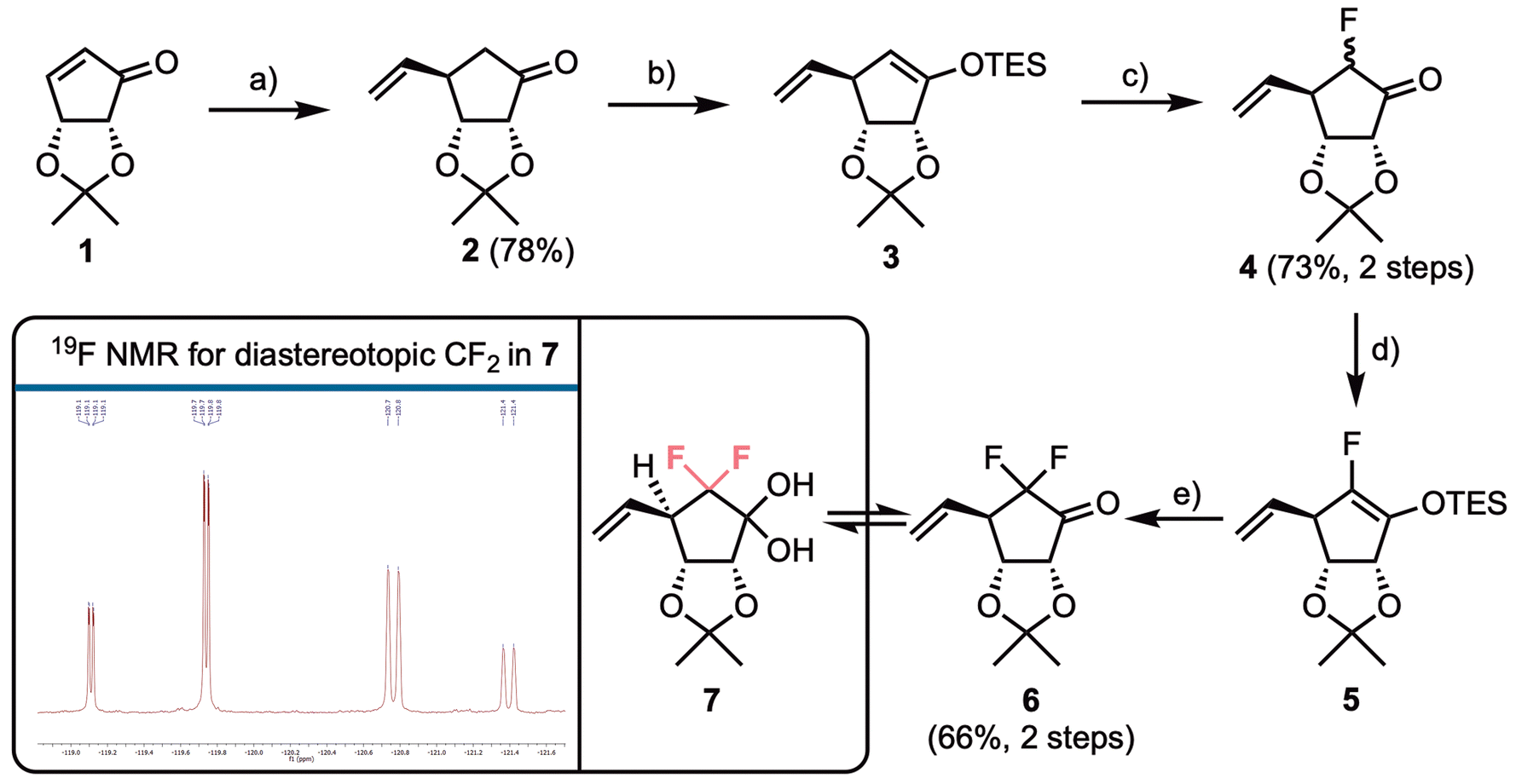 | ||
| Scheme 1 Synthesis of fluorinated cyclopentanone building blocks. (a) VinylMgBr, CuI, THF, −78 °C; (b) TESCl, Li/HMDS, THF, −78 °C; (c) Selectfluor®, DMF, 0 °C–rt, 8/1, 4-(R)/4-(S); (d) TESCl, Li/HMDS, THF, −78 °C; (e) Selectfluor®, DMF, 0 °C–rt, observed as hydrate; box illustrates 19F NMR (377 MHz, CDCl3) for 7, with splitting patterns for diastereotopic fluorines shown. | ||
α-Ketofluorination of 2 was completed using a two-step procedure of silyl enol ether formation, followed by reaction with Selectfluor®. Formation of 3 proceeded in quantitative crude yield with a significant downfield chemical shift observed for the vinyl ether proton (δH = 4.65 ppm). Silyl enol ether 3 was used immediately for electrophilic fluorination, delivering 4 in an isolated yield of 73% and with an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 preference for fluorination having occurred on the β-face of the system (i.e., giving the (R)-F diastereoisomer). Installation of fluorine α to the ketone in 2 was confirmed using 1H and 13C NMR, with large 1J and 2J couplings observed for the remaining α-proton in 4 (2JH,F = 50.5 Hz and 1JC,F = 201.2 Hz). Electrophilic fluorination of 2 with a C4-vinyl group in place has not been reported previously and compares favourably to results obtained by Jeong using a tBu protected hydroxymethyl C4 substituent [5:1, (R)/(S)] and a C4-hydroxyethyl homologue [(R)-F selective].17,31
:1 preference for fluorination having occurred on the β-face of the system (i.e., giving the (R)-F diastereoisomer). Installation of fluorine α to the ketone in 2 was confirmed using 1H and 13C NMR, with large 1J and 2J couplings observed for the remaining α-proton in 4 (2JH,F = 50.5 Hz and 1JC,F = 201.2 Hz). Electrophilic fluorination of 2 with a C4-vinyl group in place has not been reported previously and compares favourably to results obtained by Jeong using a tBu protected hydroxymethyl C4 substituent [5:1, (R)/(S)] and a C4-hydroxyethyl homologue [(R)-F selective].17,31
The diastereomeric mixture 4 was inseparable by silica gel chromatography and the mixture was used in a second fluorination via TES-enol either 5, affording difluorinated 6 in 66% yield over the two steps and reliably delivering this material in >10 g quantities. Ketone 6 was observed by 1H NMR to be in equilibrium with its hydrate form, 7. The presence of the diastereotopic CF2 group within 7 was confirmed using 19F NMR; the expected doublet of doublets observed for each fluorine, with a 19F–19F geminal coupling constant of J = 237.5 Hz, alongside smaller 3JF,H couplings to H4 of 9.0 and 21.6 Hz respectively. In addition, for one fluorine a small coupling (J = 1.8 Hz, presumably to OH) could be observed, altering the multiplicity to a doublet of doublets of doublets (Scheme 1, box).
After a diastereoselective NaBH4 reduction of monofluoroketone mixture 4 (Scheme 2), the resultant secondary alcohols 8–11 could be separated using chromatography with isolation of 10 as the major product in 70% yield. For this major (R)-F analogue, stereochemical assignment at C1 was confirmed through an X-Ray crystal structure of a derived C1-dinitrobenzoate (Scheme 2, boxes). The minor (S)-F diastereoisomer 8 was also obtained, in 3% yield, alongside the (R)-F C1 epimer 11, in 3% yield (compound 9 was not isolated). Both 8 and 11 were crystalline solids and X-ray crystallography also confirmed their stereochemical assignment at both C6 (fluorination site) and C1 (secondary alcohol, Scheme 2, boxes). Due to the small amount of 8 isolated, no further synthesis towards an (S)-F analogue was completed. Diastereoselective reduction of difluorinated 6/7 was completed in a similar manner giving alcohol 12 in 79% yield, with no evidence of a C1 epimer (Scheme 2).
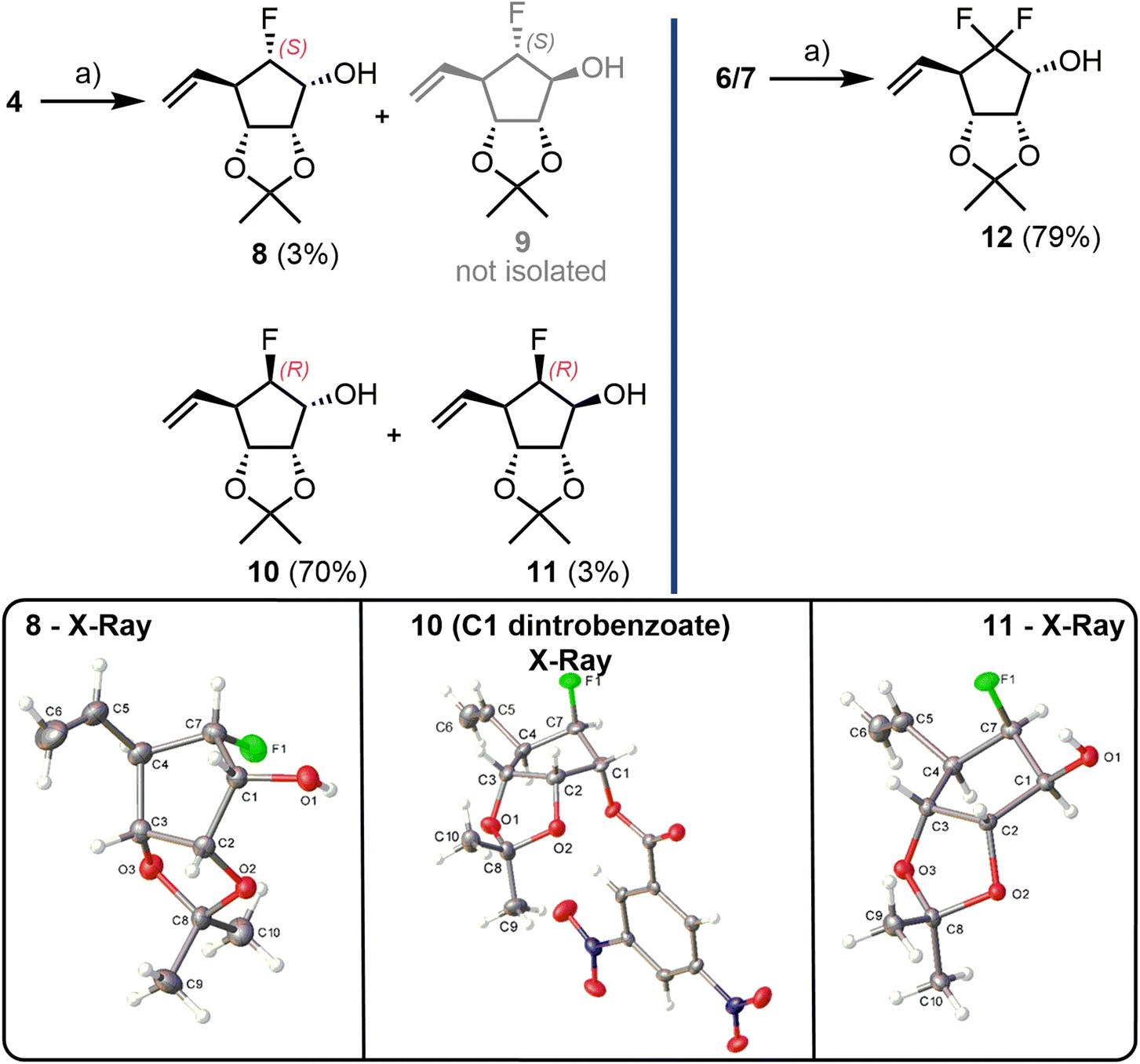 | ||
| Scheme 2 Diastereoselective reduction of fluorinated cyclopentanone building blocks. (a) NaBH4, MeOH, rt. | ||
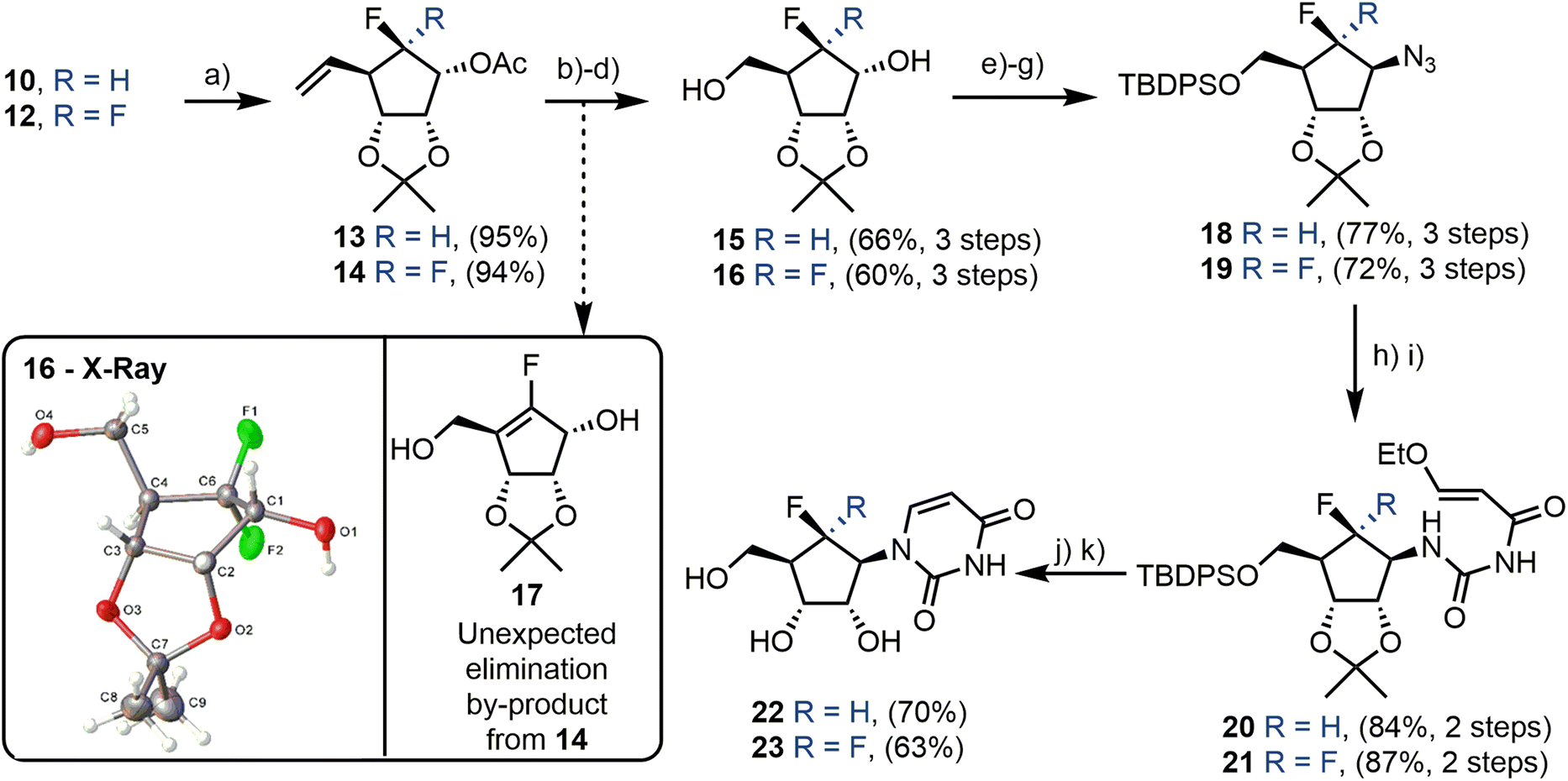
Secondary alcohols 10 and 12 were next acetylated at C1 in excellent yield (95% and 94% for 13 and 14 respectively, Scheme 3). This delivered appropriate materials for oxidative alkene cleavage, first with AD-mix β, to give a vicinal diol mixture, followed by cleavage using NaIO4. Subsequent reduction of the generated aldehyde with concomitant acetate removal furnished alcohols 15 and 16. gem-Difluoroalcohol 16 was crystalline and X-ray crystallography was again used to confirm the previous diastereoselective reduction had occurred from the top face (Scheme 3, box), to provide appropriate material for later installation of the nucleobase via stereochemical inversion at C1.
 | ||
| Scheme 3 Synthesis of mono- and gem-difluorinated uridines. (a) Ac2O, pyridine, DCM, rt; (b) AD-mix β, iPrOH, H2O, rt; (c) for 15: NaIO4 (1.7 equiv.), dioxane, H2O, rt, 1 h; for 16: NaIO4 (10 equiv.), dioxane, H2O, 0 °C, 6 h; (d) NaBH4, MeOH, rt; (e) TBDPSCl, imidazole, DMF; (f) Tf2O, pyridine; (g) NaN3, DMF, 100 °C; (h) H2, Pd/C, MeOH, rt; (i) (3-ethoxyacryloyl)isocyanate, DMF, rt; (j) H2SO4, 1,4-dioxane, reflux; (k) TBAF, THF, rt. | ||
This three-step cleavage proceeded efficiently in the case of the monofluorinated substrate, yielding 15 in 66% over three steps. A complication was however encountered with the difluorinated substrate 14, resulting in the desired material from NaIO4-mediated diol cleavage isolable only as a minor component and an elimination product (the enal) observed as the major product from this transformation. This led to the formation of 17 (Scheme 3, box) as the major product, following redcution. By lowering the reaction temperature to 0 °C and increasing the equivalents of NaIO4 five-fold, the unwanted elimination was suppressed and the desired material (16) was obtained. Following this optimisation, concomitant reduction and deprotection delivered 15 and 16, with 16 isolated in 60% yield over the three steps.
Alcohols 15 and 16 were next protected with TBDPS at the C5 position. Some additional TBDPS protection of the C1 secondary alcohol was observed in the case of 15. However, 15 was easily recoverable following global silyl-deprotection using TBAF in THF. Azidation of the remaining secondary alcohol was achieved via formation of an α-triflate, followed by stereoinversion using NaN3 to give 18 and 19 in yields of 77% and 72% respectively over three steps. Direct SN2 displacement of an α-mesylate with pyrimidine nucleobases or using Mitsunobu inversion were unsuccessful, in line with results reported for the related synthesis of fluorinated aristeromycin derivatives.30
Acryloyl urea intermediates 20 and 21 were next obtained, first by reduction of 18 and 19 to the C1 amine, which was then treated with (3-ethoxy-acryloyl)isocyanate, freshly prepared by reaction of 3-ethoxy-acryloyl chloride with AgOCN in toluene,33 to give 20 and 21 in yields of 84% and 87% over two steps. Finally, cyclisation to the desired pyrimidine and acetonide removal was achieved by treatment with H2SO4 in dioxane. A final TBAF deprotection yielded fluorinated uridines 22 and 23 in total yields of 9% and 6% respectively, and over 15 and 17 steps from 1.
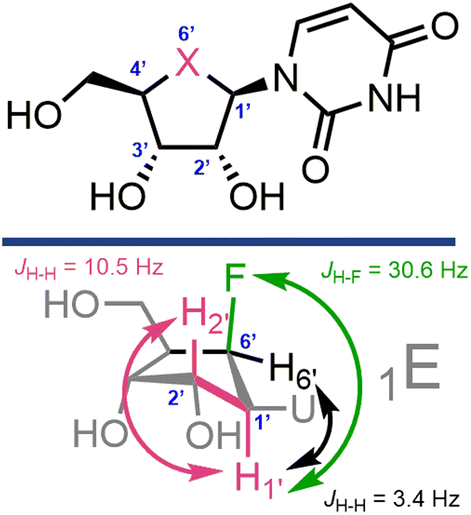
With 22 and 23 in hand we were interested to compare elements of their NMR data to canonical uridine. We noted an upfield chemical shift for the pseudo-anomeric position in 22 (δH = 4.89 ppm, δC = 62.3 ppm) relative to uridine (δH = 5.83 ppm, δC = 91.7 ppm); this effect was less pronounced in gem-difluorinated 23 (δH = 5.37 ppm, δC = 63.6 ppm). Furthermore, Table 1 illustrates selected observed 3J and 1J coupling constants for 22 and 23.
| X | 3 J (Hz) | ||||
|---|---|---|---|---|---|
| 1′-2′ | 1′-6′ | 1′-F | |||
|
O | 4.6 | — | — | |
| CHF | 10.5 | 3.4 | 30.6 | ||
| CF2 | 10.2 | — | 18.8, 10.0 | ||
| 1 J (Hz) | |||||
| X | 1′ | 2′ | 3′ | 4′ | |
| O | 171.8 | 152.4 | 149.7 | 150.1 | |
| CHF | 140.9 | 145.9 | 151.4 | 128.1 | |
| CF2 | 144.3 | 144.5 | 154.5 | 135.7 | |
First considering 3JH–H couplings, we noted changes between H1 and H2 for both the mono- and difluorinated systems. The 3JH1′–H2′ coupling in uridine is 4.6 Hz, but this increased to >10 Hz for both 22 and 23, indicating a change to this dihedral angle. Carbanucleosides tend to adopt a 1′-exo envelope conformation (1E),34 due to lack of the anomeric effect and gauche interactions between the furanose oxygen and the 2′- and 3′-OH groups. This 1E conformation is suggested to be energetically and sterically favourable, placing the bulky nucleobase in a pseudoequatorial position.14,15,34 The sizable 3JH1′–H2′ coupling constants observed here for 22 and 23 are indicative of H1–C1–C2–H2 torsion angles tending to 150°, and tentatively support this 1E conformational assignment in solution-phase. However, these couplings are larger than related values observed for non-fluorinated carbanucleosides (3JH1′–H2′ = 5.6 Hz for carbauridine in DMSO-d6,16 and 3JH1′–H2′ = 8.5 Hz for aristeromycin and carbaguanosine in D2O).35
1 J coupling constants can serve as convenient identifiers for the anomeric carbon within nucleoside rings, alongside supporting patterns of stereochemical identity (α and β anomers).36 Recording 1J C–H values for 22 and 23 (Table 1), we noted a significant decrease in this coupling constant at C1′ and C4′ when replacing furanosyl oxygen with CHF or CF2. CHF showed the largest change relative to uridine (for C1′: 1JH,C = 171.8 Hz for uridine and 140.9 Hz for 22). This effect was not pronounced for C2′ or C3′, where the coupling constants remained similar to uridine. Since 1J C–H coupling constants of carbon generally increase with the electronegativity of attached functional groups, these data exhibit the effect of the removal of oxygen from the ring, and indicate that replacing with fluorinated motifs produces coupling constants that resemble those for ring carbons bearing OH groups (i.e., 1JH,C = 149.7 Hz for C3′ in uridine and 140.9 Hz for C1′ in 22).
With established access to gram quantities of 22 and 23 we next sought to diversify these analogues, targeting modification of the pyrimidine base, ProTide forms and 2′-deoxygenation. Accordingly, cytidine analogues 26 and 27 were prepared in three steps from 22 and 23. Global hydroxyl group protection was performed to give acetate protected uridines 24 and 25 in excellent yields (Scheme 4). This was followed by treatment with 1,2,4-triazole and POCl3 to provide an intermediate 4-triazole which was subsequently aminated alongside simultaneous hydroxyl group deprotection, using 35% NH4OH, to deliver 26 and 27 in yields of 53% and 42% respectively over three steps.
 | ||
| Scheme 4 Synthesis of mono- and gem-di-fluorinated cytidines. (a) Ac2O, pyridine, DMAP, DCM, rt; (b) 1,2,4-triazole, POCl3, Et3N, MeCN, rt; (c) 35% NH4OH, H2O, rt. | ||
Uridine analogues 22 and 23 were next 3′,5′-O-protected with TIPDSiCl2 in pyridine to give 28 and 29 in 76% and 43% yields respectively (Scheme 5). Some unwanted protection of the 2′-O-hydroxyl group was also observed, however, 22 or 23 could easily be regenerated using a global TBAF deprotection. A Barton-McCombie 2′-deoxygenation was next performed via the 2′-O-thiocarbamate. This intermediate was obtained from treatment of either 28 or 29 with 1,1′-thiocarbonyldiimidazole in MeCN, followed by radical mediated deoxygenation using Bu3SnH and AIBN, delivering 3′,5′-O-protected uridines 30 and 31 in 52% and 68% yields respectively, over two steps. The target 2′-deoxy uridines 32 and 33 were subsequently obtained in 66% and 55% yields, following TIPDS-deprotection with TBAF. The 3′,5′-O-protected uridines 30 and 31 were also converted through to their cytidine derivatives using the procedures described above for 24 and 25, affording 34 and 35 in 11% and 9% yields respectively over three steps.
 | ||
| Scheme 5 Synthesis of 2′-deoxy mono- and gem-di-fluorinated uridine and cytidine. (a) TIPDSiCl2, pyridine, rt; (b) TCDI, DMAP, MeCN, 45 °C; (c) Bu3SnH, AIBN, toluene, reflux; (d) TBAF, THF, rt; (e) 1,2,4-triazole, POCl3, Et3N, MeCN, 0 °C–rt; (f) 35% NH4OH, 1,4-dioxane, rt; (g) TBAF, THF, rt. | ||
Finally, each of 22 and 23 were converted through to their ProTide phosphoramidate forms (Scheme 6).37 Firstly, a 2′,3′-O-acetonide protecting group was installed, giving 36 and 37 in yields of 81% and 71% respectively. Deprotonation of the remaining 5′-OH with tert-butylmagnesium chloride, followed by reaction with commercial phosphoramidate reagent 38,38 delivered uridine phosphoramidate analogues 39 and 40 in 24% and 27% yields respectively, following acetonide removal.
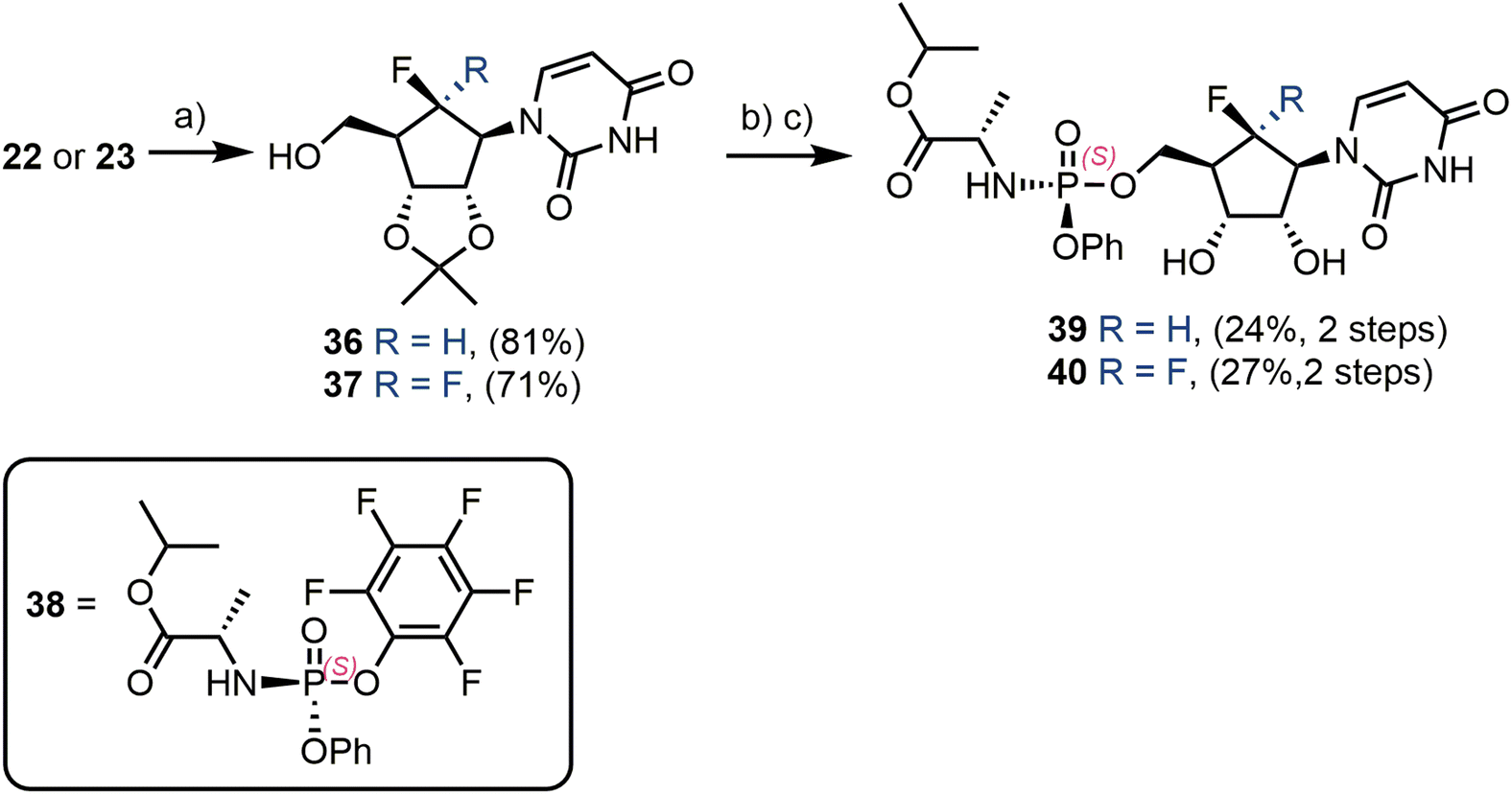 | ||
| Scheme 6 Synthesis of uridine phosphoramidates. (a) H2SO4, Me2CO, reflux; (b) 38, tBuMgCl, THF, −78 °C; (c) formic acid, H2O, rt. | ||
Compounds 22, 23, 26, 27, 32–35, 39 and 40 were evaluated in cellular viability assays against cancer cell lines U87-MG and PANC-1 (Table 2). 6-(R)-Monofluorocytidine 26 showed mild activity against U87-MG cells (Table 2, entry 5). Whilst the remaining compounds showed little activity below 100 μM, this may prove advantageous for exploring antiviral activity. In addition, whilst the ProTide strategy adopted here (compounds 39 and 40, Table 2, entries 11 & 12) did not translate to biological activity, it is possible that enzymatic release of the masked monophosphate was prevented in this assay and that alternative pronucleotide design could be considered; for example, the cycloSal approach obviates enzymatic activation of a prodrug unit.39,40 Within a wider program of work, we will explore antiviral activity for the compounds reported herein, alongside targeting nucleotide triphosphate derivatives and associated in vitro polymerase activity, to further inform potential monophosphate prodrug candidate development.
| Entry | Compound | Cell viability (%) | |||
|---|---|---|---|---|---|
| U87-MG | PANC-1 | ||||
| 100 μM | 10 μM | 100 μM | 10 μM | ||
| a Gemcitabine evaluated at top concentration of 10 μM. b Cytarabine evaluated at top concentration of 100 μM. | |||||
| 1 | Gemcitabinea | — | 32 | — | 37 |
| 2 | Cytarabineb | 22 | — | 42 | — |
| 3 | 22 | ≥100 | ≥100 | ≥100 | ≥100 |
| 4 | 23 | ≥100 | ≥100 | ≥100 | ≥100 |
| 5 | 26 | 42 | 63 | 70 | ≥100 |
| 6 | 27 | 93 | 96 | ≥100 | ≥100 |
| 7 | 32 | ≥100 | ≥100 | ≥100 | ≥100 |
| 8 | 33 | ≥100 | ≥100 | ≥100 | ≥100 |
| 9 | 34 | ≥100 | ≥100 | 89 | ≥100 |
| 10 | 35 | ≥100 | ≥100 | ≥100 | ≥100 |
| 11 | 39 | ≥100 | ≥100 | ≥100 | ≥100 |
| 12 | 40 | ≥100 | ≥100 | ≥100 | ≥100 |
Conclusion
In summary, we have developed a reliable and scalable synthesis of 6′-(R)-monofluoro and 6′-gem-difluorouridines as common building blocks for wider diversification to alternative pyrimidines, ProTide prodrug forms and 2′-deoxygentated nucleoside analogues. A robust conjugate addition using a vinyl organocuprate, followed by diastereoselective Selectfluor α-keto fluorination via the silyl enol ether delivers key scaffold materials. These are elaborated through 5-position alkene oxidation/cleavage and nucleobase installation, building up from a C1 amine using (3-ethoxyacryloyl)isocyanate. Importantly this alkene manipulation methodology lays a foundation for exploration of 4′-position modifications within this fluorocarbanucleoside scaffold. The synthesis is supported by key X-ray structures confirming cyclopentane ring stereochemistries. Additionally, key differences to canonical pyrimidine nucleoside 1H NMR data are observed and work to further understand the conformational preferences for this analogues class, alongside their wider biological evaluation is currently underway.Experimental section
General experimental methods
All chemicals were purchased from Acros Organics, Alfa Aesar, Biosynth Carbosynth, Fisher Scientific, Fluorochem, Sigma Aldrich or TCI Chemicals and were used without further purification unless otherwise stated. CuI was purified prior to use.41 The concentration of vinylmagnesium bromide was determined via iodometric titration42 prior to use. Anhydrous DMF, MeOH, pyridine and Et3N were obtained from Sure/Seal™ bottles via chemical suppliers. Anhydrous THF, DCM and toluene were obtained by passing solvent through activated alumina columns and dispensed from a PureSolv MD ASNA solvent purification system and stored over 4 Å molecular sieves. Unless otherwise stated, all reactions were conducted using anhydrous solvents, under an atmosphere of N2 which was passed through a Drierite® drying column. Thin layer chromatography (TLC) was performed using pre-coated 0.25 mm 60 F254 silica gel plates (Merck). Visualisation was achieved using UV light (λ = 254 nm), and KMnO4 staining followed by heating, or Ninhydrin staining followed by heating, or 5% H2SO4/EtOH staining followed by heating. Flash column chromatography was performed using silica gel, high purity grade, pore size 60 Å, 230–400 mesh particle size, 40–63 μm particle size (Sigma Aldrich). All final compounds were purified on an Agilent 1260 Infinity II preparative HPLC system equipped with a variable wavelength detector and a fraction collector, on a reverse phase column (Polaris 180 Å C18-A. 21.2 × 250 mm, 5 μm) to achieve a purity level >95%. Visualisation was achieved using UV detection at 254 nm. Optical rotations were recorded on a Bellingham + Stanley ADP430 (specific rotation, tube length: 50 mm, concentrations in g per 100 mL). All high-resolution mass spectra were measured at the EPSRC National Mass Spectrometry Facility at Swansea University, UK. All crystallographic data were collected on a Bruker D8 Quest ECO diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and a Photon II-C14 CPAD detector. The ADPs are rendered at 50% probability level. NMR spectra were recorded on a Bruker Avance 400 spectrometer. The chemical shift data for each signal are given as δ in units of parts per million (ppm) relative to tetramethylsilane, where δ = 0.00 ppm. The number of protons (n) for a given resonance is indicated by nH. The multiplicity of each signal is indicated by: s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), p (pentet), sep (septet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dddd (doublet of doublet of doublet of doublets), dt (doublet of triplets), tt (triplet of triplets), dqd (doublet of quartets of doublets) or m (multiplet). Coupling constants (J) are quoted in Hz and calculated to the nearest 0.1 Hz. Cell culture: PANC-1 (ATCC, catalogue number: CRL-1469) cells were cultured at 37 °C with 5% CO2 in DMEM (Corning, catalogue number: 10-013CV), supplemented with 10% heat-inactivated FBS (Corning, catalogue number: 35016CV) and 1X Non-essential amino acids [(0.1 mM per amino acid) (Corning, catalogue number: 25025CI)] and 1× Penicillin–Streptomycin Solution [Penicillin (100 IU) and Streptomycin (100 μg mL−1) (Corning, catalogue number: 30002CI)]. U87-MG (ATCC, catalogue number: HTB-14) cells were cultured at 37 °C with 5% CO2 in EMEM (Lonza, catalogue number: 12-611F), supplemented with 10% heat-inactivated FBS (Corning, catalogue number: 35016CV) and 1× non-essential amino acids (0.1 mM per amino acid) (Corning, catalogue number: 25025CI) and 1× Corning™ Penicillin–Streptomycin Solution [Penicillin (100 IU) and Streptomycin (100 μg mL−1) (Corning, catalogue number 30002CI)].:1) showed complete consumption of the starting material to a higher Rf. The reaction mixture was warmed to 0 °C and quenched with 5:1 sat. NH4Cl/sat. Na2S2O3 (v/v, 600 mL). The aqueous phase was extracted with EtOAc (3 × 500 mL), and the combined organic phases were washed with H2O (500 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (0–20% Et2O/petroleum-ether) to give 2 as a colourless oil (18.4 g, 101 mmol, 78%). Rf = 0.39 (petroleum-ether/Et2O, 4:1); 1H NMR (400 MHz, CDCl3): δ 5.83 (1H, ddd, 3JH5–H6b = 17.2 Hz, 3JH5–H6a = 10.6 Hz, 3JH5–H4 = 6.5 Hz, H5), 5.15 (1H, d, 3JH6a–H5 = 10.6 Hz, H6a), 5.10 (1H, dd, 3JH6b–H5 = 17.4 Hz, 2JH6b–H6a = 1.6 Hz, H6b), 4.64 (1H, d, 3JH3–H2 = 5.2 Hz, H3), 4.20 (1H, d, 3JH2–H3 = 5.2 Hz, H2), 3.11 (1H, dd, 3JH4–H7a = 7.9 Hz, 3JH4–H5 = 7.0 Hz, H4), 2.84 (1H, dd, 2JH7a–H7b = 18.3 Hz, 3JH7a–H4 = 8.6 Hz, H7a), 2.29 (1H, d, 2JH7b–H7a = 18.2 Hz, H7b), 1.45 (3H, s, CH3), 1.35 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 213.2 (C1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 137.2 (C5), 116.5 (C6), 112.5 [C(CH3)2], 81.4 (C3), 77.9 (C2), 39.8 (C4), 38.6 (C7), 26.9 [C(CH3)2], 24.9 [C(CH3)2]; HRMS (ESI): calculated for C10H14O3Na [M + Na]+ 205.0835, found 205.0827. These data were in good agreement with literature values.32
:1) showed complete conversion of the starting material to a higher Rf. The reaction was warmed to 0 °C and quenched with sat. NH4Cl (400 mL). The aqueous phase was extracted with EtOAc (3 × 400 mL), and the combined organic phases were washed with H2O (300 mL), brine (300 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The residue was dissolved in DMF (770 mL) and cooled to 0 °C, after which Selectfluor® (82.0 g, 231 mol, 1.2 equiv.) was added in three portions over 30 min. The reaction mixture was allowed to warm to rt and was stirred for 3 h, at which point TLC analysis (petroleum-ether/Et2O, 4:1) showed complete conversion from a higher Rf to a lower Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NH4Cl (400 mL). The aqueous phase was extracted with EtOAc (4 × 300 mL), and the combined organic phases were washed with H2O (3 × 200 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (0–50% Et2O/petroleum-ether) to give 4, as an 8:1 inseparable mixture of diastereomers and a colourless oil (28.2 g, 140 mmol, 73%). Rf = 0.09 (petroleum-ether/Et2O, 4:1); 1H NMR (400 MHz, CDCl3) (major): δ 5.58 (1H, ddd, 3JH5–H6b = 17.3 Hz, 3JH5–H6a = 10.8 Hz, 3JH5–H4 = 7.8 Hz, H5), 5.50 (1H, ddd, 2JH7–F = 50.5 Hz, 3JH7–H4 = 7.9 Hz, 4JH7–H3 = 0.9 Hz, H7), 5.31 (1H, dt, 3JH6a–H5 = 10.8 Hz, 2JH6a–H6b = 0.9 Hz, H6a), 5.26 (1H, dt, 3JH6b–H5 = 17.3 Hz, 2JH6b–H6a = 1.1 Hz, H6b), 4.74 (1H, ddd, 3JH3–H2 = 5.7 Hz, 3JH3–H4 = 4.7 Hz, 4JH3–H7 = 0.9 Hz, H3), 4.25 (1H, dd, 3JH2–H3 = 6.0 Hz, 4JH2–F = 2.6 Hz, H2), 3.42–3.38 (1H, m, H4), 1.48 (3H, s, CH3), 1.35 (3H, s, CH3); 13C NMR (101 MHz, CDCl3) (major): δ 204.5 (d, 2JC1–F = 13.6 Hz, C1, CO), 130.1 (d, 3JC5–F = 5.4 Hz, C5), 120.5 (d, 4JC6–F = 1.5 Hz, C6), 113.1 [C(CH3)2], 91.0 (d, 1JC7–F = 201.2 Hz, C7), 78.1 (d, 3JC3–F = 6.1 Hz, C3), 74.0 (d, 3JC2–F = 2.3 Hz, C2), 43.5 (d, 2JC4–F = 16.3 Hz, C4), 26.3 [C(CH3)2], 24.0 [C(CH3)2]; 19F NMR (376 MHz, CDCl3) (major): δ −216.6 (d, 2JF–H7 = 50.5 Hz); HRMS (ESI): calculated for C10H14FO3 [M + H]+ 201.0921, found 201.0919.
:1) showed complete conversion of the starting material to a higher Rf. The reaction was cooled to 0 °C and quenched with sat. NH4Cl (200 mL). The aqueous phase was extracted with EtOAc (3 × 200 mL). The combined organic phases were washed with H2O (150 mL), brine (150 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The residue was dissolved in DMF (380 mL) and cooled to 0 °C, after which Selectfluor® (32.3 g, 91.2 mmol, 1.2 equiv.) was added in two portions over 20 min. The reaction mixture was allowed to warm to rt, and stirred for 16 h, at which point TLC analysis (petroleum-ether/Et2O, 4:1) showed complete conversion from a higher Rf to a lower Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NH4Cl (200 mL), and the aqueous phase was extracted with EtOAc (4 × 200 mL). The combined organic phases were washed with H2O (3 × 100 mL), brine (150 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (0–50% Et2O/petroleum-ether) to give 6, as a colourless oil (11.8 g, 50.1 mmol, 66%). Rf = 0.10 (petroleum-ether/Et2O, 4:1); 1H NMR (400 MHz, CDCl3) (hydrate 7): δ 5.85 (1H, ddd, 3JH5–H6a = 17.3 Hz, 3JH5–H6b = 10.4 Hz, 3JH5–H4 = 8.3 Hz, H5), 5.38–5.34 (2H, m, H6a, H6b), 4.504.46 (1H, m, H2), 4.44–4.42 (1H, m, H3), 4.26 (1H, d, 4JOH–F = 1.7 Hz, OH), 3.15–3.05 (1H, m, H4), 3.02 (1H, s, OH), 1.57 (3H, s, CH3), 1.37 (3H, s, CH3); 13C NMR (101 MHz, CDCl3) (hydrate 7): δ 129.7 (d, 3JC5–F = 4.1 Hz, C5), 124.1 (dd, 1JC7–F = 271.1 Hz, 1JC7–F = 253.5 Hz, C7), 121.1 (C6), 114.2 [C(CH3)2], 95.2 (dd, 2JC1–F = 25.7 Hz, 2JC1F = 22.0 Hz, C1), 79.3 (d, 3JC3–F = 3.2 Hz, C3), 78.6 (dd, 3JC2–F = 7.7 Hz, 3JC2–F = 1.2 Hz, C2), 52.5 (dd, 2JC4–F = 20.6 Hz, 3JC4–F 20.1 Hz, C4), 26.2 [C(CH3)2], 24.8 [C(CH3)2]; 19F NMR (376 MHz, CDCl3) (hydrate 7): δ −119.4 (ddd, 2JF–F = 237.5, 3JF–H4 = 9.0 Hz, 4JF–OH = 1.8 Hz), −121.1 (dd, 2JF–F = 237.5 Hz, 3JF–H4 = 21.7 Hz); HRMS (ketone 6) (ESI): calculated for C10H13F2O3 [M + H]+ 219.0827, found 219.0826.
:1) showed complete consumption of the starting material to a higher Rf. The reaction mixture was cooled to 0 °C and H2O (200 mL) was added. The solvent was removed in vacuo, and the residue was partitioned between H2O (200 mL) and EtOAc (200 mL). The aqueous phase was extracted with EtOAc (3 × 200 mL), and the combined organic phases were washed with brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (10–20% Et2O/hexane).
:1); mp: 48–50 °C; [α]24.9D = −15.5 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.62 (1H, ddd, 3JH5–H6a = 17.6 Hz, 3JH5–H6b = 10.7 Hz, 3JH5–H4 = 7.0 Hz, H5), 5.20–5.12 (2H, m, H6a, H6b), 4.79–4.65 (1H, m, H7), 4.60–4.59 (2H, m, H2, H3), 4.11–4.00 (1H, m, H1), 3.12 (1H, dd, 3JH4–F = 18.6 Hz, 3JH4–H3/H5 = 6.4 Hz, H4), 2.76 (1H, d, 3JOH–H1 = 10.5 Hz, OH), 1.51 (3H, s, CH3), 1.34 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 133.6 (d, 3JC5–F = 6.1 Hz, C5), 117.9 (C6), 112.0 [C(CH3)2], 97.1 (d, 1JC7–F = 189.6 Hz, C7), 83.0 (C3), 77.7 (C2), 72.6 (d, 2JC1–F = 17.0 Hz, C1), 50.4 (d, 2JC4–F = 19.1 Hz, C4), 26.3 [C(CH3)2], 24.5 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −197.9 (ddd, 2JF–H = 52.0 Hz, 3JF–H1 = 26.2 Hz, 3JF–H4 = 18.4 Hz); HRMS (NSI): calculated for C10H15FO3Na [M + Na]+ 225.0897, found 225.0898.
:1); [α]24.9D = +8.6 (c 0.2, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.87 (1H, ddd, 3JH5–H6a = 17.9 Hz, 3JH5–H6b = 10.4 Hz, 3JH5–H4 = 7.7 Hz, H5), 5.30–5.24 (2H, m, H6a, H6b), 4.90 (1H, dt, 2JH7–F = 50.7 Hz, 3JH7–H1/H4 = 4.2 Hz, H7), 4.73–4.69 (1H, m, H2), 4.64–4.61 (1H, m, H3), 4.17–4.11 (1H, m, H1), 3.05–2.94 (1H, m, H4), 2.76 (1H, dd, 3JOH–H1 = 4.5 Hz, 4JOH–F = 2.4 Hz, OH), 1.54 (3H, s, CH3), 1.38 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 132.5 (d, 3JC5–F = 6.1 Hz, C5), 118.7 (C6), 113.8 [C(CH3)2], 100.6 (d, 1JC7–F = 181.0 Hz, C7), 82.8 (d, 3JC3–F = 2.0 Hz, C3), 77.7 (d, 3JC2–F = 2.7 Hz, C2), 72.0 (d, 2JC1–F = 26.3 Hz, C1), 50.0 (d, 2JC4–F = 17.6 Hz, C4), 26.3 [C(CH3)2], 24.5 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −204.2 (ddd, 2JF–H7 = 50.7 Hz, 3JF–H4 = 26.2 Hz, 3JF–H1 = 10.5 Hz); HRMS (NSI): calculated for C10H15FO3Na [M + Na]+ 225.0897, found 225.0898.
:1); mp: 86–87 °C; [α]21.4D = +33.4 (c 5, DCM); 1H NMR (400 MHz, CDCl3): δ 5.95 (1H, ddd, 3JH5–H6a = 17.4 Hz, 3JH5–H6b = 10.3 Hz, 3JH5–H4 = 8.1 Hz, H5), 5.31–5.22 (2H, m, H6a, H6b), 5.00 (1H, dt, 2JH7–F = 52.4 Hz, 3JH7–H1 = 3.8 Hz, H7), 4.63 (1H, ddd, 3JH3–H2 = 7.0 Hz, 3JH3–H4 5.2 Hz, 4JH3–F = 1.7 Hz, H3), 4.57–4.53 (1H, m, H2), 4.20 (1H, ddt, 3JH1–F = 19.6 Hz, 3JH1–OH = 6.8 Hz, 3JH1–H2/H7 = 3.4 Hz, H1), 2.81 (1H, ddt, 3JH4–F = 28.4 Hz, 3JH4–H5 = 8.5 Hz, 3JH4–H3 = 4.5 Hz, H4), 2.18 (1H, dd, 3JOH–H1 = 7.3, 4JOH–F = 2.5 Hz, OH), 1.50 (3H, s, CH3), 1.31 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 133.0 (d, 3JC5–F = 5.6 Hz, C5), 118.6 (C6), 113.0 [C(CH3)2], 98.4 (d, 1JC7–F = 181.8 Hz, C7), 84.7 (d, 3JC2–F = 1.0 Hz, C2), 82.6 (d, 3JC3–F = 1.7 Hz, C3), 78.0 (d, 2JC1–F = 16.7 Hz, C1), 51.9 (d, 2JC4–F = 17.5 Hz, C4), 27.0 [C(CH3)2], 24.4 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −210.9 (ddd, 2JF–H7 = 49.9 Hz, 3JF–H4 = 28.4 Hz, 3JF–H1 = 19.6 Hz); HRMS (NSI): calculated for C10H15FO3Na [M + Na]+ 225.0897, found 225.0898.
:1); [α]24.3D = −18.5 (c 4.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.80 (1H, ddd, 3JH5–H6a = 17.8 Hz, 3JH5–H6b = 10.2 Hz, 7.9 Hz, H5), 5.17–5.32 (1H, m, H6a, H6b), 4.63–4.59 (1H, m, H2), 4.55–4.52 (1H, m, H3), 4.04 (1H, app. p, J = 6.0 Hz, H1), 3.17–3.07 (1H, m, H4), 2.90 (1H, dd, 3JOH–H1 = 5.4 Hz, 4JOH–F = 1.4 Hz, OH), 1.55 (3H, s, CH3), 1.37 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 129.8 (dd, 3JC5–F = 4.6 Hz, 3JC5–F = 2.3 Hz, C5), 126.9 (dd, 1JC7–F = 266.6 Hz, 1JC7–F = 249.7 Hz, C7), 120.6 (C6), 113.3 [C(CH3)2], 80.4 (dd, 3JC3–F = 6.2 Hz, 3JC3–F = 1.4 Hz, C3), 75.1 (t, 3JC2–F = 3.3 Hz, C2), 70.8 (dd, 2JC1–F = 31.8 Hz, 2JC1–F = 20.0 Hz, C1), 51.8 (dd, 2JC4–F = 21.4 Hz, 2JC4–F = 19.8 Hz, C4), 26.0 [C(CH3)2], 24.5 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −112.8 (dt, 2JF–F = 245.0 Hz, 3JF–H1/H4 = 6.6 Hz), −114.7 (ddd, 2JF–F = 245.0 Hz, 3JF–H1 = 19.6 Hz, 3JF–H4 = 4.7 Hz); HRMS (ESI): calculated for C10H14F2O3Na [M + Na]+ 243.0803, found 243.0803.
:1) showed complete consumption of the starting material to a higher Rf. The reaction mixture was poured onto 1.0 M aqueous HCl (50 mL), and the aqueous phase was extracted with DCM (3 × 100 mL). The combined organic phases were washed with sat. NaHCO3 (50 mL), H2O (100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
:1); [α]24.9D = −62.5 (c 0.2, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.78 (1H, ddd, 3JH5–H6a = 17.9 Hz, 3JH5–H6b = 10.3 Hz, 3JH5–H4 = 7.8 Hz, H5), 5.30–5.25 (2H, m, H6a, H6b), 5.14 (1H, dt, 2JH7–F = 52.5 Hz, 3JH7–H4/H1 = 6.6 Hz, H7), 4.96 (1H, dt, 3JH1–F = 14.4 Hz, 3JH1–H2/H7 = 6.4 Hz, H1), 4.79 (1H, td, 3JH1–H7 = 6.0 Hz, 3JH2–H3 = 2.4 Hz, H2), 4.58 (1H, dt, 3JH3–H4 = 5.8 Hz, 3JH3–H2 = 2.2 Hz, H3), 3.11–3.05 (1H, m, H4), 2.14 (3H, s, Ac–CH3), 1.46 (3H, s, CH3), 1.30 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 170.3 (CO), 131.6 (d, 3JC5–F = 6.1 Hz, C5), 119.3 (C6), 112.1 [C(CH3)2], 95.9 (d, 1JC7–F = 188.2 Hz, C7), 81.7 (d, 3JC3–F = 4.2 Hz, C3), 75.1 (d, 2JC1–F = 22.7 Hz, C1), 74.9 (d, 3JC2–F = 6.8 Hz, C2), 47.5 (d, 2JC4–F = 17.5 Hz, C4), 26.1 [C(CH3)2], 24.5 [C(CH3)2], 20.7 (Ac-CH3); 19F NMR (377 MHz, CDCl3): δ −207.3 (dddt, 2JF–H7 = 51.6 Hz, 3JF–H1 = 14.5 Hz, 3JF–H4 = 9.5 Hz, 4JF–H2/H3 = 2.5 Hz); HRMS (ESI): calculated for C12H18FO4 [M + H]+ 245.1184, found 254.1180.
:1); [α]24.5D = −87.0 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.82 (1H, ddd, 3JH5–6a = 17.4 Hz, 3JH5–H6b = 10.5 Hz, 3JH5–H4 = 7.9 Hz, H5), 5.45–5.33 (2H, m, H6a, H6b), 5.10 (1H, dt, 3JH1–F = 8.1 Hz, 3JH1–H2 = 5.4 Hz, H1), 4.72 (1H, ddd, 3JH2–H3 = 6.8 Hz, 3JH2–H1 = 5.6 Hz, 4JH2–F = 3.8 Hz, H2), 4.53 (1H, ddd, 3JH3–H2 = 6.3 Hz, 4JH3–H4 = 3.8 Hz, 3JH3–F = 1.7 Hz, H3), 3.18–3.05 (1H, m, H4), 2.19 (3H, s, Ac–CH3), 1.50 (3H, s, CH3), 1.33 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 169.3 (CO), 129.4 (dd, 3JC5–F = 4.9 Hz, 3JC5–F = 1.5 Hz, C5), 126.1 (dd, 1JC7–F = 269.7 Hz, 1JC7–F = 248.4 Hz, C7), 121.0 (C6), 113.6 [C(CH3)2], 80.6 (dd, 3JC3–F = 6.8 Hz, 3JC3–F = 0.8 Hz, C3), 74.9 (dd, 3JC2–F = 3.3 Hz, 3JC2–F = 1.8 Hz, C2), 71.8 (dd, 2JC1–F = 33.5 Hz, 2JC1–F = 18.7 Hz, C1), 52.8 (dd, 2JC4–F = 21.0 Hz, 2JC4–F = 19.9 Hz, C4), 26.1 [C(CH3)2], 24.7 [C(CH3)2], 20.6 (Ac–CH3); 19F NMR (377 MHz, CDCl3): δ −110.7 (dddd, 2JF–F = 245.4 Hz, 3JF–H1 = 9.2 Hz, 4JF–H4 = 5.4 Hz, 4JF–H3 = 1.5 Hz), −112.9 (ddddd, 2JF–F = 245.5 Hz, 3JF–H4 = 21.4 Hz, 3JF–H1 = 8.1 Hz, 4JF–H2 = 3.6 Hz, 4JF–H3 = 1.8 Hz); HRMS (ESI): calculated for C12H16FO4Na [M + Na]+ 285.0909, found 285.0914.
O), 137.2 (C5), 116.5 (C6), 112.5 [C(CH3)2], 81.4 (C3), 77.9 (C2), 39.8 (C4), 38.6 (C7), 26.9 [C(CH3)2], 24.9 [C(CH3)2]; HRMS (ESI): calculated for C10H14O3Na [M + Na]+ 205.0835, found 205.0827. These data were in good agreement with literature values.32
:1) showed complete conversion of the starting material to a higher Rf. The reaction was warmed to 0 °C and quenched with sat. NH4Cl (400 mL). The aqueous phase was extracted with EtOAc (3 × 400 mL), and the combined organic phases were washed with H2O (300 mL), brine (300 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The residue was dissolved in DMF (770 mL) and cooled to 0 °C, after which Selectfluor® (82.0 g, 231 mol, 1.2 equiv.) was added in three portions over 30 min. The reaction mixture was allowed to warm to rt and was stirred for 3 h, at which point TLC analysis (petroleum-ether/Et2O, 4:1) showed complete conversion from a higher Rf to a lower Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NH4Cl (400 mL). The aqueous phase was extracted with EtOAc (4 × 300 mL), and the combined organic phases were washed with H2O (3 × 200 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (0–50% Et2O/petroleum-ether) to give 4, as an 8:1 inseparable mixture of diastereomers and a colourless oil (28.2 g, 140 mmol, 73%). Rf = 0.09 (petroleum-ether/Et2O, 4:1); 1H NMR (400 MHz, CDCl3) (major): δ 5.58 (1H, ddd, 3JH5–H6b = 17.3 Hz, 3JH5–H6a = 10.8 Hz, 3JH5–H4 = 7.8 Hz, H5), 5.50 (1H, ddd, 2JH7–F = 50.5 Hz, 3JH7–H4 = 7.9 Hz, 4JH7–H3 = 0.9 Hz, H7), 5.31 (1H, dt, 3JH6a–H5 = 10.8 Hz, 2JH6a–H6b = 0.9 Hz, H6a), 5.26 (1H, dt, 3JH6b–H5 = 17.3 Hz, 2JH6b–H6a = 1.1 Hz, H6b), 4.74 (1H, ddd, 3JH3–H2 = 5.7 Hz, 3JH3–H4 = 4.7 Hz, 4JH3–H7 = 0.9 Hz, H3), 4.25 (1H, dd, 3JH2–H3 = 6.0 Hz, 4JH2–F = 2.6 Hz, H2), 3.42–3.38 (1H, m, H4), 1.48 (3H, s, CH3), 1.35 (3H, s, CH3); 13C NMR (101 MHz, CDCl3) (major): δ 204.5 (d, 2JC1–F = 13.6 Hz, C1, CO), 130.1 (d, 3JC5–F = 5.4 Hz, C5), 120.5 (d, 4JC6–F = 1.5 Hz, C6), 113.1 [C(CH3)2], 91.0 (d, 1JC7–F = 201.2 Hz, C7), 78.1 (d, 3JC3–F = 6.1 Hz, C3), 74.0 (d, 3JC2–F = 2.3 Hz, C2), 43.5 (d, 2JC4–F = 16.3 Hz, C4), 26.3 [C(CH3)2], 24.0 [C(CH3)2]; 19F NMR (376 MHz, CDCl3) (major): δ −216.6 (d, 2JF–H7 = 50.5 Hz); HRMS (ESI): calculated for C10H14FO3 [M + H]+ 201.0921, found 201.0919.
:1) showed complete conversion of the starting material to a higher Rf. The reaction was cooled to 0 °C and quenched with sat. NH4Cl (200 mL). The aqueous phase was extracted with EtOAc (3 × 200 mL). The combined organic phases were washed with H2O (150 mL), brine (150 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The residue was dissolved in DMF (380 mL) and cooled to 0 °C, after which Selectfluor® (32.3 g, 91.2 mmol, 1.2 equiv.) was added in two portions over 20 min. The reaction mixture was allowed to warm to rt, and stirred for 16 h, at which point TLC analysis (petroleum-ether/Et2O, 4:1) showed complete conversion from a higher Rf to a lower Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NH4Cl (200 mL), and the aqueous phase was extracted with EtOAc (4 × 200 mL). The combined organic phases were washed with H2O (3 × 100 mL), brine (150 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (0–50% Et2O/petroleum-ether) to give 6, as a colourless oil (11.8 g, 50.1 mmol, 66%). Rf = 0.10 (petroleum-ether/Et2O, 4:1); 1H NMR (400 MHz, CDCl3) (hydrate 7): δ 5.85 (1H, ddd, 3JH5–H6a = 17.3 Hz, 3JH5–H6b = 10.4 Hz, 3JH5–H4 = 8.3 Hz, H5), 5.38–5.34 (2H, m, H6a, H6b), 4.504.46 (1H, m, H2), 4.44–4.42 (1H, m, H3), 4.26 (1H, d, 4JOH–F = 1.7 Hz, OH), 3.15–3.05 (1H, m, H4), 3.02 (1H, s, OH), 1.57 (3H, s, CH3), 1.37 (3H, s, CH3); 13C NMR (101 MHz, CDCl3) (hydrate 7): δ 129.7 (d, 3JC5–F = 4.1 Hz, C5), 124.1 (dd, 1JC7–F = 271.1 Hz, 1JC7–F = 253.5 Hz, C7), 121.1 (C6), 114.2 [C(CH3)2], 95.2 (dd, 2JC1–F = 25.7 Hz, 2JC1F = 22.0 Hz, C1), 79.3 (d, 3JC3–F = 3.2 Hz, C3), 78.6 (dd, 3JC2–F = 7.7 Hz, 3JC2–F = 1.2 Hz, C2), 52.5 (dd, 2JC4–F = 20.6 Hz, 3JC4–F 20.1 Hz, C4), 26.2 [C(CH3)2], 24.8 [C(CH3)2]; 19F NMR (376 MHz, CDCl3) (hydrate 7): δ −119.4 (ddd, 2JF–F = 237.5, 3JF–H4 = 9.0 Hz, 4JF–OH = 1.8 Hz), −121.1 (dd, 2JF–F = 237.5 Hz, 3JF–H4 = 21.7 Hz); HRMS (ketone 6) (ESI): calculated for C10H13F2O3 [M + H]+ 219.0827, found 219.0826.
:1) showed complete consumption of the starting material to a higher Rf. The reaction mixture was cooled to 0 °C and H2O (200 mL) was added. The solvent was removed in vacuo, and the residue was partitioned between H2O (200 mL) and EtOAc (200 mL). The aqueous phase was extracted with EtOAc (3 × 200 mL), and the combined organic phases were washed with brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (10–20% Et2O/hexane).
:1); mp: 48–50 °C; [α]24.9D = −15.5 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.62 (1H, ddd, 3JH5–H6a = 17.6 Hz, 3JH5–H6b = 10.7 Hz, 3JH5–H4 = 7.0 Hz, H5), 5.20–5.12 (2H, m, H6a, H6b), 4.79–4.65 (1H, m, H7), 4.60–4.59 (2H, m, H2, H3), 4.11–4.00 (1H, m, H1), 3.12 (1H, dd, 3JH4–F = 18.6 Hz, 3JH4–H3/H5 = 6.4 Hz, H4), 2.76 (1H, d, 3JOH–H1 = 10.5 Hz, OH), 1.51 (3H, s, CH3), 1.34 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 133.6 (d, 3JC5–F = 6.1 Hz, C5), 117.9 (C6), 112.0 [C(CH3)2], 97.1 (d, 1JC7–F = 189.6 Hz, C7), 83.0 (C3), 77.7 (C2), 72.6 (d, 2JC1–F = 17.0 Hz, C1), 50.4 (d, 2JC4–F = 19.1 Hz, C4), 26.3 [C(CH3)2], 24.5 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −197.9 (ddd, 2JF–H = 52.0 Hz, 3JF–H1 = 26.2 Hz, 3JF–H4 = 18.4 Hz); HRMS (NSI): calculated for C10H15FO3Na [M + Na]+ 225.0897, found 225.0898.
:1); [α]24.9D = +8.6 (c 0.2, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.87 (1H, ddd, 3JH5–H6a = 17.9 Hz, 3JH5–H6b = 10.4 Hz, 3JH5–H4 = 7.7 Hz, H5), 5.30–5.24 (2H, m, H6a, H6b), 4.90 (1H, dt, 2JH7–F = 50.7 Hz, 3JH7–H1/H4 = 4.2 Hz, H7), 4.73–4.69 (1H, m, H2), 4.64–4.61 (1H, m, H3), 4.17–4.11 (1H, m, H1), 3.05–2.94 (1H, m, H4), 2.76 (1H, dd, 3JOH–H1 = 4.5 Hz, 4JOH–F = 2.4 Hz, OH), 1.54 (3H, s, CH3), 1.38 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 132.5 (d, 3JC5–F = 6.1 Hz, C5), 118.7 (C6), 113.8 [C(CH3)2], 100.6 (d, 1JC7–F = 181.0 Hz, C7), 82.8 (d, 3JC3–F = 2.0 Hz, C3), 77.7 (d, 3JC2–F = 2.7 Hz, C2), 72.0 (d, 2JC1–F = 26.3 Hz, C1), 50.0 (d, 2JC4–F = 17.6 Hz, C4), 26.3 [C(CH3)2], 24.5 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −204.2 (ddd, 2JF–H7 = 50.7 Hz, 3JF–H4 = 26.2 Hz, 3JF–H1 = 10.5 Hz); HRMS (NSI): calculated for C10H15FO3Na [M + Na]+ 225.0897, found 225.0898.
:1); mp: 86–87 °C; [α]21.4D = +33.4 (c 5, DCM); 1H NMR (400 MHz, CDCl3): δ 5.95 (1H, ddd, 3JH5–H6a = 17.4 Hz, 3JH5–H6b = 10.3 Hz, 3JH5–H4 = 8.1 Hz, H5), 5.31–5.22 (2H, m, H6a, H6b), 5.00 (1H, dt, 2JH7–F = 52.4 Hz, 3JH7–H1 = 3.8 Hz, H7), 4.63 (1H, ddd, 3JH3–H2 = 7.0 Hz, 3JH3–H4 5.2 Hz, 4JH3–F = 1.7 Hz, H3), 4.57–4.53 (1H, m, H2), 4.20 (1H, ddt, 3JH1–F = 19.6 Hz, 3JH1–OH = 6.8 Hz, 3JH1–H2/H7 = 3.4 Hz, H1), 2.81 (1H, ddt, 3JH4–F = 28.4 Hz, 3JH4–H5 = 8.5 Hz, 3JH4–H3 = 4.5 Hz, H4), 2.18 (1H, dd, 3JOH–H1 = 7.3, 4JOH–F = 2.5 Hz, OH), 1.50 (3H, s, CH3), 1.31 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 133.0 (d, 3JC5–F = 5.6 Hz, C5), 118.6 (C6), 113.0 [C(CH3)2], 98.4 (d, 1JC7–F = 181.8 Hz, C7), 84.7 (d, 3JC2–F = 1.0 Hz, C2), 82.6 (d, 3JC3–F = 1.7 Hz, C3), 78.0 (d, 2JC1–F = 16.7 Hz, C1), 51.9 (d, 2JC4–F = 17.5 Hz, C4), 27.0 [C(CH3)2], 24.4 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −210.9 (ddd, 2JF–H7 = 49.9 Hz, 3JF–H4 = 28.4 Hz, 3JF–H1 = 19.6 Hz); HRMS (NSI): calculated for C10H15FO3Na [M + Na]+ 225.0897, found 225.0898.
:1); [α]24.3D = −18.5 (c 4.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.80 (1H, ddd, 3JH5–H6a = 17.8 Hz, 3JH5–H6b = 10.2 Hz, 7.9 Hz, H5), 5.17–5.32 (1H, m, H6a, H6b), 4.63–4.59 (1H, m, H2), 4.55–4.52 (1H, m, H3), 4.04 (1H, app. p, J = 6.0 Hz, H1), 3.17–3.07 (1H, m, H4), 2.90 (1H, dd, 3JOH–H1 = 5.4 Hz, 4JOH–F = 1.4 Hz, OH), 1.55 (3H, s, CH3), 1.37 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 129.8 (dd, 3JC5–F = 4.6 Hz, 3JC5–F = 2.3 Hz, C5), 126.9 (dd, 1JC7–F = 266.6 Hz, 1JC7–F = 249.7 Hz, C7), 120.6 (C6), 113.3 [C(CH3)2], 80.4 (dd, 3JC3–F = 6.2 Hz, 3JC3–F = 1.4 Hz, C3), 75.1 (t, 3JC2–F = 3.3 Hz, C2), 70.8 (dd, 2JC1–F = 31.8 Hz, 2JC1–F = 20.0 Hz, C1), 51.8 (dd, 2JC4–F = 21.4 Hz, 2JC4–F = 19.8 Hz, C4), 26.0 [C(CH3)2], 24.5 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −112.8 (dt, 2JF–F = 245.0 Hz, 3JF–H1/H4 = 6.6 Hz), −114.7 (ddd, 2JF–F = 245.0 Hz, 3JF–H1 = 19.6 Hz, 3JF–H4 = 4.7 Hz); HRMS (ESI): calculated for C10H14F2O3Na [M + Na]+ 243.0803, found 243.0803.
:1) showed complete consumption of the starting material to a higher Rf. The reaction mixture was poured onto 1.0 M aqueous HCl (50 mL), and the aqueous phase was extracted with DCM (3 × 100 mL). The combined organic phases were washed with sat. NaHCO3 (50 mL), H2O (100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
:1); [α]24.9D = −62.5 (c 0.2, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.78 (1H, ddd, 3JH5–H6a = 17.9 Hz, 3JH5–H6b = 10.3 Hz, 3JH5–H4 = 7.8 Hz, H5), 5.30–5.25 (2H, m, H6a, H6b), 5.14 (1H, dt, 2JH7–F = 52.5 Hz, 3JH7–H4/H1 = 6.6 Hz, H7), 4.96 (1H, dt, 3JH1–F = 14.4 Hz, 3JH1–H2/H7 = 6.4 Hz, H1), 4.79 (1H, td, 3JH1–H7 = 6.0 Hz, 3JH2–H3 = 2.4 Hz, H2), 4.58 (1H, dt, 3JH3–H4 = 5.8 Hz, 3JH3–H2 = 2.2 Hz, H3), 3.11–3.05 (1H, m, H4), 2.14 (3H, s, Ac–CH3), 1.46 (3H, s, CH3), 1.30 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 170.3 (CO), 131.6 (d, 3JC5–F = 6.1 Hz, C5), 119.3 (C6), 112.1 [C(CH3)2], 95.9 (d, 1JC7–F = 188.2 Hz, C7), 81.7 (d, 3JC3–F = 4.2 Hz, C3), 75.1 (d, 2JC1–F = 22.7 Hz, C1), 74.9 (d, 3JC2–F = 6.8 Hz, C2), 47.5 (d, 2JC4–F = 17.5 Hz, C4), 26.1 [C(CH3)2], 24.5 [C(CH3)2], 20.7 (Ac-CH3); 19F NMR (377 MHz, CDCl3): δ −207.3 (dddt, 2JF–H7 = 51.6 Hz, 3JF–H1 = 14.5 Hz, 3JF–H4 = 9.5 Hz, 4JF–H2/H3 = 2.5 Hz); HRMS (ESI): calculated for C12H18FO4 [M + H]+ 245.1184, found 254.1180.
:1); [α]24.5D = −87.0 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.82 (1H, ddd, 3JH5–6a = 17.4 Hz, 3JH5–H6b = 10.5 Hz, 3JH5–H4 = 7.9 Hz, H5), 5.45–5.33 (2H, m, H6a, H6b), 5.10 (1H, dt, 3JH1–F = 8.1 Hz, 3JH1–H2 = 5.4 Hz, H1), 4.72 (1H, ddd, 3JH2–H3 = 6.8 Hz, 3JH2–H1 = 5.6 Hz, 4JH2–F = 3.8 Hz, H2), 4.53 (1H, ddd, 3JH3–H2 = 6.3 Hz, 4JH3–H4 = 3.8 Hz, 3JH3–F = 1.7 Hz, H3), 3.18–3.05 (1H, m, H4), 2.19 (3H, s, Ac–CH3), 1.50 (3H, s, CH3), 1.33 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 169.3 (CO), 129.4 (dd, 3JC5–F = 4.9 Hz, 3JC5–F = 1.5 Hz, C5), 126.1 (dd, 1JC7–F = 269.7 Hz, 1JC7–F = 248.4 Hz, C7), 121.0 (C6), 113.6 [C(CH3)2], 80.6 (dd, 3JC3–F = 6.8 Hz, 3JC3–F = 0.8 Hz, C3), 74.9 (dd, 3JC2–F = 3.3 Hz, 3JC2–F = 1.8 Hz, C2), 71.8 (dd, 2JC1–F = 33.5 Hz, 2JC1–F = 18.7 Hz, C1), 52.8 (dd, 2JC4–F = 21.0 Hz, 2JC4–F = 19.9 Hz, C4), 26.1 [C(CH3)2], 24.7 [C(CH3)2], 20.6 (Ac–CH3); 19F NMR (377 MHz, CDCl3): δ −110.7 (dddd, 2JF–F = 245.4 Hz, 3JF–H1 = 9.2 Hz, 4JF–H4 = 5.4 Hz, 4JF–H3 = 1.5 Hz), −112.9 (ddddd, 2JF–F = 245.5 Hz, 3JF–H4 = 21.4 Hz, 3JF–H1 = 8.1 Hz, 4JF–H2 = 3.6 Hz, 4JF–H3 = 1.8 Hz); HRMS (ESI): calculated for C12H16FO4Na [M + Na]+ 285.0909, found 285.0914.
Sharpless asymmetric dihydroxylation. AD-mix β (1.4 g per 1 mmol of substrate) was suspended in 1
:1 iPrOH:H2O (v/v, 0.1 M), cooled to 0 °C and stirred vigorously for 30 min, before it was added to 13 or 14 (1.0 equiv.). The reaction mixture was allowed to warm to rt over 1 h and stirred for a further 6–16 h. TLC analysis (hexane/EtOAc, 1:1) showed complete consumption of the starting material to two spots of lower Rf. The reaction was quenched by the addition of Na2SO3 (1.5 g per 1 mmol of substrate), and the mixture was stirred for 30 min, at which point H2O (200 mL) was added. The aqueous phase was extracted with EtOAc (3 × 300 mL), and the combined organic phases were washed with H2O (400 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
Oxidative cleavage conditions A. Vicinal diol intermediate was dissolved in 1
:5 H2O/1,4-dioxane (v/v, 0.1 M) and NaIO4 (1.7 equiv.) was added. The reaction mixture was stirred at rt for 1 h, at which point TLC analysis (hexane/EtOAc, 1:1) showed complete conversion from two spots to one with a higher Rf. The reaction mixture was diluted with H2O (200 mL) and extracted with EtOAc (3 × 300 mL). The combined organic phases were washed with H2O (500 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
Oxidative cleavage conditions B. Vicinal diol intermediate was dissolved in 1
:5 H2O/1,4-dioxane (v/v, 0.1 M) and cooled to 0 °C. NaIO4 (10 equiv.) was added, and the reaction mixture was stirred at this temperature for 6 h, at which point TLC analysis (hexane/EtOAc, 1:1) showed complete conversion of two spots to two spots with a higher Rf. The reaction mixture was diluted with H2O (200 mL) and extracted with EtOAc (3 × 300 mL). The combined organic phases were washed with H2O (500 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
Reduction and acyl deprotection. Aldehyde intermediate was dissolved in MeOH (0.27 M), cooled to 0 °C and NaBH4 (1.5 equiv.) was added in two portions over 10 min. The reaction mixture was allowed to warm to rt and stirred for a further 16 h, at which point TLC analysis (hexane/EtOAc, 1
:1) showed complete conversion to a lower Rf. The reaction mixture was cooled to 0 °C and H2O (100 mL) was added. The mixture was allowed to warm to rt and stirred for a further 2 h. The solvent was removed in vacuo and the residue was partitioned between H2O (200 mL) and EtOAc (300 mL). The aqueous phase was extracted with EtOAc (3 × 300 mL) and the combined organic phases were washed with H2O (200 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography on silica gel (10–50% EtOAc/hexane).
:4); [α]24.1D = −13.8 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.05 (1H, ddd, 2JH6–F = 51.4 Hz, 3JH6–H4 = 5.4 Hz, 3JH6–H1 = 4.6 Hz, H6), 4.67–4.65 (2H, m, H2, H3), 4.23 (1H, dd, 3JH1–F = 13.3 Hz, 3JH1–H2/H6/OH = 4.5 Hz, H1), 3.90–3.82 (2H, m, H5a, H5b), 2.76 (1H, d, 3JOH–H1 = 3.8 Hz, OH), 2.53 (1H, dddd, 3JH4–F = 20.2 Hz, 3JH4–H5a/H5b = 11.4 Hz, 3JH4–H6 = 5.6 Hz, 3JH4–H3 = 2.8 Hz, H4), 1.51 (3H, s, CH3), 137 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 113.0 [C(CH3)2], 100.1 (d, 1JC6–F = 180.7 Hz, C7), 81.2 (d, 3JC3–F = 2.2 Hz, C3), 77.6 (d, 3JC2–F = 4.8 Hz, C2), 73.1 (d, 2JC1–F = 25.7 Hz, C1), 59.5 (d, 3JC5–F = 8.4 Hz, C5), 47.8 (d, 2JC4–F = 17.7 Hz, C4), 26.2 [C(CH3)2], 24.3 [C(CH3)2]; 19F NMR (376 MHz, CDCl3): δ −206.5 (ddd, 2JF–H6 = 51.5 Hz, 3JF–H4 = 20.3 Hz, 3JF–H1 = 13.7 Hz); HRMS (ESI): calculated for C9H15FO4Na [M + Na]+ 229.0847, found 229.0848.
:1); mp: 92–95 °C; [α]24.9D = −36.3 (c 4.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 4.66 (1H, ddd, 3JH2–H1 = 6.4 Hz, 3JH2–H3 = 3.1 Hz, 4JH2–OH = 1.3 Hz, H2), 4.57 (1H, td, 3JH3–H4 = 6.4 Hz, 3JH3–H2 = 3.0 Hz, H3), 4.19 (1H, dddd, 3JH1–F = 14.0 Hz, 3JH1–OH = 9.4 Hz, 3JH1–F = 8.1 Hz, 3JH1–H2 = 6.2 Hz, H1), 3.96–3.81 (2H, m, H5a, H5b), 2.82 (1H, dd, 3JOH–H1 = 9.4 Hz, 4JOH–H2 = 1.1 Hz, OH), 2.56–2.48 (1H, m, H4), 1.52 (3H, s, CH3), 1.36 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 127.2 (dd, 1JC7–F = 258.8 Hz, 1JC7–F = 256.6 Hz, C7), 111.9 [C(CH3)2], 79.0 (dd, 3JC2–F = 4.6 Hz, 3JC2–F = 2.5 Hz, C2), 75.4 (app. d, 3JC3–F = 8.8 Hz, C3), 73.1 (dd, 2JC1–F = 28.0 Hz, 2JC1–F = 18.9 Hz, C1), 58.9 (dd, 3JC5–F = 6.2 Hz, 3JC5–F = 5.8 Hz, C5), 50.0 (dd, 2JC4–F = 21.0 Hz, 2JC4–F = 20.0 Hz, C4), 26.0 [C(CH3)2], 24.3 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −107.8 – −108.7 (m), −114.6 – −115.3 (m); HRMS (ESI): calculated for C25H32FO4Si [M − H]− 223.0787, found 223.0792.
:1), [α]21.6D = +41.7 (c 2.0, MeOH); 1H NMR (400 MHz, CDCl3): δ 5.07 (1H, app t, 3JH3–H2 = 6.6 Hz, H3), 4.72 (1H, td, 3JH2–H3 = 6.1 Hz, 3JH2–H1 = 3.5 Hz, H2), 4.48 (1H, app. br s, H1), 4.41–4.28 (2H, m, H5a, H5b), 2.84 (1H, d, 3JOH–H1 = 9.0 Hz, OH), 1.48 (3H, s, CH3), 1.42 (3H, s, CH3); 13C NMR (101 MHz, CDCl3): δ 157.7 (d, 1JC6–F = 289.8 Hz, C6), 116.6 (d, 2JC4–F = 5.5 Hz, C4), 112.7 [C(CH3)2], 78.8 (d, 3JC3–F = 9.5 Hz, C3), 73.8 (d, 3JC2–F = 7.8 Hz, C2), 69.0 (d, 2JC1–F = 21.0 Hz, C1), 55.0 (C5), 27.5 [C(CH3)2], 26.2 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −129.73 – −129.79 (m).
:1) showed complete consumption of the starting material to a higher Rf. H2O (50 mL) was added and aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic phases were washed with H2O (2 × 100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography (0–10% EtOAc/hexane).
:1); [α]25.0D = −13.5 (c 0.6, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 7.70–7.67 (4H, m, ArH), 7.48–7.36 (6H, m, ArH), 5.06 (1H, dt, 2JH6–F = 52.0 Hz, 3JH6–H1/H4 = 6.0 Hz, H6), 4.62 (1H, dt, 3JH2–H1 = 6.3 Hz, 3JH2–H3 = 2.1 Hz, H2), 4.55 (1H, dt, 3JH3–H4 = 6.3 Hz, 3JH3–H2 = 2.2 Hz, H3), 4.42 (1H, app. dq, 3JH1–F = 14.6 Hz, 3JH1–H2/H6 = 6.3 Hz, H1), 3.89 (1H, dd, 2JH5a–H5b = 10.3 Hz, 3JH5a–H4 = 4.2 Hz, H5a), 3.74 (1H, ddd, 2JH5b–H5a = 10.4 Hz, 3JH5b–H4 = 5.7 Hz, 4JH5b–F = 1.9 Hz, H5b), 2.81 (1H, dd, 3JOH–H1 = 6.9 Hz, 4JOH–F = 1.2 Hz, OH), 2.56–2.48 (1H, m, H4), 1.51 (3H, s, CH3), 1.35 (3H, s, CH3), 1.08 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 135.68 (ArC), 135.66 (ArC), 133.0 (ArC), 132.8 (ArC), 129.89 (ArC), 129.85 (ArC), 127.83 (ArC), 127.81 (ArC), 112.0 [C(CH3)2], 99.3 (d, 1JC6–F = 185.0 Hz, C6), 80.8 (d, 3JC3–F = 3.5 Hz, C3), 77.0 (d, 3JC2–F = 7.1 Hz, C2), 73.8 (d, 2JC1–F = 24.4 Hz, C1), 59.9 (d, 3JC5–F = 8.3 Hz, C5), 46.9 (d, 2JC4–F = 18.0 Hz, C4), 26.9 [C(CH3)3], 26.2 [C(CH3)2], 24.2 [C(CH3)2], 19.2 [C(CH3)3]; 19F NMR (376 MHz, CDCl3): δ −208.5 (app. dt, 2JF–H6 = 52.2 Hz, 3JF–H1/H4 = 13.7 Hz); HRMS (NSI) calculated for C25H33FO4SiNa [M + Na]+ 476.2024, found 467.2021.
:1); [α]24.3D = −4.6 (c 0.2, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.65–7.61 (4H, m, ArH), 7.49–7.37 (6H, m, ArH), 4.64–4.57 (2H, m, H2, H3), 4.46–4.34 (1H, m, H1), 3.90 (1H, dd, 2JH5a–H5b = 10.7 Hz, 3JH5a–H4 = 2.5 Hz, H5a), 3.68 (1H, dddd, 2JH5b–H5a = 10.6 Hz, 3JH5b–H4 = 3.3 Hz, 4JH5b–F = 2.0 Hz, 4JH5b–F = 1.3 Hz, H5b), 2.84 (1H, dd, 3JOH–H1 = 11.0 Hz, 3JOH–F = 1.5 Hz, OH), 2.41 (1H, dt, 3JH4–F = 17.0 Hz, 3JH4–H5 = 3.0 Hz, H4), 1.49 (3H, s, CH3), 1.35 (3H, s, CH3), 1.05 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 135.6 (ArC), 135.6 (ArC), 132.3 (ArC), 132.1 (ArC), 130.1 (ArC), 130.0 (ArC), 128.0 (ArC), 127.9 (ArC), 124.2 (dd, 1JC6–F = 275.3 Hz, 1JC6–F = 252.5 Hz, C6), 111.4 [C(CH3)2], 79.2 (d, 3JC2–F = 5.5 Hz, C2), 75.5 (d, 3JC3–F = 10.2 Hz, C3), 74.0 (dd, 2JC1–F = 26.3 Hz, 2JC1–F = 19.0 Hz, C1), 60.3 (dd, 3JC5–F = 8.3 Hz, 3JC5–F = 4.9 Hz, C5), 50.2 (dd, 2JC4–F = 21.0 Hz, 2JC4–F = 20.2 Hz, C4), 26.9 [C(CH3)3], 25.9 [C(CH3)2], 24.2 [C(CH3)2], 19.1 [C(CH3)3]; 19F NMR (377 MHz, CDCl3): δ −107.4 (dt, 2JF–F = 240.7 Hz, 3JF–H1/H4 = 16.5 Hz), −114.0 – −114.9 (m); HRMS (NSI): calculated for C25H32F2O4SiNa [M + Na]+ 485.1930, found 485.1936.
Triflation. TBDPS alcohol (1.0 equiv.) was dissolved in pyridine (0.32 M) and cooled to −10 °C. Tf2O (2.0 equiv.) was added dropwise. The reaction mixture was stirred at this temperature for 10 min, at which point TLC analysis (hexane/EtOAc, 9
:1) showed complete consumption to a higher Rf. H2O (50 mL) was added and the aqueous phase was extracted with Et2O (3 × 200 mL). The combined organic phases were washed with sat. CuSO4 (200 mL), followed by H2O (200 mL), brine (200 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
Azidation. Triflate intermediate was dissolved in DMF (0.2 M), and NaN3 (3.0 equiv.) was added. The reaction was stirred at 80–100 °C for 3–6 h, at which point TLC analysis (hexane/EtOAc 9
:1) showed complete conversion from a lower Rf to higher Rf. The reaction was cooled to rt, and H2O (100 mL) was added. The aqueous phase was extracted with Et2O (3 × 200 mL), and the combined organic phases were washed with H2O (2 × 100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography (0–10% EtOAc/hexane).
:1); [α]24.9D = −8.0 (c 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 7.68–7.64 (4H, m, ArH), 7.46–7.35 (6H, m, ArH), 5.28 (1H, app. dt, 2JH6–F = 52.5 Hz, 3JH6–H1/H4 = 3.2 Hz, H6), 4.66 (1H, ddd, 3JH2–H3 = 7.3 Hz, 3JH2–H1 = 6.0 Hz, 4JH2–F = 1.3 Hz, H2), 4.38–4.34 (1H, m, H3), 3.87 (2H, d, 3JH5a/H5b–H4 = 8.3 Hz, H5a, H5b), 3.71 (1H, ddd, 3JH1–F = 28.0 Hz, 3JH1–H2 = 5.8 Hz, 3JH1–H6 = 3.0 Hz, H1), 2.46 (1H, m, H4), 1.50 (3H, s, CH3), 1.28 (3H, s, CH3), 1.06 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 135.6 (ArC), 135.6 (ArC), 133.3 (ArC), 133.1 (ArC), 129.8 (ArC), 127.8 (ArC), 127.7 (ArC), 114.26 [C(CH3)2], 97.0 (d, 1JC6–F = 184.0 Hz, C6), 82.1 (C2), 79.7 (C3), 68.1 (d, 3JC5–F = 16.0 Hz, C5), 60.3 (d, 2JC1–F = 7.4 Hz, C1), 50.9 (d, 2JC4–F = 17.7 Hz, C4), 27.1 [C(CH3)3], 26.8 [C(CH3)3], 24.6 [C(CH3)2], 19.2 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −206.64 (ddd, 2JF–H6 = 52.5 Hz, 3JF–H1 = 32.6 Hz, 3JF–H4 = 28.1 Hz); HRMS (ASAP): calculated for C25H33FNO3Si [M + H–N2]+ 442.2208, found 442.2203.
:1); [α]24.4D = −28.0 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 7.70–7.65 (4H, m, ArH), 7.47–7.37 (6H, m, ArH), 4.41–4.36 (1H, m, H3), 4.36–4.30 (1H, m, H2), 3.99–3.80 (3H, m, H1, H5a, H5b), 2.75–2.62 (1H, m, H4), 1.52 (3H, s, CH3), 1.28 (3H, s, CH3), 1.06 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 135.8 (ArC), 135.8 (ArC), 127.9 (ArC), 127.9 (ArC), 127.0 (dd, 1JC6–F = 264.0 Hz, 1JC6–F = 256.1 Hz, C6), 113.5 [C(CH3)2], 79.7 (d, 3JC2–F = 6.7 Hz, C2), 77.5 (d, 3JC3–F = 8.1 Hz, C3) 69.2 (dd, 2JC1–F = 23.9 Hz, 2JC1–F = 18.7 Hz, C1), 59.5 (d, 3JC5–F = 6.8 Hz, C5), 52.0 (t, 2JC4–F = 19.7 Hz, C4), 27.1 [C(CH3)3], 26.9 [C(CH3)2], 24.8 [C(CH3)2], 19.3 [C(CH3)3]; 19F NMR (377 MHz, CDCl3): δ −99.7 (dt, 2JF–F = 238.7 Hz, 3JF–H1/H4 = 7.9 Hz), −118.8 – −119.6 (m); HRMS (ASAP): calculated for C25H32F2N3O3Si [M + H]+ 488.2176, found 488.2182.
Reduction. Azide 18 or 19 (1.0 equiv.) was dissolved in MeOH (0.2 M) and Pd/C (0.1 equiv.) was added. The reaction vessel was evacuated and refilled with H2 seven times, and the reaction was stirred at rt for 4–18 h. TLC analysis (hexane/EtOAc, 9
:1) showed complete consumption of the starting material to a lower Rf. The reaction mixture was filtered through a Celite® pad and eluted with MeOH. This material was used without further purification.
:1); [α]24.7D = +3.2 (c 0.4, CH2Cl2); 1H NMR (400 MHz, CDCl3:) δ 7.70–7.65 (4H, m, ArH), 7.45–7.34 (6H, m, ArH), 5.10 (1H, dt, 3JH6–F = 53.0 Hz, 3JH6–H1/H4 = 3.1 Hz, H6), 4.35–4.27 (2H, m, H2, H3), 3.88–3.85 (2H, m, H5a, H5b), 3.36–3.25 (1H, m, H1), 2.50–2.34 (1H, m, H4), 1.49 (3H, s, CH3), 1.26 (3H, s, CH3), 1.05 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 135.6 (ArC), 135.6 (ArC), 133.5 (ArC), 133.6 (ArC), 129.7 (ArC), 129.7 (ArC), 127.7 (ArC), 127.7 (ArC), 113.5 [C(CH3)2], 99.2 (d, 1JC6–F = 176.3 Hz, C6), 86.8 (C2), 80.2 (C3), 61.8 (d, 2JC1–F = 17.3 Hz, C1), 60.7 (d, 3JC5–F = 7.4 Hz, C5), 51.4 (d, 2JC4–F = 18.0 Hz, C4), 27.3 [C(CH3)3], 26.8 [C(CH3)3], 24.7 [C(CH3)2], 19.3 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −210.5 (ddd, 2JF–H6 = 53.0 Hz, 3JF–H4 = 34.9 Hz, 3JF–H1 = 30.5 Hz). HRMS (ESI): calculated for C25H35FNO3Si [M + H]+ 444.2365, found 444.2365.
:4); [α]22.3D = −9.8 (c 0.5, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 7.70–7.66 (4H, m, ArH), 7.46–7.36 (6H, m, ArH), 4.35–4.31 (1H, m, H3), 4.17–4.11 (1H, m, H2), 3.96–3.86 (2H, m, H5a, H5b), 3.38 (1H, app. dt, 3JH1–F = 17.7 Hz, 3JH1–H2 = 5.9 Hz, H1), 2.69–2.56 (1H, m, H4), 1.51 (3H, s, CH3), 1.28 (3H, s, CH3), 1.06 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3) δ 135.7 (ArC), 135.6 (ArC), 133.2 (ArC), 133.1 (ArC), 129.8 (ArC), 129.8 (ArC), 112.7 (ArC), 82.8 (d, 3JC2–F = 8.3 Hz, C2), 77.2 (app. d, 3JC3–F = 7.7 Hz, C3), 62.7 (dd, 2JC1–F = 23.2 Hz, 2JC1–F = 21.0 Hz, C1), 59.9 (d, 3JC5–F = 6.8 Hz, C5), 51.9 (t, 2JC4–F = 20.5 Hz, C4), 27.1 [C(CH3)2], 26.7 [C(CH3)3], 24.7 [C(CH3)2], 19.2 [C(CH3)3]; 19F NMR (377 MHz, CDCl3): δ −104.4 (app. dt, 2JF–F = 234.9 Hz, 3JF–H1/H4 = 8.0 Hz), −123.2 (app. dt, 2JF–F = 234.9 Hz, 3JF–H1/H4 = 19.4 Hz); HRMS (ESI): calculated for C25H34F2NO3Si [M + H]+ 462.2271, found 462.2269.
Nucleobase synthesis. C1-Amine was dissolved in DMF (0.2 M) and cooled to 0 °C. (3-Ethoxy-acryloyl)isocyanate (2.0 equiv., 0.35 M in toluene,) was added dropwise, and the reaction was allowed to warm to rt and stirred for a further 16 h. TLC analysis (hexane/EtOAc, 1
:1) showed complete consumption of the starting material to a higher Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NaHCO3 (100 mL) and the aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic phases were washed with H2O (2 × 100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo. The crude material was purified via flash column chromatography (10–50% EtOAc/hexane).
:1); [α]24.9D = −9.0 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 9.68 (1H, s, H3), 9.24 (1H, d, 3JH1–H1′ = 8.5 Hz, H1), 7.69–7.65 (4H, m, ArH, H6), 7.45–7.36 (6H, m, ArH), 5.40 (1H, d, 3JH5–H6 = 12.2 Hz, H5), 5.26 (1H, dt, 2JH6′–F = 53.0 Hz, 3JH6′–H1′/H4′ = 3.0 Hz, H6′), 4.58 (1H, dd, 3JH2′–H1′ = 6.2 Hz, 3JH2′–H3′ = 7.0 Hz, H2′), 4.45 (1H, dddd, 3JH1′–F = 31.2 Hz, 3JH1′–H1 = 8.8 Hz, 3JH1′–H2′ = 6.1 Hz, 3JH1′–H6′ = 3.0 Hz, H1′), 4.32 (1H, t, 3JH3′–H2′/H4′ = 6.8 Hz, H3′), 3.98 (2H, q, J = 7.1 Hz, –OCH2CH3), 3.89 (2H, app. d, J = 8.1 Hz, H5′a, H5′b), 2.49 (1H, dddd, 2JH4′–F = 35.8 Hz, 2JH4′–H5′a = 10.3 Hz, 2JH4′–H5′b = 8.0 Hz, 2JH4′–H6′ = 2.9 Hz, H4′), 1.51 (3H, s, CH3), 1.35 (3H, t, J = 7.1 Hz, –OCH2CH3), 1.26 (3H, s, CH3), 1.05 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 168.3 (C4, CO), 163.1 (C6), 155.2 (C2, CO), 135.6 (ArC), 133.4 (ArC), 133.2 (ArC), 129.7 (ArC), 127.8 (ArC), 127.7 (ArC), 114.0 [C(CH)3], 98.1 (C5), 84.5 (C2′), 79.7 (C3′), 67.6 (-OCH2CH3), 60.5 (d, 3JC5′–F = 7.1 Hz, C5′), 58.9 (d, 2JC1′–F = 16.0 Hz, C1′), 51.5 (d, 2JC4′–F = 17.7 Hz, C4′), 27.3 (CH3), 26.8 (CH3), 24.7 (CH3), 14.6 (-OCH2CH3); 19F NMR (376 MHz, CDCl3): δ −206.4 (ddd, 1JF–H6′ = 53.2 Hz, 2JF–H4′ = 35.7 Hz, 2JF–H1′ = 31.5 Hz); HRMS (ESI): calculated for C31H41FN2O6SiNa [M + Na]+ 607.2610, found 607.2604.
:1); [α]24.5D = −21.2 (c 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 9.96 (1H, br s, H3), 9.29 (1H, d, 3JH1–H1′ = 9.0 Hz, H1, 7.74–7.63 (5H, m, ArH, H6), 7.46–7.36 (6H, m, ArH), 5.39 (1H, d, 3JH5–H6 = 12.3 Hz, H5), 4.76–4.64 (1H, m, H1′), 4.38–4.31 (2H, m, H2′, H3′), 4.04–3.93 (3H, m, H5′a, –OCH2CH3), 3.89 (1H, dd, 2JH5′b–H5′a = 10.6 Hz, 3JH5′b–H4′ = 6.3 Hz, H5′b), 2.79–2.65 (1H, m, H4′), 1.55 (1H, s, CH3), 1.34 (3H, t, J = 7.1 Hz, –OCH2CH3), 1.29 (3H, s, CH3), 1.07 (9H, s, 3 × CH3); 13C NMR (101 MHz, CDCl3): δ 168.5 (C4, CO), 163.1 (C6), 155.6 (C2, CO), 135.67 (ArC), 135.65 (ArC), 133.2 (ArC), 133.1 (ArC), 129.8 (ArC), 127.8 (ArC), 126.4 (dd, 1JC6′–F = 266.9 Hz, 1JC6′–F = 250.2 Hz, C6′), 113.3 [C(CH3)2], 98.0 (C5), 81.0 (d, 3JC2′–F = 8.5 Hz, C2′), 76.8 (d, 3JC3′–F = 8.2 Hz, C3′), 67.4 (–OCH2CH3), 59.6 (dd, 2JC1′–F = 22.2 Hz, 2JC1′–F = 18.3 Hz, C1′), 59.4 (d, 3JC5′–F = 6.7 Hz, C5′), 51.6 (t, 3JC4′–F = 19.6 Hz, C4′), 27.2 [C(CH3)2], 26.7 [C(CH3)3], 24.9 [C(CH3)2], 19.2 [C(CH3)3], 14.5 (–OCH2CH3); 19F NMR (377 MHz, CDCl3): δ −104.5 (dt, 2JF–F = 237.8 Hz, 3JF–H1′/H4′ = 5.8 Hz), −120.1 (dt, 2JF–F = 236.9 Hz, 3JF–H1′/H4′ = 23.0 Hz); HRMS (ESI): calculated for C31H40F2N2O6SiNa [M + Na]+ 625.2516, found 625.2507.
Cyclisation. Acryloyl urea 20 or 21 was dissolved in 1,4-dioxane (0.5 M), and H2SO4 (0.5 equiv., 2.0 M) was added. The reaction mixture was stirred at reflux for 3 h, at which point TLC analysis (EtOAc/hexane, 4
:1) showed complete conversion of the starting material to a lower Rf. The reaction mixture was cooled to rt and solid Na2CO3 was added to adjust the pH to 7. The mixture was filtered, and the solvent of the filtrate removed in vacuo. This material was used without further purification.
Silyl deprotection. Uridine intermediate was dissolved in THF (0.2 M) and TBAF (1.0 equiv., 1.0 M in THF) was added. The reaction mixture was stirred at rt for 3 h, at which point TLC analysis (10% MeOH/DCM) showed complete conversion from a higher Rf to a lower Rf. The solvent was removed in vacuo, and the residue was purified via flash column chromatography on silica gel (0–10% MeOH/EtOAc).
HPLC purification. A sample of 22 or 23 was dissolved in H2O (100 mg mL−1), and injected onto a reverse phase column (see general experimental) and purified at a flow rate of 20.00 ml min−1 using the following gradient system:
| Time (min) | %A (H2O) | %B (MeOH) |
|---|---|---|
| 0.0 | 95 | 5 |
| 5.0 | 95 | 5 |
| 12.0 | 0 | 100 |
| 15.0 | 0 | 100 |
| 15.1 | 95 | 5 |
| 20.0 | 95 | 5 |
O), 153.4 (C2, CO), 144.0 (d, 4JC6–F = 4.8 Hz, C6), 101.6 (C5), 90.9 (d, 1JC6′–F = 180.8 Hz, C6′), 70.2 (C2′), 69.2 (C3′), 61.6 (d, 2JC1′–F = 16.4 Hz, C1′), 58.3 (d, 3JC5′–F = 11.3 Hz, C5′), 50.6 (d, 2JC4′–F = 18.1 Hz, C4′); 19F NMR (377 MHz, D2O): δ −207.8 (dt, 2JF–H6′ = 55.0 Hz, 3JF–H1′/H4′ = 30.9 Hz); HRMS (ESI): calculated for C10H12FN2O5 [M − H]− 259.0736, found 259.0738. These data were in good agreement with the literature.17
O), 153.3 (C2, CO), 144.5 (d, 4JC6–F = 4.2 Hz, C6), 126.0 (dd, 1JC6′–F = 260.0 Hz, 1JC6′–F = 253.7 Hz, C6′), 102.6 (C5), 71.7 (d, 3JC2′–F = 8.0 Hz, C2′), 71.0 (dd, 3JC3′–F = 5.3 Hz, 3JC3′–F = 2.5 Hz, C3′), 63.6 (dd, 2JC1′–F = 24.3 Hz, 2JC1′–F = 18.3 Hz, C1′), 58.7 (d, 4JC5′–F = 10.9 Hz, C5′), 55.4 (t, 3JC4′–F = 20.2 Hz, C4′); 19F NMR (377 MHz, MeOD): δ −96.7 (ddd, 2JF–F = 238.6 Hz, 3JF–H4′ = 15.4 Hz, 3JF–H1′ = 9.9 Hz), −117.0 (app. dt, 2JF–F = 238.6 Hz, 3JF–H1′/H4′ = 15.6 Hz). HRMS (ESI): calculated for C10H11F2N2O5 [M − H]− 277.0642, found 277.0643. These data are in good agreement with literature values.17
:4); [α]22.1D = −10.7 (c 0.5, MeOH); 1H NMR (400 MHz, CDCl3): δ 8.87 (1H, br s, NH), 7.32 (1H, dd, 3JH6–H5 = 8.2 Hz, 5JH6–F = 1.8 Hz, H6), 5.77 (1H, dd, 3JH5–H6 = 8.2 Hz, 6JH5–F = 2.1 Hz, H5), 5.64 (1H, dd, 3JH2′–H1′ = 9.9 Hz, 3JH2′–H3′ = 7.0 Hz, H2′), 5.32–5.22 (2H, m, H1′, H3′), 5.13 (1H, dt, 2JH6′–F = 50.7 Hz, H6′), 4.34 (1H, ddd, 2JH5′a–H5′b = 11.2 Hz, 3JH5′a–H4′ = 6.3 Hz, 4JH5′a–F = 1.4 Hz, H5′a), 4.26 (1H, dd, 2JH5′b–H5′a = 11.3 Hz, 3JH5′b–H4′ = 8.8 Hz, H5′b), 2.83–2.68 (1H, m, H4′), 2.11 (3H, s, Ac–CH3), 2.07 (3H, s, Ac–CH3), 2.05 (3H, s, Ac–CH3); 13C NMR (101 MHz, CDCl3): δ 170.5 (CO), 169.8 (CO), 169.7 (CO), 162.4 (C2, CO), 151.2 (C4, CO), 141.2 (d, 4JC6–F = 5.2 Hz, C6), 102.8 (C5), 90.3 (d, 1JC6′–F = 184.5 Hz, C6′), 69.6 (C2), 69.4 (C3), 59.8 (d, 3JC5′–F = 10.1 Hz, C5′), 59.7 (d, 2JC1′–F = 16.4 Hz, C1′), 46.0 (d, 2JC4′–F = 18.2 Hz, C4′), 20.7 (Ac–CH3), 20.5 (Ac–CH3), 20.4 (Ac–CH3); 19F NMR (377 MHz, CDCl3) δ −206.2 (dt, 2JF–H6′ = 54.5 Hz, 3JF–H1′/H4′ = 30.2 Hz); HRMS (NSI): calculated for C16H20FN2O8 [M + H]+ 387.1198, found 387.1200.
:4); [α]24.6D = −33.2 (c 0.4, MeOH); 1H NMR (400 MHz, CDCl3): δ 9.58 (1H, br s, NH), 7.24 (1H, dd, 3JH6–H5 = 8.2 Hz, 5JH6–F = 2.5 Hz, H6), 5.80 (1H, d, 3JH5–H6 = 8.2 Hz, H5), 5.59–5.51 (2H, m, H1′, H2′), 5.32–5.29 (1H, m, H3′), 4.37–4.33 (2H, m, H5′a, H5′b), 3.03–2.90 (1H, m, H4′), 2.14 (3H, s, Ac–CH3), 2.09 (3H, s, Ac–CH3), 2.05 (3H, s, Ac–CH3); 13C NMR (101 MHz, CDCl3): δ 170.3 (CO), 169.7 (CO), 169.7 (CO), 162.7 (C2, CO), 151.1 (C4, CO), 141.0 (d, 4JC6–F = 4.8 Hz, C6), 122.5 (dd, 1JC6′–F = 262.9 Hz, 1JC6′–F = 253.7 Hz, C6′), 103.3 (C5), 68.8 (d, 3JC3′–F = 5.5 Hz, C3′), 68.6 (d, 3JC2′–F = 8.1 Hz, 3JC2′–F), 60.4 (dd, 2JC1′–F = 25.9 Hz, 2JC1′–F = 18.6 Hz, C1′), 58.6 (d, 3JC5′–F = 9.0 Hz, C5′), 48.3 (dd, 2JC4′–F = 23.0 Hz, 2JC4′–F 20.7 Hz, C4′), 20.7 (Ac–CH3), 20.6 (Ac–CH3), 20.4 (Ac–CH3); 19F NMR (377 MHz, CDCl3): δ −100.8 (d, 2JF–F = 240.8 Hz), −115.5 (app. dt, 2JF–F = 240.5 Hz, 3JF–H1′/H4′ = 17.5 Hz); HRMS (NSI) calculated for C16H19F2N2O8 [M + H]+ 405.1104, found 405.1104.
Installation of 1,2,4-triazole. Triacetate 24 or 25 (1.0 equiv.) was dissolved in MeCN (0.1 M) and cooled to 0 °C. 1,2,4-Triazole (23 equiv.), POCl3 (2.4 equiv.) and Et3N (23 equiv.) were added and the reaction mixture was allowed to slowly warm to rt and stirred for 18 h. TLC analysis (EtOAc/hexane, 4
:1) showed majority conversion of the starting material to a lower Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NaHCO3 (5 mL). The aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic phases were washed with H2O (10 mL), brine (10 mL), dried over MgSO4, filtered and the solvent removed in vacuo.
Amination and deprotection. 1,2,4-Triazole derivative was dissolved in 35% NH4OH/1,4-dioxane (1
:1, v/v, 0.1 M) and the reaction vessel was sealed. The reaction mixture was stirred at rt for 18 h, at which point TLC analysis (5% MeOH/DCM) showed complete conversion from a higher Rf to a lower Rf. The solvent was removed in vacuo, and the residue was purified via flash column chromatography on silica gel (10% MeOH/EtOAc).
HPLC purification. A sample of 26 or 27 was dissolved in H2O (100 mg mL−1), and injected onto a reverse phase column (see general experimental) and purified at a flow rate of 20.00 ml min−1 using the following gradient system:
| Time (min) | %A (H2O) | %B (MeOH) |
|---|---|---|
| 0.0 | 96 | 4 |
| 10.0 | 96 | 4 |
| 12.0 | 0 | 100 |
| 16.0 | 0 | 100 |
| 16.5 | 96 | 4 |
| 20.0 | 96 | 4 |
O), 158.7 (C4), 143.8 (d, 4JC6–F = 4.2 Hz, C6), 95.7 (C5), 91.0 (d, 1JC6′–F = 180.6 Hz, C6′), 70.3 (C2), 69.3 (C3), 62.3 (d, 2JC1′–F = 16.5 Hz, C1′), 58.3 (d, 3JC5′–F = 11.3 Hz, C5′), 50.6 (d, 2JC4′–F = 18.1 Hz, C4′); 19F NMR (377 MHz, D2O): δ −208.2 (dt, 2JF–H6′ = 55.0 Hz, 3JF–H1′/H4′ = 31.0 Hz); HRMS (NSI): calculated for C10H13FN3O4 [M − H]− 258.0896, found 258.0895. These data are in good agreement with literature.17
O), 158.7 (C4), 143.5 (d, 4JC6–F = 4.0 Hz, C6), 124.2 (dd, 1JC6′–F = 259.8 Hz, 1JC6′–F 253.5 Hz, C6′), 96.4 (C5), 69.9 (d, 3JC2′–F = 8.0 Hz, C2′), 68.9 (dd, 3JC3′–F = 6.2 Hz, 3JC3′–F = 2.1 Hz, C3′), 62.7 (dd, 2JC1′–F = 23.8 Hz, 2JC1′–F = 18.7 Hz, C1′), 57.3 (d, 4JC5′–F = 10.2 Hz, C5′), 52.9 (t, 3JC4′–F = 20.3 Hz, C4′); 19F NMR (377 MHz, D2O): δ −97.9 (ddd, 2JF–F = 236.5 Hz, 3JF–H4′ = 14.0 Hz, 3JF–H1′ = 8.7 Hz), −117.2 (app. dt, 2JF–F = 236.7 Hz, 3JF–H1′/H4′ = 16.9 Hz); HRMS (NSI): calculated for C10H12F2N3O4 [M − H]− 276.0801, found 276.0797. These data were in good agreement with literature.17
:1); [α]23.4D = −73.4 (c 1.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 9.32 (1H, br s, NH), 7.41 (1H, d, 3JH6–H5 = 7.6 Hz, H6), 5.75 (1H, d, 3JH5–H6 = 8.1 Hz, H5), 5.09 (1H, ddd, 2JH6′–F = 55.2 Hz, 3JH6′–H4′ = 4.1 Hz, 3JH6′–H1′ = 3.1 Hz, H6′), 4.74 (1H, ddd, 3JH1′–F = 30.4 Hz, 3JH1′–H2′ = 9.2 Hz, 3JH1′–H6′ = 2.7 Hz, H1′), 4.55–4.45 (2H, m, H2′, H3′), 4.14 (1H, dd, 2JH5′a–H5′b = 12.2 Hz, 3JH5′a–H4′ = 4.0 Hz, H5′a), 3.93 (1H, dd, 2JH5′b–H5′a = 12.0 Hz, 3JH5′b–H4′ = 10.4 Hz, H5′b), 3.03 (1H, d, 3JOH–H1′ = 8.6 Hz, OH), 2.46 (1H, m, H4′), 1.11–1.01 (28H, m, [–OSi(CH)2(CH3)4]2); 13C NMR (101 MHz, CDCl3): δ 163.1 (C2, CO), 151.8 (C4, CO), 141.7 (d, 4JC6–F = 4.1 Hz, C6), 102.4 (C5), 92.2 (d, 1JC6′–F = 180.6 Hz, C6′), 71.3 (d, 3JC3′–F = 0.6 Hz, C3′), 70.4 (d, 3JC2′–F = 1.7 Hz, C2′), 63.0 (d, 2JC1′–F = 16.4 Hz, C1′), 61.9 (d, 3JC5′–F = 11.8 Hz, C5′), 53.3 (d, 2JC4′–F = 17.6 Hz, C4′), 17.7 {[–OSi(CH)2(CH3)4]2}, 17.5 {[–OSi(CH)2(CH3)4]2}, 17.4 {[–OSi(CH)2(CH3)4]2}, 17.24 {[–OSi(CH)2(CH3)4]2}, 17.22 {[–OSi(CH)2(CH3)4]2}, 17.19 {[–OSi(CH)2(CH3)4]2}, 17.15 {[–OSi(CH)2(CH3)4]2}, 17.11 {[–OSi(CH)2(CH3)4]2}, 17.05 {[–OSi(CH)2(CH3)4]2}, 13.50 {[–OSi(CH)2(CH3)4]2}, 13.47 {[–OSi(CH)2(CH3)4]2}, 13.19 {[–OSi(CH)2(CH3)4]2}, 13.18 {[–OSi(CH)2(CH3)4]2}, 12.6 {[–OSi(CH)2(CH3)4]2}; 19F NMR (377 MHz, CDCl3) δ −207.0 (ddd, 2JF–H6′ = 55.3 Hz, 3JF–H4′ = 35.2 Hz, 3JF–H1′ = 30.6 Hz); HRMS (NSI): calculated for C22H40FN2O6Si2 [M + H]+ 503.2403, found 503.2402.
:1); [α]20.3D = −65.9 (c 0.5, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 9.22 (1H, s, NH), 7.17 (1H, dd, 3JH6–H5 = 8.2 Hz, 5JH6–F = 1.5 Hz, H6), 5.76 (1H, d, 3JH5–H6 = 8.1 Hz, H5), 5.08 (1H, dt, 3JH1′–F = 19.4 Hz, 3JH1′–H2′ = 7.2 Hz, H1′), 4.57 (1H, app. t, 3JH3′–H2′/H4′ = 6.5 Hz, H3′), 4.37 (1H, app. q, 3JH2′–H1′/H3′/OH = 6.0 Hz, H2′), 4.15 (1H, dd, 2JH5′a–H5′b = 12.3 Hz, 3JH5′a–H4′ = 4.2 Hz, H5′a), 4.02 (1H, dd, 2JH5′b–H5′a = 12.3 Hz, 3JH5′b–H4′ = 7.5 Hz, H5′b), 3.20 (1H, d, 3JOH–H2′ = 6.0 Hz, OH), 2.72–2.58 (1H, m, H4′), 1.10–1.04 (28H, m, [–OSi(CH)2(CH3)4]2); 13C NMR (101 MHz, CDCl3): δ 162.8 (C2, CO), 151.0 (C4, CO), 141.9 (d, 4JC6–F = 4.3 Hz, C6), 123.5 (dd, 1JC6′–F = 262.3 Hz, 1JC6′–F = 251.3 Hz, C6′), 103.0 (C5), 70.1 (d, 3JC3′–F = 6.8 Hz, C3′), 69.6 (d, 3JC2′–F = 8.7 Hz, C2), 64.4 (dd, 2JC1′–F = 22.3 Hz, 2JC1′–F = 17.7 Hz, C1′), 58.1 (d, 3JC5′–F = 8.1 Hz, C5′), 52.9 (t, 2JC4′–F = 19.5 Hz, C4′), 17.4 {[–OSi(CH)2(CH3)4]2}, 17.3 {[–OSi(CH)2(CH3)4]2}, 17.2 {[–OSi(CH)2(CH3)4]2}, 17.1 {[–OSi(CH)2(CH3)4]2}, 17.00 {[–OSi(CH)2(CH3)4]2}, 16.99 {[–OSi(CH)2(CH3)4]2}, 16.9 {[–OSi(CH)2(CH3)4]2}, 13.3 {[–OSi(CH)2(CH3)4]2}, 13.2 {[–OSi(CH)2(CH3)4]2}, 12.9 {[–OSi(CH)2(CH3)4]2}, 12.5 {[–OSi(CH)2(CH3)4]2}; 19F NMR (377 MHz, CDCl3): δ −106.5 (d, 2JF–F = 237.5 Hz), −118.7 (dt, 2JF–F = 237.2 Hz, 3JF–H1′/H4′ = 21.5 Hz); HRMS (NSI): calculated for C22H38F2N2O6Si2Na [M + Na]+ 543.2129, found 543.2121.
Synthesis of thiocarbamate. Uridine derivative 28 or 29 (1.0 equiv.) was dissolved in MeCN (0.2 M). TCDI (1.1 equiv.) was added followed by DMAP (0.1 equiv.) and the reaction mixture was heated to 45 °C and stirred for 4 h. TLC analysis (hexane/EtOAc, 1
:1) showed complete consumption of the starting material to a lower Rf. The reaction mixture was cooled to rt, and the solvent was removed in vacuo. The residue was partitioned between H2O (10 mL) and EtOAc (10 mL), and the aqueous phase was extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine, dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
Barton McCombie deoxygenation. Thiocarbamate intermediate was charged to an oven dried multinecked RB flask equipped with a magnetic stirrer, and the reaction vessel was evacuated and refilled with N2 × 3. The crude material was dissolved in degassed toluene (0.2 M), and Bu3SnH (1.5 equiv.) and AIBN (0.16 equiv.) were added. The reaction mixture was stirred at reflux for 30 min, at which point TLC analysis (1
:1, hexane/EtOAc) showed complete conversion from a lower Rf to a higher Rf. The reaction mixture was cooled to rt, and the solvent removed in vacuo. The residue was purified via flash column chromatography on silica gel (20–50% EtOAc/hexane).
:1); [α]24.3D = −52.3 (c 0.5, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 9.70 (1H, br s, NH), 7.23 (1H, dd, 3JH6–H5 = 8.1 Hz, 5JH6–F = 0.9 Hz, H6), 5.66 (1H, d, 3JH5–F = 8.1 Hz, H5), 5.22 (1H, dddd, 3JH1′–F = 30.6 Hz, 3JH1′–H2′a = 11.3 Hz, 3JH1′–H2′b = 9.2 Hz, 3JH1′–H6′ = 2.4 Hz, H1′), 5.01 (1H, dt, 2JH6′–F = 56.0 Hz, 3JH6′–H1′/H4′ = 3.0 Hz, H6′), 4.60 (1H, ddd, 3JH3′–H2′a = 7.7 Hz, 3JH3′–H4′ = 5.2 Hz, 3JH3′–H2′b = 2.3 Hz, H3′), 4.08 (1H, dd, 2JH5′a–H5′b = 12.0 Hz, 3JH5′a–H4′ = 3.7 Hz, H5′a), 3.88 (1H, dd, 2JH5′b–5′a = 11.9 Hz, 3JH5′b–H4′ = 9.4 Hz, H5′b), 2.38 (1H, dt, 2JH2′a–H2′b = 12.8 Hz, 3JH2′a–H1′ = 12.8 Hz, 3JH2′a–H3′ = 8.3 Hz, H2′a), 2.36–2.20 (1H, m, H4′), 2.11 (1H, ddd, 3JH2′b = 11.8 Hz, 3JH2′b–H1′ = 8.7 Hz, 3JH2′b–H3′ = 2.0 Hz, H2′b), 1.05–0.93 (28H, m, [–OSi(CH)2(CH3)4]2); 13C NMR (101 MHz, CDCl3): δ 162.3 (C2, CO), 150.4 (C4, CO), 140.7 (d, 4JC6–F = 5.5 Hz, C6), 101.0 (C5), 95.3 (d, 1JC6′–F = 181.5 Hz, C6′), 71.4 (C3′), 60.8 (d, 3JC5′–F = 10.8 Hz, C5′), 54.3 (d, 2JC1′/C4′–F = 17.1 Hz, C1′, C4′), 35.0 (C2′), 16.6 {[–OSi(CH)2(CH3)4]2}, 16.4 {[–OSi(CH)2(CH3)4]2}, 16.4 {[–OSi(CH)2(CH3)4]2}, 16.4 {[–OSi(CH)2(CH3)4]2}, 16.4 {[–OSi(CH)2(CH3)4]2}, 16.2 {[–OSi(CH)2(CH3)4]2}, 15.98 {[–OSi(CH)2(CH3)4]2}, 15.95 {[–OSi(CH)2(CH3)4]2}, 12.33 {[–OSi(CH)2(CH3)4]2}, 12.30 {[–OSi(CH)2(CH3)4]2}, 11.9 {[–OSi(CH)2(CH3)4]2}, 11.5 {[–OSi(CH)2(CH3)4]2}; 19F NMR (377 MHz, CDCl3) δ −209.2 (ddd, 2JF–H6′ = 55.9 Hz, 3JF–H4′ = 36.6 Hz, 3JF–H1′ = 30.7 Hz).
:1); [α]25.9D = −46.5 (c 2.0, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 8.29 (1H, br s, NH), 7.17 (1H, dd, 3JH6–H5 = 8.2 Hz, 5JH6–F = 2.5 Hz, H6), 5.76 (1H, dd, 3JH5–H6 = 8.2 Hz, 6JH5–F = 1.9 Hz, H5), 5.58 (1H, dtd, 3JH1′–F = 16.7 Hz, 3JH1′–H2′a = 9.9 Hz, 3JH1′–H2′b = 6.5 Hz, H1′), 4.58 (1H, dd, 3JH3′–H4 = 13.4 Hz, 3JH3′–H2′a/H2′b = 6.3 Hz, H3′), 4.17–3.98 (2H, m, H5′a, H5′b), 2.51–2.39 (1H, m, H4′), 2.38–2.24 (2H, m, H2′a, H2′b), 1.11–1.02 (28 H, m, [–OSi(CH)2(CH3)4]2); 13C NMR (101 MHz, CDCl3): δ 162.0 (C2, CO), 150.7 (C4, CO), 141.1 (d, 4JC6–F = 5.9 Hz, C6), 102.8 (C5), 68.8 (C3′), 58.1 (d, 3JC5′–F = 6.8 Hz, C5′), 55.0 (C1′), 54.7 (C4′), 34.7 (d, 3JC2′–F = 5.2 Hz, C2′), 17.44 {[–OSi(CH)2(CH3)4]2}, 17.36 {[–OSi(CH)2(CH3)4]2}, 17.32 {[–OSi(CH)2(CH3)4]2}, 17.29 {[–OSi(CH)2(CH3)4]2}, 17.1 {[–OSi(CH)2(CH3)4]2}, 17.0 {[–OSi(CH)2(CH3)4]2}, 16.9 {[–OSi(CH)2(CH3)4]2}, 13.3 {[–OSi(CH)2(CH3)4]2}, 13.1 {[–OSi(CH)2(CH3)4]2}, 12.8 {[–OSi(CH)2(CH3)4]2}, 12.5 {[–OSi(CH)2(CH3)4]2}; 19F NMR (377 MHz, CDCl3): δ −108.8 (dt, 2JF–F = 232.4 Hz, 3JF–H4′ = 7.3 Hz), −121.6 (ddd, 2JF–F = 232.2 Hz, 3JF–H4′ = 26.0 Hz, 3JF–H1′ = 20.3 Hz); HRMS (NSI): calculated for C22H39F2N2O5Si2 [M + H]+ 505.2360, found 505.2360.
HPLC purification. A sample of 32 or 33 was dissolved in 9
:1 H2O/MeOH (v/v, 100 mg mL−1), and injected onto a reverse phase column (see general experimental) and purified at a flow rate of 20.00 ml min−1 using the following gradient system:
| Time (min) | %A (H2O) | %B (MeOH) |
|---|---|---|
| 0.0 | 90 | 10 |
| 5.0 | 90 | 10 |
| 12.0 | 0 | 100 |
| 15.0 | 0 | 100 |
| 15.1 | 90 | 10 |
| 20.0 | 90 | 10 |
O), 152.5 (C4, CO), 144.6 (d, 4JC6–F = 5.3 Hz, C6), 101.1 (C5), 94.4 (d, 1JC6′–F = 181.3 Hz, C6′), 70.2 (C3′), 58.1 (d, 3JC5′–F = 9.7 Hz, C5′), 55.7 (d, 2JC1′–F = 16.4 Hz, C1′), 53.2 (d, 2JC4′–F = 17.7 Hz, C4′), 34.4 (C2′); 19F NMR (377 MHz, D2O) δ −209.7 (app. dt, 2JF–H6′ = 54.5 Hz, 3JF–H1′/H4′ = 32.2 Hz); HRMS (NSI): calculated for C10H12FN2O4 [M − H]− 243.0787, found 243.0783.
O), 153.2 (C4, CO), 143.9 (d, 4JC6–F = 4.5 Hz, C6), 127.1 (dd, 1JC6′–F = 260.7 Hz, 1JC6′–F = 254.0 Hz, C6′), 102.0 (C5), 67.7 (d, 3JC3′–F = 8.4 Hz, C3′), 57.1 (d, 3JC5′–F = 8.8 Hz, C5′), 56.3 (dd, 2JC1′–F = 25.2 Hz, 2JC1′–F = 18.4 Hz, C1′), 53.6 (t, 2JC4′–F = 19.5 Hz, C4′), 33.5 (d, 3JC2′–F = 6.3 Hz, C2′); 19F NMR (377 MHz, D2O): δ −102.8 (app. dt, 2JF–F = 232.4 Hz, 3JF–H4′/H1′ = 9.2 Hz), −120.5 (app. dt, 2JF–F = 232.4 Hz, 3JF–H1′/H4′ = 19.6 Hz); HRMS (NSI): calculated for C10H11F2N2O4 [M-H]− 261.0692, found 261.0692
Installation of 1,2,4-triazole. Uridine 30 or 31 (1.0 equiv.) was dissolved in MeCN (0.1 M) and cooled to 0 °C. 1,2,4-Triazole (23 equiv.), POCl3 (2.4 equiv.) and Et3N (23 equiv.) were added and the reaction mixture was allowed to slowly warm to rt and stirred for 18 h. TLC analysis (4
:1 EtOAc/hexane) showed majority conversion of the starting material to a lower Rf. The reaction mixture was cooled to 0 °C and quenched with sat. NaHCO3 (5 mL). The aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic phases were washed with H2O (10 mL), brine (10 mL), dried over MgSO4, filtered and the solvent removed in vacuo.
Amination. 1,2,4-Triazole derivative was dissolved in 35% NH4OH/1,4-dioxane (1
:1, v/v, 0.1 M) and the reaction vessel was sealed. The reaction mixture was stirred at rt for 18 h, at which point TLC analysis (5% MeOH/DCM) showed complete conversion from a higher Rf to a lower Rf. The solvent was removed in vacuo, and the residue was passed through a silica plug (eluting first with 1:1 hexane/EtOAc, then 5% MeOH/EtOAc).
:1); [α]24.5D = −55.8 (c 1.0, MeOH); 1H NMR (400 MHz, MeOD): δ 8.30 (2H, br s, NH2), 7.62 (1H, dd, 3JH6–H5 = 7.5 Hz, 5JH6–F = 1.3 Hz, H6), 5.87 (1H, d, 3JH5–H6 = 7.5 Hz, H5), 5.27 (1H, dddd, 3JH1′–F = 30.5 Hz, 3JH1′–H2′a = 11.6 Hz, 3JH1′–H2′b = 8.2 Hz, 3JH1′–H6′ = 2.5 Hz, H1′), 5.21 (1H, dt, 2JH6′–F = 56.1 Hz, 3JH6′–H1′/H4′ = 3.1 Hz, H6′), 4.71 (1H, ddd, 3JH3′–H2′a = 7.3 Hz, 3JH3′–H4′ = 4.9 Hz, 3JH3′–H2′b = 2.1 Hz, H3′), 4.17 (1H, dd, 2JH5′a–H5′b = 11.9 Hz, 3JH5′a–H4′ = 3.8 Hz, HH5′a), 3.97 (1H, dd, 2JH5′b–H5′a = 11.8 Hz, 3JH5′b–H4′ = 9.8 Hz, H5′b), 2.64 (1H, td, 2JH2′a–2′b = 12.8 Hz, 3JH2′a–H1′ = 12.8 Hz, 3JH2′a–H3′ = 7.9 Hz, H2′a), 2.40 (1H, dtd, 3JH4′–F = 35.8 Hz, 3JH4′–H5′b = 8.6 Hz, 3JH4′–H3′/H5′a/H6′ = 4.0 Hz, H4′), 2.10 (ddd, 2JH2′b–H2′a = 13.1 Hz, 3JH2′b–H1′ = 8.3 Hz, 3JH2′b–H3′ = 1.9 Hz, H2′b), 1.14–1.02 (28H, m, [–OSi(CH)2(CH3)4]2); 13C NMR (101 MHz, MeOD): δ 167.3 (C2, CO), 159.0 (C4), 144.6 (d, 4JC6–F = 4.3 Hz, C6), 97.1 (d, 1JC6′–F = 180.7 Hz, C6′), 95.5 (C5), 74.1 (C3′), 63.3 (d, 3JC5′–F = 11.5 Hz, C5′), 58.0 (d, 2JC1′–F = 16.7 Hz, C1′), 57.0 (d, 2JC4′–F = 17.1 Hz, C4′), 36.8 (C2′), 18.1 {[–OSi(CH)2(CH3)4]2}, 18.0 {[–OSi(CH)2(CH3)4]2}, 17.94 {[–OSi(CH)2(CH3)4]2}, 17.92 {[–OSi(CH)2(CH3)4]2}, 17.85 {[–OSi(CH)2(CH3)4]2}, 17.7 {[–OSi(CH)2(CH3)4]2}, 17.54 {[–OSi(CH)2(CH3)4]2}, 17.51 {[–OSi(CH)2(CH3)4]2}, 14.6 {[–OSi(CH)2(CH3)4]2}, 14.5 {[–OSi(CH)2(CH3)4]2}, 14.2 {[–OSi(CH)2(CH3)4]2}, 13.8 {[–OSi(CH)2(CH3)4]2}; 19F NMR (377 MHz, CDCl3): δ −207.68 (ddd, 2JF–H6′ = 56.0 Hz, 3JF–H4′ = 35.6 Hz, 3JF–H1′ = 30.6 Hz); HRMS (NSI): calculated for C22H40FN3O4Si2 [M + H]+ 486.2614, found 486.2614.
O), 158.6 (C4), 144.6 (d, 4JC6–F = 5.0 Hz, C6), 127.9 (dd, 1JC6′–F = 261.5 Hz, 1JC6′–F = 252.9 Hz, C6′), 96.3 (C5), 70.8 (d, 3JC3′–F = 9.2 Hz, C3′), 59.6 (d, 3JC5′–F = 7.8 Hz, C5′), 57.2 (dd, 2JC1′–F = 24.2 Hz, 2JC1′–F = 17.3 Hz, C1′), 56.6 (t, 2JC4′–F = 19.0 Hz, C4′), 35.7 (d, 3JC2′–F = 5.7 Hz, C2′), 18.0 {[–OSi(CH)2(CH3)4]2}, 17.9 {[–OSi(CH)2(CH3)4]2}, 17.83 {[-OSi(CH)2(CH3)4]2}, 17.80 {[–OSi(CH)2(CH3)4]2}, 17.7 {[–OSi(CH)2(CH3)4]2}, 17.52 {[-OSi(CH)2(CH3)4]2}, 17.49 {[–OSi(CH)2(CH3)4]2}, 14.54 {[-OSi(CH)2(CH3)4]2}, 14.47 {[–OSi(CH)2(CH3)4]2}, 14.4 {[–OSi(CH)2(CH3)4]2}, 13.8 {[–OSi(CH)2(CH3)4]2}; 19F NMR (377 MHz, MeOD) δ −109.35 (dt, 2JF–F = 231.6 Hz, 3JF–H4′ = 6.8 Hz), −122.6 (dt, 2JF–F = 231.0 Hz, 3JF–H1′/H4′ = 23.1 Hz); HRMS (NSI): calculated for C22H39F2N3O4Si2 [M + H]+ 504.2520, found 504.2519.
Desilylation. 2′-Deoxycytidine derivative (1.0 equiv.) was dissolved in THF (0.2 M), and TBAF (2.0 equiv., 1.0 M in THF) was added. The reaction was stirred at rt for 16 h, at which point TLC analysis (10% MeOH/DCM) showed complete consumption of the starting material to a lower Rf. DOWEX 50WX8 (H+) resin (400 mg), CaCO3 (160 mg) and MeOH (1.0 mL) were added, and the reaction mixture was stirred for a further 1 h. The reaction mixture was filtered through a Celite® plug and eluted with MeOH. The solvent as removed in vacuo, and the residue was purified via flash column chromatography (0–10% MeOH/EtOAc).
HPLC purification. A sample of 34 or 35 was dissolved in H2O (100 mg mL−1), and injected onto a reverse phase column (see general experimental) and purified at a flow rate of 20.00 ml min−1 using the following gradient system:
| Time (min) | %A (H2O) | %B (MeOH) |
|---|---|---|
| 0.0 | 96 | 4 |
| 12.0 | 96 | 4 |
| 15.0 | 0 | 100 |
| 18.0 | 0 | 100 |
| 18.5 | 96 | 4 |
| 22.0 | 96 | 4 |
O), 158.5 (C4), 143.8 (d, 4JC6–F = 4.6 Hz, C6), 126.2 (m, C6′), 96.0 (C5), 67.8 (d, 4JC3′–F = 8.5 Hz, C3′), 57.1 (d, 4JC5′–F = 8.5 Hz, C5′), 56.8 (dd, 3JC1′–F = 25.3 Hz, 3JC1′–F = 18.3 Hz, C1′), 53.8 (t, 3JC4′–F = 19.6 Hz, C4′), 33.9 (d, 4JC2′–F = 6.5 Hz, C2′); 19F NMR (377 MHz, D2O): δ −102.8 (app. dt, 2JF–F = 231.5 Hz, 3JF–H1′/H4′ = 9.2 Hz), −120.7 (app. dt, 2JF–F = 231.5 Hz, 3JF–H1′/H4′ = 19.6 Hz); HRMS (NSI): calculated for C10H12F2N3O3 [M − H]− 260.0852, found 260.0848.
O), 151.1 (C4, CO), 141.6 (C6), 115.2 [C(CH3)2], 102.6 (C5), 97.8 (d, 1JC6′–F = 181.0 Hz, C6′), 80.2 (C2′), 79.7 (C3′), 62.7 (d, 2JC1′–F = 15.7 Hz, C1′), 59.6 (d, 3JC5′–F = 8.4 Hz, C5), 50.6 (d, 2JC4′–F = 17.5 Hz, C4′), 27.3 [C(CH3)2], 24.9 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −205.2 (app. dt, 2JF–H6′ = 54.3 Hz, 3JF–H1′/H4′ = 34.8 Hz); HRMS (NSI) calculated for C13H17FN2O5Na [M + Na]+ 323.1014, found 232.1014. These data were in good agreement with literature.17
O), 151.1 (C4, CO), 141.8 (app. d, 4JC6–F = 5.2 Hz, C6), 127.1 (dd, 1JC6′–F = 266.1 Hz, 1JC6′–F = 253.3 Hz, C6′), 114.2 [C(CH3)2], 103.2 (C5), 77.9 (d, 3JC2′–F = 7.8 Hz, C2′), 77.2 (app. d, 3JC3′–F = 8.1 Hz, C3′), 64.2 (dd, 2JH1′–F = 24.2 Hz, 2JH1′–F = 17.8 Hz, C1′), 58.1 (app. d, 3JC5′–F = 7.9 Hz, C5′), 52.1 (app. t, 2JC4′–F = 19.7 Hz, C4′), 27.2 [C(CH3)2], 25.0 [C(CH3)2]; 19F NMR (377 MHz, CDCl3): δ −100.0 (app. d, 2JF–F = 237.2 Hz), −116.4 (app. dt, 2JF–F = 237.4, 3JF–H1′/H4′ = 21.8 Hz); HRMS (NSI) calculated for C13H16F2N2O5Na [M + Na]+ 341.0919, found 341.0921. These data were in good agreement with literature.17
Synthesis of phosphoramidate intermediate. Uridine derivative 36 or 37 (1.0 equiv.) was suspended in THF (0.2 M) and cooled to 0 °C. tBuMgCl (2.0 equiv., 1.0 M in THF) was added dropwise and the reaction mixture was stirred for 1 h. Isopropyl [(S)-(perfluorophenoxy)(phenoxy)phosphoryl]-L-alaninate 38 (1.1 equiv.) was added in one portion and the reaction mixture was allowed to warm to rt and stirred for 4 h. TLC analysis showed complete consumption of the starting material to a higher Rf. The reaction was quenched with MeOH (1 mL), and the solvent removed in vacuo. The residue was partitioned between H2O (5 mL) and EtOAc (5 mL), and the aqueous phase was extracted with EtOAc (2 × 5 mL), washed with brine (5 mL), dried over MgSO4, filtered and the solvent removed in vacuo. This material was used without further purification.
Deprotection. Phosphoramidate intermediate was dissolved in 1
:1 formic acid/H2O (v/v, 0.1 M) and stirred at rt for 16 h. The solvent was removed in vacuo and the crude material was purified via flash column chromatography on silica gel (0–5% MeOH/EtOAc).
HPLC purification. A sample of 39 or 40 was dissolved in 1
:1 H2O/MeOH (v/v, 100 mg mL−1), and injected onto a reverse phase column (see general experimental) and purified at a flow rate of 20.00 ml min−1 using the following gradient system:
| Time (min) | %A (H2O) | %B (MeOH) |
|---|---|---|
| 0.0 | 50 | 50 |
| 5.0 | 50 | 50 |
| 12.0 | 0 | 100 |
| 15.0 | 0 | 100 |
| 15.1 | 50 | 50 |
| 20.0 | 50 | 50 |
O–P = 5.4 Hz, Ala CO), 166.3 (C4, CO), 153.4 (C2, CO), 152.3 (d, 2JArC–P = 7.0 Hz, ArC), 144.6 (d, 4JC6–F = 3.9 Hz, C6), 130.8 (d, 4JArC–P = 0.6 Hz, ArC), 126.1 (d, 5JArC–P = 1.2 Hz, ArC), 121.5 (d, 3JArC–P = 4.7 Hz, ArC), 102.0 (C5), 91.8 (d, 1JC6′–F = 182.3 Hz, C6′), 71.6 (C2′), 70.4 (C3′), 70.1 [CH(CH3)2], 64.6 (dd, 2JC5′–P = 11.3 Hz, 3JC5′–F = 5.6 Hz, C5′), 63.5 (d, 2JC1′–F = 16.3 Hz, C1′), 51.7 (Ala–CH), 51.0 (dd, 2JC4′–F = 17.5 Hz, 3JC4′–P = 7.5 Hz, C4′), 22.0 [CH(CH3)2], 21.9 [CH(CH3)2], 20.5 (d, 3JAlaCH3–P = 6.5 Hz, Ala–CH3); 19F NMR (377 MHz, MeOD): δ −208.4 (dt, 2JF–H6′ = 55.4 Hz, 3JF–H1′/H4′ = 30.3 Hz); 31P {1H} NMR (162 MHz, MeOD): δ 3.46 (s); HRMS (NSI): calculated for C22H28FN3O9P [M − H]− 528.1553, found 528.1547. These data are in good agreement with literature.17
O–P = 5.3 Hz, CO Ala), 164.4 (CO, C2), 151.8 (CO, C4), 150.8 (d, 2JArC–P = 7.0 Hz, ArC), 142.9 (d, 4JC6–F = 4.1 Hz, C6), 129.4 (d, 4JArC–P = 0.6 Hz, ArC), 124.8 (d, 5JArC–P = 1.0 Hz, ArC), 123.8 (dd, 1JC6′–F = 261.1, 1JC6′–F = 253.7 Hz, C = 6′), 120.0 (d, 3JArC–P = 4.8 Hz, ArC), 101.3 (C5), 69.8 (d, 3JC2′–F = 7.8 Hz, C2′), 68.9 (dd, 3JC3′–F = 5.6 Hz, 3JC3′–F = 1.7 Hz, C3), 68.8 [CH(CH3)2], 62.2 (dd, 2JC1′–F = 24.2 Hz, 2JC1′–F = 18.2 Hz, C1′), 61.9 (dd, 3JC5′–P = 11.0 Hz, 3JC5′–F = 5.4 Hz, C5′), 51.9 (ddd, 3JC4′–F = 21.6 Hz, 3JC4′–F = 19.0 Hz, 3JC4′–P = 8.3 Hz, C4′) 50.2 (Ala–CH), 20.6 [CH(CH3)2], 20.5 [CH(CH3)2], 19.1 (d, 3JCH3–P = 6.5 Hz, Ala–CH3); 19F NMR (377 MHz, MeOD): δ −98.6 (ddd, 2JF–F = 238.7 Hz, 3JF–H4′ = 13.0 Hz, 3JF–H1′ = 9.7 Hz), −117.0 (app. dt, 2JF–F = 238.6 Hz, 3JF–H1′/H4′ = 16.7 Hz); 31P {1H} NMR (162 MHz, MeOD): δ 3.29 (s); HRMS (NSI): calculated for C22H27F2N4O9P [M − H]− 546.1458, found 541.1460. These data were in good agreement with literature.17
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Keele University, Riboscience LLC and the Royal Society of Chemistry (Research Enablement Grant E21-7055928079) are thanked for funding to C. M. M. B. UK Research and Innovation (UKRI, Future Leaders Fellowship, MR/T019522/1) are thanked for project grant funding to G. J. M. We also thank the EPSRC UK National Mass Spectrometry Facility (NMSF) at Swansea University.References
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef PubMed.
- M. Guinan, C. Benckendorff, M. Smith and G. J. Miller, Molecules, 2020, 25, 2050 CrossRef PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef PubMed.
- R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, ACS Cent. Sci., 2020, 6, 672–683 CrossRef.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS.
- A. J. Burke, W. R. Birmingham, Y. Zhuo, T. W. Thorpe, B. Z. da Costa, R. Crawshaw, I. Rowles, J. D. Finnigan, C. Young, G. M. Holgate, M. P. Muldowney, S. J. Charnock, S. L. Lovelock, N. J. Turner and A. P. Green, J. Am. Chem. Soc., 2022, 144, 3761–3765 CrossRef CAS PubMed.
- M. Meanwell, S. M. Silverman, J. Lehmann, B. Adluri, Y. Wang, R. Cohen, L.-C. Campeau and R. Britton, Science, 2020, 369, 725–730 CrossRef CAS PubMed.
- S. C. Cosgrove and G. J. Miller, Expert Opin. Drug Discovery, 2022, 17, 355–364 CrossRef CAS.
- G. A. Kicska, L. Long, H. Hörig, C. Fairchild, P. C. Tyler, R. H. Furneaux, V. L. Schramm and H. L. Kaufman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4593–4598 CrossRef CAS.
- M. Guinan, N. Huang, C. S. Hawes, M. A. Lima, M. Smith and G. J. Miller, Org. Biomol. Chem., 2022, 20, 1401–1406 RSC.
- M. Guinan, N. Huang, M. Smith and G. J. Miller, Bioorg. Med. Chem. Lett., 2022, 61, 128605 CrossRef CAS PubMed.
- Z. W. Dentmon, T. M. Kaiser and D. C. Liotta, Molecules, 2020, 25, 5165 CrossRef PubMed.
- A. C. Ojeda-Porras, V. Roy and L. A. Agrofoglio, Chem. Rec., 2022, 22, e202100307 CrossRef PubMed.
- A. Kálmán, T. Yoritsánszky, J. Béres and G. Sági, Nucleosides Nucleotides, 1990, 9, 235–243 CrossRef.
- T. Kishi, M. Muroi, T. Kusaka, M. Nishikawa, K. Kamiya and K. Mizuno, Chem. Pharm. Bull., 1972, 20, 940–946 CrossRef.
- M. Akabane-Nakata, T. Chickering, J. M. Harp, M. K. Schlegel, S. Matsuda, M. Egli and M. Manoharan, Org. Lett., 2022, 24, 525–530 CrossRef CAS PubMed.
- J. Yoon, G. Kim, D. B. Jarhad, H.-R. Kim, Y.-S. Shin, S. Qu, P. K. Sahu, H. O. Kim, H. W. Lee, S. Bin Wang, Y. J. Kong, T.-S. Chang, N. S. Ogando, K. Kovacikova, E. J. Snijder, C. C. Posthuma, M. J. van Hemert and L. S. Jeong, J. Med. Chem., 2019, 62, 6346–6362 CrossRef CAS.
- G. M. Blackburn, D. A. England and F. Kolkmann, J. Chem. Soc., Chem. Commun., 1981, 930–932 RSC.
- G. V. B. Madhavan, D. P. C. McGee, R. M. Rydzewski, R. Boehme, J. C. Martin and E. J. Prisbe, J. Med. Chem., 1988, 31, 1798–1804 CrossRef CAS PubMed.
- K. Biggadike, A. D. Borthwick, A. M. Exall, B. E. Kirk, S. M. Roberts, P. Youds, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 255–256 RSC.
- A. D. Borthwick, D. N. Evans, B. E. Kirk, K. Biggadike, A. M. Exall, P. Youds, S. M. Roberts, D. J. Knight and J. A. v. Coates, J. Med. Chem., 1990, 33, 179–186 CrossRef CAS.
- M. D. Kosobokov, V. v. Levin, A. A. Zemtsov, M. I. Struchkova, A. A. Korlyukov, D. E. Arkhipov and A. D. Dilman, Org. Lett., 2014, 16, 1438–1441 CrossRef CAS PubMed.
- S. Pal, G. Chandra, S. Patel and S. Singh, Chem. Rec., 2022, 22, e202100335 CrossRef PubMed.
- H. Kumamoto, K. Haraguchi, M. Ida, K. T. Nakamura, Y. Kitagawa, T. Hamasaki, M. Baba, S. S. Matsubayashi and H. Tanaka, Tetrahedron, 2009, 65, 7630–7636 CrossRef.
- Y.-Y. Yang, W.-D. Meng and F.-L. Qing, Org. Lett., 2004, 6, 4257–4259 CrossRef PubMed.
- C. F. Bennett and E. E. Swayze, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259–293 CrossRef PubMed.
- Y. P. Yurenko, J. Novotný, V. Sklenář and R. Marek, Chem. – Eur. J., 2015, 21, 17933–17943 CrossRef CAS PubMed.
- R. Mondal, M. Agbaria and Z. Nairoukh, Chem. – Eur. J., 2021, 27, 7193–7213 CrossRef CAS.
- K. Huonnic and B. Linclau, Chem. Rev., 2022, 122, 15503–15602 CrossRef CAS.
- G. Kim, J. Yoon, D. B. Jarhad, Y. S. Shin, M. S. Majik, V. A. Mulamoottil, X. Hou, S. Qu, J. Park, M.-H. Baik and L. S. Jeong, Org. Lett., 2017, 19, 5732–5735 CrossRef CAS.
- Y. S. Shin, D. B. Jarhad, M. H. Jang, K. Kovacikova, G. Kim, J. Yoon, H.-R. Kim, Y. E. Hyun, A. S. Tipnis, T.-S. Chang, M. J. van Hemert and L. S. Jeong, Eur. J. Med. Chem., 2020, 187, 111956–111956 CrossRef CAS.
- M. Yang, W. Ye and S. W. Schneller, J. Org. Chem., 2004, 69, 3993–3996 CrossRef.
- F. Fernández, X. García-Mera, M. Morales and J. E. Rodríguez-Borges, Synthesis, 2001, 2001, 0239–0242 CrossRef.
- V. E. Marquez, in Advances in Antiviral Drug Design, 1996, vol. 2, pp. 89–146 Search PubMed.
- Y. Xu, K. Kino and H. Sugiyama, J. Biomol. Struct. Dyn., 2002, 20, 437–446 CrossRef PubMed.
- F. Seela and W. Bussmann, Nucleosides Nucleotides, 1985, 4, 391–394 CrossRef.
- Y. Mehellou, J. Balzarini and C. McGuigan, ChemMedChem, 2009, 4, 1779–1791 CrossRef CAS PubMed.
- B. S. Ross, P. Ganapati Reddy, H.-R. Zhang, S. Rachakonda and M. J. Sofia, J. Org. Chem., 2011, 76, 8311–8319 CrossRef CAS PubMed.
- C. Meier, Antiviral Chem. Chemother., 2017, 25, 69–82 CrossRef PubMed.
- C. Meier, Eur. J. Org. Chem., 2006, 1081–1102 CrossRef CAS.
- G. B. Kauffman, R. P. Pinnell, J. Peterson, D. Williams and A. L. Allred, in Inorganic Syntheses, 1960, vol. 6, pp. 3–6 Search PubMed.
- A. Krasovskiy and P. Knochel, Synthesis, 2006, 5, 890–891 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Relevant spectral NMR, X-ray crystallography and HPLC data for compounds 2–40 alongside cell viability assay data. CCDC 2193041–2193043 and 2218823. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01761j |
| This journal is © The Royal Society of Chemistry 2022 |