
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence5-Fluoro-1,2,3-triazole motif in peptides and its electronic properties†
José
Laxio Arenas
a,
Pascal
Retailleau
b,
Jean-Michel
Gillet
 c,
Nour-Eddine
Ghermani
*cd,
Sandrine
Ongeri
a and
Benoît
Crousse
*a
c,
Nour-Eddine
Ghermani
*cd,
Sandrine
Ongeri
a and
Benoît
Crousse
*a
aUMR 8076, BioCIS, CNRS, Université Paris-Saclay, 92290 Châtenay-Malabry, France. E-mail: benoit.crousse@universite-paris-saclay.fr
bUniversité Paris-Saclay, CNRS, Institut de Chimie des Substances Naturelles, 91198 Gif-sur-Yvette, France
cUMR CNRS 8580, Ecole Centrale Paris, Grande Voie des Vignes, 92290 Châtenay-Malabry, France. E-mail: noureddine.ghermani@universite-paris-saclay.fr
dUMR CNRS 8612, Université Paris-Saclay, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, 92296 Châtenay-Malabry, France
First published on 17th October 2022
Abstract
The 5-fluoro triazole amino acid scaffold prepared by halogen exchange has been incorporated into peptides. From the X-ray diffraction of the 5-fluoro triazole motif, the main observation was an important localization on one side of the negative potential surface. The fluorine atom reveals a cylindrical shape in its deformation electron density.
Over the last decades, applications of 1,2,3-triazoles have gained interest thanks to the discovery of regioselective Cu(I) or Ru(II)-catalyzed azide–alkyne cycloaddition providing 5- or 4-disubstituted-1,2,3-triazoles, respectively.1–3 Their structural and electronic properties are very close to the peptidic bond, even if the distance between side chains is slightly longer than an amide bond.4–6 Moreover, 5-H-1,2,3-triazoles are known as trans-amide surrogates with higher stability to hydrolysis. Therefore, replacing the amide bond in a peptide with the 1,2,3-triazole ring can be considered an effective solution for synthesising biologically relevant peptidomimetics and peptides.4 In previous work, our team has shown fluorinated peptidomimetic amino-acid based on triazole scaffolds such as 2-amino-trifluoropropyl-1,4-triazolyl-acetic acid, also named trifluoromethyl homo-1,4-Tz amino acid (Fig. 1, A) adopting folded ladder-like structures mimicking short multi-stranded β-sheet. Also (aminomethyl-1,4-triazolyl)-difluoroacetic acid called N-difluoromethyl 1,4-Tz (Fig. 1, B) adopts extended or worm-like chain structures.7 Furthermore, fluorine introduction into amino acids or peptides has become a widely used technique to modulate both conformation and activity of compounds due to the capacity of fluorine to modify biological factors such as hydrophobicity, acidity, chemical and enzymatic stability.8,9 In addition, the fluorine can participate in multipolar C–F⋯H–N, C–F⋯C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–F⋯H–Cα interactions and the side-chain amide moieties of Asn and Gln.10 For these reasons developing new access for fluorinated moiety has become a leading strategy. Then we focus on the incorporation of the 5-fluoro-1,2,3-triazole scaffold in peptides (Fig. 1, C). Unlike the 5-halo-1,2,3 triazoles containing iodine, bromine or chlorine, the 5-fluoro-1,2,3-triazoles are not so much described in the literature for synthesis, properties, or applications.11 Herein, we describe the incorporation of the 5-fluoro-1,2,3-triazoles in peptides and their conformational and electronic properties thanks to X-ray crystallography and ab initio calculations.
O, and C–F⋯H–Cα interactions and the side-chain amide moieties of Asn and Gln.10 For these reasons developing new access for fluorinated moiety has become a leading strategy. Then we focus on the incorporation of the 5-fluoro-1,2,3-triazole scaffold in peptides (Fig. 1, C). Unlike the 5-halo-1,2,3 triazoles containing iodine, bromine or chlorine, the 5-fluoro-1,2,3-triazoles are not so much described in the literature for synthesis, properties, or applications.11 Herein, we describe the incorporation of the 5-fluoro-1,2,3-triazoles in peptides and their conformational and electronic properties thanks to X-ray crystallography and ab initio calculations.
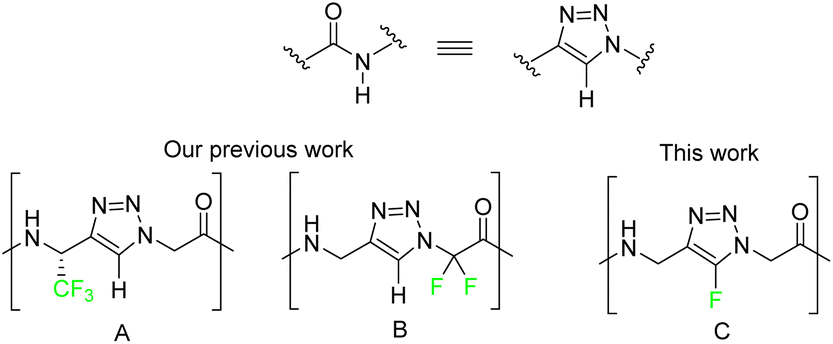 | ||
| Fig. 1 1,2,3-Triazole isostere of amide bond. Fluorinated scaffolds developed in our team. (A) trifluoromethyl homo-1,4-Tz amino acid and (B) N-difluoromethyl 1,4-Tz. | ||
Due to the low stability and difficulty obtaining fluoro-alkynes, access to 5-fluoro triazoles is very limited. Currently, there is only one access route to 5-fluoro triazoles through the halogen exchange reaction (Halex reaction) between 5-iodo triazoles and fluoride salts. In 2012,12 Fokin et al. first reported the Halex reaction in a biphasic acetonitrile/water medium with potassium fluoride (KF) or potassium bifluoride (KHF2) from 5-iodo triazoles under microwave in a short time (10 min) at 180 °C. The silver-mediated fluorination described in 2015 by Chu et al.13 combines silver fluoride (salts AgF) with tetramethylethylenediamine (TMEDA) in toluene at 120 °C. Silver fluorination conditions are applicable in a wide range of 5-iodotriazoles bearing electron-rich and electron-poor substituents, including substituted aromatics, heteroaromatics, and alkyls with mostly moderate to good yield. Although the above conditions are primarily effective for aromatic and alkyl groups, we attempted the access of the 5-fluorotriazole amino acid scaffold C and its incorporation into peptides.
Results and discussion
First, we synthesized the 5-iodo-1,2,3-triazole amino acid 1a–b by click chemistry between the N-Boc iodoalkyne synthesized using KOH solution and iodine in methanol overnight14 and azide acetates15 with copper iodide and triethylamine in THF (Scheme 1).16,17tert-Butyl carbamate and benzyl/ethyl ester are used as protecting groups to have orthogonal and easily deprotected groups. | ||
| Scheme 1 Synthesis of 5-iodo-1,2,3-triazoles. | ||
Having in hand the 5-iodo triazoles, the Halex reaction has been investigated according to the two conditions. First, we started with the conditions described by Chu et al. with 5 equivalents of silver fluoride, 0.5 equivalent of the tetramethyl ethyl diamine (TMEDA) in toluene for 18 hours at 120 °C (Scheme 2).13 Unfortunately, only fluorinated side products are detected. The excess fluoride seemed to destroy our substrate. So, we screened different conditions by reducing the amount of reagents (fluoride source, ligand) and changing the solvent (THF, dioxane), the temperature and the reaction time. The best conditions are using 1.2 equivalent of AgF and 20% of TMEDA in toluene at 120 °C to afford compounds 2a and 2b in 56% and 53% yield, respectively (Scheme 3). In the same line, the conditions described by Fokin et al.,12 using the microwave for 10 minutes at 180 °C with 5 equivalents of KF, led to the 5-fluorotriazole 2a in 56% yield (Scheme 2). These results show that it is unnecessary to have an aromatic group in position 4 and an aliphatic substituent in position N1 of the 5-iodotriazoles for an efficient transformation into 5-fluorotriazoles.
 | ||
| Scheme 2 Synthesis of 5-fluoro-1,2,3-triazoles by silver-mediated fluorination reaction by Halex reaction. | ||
 | ||
| Scheme 3 Incorporation of amino acids. | ||
Given these good results, we undertook to carry out both fluorination conditions on peptides with 5-iodo triazole. Unfortunately, complete decomposition of starting material was observed from the 5-iodo triazole having amino acids on either side. Therefore, we turned directly to amino acid coupling with 5-fluorotriazole motifs 2a and 2b. Initially, after deprotection of the tert-butylcarbamate function of 2a and 2b with a hydrochloric acid solution, the amine was coupled to phenylalanine acid to give the products 3a and 3b with yields of 56 and 53%, respectively. Then the carboxylic acid was obtained by saponification of 2a with lithium hydroxide (LiOH·H2O). On the other hand, the hydrogenolysis of the benzyl group is effective with Pd/C and the triethylsilane of 2b. Unfortunately and surprisingly, hydrogenolysis under hydrogen failed. Finally, the corresponding carboxylic acid was placed under coupling conditions with the desired amino acid to yield the related peptide 4 (Scheme 3).
In the literature, it is important to note that the reactivity of 5-fluoro-1,2,3-triazoles toward SNAr type reaction has been observed and described.12 The fluorine is a good leaving group and can be substituted under mild conditions by amide, alcohols, azoles and thiols. In coupling amino acids, no substitution of the fluorine atom has been observed. Here the 5-fluorotriazole scaffold is compatible with the peptide conditions. Interestingly, the reduction has been carried out with NaBH4 in methanol to give the corresponding alcohol in 59% yield. The fluorine is also resistant to the reduction conditions (Scheme 4).
 | ||
| Scheme 4 Reduction of the ester group. | ||
Next, we investigated the helpful information about the structure of molecules 2a and 4 derived from the NMR spectra. Following the HOESY 2D 19F–1H spectra, both products showed strong short-range correlations involving the fluorine atom and protons in α position of the triazole. Weak proton-fluorine interactions are observed for 2a with the tert-butyl carbamate group and 4 with a phenyl group (see ESI†). These results suggest a folded conformation in the solution.
Also, what interests us mainly is the effect of fluorine on the aromatic nucleus of triazole. Indeed, both fluorine's physical and electronic impact on triazole is unknown. So, we focused on an in-depth study of 5-F triazole in physical and electronic properties. Thanks to the high-resolution X-ray crystal diffraction (CCDC 2141412,†Fig. 2), the solid-state structure, the charge distribution, and the electrostatic potential of molecule 2a have been investigated. The conventional atomic positions and thermal parameters were determined from the multipole refinements using the MOPRO software.18,19 The numbering schemes for molecule 2a are given in Fig. 2.
 | ||
| Fig. 2 X-ray crystal structure of 2a. Thermal ellipsoids are shown at the 50% probability level. View of the crystal packing showing the intermolecular hydrogen bonds involving the fluorine. | ||
The central fluoro triazole ring is characterized by a short C5–F bond distance of 1.3163(3) Å, comparable to those of N1–N2 (1.3443(3) Å) and N2–N3 (1.3129(3) Å). Carboxyl groups exhibit C–O bond lengths and O–C–O angles as follows: C3–O2 = 1.2070(3) Å, C8–O4 = 1.2219(3) Å, C3–O1 = 1.3286(3) Å, C8–O3 = 1.3435(3) Å and O1–C3–O2 = 125.472(19)°, O3–C8–O4 = 125.898(17)°, respectively. The main torsion angles of molecule 2a in the solid state are C5–N1–C4–C3 = 82.230(11)° and N4–C7–C6–C5 = 47.441(14)°, which show a particular extended structure in the crystal. For isolated molecule 2a with the optimized theoretical geometry, these torsion angles are significantly different: C5–N1–C4–C3 = 75.75° and N4–C7–C6–C5 = 110.17°. From the experiment, molecules 2a related by translation along the direction [1 0 1] are interacting in the crystal lattice via a hydrogen bond network involving the N4–H0 group and the O4 oxygen atom (N4–H0⋯O4 = 1.010(4) Å and N4–H0⋯O4 angle of 152.9(2)°) to form a C11(4) chain.20 The fluorine is only involved in longer intermolecular contacts with one H atom of the tert-butyl group (F⋯H102 = 2.607(4) Å) in position x − ½, ½ − y, z + ½ and to some extent, one atom H atom of the methene group (F⋯H71 = 2.847(5) Å) in position 1 + x, y, z (Fig. 3). The theoretical and experimental static deformation densities in different parts of molecule 2a have been displayed in Fig. 3. Lone pair densities and electron polarizations for O and N atoms are well revealed by the multipole refinements based on experimental structure factors. In contrast, electron density depletion is found in the vicinity of the fluorine atom (image on the top). The peak heights of the lone pair densities are as follows: 0.8 e Å−3 for N atoms and even higher than 0.9 e Å−3 for the O lone pairs (1 e Å−3 = 0.15 e bohr−3). The covalent character of C–C, C–N, C–O, N–N and N–H bonds are well defined, displaying a concentration of electrons higher than 0.8 e Å−3 between the atoms. The C5–F bond is, in contrast, less charged in electrons (0.3 e Å−3). However, the fluorine atom exhibits a large peak of electrons (0.6 e Å−3) in the perpendicular plan of the C5–F bond (image on the top). This polarization of the electron density is directed towards the tert-butyl H102 and H111 hydrogen atoms of an adjacent molecule. The theoretical lone pair densities are similar to those displayed on the experimental maps. However, the fluorine atom reveals a cylindrical shape in its deformation electron density. In contrast, the experiment reports the F⋯H102 interaction (see the parallel and perpendicular plane of the fluorinated triazole in Fig. 3).
 | ||
| Fig. 3 Experimental static electron deformation density (top) and theoretical ones (from TSF refinement, middle and isolated molecule, bottom) in chosen planes of molecule 2a. Contours are 0.05 e Å−3, negative contours are in red. | ||
The experimental and theoretical charges for 2a using the integration of the electron density over Bader's atomic basins are listed in Table 1.21 Both approaches lead to similar features. All electronegative atoms carry negative charges as can be expected: 9.7 e for F, 9.0 e for O atoms, and 7.5 e for N atoms on average, except the donor N4 atom (8.2 e from both experiments and theory) involved in the N4–H0⋯O4 hydrogen bond. Consequently, the H0 atom carries the most positive charge (0.5 e) compared to the other H′s, which are almost neutral in the theoretical approach. While ab initio calculations have only involved an isolated molecule, the striking charge similarities of both determinations on N4 (8 e) and H0 (0.6 e vs. 0.5 e from the experiment) atoms seem not influenced by the crystal environment.
| Atom | Experiment | Theory(isolated) | Theory (periodic) |
|---|---|---|---|
| F | 9.65 | 9.58 | 9.63 |
| O1 | 8.99 | 9.12 | 8.84 |
| O2 | 9.02 | 9.00 | 8.89 |
| O3 | 8.93 | 9.02 | 8.81 |
| O4 | 9.01 | 9.06 | 8.96 |
| N1 | 7.69 | 7.72 | 7.55 |
| N2 | 7.06 | 7.08 | 7.11 |
| N3 | 7.36 | 7.51 | 7.42 |
| N4 | 8.17 | 8.11 | 7.96 |
| C4 | 5.86 | 5.54 | 5.87 |
| C5 | 3.27 | 4.13 | 5.32 |
| C6 | 5.71 | 5.83 | 5.69 |
| C7 | 5.88 | 5.47 | 3.58 |
| H0 | 0.48 | 0.56 | 0.53 |
Next, the features of the electrostatic potential of 2a were obtained from the experimental and theoretical methods. The chosen representation for the electrostatic potential is the isovalue surfaces giving the extension of this property at a particular cut-off (here + 0.1 e Bohr−1 for the positive potential and −0.05 e Bohr−1 for the negative one). The most interesting part of the electrostatic potential for molecular compounds is the negative region (nucleophilic), where interactions with positive cations or hydrogenated species can occur. At first glance, theory and experiment yield similar electrostatic potential for an isolated molecule 2a emphasizing the intrinsic chemical character of the molecule even in the crystal lattice. From both approaches, the negative potential surface is mainly generated by the most electronegative atoms (O, N and F) of molecule 2a and is extended on only one side of the central fluorinated triazole ring. It is, however, worthy to note that the negative potential surface is less extensive in the theoretical figures (bottom of Fig. 4) for molecule 2a in a vacuum.
 | ||
| Fig. 4 Experimental and theoretical electrostatic potential for two orientations of molecule 2a. Isovalue surface cut-off is +0.1 e Bohr−1 (in grey) and −0.05 e Bohr−1 (in red). | ||
From the experimental electron density refinement, the total dipole magnitude of 2a was equal to 10.3 D. This property reflects the electron polarization of the molecule in the crystal lattice since, for an isolated molecule with its optimized molecular geometry, only a dipole value of 3.8 D was found from the theoretical calculations (the total molecular energies are also given for the four molecules in Table 3 in ESI†). For the experimental geometry, the theoretical molecular dipole was equal to 4.8 D. From the refinement of the Theoretical Structure Factor (TSF), the dipole moment of molecule 2a has a value of 7.6 D. These respective results are in accord with the experimental and theoretical negative potential surfaces, which are different in shape and extension (Table 3 in ESI†). Furthermore, we have performed ab initio calculations starting from the initial fluorinated triazole ring A to highlight the modifications of the electrostatic potential and dipole moment values during the chemical construction of molecule 2a. The dipole magnitudes for the four constructed molecules A–C, 2a are in the range of 2.9 to 3.8 D, meaning that the long-range reactivity (van der Waals interaction) of molecule 2a remains mainly governed by the central fluorinated triazole ring electrostatic properties (Table 3, ESI†). Interestingly, the 5H-triazole molecules, compared to the 5-fluoro triazole derivatives, always have a higher dipole moment (Table 4 in ESI†).
Following all experimental and theoretical results, it can be concluded that the presence of fluorine on the triazole does not considerably modify the properties of the triazole nucleus. We can notice, on the one hand, the extended negative potential surface around 5-fluorotriazole and carbonyls, on the other hand, the fluorine atom exhibits a large peak of electrons. If we compare the different data of the 5-fluorotriazole unit with the amide bond and 5-H triazole, the dipole moment, the distance between residues, and the angles are close. Also, 5-fluoro triazole can form a hydrogen bond at the azote N3. The main highlighted difference is the new potential interactions that fluorine can generate, such as that C–F⋯H–N, C–F⋯CO, and C–F⋯H–Cα (Scheme 5).
 | ||
| Scheme 5 5-F triazole in comparison to amide bond and 5-H triazole. | ||
In conclusion, the 5-fluoro triazole amino acid has been prepared easily by halogen exchange between the 5-iodo triazole and fluoride salts. The incorporation of this scaffold has been carried out in peptides. Interestingly, the different data obtained from the X-ray diffraction of compound 2a confirmed that the 5-fluoro triazole possessed, in comparison to the 5H-triazole and the amide bond, a dipolar moment, the distance between residues and angles close. The significant differences are the localization on one side of the negative potential surface, and the fluorine atom reveals a cylindrical shape in its deformation electron density. The specific interactions involving fluorine and CO, N–H, and C–H with peptides are under investigation in our laboratory.
Author contributions
J. Laxio-Arenas conducting the research and investigation process in the chemical synthesis, specifically performing the experiments, and data collection; P. Retailleau obtained X-ray structure; N.-E. Ghermani and J. M. Gillet performed electronic properties and ab initio calculations. S. Ongeri: supervision and verification of the overall reproducibility of results and experiments in chemical synthesis; B. Crousse: oversight and leadership responsibility for the research activity planning and execution.Conflicts of interest
There are no conflicts to declareAcknowledgements
The Ministère de l'Enseignement Supérieur et de la Recherche (MESR) is thanked for financial support for José Laxio Arenas. The French Fluorine Network (GIS-FLUOR) is also acknowledged for its support.References
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed.
- (a) N. Agouram, E. M. E. Hadrami and A. Bentama, Molecules, 2021, 26, 2937 CrossRef CAS PubMed; (b) W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013 RSC.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- (a) J. Laxio Arenas, Y. Xu, T. Milcent, C. Van Heijenoort, F. Giraud, T. Ha-Duong, B. Crousse and S. Ongeri, ChemPlusChem, 2021, 86, 241 CrossRef CAS PubMed; (b) M. Mamone, R. S. B. Gonçalves, F. Blanchard, G. Bernadat, S. Ongeri, T. Milcent and B. Crousse, Chem. Commun., 2017, 53, 5024 RSC; (c) J. Engel-Andreasen, I. Wellhöfer, K. Wich and C. A. Olsen, J. Org. Chem., 2017, 82, 11613 CrossRef CAS PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed.
- E. N. G. Marsh, Fluorinated Proteins: From Design and Synthesis to Structure and Stability, 2014 Search PubMed.
- (a) B. Kuhn, E. Gilberg, R. Taylor, J. Cole and O. Korb, J. Med. Chem., 2019, 62, 10441 CrossRef CAS PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) S. A. Harry, M. Kazim, P. Minh Nguyen, A. Zhu, M. R. Xiang, J. Catazaro, M. Siegler and T. Lectka, Angew. Chem., Int. Ed., 2022, e202207966 CAS.
- J. Laxio Arenas and B. Crousse, Eur. J. Org. Chem., 2021, 18, 2665 CrossRef.
- B. T. Worrell, J. E. Hein and V. V. Fokin, Angew. Chem., Int. Ed., 2012, 51, 11791 CrossRef CAS PubMed.
- D. Wang, W. Sun and T. Chu, Eur. J. Org. Chem., 2015, 19, 4114 CrossRef.
- M. Kohr and U. Kazmaier, Synthesis, 2018, 50, 4690 CrossRef CAS.
- W. Zhao, H. Li, J. Zhang and S. Cao, Chin. J. Chem., 2011, 29, 2763 CrossRef CAS.
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018 CrossRef CAS PubMed.
- L. Li, G. Hao, A. Zhu, S. Liu and G. Zhang, Tetrahedron Lett., 2013, 54, 6057 CrossRef CAS.
- B. Guillot, L. Viry, R. Guillot, C. Lecomte and C. Jelsch, J. Appl. Crystallogr., 2000, 34, 214 CrossRef.
- C. Jelsch, B. Guillot, A. Lagoutte and C. Lecomte, J. Appl. Crystallogr., 2005, 38, 38 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules - A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2141412. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01716d |
| This journal is © The Royal Society of Chemistry 2022 |