
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-mediated aerobic Ritter-type C–H amination of diarylmethanes using DDQ/tert-butyl nitrite†
Tianci
Li
a,
Jiangyu
Yang
ab,
Xin
Yin
ab,
Jinhua
Shi
a,
Qun
Cao
 *c,
Miaomiao
Hu
a,
Xiaowen
Xu
a,
Meichao
Li
*a and
Zhenlu
Shen
*a
*c,
Miaomiao
Hu
a,
Xiaowen
Xu
a,
Meichao
Li
*a and
Zhenlu
Shen
*a
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, 310014, China. E-mail: limc@zjut.edu.cn; zhenlushen@zjut.edu.cn
bZhejiang Jitai New Materials Co., Ltd, Shaoxing 312369, China
cSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Belfast, BT9 5AG, UK. E-mail: qcao01@qub.ac.uk
First published on 25th October 2022
Abstract
A metal-free photocatalytic Ritter-type C–H amination of unactivated sp3 carbons using molecular oxygen as a terminal oxidant has been developed. By employing a co-catalytic system of 3-dichloro-4,5-dicyano-1,4-benzoquinone (DDQ) and tert-butyl nitrite (TBN), this novel strategy provides a low cost, sustainable and scalable way to synthesise a broad range of secondary amides in moderate to excellent yields under mild conditions.
Introduction
Catalytic transformation of omnipresent C–H bonds to C–N bonds offers an efficient synthetic strategy for the construction of valuable nitrogen-containing compounds.1–3 As one of the most popular strategies to construct C–N bonds, the Ritter reaction has been established as a powerful tool for the synthesis of amides,4–6 which widely exist in modern pharmaceutical molecules.7,8 The classic Ritter reaction involves the generation of a carbocation from a functionalized substrate (e.g., alcohols or alkenes), which then reacts with a nitrile leading to the formation of an amide product (Fig. 1A).4–6 Recently, much effort has been directed towards the development of catalytic Ritter-type C–H amination, which allows amides to be selectively synthesised from non-functionalized alkanes, with the carbocation intermediate generated from C–H bond oxidation using a variety of oxidants such as ceric ammonium nitrate (CAN),9 Selectfluor,10 hypervalent iodine,11 iodic acid,12 3-dichloro-4,5-dicyano-1,4-benzoquinone (DDQ)13 and sodium persulfate14 (Fig. 1B). However, these methods still suffer from high loadings of transition metals, high temperature and low sustainability due to unwanted by-products derived from the use of stoichiometric oxidants. Considering the strive for greener chemical production, molecular oxygen (O2) is the most ideal oxidant as it is inexpensive and environmentally friendly.15,16 In the past 10 years, important advances have been made in the catalytic aerobic oxidation of C(sp3)–H bonds;17–21 however, to the best of our knowledge, there is no aerobic Ritter type C–H amination method reported to date.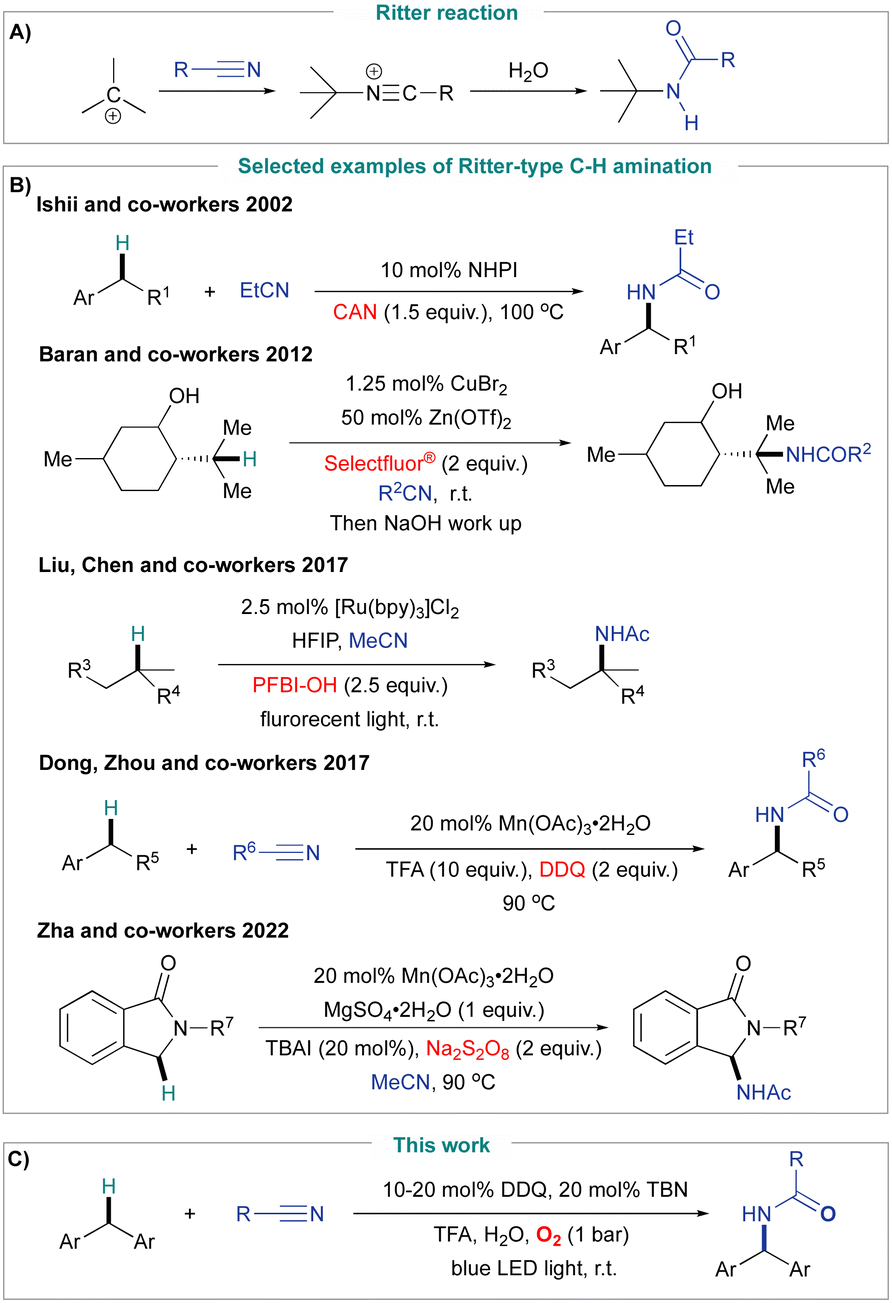 | ||
| Fig. 1 (A) Ritter reaction with a carbocation as an intermediate. (B) Representative examples of catalytic Ritter-type amination methods with stoichiometric oxidants. (C) This work: metal-free aerobic photocatalytic Ritter-type C–H amination. | ||
Therefore, developing an environmentally friendly and low-cost metal-free catalytic system for C–H amination is highly desirable. Herein, we report the first photocatalytic aerobic Ritter-type C–H amination for the synthesis of various amides under mild and metal-free conditions using a DDQ/tert-butyl nitrite (TBN) catalytic system (Fig. 1C).
Results and discussion
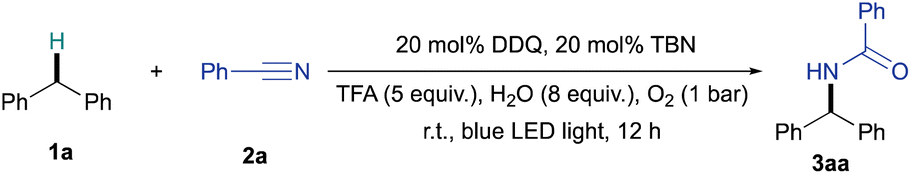
We began our studies by using the DDQ/TBN/O2 catalytic system, which has previously been successfully used for the oxidation of C(sp3)–H bonds,22–25 oxidative C–O coupling26 and C–N coupling reactions.27–29 Under the optimal conditions, diphenylmethane (1a, 0.5 mmol), DDQ (20 mol%), TBN (20 mol%), H2O (4 mmol, 8 equiv.) and trifluoroacetic acid (TFA, 5 equiv.) in 2 mL of benzonitrile (2a) under an oxygen atmosphere (1 bar) led to the desired product 3aa in 76% yield under the irradiation of blue LED light for 12 h at room temperature (Table 1, entry 1). When the loading of DDQ was decreased from 20 mol% to 10 mol%, the yield of 3aa decreased slightly to 74% (Table 1, entry 2). Further decreasing the loading of DDQ or the co-catalyst TBN led to unsatisfactory yields of 3aa (41–61%, for details see ESI Table S1†). It was found that the solvent played a key role in this catalytic reaction. Attempts to replace benzonitrile with other organic solvents (e.g., dichloroethane, toluene, THF, dioxane) led to poor yields (Table 1, entries 3–6), and no amide product was observed when the reaction was carried out in dimethylformamide (DMF, Table 1, entry 7) and dimethoxyethane (DME, Table 1, entry 8). Both DDQ and TBN were essential for this catalytic system. No desired product was formed in the absence of DDQ (Table 1, entry 9), whereas a lower yield of 3aa (32%) was obtained when TBN was omitted or replaced by other NO donors (Table 1, entry 10 and Table S2 in the ESI†). Control experiment also confirmed that the absence of TFA or replacing TFA with other acids (e.g., acetic acid, aq. HCl, H2SO4) resulted in lower yields of 3aa (11–21%, Table 1, entries 11–14), which clearly shows the importance of TFA as an additive. Attempts to carry out the reaction under a N2 atmosphere or without blue LED light led to a yield of 16% and 8% of 3aa, respectively (Table 1, entries 15 & 16; for further light source and temperature screening, see Table S2 in the ESI†).|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldsa [%] |
| Standard conditions: the reaction was performed with 1a (0.5 mmol), 2a (2 mL, as a reagent/solvent), DDQ (20 mol%, 0.10 mmol), TBN (20 mol%, 0.10 mmol), TFA (5 equiv., 2.5 mmol), and H2O (4 mmol, 8 equiv.) with blue LED light and an oxygen balloon at room temperature for 12 h.a Yields were determined by GC using biphenyl as an internal standard.b 10 mol% DDQ with a specified solvent (2 mL) was used.c N.D. implies that no product could be detected by GC-MS and GC.d 35 wt% aq. HCl was used.e 98 wt% H2SO4 was used. | ||
| 1 | None | 76 |
| 2 | 10 mol% DDQ | 74 |
| 3b | DCE as the solvent + 15 equiv. of 2a | 61 |
| 4b | Toluene as the solvent + 15 equiv. of 2a | 42 |
| 5b | THF as the solvent + 15 equiv. of 2a | 13 |
| 6b | Dioxane as the solvent + 15 equiv. of 2a | <1 |
| 7b | DMF as the solvent + 15 equiv. of 2a | N.D.c |
| 8b | DME as the solvent + 15 equiv. of 2a | N.D.c |
| 9 | No DDQ | N.D.c |
| 10 | No TBN | 32 |
| 11 | No TFA | 21 |
| 12 | CH3COOH instead of TFA | 52 |
| 13d | HCl instead of TFA | 28 |
| 14e | H2SO4 instead of TFA | 11 |
| 15 | Under N2 instead of O2 | 16 |
| 16 | Dark environment | 8 |
With the optimized reaction conditions in hand, the applicability of these conditions to a range of diarylmethanes was explored (Fig. 2). We found that with 10–20 mol% DDQ and 20 mol% TBN, we could obtain the desired amides in good yields. Having methyl/tert-butyl substituents at the ortho-, para-, and meta-positions of diphenylmethane led to the corresponding amide in 64–85% yields (3ba–3ga). Unfortunately, diphenylmethane bearing a methoxy group did not participate in the reaction (3ha), as the substrate was preferentially oxidized to its corresponding ketone under the optimized conditions. It was found that fluoro-, chloro-, and bromo-substituted diphenylmethanes were successfully transformed to their desired amides in fair to good yields (58–76%, 3ia–3oa). Notably, strong electron withdrawing substituents (e.g., CF3– and NO2–) were well tolerated in the catalytic system and afforded amides in moderate yields (3pa, 3qa). Although benzylheteroarene was not suitable in our reaction system (3ra), benzylnaphthalenes showed good activities, affording the desired products (3sa, 3ta) in 67% and 64% yields, respectively.
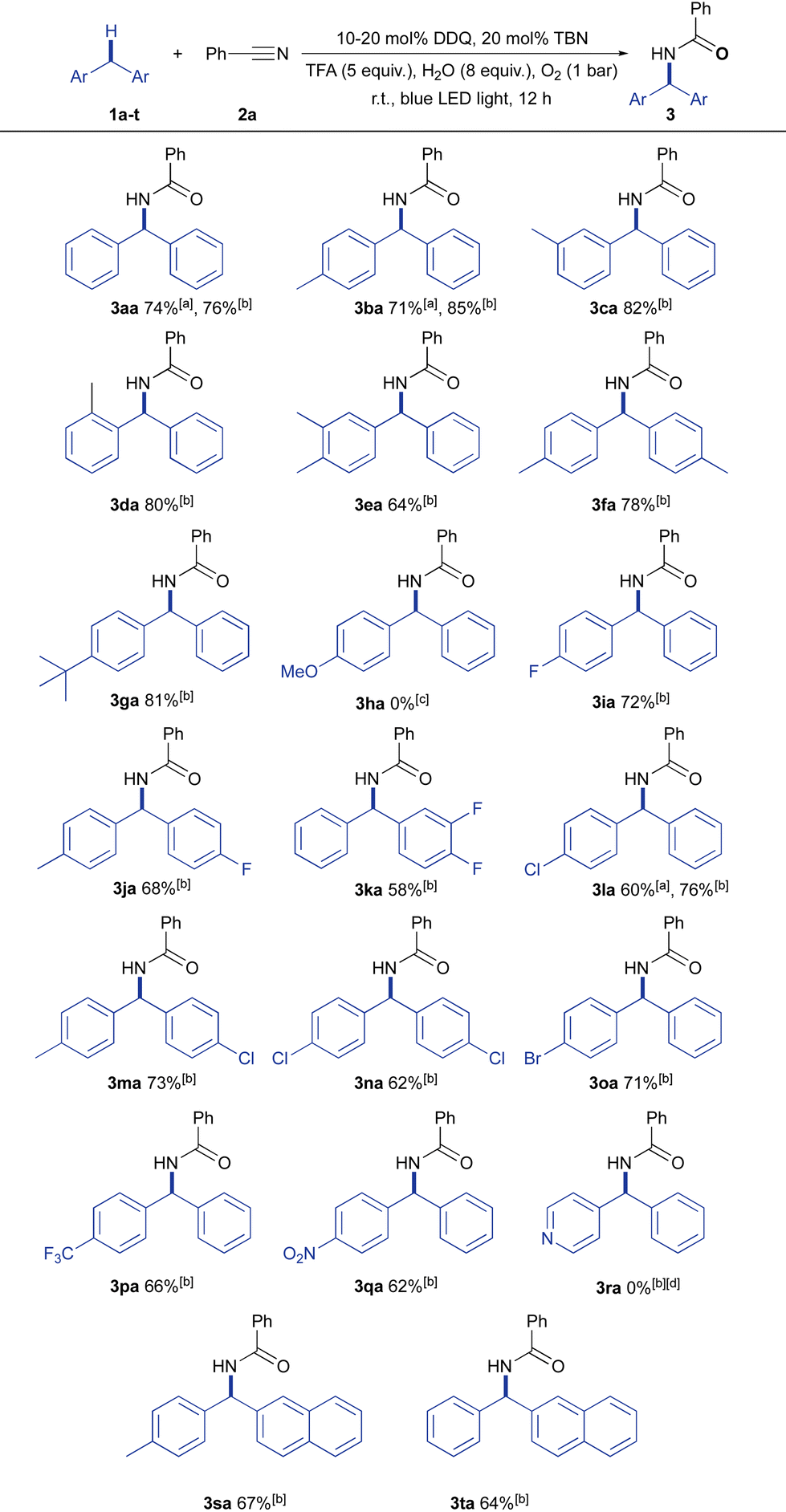 | ||
Fig. 2 Scope of diarylmethanes for the aerobic photocatalytic Ritter-type C–H amination reaction using the DDQ/TBN system. Reaction conditions: 1 (0.5 mmol), 2a (2 mL, as a reagent/solvent), DDQ (10–20 mol% as specified), TBN (20 mol%, 0.10 mmol), TFA (5 equiv., 2.5 mmol) and H2O (8 equiv., 4 mmol) with blue LED light and an oxygen balloon at room temperature for 12 h. Unless otherwise noted, isolated yields are shown. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 mol% DDQ was used. b20 mol% DDQ was used. c(4-Methoxyphenyl)(phenyl)methanone was isolated in 89% yield. d87% 1r was recovered. 10 mol% DDQ was used. b20 mol% DDQ was used. c(4-Methoxyphenyl)(phenyl)methanone was isolated in 89% yield. d87% 1r was recovered. | ||
In previous related Ritter-type C–H amination reports,4–9,30 the scope of nitriles was less studied and often limited to acetonitrile and benzonitrile. With a broad range of diarylmethanes examined, we tested the system on a variety of nitriles. Excellent functional group tolerance was exhibited by a wide range of nitriles as shown in Fig. 3. We were delighted that benzonitriles bearing an electron donating group (CH3–) and halogens (F–, Cl–, Br–) could participate in the reaction to furnish their corresponding amides in good to very good yields (3ab–3ad, 3ae–3ag). Furan and thiophene derivatives (e.g., 3ah and 3ai) could also be prepared, albeit in moderate yields, highlighting the applicability of this method to synthesize substituted heterocycles. It was found that α,β-unsaturated nitriles could also react successfully, as exemplified by the preparation of 3aj and 3ak. Additionally, this system was applied to aliphatic nitriles; they were transformed into their corresponding N-benzhydryl amides (3al, 3am, 3an) in good yields.
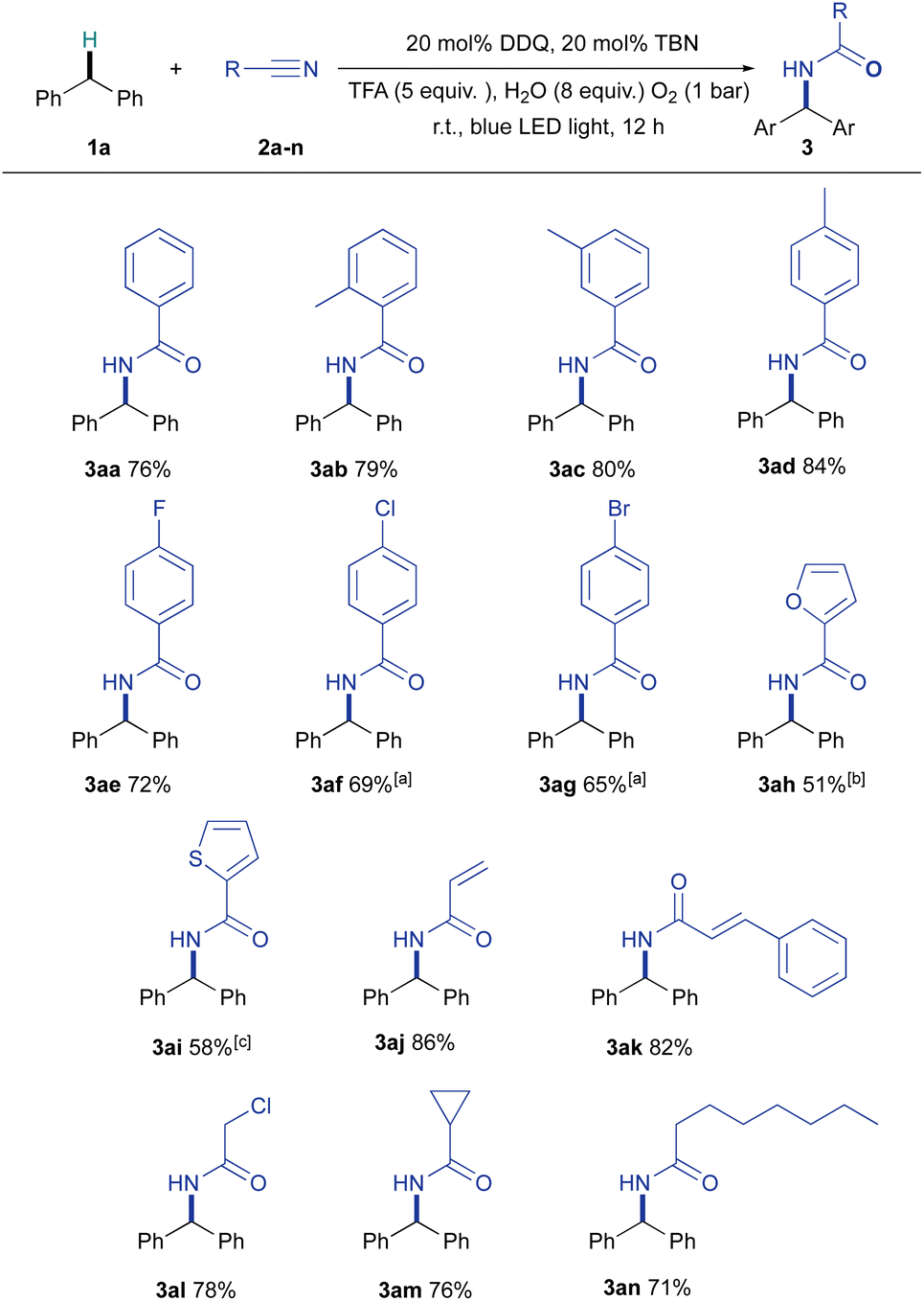 | ||
| Fig. 3 Scope of nitriles for the aerobic photocatalytic Ritter-type C–H amination reaction using the DDQ/TBN system. Standard conditions: 1 (0.5 mmol), 2 (2 mL, as a reagent/solvent), DDQ (20 mol%, 0.10 mmol), TBN (20 mol%, 0.10 mmol), TFA (5 equiv., 2.5 mmol) and H2O (8 equiv., 4 mmol) with blue LED light and an oxygen balloon at room temperature for 12 h. Unless otherwise noted, isolated yields are shown. aReaction was carried out with 15 equiv. of the corresponding nitrile in 2 mL of DCE. b42% 1a was recovered. c40% 1a was recovered. | ||
To further explore the utility of our Ritter-type amination method, we expanded the reaction to a gram scale under the optimized conditions with 10 mol% DDQ and 20 mol% TBN as the co-catalyst (Fig. 4). This work thus far highlights the improved performance of the system compared with the previous system, given the low cost of the catalyst, sustainable mild reaction conditions, and the absence of any metal salts and stoichiometric oxidants.
 | ||
| Fig. 4 Gram scale reaction. | ||
To gain insights into the reaction mechanism, several control experiments were carried out (Fig. 5). When 2 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) were added as a radical scavenger, the photocatalytic oxidation reaction was completely inhibited and an adduct (4) of the N-radical from 1a with TEMPO was observed by HPLC-MS analysis (Fig. 5a and Fig. S1 in the ESI†). These results suggested that the reaction involved a radical pathway with a carbon centered radical being involved as a key intermediate, which agrees with the mechanism proposed for previously reported Ritter type C–H amination methods.9–14
 | ||
| Fig. 5 Control experiments. aYields were determined by GC using biphenyl as an internal standard. bReaction was carried out using 3 Å molecular sieves pre-dried 1a, 2a and TBN without the addition of H2O. cWater content was determined by Karl-Fischer titration. | ||
It was reported that the DDQ/TBN system could be employed for visible-light photocatalytic aerobic benzylic C(sp3)–H oxygenations with alcohols being formed as either intermediates or final products,31 which could then serve as substrates for the Ritter reaction.4–6 In addition, it was found that a small amount of 2a could be hydrolysed to benzylamide (7) under the standard reaction conditions (Fig. S2 in the ESI†). To investigate the roles of alcohols and amides, 1a was first replaced with diphenylmethanol (5) as the starting material, which led to only 14% yield of 3aa with benzophenone (6) formed as the major product in 57% yield (Fig. 5b). In addition, the catalytic reaction with benzamide (7) instead of 2a as the starting material led to a sluggish reaction with 21% yield of 3aa obtained (Fig. 5c). These observations indicate that alcohols or amides generated in situ are not involved as the major intermediates during the catalytic cycle. Consistent with previous reports,14 it was found that the amount of water also played an important role in the catalytic cycle. When the amount of water in the catalytic system was decreased from 8 equiv. to 0.32 equiv., the yield of 3aa decreased to 55% (Fig. 5d).
According to the above experimental results and literature reports,14,28,31,32 a mechanism exemplified by the reaction of diphenylmethane (1a) with benzonitrile (2a) is proposed as shown in Fig. 6. Initial photolysis and acidolysis of TBN liberate NO, which can be readily oxidised to NO2 by O2.31,33,34 Under visible light irradiation, DDQ is excited to its triplet excited state (3DDQ*).35 The hydrogen atom transfer (HAT) reaction between 1a and 3DDQ* generates DDQH˙ and an alkyl radical (I),36,37 which undergoes subsequent single electron transfer (SET) and leads to the formation of diphenylmethylium (II) and DDQH−.31,37 The carbocation (II) then reacts with 2a to give the nitrilium ion intermediate (III), which undergoes hydrolysis to the corresponding amide (3a). Meanwhile, DDQH− is protonated to the hydroquinone DDQH2, which can be re-oxidized to DDQ by NO2.38,39
 | ||
| Fig. 6 The proposed mechanism of the aerobic photocatalytic Ritter-type C–H amination using the DDQ/TBN system. | ||
Conclusions
In summary, by using the DDQ/TBN catalytic system, we have developed the first photocatalytic metal-free Ritter-type C–H amination method with molecular oxygen as a green oxidant. With high functional group tolerance and improved nitrile substrate scope, this protocol provides a simple, sustainable, and scalable method for the construction of secondary C–N bonds under mild conditions with moderate to excellent selectivities. We expect this new protocol to complement existing amination methods and to expand the toolbox for C–H functionalization of unreactive C(sp3)–H bonds.Author contributions
ZS and ML conceived the project. ZS, ML and QC designed the methodology and supervised the project. TL, JY, XY, JS, MH and XX performed synthetic and catalytic experiments. TL, QC and ZS analysed the data and wrote the manuscript.Conflicts of interest
CN Patent CN113943199A by Z. Shen et al. contains intellectual property described in this article. The other authors (e.g., J. Yang, X. Yin, J. Shi, Q. Cao, M. Hu and X. Xu) have no conflict of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22178321, 21773211 and 21776260) and the Key Research and Development Project of Zhejiang Province (no. 2021C01079).Notes and references
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef PubMed.
- J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef.
- M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC.
- G. Mohammadi Ziarani, F. Soltani Hasankiadeh and F. Mohajer, ChemistrySelect, 2020, 5, 14349–14379 CrossRef.
- D. Jiang, T. He, L. Ma and Z. Wang, RSC Adv., 2014, 4, 64936–64946 RSC.
- A. Guérinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 19–28 CrossRef.
- X. Wang, Nat. Catal., 2019, 2, 98–102 CrossRef.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- S. Sakaguchi, T. Hirabayashi and Y. Ishii, Chem. Commun., 2002, 516–517 RSC.
- Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547–2550 CrossRef.
- G.-X. Li, C. A. Morales-Rivera, F. Gao, Y. Wang, G. He, P. Liu and G. Chen, Chem. Sci., 2017, 8, 7180–7185 RSC.
- K. Kiyokawa, K. Takemoto and S. Minakata, Chem. Commun., 2016, 52, 13082–13085 RSC.
- Y. Zhang, J. Dong, L. Liu, L. Liu, Y. Zhou and S.-F. Yin, Org. Biomol. Chem., 2017, 15, 2897–2901 RSC.
- H. Lai, J. Xu, J. Lin, B. Su and D. Zha, Org. Chem. Front., 2022, 9, 1541–1549 RSC.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2364 CrossRef.
- H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef.
- H. Y. Kim and K. Oh, Org. Biomol. Chem., 2021, 19, 3569–3583 RSC.
- J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245–5256 RSC.
- Y. Bian, Y. Gu, X. Zhang, H. Chen and Z. Li, Chin. Chem. Lett., 2021, 32, 2837–2840 CrossRef.
- Y. Ma, F. Gao, W. Xiao, N. Li, S. Li, B. Yu and X. Chen, Chin. Chem. Lett., 2022, 33, 4395–4399 CrossRef.
- Z. Shen, J. Dai, J. Xiong, X. He, W. Mo, B. Hu, N. Sun and X. Hu, Adv. Synth. Catal., 2011, 353, 3031–3038 CrossRef.
- J. Ma, Z. Hu, M. Li, W. Zhao, X. Hu, W. Mo, B. Hu, N. Sun and Z. Shen, Tetrahedron, 2015, 71, 6733–6739 CrossRef.
- D. Pan, Y. Wang, M. Li, X. Hu, N. Sun, L. Jin, B. Hu and Z. Shen, Synlett, 2019, 218–224 Search PubMed.
- Z. Shen, T. Li, J. Ma and M. Li, CN113943199A, 2021.
- D. Pan, Z. Pan, Z. Hu, M. Li, X. Hu, L. Jin, N. Sun, B. Hu and Z. Shen, Eur. J. Org. Chem., 2019, 5650–5655 CrossRef.
- C. Song, X. Dong, H. Yi, C.-W. Chiang and A. Lei, ACS Catal., 2018, 8, 2195–2199 CrossRef.
- C. Song, H. Yi, B. Dou, Y. Li, A. K. Singh and A. Lei, Chem. Commun., 2017, 53, 3689–3692 RSC.
- S. Das, P. Natarajan and B. König, Chem. – Eur. J., 2017, 23, 18161–18165 CrossRef PubMed.
- T. Shen and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 8597–8602 CrossRef PubMed.
- F. Rusch, J.-C. Schober and M. Brasholz, ChemCatChem, 2016, 8, 2881–2884 CrossRef.
- Y. Zhang, W. Chen, L. Wang and P. Li, Org. Chem. Front., 2018, 5, 3562–3566 RSC.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef.
- T. Ting, R. Stanger and T. Wall, Int. J. Greenhouse Gas Control, 2013, 18, 15–22 CrossRef.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS.
- X. Guo, H. Zipse and H. Mayr, J. Am. Chem. Soc., 2014, 136, 13863–13873 CrossRef CAS.
- V. S. Batista, R. H. Crabtree, S. J. Konezny, O. R. Luca and J. M. Praetorius, New J. Chem., 2012, 36, 1141–1144 RSC.
- E. Bosch, R. Rathore and J. K. Kochi, J. Org. Chem., 1994, 59, 2529–2536 CrossRef.
- R. Rathore, E. Bosch and J. K. Kochi, Tetrahedron Lett., 1994, 35, 1335–1338 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01713j |
| This journal is © The Royal Society of Chemistry 2022 |