
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic enantioselective Mannich reaction of isoxazol-5(4H)-ones to isatin-derived ketimines†‡
Ricardo
Torán
 ,
Dario
Puchán
,
Amparo
Sanz-Marco
,
Carlos
Vila
,
José R.
Pedro
and
Gonzalo
Blay
*
,
Dario
Puchán
,
Amparo
Sanz-Marco
,
Carlos
Vila
,
José R.
Pedro
and
Gonzalo
Blay
*
Departament de Química Orgànica, Facultat de Química, Universitat de València, 46100-Burjassot, Spain. E-mail: gonzalo.blay@uv.es
First published on 11th October 2022
Abstract
An efficient organocatalytic asymmetric Mannich reaction between isoxazol-5(4H)-ones and isatin-derived ketimines has been developed. A bifunctional squaramide/Brønsted base organocatalyst catalyzed the enantioselective Mannich addition to afford chiral 3-aminooxindoles bearing a tetrasubstituted stereocenter at C3 decorated with an isoxazole moiety in good yields and with excellent enantioselectivities. Additionally, several synthetic transformations were described showing the versatility of the prepared compounds.
The isoxazol-5(4H)-one is a privileged heterocycle which is found in a wide range of drugs, natural products and biological active compounds.1 Moreover, this heterocycle is a versatile building block for the synthesis of β-amino acids or alkynes, among others.2 Additionally, isoxazol-5-ones have found applications in materials science due to their photonic properties.3 Based on the presence of the isoxazol-5(4H)-one moiety in bioactive compounds and on the potential applications of their derivatives, the interest in developing new procedures for the synthesis and functionalization of this kind of heterocycles is understandable, particularly in an enantioselective fashion.
Isoxazol-5(4H)-ones are methylene active compounds on account of the relatively acidic character of the C4–H;4 with three potential nucleophilic sites in the ring: the N2, the C4, and the exocyclic carbonyl O atom although they normally react at the N2 and C4 positions (Scheme 1a). Despite recent advances, asymmetric methods for the electrophilic functionalization of isoxazol-5(4H)-ones at either N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 or C46 are still scarce, compared with other related five-membered heterocycles. Focusing on organocatalytic procedures,7 nitroolefins,8N-fluorobenzenesulfonimide,9 α′-alkylidene-2-cyclopentenones10 and β,γ-alkynyl-α-imino esters11 have been efficiently used as electrophiles in the asymmetric C4-regioselective functionalization of isoxazol-5-ones. Despite these examples, a successful aminalkylation (Mannich) reaction of isoxazol-5(4H)-ones is still missing in the literature.
5 or C46 are still scarce, compared with other related five-membered heterocycles. Focusing on organocatalytic procedures,7 nitroolefins,8N-fluorobenzenesulfonimide,9 α′-alkylidene-2-cyclopentenones10 and β,γ-alkynyl-α-imino esters11 have been efficiently used as electrophiles in the asymmetric C4-regioselective functionalization of isoxazol-5-ones. Despite these examples, a successful aminalkylation (Mannich) reaction of isoxazol-5(4H)-ones is still missing in the literature.
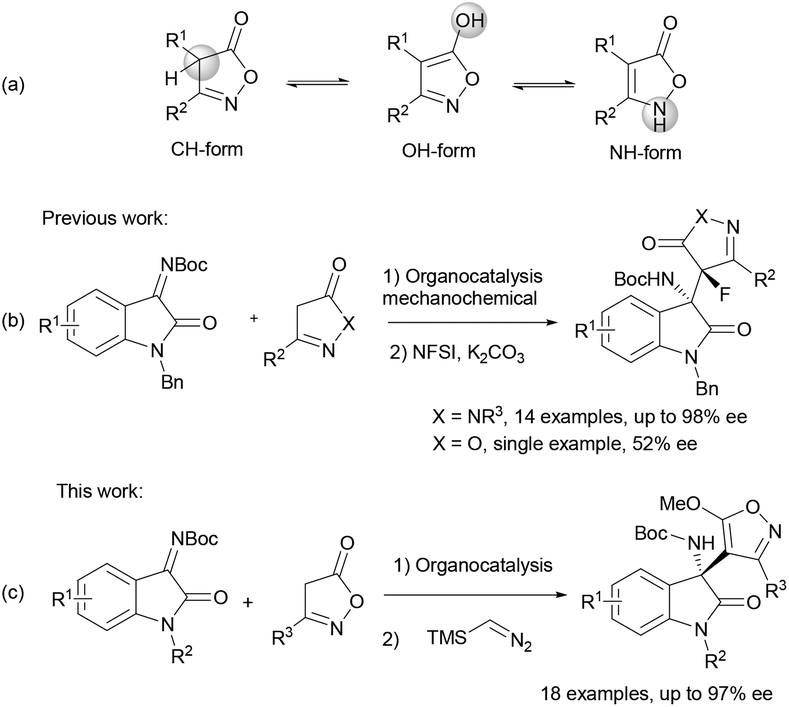 | ||
| Scheme 1 Tautomeric forms of isoxazol-5-(4H)-ones and asymmetric Mannich reaction with isatin-derived ketimines. | ||
The asymmetric Mannich addition is an extremely useful strategy to obtain chiral molecules bearing amino substituents in the stereogenic center.12 Among the variety of imines that have been employed in this reaction, the isatin-derived ketimines are one of the most interesting substrates.13 These compounds are very popular in Mannich reactions as they allow obtaining 3-aminooxindoles with a fully substituted C3. This structure has an enormous interest in medicinal and organic chemistry and can be found in numerous bioactive and natural products.14
The addition of pyrazolones to isatin-derived ketimines has been previously reported by several groups,15 including ours.15b However, to the best of our knowledge, there is only a single example of Mannich reaction involving an isoxazol-5(4H)-one as nucleophile that has been reported as an extension of one of these previous studies carried out with pyrazolones (Scheme 1b).15c This reaction consisted of an asymmetric organocatalytic domino Mannich reaction-fluorination with isatin-derived ketimines with mechanochemical activation. Although high yields and enantioselectivities were afforded for the products obtained from pyrazolones, the product obtained from isoxazol-5(4H)-one (single example) was achieved in low yield and moderate enantioselectivity. Following our interest on the use of ketimines16 and isoxazol-5(4H)-ones5a,17 in asymmetric reactions, herein we describe an efficient asymmetric Mannich reaction between isatin-derived ketimines and isoxazol-5(4H)-ones catalyzed by bifunctional squaramide/Brønsted base organocatalyst (Scheme 1c).
To develop this methodology, we began our study to examine the model reaction of 3-phenyl-4(H)-isoxazol-5-one (2a) and isatin-derived N-Boc ketimine 1a in the presence of 10 mol% organocatalyst I in dichloromethane at room temperature (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), Cat. (0.02 mmol), solvent (2 mL), rt, 20 h. b Isolated yield of 3aa. c Determined by chiral HPLC analysis. d Reaction performed at 0 °C. e Reaction performed with 0.005 mmol of IV. | ||||
| 1 | I | CH2Cl2 | 72 | 76 |
| 2 | II | CH2Cl2 | 67 | 83 |
| 3 | III | CH2Cl2 | 67 | 91 |
| 4 | IV | CH2Cl2 | 70 | 93 |
| 5 | V | CH2Cl2 | 62 | 89 |
| 6 | VI | CH2Cl2 | 63 | 83 |
| 7 | IV | Toluene | 6 | 87 |
| 8 | IV | CHCl3 | 55 | 85 |
| 9d | IV | CH2Cl2 | 57 | 92 |
| 10e | IV | CH2Cl2 | 60 | 90 |

To our delight, in the presence of catalyst I the reaction proceeded cleanly to give the expected addition product as a tautomer. To determine the enantioselectivity of the reaction, the diastereomer mixture was subjected to O-alkylation by treatment with trimethylsilyldiazomethane to give compound 3aa in 70% yield and 76% ee (Table 1, entry 1). Next, different bifunctional squaramide and thiourea organocatalysts were tested (Table 1, entries 2–6). Remarkably, using the bifunctional squaramide IV the desired aminooxindole 3aa was obtained in 70% yield and with higher 93% ee (Table 1, entry 4). Moreover, other solvents were studied (Table 1, entries 7 and 8) but none of them improved the results obtained in dichloromethane. Lowering the reaction temperature (Table 1, entry 9) or reducing the catalyst load (Table 1, entry 10) compromised the yield, although the enantioselectivity was not seriously affected.
Having established the optimal reaction conditions (Table 1, entry 4),§ the generality of the developed methodology was evaluated (Table 2). A range of isatin-derived N-Boc ketimines 1b–1l bearing a variety of substituents with different electronic properties or different N1-protecting groups were successfully tested with isoxazolone 2a, showing the robustness and efficiency of our protocol. The desired products 3ba–3la were obtained with excellent enantioselectivities (>90% ee), and in fair to good yields for the overall Mannich addition/methylation process. Electron-withdrawing (NO2, Cl) or electron-donating groups (OMe, Me) at the 5 position of the isatin derived ketimines (1b–1f) were tolerated and the corresponding aminooxindole products 3ba–3fa were obtained with good results (95–99% ee). Additionally, isatin-derived ketimines bearing substituents at the positions 6 or 7 of the homoaromatic ring 1g–1i as well as disubstituted substrate 1j were evaluated affording the products 3ga–3ja in moderate yields and high enantioselectivities (92–94%). Moreover, other isatin ketimines derivatives bearing different substituents at the N1-atom were studied (1k–1l). Methyl and methoxymethyl were appropriate groups affording the desired product with high enantioselectivities.

A variety of isoxazol-5(4H)-ones 2 was then explored. The reaction with isoxazolinones bearing para-substituted phenyl rings at C3 (2b–2c) afforded the desired products 3ab, 3ac and 3fb with high enantioselectivities. However, when aliphatic substituents at C3 of compound 2 were used (2d–2e), the corresponding products were obtained with somehow lower enantioselectivity.
Finally, we faced the reaction with isoxazolinones substituted at C4 to obtain oxindoles featuring two consecutive tetrasubstituted stereogenic centers, a challenging goal due to steric congestion.18 Under the reaction conditions, isoxazolone 2f reacted with ketimine 2a to give oxindole 3af in 85% yield, 41:59 dr favoring the anti diastereomer and high enantioselectivity (99% and 72% ee, respectively). Similarly, isoxazolinone 2g, bearing a Ph group at C3 and a Me group at C4, reacted with 1a to give compound 3ag with excellent yield, similar dr and higher enantiomeric excess for the major anti diasteromer. On the other hand, isoxazolinone 2h featuring a C3 methyl group reacted with lower stereoselectivity, in a similar way as the related 3-methyl isoxazolinone 2d, although still with good yield.
Next, we achieved some modifications to show the synthetic potential of the developed Mannich reaction. First, we demonstrated the scalability and applicability of this methodology by performing the asymmetric Mannich reaction of 1a and 2a at one mmol scale (Scheme 2A). The reaction afforded oxindole 3aa in acceptable yield without alteration of the enantioselectivity. On the other hand, a modification of the work up treating the Mannich reaction mixture with NFSI in basic medium provided oxazolinone 4 bearing a fluorinated tetrasubstituted stereocenter in good yield as a single diastereomer in 94% ee (Scheme 2B). Finally, a decarboxylative protocol of the isoxazol-5-one moiety provided the known β-chiral branched carbonyl compound 5 in high yield and enantioselectivity for the overall two-step procedure (Scheme 2C). This strategy shows that isoxazol-5(4H)-ones can act as suitable surrogates for aryl methyl ketones in the asymmetric catalytic Mannich reaction with N-alkoxycarbonyl isatin imines.
 | ||
| Scheme 2 One mmol scale reaction and synthetic transformations. Overall isolated yields. | ||
A comparison of the optical rotation of compound 5 obtained in this way with that reported in the literature19 allowed us to assign the configuration of C3 in the aminooxindole skeleton as S. Accordingly, the absolute stereochemistry of compounds 3 and 4 was assigned by chemical correlation.
To explain the observed stereochemistry, we propose the transition-state model outlined in Scheme 3, where the bifunctional catalyst is responsible for the activation and pre-orientation of the reacting partners.15b Thus, the isoxazolinone is deprotonated by the tertiary amine of the catalyst while the ketimine is activated upon formation of hydrogen bonds between the N-Boc group and the squaramide moiety. The isoxazolinone will approach the Re face of the ketimine double bond, thus accounting for the observed stereochemistry.
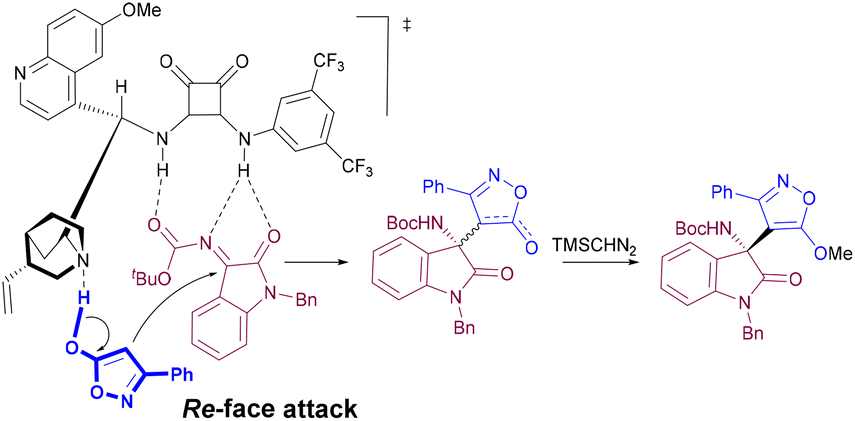 | ||
| Scheme 3 Plausible transition-state model for the reaction of isoxazol-5(4H)-ones 2 and isatin-derived ketimines 3 catalyzed by bifunctional catalyst IV. | ||
Conclusions
In conclusion, we have developed an enantioselective organocatalytic asymmetric Mannich reaction between isatin-derived ketimines 1 and isoxazol-5(4H)-ones 2. The reaction is catalyzed by a bifunctional squaramide derived from quinine and provides 3-aminooxindoles with an isoxazolinone ring in their structure which make them interesting compounds for medicinal and organic chemistry. Moreover, this methodology has been extended to synthetized 3-aminooxindoles featuring two consecutive tetrasubstituted stereogenic centers. Finally, several synthetic transformations have been carried out in order to prove the versatility and synthetic potential of this methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge grant PID2020-116944GB-100 funded by MCIN/AEI/10.13039/501100011033 and by the “European Union Next Generation EU/PRTR”. Grant CIAICO/2021/147 funded by Conselleria d'Innovació, Universitats, Ciència i Societat Digital. Grant RyC-2016-20187 funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future” to C. V., R. T. thanks the Universitat de València for a pre-doctoral“Atracció de Talent” grant. We acknowledge access to the NMR and MS facilities from the Servei Central de Suport a la Investigació Experimental (SCSIE-UV).References
- (a) M. Gordaliza, G. T. Faircloth, M. A. Castro, J. M. M. del Corral, L. López-Vázquez and A. S. Feliciano, J. Med. Chem., 1996, 39, 2865–2868 CrossRef CAS PubMed; (b) M. González, J. L. Segura, C. Seoane and N. Martín, J. Org. Chem., 2001, 66, 8872–8882 CrossRef PubMed; (c) P. Rozan, Y.-H. Kuo and F. Lambein, Phytochemistry, 2001, 58, 281–289 CrossRef CAS PubMed; (d) E. Hedner, M. Sjögren, S. Hodzic, R. Andersson, U. Göransson, P. R. Jonsson and L. Bohlin, J. Nat. Prod., 2008, 71, 330–333 CrossRef CAS PubMed; (e) S. A. Laufer and S. Margutti, J. Med. Chem., 2008, 51, 2580–2584 CrossRef CAS PubMed; (f) S. S. Mahajan, M. Scian, S. Sripathy, J. Posakony, U. Lao, T. K. Loe, V. Leko, A. Thalhofer, A. D. Schuler, A. Bedalov and J. A. Simon, J. Med. Chem., 2014, 57, 3283–3294 CrossRef CAS PubMed; (g) B. Kafle, N. G. Aher, D. Khadka, H. Park and H. Cho, Chem. – Asian J., 2011, 6, 2073–2079 CrossRef CAS PubMed.
- (a) A. F. da Silva, A. A. G. Fernandes, S. Thurow, M. L. Stivanin and I. D. Jurberg, Synthesis, 2018, 2473–2489 CAS; (b) I. D. Jurberg, Chem. – Eur. J., 2017, 23, 9716–9720 CrossRef CAS PubMed; (c) M. L. Stivanin, M. Duarte, C. Sartori, N. M. R. Capreti, C. F. F. Angolini and I. D. Jurberg, J. Org. Chem., 2017, 82, 10319–10330 CrossRef CAS PubMed.
- (a) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357–2368 CrossRef CAS PubMed; (b) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (c) M. Tonga, Tetrahedron Lett., 2020, 61, 152205 CrossRef CAS.
- A. J. Boulton and A. R. Katritzky, Tetrahedron, 1961, 12, 41–50 CrossRef CAS.
- For enantioselective N-alkylation reactions see: (a) R. Torán, C. Vila, A. Sanz-Marco, M. C. Muñoz, J. R. Pedro and G. Blay, Eur. J. Org. Chem., 2020, 627–630 CrossRef; (b) S.-S. Qi, Z.-H. Jiang, M.-M. Chu, Y.-F. Wang, X.-Y. Chen, W.-Z. Ju and D.-Q. Xu, Org. Biomol. Chem., 2020, 18, 2398–2404 RSC; (c) F. Li, S. Liang, Y. Luan, X. Chen, H. Zhao, A. Huang, P. Li and W. Li, Org. Chem. Front., 2021, 8, 1243–1248 RSC.
- Metal-catalyzed functionalization of C4: alkylation: (a) T. Hellmuth, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2015, 54, 2788–2791 CrossRef CAS PubMed Allylation: (b) S. Rieckhoff, J. Meisner, J. Kästner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2018, 57, 1404–1408 CrossRef CAS PubMed Double alkylation: (c) L. Li, P. Luo, Y. Deng and Z. Shao, Angew. Chem., Int. Ed., 2019, 58, 4710–4713 CrossRef CAS PubMed Fluorination: (d) N. Wannenmacher, C. Pfeffer, W. Frey and R. Peters, J. Org. Chem., 2022, 87, 670–682 CrossRef CAS PubMed.
- (a) Asymmetric Organocatalysis, ed. B. List, Springer, Heidelberg, 2010 Search PubMed; (b) Enantioselective Organocatalyzed Reactions, ed. R. Mahrwald, Springer, Heidelberg, 2011 Search PubMed; (c) Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS PubMed.
- W.-T. Meng, Y. Zheng, J. Nie, H.-Y. Xiong and J.-A. Ma, J. Org. Chem., 2013, 78, 559–567 CrossRef CAS PubMed.
- H. Zhang, B. Wang, L. Cui, X. Bao, J. Qu and Y. Song, Eur. J. Org. Chem., 2015, 2143–2147 CrossRef CAS.
- W. Xiao, Z. Zhou, Q.-Q. Yang, W. Du and Y.-C. Chen, Adv. Synth. Catal., 2018, 360, 3526–3533 CrossRef CAS.
- F. Li, S. Liang, Y. Luan, X. Chen, H. Zhao, A. Huang, P. Li and W. Li, Org. Chem. Front., 2021, 8, 1243–1248 RSC.
- (a) B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS; (b) J. Rostoll-Berenguer, G. Blay, J. R. Pedro and C. Vila, Adv. Synth. Catal., 2021, 363, 602–628 CrossRef CAS; (c) I. Bagheri, L. Mohammadi, V. Zadsirjan and M. M. Heravi, ChemistrySelect, 2021, 6, 1008–1066 CrossRef CAS; (d) C. Xu, C. Reep, J. Jarvis, B. Nauman, B. Captain and N. Takenaka, Catalysts, 2021, 11, 712 CrossRef CAS PubMed.
- Reviews: (a) J. Kaur and S. S. Chimni, Org. Biomol. Chem., 2018, 16, 3328–3347 RSC; (b) A. Laviós, A. Sanz-Marco, C. Vila, G. Blay and J. R. Pedro, Eur. J. Org. Chem., 2021, 2268–2284 CrossRef Selected examples: (c) D. Wang, J. Liang, J. Feng, K. Wang, Q. Sun, L. Zhao, D. Li, W. Yan and R. Wang, Adv. Synth. Catal., 2013, 355, 548–558 CAS; (d) J. Zhao, B. Fang, W. Luo, X. Hao, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 241–244 CrossRef CAS PubMed; (e) M. Montesinos-Magraner, C. Vila, A. Rendón-Patiño, G. Blay, I. Fernandez, M. C. Muñoz and J. R. Pedro, ACS Catal., 2016, 6, 2689–2693 CrossRef CAS; (f) X. Xia, Q. Zhu, J. Wang, J. Chen, W. Cao, B. Zhu and X. Wu, J. Org. Chem., 2018, 83, 14617–14625 CrossRef CAS PubMed; (g) Q. Huang, L. Zhang, Y. Cheng, P. Li and W. Li, Adv. Synth. Catal., 2018, 360, 3266–3270 CrossRef CAS; (h) S. Ričko, A. Meden, L. Ciber, B. Štefane, F. Požgan, J. Svete and U. Grošelj, Adv. Synth. Catal., 2018, 360, 1072–1076 CrossRef; (i) J.-S. Yu, H. Noda and M. Shibasaki, Chem. – Eur. J., 2018, 24, 15796–15800 CrossRef CAS PubMed; (j) M. Franc, M. Urban, I. Císařová and J. Veselý, Org. Biomol. Chem., 2019, 17, 7309–7314 RSC; (k) Z. Li, J. Peng, C. He, J. Xu and H. Ren, J. Org. Chem., 2020, 85, 3894–3901 CrossRef CAS PubMed; (l) L. Carceller-Ferrer, C. Vila, G. Blay, M. C. Muñoz and J. R. Pedro, Org. Lett., 2021, 23, 7391–7395 CrossRef CAS PubMed; (m) L.-J. Zhou, Y.-C. Zhang, F. Jiang, G. He, J. Yan, H. Lu, S. Zhang and F. Shi, Adv. Synth. Catal., 2016, 358, 3069–3083 CrossRef CAS; (n) F.-T. Sheng, Z.-M. Li, Y.-Z. Zhang, L.-X. Sun, Y.-C. Zhang, W. Tan and F. Shi, Chin. J. Chem., 2020, 38, 538–589 CrossRef.
- (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed; (b) K. Bernard, S. Bogliolo and J. B. Ehrenfeld, Br. J. Pharmacol., 2005, 144, 1037–1050 CrossRef CAS PubMed; (c) T. Shimazaki, M. Iijima and S. Chaki, Eur. J. Pharmacol., 2006, 543, 63–67 CrossRef CAS PubMed; (d) T. Oost, G. Backfisch, S. Bhowmik, M. M. van Gaalen, H. Geneste, W. Hornberger, W. Lubisch, A. Netz, L. Unger and W. Wernet, Bioorg. Med. Chem. Lett., 2011, 21, 3828–3831 CrossRef CAS PubMed; (e) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327–334 RSC.
- (a) X. Bao, B. Wang, L. Cui, G. Zhu, Y. He, J. Qu and Y. Song, Org. Lett., 2015, 17, 5168–5171 CrossRef CAS PubMed; (b) F. I. Amr, C. Vila, G. Blay, M. C. Muñoz and J. R. Pedro, Adv. Synth. Catal., 2016, 358, 1583–1588 CrossRef CAS; (c) D. Krištofíková, M. Mečiarová, E. Rakovský and R. Šebesta, ACS Sustainable Chem. Eng., 2020, 8, 14417–14424 CrossRef.
- (a) M. Holmquist, G. Blay, M. C. Muñoz and J. R. Pedro, Adv. Synth. Catal., 2015, 357, 3857–3862 CrossRef CAS; (b) M. Montesinos-Magraner, C. Vila, R. Cantón, G. Blay, I. Fernández, M. C. Muñoz and J. R. Pedro, Angew. Chem., Int. Ed., 2015, 54, 6320–6324 CrossRef CAS PubMed; (c) C. Vila, A. Rendón-Patiño, M. Montesinos-Magraner, G. Blay, M. C. Muñoz and J. R. Pedro, Adv. Synth. Catal., 2018, 360, 859–864 CrossRef CAS.
- P. Martínez-Pardo, A. Laviós, A. Sanz-Marco, C. Vila, J. R. Pedro and G. Blay, Adv. Synth. Catal., 2020, 362, 3564–3569 CrossRef.
- (a) E. A. Peterson and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943–11948 CrossRef CAS PubMed; (b) J. Christoffers and A. Baro, in Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, Wiley-VCH, Weinheim, 2005 CrossRef; (c) Q. Huang, L. Zhang, Y. Cheng, P. Li and W. Li, Adv. Synth. Catal., 2018, 360, 3266–3270 CrossRef CAS; (d) S. Ričko, A. Meden, L. Ciber, B. Štefane, F. Požgan, J. Svete and U. Grošelj, Adv. Synth. Catal., 2018, 360, 1072–1076 CrossRef; (e) J.-S. Yu, H. Noda and M. Shibasaki, Chem. – Eur. J., 2018, 24, 15796–15800 CrossRef CAS PubMed; (f) M. Franc, M. Urban, I. Císařová and J. Veselý, Org. Biomol. Chem., 2019, 17, 7309–7314 RSC; (g) Z. Li, J. Peng, C. He, J. Xu and H. Ren, J. Org. Chem., 2020, 85, 3894–3901 CrossRef CAS PubMed.
- S enantiomer: (a) J. Kaur, A. Kumari, V. K. Bhardwaj and S. S. Chimni, Adv. Synth. Catal., 2017, 359, 1725–1734 CrossRef CAS R enantiomer: (b) C. Cheng, X. Lu, L. Ge, J. Chen, W. Cao, X. X. Wu and G. Zhao, Org. Chem. Front., 2017, 4, 101–114 RSC.
Footnotes |
| † Dedicated to Professor Joan Bosch, Universitat de Barcelona, on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01692c |
| § General procedure for the enantioselective Mannich reaction/O-alkylation: an oven dried test tube was charged with isatin-derived ketimine 1 (0.2 mmol), isoxazol-5-one 2 (0.2 mmol) and squaramide IV (12.0 mg, 0.02 mmol). The test tube was purged with N2 10 minutes. Dichloromethane (2 mL) was added and the mixture was stirred at room temperature until completion (TLC). A 1 M solution of TMS-diazomethane in diethyl ether (0.4 mL, 0.4 mmol) was added via a syringe and the reaction was stirred 2 hours at room temperature. The reaction mixture was chromatographed on silica gel eluting with hexane:EtOAc mixtures to give compound 3. |
| This journal is © The Royal Society of Chemistry 2022 |