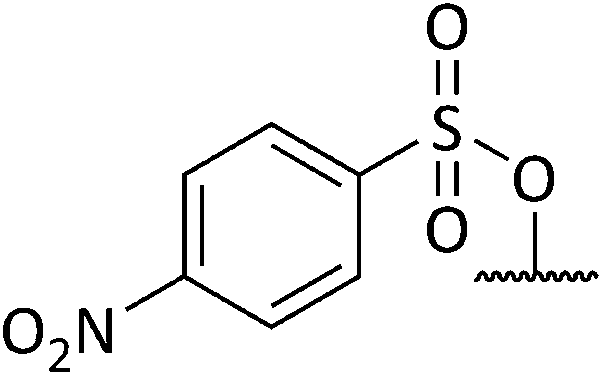
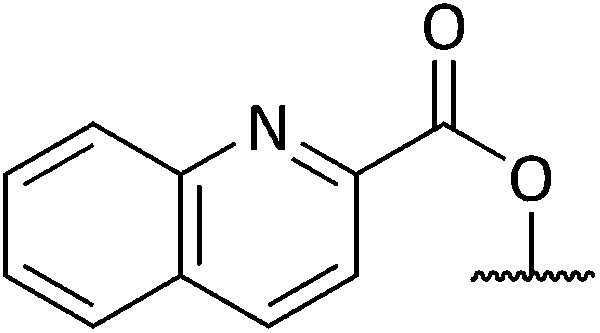
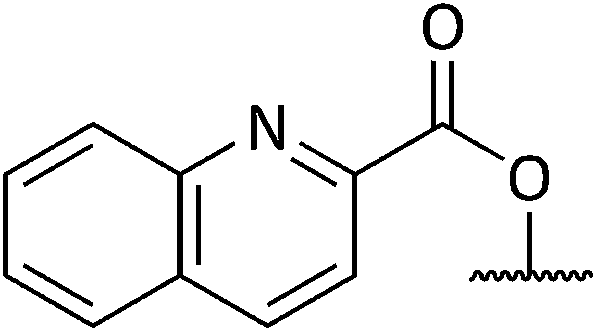
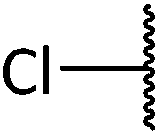Chemical synthesis of a library of natural product-like derivatives based on pinnaic acid and initial evaluation of their anti-cancer activity†
Received
6th September 2022
, Accepted 23rd October 2022
First published on 26th October 2022
Abstract
Halichlorine and pinnaic acid have been shown previously to be potent inhibitors of the inflammatory enzymes cPLA2 and VCAM-1 and have also demonstrated some anti-cancer activity. They possess an almost identical azaspirocyclic core consisting of a unique 3-dimensional geometry with four stereocentres, making them compounds of interest for further study to reveal any bioactivity not yet discovered. The azaspirocyclic core was synthesised from an established protocol, from which a small library of novel analogues were synthesised and tested for activity against two cancer cell lines, HeLa and CaCo-2, along with the non-cancerous cell line HaCaT. Eleven compounds were found to be selective for CaCo-2 cells.
Introduction
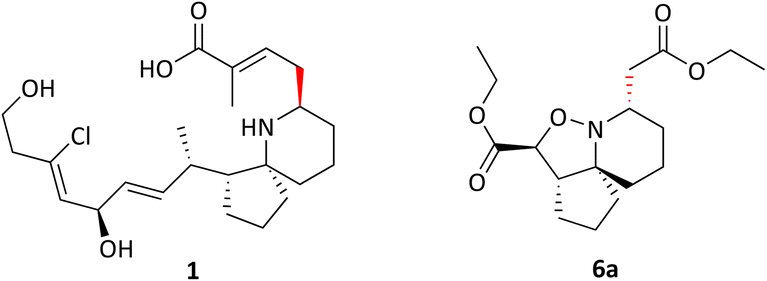
Pinnaic acid (PA) 1 and halichlorine (HC) 2 are natural products that were isolated from marine organisms in the 1990s and were shown to be selective inhibitors of the inflammatory enzymes cytosolic phospholipase A2 (cPLA2) and vascular cell adhesion moleclule-1 (VCAM-1), with IC50 values of 0.2 mM and 7 μg mL−1, (in vitro) respectively.1–4 cPLA2 is responsible for the initiation of the arachidonic cascade pathway by catalysing the cleavage of membrane phospholipids via hydrolysis releasing arachidonic acid as a result, leading to the production of eicosanoids which play a significant role in inflammatory response.5 VCAM-1 on the other hand is expressed upon the stimulation of endothelial cells which in turn occurs via the release of inflammatory mediators. Leukocytes may then bind to VCAM-1, thereby facilitating cell migration.6 The ability of pinnaic acid and halichlorine to inhibit these enzymes have made them biological targets of interest with the potential to treat a variety of diseases including atherosclerosis and non-cardiovascular inflammatory diseases.6–8 Furthermore, VCAM-1 has also been shown to play a role in metastases, as tumour cells may be able to utilise the adhesive property of VCAM-1 to break away from the primary tumour site, facilitating metastatic spread therefore making VCAM-1 a potential cancer target.9,10 The structure of pinnaic acid and halichlorine are similar with both containing a unique 3-dimensional azaspirocyclic core containing multiple stereocentres and many examples of the total synthesis, studies and reviews of these compounds may be found in the literature.2–4,10–17 One such example reported by the Stockman group focused on the synthesis of azaspirocycle of type 3 from symmetric chain 5, involving [3 + 2] cycloadditions, incorporating two directional synthesis.18 The synthetic pathway was later refined by Stockman leading to the formation of 6a (Scheme 1).19 This expanded on the work carried out by Tufariello in 1974 into [3 + 2] cycloadditions and the later work by Grigg in 1991 who synthesised several fused, bridged and spirocyclic compounds via the diastereoselective [3 + 2] cycloaddition of nitrones generated from oximes.20,21 With so much importance now placed on the synthesis of small molecules with a diverse 3-dimensional structure, Stockman had provided a versatile synthetic route, with 6a containing four of the five chiral centres found in pinnaic acid.22–25 The drawback however, was that the carbon α to the isoxazolidine nitrogen exhibited the wrong relative stereochemistry compared with the natural product (PA) (Fig. 2). Attempts at epimerisation by Stockman were only successful upon reductive cleavage of the N–O bond.26 Despite this, it was anticipated 6a and variations thereof could constitute a library of novel natural product-like molecules that may then be screened for bioactivity (Fig. 1).
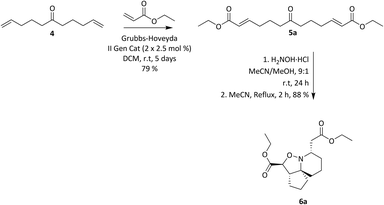 |
| | Scheme 1 Stockman's optimised route to isoxazolidine 6a. | |
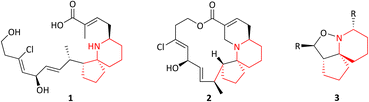 |
| | Fig. 1 Pinnaic acid 1, halichlorine 2 and azaspirocycle 3. | |
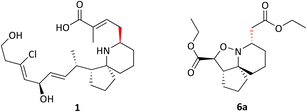 |
| | Fig. 2 Pinnaic acid 1 and Isoxazolidine 6a showing the correct and incorrect relative stereochemistry. | |
Results and discussion
Initially a sufficient quantity of 6a was synthesised to use as a starting material for library synthesis, via the established route in Scheme 1. It was also decided to synthesise two other variations of the isoxazolidine 6a with different substituents which could be introduced via cross-metathesis (Scheme 2) with products shown in Fig. 3.
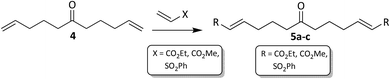 |
| | Scheme 2 Cross-matathesis. | |
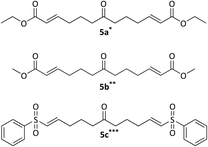 |
| | Fig. 3 Cross-metathesis products 5a–c. Reagents and conditions: alkene, Grubbs–Hoveyda II gen cat, DCM, rt, *48 h, **72 h, ***50 °C, 7 days. | |
During the course of the cross-metathesis work, it was decided to investigate both Hoveyda–Grubbs II gen and Grubbs II gen catalysts and it was found that the Hoveyda–Grubbs II gen performed best, consistent with the previously reported work by the Stockman group.19 It was observed that if the catalyst was left to age at rt for 4 weeks, a mixture of the mono (Fig. 4) and bis-homologated products (Fig. 3) would form. This could be exploited as it gave another variation of the azaspirocycle which could be further functionalised. Grubbs II gen performed the best, giving the highest yields for the mono-homologated products. The cross-metathesis products 5a–c were transformed into their corresponding isoxazolidines (Fig. 5) via Stockman's route.
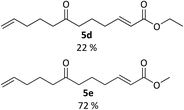 |
| | Fig. 4 Cross metathesis products 5d and 5e. Reagents and conditions: alkene, Grubbs II gen cat, DCM, rt, 48 h, (5d), 72 h (5e). | |
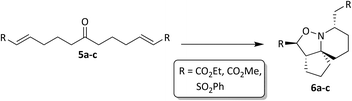 |
| | Fig. 5 Synthesis of isoxazolidines 6a–c. | |
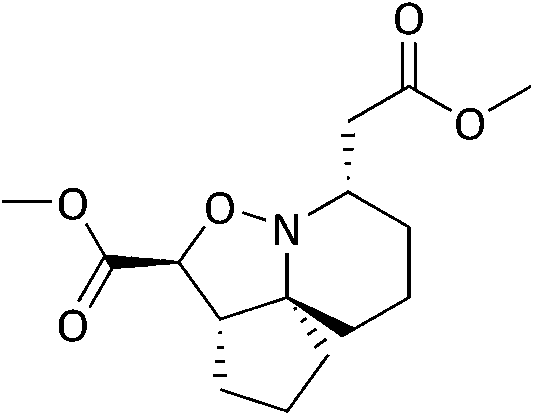
It was observed however, that the yields of the mono-functionalised isoxazolidines were poor when 5d and 5e were treated under standard cyclisation conditions (Fig. 6). Consultation of the literature led to an alternative procedure using ethanol as the solvent, resulting in a significant increase in yield.27 The products 6d and 6e are given in Fig. 7. Compounds 6d, 6e, 11 and 12 (along with 6a) although reported previously, were subject to further transformation resulting in novel analogues (Schemes 3–5). It was decided to reduce the ester of 6d to the corresponding aldehyde as this would give a handle for further functionalisation. 6d was treated with DIBAL-H at −78 °C but did not show the presence of any product at 1 hour when analysed via TLC. An increase in temperature to 0 °C however, resulted in over-reduction to the primary alcohol 7 (Scheme 3).
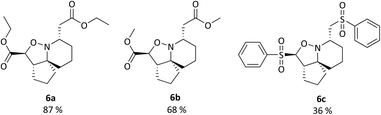 |
| | Fig. 6 Isoxazolidines 6a–c. Reagents and conditions: H2NOH·HCl, NaOAc, MeCN/MeOH 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, r.t.-reflux, 12 h. :1, r.t.-reflux, 12 h. | |
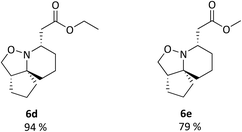 |
| | Fig. 7 Isoxazolidines 6d and 6e. Reagents and conditions: H2NOH·HCl, NaOAc, EtOH, reflux, 6 h. | |
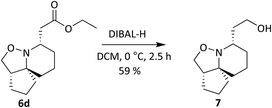 |
| | Scheme 3 Reduction of ester 6d with DIBAL-H. | |
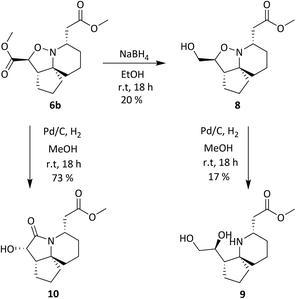 |
| | Scheme 4 Ester reduction with NaBH4 and N–O Bond cleavage. | |
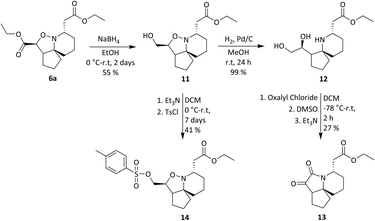 |
| | Scheme 5 Synthesis of intermediates to novel analogues. | |
Compound 6b was reduced with NaBH4 (Scheme 4) as this reducing agent had been shown previously to selectively reduce the ethyl ester α to the isoxazolidine oxygen of 6a.19 Hydroxyester 8 then underwent a reductive cleavage of the N–O bond to give 9, and 6b was also hydrogenated over Pd/C to give hydroxylactam 10 in 73% yield.
A tosylate group was introduced via the previously reported selective ester reduction with NaBH4, giving 14 (Scheme 5) to facilitate further functionalisation. Compound 13 was obtained unexpectedly whilst attempting the selective oxidation of the primary alcohol of 12. Two compounds were isolated from the reaction and analysis of 1H NMR data for the first compound showed the presence of a peak at δ 9.87 corresponding to the aldehyde proton, confirming the oxidation of the primary alcohol had occurred. Analysis of 1H NMR data for the second compound isolated confirmed the absence of protons α to both hydroxyl groups and of the proton α to the isoxazolidine nitrogen. A new signal was observed at δ 5.02–4.96, integrating for one proton and three carbonyl peaks were seen in the 13C NMR spectrum. There was a sense of familiarity with this data and so previous work reported by the Stockman group was reviewed. Their attempts to epimerise the carbon α to the isoxazolidine nitrogen of 6avia an elimination/Michael addition was unsuccessful but did lead to the formation of a very polar product instead.28 Their analysis of NMR data for this polar product confirmed the loss of peaks corresponding to one of the esters and of the appearance of three carbonyl peaks in the 13C NMR spectrum. Comparison of our data to the data obtained by Stockman was a complete match. In our oxidation attempt we had also inadvertently synthesised ketolactam 13 but via an alternative synthetic route. The mechanism is postulated in Scheme 6.
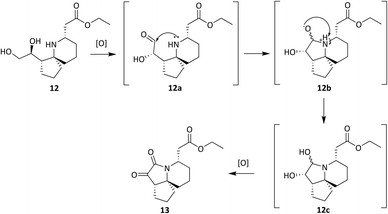 |
| | Scheme 6 Proposed mechanism leading to ketolactam 13. | |
Oxidation of the primary alcohol of 12 to the corresponding aldehyde 12a is followed by nucleophilic addition of the secondary amine to the carbonyl carbon resulting in diol 12b. This spontaneous cyclisation is in turn followed by oxidation of the secondary alcohols, giving ketolactam 13. All the compounds synthesised in this series of reactions were carried forward for biological screening against two cancer cell lines, which had not been investigated previously. To further diversify the structure of 6a whilst keeping the tricyclic ring system intact and thereby retaining the 3-dimensional geometry, attempts were made to transform the esters sequentially. Efforts to selectively reduce the ester α to the isoxazolidine oxygen with NaBH4 found that a reaction time of 48 hours was required, and yields were moderate (Scheme 5). After consideration it was decided to try sodium bis(2-methoxyethoxy) aluminium hydride (Red-Al) due to its milder nature (Scheme 7).29,30 Despite adjusting reaction conditions, no selectivity was observed and both esters were reduced to the corresponding primary alcohols 15. With diol 15 in hand, it was decided to acylate 15 with several acid chlorides to give a small library of analogues. A range of acid chlorides incorporating sulfonyl chlorides and a variety of functional groups, with some containing bulkier groups were selected to determine if control of the selectivity either sterically and/or electronically would be possible. A test reaction was carried out by exposure of diol 15 to benzoyl chloride in DCM (Scheme 8).
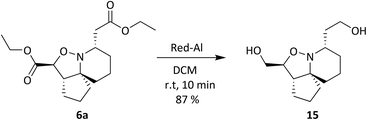 |
| | Scheme 7 Ester reduction with Red-Al. | |
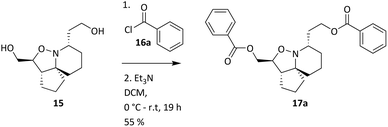 |
| | Scheme 8 Acylation of 15 with benzoyl chloride. | |
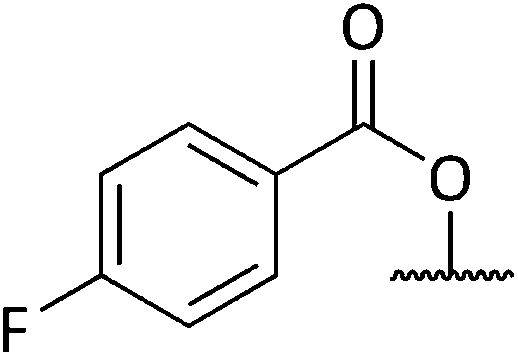
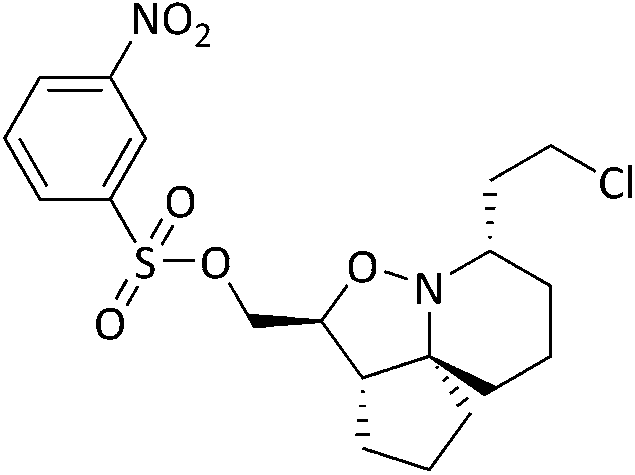
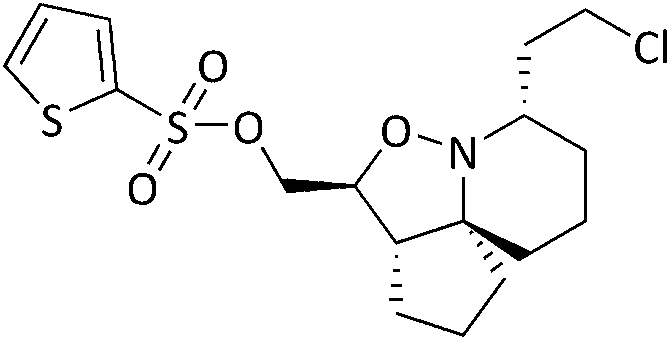
Analysis of spectroscopic data of 17a revealed the presence of double the number of aromatic signals in the 1H NMR spectrum, with two carbonyl peaks observed in the 13C NMR spectrum, corresponding to the ester carbonyls. Visual inspection of the IR spectrum confirmed the absence of a peak corresponding to the hydroxyl groups, confirming that a double acylation had occurred. Unperturbed this series of reactions continued, and the results are given in Table 1. Selectivity was observed when sulfonyl chlorides or acid chlorides with biphenyl groups were used. The first of these analogues where selectivity was observed was 17e. Diol 15 was treated with 3-nitrobenzenesulfonyl chloride and Et3N in DCM. Spectroscopic analysis of the purified product showed the presence of only one set of aromatic signals in the 1H NMR spectrum integrating for a total of 4 protons. Closer examination of 1H NMR data revealed a shift downfield for protons 5-H and only a very slight shift seen for 1-H. 13C NMR data showed the presence of only one carbonyl peak. These data suggested that acylation had occurred at the hydroxyl group α to C-5. Upon analysis of mass spectrometry data, the isotopic pattern of chlorine was observed. A base peak with an m/z of 445 was seen along with another peak at m/z 446, in the ratio of 3:1. The expected mass for the mono acylated product would have been 426. This indicated that the molecule contained a chlorine atom and if the hydroxyl group assumed to be present on the right-hand side of the molecule α to the isoxazolidine nitrogen was substituted with Cl, this would result in a mass of 444 and 446 consistent with M+ and M+2. Observed in the mass spectrum was a base peak at m/z 445 and 446 corresponding to [M + H]+ and M+2. This data suggested that in addition to acylation a chlorination had also occurred, resulting in the formation of 17e. The absence of a hydroxyl peak from the IR spectrum was also noted. The next product compound to display selectivity was 17f. Diol 15 was treated with 4-nitrobenzenesulfonyl chloride and Et3N in DCM and analysis of spectroscopic data mirrored that of 17e and was expected as the only difference between 17e and 17f being the nitro group of the aromatic ring positioned meta and para respectively. The next reaction to show selectivity resulted in the formation of 17gb and 17ga, when diol 15 was treated with 2-thiophenesulfonyl chloride and Et3N in DCM. 1H NMR data showed a significant shift downfield for 5-H and a slight shift upfield for 1-H. 13C NMR data indicated a shift downfield for 5-C from and a shift upfield for 1-C. These data indicated acylation had occurred at the hydroxyl group α to C-5. Analysis of the mass spectrum once again showed the isotopic pattern for chlorine with lines separated by an m/z of 2 in a 3:1 ratio. [M + H]+ was present at m/z 406 with a second peak present at m/z 407, corresponding to M+2. As with 17e and 17f, this data indicated that 17gb resulted from an acylation and a chlorination. The absence of a hydroxyl peak was also noted in the IR spectrum. Analysis of 1H NMR data for compound 17ga showed a significant shift downfield for 1-H with the shift for 5-H similar to diol 15. 13C NMR data showed a shift upfield for 1-C only a small change in shift for 5-C. Analysis of mass spectrometry data confirmed the presence of the [M + H]+ as the base peak at m/z 388 consistent with the mass of 17ga, with no evidence of the chlorine isotopic pattern within the mass spectrum. There was now enough evidence to postulate a mechanistic pathway giving rise to these structures (Scheme 9). Sulfonylation occurs initially at the right-hand hydroxyl group as it is more easily accessible because it is one carbon further away from the ring system than the left-hand hydroxyl group. This is immediately followed by an SN2 reaction giving chlorinated compound 20. A sulfonylation can now take place at the left-hand hydroxyl group, resulting in the formation of 22. It was fortunate that a quantity of 17ga was isolated as this is an intermediate to 17gb. The mechanism proposed also helps to explain why chlorination is not seen to occur at the left-hand hydroxyl, because this site is more sterically congested of the two hydroxyls so the right-hand hydroxyl would always be the first preferential site of reaction. The next reaction that showed selectivity resulted in a mixture of 17ka and 17kb, by treatment of diol 15 with 1-naphthoyl chloride in DCM. After isolation of the two compounds, spectroscopic analysis of 1H NMR data for the first compound 17kb confirmed a double set of aromatic signals integrating for 14 protons. Further analysis showed a significant shift downfield for 1-H and 5-H. Two carbonyl peaks were also present in the 13C NMR spectrum. Mass spectrometry data confirmed the presence of a base peak at m/z 550, consistent with [M + H]+, therefore confirming the double acylation of 15. The absence of a hydroxyl peak from the IR spectrum was also noted. Analysis of 1H NMR data for the second product, 17ka revealed a significant shift downfield for 5-H, whereas the shift for 1-H was very similar to diol 15. Also diagnostically significant was the presence of only one carbonyl peak in the 13C NMR spectrum. Mass spectrometry data confirmed the presence of the [M + H]+ at m/z 396, along with the base peak at m/z 418, corresponding to [M + Na]+. Also present was a peak in the IR spectrum consistent with a hydroxyl group. This data confirmed that a mono acylation had occurred at the hydroxyl group α to C-5. The last reaction where selectivity was observed resulted in the formation of three compounds 17la, 17lb and 17lc (Scheme 10). 1H NMR data of the first compound isolated confirmed a significant shift downfield for protons 2-H and 6-H and 13C NMR data confirmed the presence of two carbonyl peaks. Mass spectrometry data confirmed the presence of the base peak at m/z 552 corresponding to [M + H]+. This data confirmed that a double acylation had occurred giving 17la. 1H NMR data for the second component isolated (17lb and 17lc) although very clean, showed double the number of signals expected. Signals in the aromatic region integrated for 12 protons however, it did not represent a double acylation of the same molecule as all signals had been duplicated and the shifts were different to that seen in 17la. Diagnostically significant was the presence of a doublet at δ 4.65 integrating for 2 protons which couples to a multiplet at δ 4.09–4.05 integrating for 1 proton, corresponding to 5-H and 4-H in 17lc respectively. A pair of diastereotopic protons was observed at δ 4.79–4.74 and δ 4.59–4.53 integrating for one proton each corresponding to 2-H in 17lb. These protons were seen to couple to the multiplet at δ 2.35–2.27 and to two signals in the multiplet at δ 2.11–1.41. These signals correspond to 3-H in 17lb and this would be logical as the shift for protons immediately adjacent to the acyl group would be further downfield than protons α to the primary alcohol. 13C NMR data confirmed the presence of two carbonyl peaks corresponding to the ester carbonyls. Analysis of mass spectrometry data confirmed the presence of a peak at m/z 397, consistent with a mono acylation of diol 15 with quinaldoyl chloride. There was now enough evidence to suggest that the second component
isolated in this experiment was a mixture of regioisomers 17lb and 17lc and this would explain the duplicate signals seen in the NMR data. Although this mixture appeared as one spot when the reaction mixture was analysed by TLC, the polarities of these compounds would be very similar if not identical therefore appearing as one spot, making it difficult to separate out the two compounds. We wanted to gain a greater understanding of the selectivity seen as the use of sulfonyl chlorides resulted in reactions occurring at the right-hand hydroxyl oxygen (O2) followed by the left-hand hydroxyl oxygen (O1) (Fig. 8), but only after the sulfonate ester at O1 had been displaced with Cl. The second type of selectivity observed involved the use of acid chlorides with biphenyl groups which resulted in the reverse, with a reaction occurring at O1 followed by O2. Steric constraints clearly played a significant role in the selectivity, as the approach of a bulky electrophile to O1 would be met with a great degree of steric hindrance from the rigid and congested nature of the ring system relative to O2, as O2 is one carbon further away from the ring system. However, the nature of the selectivity observed cannot be explained by sterics alone and therefore there must be another factor operating. On that basis it was decided to conduct a computational study and DFT calculations were performed looking at the frontier molecular orbitals (FMO) of the HOMO of 15. Additionally, the relative nucleophilicities of the lone pairs of the oxygen atoms of the two hydroxyl groups were to be compared to determine if this may play a role in any potential selectivity that may be exhibited. The reactivity indexes defined within conceptual DFT can be powerful tools in understanding polar reactions31 and from this it is their local electrodonating power (ω−fj−) which can provide an insight into the comparison of the relative reactivities of the two oxygen atoms as nucleophiles.32 Examination of the HOMO of 15 (Fig. 8, left) revealed that electron density was found to be localised over the tricyclic ring system and O1, with none seen on O2. This was unexpected and based on this factor alone suggested that a reaction should not occur at O2, but this was in complete contradiction of experimental observations. The next highest molecular orbital (N-HOMO) was then considered, as it has been shown previously that the N-HOMO can contribute as a reactive FMO.33–35 Examination of the N-HOMO of 15 confirmed the presence of a high degree of electron density localised over O2 (Fig. 8, far right) with none seen on O1, therefore suggesting that the N-HOMO is contributing as an electron donating orbital to the FMO's and in so doing is facilitating a reaction at O2. Attention now turned to the examination of the nucleophilicity (. ω−fj−) of the two oxygen atoms (O1 and O2) of 15. ω−fj− of an atom in a molecule gives an indication of its tendency to act as a nucleophile and the ω−fj−- of the two oxygen reactive sites is given in Table 2. Though both Hirschfield-CM5 and NBO methods demonstrated a similar electron donation ability for both, the value seen for O2 was slightly greater for both methods, suggesting that the O2 has a greater tendency to donate electrons, despite the fact the HOMO confirmed a complete lack of electron density present at O2. Calculation of the Gibbs free energy of the reaction (ΔGr) for sulfonylation at O2 and O1 confirmed the difference in ΔGr between each site (ΔΔG) was found to be ∼2–3 kJ mol−−1 higher in energy at O1 confirming that sulfonylation is more energetically favourable to occur at O2, likely as a result of the inherent steric constraints of the ring system and the steric nature of the electrophilic sulfonyl chloride itself. These data allowed us to propose a compelling case for the selectivity seen. Smaller less sterically hindered electrophiles would be able to approach O1 with the HOMO accessible and reactions occurring via this orbital. Electrophiles more sterically congested, such as the sulfonyl chlorides would experience difficulty on their approach to O1 due to the proximity of O1 to the sterically crowded nature of the ring system. Therefore, reaction would be favoured at O2, occurring via the N-HOMO as it is more easily accessible and due to its greater tendency to donate electrons. Although higher in energy (ΔGr) a second reaction is still able to proceed at the less favourable O1 where molecules are already substituted at O2. Acid chlorides consisting of a biphenyl group can react with 15 at O1 via the HOMO as the electrophilic centre of the acid chloride is less sterically congested relative to the sulfonyl chlorides and therefore more accessible. A second acylation reaction may then follow at O2 via the N-HOMO as discussed above.
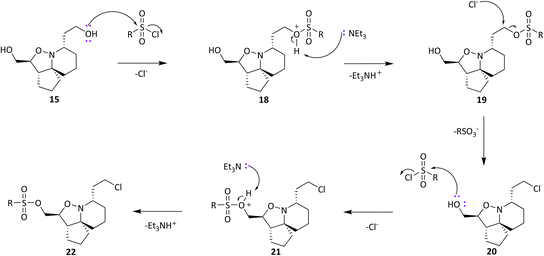 |
| | Scheme 9 Proposed mechanism of sulfonylation reactions. | |
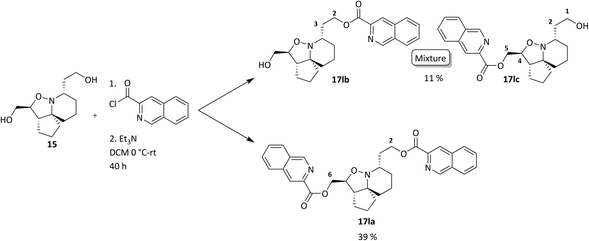 |
| | Scheme 10 Acylation of 15 with quinaldoyl chloride. | |
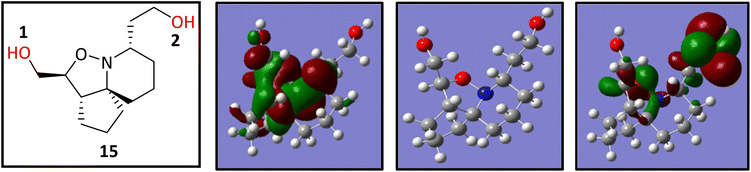 |
| | Fig. 8 The FMO of 15 showing the structure of 15 (far left) HOMO (left), optimised structure of 15 (right) and the N-HOMO (far right). | |
Table 1 Novel product analogues 17a–l of acylation and sulfonylation reactions
Table 2 The local electro donating powers (ω − fj−) of the two oxygen reactive sites using the Hirshfeld-CM5 and NBO population methods calculated at the B3LYP/6-31G(p,d) level of theory. Values given are in eV
| Atom |
Population method |
| Hirschfield-CM5 |
NBO |
|
ω
−
f
j
−
|
ω
−
f
j
−
|
| O2 |
−0.1221 |
−0.073 |
| O1 |
−0.1192 |
−0.0593 |
Biological screening
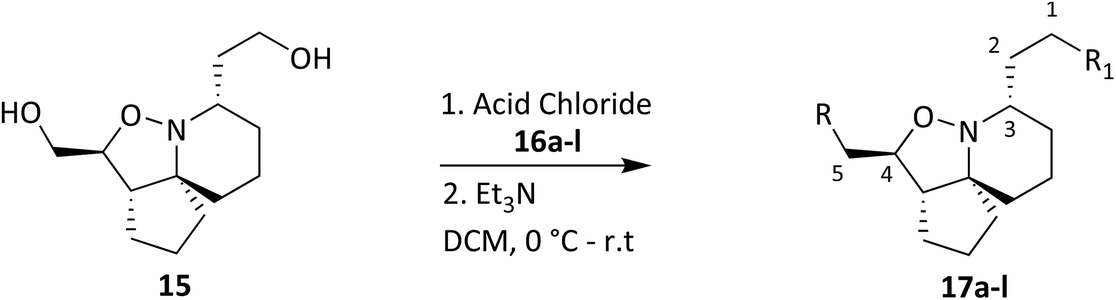
Tumour cells can escape T-cell immunity by overexpressing the endothelial cell adhesion molecule VCAM-1, which normally mediates leukocyte extravasation to sites of tissue inflammation.2 VCAM-1 in tumours may promote T-cell migration away from tumours, resulting in decreased accumulation of T cells in the tumour microenvironment. This decreased accumulation of T cells around tumour cells may contribute to the ability of VCAM-1-expressing tumour cells to escape immune attack. PLA2 expression and activity on the other hand is increased in numerous cancers including breast, pancreatic, prostate, liver and skin and PLA2 has been shown to be implicated in carcinogenesis. One of the proposed reasons is that arachidonic acid and lysophospholipids formed by the action of PLA2, can be metabolized to several molecules that induce cancer cell growth.7,36,37 A collaboration was established with our in-house anti-cancer research group who were working with HeLa and CaCo-2 cell lines and our whole compound library was tested for anti-cancer activity in vitro accordingly. Unfortunately, no activity was observed when tested against HeLa cells, but activity was observed when tested against CaCo-2 cells. Cell viability data for CaCo-2 is given in Fig. 9–12. Twelve compounds demonstrated anti-cancer activity when exposed to CaCo-2 cells. All treatments demonstrated excellent activity at 100 μM concentration with cell viability ranging between 30 and 65% over all treatment exposure times. Generally, the effect on cell viability was less pronounced at 30 μM but with some treatments still showing excellent activity (Fig. 10).
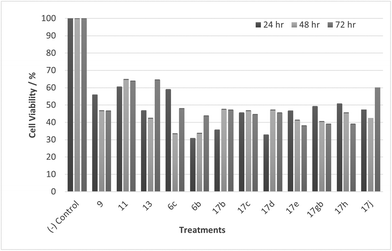 |
| | Fig. 9 Cell % viability of CaCo-2 cells exposed to treatments shown at 100 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for all treatments. | |
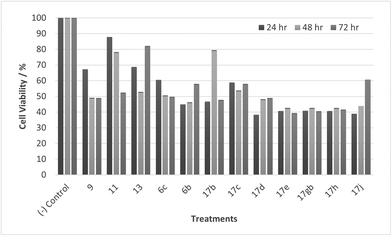 |
| | Fig. 10 Cell % viability of CaCo-2 cells exposed to treatments shown at 30 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for all treatments. | |
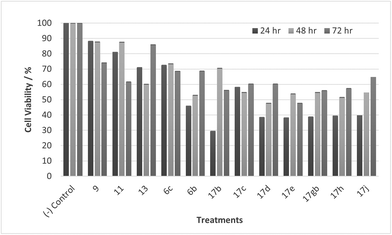 |
| | Fig. 11 Cell % viability of CaCo-2 cells exposed to treatments shown at 10 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for all treatments. | |
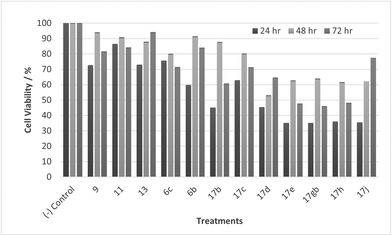 |
| | Fig. 12 Cell % viability of CaCo-2 cells exposed to treatments shown at 1 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for all treatments. | |
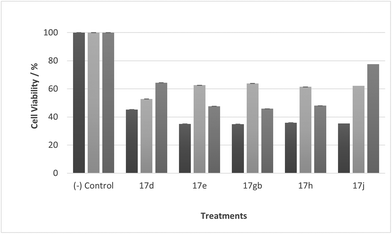 |
| | Fig. 13 Cell % viability of CaCo-2 cells exposed to treatments shown at 1 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for all treatments. | |
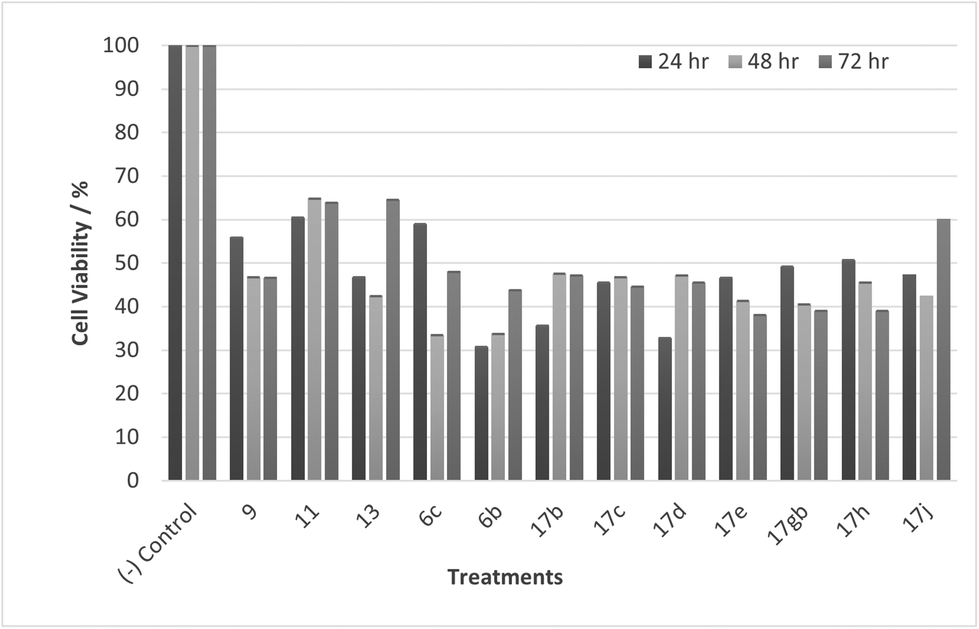
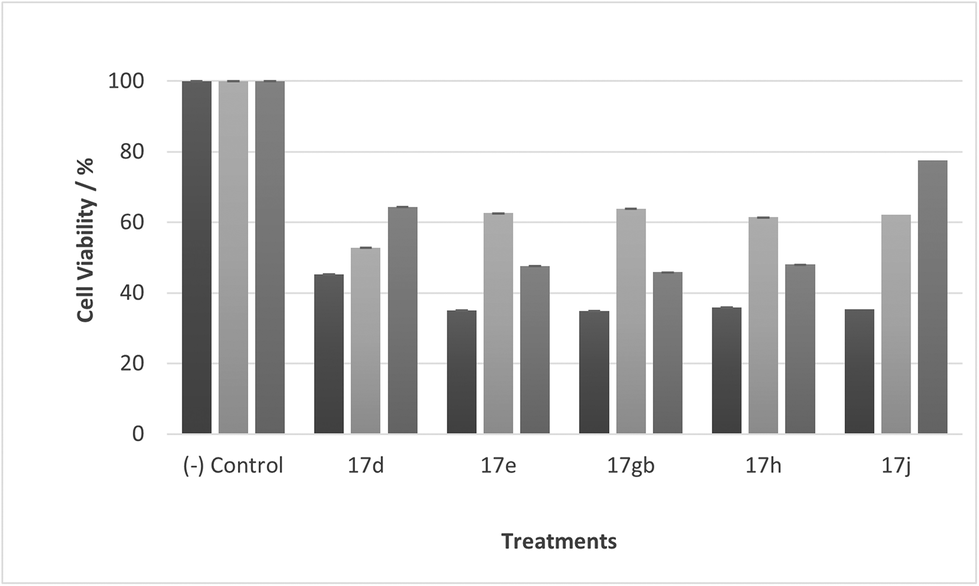
Most noticeable were treatments 17d, 17e, 17gb, 17h, and 17j which reduced cell viability significantly at all concentrations over all exposure times. Results for these treatments showed that at 100 μM cell viability was reduced to 33%–51% (24 h), 41%–47% (48 h) and 39%–60% (72 h). At 30 μM cell viability was reduced to 38%–41% (24 h), 42%–54% (48 h) and 39%–60% (72 h). At 10 μM cell viability was reduced to 38%–40% (24 h), 48%–55% (48 h) 48%–65% (72 h). At 1 μM cell viability was reduced to 35%–45% (24 h), 53%–64% (48 h) and 46%–77% (72 h). Treatments 17d, 17e, 17gb, 17h, and 17j generally performed very well with some encouraging results from 1 μM concentration at 72 hours exposure time (Fig. 13). These data suggest that lower concentrations have a greater cytotoxic effect than higher concentrations. The increase and subsequent decrease in cell viability observed with increasing time observed from the CaCo-2 was difficult to interpret. The data suggests that rather than the treatments causing cell death they are inhibiting a cellular function(s). This increase and subsequent decrease with increasing treatment time may arise from a competing cellular process preventing the inhibition of the treatment resulting in cell recovery but with further prolonged treatment time the inhibitory effect of the treatment becomes the dominant factor, resulting in the subsequent reduction in viability observed. Consultation of the literature with reference to treatments for colorectal cancer (CRC) led to research into the development of methods to overcome resistance to methotrexate (MTX), a chemotherapeutic drug used to treat CRC.38 The gene CD44 is related to the resistance of treatment involving MTX and its expression is linked to tumour cell migration, angiogenesis and cell adhesion contributing to tumour growth, with its over-expression seen in several cancers including CRC. CD44 belongs to the cell adhesion molecule (CAM) group and its significant role in cell adhesion contributes to its involvement in intracellular signalling leading to the growth of cancerous cells.38 Halichlorine also belongs to the CAM group and as a known inhibitor of VCAM-11,2 it may be possible that the anti-cancer activity seen in the treatment compounds arise from their structural similarity to the halichlorine 3-dimensional core, enabling them to interfere with CD44 ultimately leading to the reductions in cell viability observed. The similarity in dose response for treatments 17e and 17gb suggest their mechanism of action may be the same and with both treatments capable of undergoing alkylation they may be acting as alkylating agents targeting DNA leading to DNA disruption and damage.39,40 IC50 values were also calculated and are given in Table 3. The most potent treatments observed were 17e, 17h, 17gb, 17d and 17j, consistent with cell viability data. Although the mechanism/s of action here are unknown, the data shows that compounds are reducing cell density, with four compounds showing excellent anti-cancer activity at 1 μM concentration. The twelve compounds were also exposed to human immortalised keratinocytes (HaCaT cells) for cell viability to check if the compounds exhibited a cytotoxic effect on non-cancerous cells. Cell viability data is given in Fig. 14–17.
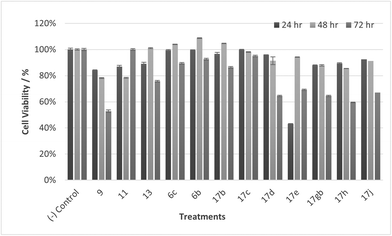 |
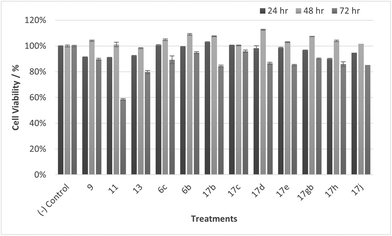
| | Fig. 14 Cell % viability of HaCaT cells exposed to treatments shown at 100 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for 17c and 9 and 11, 17d, 17gb, 17h, 17j at 48 & 72 h and 13, 17e at 72 hours. | |
 |
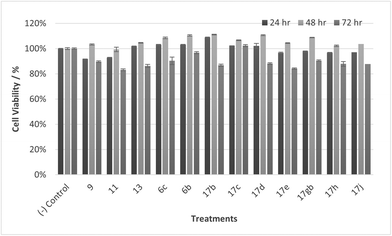
| | Fig. 15 Cell % viability of HaCaT cells exposed to treatments shown at 30 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for 17c and 9 and 11, 17d, 17gb, 17h, 17j at 48 & 72 h and 13, 17e at 72 hours. | |
 |
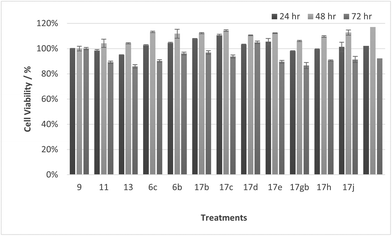
| | Fig. 16 Cell % viability of HaCaT cells exposed to treatments shown at 10 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for 17c and 9 and 11, 17d, 17gb, 17h, 17j at 48 & 72 h and 13, 17e at 72 hours. | |
 |
| | Fig. 17 Cell % viability of HaCaT cells exposed to treatments shown at 1 μM, 24, 48, 72 h, each data point represents n = 3, obtained from three independent experiments, p < 0.05 for 17c and 9 and 11, 17d, 17gb, 17h, 17j at 48 & 72 h and 13, 17e at 72 hours. | |
Table 3 IC50 values of compounds tested against CaCo-2 cells
| Entry |
Treatment/compound |
IC50/μM |
Entry |
Treatment/compound |
IC50/μM |
|
9
|

|
84.6 |
17c
|

|
74.3 |
|
11
|

|
86.9 |
17d
|
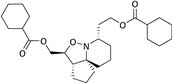
|
62.8 |
|
13
|

|
85.3 |
17e
|
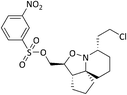
|
58.6 |
|
6c
|
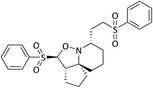
|
79.4 |
17gb
|

|
59.6 |
|
6b
|

|
75.2 |
17h
|

|
59.4 |
|
17b
|

|
69.9 |
17j
|

|
65.2 |
It was observed that at lower concentrations, only one treatment (11) led to a decrease in cell viability, showing a cytotoxic effect at 30 μM at 72 hours exposure time. Otherwise, at 10 and 1 μM concentrations all compounds generally do not reduce cell viability, confirming the treatments are well tolerated and selective for CaCo-2 cells.
Conclusion
In summary, a small library of novel natural product like analogues were synthesised, with computer modelling providing evidence to explain the selectivity observed experimentally. Twelve compounds demonstrated anti-cancer activity in vitro with only one compound reducing cell viability when exposed to HaCaT cells. Our work continues to establish the mechanism of action operating leading to the anti-cancer activity observed.
Experimental section
Cell lines and cell culture conditions
Colorectal cancer cell line CaCo-2 cells (ATCC® HTB-37) and human immortalised keratinocytes HaCaT (ATCC® PCS-200-011) cells were obtained from American Type Culture Collection (ATCC). These cell lines were cultured in Dulbecco's Modified Eagle's Medium (DMEM [Sigma Aldrich]) supplemented with 10% fetal bovine serum (FBS [Sigma Aldrich]) and 1% penicillin–streptomycin (P/S [Sigma Aldrich]) and incubated in a humidified atmosphere at 37 °C with 5% CO2.
Cell subculturing
Cell lines were sub-cultured once the cells had reached 80% confluency in a T75 flask. The medium was removed from the flask and the cells washed with Dulbecco's phosphate buffered saline (DBPS) (1×) (pH 7.4) containing 10 mM sodium dibasic phosphate, 2.7 mM potassium chloride, 2 mM potassium phosphate, 137 mM sodium chloride. After washing, the cells were detached from the flask with 1× trypsin − 5 mM EDTA in phosphate buffered saline (PBS) solution and incubated for five minutes. Dissociated cells were resuspended with DMEM containing 10% Fetal Bovine Serum (FBS) to inhibit trypsin activity. After incubation, cells underwent centrifugation at 1200 rpm for five minutes. Following centrifugation, the supernatant was discarded, and cells were resuspended with medium for further assays.
Cell counting
The number of cells were determined by using a haemocytometer. 10 μL of harvested cells and trypan blue suspension were added to the haemocytometer and then placed in the inverted microscope with 10× objective. The number of cells were calculated by counting the cells in the central grid and number of cells estimated by use of the following formula:
| Number of cells = (no of cells × 10000, dilution factor)/no of squares. |
Sulforhodamine B (SRB) assay
CaCo-2 cells and HaCaT cells were seeded in 96-well plates with approximately 5 × 103 cells per well. Cells were incubated and grown for 24 hours. Treatments were added at 1 μM, 10 μM, 30 μM or 100 μM concentration and incubated for 24, 48 and 72 hours. Cells were fixated with 10% trichloroacetic acid solution and incubated for 1 hour. The solution was removed, wells were washed with 200 μL of dH2O three times and the plate tapped gently to remove excess water and then left to air dry. 45 μL of Sulforholdamine B (SRB) staining solution was added to each well and stained for 15 minutes in the dark. Excess SRB solution was removed, and wells washed with 4 × 200 μL of 1% AcOH washing solution. After air drying the plate, the stain was solubilised with the addition of 200 μL to each well and the plate placed on the agitator for 10 minutes. Absorbance was measured at 540 nm.
Statistical analysis
Cell viability data is given as the mean ± SD for three independent experiments, where each data point represents n = 3. An ANOVA one-way test was used to analyse the data for significance (p ≤ 0.05).
IC50 values were obtained by linear regression approximation of cell survival vs. the treatment concentration. The cell survival rate was calculated as (a − c)/(b − c) × 100 (where a = absorbance of the anti-cancer treatment, where b = absorbance at 0 μM of the anti-cancer treatment and where c is absorbance of the blank).
General methods (chemical synthesis)
Flash chromatography was performed using Merck 230–400 mesh silica gel. TLC analysis was performed using Merck TLC silica gel 60 F254 aluminium backed plates. TLC plates were visualised under UV light (254 nm) or stained with KMnO4.
Infrared spectra were recorded on a PerkinElmer 100 FT-IR spectrometer Liquid samples were recorded neat.
1H NMR and 13C NMR spectra were obtained using a Bruker Avance III 400 two channel FT-NMR spectrometer operating at 400 MHz for 1H NMR and 125 MHz for 13C NMR. Spectra were recorded at room temperature in CDCl3 as solvent unless otherwise stated. GC-LRMS spectra were recorded on an Agilent Technologies 5973 (EI) electron ionisation mass spectrometer (MS) and Agilent 6890N GC at 80–380 °C (1 μl injection volume, screening range m/z 50–550). HRMS spectra were obtained via the National Mass Spectrometry Facility at Swansea University. Only molecular ions, the base peak and fragments of diagnostic significance were reported, along with their relative intensities.
Microwave reactions were carried out in a Biotage Initiator Sixty microwave reactor. Hydrogenation reactions were carried out either at 1 atm or at higher pressure in an Asynt PressureSyn reactor.
Reaction conditions, chemical reagents and solvents
All reactions were carried out in anhydrous solvents, under an inert atmosphere of N2 and were moisture free – unless carried out under aqueous conditions and stated as such.
Chemical reagents were purchased from commercial suppliers and were used without any further purification. Anhydrous solvents were purchased from Acros Organics via Fischer Scientific or Merck in SureSeal packaged bottles over molecular sieves under an atmosphere of argon. Other solvents were purchased from commercial suppliers, of standard laboratory grade and were used without any further purification.
DFT calculations
All calculations were carried out in the gas phase using the Gaussian 16 package with density functional theory (DFT) calculations performed at the B3LYP/6-31G(p,d) level of theory.41 The ground state optimized geometry of 16 was determined by vibrational analysis and this was then used in all subsequent calculations. The frontier molecular orbitals (FMOs) were visualised using GaussView. In the reaction, the electron density is moving from the OH oxygen atoms towards the electrophiles therefore interest lies in their propensity to donate charge or electrodonating power (as they act as nucleophiles in this reaction). NBO and Hirshfeld-CM5 population analysis were used in calculating the local eletrodonating power (ωj−) which can be calculated using Gazquez's method:32 Where fj− is the condensed Fukui electrophilicity and ω− is the electrodonating power of the molecule which Gazquez approximated as:
Here IP and AE are the ionization potential and the electron affinity respectively of the molecule.
The condensed Fukui electrophilicity (fj−) is calculated using:42
Where qj(N) is the gross charge of atom j in the molecule of N electrons (in this case the neutral molecule) and qj(N − 1) is the gross charge of atom j in the molecule of N − 1 electrons (in this case the cationic state of the molecule).
Experimental data
-[(3aS,7S,10aS)-Octahydro-1H-cyclopenta[3,4][1,2]oxazolo[2,3-a]pyridin-7-yl]ethan-1-ol (7).
To a solution of 6d (0.051 g, 0.20 mmol) in anhydrous DCM (5 mL) pre-cooled to 0 °C was added DIBAL-H (0.23 mL, 0.221 mmol) in one portion and the resulting mixture left to stir at 0 °C for 2.5 hours. The reaction was quenched with MeOH (5 mL), diluted with dH2O (5 mL) and left to stir for 10 minutes. The solution was added to HCl (5 mL, 10% sol'n) and the pH raised to 10 with the addition of NaOH (1 M). The mixture was diluted with EtOAc (15 mL), extracted with EtOAc (3 × 15 mL), organic fractions combined, dried over MgSO4, filtered and concentrated to give 7 as a brown oil 0.025 g, 59%. IRνmax (thin film)/cm−1 3360-OH, 2932, 2863, 1444, 1054; NMRδH (400 MHz, CDCl3) 4.25 (1H, t J 8.80, 4H), 3.92–3.86 (1H, m, 1H), 3.67–3.62 (1H, m, 1H), 3.43 (1H, dd, J 8.62, 4.37, 4H), 2.82–2.77 (1H, m, 3-H), 2.67–2.62 (1H, m, 5-H), 2.10–2.02 (1H, m, 2H). 1.97–1.83 (2H, m, 6-H, 11-H), 1.77–1.30 (12H, m, 2-H, 6-H, 7-H2, 8-H2, 9-H2, 10-H2, 11-H2); δC (75 MHz, CDCl3), 76.7 (12-C), 71.5 (4-C), 60.4 (1-C), 58.9 (3-C), 47.8 (5-C), 41.8 (6-C), 35.6 (2-C), 33.8 (11-C), 31.5, 28.9, 22.9, 21.1 (7-C to 10-C); MSm/z (EI) 211 (M+, 8), 166 (100); HRMS found 212.1642, C12H22NO2, [M + H]+, requires 212.1645.
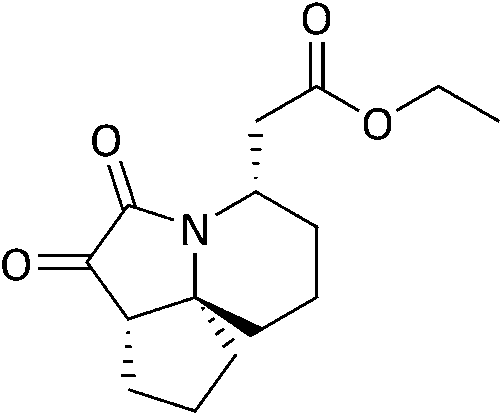
Methyl (3aS,4S,7S,10aS)-7-(2-methoxy-2-oxoethyl)octahydro-1H-cyclopenta[3,4][1,2]oxazolo[2,3-a]pyridine-4-carboxylate (6b).
To a solution of NH2OH·HCl (0.80 g, 11.5 mmol) and NaOAc (2.0 g, 24 mmol) in anhydrous MeOH (80 mL) and anhydrous MeCN (30 mL) was added a solution of 5b (2.70 g, 9.6 mmol) in anhydrous MeCN (10 mL) and the resulting mixture left to stir at r.t. for 19.5 hours. The solvent was evaporated and the resulting brown solid diluted with DCM (50 mL), filtered under suction and washed continuously with DCM until the solid became white in colour. The filtrate was concentrated, and the oil obtained dissolved in MeCN (50 mL). This solution was stirred at r.t. and upon complete consumption of the intermediate nitrone the reaction was stopped, concentrated in-vacuo, refluxed in hexane for 30 minutes and filtered hot to give 6b as a yellow oil 1.9 g, 68%. IRνmax (neat)/cm−1 2948, 1730, 1436, 1262, 1163; NMRδH (400 MHz, CDCl3) 4.12 (1H, d, J 6.42, 4-H), 3.72 (3H, s, 6-H3), 3.63 (3H, s, 15-H3), 3.13 (1H, dd, J 18.6, 12.3, 3.20, 2-H), 3.02–2.99 (1H, m, 7-H), 2.88–2.82 (1H, m, 3-H), 2.28 (1H, dd, J 5.86, 2-H), 1.95–1.21 (12H, m, 8-H2, 9-H2, 10-H2, 11-H2, 12-H2, 13-H2); δC (75 MHz, CDCl3), 171.5 (1-C), 170.7 (5-C), 81.4 (4-C), 75.7 (14-C), 58.6 (3-C), 50.5 (6-C), 49.5 (15-C), 49.2 (7-C), 37.3 (13-C), 37.0 (2-C), 31.0, 28.1, 27.3, 19.2, 18.7 (8-C to 12-C); MSm/z (EI) 297 (M+ 16), 266 (5), 238 (25); HRMS: found 373.2208 C22H29O3N, [M + H]+ requires 373.2203.
(3aS,7S,10aS)-4-(Benzenesulfonyl)-7-[(benzenesulfonyl)methyl]octahydro-1H-cyclopenta[3,4][1,2]oxazolo[2,3-a]pyridine (6c).
To a solution of NH2OH·HCl (0.037 g, 0.54 mmol) and NaOAc (0.092 g, 1.1 mmol) in anhydrous MeCN (9 mL) and anhydrous MeOH (7 mL) was added a solution of 5c (0.20 g, 0.45 mmol) in anhydrous MeCN (1 mL) and the resulting mixture left to stir at r.t. overnight. Upon complete consumption of the starting material the solvent was evaporated and the resulting brown solid diluted with DCM (50 mL), filtered under suction and washed continuously with DCM until the solid became white in colour. The filtrate was concentrated, dissolved in MeCN (20 mL) and stirred at 50 °C for 3 hours. The crude mixture was concentrated and purified via flash chromatography eluting with hexane/EtOAc 1:2 to give 6c as a yellow oil, 0.077 g, 37%. IRνmax (thin film)/cm−1 2927, 1629, 1305, 1146, 1085; NMRδH (400 MHz, CDCl3) 7.95–7.87 (4H, m, 5-H2, 17-H2), 7.68–7.59 (2H, m, 7-H, 19-H), 7.56–7.52 (4H, m, 6-H2, 18-H2), 4.47 (1H, d, J 7.41, 3-H), 3.85–3.80 (1H, m, 2-H), 3.53 (1H, dd, J 14.2, 2.61, 1-H), 3.32 (1H, t, J 6.62, 8-H), 3.20 (1H, dd, J 14.2, 9.41, 1-H), 2.35–2.30 (1H, m, 14-H), 1.97–1.39 (11H, m, 9-H2, 10-H2, 11-H2, 12-H2, 13-H2, 14-H); δC (75 MHz, CDCl3) 141.0 (4-C), 137.6 (16-C), 134.2 (7-C), 133.6 (19-C), 129.3 (6-C), 129.2 (18-C), 129.0 (5-C), 127.8 (17-C), 99.7 (3-C), 77.3 (15-C), 60.4 (1-C), 60.3 (2-C), 48.6 (8-C), 38.5, 32.0, 29.7, 28.0, 20.3, 19.4 (9-C to 14-C); MSm/z (FT) 462 ([M + H]+ 100), 320 (100), HRMS found 462.1396. C23H27O5S2N [M + H]+, requires 462.1403.
Methyl[(3aS,4S,7S,10aS)-4-(hydroxymethyl)octahydro-1H-cyclopenta[3,4][1,2]oxazolo[2,3-a]pyridin-7-yl]acetate (8).
A solution of 11 (0.53 g, 1.9 mmol) in anhydrous MeOH (40 mL) was added to Pd/C (0.53 g, 100 wt%) and left to stir under an atmosphere of H2 at r.t. for 18 hours. The catalyst was filtered off and the solvent evaporated under reduced pressure. The crude was purified via flash chromatography eluting with DCM/MeOH 9:1 to give 8 as a colourless oil, 0.085 g, 20%. IRνmax (thin film)/cm−1 3436-OH, 2937, 1732, 1436, 1195, 1114; NMR δH (400 MHz, CDCl3), 4.60 (1H, s (br), 15-H), 4.51 (1H, d, J 9.80, 5-H), 4.39 (1H, d, J 9.90, 5-H), 3.66 (3H, s, 14-H3), 3.54–3.48 (1H, m, 4-H), 3.32–3.28 (1H, m, 3-H), 2.48 (1H, dd, J 14.3, 9.86, 2-H), 2.34 (1H, dd, J 14.3, 8.85, 2-H), 2.14–2.07 (1H, m, 12-H), 1.85–1.38 (12H, m, 6-H, 7-H2, 8-H2, 9-H2, 10-H2, 11-H2, 12-H); δC (75 MHz, CDCl3) 172.9 (1-C), 78.5 (4-C), 77.2 (5-C), 77.1 (13-C), 52.7 (3-C), 51.7 (13-C), 40.7 (6-C), 37.7 (2-C), 36.1, 30.8, 28.2, 23.7, 19.9, 17.6 (7-C to 12-C); MSm/z (EI) 252 (24), 224 (19), 210 (69), 196 (31); HRMS found 270.1698, C14H24NO4, [M + H]+, requires 270.1700.
Methyl{(1S,5S,7S)-1-[(1S)-1,2-dihydroxyethyl]-6-azaspiro[4.5]decan-7-yl}acetate (9).
A solution of 8 (0.76 g, 2.7 mmol) in anhydrous MeOH (40 mL) followed by Pd/C (0.76 g, 100 wt%) was added to a pressure reactor and stirred at r.t. for 18 hours under an atmosphere of H2 at 5 bar pressure. The catalyst was filtered off and the solvent evaporated under reduced pressure. The crude mixture was purified via flash chromatography eluting with DCM/MeOH 7:3 to give 9 as a white residue 0.13 g, 17%. IRνmax (thin film)/cm−1 3311, 2931, 1728, 1195, 1173, 1116, 1070; NMRδH (400 MHz, CDCl3), 3.71–3.67 (2H, m, 4-H, 5-H), 3.65 (3H, s, 14-H3), 3.46–3.42 (1H, m, 5-H), 3.32–3.26 (1H, m, 3-H3), 2.46–2.42 (2H, m, 2-H2), 2.13–2.09 (1H, m, 6-H), 1.87–1.29 (12H, m, 7-H2, 8-H2, 9-H2, 10-H2, 11-H2, 12-H2); δC (75 MHz, CDCl3) 172.1 (1-C), 77.3 (13-C), 74.0 (4-C), 65.3 (5-C), 51.8 (14-C), 50.3 (3-C), 41.0 (6-C), 40.3 (2-C), 38.6, 36.0, 30.9, 26.6, 20.0, 19.6 (7-C to 12-C); MSm/z (EI), 271 (M+ 3), 254 (34), 240 (67), 224 (13), 198 (40); HRMS found 272.1858 C4H25NO4, [M + H]+, requires 272.1856.
Ethyl[(3aS,4S,7S,10aS)-4-{[(4-methylbenzene-1-sulfonyl)oxy]methyl}octahydro-1H-cyclopenta[3,4][1,2]oxazolo [2,3-a]pyridin-7-yl]acetate (14).
To a solution of 11 (0.10 g, 0.39 mmol) in anhydrous DCM (6 mL) was added Et3N (0.051 g, 0.070 mL, 0.51 mmol) and the resulting mixture was left to stir at r.t. for 30 minutes. The temperature was lowered to 0 °C, followed by the addition of TsCl (0.071 g, 0.37 mmol) and the mixture left to stir at the same temperature for 5 minutes, then warmed to r.t. and left to stir for 2 weeks. The reaction was stopped and quenched with brine (10 mL) at 0 °C, extracted with DCM (2 × 30 mL), organics combined, dried over MgSO4, filtered and concentrated to give a yellow residue. The crude was purified via flash chromatography eluting with hexane/EtOAc 1:1 to give 14 as a colourless oil, 0.60 g, 41%. IRνmax (thin film)/cm−1 2938, 1728, 1364, 1189, 1175, 1096; NMRδH (400 MHz, CDCl3), 7.81–7.78 (2H, m, 7-H2), 7.35–7.32 (2H, m, 8-H2), 4.16 (2H, d, J 6.08, 5-H2), 4.08 (2H, q, J 7.14, 19-H2), 3.81–3.76 (1H, m, 4-H), 2.95–2.88 (1H, m, 3-H), 2.75 (1H, dd, J 15.1, 2-H), 2.46–2.41 (4H, m, 10-H2, 11-H2), 2.20 (1H, dd, J 15.1, 8.30, 2-H), 1.96–1.45 (12H, m, 12-H2, 13-H2, 14-H2, 15-H2, 16-H2, 17-H2), 1.23 (3H, t, J 7.14, 20-H3); δC (75 MHz, CDCl3) 172.2 (1-C), 144.9 (6-C), 133.0 (9-C), 129.9 (8-C), 128.0 (7-C), 82.5 (4-C), 76.7 (18-C), 71.6 (5-C), 61.4 (3-C), 60.3 (19-C), 51.2 (11-C), 40.1 (2-C), 38.3, 31.6, 29.2, 27.7, 21.7, 21.3, 19.6 (10-C and 12-C to 14-C), 14.2 (20-C); MSm/z (EI) 237 (M+ 12), 208 (14), 194 (100), HRMS found 438.1938, C22H32NO6S, [M + H]+ requires 438.1945.
2-[(3aS,4S,7S,10aS)-4-(Hydroxymethyl)octahydro-1H-cyclopenta[3,4][1,2]oxazolo[2,3-a]pyridin-7-yl]ethan-1-ol (15).
To a solution of 6a (0.90 g, 2.8 mmol) in anhydrous DCM (25 mL) pre-cooled to 0 °C was added a solution of Red-Al in PhMe (60 wt%, 1.6 mL, 8.3 mmol) dropwise. Upon complete addition, the cooling bath was removed and the mixture left to stir for 10 minutes. The mixture was quenched with Rochelle's salt (10% sol'n, 40 mL) and left to stir overnight. The organic phase was extracted with DCM (7 × 50 mL), combined, dried over MgSO4, filtered and concentrated to give a yellow oil. The crude mixture was purified via flash chromatography, eluting with DCM/MeOH 8:2 to give 15 as an opaque oil, 0.40 g, 55%. IRνmax (thin film)/cm−1, 3330-OH, 2929, 2868, 1445, 1124, 1056; NMRδH (400 MHz, CDCl3), 3.95–3.90 (1H, m, 1-H), 3.82–3.79 (1H, dd, J 9.51, 3.81, 5-H), 3.74–3.70 (1H, m, 4-H), 3.66–3.57 (2H, m, 1-H, 5-H), 3.05–2.99 (1H, m, 3-H), 2.53–2.50 (1H, m, 6-H), 2.04–1.97 (1H, m, 12-H), 1.94–1.34 (13H, m, 2-H2, 7-H, 8-H2, 9-H2, 10-H2, 11-H2, 12-H); δC (75 MHz, CDCl3) 86.1 (4-C), 76.6 (13-C), 62.9, (5-C) 60.9 (3-C), 60.3 (1-C), 49.6 (6-C), 38.3 (12-C), 37.0 (2-C), 31.2, 28.5, 27.5, 21.8, 19.4 (7-C to 11-C), HRMS found 264.1567 C13H23NO3Na, [M + Na]+, requires 264.1570.
{(3aS,4S,7S,10aS)-7-[2-(Benzoyloxy)ethyl]octahydro-1H-cyclopenta[3,4][1,2]oxazolo[2,3-a]pyridin-4-yl}methyl benzoate (17a) (representative example for acylation reactions. Full details in ESI†).
To a solution of 15 (0.23 g, 0.95 mmol) in anhydrous DCM (4 mL) cooled to 0 °C was added Et3N (0.19 g, 0.27 mL, 1.9 mmol) followed by a solution of benzoyl chloride (0.12 mL, 1.1 mmol) in anhydrous DCM (2 mL) dropwise. Once the addition was complete the cooling bath was removed and the mixture left to stir at r.t. Upon complete consumption of the starting material the reaction was quenched with dH2O (20 mL) and washed with dH2O (3 × 20 mL). The crude was extracted with DCM (3 × 20 mL), the organic fractions combined, dried over MgSO4, filtered and concentrated to give a yellow oil. The crude mixture was purified via flash chromatography eluting with hexane/EtOAc 1:1 to give 15a as an opaque oil, 0.18 g, 54%. IRνmax (thin film)/cm−1, 2938, 2866, 1714, 1601, 1450, 1176, 1111; NMRδH (400 MHz, CDCl3) 8.05–7.99 (4H, m, 9-H, 21-H), 7.55–7.47 (2H, m, 1-H, 23-H), 7.43–7.36 (4H, m, 8-H, 22-H), 4.55–4.37 (4H, m, 6-H2, 2-H2), 4.00–3.95 (1H, m, 5-H), 2.97–2.90 (1H, m, 4-H), 2.52–2.48 (1H, m, 12-H), 2.31–2.23 (1H, m, 18-H), 1.97–1.33 (13H, m, 13-H2, 14-H2, 15-H2, 16-H2, 17-H2, 18-H); δC (75 MHz, CDCl3) 196.6 (1-C), 166.6 (7-C), 133.1 (11-C), 132.7 (23-C), 130.5 (8-C), 129.9 (20-C), 129.7 (9-C), 129.5 (21-C), 128.4 (10-C), 128.3 (22-C), 82.6 (5-C), 76.3 (19-C), 66.9 (6-C), 62.8 (2-C), 60.8 (4-C), 52.6 (12-C), 38.6, 34.4, 31.5, 28.1, 22.3, 19.5 (13-C to 18-C); MSm/z (FT) 472 ([M + Na]+ 13), 450 (100), 328 (100); HRMS found 450.2269 C27H31NO5, [M + H]+, requires 450.2275.
Conflicts of interest
There are no conflicts to declare.
References
- M. Kuramoto, C. Tong, K. Yamada, T. Chiba, Y. Hayashi and D. Uemura, Tetrahedron Lett., 1996, 37(22), 3867–3870 CrossRef CAS.
- D. Trauner, J. B. Schwarz and S. J. Danishefsky, Angew. Chem., Int. Ed., 1999, 38(23), 3542–3545 CrossRef CAS PubMed.
- T. Chou, M. Kuramoto, Y. Otani, M. Shikano, K. Yazawa and D. Uemura, Tetrahedron Lett., 1996, 37(22), 3871–3874 CrossRef CAS.
- H. S. Christie and C. H. Heathcock, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(33), 12079–12084 CrossRef CAS PubMed.
- D. T. Stephenson, C. A. Lemere, D. J. Selkoe and J. A. Clemens, Neurobiol. Dis., 1996, 3, 51–63 CrossRef CAS PubMed.
- C. A. Foster, Allergy Clin. Immunol., 1996, 98, S270–SS277 CrossRef CAS PubMed.
- K. A. Talvinen, E. A. Kemppainen and T. J. Nevalainen, Scand. J. Gastroenterol., 2001, 36(11), 1217–1221 CrossRef CAS PubMed.
- M. Kuramoto, H. Arimoto and D. Uemura, Mar. Drugs, 2004, 2, 39–54 CrossRef CAS.
- F. Vidal-Vanacloncha, G. Fantuzzi, L. Mendoza and C. A. Dinarello, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 734–739 CrossRef PubMed.
- D. L. J. Clive, M. Yu, J. Wang, V. S. C. Yeh and S. Kang, Chem. Rev., 2005, 105(12), 4483–4514 CrossRef CAS PubMed.
- M. W. Carson, G. Kim, M. F. Hentemann, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40(23), 4450–4452 CrossRef CAS PubMed.
- M. W. Carson, G. Kim and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40(23), 4453–4456 CrossRef CAS PubMed.
- H. Arimoto, I. Hayakawa and D. Uemura, Heterocycles, 2003, 59(2), 441–444 CrossRef.
- R. B. Andrade and S. F. Martin, Org. Lett., 2005, 7(25), 5733–5735 CrossRef CAS PubMed.
- H. Wu, H. Zhang and G. Zhao, Tetrahedron, 2007, 63(28), 6454–6461 CrossRef CAS.
- D. Liu, H. P. Acharya, M. Yu, J. Wang, V. S. C. Yeh, S. Kang, C. Chiruta, S. M. Jachak and D. L. J. Clive, J. Org. Chem., 2009, 74(19), 7417–7428 CrossRef CAS PubMed.
- S. Xu, D. Unabara, D. Uemura and H. Arimoto, Chem. – Asian J., 2014, 9(1), 367–375 CrossRef CAS PubMed.
- L. Arini, P. Szeto, D. Hughes and R. Stockman, Tetrahedron Lett., 2004, 45, 8371–8374 CrossRef CAS.
- C. Gignoux, A. F. Newton, A. Barthelme, W. Lewis, M. L. Alcaraz and R. L. Stockman, Org. Biomol. Chem., 2012, 10, 67–69 RSC.
- J. J. Tufariello and E. J. Trybulski, J. Org. Chem., 1974, 39(23), 3378–3384 CrossRef CAS PubMed.
- R. Grigg, J. Markandu, S. Surendrakumar, M. Thornton-Pett and W. J. Warnock, Tetrahedron, 1992, 48(47), 10399–10422 CrossRef CAS.
- J. Meyers, M. Carter, N. Yi Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef PubMed.
- W.-W. Ni, H.-L. Fang, Y.-X. Ye, W.-Y. Li, L. Liu, Z.-J. Fu, Dawalamu, W.-Y. Zhu, K. Li, F. Li, X. Zou, H. Ouyang, Z.-P. Xiao and H.-L. Zhu, Med. Chem., 2021, 17, 1046–1059 CrossRef CAS PubMed.
- W.-Q. Song, M.-L. Liu, S.-Y. Li and Z.-P. Xiao, Curr. Top. Med. Chem., 2022, 22(2), 95–107 CrossRef CAS PubMed.
- X. Zhang, L. Sun, S. Xu, T. Huang, S. Cherukupalli, X. Ding, Y. Tao, D. Kang, E. de Clercq, C. Pannecouque, A. Dick, S. Cocklin, X. Liu and P. Zhan, Eur. J. Med. Chem., 2021, 221, 113848 CrossRef PubMed.
- L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371–8374 CrossRef CAS.
-
M. Shindo, Y.-I. Fukuda and K. Shishido, The efficient entry into the tricyclic core of halichlorine, 2000, vol. 41 Search PubMed.
- A. Sinclair, L. Arini, M. Rejzek, P. Szeto and R. Stockman, Synlett, 2006, 14, 2321–2324 Search PubMed.
- W. Dauben and R. Bozak, J. Org. Chem., 1959, 24, 1596–1597 CrossRef CAS.
- N. Bajwa and M. P. Jennings, J. Org. Chem., 2008, 73, 3638–3641 CrossRef CAS PubMed.
- N. Kerru,
et al.
, Sci. Rep., 2016, 9, 1–17 Search PubMed.
- J. L. Gázquez, A. Cedillo and A. Vela, J. Phys. Chem. A, 2007, 111, 1966–1970 CrossRef PubMed.
- D.-C. Fang and X.-Y. Fu, J. Mol. Struct., 1999, 459, 15–21 CrossRef CAS.
- M. Arbelot, A. Allouche, K. F. Purcell and M. Chanon, J. Org. Chem., 1995, 60, 2330–2343 CrossRef CAS.
- D.-C. Fang, Z.-F. Xu and X.-Y. Fu, J. Mol. Struct., 1995, 333, 159–163 CrossRef CAS.
- M. I. Patel, J. Singh, M. Niknami, C. Kurek, M. Yao, S. Lu, F. Maclean, N. J. C. King, M. H. Gelb, K. F. Scott, P. J. Russell, J. Boulas and Q. Dong, Clin. Cancer Res., 2008, 14, 8070–8079 CrossRef CAS PubMed.
- L. Vecchi, T. G. Araújo, F. V. Petten de Vasconcelos Azevedo, S. T. Soares Mota, VdM. R. Ávila, M. A. Ribeiro and L. R. Goulart, Cell, 2021, 10, 1472 CrossRef CAS PubMed.
- Q. Su, J. Song, X. Zhang, Y. Jiang and H. Gao, Pathol., Res. Pract., 2021, 229, 2–10 Search PubMed.
- B. Biersach, Cancer Drug Resist., 2019, 2, 1–17 Search PubMed.
- W. Grady and C. Ulrich, Gut, 2007, 56, 318–320 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2016, 21, 1–22 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Okan M.
Cakir
,
Yousaf
Khan
,
Okan M.
Cakir
,
Yousaf
Khan