Synthesis and properties of oligodiaminogalactoses that bind to A-type oligonucleotide duplexes†
Received
30th August 2022
, Accepted 28th September 2022
First published on 30th September 2022
Abstract
Recently, double-stranded oligonucleotide therapeutics with A-type duplex structures such as short interfering RNAs have gained considerable attention. We have reported the synthesis of cationic oligosaccharides that selectively bind to A-type oligonucleotide duplexes. In particular, oligodiaminogalactose (ODAGal) has a strong stabilizing effect on A-type oligonucleotide duplexes. However, an efficient synthetic method has not been established for ODAGals and the properties of ODAGals have been investigated only up to 4mer. The most crucial problem of the synthesis was side reactions on a p-methoxybenzyl (PMB) protecting group of a 3-hydroxy group. In this paper, the benzyl (Bn) group was chosen as a protecting group of the 3-hydroxy group to suppress the side reactions on protecting groups, and the yields of glycosylation reactions were significantly improved. Moreover, optimization of the conditions for the deprotection of the Bn groups allowed the efficient synthesis of fully deprotected ODAGals, and ODAGal 5mer and 6mer were synthesized for the first time. In addition, we systematically investigated the effects of these ODAGals on the properties of several oligonucleotide duplexes. It was found that ODAGal 4–6mers stabilized the A-type oligonucleotide duplexes thermally and biologically, typically without their structural changes and the effect was notable with longer ODAGals.
Introduction
In recent years, oligonucleotide therapeutics have gained attention as next-generation drugs following small molecule and antibody drugs. Double-stranded RNA molecules are one of the intensively studied modalities since the discovery of “RNA interference (RNAi)”.1–3 RNAi is an intrinsic gene regulatory mechanism; the expression of genes is knocked down by internal double-stranded RNAs or by exogenous short interfering RNAs (siRNAs) comprising 21–23 bases. Up to now, five siRNAs have been approved as RNAi drugs: patisiran,4 givosiran,5 lumasiran,6 inclisiran,7 and vutrisiran.8
Meanwhile, antisense oligonucleotides (ASOs) are the most studied oligonucleotide therapeutics. One of the most effective ASOs is a gapmer-type ASO, which has a central gap region of contiguous DNA and a wing region consisting of chemically modified ribonucleotides with high affinity for its complementary RNAs such as 2′-OMe, 2′-O-MOE RNA,9 and LNA.10 Furthermore, a new type of double-stranded oligonucleotide therapeutics called heteroduplex oligonucleotides (HDOs) has been reported recently.11 An HDO is composed of a DNA/LNA gapmer as an ASO and its complementary RNA and is a promising modality for antisense drugs.
The key factors for developing potent oligonucleotide therapeutics include the high biological stability of oligonucleotides and the efficiency of the delivery. For the former issue, most RNAi and antisense drugs and their candidates have at least one type of chemical modification such as phosphorothioate, 2′-OMe, and 2′-O-MOE sugar modifications, to gain nuclease resistance.12 For the effective delivery of oligonucleotide therapeutics to intended organs, small molecule ligands are conjugated to oligonucleotides in some cases. In particular, siRNAs such as givosiran, lumasiran, and inclisiran have GalNAc modifications that enable a selective delivery to the liver.13 Yokota et al. found that a cholesterol-conjugated HDO could cross the blood–brain barrier and be delivered to the brain.14
Under these circumstances, our group has developed artificial molecules that bind to oligonucleotide duplexes through non-covalent interactions, enabling thermal and biological stabilization of these duplexes and selective delivery to specific organs. One of the artificial molecules is oligodiaminosaccharide, where 2 and 6-hydroxy groups of a pyranose were substituted with amino groups. So far, we have achieved the synthesis of three types of oligodiaminosaccharides; oligodiaminoglucoses (ODAGlcs, Fig. 1A),15 oligodiaminomannoses (ODAMans, Fig. 1B),16 and oligodiaminogalactoses (ODAGals, Fig. 1C).17 The most characteristic feature of these oligodiaminosaccharides is that they can bind to A-type nucleic acid duplexes, such as RNA/RNA and DNA/RNA, with high affinity and selectivity. This feature is attributed to the multiple cation–anion interactions between phosphate groups in a major groove of an A-type duplex and positively charged amino groups of oligodiaminosaccharides. In particular, ODAGals have a significant stabilizing effect on A-type double-stranded nucleic acids, including siRNAs and HDOs.18 In addition, ODAGals bind more strongly to nucleic acid duplexes than existing aminoglycosides such as neomycin, and ODAGal4 (n = 4) stabilizes the duplexes more than ODAGal3 (n = 3).17 Most recently, we have reported that ODAGal4 is effective for modulating the mismatch discrimination by the RNase H cleavage.19 Moreover, ODAGal is useful for the selective delivery of a siRNA to the liver cell by conjugating vitamin E to ODAGal.20 Notably, it has been suggested that the presence of ODAGals does not disturb the gene silencing effect of siRNAs.18
 |
| | Fig. 1 (A) Structure of ODAGlc. (B) Structure of ODAMan. (C) Structure of ODAGal. | |
As described above, ODAGals are promising candidates for improving the properties of siRNAs and HDOs. However, an efficient method for synthesizing ODAGals has not been established, and the properties of ODAGals have been investigated only up to tetrasaccharides. Therefore, in this study, we established an efficient method for synthesizing ODAGals, and obtained 5mer (ODAGal5) and 6mer (ODAGal6). We herein report the effect of the obtained ODAGals on the properties of oligonucleotide duplexes.
Synthesis of ODAGals
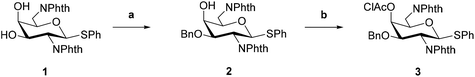
We previously reported the synthesis of ODAGal2, ODAGal3, and ODAGal4.17 In the report, the major problem with the synthesis of ODAGals was the reduced yield in glycosylation reactions as the number of repeats increased. In the reactions, a thiophenyl glycoside, where the amino groups at 2- and 6-positions were protected by phthaloyl groups and the 3-hydroxy group was protected by p-methoxybenzyl (PMB) group, was used as the glycosyl donor, and commonly used activation reagents, namely, N-iodosuccinimide (NIS) and trifluoromethanesulfonic acid (TfOH), were used for activation. However, partial removal or iodination of the PMB groups was observed when CH2Cl2 or a mixture of CH2Cl2 and Et2O was used as the solvent. Furthermore, the iodinated PMB groups were more resistant to acidic conditions than the unmodified PMB groups; thus, the removal of the iodinated PMB groups was challenging. To prevent the side reactions, increasing the ratio of Et2O in the reaction solvent was effective, but the low solubility of the glycosyl acceptors, particularly the trisaccharide, to the reaction solvent decreased the yields. We attempted other conditions for activating the thioglycoside, but it was difficult to activate the glycosyl donor efficiently (data not shown). In this study, we investigated the synthesis method of ODAGals by using the benzyl (Bn) group as a protecting group of the 3-hydroxy group in place of the PMB group and attempted to synthesize longer ODAGals.
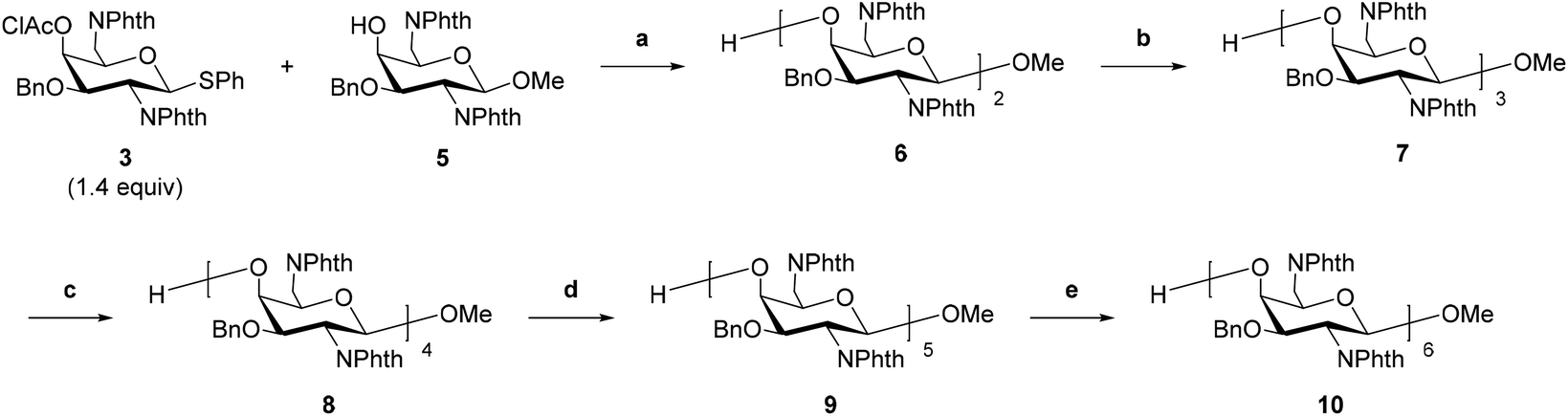
The glycosyl donor 3 was synthesized according to a similar procedure described in our previous report from 1 (Scheme 1).20 Next, to synthesize a glycosyl acceptor, the glycosyl donor 3 was condensed with methanol using NIS and TfOH in CH2Cl2, followed by removal of the chloroacetyl (ClAc) group on the 4-position in pyridine–water to afford 5 (Scheme 2). Then, the 4-hydroxy group of compound 5 was allowed to react with the glycosyl donor 3 (Scheme 3). The glycosylation reaction was conducted using NIS and TfOH in CH2Cl2, and after work-up, the crude product was roughly purified by silica gel column chromatography. Then, the mixture was subject to removing the ClAc group. The disaccharide 6 was isolated in an 82% yield over two steps. These synthetic cycles, including glycosylation and 4-O-deprotection, were repeated, affording the protected 3–6mers (7, 8, 9, and 10). In the previous report, the longer the sugar chain was, the lower the yield of the glycosylation reaction became, and the yields of the trisaccharide and tetrasaccharide were 64% and 10%, respectively.17 On the other hand, in this method, the yield did not decrease with the elongation of the sugar chain, and the yields of the trisaccharide 7 and tetrasaccharide 8 were higher than 90%, and the pentasaccharide 9 was also obtained in a good yield (77% over two steps). The low yield (21%) of the hexasaccharide 10 is attributed to a loss during multiple purifications by silica gel column chromatography and preparative thin-layer chromatography (PTLC). The use of the Bn group as a hydroxy-protecting group enabled glycosylation in CH2Cl2, a good solvent of the glycosyl donors and acceptors, which contributed to the high efficiency of the reactions. Notably, there were no side reactions such as removal of Bn groups or iodination of aromatic rings observed with PMB groups.
 |
| | Scheme 1 Synthesis of the glycosyl donor 3. Reagents and conditions: (a), (i) Bu2SnO (1.3 equiv.), toluene, reflux, 5.5 h; (ii) BnBr (1.3 equiv.), TBAI (1.1 equiv.), toluene, reflux, 17.5 h, 95% over two steps. (b) ClAcCl (2.0 equiv.), CH2Cl2–pyridine (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), 0 °C, 1 h, 89% over three steps. :1, v/v), 0 °C, 1 h, 89% over three steps. | |
 |
| | Scheme 2 Synthesis of the glycosyl acceptor 5. Reagents and conditions: (a) (i) NIS (3.9 equiv.), TfOH, MeOH (2.3 equiv.), CH2Cl2, 0 °C, 30 min; (ii) H2O–pyridine (1:1, v/v), rt, 20 h, 82% over two steps. | |
 |
| | Scheme 3 Sugar chain elongation. Reagents and conditions: (a), (i) NIS (2.5 equiv.), TfOH, CH2Cl2, 0 °C, 30 min; (ii) H2O–pyridine (1:1, v/v), rt, 12 h, 85% over two steps. (b), (i) 3 (1.3 equiv.), NIS (2.5 equiv.), TfOH, CH2Cl2, 0 °C, 40 min; (ii) H2O–pyridine (2:3, v/v), rt, 19 h, 92% over two steps. (c), (i) 3 (1.6 equiv.), NIS (2.5 equiv.), TfOH, CH2Cl2, 0 °C, 60 min; (ii) H2O–pyridine (1:2, v/v), rt, 17 h, 91% over two steps. (d) (i) 3 (1.6 equiv.), NIS (2.6 equiv.), TfOH, CH2Cl2, 0 °C, 45 min; (ii) H2O–pyridine (1:2, v/v), rt, 9 h, 74% over two steps. (e), (i) 3 (1.9 equiv.), NIS (2.9 equiv.), TfOH, CH2Cl2, 0 °C, 45 min; (ii) H2O–pyridine (1:2, v/v), rt, 16 h, 21% over two steps. | |
Then, the conditions for removing the protecting groups were studied. The N-phthaloyl groups at the C-2 and C-6 positions were removed by treatment with hydrazine, and the resulting oligosaccharides were isolated by reversed-phase high-performance liquid chromatography (RP-HPLC) using 0.05% TFA in water and 0.05% TFA in CH3CN as an eluent. After treatment with hydrochloric acid to convert trifluoroacetic acid salts to hydrochloride counterparts, reductive cleavage of the Bn groups was performed using palladium on carbon and hydrogen under acidic and high-pressure conditions (Scheme 4). When we attempted to remove the Bn groups by bubbling hydrogen at room temperature, the reaction was very slow and took 3 days to complete for trisaccharide. Therefore, it was necessary to remove the Bn group at high temperatures and pressures after converting the hydrochloride salts, as shown in Scheme 4. The final products were purified by reprecipitation, and the purities were confirmed by 1H and 13C nuclear magnetic resonance (NMR) analysis. By using the deprotection reactions described above, we successfully obtained the tri-, tetra-, penta-, and hexa-saccharides (ODAGal3, ODAGal4, ODAGal5, and ODAGal6). The total yields from the compound 3 to ODAGals were 44% (ODAGal3), 26% (ODAGal4), 6% (ODAGal5), and 0.5% (ODAGal6), respectively. In the previous study, these yields were 18% (ODAGal3), and 2% (ODAGal4),17 suggesting that the present strategy enables a more efficient synthesis of ODAGals. This method would accelerate access to not only ODAGals but also their analogs.21
 |
| | Scheme 4 Synthesis of ODAGals. Reagents and conditions: (a), (i) NH2NH2·H2O, EtOH, 70 °C, 14–23 h. (ii) HClaq, 56% (12, n = 4), 48% (13, n = 5), and 20% (14, n = 6) over two steps. (b), (i) 10% Pd/C, H2, 0.05 M HCl, H2O–MeOH (1:1, v/v), 60 °C, 400–950 kPa, 8–10 h, 68% from 7 (15, n = 3), 78% from 12 (16, n = 4), 30% from 13 (17, n = 5), and 26% from 14 (18, n = 6). | |
Thermal denaturation study
First, we conducted thermal denaturation tests to investigate the effect of ODAGal4, ODAGal5, and ODAGal6 on the thermal stability of various double-stranded nucleic acids. All measurements were conducted under physiological conditions using a 10 mM phosphate buffer containing 100 mM NaCl at pH 7.0. The UV melting curves of the duplexes were measured using 5 μM of 12mers and 16mers or 2.5 μM of 20mers with and without 1 or 2 equivalents of ODAGal4, ODAGal5, and ODAGal6. Analytical samples were prepared by annealing the oligonucleotides, followed by adding an aqueous solution of ODAGals. The melting temperature (Tm) value was determined from the peak value of the first derivative of the thermal melting curve. In all cases, Tm values were analyzed from the UV melting curves at 260 nm. Fig. 2 and 3 and ESI† show the representative UV melting curves, and Tables 1–4 show Tm values for the various duplexes.
 |
| | Fig. 2 (A) UV melting curves of AP16RR in the absence and presence of ODAGals. (B) UV absorbance value at 320 nm. | |
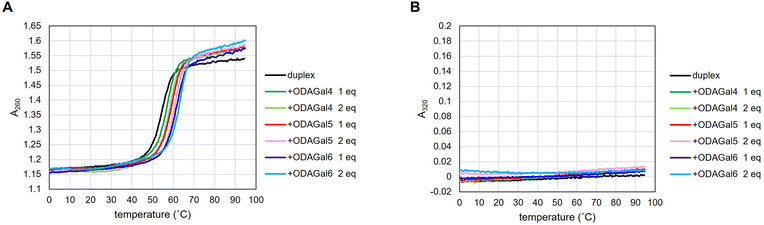 |
| | Fig. 3 (A) UV melting curves of AP16DR in the absence and presence of ODAGals. (B) UV absorbance value at 320 nm. | |
Table 1
T
m values of RNA/RNA duplexes
| Entry |
Sequence |
|
T
m/°C |
ΔTm/°C |
| ODAGal4 |
ODAGal5 |
ODAGal6 |
| 1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
Absorbance at 320 nm was detected.
Deformation of melting curve was observed; —: cannot be calculated because of the deformation of melting curve.
|
| 1 |
CAGURR |
12mer |
61.0 |
3.4 |
4.1 |
3.7 |
5.6 |
7.0a,b |
—a |
| 2 |
AP12RR |
|
53.7 |
5.2 |
6.9 |
7.7 |
7.5 |
9.1 |
—a |
| 3 |
AP16RR |
16mer |
64.9 |
4.9 |
5.1 |
6.7 |
8.2 |
9.4 |
11.0a,b |
| 4 |
ML16RR |
|
64.5 |
3.8 |
6.3 |
7.9 |
8.9 |
9.8 |
12.8a,b |
| 5 |
AP20RR |
20mer |
69.9 |
2.2 |
3.8 |
4.5 |
6.3 |
6.7 |
9.1 |
| 6 |
MP20RR |
|
83.0 |
0.0 |
0.0 |
0.0 |
0.0 |
−0.3 |
0.1a |
Table 2
T
m values of DNA/RNA duplexes
| Entry |
Sequence |
|
T
m/°C |
ΔTm/°C |
| ODAGal4 |
ODAGal5 |
ODAGal6 |
| 1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
Absorbance at 320 nm was detected.
Deformation of melting curve was observed; —: cannot be calculated because of the deformation of melting curve.
|
| 1 |
CAGTDR |
12mer |
48.6 |
1.2 |
1.6 |
3.0 |
4.4 |
4.6a,b |
—a |
| 2 |
AP12DR |
|
44.7 |
2.5 |
2.5 |
3.0 |
5.2 |
5.7a,b |
—a |
| 3 |
AP16DR |
16mer |
54.4 |
2.5 |
3.8 |
5.8 |
6.9 |
6.7 |
9.9 |
| 4 |
ML16DR |
|
55.0 |
0.4 |
2.2 |
3.2 |
2.9 |
4.2 |
7.2a,b |
| 5 |
AP20DR |
20mer |
59.1 |
1.5 |
2.4 |
3.7 |
3.8 |
5.2 |
6.4a,b |
| 6 |
MP20DR |
|
74.2 |
−0.4 |
−0.6 |
0.2 |
1.8 |
2.3 |
3.5 |
Table 3
T
m values of DNA/DNA duplexes
| Entry |
Sequence |
|
T
m/°C |
ΔTm/°C |
| ODAGal4 |
ODAGal5 |
ODAGal6 |
| 1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
Absorbance at 320 nm was detected.
Deformation of melting curve was observed; —: cannot be calculated because of the deformation of melting curve.
|
| 1 |
AP16DD |
16mer |
57.5 |
−1.0 |
0.2 |
0.5 |
−0.2a,b |
−0.1a,b |
0.7a,b |
| 2 |
MP20DD |
20mer |
70.4 |
−0.1 |
−0.3 |
−0.1 |
0.9 |
—a |
—a |
Table 4
T
m values of gapmer/RNA and 2′-OMe RNA/RMA duplexes
| Entry |
Sequence |
|
T
m/°C |
ΔTm/°C |
| ODAGal4 |
ODAGal5 |
ODAGal6 |
| 1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
1 equiv. |
2 equiv. |
Absorbance at 320 nm was detected.
Deformation of melting curve was observed; —: cannot be calculated because of the deformation of melting curve.
|
| 1 |
AP12G2 |
12mer |
46.5 |
3.2 |
5.2 |
4.6 |
5.6 |
7.0a,b |
—a |
| 2 |
AP12G4 |
|
49.7 |
4.5 |
5.7 |
6.0 |
7.4 |
8.2a |
—a |
| 3 |
AP12MR |
|
61.2 |
3.7 |
4.0 |
5.2 |
6.5 |
—a |
—a |
| 4 |
AP16G2 |
16mer |
56.2 |
2.8 |
2.7 |
2.7 |
5.2 |
6.1a |
—a |
| 5 |
ML16G2 |
|
55.9 |
2.4 |
3.9 |
3.4 |
5.3 |
5.9a |
—a |
| 6 |
AP16G4 |
|
60.7 |
2.7 |
4.0 |
3.3 |
5.6 |
6.1a,b |
—a |
| 7 |
ML16G4 |
|
61.7 |
1.9 |
3.3 |
2.6 |
4.2 |
5.2a |
—a |
| 8 |
AP16MR |
|
70.8 |
4.0 |
4.6 |
6.0 |
5.6 |
6.5a |
—a |
| 9 |
ML16MR |
|
71.4 |
2.1 |
3.2 |
3.0 |
3.6 |
5.9 |
—a |
We initially conducted thermal denaturation tests with RNA/RNA duplexes, which form A-type duplexes. Sequences of RNA/RNA duplexes were a repeated structure (rCAGUCAGUCAGU), an antisense sequence of ApoB mRNA and Malat1 non-coding RNA, and that of Mipomersen22 and their complementary RNAs (Tables 1–4). The results are shown in Table 1 and Fig. 2A. The addition of ODAGals increased the Tm values of most RNA/RNA duplexes, except for the sequence MP20RR. The inability of ODAGals to stabilize MP20RR can be attributed to the high Tm value of MP20RR (83.0 °C). Besides MP20RR, 1 equivalent of ODAGal4 increased Tm values of the duplexes by more than 2 °C, and the extent of the increase was greater with 2 equivalents of ODAGal4. ODAGal5 exhibited a more prominent ability to stabilize the duplexes than ODAGal4. Furthermore, it was confirmed that 1 equivalent of ODAGal6 is more effective in stabilizing the duplexes than 2 equivalents of ODAGal5. Therefore, the order of the ability for the stabilization of RNA/RNA duplexes would be ODAGal6 > ODAGal5 > ODAGal4.
Next, the effect of ODAGals on the thermal stability of DNA/RNA duplexes, which form A-type duplexes, was studied (Table 2, Fig. 3A, and Fig. S2†). A similar tendency was observed with the results of RNA/RNA duplexes. Meanwhile, ΔTm values were less significant with DNA/RNA duplexes. However, the Tm values of DNA/RNA duplexes were lower than those of RNA/RNA duplexes. These facts indicate that ODAGals stabilize RNA/RNA duplexes more than DNA/RNA duplexes.
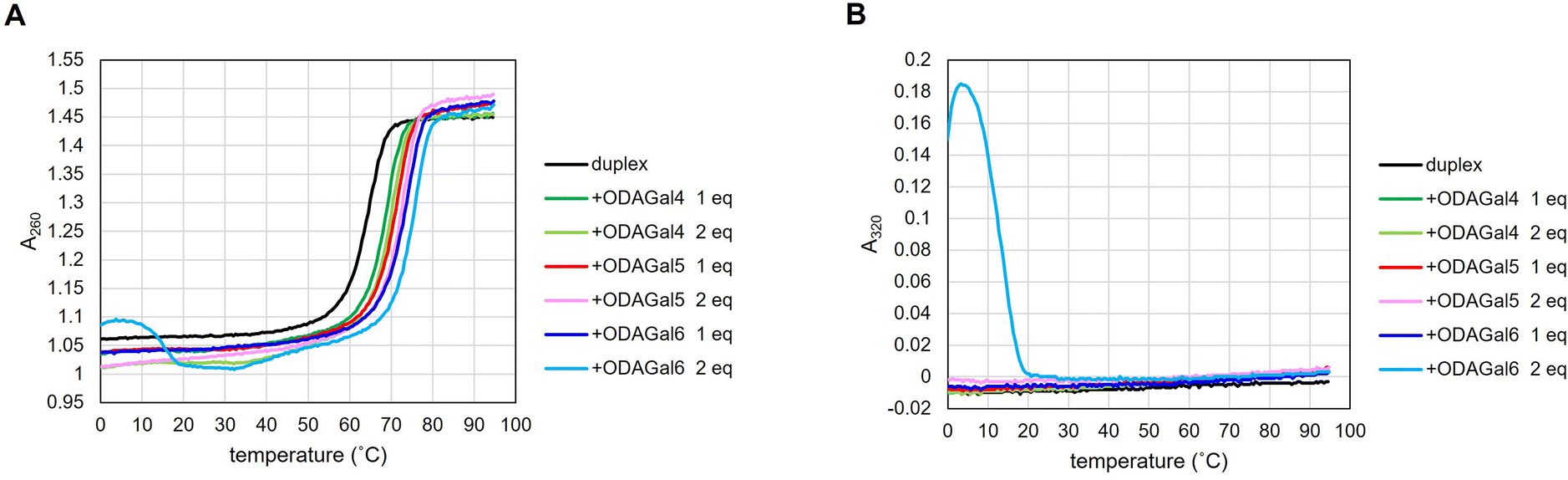
On the other hand, although the presence of ODAGal6 significantly increased the Tm values of RNA/RNA and DNA/RNA duplexes, 2 equivalents of ODAGal6 deformed the melting curve in a particular instance. Moreover, in some cases, the addition of 2 equivalents of ODAGal6 to the solution of an oligonucleotide duplex resulted in turbidity and/or precipitation. These phenomena suggest that nucleic acid duplexes and ODAGal6 can form an aggregate. This may be due to an unintended intermolecular binding of multiple oligonucleotide chains with ODAGal6 through cation–anion interactions. Note that DNA aggregation in the presence of multivalent cations has been reported.23 Typically, oligonucleotides exhibit negligible UV absorbance at 300 nm or longer wavelengths, and a study has used absorbance at 320 nm as a barometer of nucleosome aggregation.24 For these reasons, it was decided to measure absorbance at 320 nm to evaluate the formation of an aggregate in the thermal denaturation experiments. As a result, in a particular instance, absorption at 320 nm was detected in the presence of 2 equivalents of ODAGal6, and as the temperature increased, the absorbance decreased (Fig. 2B and 3B and ESI†). These results indicate that excessive equivalents of ODAGal6 are prone to cause aggregation of the duplexes.
We also conducted thermal denaturation tests with DNA/DNA duplexes, which form B-type duplexes (Table 3 and Fig. S3†). There were no significant changes in the Tm values of DNA/DNA duplexes upon the addition of ODAGals, suggesting that ODAGals preferentially stabilize A-type duplexes. These results agree with previous reports.17 On the other hand, significant absorbance at 320 nm was observed in the presence of ODAGal6 (see ESI†). These results indicate that ODAGals distinctly interact with DNA/DNA duplexes from RNA/RNA and DNA/RNA duplexes.
In addition, we investigated the stabilization effect of ODAGals on 2′-OMe-modified oligonucleotide duplexes. Duplexes of an RNA strand and a gapmer-type strand with a DNA gap and 2′-OMe modifications in wing regions or a fully 2′-OMe-modified strand were used for thermal denaturation tests. The results are presented in Table 4. In the absence of ODAGals, there was a clear correlation between Tm values and the content of 2′-OMe modifications, which agrees well with a previous report.25 Upon the addition of ODAGal4 and ODAGal5, Tm values increased and ODAGal5 had a more potent ability to stabilize 2′-OMe-modified duplexes as well as RNA/RNA and DNA/RNA duplexes. In the presence of ODAGal6, the thermal stability of the duplexes was significantly improved, but the samples exhibited absorbance at 320 nm, and 2 equivalents of ODAGal6 deformed the melting curves of the duplexes, as in the case of RNA/RNA and DNA/RNA duplexes. These results indicate that 2′-OMe-modified duplexes and ODAGal6 also form an aggregate. With the duplexes tested in this study, no aggregation phenomenon was observed in the presence of up to 2 equivalents of ODAGal4 and ODAGal5. Combining these results, it was suggested that ODAGals effectively stabilized RNA/RNA, DNA/RNA, and 2′-OMe-modified duplexes and the order of magnitude of the stabilization effect was ODAGal6 > ODAGal5 > ODAGal4 even though ODAGal6 tended to result in the formation of an aggregate. Although the existence of an aggregate should be carefully monitored, ODAGals improved the thermal stability of A-type oligonucleotide duplexes, and this effect was notable with longer ODAGals.
UV spectroscopy
In the above thermal denaturation study, absorption at 320 nm was observed with some combinations of nucleic acid duplexes and ODAGals. Therefore, UV spectra of 16mer DNA/RNA, RNA/RNA, and 2′-OMe-modified duplexes at 200–400 nm were measured in the presence and absence of ODAGals. A 10 mM phosphate buffer containing 100 mM NaCl was used as the medium. The samples were prepared in the following order: a solution of pairs of complementary strands was annealed, and then, an aqueous solution of ODAGal was added to the mixture. The spectra are shown in Fig. 4 and Fig. S5–S7.† Almost no changes were observed upon the addition of 2 equivalents of ODAGal4 or ODAGal5. On the other hand, an increase in absorbance around 300 nm was detected in the presence of 2 equivalents ODAGal6. As the temperature increased, absorbance near 300 nm decreased (Fig. S6 and S7†). These results indicate that ODAGal6 is prone to cause aggregation of the duplexes, which agrees well with the results from thermal denaturation experiments. Therefore, the UV spectrum, particularly absorbance at 320 nm may be effective for observing structural changes in oligonucleotide duplexes caused by the formation of an aggregate.
 |
| | Fig. 4 (A) UV spectra of 5 μM DNA/RNA (AP16DR) in phosphate buffer at room temperature (24 ± 1 °C). (B) UV spectra of 5 μM RNA/RNA (AP16RR) in phosphate buffer at room temperature (24 ± 1 °C). | |
Circular dichroism spectroscopy
The circular dichroism (CD) spectra of 16mer DNA/RNA, RNA/RNA, and 2′-OMe-modified duplexes were measured to analyze their structures in the presence and absence of ODAGals at 37 °C. The samples were prepared in the same way as in the UV spectrum measurement. The spectra are shown in Fig. 5 and Fig. S8–S9.† In the absence of ODAGals, the spectra of both DNA/RNA and RNA/RNA duplexes exhibited a positive peak around 265 nm and a negative peak around 210 nm, indicating that these duplexes formed A-type duplex structures. On the other hand, an increase in the intensities of the positive peak was observed in the presence of 1 equivalent of ODAGal4, ODAGal5, or ODAGal6 (Fig. 5). However, in the presence of 2 equivalents of ODAGal6, a decrease in the intensity of the positive peak around 265 nm was occasionally observed (Fig. S9,† RNA/RNA, light blue line), but a decrease was not always observed when the aggregation was observed by UV measurements. The changes in the spectra caused by the addition of ODAGals were marginal, suggesting that ODAGals interact with DNA/RNA and RNA/RNA duplexes without alteration of their A-type duplex structures. A similar tendency was found concerning 2′-OMe RNA/RNA and 2′-OMe-modified gapmer/RNA duplexes (Fig. S8†).
 |
| | Fig. 5 (A) CD spectra of DNA/RNA (AP16DR) in the absence or presence of 1 equivalent of ODAGals at 37 °C. (B) CD spectra of RNA/RNA (AP16RR) in the absence or presence of 1 equivalent of ODAGals at 37 °C. | |
In addition to this, the CD spectra at 10 °C and 50 °C were recorded. Interestingly, at 10 °C, a notable increase in the negative Cotton effect around 210 nm was observed in the presence of all ODAGals with DNA/RNA or RNA/RNA duplexes (Fig. S10 and S11†). The extent of the change was more significant at 10 °C than at 37 °C. These results suggested that the addition of ODAGals causes a structural change in the nucleic acid duplexes at low temperatures, but the change was less noticeable at 37 °C.
RNase A resistance experiment
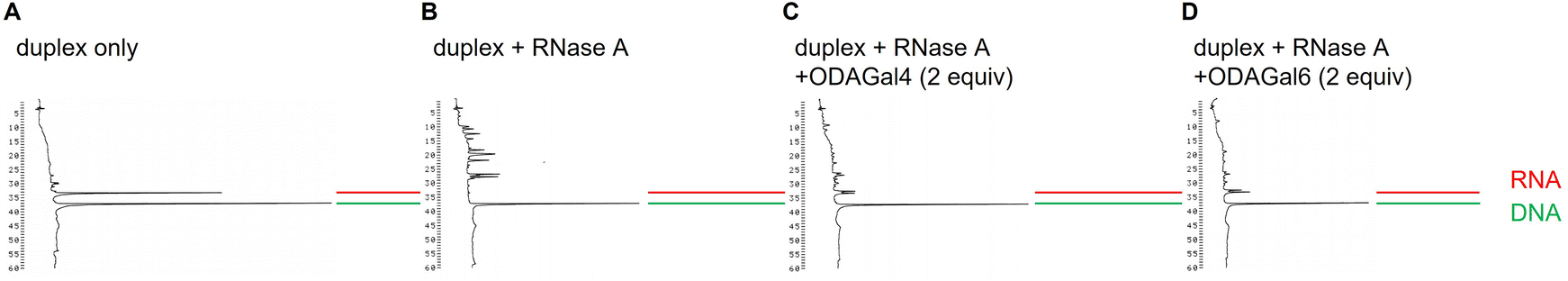
Next, we investigated the effect of ODAGal4 and ODAGal6 on the resistance of the duplex against RNase A digestion. The 16mer DNA/RNA duplex sequence AP16DR and RNase A from the bovine pancreas were used for the experiments. Solutions of 1 μM duplex and 0 or 2 equivalents of ODAGal in a 10 mM Tris buffer containing 100 mM NaCl with pH 7.1 were treated with RNase A at 30 °C. After the reaction, the solution of RNase inhibitor was added to deactivate RNase A. Then, the mixtures were analyzed by RP-HPLC. Under these conditions, no aggregate was observed before and after the reaction.
The HPLC profiles are shown in Fig. 6 and 7, and ESI.†Fig. 6 and 7 show the results of 3 and 24 h after the treatment with 0.5 μg mL−1 RNase A, respectively. It has been reported that the degradation pattern of a siRNA after treatment with 0.5 μg mL−1 RNase A is similar to the degradation pattern in serum.26Fig. 6 indicates that the RNA 16mer is completely digested after the treatment of RNase A for 3 h in the absence of ODAGals (Fig. 6B), whereas intact RNAs are observed in the presence of ODAGal4 and ODAGal6 (Fig. 6C and D). On the other hand, after 24 h, intact RNA was not detected, and numerous peaks with short retention times, probably derived from degraded RNAs, were observed in the presence of 2 equivalents of ODAGal4 (Fig. 7C), whereas the intensities of these peaks were small in the presence of ODAGal6 (Fig. 7D). These results suggested that the cleavage of the RNA was inhibited in the presence of 2 equivalents of ODAGal4 and ODAGal6, and the latter has a more prominent ability to suppress the degradation. Furthermore, the reactions were conducted under harsher conditions (10 μg mL−1 RNase A, Fig. 8). The RNA was digested within 5 min in the absence of ODAGal (Fig. 8B). In the presence of 2 equivalents of ODAGal4 and ODAGal6, the RNA was partially degraded, but it seems to withstand decomposition in a few minutes (Fig. 8C and D).
 |
| | Fig. 6 RP-HPLC profiles of DNA/cRNA before (A) and after (B–D) the treatment with RNase A (0.5 μg mL−1) for 3 h at 30 °C in the absence or presence of ODAGal4 or ODAGal6; (A) RNase A (−), ODAGal (−); (B) RNase A (+), ODAGal (−); (C) RNase A (+), ODAGal4 (+); (D) RNase A (+), ODAGal6 (+). RP-HPLC analyses (UV detection at 260 nm) were performed using a linear gradient of 0%–20% CH3CN in 0.1 M triethyl ammonium acetate (TEAA) buffer (pH 7.0) at 50 °C for 60 min with a flow rate of 0.5 mL min−1 using a C18 column. | |
 |
| | Fig. 7 RP-HPLC profiles of DNA/cRNA before (A) and after (B–D) the treatment with RNase A (0.5 μg mL−1) for 24 h at 30 °C in the absence or presence of ODAGal4 or ODAGal6; (A) RNase A (−), ODAGal (−); (B) RNase A (+), ODAGal (−); (C) RNase A (+), ODAGal4 (+); (D) RNase A (+), ODAGal6 (+). RP-HPLC analyses are performed under the same conditions as in Fig. 6. | |
 |
| | Fig. 8 RP-HPLC profiles of DNA/cRNA before (A) and after (B–D) the treatment with RNase A (10 μg mL−1) for 5 min at 30 °C in the absence or presence of ODAGal4 or ODAGal6; (A) RNase A (−), ODAGal (−); (B) RNase A (+), ODAGal (−); (C) RNase A (+), ODAGal4 (+); (D) RNase A (+), ODAGal6 (+). RP-HPLC analyses are performed under the same conditions as in Fig. 6. | |
These results indicated that ODAGals not only improved the thermal stability of A-type duplexes but also protected DNA/RNA duplexes against the cleavage by RNase A, and ODAGal6 seems to be more potent than ODAGal4.
RNase H1 experiment
Finally, the RNase H1 activity in the absence and presence of the ODAGals was evaluated using a DNA/RNA duplex. The 16mer DNA/RNA duplex sequence ML16DR and human RNase H1 were used for the experiments. Solutions of 4 μM duplex and 1 equivalent of ODAGal in a 60 mM Tris buffer containing 60 mM KCl, 2.5 mM MgCl2, and 2.0 mM DTT with pH 7.5 were treated with RNase H1 for 30 min at 37 °C.27 After the reaction, the mixtures were analyzed by RP-HPLC. The results are shown in Fig. 9. In the absence of ODAGals, the RNA strand was completely digested after the treatment of RNase H1 (Fig. 9B). In the presence of ODAGal, although peaks corresponding to cleaved fragments were detected, the peak corresponding to the intact RNA strand was observed, suggesting that RNA cleavage by RNaseH1 was partially suppressed (Fig. 9C–E). The amount of the intact RNA was increased in the presence of ODAGal5 or ODAGal6 than in the presence of ODAGal4, indicating that the extent of RNase H1 activity suppression by former two was more pronounce than ODAGal4. However, it was found that ODAGals did not completely inhibit the RNase H1 activity. On the other hand, there was no significant difference in cleavage sites in the presence or absence of ODAGals. Nowotny et al. have reported the crystal structure of complexes of a human RNase H1 and RNA/DNA duplexes, and it was found that RNase H1 bound to the minor groove of the duplex.28 In contrast, NOE correlation analysis revealed that ODAGal4 was located in the major groove of A-type oligonucleotide duplexes.17 The difference in binding sites might be contributed to partial, not complete, inhibition of RNA cleavage by OGAGals.
 |
| | Fig. 9 RP-HPLC profiles of DNA/cRNA before (A) and after (B–E) the treatment with RNase H1 for 30 min at 37 °C in the absence or presence of 1 equivalent of ODAGal4, ODAGal5, or ODAGal6; (A) RNase H1 (−), ODAGal (−); (B) RNase H1 (+), ODAGal (−); (C) RNase H1 (+), ODAGal4 (+); (D) RNase H1 (+), ODAGal5 (+); (E) RNase H (+), ODAGal6 (+). RP-HPLC analyses (UV detection at 260 nm) were performed using a linear gradient of 0%–12% CH3CN in 0.1 M TEAA buffer (pH 7.0) at 50 °C for 60 min with a flow rate of 0.5 mL min−1 using a C18 column. | |
Conclusion
In this study, we have reported an improved synthesis method and properties of ODAGals that bind to A-type oligonucleotide duplexes. The use of the Bn-protecting group for the 4-hydroxy group of diaminogalactose derivatives enabled the successful synthesis of pentasaccharide (ODAGal5) and hexasaccharide (ODAGal6) derivatives. From the thermal denaturation studies, UV and CD spectroscopy, and RNase A resistance experiments, ODAGal4–6 stabilizes the A-type oligonucleotide duplexes thermally and biologically, typically without changes in their structure, and the effect was notable with longer ODAGals. On the other hand, the addition of ODAGal6 resulted in the formation of aggregates to some extent. These results would contribute to the rational design of ODAGal derivatives as modulators of the properties of oligonucleotide therapeutics such as siRNAs and HDOs.
General methods and materials
All reactions were conducted under an argon atmosphere unless otherwise stated. 1H NMR spectra were obtained using tetramethylsilane as an internal standard (δ 0.00) in CDCl3 and with acetonitrile as an internal or external standard (δ 2.06) in D2O. 13C NMR spectra were obtained using CDCl3 as an internal standard (δ 77.0) in CDCl3 and acetonitrile as an internal or external standard (δ 1.5) in D2O. Silica gel column chromatography was performed using silica gel 60 N (63–210 μm). PTLC was performed using PLC silica gel 60F254, 1 mm. RP-HPLC for purification was performed using a SunFire™ Prop. column (5 μm, C18, 100 Å, 19 mm × 150 mm, Waters). The organic solvents were purified and dried through an appropriate procedure. The yields of the compounds were calculated from their dry weights, except for 15, 16, 17, and 18, which were estimated from the integral values in the NMR spectra compared with that of the internal acetonitrile (internal standard).
DNA and RNA oligomers were purchased from Japan Bio Services Co., Ltd A PROTEOSAVE™SS 0.5 mL Microtube (SumitomoBakelite Co., Ltd) was used as the reaction container for the RNase A and RNase H1 assay. RP-HPLC was performed using μBondasphere or Delta-Pak column (5 μm, C18, 100 Å, 3.9 × 150 mm, Waters), and detection at 260 nm.
Thermal denaturation study
Thermal denaturation tests were conducted using a Shimadzu UV-1900i spectrometer (Shimadzu, Kyoto, Japan) equipped with a Tm analysis accessory and an eight-sample cell changer in quartz cells with a path length of 1 cm. The experiments were conducted in a 10 mM NaH2PO4-Na2HPO4 buffer, pH 7.0, containing 100 mM NaCl. A solution containing a pair of complementary strands was heated for 5 min at 95 °C and cooled to 4 °C at a rate of 0.5 °C min−1. 1 or 2 equivalents of ODAGal were added to the solution, and the final concentration of the duplex was 2.5 (for 20mer duplexes) or 5 μM (for 12mer and 16mer duplexes). After ODAGal was added to the solution, these samples were additionally cooled to 0 °C and left for 10 min. Finally, absorbance at 260 and 320 nm was recorded in the forward direction from 0 °C to 95 °C at a rate of 0.5 °C min−1. Tm values were calculated from the peak value of the first derivative of the thermal melting curve.
UV spectroscopy
UV spectrum measurements were conducted using a JASCO V-550 UV/VIS spectrophotometer (JASCO Corporation, Tokyo, Japan) and Shimadzu UV-1900i spectrometer (Shimadzu, Kyoto, Japan). The experiments were conducted in a 10 mM NaH2PO4–Na2HPO4 buffer, pH 7.0, containing 100 mM NaCl. A solution containing a pair of complementary strands was heated for 5 min at 95 °C and cooled to 4 °C at a rate of 0.5 °C min−1. 1 or 2 equivalents of ODAGal were added to the solution, and the final concentration of the duplex was 4 or 5 μM. All UV spectra were recorded in a wavelength range of 200–400 nm with a resolution of 0.5 nm.
Circular dichroism spectroscopy
CD spectroscopy was conducted using a JASCOJ-820 spectrometer (JASCO Corporation, Tokyo, Japan). The experiments were conducted in a 10 mM NaH2PO4–Na2HPO4 buffer, pH 7.0, containing 100 mM NaCl. A solution containing a pair of complementary strands was heated for 5 min at 95 °C and cooled to 4 °C at a rate of 0.5 °C min−1. 1 equivalent of ODAGal was added to the solution, and the final concentration of the duplex was 5 μM. All CD spectra were recorded in a wavelength range of 200–320 nm at 37 °C using 250 μL of the solution. The following instrument settings were used: resolution, 0.1, 0.5, or 1 nm; sensitivity, 5 mdeg; response, 1 s; speed, 100 nm min−1; accumulation, 10.
RNase A resistance experiment
The experiments were conducted in a 10 mM Tris buffer containing 100 mM NaCl, pH 7.1 at 30 °C. A solution containing a pair of complementary strands was heated for 5 min at 95 °C and cooled to 4 °C at a rate of 0.5 °C min−1. 1 or 2 equivalents of ODAGal were added to the solution, and the final concentration of the duplex was 1 μM. A solution of RNase A from the bovine pancreas in Tris buffer was added to the solution of the duplex with or without ODAGal to yield a final concentration of 0.50 μg mL−1 or 10 μg mL−1. After treatment with RNase A, the reaction was quenched by adding an RNase A inhibitor and then rapidly cooled to 4 °C. The RNase A inhibitor was purchased from TOYOBO Co., Ltd. The mixture was analyzed by RP-HPLC.
RNase H1 experiment
The experiments were performed in a 60 mM Tris buffer containing 60 mM KCl, 2.5 mM MgCl2, and 2.0 mM DTT pH 7.5 at 37 °C. A solution containing a pair of complementary strands was heated for 5 min at 95 °C and cooled to 4 °C at a rate of 0.5 °C min−1. 1 equivalent of ODAGal was added to the solution and the final concentration of the duplex was 4 μM. To the solution of the duplex with or without ODAGal, 0.02 equivalent of human RNase H1 (abcam, ab153634) was added. After 30 min, the mixture was rapidly heated to 95 °C, left for 5 min, and then rapidly cooled to 4 °C to denature RNase H1. The mixture was analyzed by RP-HPLC.
Phenyl 3-O-benzyl-2,6-dideoxy-2,6-diphthalimimdo-1-thio-β-D-galactopyranoside (2)
Compound 1 (8.69 g, 16.3 mmol) was coevaporated with toluene and dissolved in dry toluene (280 mL). Dibutyltin oxide (5.26 g, 21.1 mmol) was added to the solution, and it was refluxed for 5.5 h. Tetrabutylammonium iodide (6.67 g, 18.0 mmol) and benzyl bromide (2.4 mL, 20.9 mmol) were added. After 17.5 h, the mixture was cooled to room temperature and diluted with toluene (300 mL). The solution was washed with water (150 mL), a saturated NaHCO3 aqueous solution (150 mL), and then water (150 mL). The aqueous layers were collected, filtrated, and extracted with toluene (3 × 100 mL). The organic layers were combined, dried over MgSO4, filtrated, and concentrated to dryness. The crude product was purified by silica gel column chromatography using CH2Cl2–MeOH (100:0–100:1.5, v/v) as an eluent, and 2 was obtained as a colorless foam (9.66 g, 15.6 mmol, 95%). 1H NMR (CDCl3, 400 MHz) δ 7.88–7.85 (m, 3H), 7.77–7.65 (m, 5H), 7.24–7.21 (m, 2H), 7.04–6.93 (m, 6H), 6.88–6.84 (m, 2H), 5.36 (d, J = 10.5 Hz, 1H), 4.62–4.55 (m, 2H), 4.39 (dd, J = 14.2, 8.9 Hz, 1H), 4.32 (d, J = 12.1 Hz, 1H), 4.27 (dd, J = 10.5, 3.4 Hz, 1H), 4.09 (s, 1H), 3.98 (dd, J = 9.0, 4.2 Hz, 1H), 3.80 (dd, J = 14.2, 4.3 Hz, 1H), 2.72–2.70 (m, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 168.1, 168.0, 136.9, 134.0, 133.8, 132.9, 132.0, 131.8, 131.6, 128.4, 128.3, 127.9, 127.9, 127.6, 123.6, 123.5, 123.3, 83.7, 75.4, 75.0, 71.4, 65.9, 50.7, 38.8. HRMS (ESI/Q-TOF) m/z: calcd for C35H32N3O7S+ [M + NH4]+, 638.1955; found 638.1947.
Phenyl 3-O-benzyl-4-O-chloroacetyl-2,6-dideoxy-2,6-diphthalimido-1-thio-β-D-galactopyranoside (3)
Compound 2 (7.82 g, 12.6 mmol) was dissolved in a mixed solvent of dry pyridine–dry CH2Cl2 (1:20, v/v, 126 mL) and cooled to 0 °C. Chloroacetyl chloride (2.0 mL, 25.2 mmol) was added to the solution, and the mixture was allowed to stir for 1 h at 0 °C. The solution was diluted with toluene (50 mL) and concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (100 mL). The solution was washed with saturated NaHCO3 aqueous solutions (3 × 50 mL), and the combined aqueous layers were extracted with CH2Cl2 (2 × 100 mL). The organic layers were dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was purified by silica gel column chromatography using ethyl acetate–hexane (1:2, v/v) as an eluent, affording 3 as a yellow foam (7.82 g, 11.2 mmol, 89%). 1H NMR (CDCl3, 400 MHz) δ 7.87–7.84 (m, 3H), 7.77–7.68 (m, 4H), 7.63 (dd, J = 6.2, 1.4 Hz, 1H), 7.29–7.27 (m, 2H), 7.07–7.03 (m, 1H), 7.01–6.95 (m, 3H), 6.93–6.86 (m, 4H), 5.58 (d, J = 2.7 Hz, 1H), 5.47 (d, J = 10.5 Hz, 1H), 4.54–4.45 (m, 2H), 4.40–4.25 (m, 4H), 4.18 (d, J = 12.3 Hz, 1H), 4.02 (dd, J = 14.0, 8.2 Hz, 1H), 3.85 (dd, J = 14.0, 5.7 Hz, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 167.8, 167.7, 167.3, 167.1, 136.7, 134.1, 134.0, 133.8, 133.0, 131.7, 131.5, 131.4, 128.5, 128.1, 127.8, 127.7, 123.5, 123.3, 83.9, 73.4, 73.3, 71.3, 67.8, 51.1, 41.0, 38.0. HRMS (ESI/Q-TOF) m/z: calcd for C37H3335ClN3O8S+ [M + NH4]+, 714.1672; found 714.1671.
Methyl 3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranoside (5)
The glycosyl donor 3 (0.381 g, 0.55 mmol) was coevaporated with toluene (6 × 2 mL) and dissolved in dry CH2Cl2 (5.5 mL). Dry MeOH (50 μL, 1.23 mmol) and NIS (0.476 g, 2.12 mmol) were added to the solution, which was then cooled to 0 °C. Trifluoromethanesulfonic acid (TfOH, 100 μL) was subsequently added, and the solution was stirred for 30 min at 0 °C. The reaction was quenched by adding a saturated NaHCO3 aqueous solution (5 mL), and the mixture was diluted with CHCl3 (40 mL). The solution was washed with saturated NaHCO3 aqueous solutions (3 × 5 mL), and the combined aqueous layers were extracted with CHCl3 (3 × 5 mL). The organic layers were combined and washed with 10% Na2S2O3 aqueous solutions (3 × 5 mL), and the organic layers were extracted with CHCl3 (3 × 5 mL). The organic layers were combined, dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was dissolved in H2O–pyridine (1:1, v/v, 60 mL), and the resultant solution was stirred at room temperature. After 20 h, the solution was concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography using ethyl acetate–hexane (2:3, v/v) as an eluent to afford 5 as a colorless foam (0.241 g, 0.445 mmol, 82%, 2 steps). 1H NMR (CDCl3, 400 MHz) δ 7.91–7.70 (m, 8H), 7.04–7.02 (m, 2H), 7.00–6.93 (m, 3H), 4.98 (d, J = 8.7 Hz, 1H), 4.62 (d, J = 12.3 Hz, 1H), 4.54–4.50 (m, 1H), 4.35–4.25 (m, 3H), 4.06–3.95 (m, 3H), 3.27 (s, 3H), 3.04 (s, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 168.1, 167.6, 137.1, 134.1, 133.8, 133.6, 131.7, 131.6, 128.1, 127.7, 127.6, 123.3, 123.0, 98.8, 74.8, 71.5, 71.3, 65.5, 55.9, 51.8, 38.3. HRMS (ESI/Q-TOF) m/z: calcd for C30H30N3O8+ [M + NH4]+, 560.2027; found 560.2026.
Methyl (3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranoside (6)
The glycosyl donor 3 (0.293 g, 0.42 mmol) and the acceptor 5 (0.165 g, 0.30 mmol) were coevaporated with dry toluene (6 × 2 mL) and then dissolved in dry CH2Cl2 (6.0 mL). NIS (0.172 g, 0.76 mmol) was added to the solution, and the resultant mixture was cooled to 0 °C. A solution of 1 M TfOH in dry CH2Cl2 (0.3 mL) was subsequently added to the solution and stirred for 30 min at 0 °C. The reaction was quenched by adding a saturated NaHCO3 aqueous solution (4 mL), and the mixture was diluted with CHCl3 (30 mL). The solution was washed with saturated NaHCO3 aqueous solutions (3 × 4 mL), and the combined aqueous layers were extracted with CHCl3 (3 × 4 mL). The organic layers were combined and washed with 10% Na2S2O3 aqueous solutions (3 × 4 mL). The combined aqueous layers were extracted with CHCl3 (3 × 4 mL). The organic layers were combined, dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was roughly purified by silica gel column chromatography using ethyl acetate–hexane (1:1, v/v) as an eluent to afford the glycosylation product as a yellow foam (0.331 g, crude). The product was dissolved in H2O–pyridine (1:2, v/v, 30 mL), and the resultant solution was stirred at room temperature. After 12 h, the solution was concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography using ethyl acetate–hexane (1:1, v/v) as an eluent to afford 6 as a colorless foam (0.273 g, 0.26 mmol, 85%, 2 steps). 1H NMR (CDCl3, 400 MHz) δ 8.11 (d, J = 7.5 Hz, 1H), 7.92–7.50 (m, 13H), 7.37 (d, J = 7.3 Hz, 2H), 7.09–7.07 (m, 2H), 7.02–6.89 (m, 3H), 6.80 (t, J = 7.3 Hz, 1H), 6.67 (t, J = 7.5 Hz, 2H), 6.50 (d, J = 7.3 Hz, 2H), 5.27 (d, J = 8.5 Hz, 1H), 4.74–4.65 (m, 3H), 4.49 (dd, J = 10.9, 3.3 Hz, 1H), 4.41–4.22 (m, 4H), 4.17 (s, 1H), 4.09–4.04 (m, 2H), 3.93 (dd, J = 11.1, 2.6 Hz, 1H), 3.87–3.80 (m, 3H), 3.71 (dd, J = 14.9, 3.0 Hz, 1H), 3.63 (dd, J = 8.8, 2.5 Hz, 1H), 3.19 (s, 1H), 3.10 (s, 3H); 13C {1H} NMR (CDCl3, 101 MHz) δ 168.7, 167.9, 167.9, 167.7, 167.4, 166.0, 137.2, 137.1, 133.9, 133.7, 133.3, 133.1, 132.8, 132.7, 131.9, 131.6, 131.3, 131.3, 128.0, 127.9, 127.6, 127.4, 127.1, 124.4, 123.4, 123.2, 123.0, 122.7, 122.5, 122.0, 99.7, 97.7, 74.9, 74.2, 71.9, 71.5, 71.4, 71.0, 66.0, 54.3, 51.9, 50.9, 39.9, 39.2. HRMS (ESI/Q-TOF) m/z: calcd for C59H52N5O15+ [M + NH4]+, 1070.3454; found 10703454.
Methyl (3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranoside (7)
The glycosyl donor 3 (0.771 g, 1.11 mmol) and the acceptor 6 (0.864 g, 0.82 mmol) were coevaporated with dry toluene (6 × 1 mL) and then dissolved in dry CH2Cl2 (16.0 mL). NIS (0.458 g, 2.04 mmol) was added to the solution, and the mixture was cooled to 0 °C. A solution of 1 M TfOH in CH2Cl2 (0.82 mL) was subsequently added to the solution, and the mixture was stirred for 40 min at 0 °C. The reaction was quenched by adding a saturated NaHCO3 aqueous solution (2 mL), and the resultant mixture was diluted with CHCl3. The solution was washed with saturated NaHCO3 aqueous solutions (3 times), and the combined aqueous layers were extracted with CHCl3 (6 times). The organic layers were combined and washed with 10% Na2S2O3 aqueous solutions (3 times). The combined aqueous layers were extracted with CHCl3 (3 times). The combined organic layers were dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was roughly purified by silica gel column chromatography using ethyl acetate–hexane (3:2, v/v) as an eluent to afford the glycosylation product as a yellow foam. The product was dissolved in H2O–pyridine (2:3, v/v, 100 mL), and the resultant solution was stirred at room temperature. After 19 h, the solution was concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography using ethyl acetate–hexane (3:1, v/v) as an eluent to afford 7 as a colorless foam (1.18 g, 0.75 mmol, 92%, 2 steps). 1H NMR (CDCl3, 400 MHz) δ 8.40 (d, J = 7.3 Hz, 1H), 7.88–7.65 (m, 14H), 7.62–7.44 (m, 6H), 7.38–7.31 (m, 2H), 7.17–7.14 (m, 2H), 7.12 (d, J = 7.3 Hz, 1H), 7.08–7.04 (m, 3H), 6.83–6.80 (m, 1H), 6.74–6.69 (m, 3H), 6.60–6.55 (m, 4H), 6.41 (d, J = 7.1 Hz, 2H), 5.28 (d, J = 8.5 Hz, 1H), 4.95 (d, J = 8.2 Hz, 1H), 4.77–4.67 (m, 2H), 4.61–4.57 (m, 2H), 4.46 (d, J = 12.3 Hz, 1H), 4.32–4.20 (m, 4H), 4.14–4.10 (m, 3H), 4.04 (dd, J = 11.0, 8.2 Hz, 1H), 3.94 (dd, J = 9.0, 2.9 Hz, 1H), 3.89 (d, J = 2.3 Hz, 1H), 3.85 (d, J = 12.6 Hz, 1H), 3.80–3.67 (m, 8H), 3.49 (dd, J = 8.0, 1.4 Hz, 1H), 3.11 (s, 3H), 3.03 (d, J = 3.4 Hz, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 169.3, 168.6, 168.1, 168.0, 167.7, 167.4, 166.1, 165.7, 137.6, 137.4, 137.4, 134.0, 133.7, 133.6, 133.2, 133.0, 132.9, 132.4, 132.4, 132.2, 132.0, 131.9, 131.8, 131.6, 131.5, 128.2, 127.7, 127.7, 127.6, 127.5, 127.1, 127.1, 125.3, 123.9, 123.3, 123.2, 123.1, 122.8, 122.4, 122.3, 121.8, 99.4, 99.1, 97.7, 74.9, 74.3, 72.9, 72.7, 71.9, 71.5, 71.3, 71.3, 71.2, 66.5, 54.4, 52.1, 51.7, 50.9, 40.3, 39.3. HRMS (ESI/Q-TOF) m/z: calcd for C88H78N8O222+ [M + 2NH4]2+, 799.2610; found 799.2606.
Methyl (3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranoside (8)
The glycosyl donor 3 (0.641 g, 0.92 mmol) and the acceptor 7 (0.919 g, 0.59 mmol) were coevaporated with dry toluene (6 × 1 mL) and then dissolved in dry CH2Cl2 (11.0 mL). NIS (0.331 g, 1.47 mmol) was added to the solution, and the mixture was cooled to 0 °C. A solution of 1 M TfOH in dry CH2Cl2 (0.59 mL) was subsequently added to the solution, and the mixture was stirred for 1 h at 0 °C. The reaction was quenched by adding a saturated NaHCO3 aqueous solution (2 mL), and the resultant mixture was diluted with CHCl3. The solution was washed with saturated NaHCO3 aqueous solutions (3 times), and the combined aqueous layers were extracted with CHCl3 (3 times). The organic layers were combined and washed with 10% Na2S2O3 aqueous solutions (3 times). The combined aqueous layers were extracted with CHCl3 (3 times). The combined organic layers were dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was roughly purified by silica gel column chromatography using ethyl acetate–hexane (3:1, v/v) as an eluent to afford the glycosylation product as a yellow foam. The product was dissolved in H2O–pyridine (1:2, v/v, 60 mL), and the resultant solution was stirred at room temperature. After 17 h, the solution was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using ethyl acetate–hexane (3:1–1:0, v/v) as an eluent to afford 8 as a yellow foam (1.10 g, 0.531 mmol, 91%, 2 steps). 1H NMR (CDCl3, 400 MHz) δ 8.46 (d, J = 7.5 Hz, 1H), 8.17 (d, J = 7.3 Hz, 1H), 8.00 (t, J = 7.5 Hz, 1H), 7.91–7.88 (m, 2H), 7.83–7.47 (m, 22H), 7.41 (td, J = 7.5, 0.8 Hz, 1H), 7.35–7.30 (m, 2H), 7.25–7.23 (d, J = 7.5 Hz, 1H), 7.16–7.12 (m, 3H), 7.09–7.06 (m, 3H), 6.89–6.75 (m, 4H), 6.70–6.64 (m, 5H), 6.54 (t, J = 7.7 Hz, 2H), 6.46 (d, J = 7.1 Hz, 2H), 6.32 (d, J = 7.1 Hz, 2H), 5.26 (d, J = 8.0 Hz, 1H), 4.99 (d, J = 8.5 Hz, 1H), 4.93 (d, J = 8.2 Hz, 1H), 4.70–4.63 (m, 4H), 4.46 (d, J = 12.1 Hz, 1H), 4.31–4.18 (m, 6H), 4.12–3.98 (m, 6H), 3.91–3.62 (m, 15H), 3.50 (dd, J = 6.9, 3.9 Hz, 1H), 3.14 (s, 3H), 2.92 (brs, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 169.6, 169.0, 168.4, 168.1, 168.0, 167.9, 167.7, 166.2, 165.8, 165.6, 137.6, 137.5, 137.5, 137.4, 134.4, 134.0, 133.7, 133.4, 133.3, 133.2, 133.0, 133.0, 132.4, 132.3, 132.3, 132.1, 132.1, 132.0, 131.9, 131.8, 131.6, 131.5, 128.2, 127.8, 127.7, 127.6, 127.6, 127.5, 127.5, 127.4, 127.2, 127.0, 127.0, 125.8, 124.7, 123.7, 123.4, 123.3, 123.2, 123.0, 122.8, 122.5, 122.3, 122.0, 121.9, 99.5, 98.8, 98.7, 97.9, 75.0, 74.7, 74.7, 74.6, 74.2, 73.1, 73.0, 72.6, 71.9, 71.9, 71.4, 71.3, 71.0, 66.5, 54.6, 52.2, 52.1, 51.6, 51.1, 40.8, 40.0, 39.2. HRMS (ESI/Q-TOF) m/z: calcd for C117H100N10O292+ [M + 2NH4]2+, 1054.3323; found 1054.3313.
Methyl (3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranoside (9)
The glycosyl donor 3 (0.534 g, 0.77 mmol) and the acceptor 8 (0.970 g, 0.47 mmol) were coevaporated with dry toluene (6 × 1 mL) and then dissolved in dry CH2Cl2 (9.0 mL). NIS (0.275 g, 1.22 mmol) was added to the solution, and the mixture was cooled to 0 °C. A solution of 1 M TfOH in dry CH2Cl2 (0.45 mL) was subsequently added to the solution, and the mixture was stirred for 45 min at 0 °C. The reaction was quenched by adding a saturated NaHCO3 aqueous solution (2 mL), and the resultant mixture was diluted with CHCl3. The solution was washed with saturated NaHCO3 aqueous solutions (3 times), and the combined aqueous layers were extracted with CHCl3 (3 times). The organic layers were combined and washed with 10% Na2S2O3 aqueous solutions (3 times). The combined aqueous layers were extracted with CHCl3 (3 times). The combined organic layers were dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was roughly purified by silica gel column chromatography using ethyl acetate as an eluent to afford a glycosylation product as a yellow foam. The product was dissolved in H2O–pyridine (1:2, v/v, 60 mL), and the resultant solution was stirred at room temperature. After 9 h, the solution was concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography twice using ethyl acetate for the first time and ethyl acetate–hexane (9:1, v/v) for the second time as an eluent to afford 9 as a yellow foam (0.895 g, 0.35 mmol, 74%, 2 steps). 1H NMR (CDCl3, 400 MHz) δ 8.26–8.20 (m, 2H), 8.02 (d, J = 7.3 Hz, 1H), 7.90–7.42 (m, 33H), 7.35–7.30 (m, 4H), 7.13–7.07 (m, 4H), 7.04–7.01 (m, 3H), 6.88–6.85 (m, 3H), 6.80–6.67 (m, 6H), 6.60–6.48 (m, 5H), 6.41 (d, J = 7.3 Hz, 2H), 6.35 (d, J = 7.3 Hz, 2H), 5.16 (d, J = 8.0 Hz, 1H), 4.94 (d, J = 8.2 Hz, 2H), 4.88 (d, J = 8.2 Hz, 1H), 4.68 (d, J = 8.2 Hz, 1H), 4.64–4.58 (m, 3H), 4.42 (d, J = 12.1 Hz, 1H), 4.33–3.58 (m, 35H), 3.50 (dd, J = 6.4, 3.9 Hz, 1H), 3.17 (s, 3H), 2.90 (d, J = 4.1 Hz, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 177.1, 169.4, 168.8, 168.5, 168.0, 168.0, 167.7, 166.2, 165.8, 165.6, 137.6, 137.5, 137.4, 134.0, 133.9, 133.7, 133.5, 133.4, 133.2, 133.0, 132.8, 132.6, 132.5, 132.4, 132.3, 132.2, 132.2, 132.1, 132.0, 131.8, 131.7, 131.5, 131.5, 128.2, 127.9, 127.7, 127.6, 127.6, 127.5, 127.4, 127.3, 127.0, 126.9, 125.0, 124.4, 123.7, 123.4, 123.2, 123.1, 123.0, 122.8, 122.5, 122.1, 121.9, 100.0, 99.5, 99.1, 98.5, 97.9, 77.2, 75.4, 75.0, 74.6, 74.5, 74.4, 74.2, 73.3, 73.0, 72.4, 72.1, 71.8, 71.6, 71.5, 71.3, 71.1, 71.1, 70.8, 66.5, 54.6, 52.3, 52.1, 51.6, 51.5, 51.1, 40.7, 40.2, 40.1, 39.3. HRMS (ESI/Q-TOF) m/z: calcd for C146H122N12O362+ [M + 2NH4]2+, 1309.4037; found 1309.4037.
Methyl (3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-dideoxy-2,6-diphthalimido-β-D-galactopyranoside (10)
The glycosyl donor 3 (0.123 g, 0.18 mmol) and the acceptor 9 (0.240 g, 93 μmol) were coevaporated with toluene (6 × 1 mL) and then dissolved in CH2Cl2 (2.0 mL). NIS (60.8 mg, 0.27 mmol) was added to the solution, and the mixture was cooled to 0 °C. A solution of 1 M TfOH in CH2Cl2 (90 μL) was subsequently added to the solution, and the mixture was stirred for 45 min at 0 °C. The reaction was quenched by adding a saturated NaHCO3 aqueous solution (2 mL), and the resultant mixture was diluted with CHCl3. The solution was washed with saturated NaHCO3 aqueous solutions (3 times), and the combined aqueous layers were extracted with CHCl3 (3 times). The organic layers were combined and washed with 10% Na2S2O3 aqueous solutions (3 times). The combined aqueous layers were extracted with CHCl3 (3 times). The combined organic layers were dried over Na2SO4, filtrated, and concentrated to dryness. The crude product was roughly purified by silica gel column chromatography using ethyl acetate as an eluent and preparative TLC using ethyl acetate–methanol (4:1, v/v) as an eluent to afford the glycosylation product as a yellow foam. The product was dissolved in H2O–pyridine (1:2, v/v, 15 mL), and the resultant solution was stirred at room temperature. After 24 h, the solution was concentrated under reduced pressure. The crude product was purified by silica gel column chromatography using ethyl acetate and preparative TLC using ethyl acetate–methanol (2:1, v/v) as an eluent to afford 10 as a yellow foam (59.9 mg, 19.3 μmol, 21%, 2 steps). 1H NMR (CDCl3, 400 MHz) δ 8.14 (d, J = 7.1 Hz, 1H), 8.05–7.98 (m, 3H), 7.93–7.89 (m, 2H), 7.83–7.45 (m, 34H), 7.39–7.29 (m, 5H), 7.22–7.15 (m, 2H), 7.12–7.07 (m, 4H), 7.03–6.99 (m, 3H), 6.78–6.48 (m, 20H), 6.44–6.37 (m, 4H), 5.12 (d, J = 8.0 Hz, 1H), 4.96 (d, J = 8.2 Hz, 1H), 4.90–4.84 (m, 3H), 4.65 (d, J = 8.0 Hz, 1H), 4.61–4.55 (m, 3H), 4.38 (d, J = 12.1 Hz, 1H), 4.28–3.50 (m, 44H), 3.12 (s, 3H), 2.74 (d, J = 3.7 Hz, 1H); 13C {1H} NMR (CDCl3, 101 MHz) δ 169.5, 169.4, 169.1, 168.8, 168.4, 168.0, 168.0, 167.9, 167.8, 166.2, 165.8, 165.7, 137.7, 137.6, 137.4, 133.9, 133.7, 133.6, 133.4, 133.2, 133.0, 132.7, 132.6, 132.5, 132.4, 132.3, 132.1, 132.1, 131.9, 131.7, 131.4, 128.2, 127.8, 127.7, 127.6, 127.3, 127.2, 127.0, 127.0, 126.9, 124.6, 123.6, 123.4, 123.3, 123.2, 123.2, 122.8, 122.5, 122.0, 121.9, 121.7, 119.3, 119.1, 100.0, 99.7, 99.4, 99.2, 98.7, 97.9, 77.2, 76.2, 75.0, 74.6, 74.1, 73.3, 73.2, 73.0, 72.5, 71.7, 71.4, 71.3, 71.1, 67.8, 66.4, 54.6, 52.3, 52.2, 52.0, 51.7, 51.6, 51.0, 45.6, 41.0, 40.7, 40.1, 39.3. HRMS (ESI/Q-TOF) m/z: calcd for C175H144N14O432+ [M + 2NH4]2+, 1564.4750; found 1564.4755.
Methyl (3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside octahydrochloride (12)
Compound 8 (44.7 mg, 22 μmol) was dissolved in EtOH (2.0 mL) and hydrazine monohydrate (25 μL) was subsequently added to the solution. The mixture was sealed, heated to 70 °C, and stirred for 22 h. The solution was cooled to room temperature and concentrated under reduced pressure. The crude mixture was diluted with water and filtrated. Then 0.05% TFA in water was added, and lyophilization was performed. One-sixth of the crude product was purified by RP-HPLC using a linear gradient of 0%–35% CH3CN/0.05% TFA in water/0.05% TFA for 70 min at room temperature, a flow rate of 6.0 mL min−1, a C18 column, and detection at 255 nm. After the HPLC purification, the amino saccharide was dissolved in 0.1 M hydrochloric acid (1.0 mL) and concentrated under reduced pressure. The product was coevaporated with water (5 times) and lyophilized to afford 12 as a colorless solid (2.0 μmol, 56%). 1H NMR (D2O, 400 MHz) δ 7.54–7.45 (m, 20H), 4.95–4.69 (m, 10H), 4.66 (s, 1H), 4.62–4.59 (m, 3H), 4.56 (s, 1H), 4.29 (d, J = 2.3 Hz, 1H), 4.04 (t, J = 6.4 Hz, 1H), 3.94 (dd, J = 11.3, 2.2 Hz, 1H), 3.90–3.84 (m, 2H), 3.78 (m, 5H), 3.67–3.63 (dd, J = 11.2, 2.8 Hz, 1H), 3.53–3.59 (m, 4H), 3.47–3.31 (m, 9H); 13C {1H} NMR (D2O, 101 MHz) δ 126.4, 126.2, 119.7, 119.6, 119.5, 119.3, 119.2, 90.3, 89.2, 89.2, 89.0, 65.7, 65.4, 65.3, 62.8, 62.7, 62.4, 61.9, 61.5, 61.2, 60.8, 60.5, 54.4, 48.0, 42.1. HRMS (ESI/Q-TOF) m/z: calcd for C53H77N8O13+ [M + H]+, 1033.5605; found 1033.5607.
Methyl (3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl) -(1 → 4)- (3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)- (3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside decahydrochloride (13)
Compound 9 (45.0 mg, 17 μmol) was dissolved in EtOH (2.0 mL) and hydrazine monohydrate (50 μL) was subsequently added to the solution. The mixture was sealed, heated to 70 °C, and stirred for 22 h. The solution was cooled to room temperature and concentrated. The crude mixture was diluted with water and filtrated. Then 0.05% TFA in water was added, and lyophilization was performed. One-eighth of the crude product was purified by RP-HPLC using a linear gradient of 0%–35% CH3CN/0.05% TFA in water/0.05% TFA for 70 min at room temperature, a flow rate of 6.0 mL min−1, a C18 column, and detection at 255 nm. After the HPLC purification, the amino saccharide was dissolved in 0.1 M hydrochloric acid (1.0 mL) and concentrated under reduced pressure. The product was coevaporated with water (5 times) and lyophilized to afford 13 as a colorless solid (1.0 μmol, 48%). 1H NMR (D2O, 400 MHz) δ 7.55–7.49 (m, 25H), 4.95–4.68 (m, 12H), 4.67 (d, J = 1.8 Hz, 1H), 4.62–4.59 (m, 4H), 4.55 (s, 1H), 4.29 (d, J = 2.5 Hz, 1H), 4.04 (t, J = 6.4 Hz, 1H), 3.95 (dd, J = 11.1, 2.6 Hz, 1H), 3.90–3.72 (m, 10H), 3.65–3.53 (m, 5H), 3.50–3.35 (m, 12H); 13C {1H} (DEPT 135) NMR (D2O, 101 MHz) δ 129.4, 129.2, 129.0, 128.9, 100.0, 99.1, 98.8, 75.4, 75.3, 75.1, 72.6, 72.5, 72.4, 72.1, 71.6, 71.3, 71.0, 70.8, 70.5, 70.3, 64.1, 57.7, 51.8, 51.7, 51.6, 40.4, 40.2. HRMS (ESI/Q-TOF) m/z: calcd for C66H96N10O162+ [M + 2H]2+, 642.3497; found 642.3496.
Methyl (3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-3-O-benzyl-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside dodecahydrochloride (14)
Compound 10 (24.5 mg, 7.9 μmol) was dissolved in EtOH (3.0 mL), and hydrazine monohydrate (25 μL) was subsequently added to the solution. The mixture was sealed, heated to 70 °C, and stirred for 14 h. The solution was cooled to room temperature and concentrated under reduced pressure. The crude mixture was diluted with 0.05% TFA in water and filtrated. Two-fifth of the crude product was purified by RP-HPLC using a linear gradient of 0%–35% CH3CN/0.05% TFA in water/0.05% TFA for 70 min at room temperature, a flow rate of 6.0 mL min−1, a C18 column, and detection at 255 nm. After the HPLC purification, the amino saccharide was dissolved in 0.1 M hydrochloric acid (1.0 mL) and concentrated. The product was coevaporated with water (5 times) and lyophilized to afford 14 as a colorless solid (0.63 μmol, 20%). 1H NMR (D2O 400 MHz) δ 7.52–7.49 (m, 30H), 4.95–4.68 (m, 14H), 4.64–4.59 (m, 5H), 4.56 (s, 1H), 4.29 (d, J = 2.5 Hz, 1H), 4.05 (t, J = 6.4 Hz, 1H), 3.95 (dd, J = 11.3, 2.6 Hz, 1H), 3.91–3.74 (m, 14H), 3.64 (dd, J = 11.1, 2.9 Hz, 1H), 3.59–3.55 (m, 4H), 3.53–3.35 (m, 15H); 13C {1H} (DEPT 135) NMR (D2O, 101 MHz) δ 130.0, 129.9, 129.8, 129.6, 129.5, 100.6, 99.6, 99.4, 76.0, 75.9, 75.6, 75.7, 73.2, 73.1, 73.0, 72.7, 72.2, 71.9, 71.6, 71.6, 71.5, 71.4, 71.1, 70.8, 64.7, 58.3, 52.4, 52.4, 52.3, 52.2, 41.0, 40.8. HRMS (ESI/Q-TOF) m/z: calcd for C79H115N12O193+ [M + 3H]3+, 511.9462; found 511.9468.
Methyl (2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside hydrochloride (15)
Compound 7 (9.0 mg, 6.0 μmol) was dissolved in EtOH (2.0 mL), and hydrazine monohydrate (25 μL) was subsequently added to the solution. The mixture was sealed, heated to 70 °C, and stirred for 23 h. The solution was cooled to room temperature and concentrated under reduced pressure. The crude mixture was diluted with water and filtrated. Then 0.1 M hydrochloric acid (1.0 mL) was added and concentrated. The product was coevaporated with water (8 times) and lyophilized to afford crude 11. The mixture was dissolved in a mixed solvent of 0.1 M hydrochloric acid (2.0 mL) and methanol (2.0 mL). Pd/C (10%, 0.10 g) was added to the solution, and the mixture was stirred under 200–950 kPa of H2 gas at 60 °C. After 6.5 h, the mixture was filtrated, and the resulting filtrate was concentrated under reduced pressure. The crude product was dissolved in methanol (200 μL), reprecipitated from acetone (3.0 mL), and washed with acetone. The precipitate was dissolved in water (1 mL), filtrated, and lyophilized to afford ODAGal3 (15) as a colorless solid (4.0 μmol, 68%, 2 steps). 1H NMR spectra corresponded to the data in the previous report.17
Methyl (2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside hydrochloride (16)
Compound 12 (0.39 μmol) was dissolved in a mixed solvent of 0.1 M hydrochloric acid (2.4 mL) and methanol (2.4 mL). Pd/C (10%, 55 mg) was added to the solution, and the mixture was stirred under 400–920 kPa of H2 gas at 60 °C. After 10.5 h, the mixture was filtrated, and the resulting filtrate was concentrated under reduced pressure. The crude product was dissolved in methanol (50 μL), reprecipitated from acetone (2.0 mL), and washed with acetone. The precipitate was coevaporated with water (3 × 1.0 mL) and lyophilized to afford ODAGal4 (16) as a colorless solid (0.30 μmol, 78%). 1H NMR spectra corresponded to the data in the previous report.17
Methyl (2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside hydrochloride (17)
Compound 13 (0.67 μmol) was dissolved in a mixed solvent of 0.1 M hydrochloric acid (2.0 mL) and methanol (2.0 mL). Pd/C (10%, 0.152 g) was added to the solution, and the mixture was stirred under 400–950 kPa of H2 gas at 60 °C. After 9 h, the mixture was filtrated, and the resulting filtrate was concentrated under reduced pressure. The crude product was dissolved in methanol (100 μL), reprecipitated from acetone (2.0 mL), and washed with acetone. The precipitate was coevaporated with water (5 × 1.0 mL) and lyophilized to afford ODAGal5 (17) as a colorless solid (0.20 μmol, 30%). 1H NMR (D2O, 400 MHz) δ 5.11–5.03 (m, 4H), 4.73–4.68 (d, 1H), 4.39–4.37 (m, 4H), 4.20–4.08 (m, 8H), 4.03–4.00 (m, 2H), 3.96 (dd, J = 10.9, 3.1 Hz, 1H), 3.70–3.63 (m, 6H), 3.53–3.42 (m, 9H), 3.38–3.32 (m, 3H); 13C {1H} (DEPT 135) NMR (D2O, 101 MHz) δ 103.4, 103.2, 103.1, 103.1, 76.9, 72.1, 71.5, 71.4, 71.2, 71.1, 68.8, 58.1, 54.2, 53.8, 51.3, 41.1, 41.0, 40.8. HRMS (ESI/Q-TOF) m/z: calcd for C31H66N10O162+ [M + 2H]2+, 417.2324; found:417.2328.
Methyl (2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-(2,6-diamino-2,6-dideoxy-β-D-galactopyranosyl)-(1 → 4)-2,6-diamino-2,6-dideoxy-β-D-galactopyranoside hydrochloride (18)
Compound 14 (0.63 μmol) was dissolved in a mixed solvent of 0.1 M hydrochloric acid (2.0 mL) and methanol (2.0 mL). Pd/C (10%, 0.230 g) was added to the solution, and the mixture was stirred under 300–950 kPa of H2 gas at 60 °C. After 8 h, the mixture was filtrated, and the resulting filtrate was concentrated under reduced pressure. The crude product was dissolved in methanol (100 μL), reprecipitated from acetone (2.0 mL), and washed with acetone. The precipitate was coevaporated with water (3 × 1.0 mL) and lyophilized to afford ODAGal6 (18) as a colorless solid (0.17 μmol, 26%). 1H NMR (D2O, 400 MHz) δ 5.11–5.02 (m, 5H), 4.39–4.36 (m, 5H), 4.20–4.08 (m, 10H), 4.03–4.00 (m, 2H), 3.96 (dd, J = 10.9, 3.3 Hz, 1H), 3.73–3.58 (m, 7H), 3.56–3.32 (m, 15H); 13C {1H} (DEPT 135) NMR (D2O, 101 MHz) δ 101.1, 101.0, 100.9, 100.7, 100.5, 76.9, 76.8, 76.6, 76.5, 72.2, 71.4, 71.0, 69.3, 69.0, 68.9, 68.7, 68.6, 58.2, 53.9, 53.7, 42.1, 40.9, 40.7, 39.7. HRMS (ESI/Q-TOF) m/z: calcd for C37H78N12O192+ [M + 2H]2+, 497.2748; found 497.2758.
Author contributions
All authors established the research plans. T. S. conducted the experiments. All authors analyzed the data. T. S. wrote the manuscript. K. S., R. I. H., and T. W. revised the manuscript. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Dr Yayoi Yoshimura (Tokyo University of Science) for the mass spectrum measurements. We also thank Mr Yamato Harada and Mr Kazuhiro Nagashima (Tokyo University of Science) for the synthesis of some compounds. We would like to thank Enago (https://www.enago.jp) for the English language review. This work was partially supported by JSPS KAKENHI (19K06993).
References
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- A. Wittrup and J. Lieberman, Nat. Rev. Genet., 2015, 16, 543–552 CrossRef CAS PubMed.
- I. Urits, D. Swanson, M. C. Swett, A. Patel, K. Berardino, A. Amgalan, A. A. Berger, H. Kassem, A. Kaye and O. Viswanath, Neurol. Ther., 2020, 9, 301–315 CrossRef PubMed.
- Y. Y. Syed, Drugs, 2021, 81, 841–848 CrossRef CAS PubMed.
- L. J. Scott and S. J. Keam, Drugs, 2021, 81, 277–282 CrossRef CAS.
- Y. N. Lamb, Drugs, 2021, 81, 389–395 CrossRef CAS PubMed.
-
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215515Orig1s000ltr.pdf
.
- T. P. Prakash and B. Bhat, Curr. Top. Med. Chem., 2007, 7, 641–649 CrossRef CAS PubMed.
- S. K. Singh, P. Nielsen, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455–456 RSC.
- K. Nishina, W. Y. Piao, K. Yoshida-Tanaka, Y. Sujino, T. Nishina, T. Yamamoto, K. Nitta, K. Yoshioka, H. Kuwahara, H. Yasuhara, T. Baba, F. Ono, K. Miyata, K. Miyake, P. P. Seth, A. Low, M. Yoshida, C. F. Bennett, K. Kataoka, H. Mizusawa, S. Obika and T. Yokota, Nat. Commun., 2015, 6, 7969–7981 CrossRef CAS PubMed.
- X. L. Shen and D. R. Corey, Nucleic Acids Res., 2018, 46, 1584–1600 CrossRef CAS.
- A. D. Springer and S. F. Dowdy, Nucleic Acid Ther., 2018, 28, 109–118 CrossRef CAS PubMed.
- T. Nagata, C. A. Dwyer, K. Yoshida-Tanaka, K. Ihara, M. Ohyagi, H. Kaburagi, H. Miyata, S. Ebihara, K. Yoshioka, T. Ishii, K. Miyata, B. Powers, T. Igari, S. Yamamoto, N. Arimura, H. Hirabayashi, T. Uchihara, R. I. Hara, T. Wada, C. F. Bennett, P. P. Seth, F. Rigo and T. Yokota, Nat. Biotechnol., 2021, 39, 1529–1536 CrossRef CAS PubMed.
- R. Iwata, M. Sudo, K. Nagafuji and T. Wada, J. Org. Chem., 2011, 76, 5895–5906 CrossRef CAS PubMed.
- R. Iwata, A. Doi, Y. Maeda and T. Wada, Org. Biomol. Chem., 2015, 13, 9504–9515 RSC.
- R. I. Hara, Y. Maeda, T. Sakamoto and T. Wada, Org. Biomol. Chem., 2017, 15, 1710–1717 RSC.
- A. Irie, K. Sato, R. I. Hara, T. Wada and F. Shibasaki, Sci. Rep., 2020, 10, 14845–14859 CrossRef PubMed.
- R. I. Hara and T. Wada, Org. Biomol. Chem., 2021, 19, 6865–6870 RSC.
- R. Iwata, K. Nishina, T. Yokota and T. Wada, Bioorg. Med. Chem., 2014, 22, 1394–1403 CrossRef CAS PubMed.
- R. I. Hara, Y. Hisada, Y. Maeda, T. Yokota and T. Wada, Sci. Rep., 2018, 8, 4323–4331 CrossRef PubMed.
- P. Hair, F. Cameron and K. McKeage, Drugs, 2013, 73, 487–493 CrossRef CAS PubMed.
- M. Saminathan, T. Antony, A. Shirahata, L. H. Sigal, T. Thomas and T. J. Thomas, Biochemistry, 1999, 38, 3821–3830 CrossRef CAS PubMed.
- A. Iihara, K. Sato, K. Hozumi, M. Yamada, H. Yamamoto, M. Nomizu and N. Nishi, Polym. J., 2002, 34, 184–193 CrossRef CAS.
- H. Inoue, Y. Hayase, A. Imura, S. Iwai, K. Miura and E. Ohtsuka, Nucleic Acids Res., 1987, 15, 6131–6148 CrossRef CAS PubMed.
- J. Haupenthal, C. Baehr, S. Kiermayer, S. Zeuzem and A. Piiper, Biochem. Pharmacol., 2006, 71, 702–710 CrossRef CAS PubMed.
-
https://www.abcam.co.jp/recombinant-human-rnase-h1rnh1-protein-ab153634/reviews/52111
.
- M. Nowotny, S. A. Gaidamakov, R. Ghirlando, S. M. Cerritelli, R. J. Crouch and W. Yang, Mol. Cell, 2007, 28, 264–276 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Rintaro Iwata
Hara
a,
Rintaro Iwata
Hara