
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Algorithm-driven activity-directed expansion of a series of antibacterial quinazolinones†
Daniel
Francis
ab,
Sannia
Farooque
ab,
Archie
Meager
ab,
Didi
Derks
a,
Abbie
Leggott
ab,
Stuart
Warriner
ab,
Alex J.
O'Neill
bc and
Adam
Nelson
 *ab
*ab
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: a.s.nelson@leeds.ac.uk
bAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
cSchool of Molecular and Cellular Biology, University of Leeds, Leeds, LS2 9JT, UK
First published on 30th November 2022
Abstract
Activity-directed synthesis (ADS) is a structure-blind, function driven approach that can drive the discovery of bioactive small molecules. In ADS, arrays of reactions are designed and executed, and the crude product mixtures are then directly screened to identify reactions that yield bioactive products. The design of subsequent reaction arrays is then informed by the hit reactions that are discovered. In this study, algorithms for reaction array design were developed in which the reactions to be executed were selected from a large set of virtual reactions; the reactions were selected on the basis of similarity to reactions known to yield bioactive products. The algorithms were harnessed to design arrays of photoredox-catalysed alkylation reactions whose crude products were then screened for inhibition of growth of S. aureus ATCC29213. It was demonstrated that the approach enabled expansion of a series of antibacterial quinazolinones. It is envisaged that such algorithms could ultimately enable fully autonomous activity-directed molecular discovery.
Introduction
The discovery of bioactive small molecules is a central challenge in both medicinal chemistry and chemical biology. Within this context, we have developed activity-directed synthesis (ADS),1 a structure-blind molecular discovery approach2 in which synthetic routes emerge on the basis of the function of the resulting products. In ADS, arrays of reactions are designed and executed in which different combinations of components (e.g. substrates, co-substrates and catalysts) are explored. Here, “promiscuous” reactions – with multiple possible outcomes – may be deliberately harnessed in order to explore diverse chemical space. After evaporation, the crude reaction mixtures are screened directly for biological function. Hit reactions – that yield bioactive products – then inform the design of subsequent reaction arrays. To date, reaction array design has been human-driven (rather than algorithm-driven). A range of different underpinning chemistries and assay types have been harnessed to drive ADS (Scheme 1).3–6 For example, metal-catalysed diazo chemistry was exploited in the activity-directed discovery of diverse modulators of androgen receptor (panel A).3,4 More recently, Pd-catalysed carbonylation chemistry, in conjunction with a phenotypic assay (growth inhibition of S. aureus), enabled the expansion of a series of antibacterial quinazolinones (panel B);5 this series had been developed to target penicillin-binding proteins, and displays promising activity (minimum inhibitory concentrations [MICs] as low as 0.003 μg ml−1) against multiple strains of S. aureus.7 | ||
| Scheme 1 Chemistry for activity-directed synthesis. Panel A: Rh-catalysed diazo chemistry exploited in the activity-directed synthesis of androgen receptor modulators; groups derived from a diazo substrate (black) and co-substrates (blue) are indicated. Panel B: Pd-catalysed carbonylation chemistry exploited in the activity-directed expansion of a series of antibacterials (B1); groups derived from an amine co-substrate (blue) and a carbon monoxide source (red) are indicated. The structure of an antibacterial quinazolinone that was discovered is shown, together with its activity against selected S. aureus strains (B2). Panel C: Envisaged photoredox-catalysed alkylation involving aldehyde/ketone co-substrates (blue). | ||
We have previously noted1,8 that the integrated and parallel nature of ADS workflows may facilitate the realisation of fully autonomous molecular discovery. Such a workflow would require the design of reaction arrays to be algorithm-driven, and all experimental activities to be automated and integrated. In this study, we describe the application of algorithms to design reaction arrays on the basis of (dis)similarity to reactions that yield known bioactive molecules. It was envisaged that the reactions would be selected from a large virtual reaction space defined by all possible combinations of heteroaromatic substrates 4 (quinazolinones and related substrates) and aldehyde or ketone co-substrates 5 (Panel C). A bacterial growth inhibition assay would then be used to identify photocatalysed alkylations9 that yield products (e.g.6) with antibacterial activity. The photocatalysed alkylation9a of quinazolinones with both aldehydes and ketones is well precedented. It was therefore envisaged that, by using aryl-substituted co-substrates, this reaction may enable variation of the linker between the heterocyclic core and an appended aromatic ring.
Results and discussion
Establishment of a workflow for activity-directed synthesis
Initially, we established a workflow for the activity-directed synthesis of antibacterials based on photoredox-catalysed alkylation reactions9a conducted on a 300 μl scale (3 μmol limiting substrate) (Fig. 1). Accordingly, combinations of a quinazolinone substrate (S1 or S2; final concentration: 10 mM), an aldehyde/ketone co-substrate (7, 8 or 9; final concentration: 300 mM), tris(trimethylsilyl)silane (TTMS) (final concentration: 20 mM), TFA (final concentration: 20 mM) and Ir[dF(CF3)ppy]2(dtbpy)PF6 (final concentration: 0.1 mM) were assembled in vials from stock solutions in MeCN. The sealed vials were irradiated (36 W blue LED) at room temperature for 24 h, and the crude reaction mixtures were evaporated and redissolved in DMSO to give stock solutions (total product concentration: 10 mM). Adapted5 standard susceptibility testing methodology10 was used to assess antibacterial activity against two independent cultures of S. aureus ATCC2921311a at 10 μM total product concentration in 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Iso-Sensitest Broth12 (ISB)−DMSO. Crucially, the combination of S2 and aldehyde 8 (which would yield 3a5) resulted in >99% growth inhibition, whereas the other five substrate/co-substrate combinations did not inhibit bacterial growth. In addition, the individual substrates (S1 and S2), co-substrates (7–9), catalyst, TTMS and TFA were all subjected to the workflow at the appropriate concentrations, and the resulting crude mixtures were shown to not inhibit (<2%) bacterial growth (see ESI†). The workflow thus enabled both the synthesis of 3a, and the detection of its antibacterial activity in a crude product mixture; and was deemed to be suitable for the activity-directed synthesis of antibacterials.
:1 Iso-Sensitest Broth12 (ISB)−DMSO. Crucially, the combination of S2 and aldehyde 8 (which would yield 3a5) resulted in >99% growth inhibition, whereas the other five substrate/co-substrate combinations did not inhibit bacterial growth. In addition, the individual substrates (S1 and S2), co-substrates (7–9), catalyst, TTMS and TFA were all subjected to the workflow at the appropriate concentrations, and the resulting crude mixtures were shown to not inhibit (<2%) bacterial growth (see ESI†). The workflow thus enabled both the synthesis of 3a, and the detection of its antibacterial activity in a crude product mixture; and was deemed to be suitable for the activity-directed synthesis of antibacterials.
 | ||
| Fig. 1 Establishment of an activity-directed synthesis workflow. Panel A: substrates and aldehyde/ketone co-substrates used. Panel B: inhibition of growth of two independent cultures of S. aureus ATCC29213 (see Fig. 3 for colour scale) by reaction mixtures derived from specific substrate/co-substrate combinations (total product concentration: 10 μM). Reactions were performed in microscale vials and involved a substrate (S1 or S2; 10 mM), a co-substrate (7, 8 or 9; 30 eq.), TTMS (2 eq.), TFA (2 eq.) and Ir[dF(CF3)ppy]2(dtbpy)PF6. | ||
Algorithm-driven activity-directed synthesis: Round 1
Initially, we defined a set of 20660 virtual reactions based on all possible combinations of 20 substrates and 1033 co-substrates (Fig. 2). The substrates S1–S20 included analogues of quinazolinones in which the benzo ring had been substituted or replaced with a heterocyclic variant (see ESI† and Fig. 4); 16 of the substrates were prepared from four commercially-available isotoic anhydrides, and the remaining four substrates from 2-amino nicotinic acid.13 The possible aldehyde and ketone co-substrates were extracted from a set of commercially-available compounds, and were filtered to have 0–2 rotatable bonds, 8–15 heavy (non-hydrogen) atoms, 1–2 aromatic rings and no undesired functionality. | ||
| Fig. 2 Overview of the definition of a large virtual reaction space, and the algorithm used to design the Round 1 reaction array based on the hit reaction (substrate S2 + co-substrate 8). | ||
 | ||
| Fig. 3 Difference in growth inhibition by crude products of the designed reaction arrays, and the corresponding reactions in which the substrate was omitted (see ESI†). Reactions were performed in microscale vials and involved a substrate (S1 or S2; 10 mM; omitted in control reaction arrays), a co-substrate (7, 8 or 9; 30 eq.), TTMS (2 eq.), TFA (2 eq.) and Ir[dF(CF3)ppy]2(dtbpy)PF6. Crude products were assayed against three independent colonies of S. aureus ATCC29213 at 10 μM total product concentration to yield hit reactions (yellow). Reaction products that displayed activity even in the absence of a substrate (pink, see ESI†) and combinations that were not investigated due to co-substrate unavailability (grey) are indicated. Panel A: reaction array in Round 1 designed on the basis of the hit reaction S2/8 (see Fig. 1). Panel B: reaction array in Round 2 designed on the basis of hit reactions from Round 1. | ||
 | ||
| Fig. 4 Structures of substrates and co-substrates used in reaction arrays based on hit reactions (see Fig. 3). | ||
Next, we subjected the 20 substrates (S1–S20) to our activity-directed synthesis workflow. Here, “mock” reactions were performed in the absence of any co-substrate, and the crude reaction mixtures were evaluated at 10 μM total product concentration; crucially,<2% bacterial growth inhibition was observed in all cases, which gave confidence that any observed activities would be dependent on the presence of the specific co-substrate used.
An algorithm was then harnessed to design the reaction array that was executed in Round 1. Substrate combinations that were similar (Tanimoto coefficient based the ECFP4 fingerprints14 for the substrate combinations >0.68) to the hit reaction (substrate S2 + co-substrate 8, see Fig. 1) were extracted from the large virtual reaction space. This yielded 357 reactions that could, in principle, be executed in Round 1. To identify 20 possible arrays of 30 reactions for execution, Pareto optimisation15 was undertaken with two (conflicting) objectives: (a) to minimise the number of unique co-substrates that would need to be purchased; and (b) to maximise the diversity of the substrate/co-substrate combinations. It was decided to execute the designed reaction array that had the fewest unique co-substrates (Fig. 3, panel A).
A liquid handling robot was used to assemble the required combinations of substrates and co-substrates in the designed reaction array. Here, the appropriate substrate (100 μl of a 30 mM solution in MeCN) and co-substrate (100 μl of a 0.90 M solution in MeCN) were combined. Subsequently, TFA (25 μl of a 0.24 M solution in MeCN), TTMS (25 μl of a 0.24 M solution in MeCN) and Ir[dF(CF3)ppy]2(dtbpy)PF6 (50 μl of a 0.6 mM solution in MeCN) were added. The final concentration of the components in each 300 μl reaction were therefore: substrate (10 mM), co-substrate (300 mM), TFA (20 mM), TTMS (20 mM) and catalyst (0.1 mM). In parallel, a control reaction array was also assembled in which the substrates (but not the co-substrates) were omitted. The sealed vials were irradiated at room temperature for 24 h, and the crude reaction mixtures were evaporated and redissolved in 300 μl DMSO to give stock solutions with 10 mM total product concentration. The crude reaction mixtures were assayed against three independent cultures of S. aureus ATCC29213 at 10 μM total product concentration in 99:1 ISB–DMSO. The difference in growth inhibition between the crude products of the designed reactions, and the corresponding control reactions that lacked a substrate, is shown in Fig. 3, panel A.
The crude products of eight reactions resulted in >70% growth inhibition for all three cultures. However, for six of these reactions (that involved the co-substrates C13, C494, C504, C661, C673 and C933), growth inhibition was also observed in the corresponding control reactions that lacked a substrate. On this basis, two hit reactions were taken forward: the reactions between either S2 and S3 and C981 (phenyl acetaldehyde, also labelled 8). Notably, the combination of S2 and phenyl acetaldehyde had previously been identified as a hit combination during the establishment of the assay.
In parallel, we harnessed an algorithm to design a diverse array of reactions that were dissimilar to the combination of S2 and 8 (= C981). These reactions were chosen from a virtual set of 1300 virtual reactions based on all possible combinations of the 20 substrates and 65 cheap, yet diverse, co-substrates (see ESI†). Unfortunately, no validated hit combinations were identified from this array of diverse reactions (see ESI†).
Algorithm-driven activity-directed synthesis: Round 2
Both of the validated hit combinations from Round 1 had involved phenyl acetaldehyde. At this stage, we noted that only five of the 1033 co-substrates represented in the virtual reaction space defined for Round 1 were aliphatic aldehydes. In the virtual reaction space for Round 2, we therefore included additional aliphatic aldehydes that would be likely preparable from commercially-available primary alcohols (numbered C10001-). Overall, the virtual reaction space comprised 31340 virtual reactions based on all possible combinations of the same 20 substrates and an expanded number (1567) of co-substrates (Fig. 5). | ||
| Fig. 5 Overview of the definition of a large virtual reaction space, and the algorithm used to design the Round 2 reaction array based on the hit reactions from Round 1. | ||
The same algorithm was then harnessed to design the reaction array that was executed in Round 2. Substrate combinations that were similar (Tanimoto coefficient for the substrate combinations >0.8) to either of the hit reactions from Round 1 (S2/C981 and S3/C981; see panel A, Fig. 3) were extracted from the virtual set of 31340 reactions. This yielded 127 reactions that could, in principle, be executed in Round 2. We then identified 10 possible arrays of 30 reactions for execution through Pareto optimisation with the following objectives: (a) maximisation of the diversity of the substrate/co-substrate combinations; (b) minimisation of the number of unique co-substrates; (c) having the same number of reactions that were most similar to both of the hit reactions from Round 1; and (d) minimising similarity to the non-hit reactions that had been performed in Round 1. It was decided to execute the reaction array with the fewest unique co-substrates.
Unfortunately, two of the required co-substrates (C10900 and C12019) could not be readily prepared by oxidation of the corresponding primary alcohol, meaning that only 27 of the 30 designed reactions could actually be executed. Following execution of the reaction array, the crude reaction mixtures were assayed against three independent cultures of S. aureus ATCC29213 at 10 μM total product concentration in 99:1 ISB–DMSO. The difference in growth inhibition between the crude products of the designed reactions, and the corresponding control reactions that lacked a substrate, is shown in Fig. 3, panel B. The crude products of six reactions resulted in >70% growth inhibition for all three independent cultures. However, for two of these reactions, involving the same co-substrate C10957, growth inhibition was also observed in the corresponding reactions in the control array that lacked a quinazolinone substrate. Thus, four new hit reactions were identified: the reaction between the quinazolinone S2 and aldehydes C10257, C10258 or C11371; and the reaction between the quinazolinone S3 and the aldehyde C10257. A total of six hit reactions was thus identified across both rounds of activity-directed synthesis.
Evaluation of the activity of purified products
For each of the identified hit reactions from Round 1 and 2, we prepared and purified samples of the anticipated alkylated products (Fig. 6 and Table 1). In addition, inspired by the hit combinations S2/C10258, S3/C981 and S3/C10257, we also prepared the quinazolinone 3f which would have been formed from S3 and C10258, a combination that had not been explored. As controls, we prepared 3h and 3i, which would have been derived from two investigated combinations that were not identified as hits. The quinazolinones 3a, 3d, 3g, 3h and 3i were prepared by scale-up of the original photoredox-catalysed alkylation reaction, whereas 3b, 3c, 3e and 3f were prepared by an independent synthetic route.16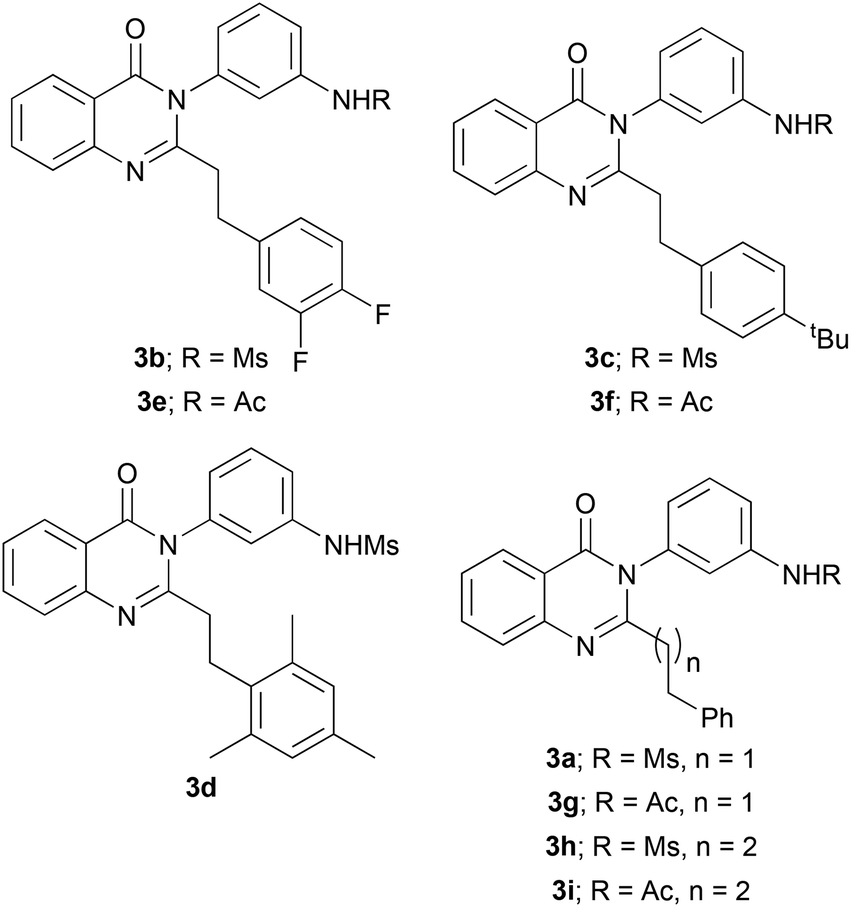 | ||
| Fig. 6 Structures of compounds for which MICs were determined (see Table 1). | ||
| Compound | Hit? | MIC (μg ml−1) | ||
|---|---|---|---|---|
| ATCC29213 | USA300 JE2 | SH1000 | ||
| a Prepared by scale-up of the photoredox-catalysed alkylation reaction (see ESI†). b Prepared by an independent synthetic route (see ESI†). c The substrate and co-substrate were exploited in other hit reactions, but this specific combination was not explored in either array. | ||||
| 3a | √ (R1) (S2, C981) | 0.016 | 0.016 | 1–2 |
| 3b | √ (R2) (S2, C10257) | 0.5–1 | 0.5–1 | 4 |
| 3c | √ (R2) (S2, C10258) | 4 | 4 | 8 |
| 3d | √ (R2) (S2, C11371) | 0.016 | 0.016 | 1–2 |
| 3e | √ (R2) (S3, C10257) | 1 | 1 | 4 |
| 3f | c (S3, C10258) | 8 | 8 | 16 |
| 3g | √ (R1) (S3, C981) | 0.5–1 | 0.5–1 | 4 |
| 3h | x (R2) (S2, C11313) | >256 | >256 | >256 |
| 3i | x (R2) (S3, C11313) | >256 | >256 | >256 |
The quinazolinone products that corresponded to identified hit combinations had MIC values against S. aureus ATCC29213 ranging from 0.016 μg ml−1 (for 3a and 3d) to 4 μg ml−1 (for 3c). Reassuringly, 3h and 3i, which corresponded to investigated combinations that were not identified as hits, did not inhibit bacterial growth, even at 256 μg ml−1. 3f, which corresponded to a substrate/co-substrate combination that was not explored (but, instead was inspired by three other hit combinations, see above) had an MIC value of 8–16 μg ml−1. All of the analogues 3a–3i were also screened against the methicillin-resistant USA300 JE2 strain11b and the laboratory strain11c,d SH1000 (see Table 1 and Discussion). They were also evaluated against yeast (Candida albicans Ca6) and were found to be inactive at 16 μg ml−1.
Discussion
We had previously hypothesised that the integrated and parallel nature of its workflows might enable activity-directed synthesis to be algorithm-driven.1,8 We have now demonstrated that algorithms can indeed drive the activity-directed synthesis of bioactive small molecules. The use of these algorithms does mean that designed reaction arrays no longer involve all combinations of components; this means that it is more convenient to use a liquid handling robot to assemble the designed arrays, rather than manually using multi-channel pipettes.We have also shown that it was feasible for a photoredox-catalysed reaction to underpin activity-directed synthesis, a reaction class that has potential to explore diverse regions of chemical space.17 As with other activity-directed synthesis workflows,3–6 it was critical that appropriate control reactions were performed to give confidence that observed activities stemmed from the specific substrate/co-substrate combination used. In this study, the use of control arrays enabled identification of co-substrates whose reactions yielded apparently antibacterial products even in the absence of a substrate. In addition, it was, of course, also important to purify, elucidate and characterise the products of identified hit reactions.
Our approach enabled expansion of a series7 of antibacterial quinazolinones. These compounds had comparable activity against S. aureus ATCC29213 and the methicillin-resistant USA300 JE2 strain, but were generally significantly less active against the laboratory strain SH1000. It was demonstrated that limited variation of the meta-substituted phenyl ring of 3a5 was possible: the corresponding m-acetamidophenyl-substituted analogue (3g) retained significant antibacterial activity. However, none of the reactions involving substrates other than S2 or S3 – for example, quinazolinones with replaced/substituted benzo rings or other phenyl substituents – resulted in any growth inhibition at the concentration tested (total product concentration: 10 μM). Some variation of the substitution of the phenyl ring of the 2-phenylethyl group of 3a was, however, possible: the quinzolinones 3c and 3f (with a 4-tert-butylphenyl group); 3b and 3e (with a 3,4-difluorophenyl group); and 3d (with a 2,4,6-trimethylphenyl group) displayed significant antibacterial activity. Indeed, the activity of 3d (with its 2,4,6-trimethylphenyl group) (MIC against ATCC 29213:0.016 μg ml−1) was comparable with that of 3a. It was notable that no reactions that involved any of the other seven substituted phenyl acetaldehydes used, nor the homologated aldehyde C11313, resulted in the formation of antibacterial products. Although two reactions of the homologated aldehyde C11313 were shown to shown to be productive, yielding the corresponding homologated products (3h and 3i), neither of these products displayed detectable antibacterial activity (MIC against S. aureus ATCC 29213:>256 μg ml−1). It was therefore concluded that the length of the 2-phenylethyl side chain was critical, and that only limited range of substituents on the phenyl ring of this side chain was tolerated.
A summary of the structure–activity-relationships of the antibacterial quinazolinones is provided in Fig. 7. Whilst the approach was certainly valuable for the rapid generation of these relationships, it is, perhaps, disappointing that it was not possible to expand the series of antibacterial quinaozolinones7 more extensively, perhaps because this series has already been largely optimised. Within an ADS workflow, the choice of screening concentration(s) is critical. Here the crude products were screened at 10 μM total product concentration.‡ This choice of screening concentration meant that the least active product identified was 3c (whose MIC against S. aureus ATCC29213 is 4 μg ml−1i.e. ∼8 μM). At this screening concentration, less active products would not have been detected, even if formed in high yield. In retrospect, it may have been helpful to have screened at higher total product concentration(s) in Round 1. This may have enabled the identification of more diverse, yet synthetically accessible, products with significant (albeit lower) antibacterial activity. These hit reactions could then have informed the design of a more diverse reaction array in Round 2.
 | ||
| Fig. 7 Summary of expanded structure–activity relationships. | ||
Conclusions
We have shown that the design of reaction arrays within ADS workflows may be algorithmically-driven. Here, the execution of reaction arrays, whose design was based on the similarity of substrate/co-substrate combinations to those known to yield bioactive products, enabled expansion of a series of antibacterial quinazolinones. The approach enabled efficient definition of the structure–activity relationships for this compound series. The compounds had comparable activity against S. aureus ATCC29213 and the methicillin-resistant USA300 JE2 strain, though were generally significantly less active against the laboratory strain SH1000; crucially, they displayed selective toxicity against bacteria, and were not active against yeast. In addition, it was demonstrated that ADS could be underpinned by photoredox-catalysed reactions, a reaction class that can enable diverse chemical space to be explored. Overall, it is envisaged that algorithms may ultimately enable the fully autonomous activity-directed discovery of small molecules with a wide range of biological functions.Author contributions
The research was conceptualised by AN, AJO'N and SW; supervised by AN, AJO'N, SW and AL; and performed by DF, SF, AM, DD and AN. This paper was drafted by AN, and reviewed and edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC (EP/N025652/1) and University of Leeds for support; Eduardo Rico for discussions and assistance; and Sam Liver, Sam Griggs, Chris Arter and George Karageorgis for helpful discussions. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.References
- G. Karageorgis, S. Liver and A. Nelson, ChemMedChem, 2020, 15, 1776 CrossRef CAS PubMed.
- For a conceptually-related approach known as synthetic fermentation, see: (a) I. A. Stepek and J. W. Bode, Curr. Opin. Chem. Biol., 2018, 46, 18 CrossRef CAS PubMed; (b) Y.-L. Huang and J. W. Bode, Nat. Chem., 2014, 6, 877 CrossRef CAS PubMed; (c) J. G. Hubert, I. A. Stepek, H. Noda and J. W. Bode, Chem. Sci., 2018, 9, 2159 RSC; (d) I. A. Stepek, T. Cao, A. Koetemann, S. Shimara, B. Wollscheid and J. W. Bode, ACS Chem. Biol., 2019, 14, 1030 CrossRef CAS PubMed.
- G. Karageorgis, S. Warriner and A. Nelson, Nat. Chem., 2014, 6, 872 CrossRef CAS PubMed.
- G. Karageorgis, M. Dow, A. Aimon, S. Warriner and A. Nelson, Angew. Chem., Int. Ed., 2015, 54, 13538 CrossRef CAS PubMed.
- A. Leggott, J. E. Clarke, S. Chow, S. L. Warriner, A. J. O'Neill and A. Nelson, Chem. Commun., 2020, 56, 8047 RSC.
- A. Green, F. Hobor, C. Tinworth, S. Warriner, A. Wilson and A. Nelson, Chem. – Eur. J., 2020, 26, 10682 CrossRef CAS PubMed.
- (a) R. Bouley, D. Ding, Z. Peng, M. Bastian, E. Lastochkin, W. Song, M. A. Suckow, V. A. Schroeder, W. R. Wolter, S. Mobashery and M. Chang, J. Med. Chem., 2016, 59, 5011 CrossRef CAS PubMed; (b) S. Gatadi, T. V. Lakshmi and S. Nanduri, Eur. J. Med. Chem., 2019, 170, 157 CrossRef CAS PubMed; (c) Y. Qian, et al. , J. Med. Chem., 2020, 63, 5287 CrossRef CAS PubMed.
- S. Chow, S. Liver and A. Nelson, Nat. Rev. Chem., 2018, 2, 174 CrossRef CAS.
- (a) J. Dong, Z. Wang, X. Wang, H. Song and Q. Wang, Sci. Adv., 2019, 5, eaax9955 CrossRef CAS PubMed; (b) X. Ji, Q. Liu, Z. Wang, P. Wang, G. Deng and H. Huang, Green Chem., 2020, 22, 8233 RSC; (c) Z. Wang, Q. Liu, X. Ji, G. Deng and H. Huang, ACS Catal., 2019, 10, 154 CrossRef; (d) Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512 RSC.
- F. R. Cockerill III, M. A. Wikler, J. Alder, M. N. Dudley, G. M. Eliopoulos, M. J. Ferraro, D. J. Hardy, D. W. Hecht, J. A. Hindler, J. B. Patel, M. Powell, J. M. Swenson, R. B. Thomson Jr., M. Traczewski, J. D. Turnidge, M. P. Weinstein and B. L. Zimmer, CLSI, M07-A9: Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, Clinical and Laboratory Standards Institute, 2012, 9th edn Search PubMed.
- (a) I. Soni, H. Chakapani and S. Chopra, Genome Announc., 2015, 3, e01095-15 CrossRef PubMed; (b) P. D. Fey, J. L. Endres, V. K. Yajjala, T. J. Widhelm, R. J. Boissy, J. L. Bose and K. W. Bayles, mBio, 2013, 4, e00537-12 CrossRef PubMed; (c) M. J. Horsburgh, J. L. Aish, I. J. White, L. Shaw, J. K. Lithgow and S. J. Foster, J. Bacteriol., 2002, 184, 5457 CrossRef CAS PubMed; (d) A. J. O'Neill, Lett. Appl. Microbiol., 2010, 51, 358 CrossRef PubMed.
- D. J. Farrell, M. Robbins, W. Rhys-Williams and W. G. Love, Antimicrob. Agents Chemother., 2010, 55, 1177 CrossRef PubMed.
- (a) D. Kumar, P. Jadhavar, M. Nautiyal, H. Sharma, P. Meena, L. Adane, S. Pancholia and A. Chakraborti, RSC Adv., 2015, 5, 30819 RSC; (b) P. Jadhavar, T. Dhameliya, M. Vaja, D. Kumar, J. Sridevi, P. Yogeeswari, D. Sriram and A. Chakraborti, Bioorg. Med. Chem. Lett., 2016, 26, 2663 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742 CrossRef CAS PubMed.
- N. Gunantara and Q. Ai, Cogent Eng., 2018, 5, 1502242 CrossRef.
- S. Gatadi, J. Gour, M. Shukla, G. Kaul, S. Das, A. Dasgupta, S. Malasala, R. Borra, Y. Madhavi, S. Chopra and S. Nanduri, Eur. J. Med. Chem., 2018, 157, 1056 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6989 CrossRef PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (e) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2021, 122, 1875 CrossRef PubMed; (f) P. Murray, J. Cox, N. Chiappini, C. Roos, E. McLoughlin, B. Hejna, S. Nguyen, H. Ripberger, J. Ganley, E. Tsui, N. Shin, B. Koronkiewicz, G. Qiu and R. Knowles, Chem. Rev., 2021, 122, 2017 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01404a |
| ‡ We did also investigate screening at 0.1 and 1 (M total product concentration, but no new hits were detected at these lower concentrations. |
| This journal is © The Royal Society of Chemistry 2022 |