
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular self-associating amphiphiles as aqueous pollutant scavengers†
Rebecca J.
Ellaby
,
Lisa J.
White
,
Jessica E.
Boles
,
Sena
Ozturk
and
Jennifer R.
Hiscock
 *
*
School of Chemistry and Forensics, University of Kent, Canterbury, CT2 7NH, UK. E-mail: J.R.Hiscock@Kent.ac.uk
First published on 15th September 2022
Abstract
We present a series of supramolecular self-associated amphiphiles, which spontaneously self-assemble into aggregated species. These aggregates are shown to absorb a variety of (polar) micropollutants from aqueous mixtures and as a result we determine the suitability for this technology to be developed further as aqueous environmental clean-up agents.
The increasing global occurrence of organic micropollutants such as dyes, pesticides and pharmaceuticals in water sources has raised many concerns, due to the detrimental effects that these agents elicit on both aquatic ecosystems and human health.1–5 Typically, cheap adsorbent materials such as activated charcoal are used to remove organic pollutants from aqueous systems. However, the use of these environmental clean-up methods is limited by slow adsorption times6,7 and decreased affinity towards hydrophilic pollutants.8 In addition, regeneration of these environmental clean-up materials is energy intensive and inefficient as current processes do not fully restore original material performance.9
The field of supramolecular chemistry has produced a number of novel materials based innovations associated with the removal of organic pollutants from aqueous systems.3 These innovations include the use of absorbent materials such as organogelators for the remediation of oil spills;10–13 and the incorporation of low molecular weight ‘host’ species within a material to selectively coordinate specific micropollutant ‘guest’ species in an aqueous environment, as exemplified by Alsbaiee and co-workers.4 Here, mesoporous polymers, produced through the crosslinking of β-cyclodextrin with tetrafluoroterephthalonitrile, are shown to adsorb a range of common organic pollutants such as plastic components, pharmaceuticals, pesticides and aromatic carcinogens. The result of this innovation was a material demonstrating adsorption rate constants 15–200 times greater than other non-porous β-cyclodextrins and the industry standard, activated charcoal. In addition, mild washing processes were found to fully regenerate material activity. Furthermore, Fernando, Mako and co-workers have shown water-soluble pillar[5]arenes to coordinate a variety of organic analytes, with association constants of 103–1010 M−1 in aqueous conditions. These complexes were then effectively removed from the aqueous bulk using a cationic exchange resin.14
Herein, we combine the principles of supramolecular material formation, host–guest complex formation and the hydrophobic/hydrophilic effect to highlight the potential of supramolecular self-associating amphiphiles (SSAs), Fig. 1–1–5, as next-generation polar (6–7) and non-polar (8) organic micropollutant aqueous environmental clean-up agents. Compounds 6–8 were chosen as a representative group of (polar) organic micropollutants due to: (i) their warranted removal from aqueous environments; (ii) the combination of basic, acidic and anionic substituents and; (iii) their representative industrial uses. Here 6 acts as a representative coumarin derivative,15,167 acts as a commonly used fluorescent probe,17,18 and 8 is a commonly used insect repellent.19
 | ||
| Fig. 1 Chemical structures of SSAs 1–5 and co-formulants 6–8. | ||
To date the anionic component of an SSA has been shown to self-associate in DMSO-d6/H2O mixtures and within the solid state to form hydrogen bonded dimers, as exemplified within Fig. 2.20–22 However, in aqueous or H2O/EtOH 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solutions these same compounds have been shown to self-associate producing spherical aggregates,21,22 while the addition of inorganic salts, such as NaCl, followed by an annealing process can result in the formation of hydrogel fibres.23 In addition, these SSAs have been shown to form stable aggregate structures when in the presence of one molar equivalent of drug(like) species, highlighting the potential of this molecular technology to be developed as a novel class of drug delivery agent.24 We now extend this work to show that SSA technology can be adapted towards the uptake of (polar) organic micropollutants from an aqueous mixture while retaining aggregate stability. This highlights the potential for this molecular technology to be developed as next-generation aqueous environment clean-up agents. The anionic components of SSAs 1, 2, 3 and 5 were designed to enable the incorporation of either a basic (1–3) or acidic (5) protonatable site to enable the coordination of complimentary acidic (6, 7) or basic (8) organic (polar) species within the resultant SSA aggregate structure, driving pollutant uptake.
:1 solutions these same compounds have been shown to self-associate producing spherical aggregates,21,22 while the addition of inorganic salts, such as NaCl, followed by an annealing process can result in the formation of hydrogel fibres.23 In addition, these SSAs have been shown to form stable aggregate structures when in the presence of one molar equivalent of drug(like) species, highlighting the potential of this molecular technology to be developed as a novel class of drug delivery agent.24 We now extend this work to show that SSA technology can be adapted towards the uptake of (polar) organic micropollutants from an aqueous mixture while retaining aggregate stability. This highlights the potential for this molecular technology to be developed as next-generation aqueous environment clean-up agents. The anionic components of SSAs 1, 2, 3 and 5 were designed to enable the incorporation of either a basic (1–3) or acidic (5) protonatable site to enable the coordination of complimentary acidic (6, 7) or basic (8) organic (polar) species within the resultant SSA aggregate structure, driving pollutant uptake.
 | ||
| Fig. 2 Single crystal X-ray structure obtained for SSA 3 showing: (a) an anionic SSA dimer, stabilised through the formation of four intermolecular hydrogen bonds; and (b) the extended hydrogen bonded network where the SSA anion dimers are connected through the presence of intermolecular hydrogen bonded water molecules. Where appropriate the H2O and TBA counter-cations have been omitted for clarity. Grey = carbon, blue = nitrogen, red = oxygen, yellow = sulfur, white = hydrogen, red dashed lines = hydrogen bonds.‡ The presence of the urea-sulfonate hydrogen bonded dimer is promoted by the presence of the weakly coordinating, comparatively hydrophobic TBA counter-cation.20 | ||
SSAs 1 and 2 were synthesised through previously published methods.21,24 SSA 3 and 4 were synthesised through the reaction of tetrabutylammonium (TBA) aminomethane sulfonate with triphosgene and either 4-aminopyridine or tert-butyl-4-aminobenzoate as appropriate to afford the final product as a white solid in a yield of 64%, or a brown oil in a yield of 81% respectively. SSA 5 was obtained through the deprotection of 4 using trifluoroacetic acid in DCM to afford the final product as a white solid in a yield of 80%. Although at present these SSAs themselves are produced through environmentally expensive processes, e.g. the use of triphosgene, it is possible to substitute these reactants with agents such as 1,1′-carbonyldiimidazole.
Initially, the self-association properties of 1–5 alone were explored in an aqueous solution standardised with 5% EtOH. TBA was used as the counter-cation throughout, as the presence of this cation has previously been shown to stabilise SSA higher-order aggregate formation at lower concentrations, while retaining solubility/self-associated aggregate distribution under these experimental conditions.21
The results from initial quantitative 1H NMR studies, conducted in a D2O/EtOH 19:1 solution, showed that when the signals for the SSA are comparatively integrated against those of the EtOH standard, all SSAs apart from 1 demonstrated an apparent ‘loss’ of material from the solution, confirming the presence of solid-like higher-order aggregated species with SSAs 2–5 (Table 1). Here we believe the hydrophilic nature of the primary amine functionality, contained within the structure of 1 decreases the amphiphilic nature of this SSA, limiting aggregate formation under these conditions. For SSAs 2–5, the percentage of SSA ‘lost’ from solution indicates the percentage of the SSA anionic or cationic component to become coordinated within a solid-like, higher-order self-associated aggregate species at this concentration.
Estimating ± 10% error for the experimental values obtained via quantitative 1H NMR studies, both SSAs 2 and 3 exhibit a disproportionate ratio of the SSA anionic:cationic component to become coordinated within these higher-order self-associated structures. We believe this is due to the presence of basic amino functionalities (tertiary amino and pyridinium groups present within SSAs 2 and 3 respectively) becoming protonated and thus forming a zwitterion, irradicating the need for the TBA to produce a charge balanced aggregate structure. Comparing the pKa of the SSA substituents diethylaniline (6.57)25 and pyridinium (5.23),25 a greater proportion of the anionic component of 2 is likely to exist in a zwitterionic form then 3, under these experimental conditions. This correlates with the degree of deviation for the SSA anion:cation, higher-order aggregate composition, away from the desired 1:1 ratio. However, we hypothesise that it is the small difference in SSA substituent logP value (pyridine = 0.73,26 diethylaniline = 0.9527), combined with monomeric unit molecular packing constraints, which contribute to the increased quantities of 2 over 3 to be incorporated into the SSA aggregated structures at 5.56 mM. In addition, we believe that it is the increase in SSA substituent hydrophilicity that is responsible for driving down the proportion of 5 to be coordinated into these higher-order aggregates, when compared to 4, under analogous experimental conditions.
The proportion of an SSA to be coordinated into higher-order self-associated structures at 5.56 mM was found to correlate with decreasing critical aggregation concentration (CAC) values, where the proportion of SSA anionic and cationic components were maintained at a 1:1 ratio (Table 1 – SSAs 4 and 5). It should be noted here that the definition of CAC is the concentration at which and additional SSA added to a solution will result in the formation of higher-order self-associated structures. However, this does not mean that at concentrations below the CAC that higher-order self-associated aggregated species do not exist in equilibrium with those forming lower order species, or those coordinating at the interface of the solution.
Comparing the hydrodynamic diameter (dH) of those self-associated species that exist at an SSA concentration of 5.56 mM, structures of similar sizes were identified for SSAs 2–4 (Table 2). However, SSA 5 produced aggregated species that are approximately double this size. This is the only SSA that contains a carboxylic acid functionality and therefore we believe that it is the presence of this functionality that is driving the formation of these larger SSA aggregates. The elucidation of SSA aggregate stability was achieved through the determination of zeta potential values (Table 2). Here the presence of the pyridinium functionality (3) was found to drive down the stability of those nanostructures formed, in comparison to those structures produced at this concentration by SSAs 2, 4 and 5.
:1 solution of 1–5 (5.56 mM) at 298 K, following an annealing process
To explore the potential of SSAs substituted with either a basic (1–3) or acidic (5) functionality to absorb the representative complimentary organic micropollutants 6–8, 1:1 molecular co-formulations a–l (Table 3) were prepared in aqueous (or D2O) mixtures.
| Co-formulation | Molecular components | Co-formulation | Molecular components |
|---|---|---|---|
| a | 1 + 6 | g | 3 + 7 |
| b | 2 + 6 | h | 5 + 7 |
| c | 3 + 6 | i | 1 + 8 |
| d | 5 + 6 | j | 2 + 8 |
| e | 1 + 7 | k | 3 + 8 |
| f | 2 + 7 | l | 5 + 8 |
Here again quantitative 1H NMR studies, using EtOH as the internal standard, except for co-formulations i–l, enabled the proportion of each SSA co-formulation component incorporated into higher-order SSA aggregated species to be calculated (Table 4). Quantitative 1H NMR experiments conducted with co-formulations i–l were optimised towards a D2O/1.0% acetonitrile solution, where acetonitrile was used as the internal standard due to the presence of NMR signal overlap. Therefore, these data should not be directly compared with the rest of the data set.
| Co-formulation | Quantitative 1H NMR (%) | CAC (mM) | Surface tension at CAC (mN m−1) | ||
|---|---|---|---|---|---|
| Anion | Cation | Co-formulant | |||
| a Could not be determined due to solubility constraints. | |||||
| a | 5224 |
5524 |
8624 |
24 |
N/A |
| b | 4124 |
4324 |
8124 |
19.8724 |
37.9524 |
| c | 3 | 3 | 14 | 16.10 | 56.89 |
| d | 2 | 18 | 3 | N/A | |
| e | 0 | 0 | 0 | N/A | |
| f | 0 | 0 | 0 | 44.13 | 51.60 |
| g | 10 | 11 | 25 | N/A | |
| h | 38 | 40 | 23 | 21.38 | 44.69 |
| i | 0 | 0 | 0 | 22.30 | 44.90 |
| j | 0 | 0 | 0 | 22.04 | 44.83 |
| k | 7 | 7 | 2 | 22.45 | 51.63 |
| l | 31 | 30 | 4 | N/A | |
The presence of co-formulant 6, which contains a carboxylic acid functionality, was found to drive the formation of SSA aggregates containing complimentary primary amine substituted 1. Here approximately 50% of the SSA is now incorporated into these aggregates, which were also found to incorporate 86% of this co-formulant. However, the presence of basic co-formulants 7 and 8 did not induce SSA aggregate formation.
The presence of basic co-formulants 7 and 8 also prevented the formation of aggregated species with SSA 2. However, although the presence of complimentary acidic co-formulant 6 was found to decrease the proportion of SSA anion within those higher order aggregated species of 2, those aggregates formed were now found to contain a 1:1 ratio of SSA anion:cation, while also absorbing 81% of the co-formulant.
SSAs 3 and 5 were found to be the most effective at absorbing the tertiary amine co-formulant 7 into any self-associated structure, with 25% and 23% of this co-formulant absorbed respectively. As previously hypothesised, tertiary amino co-formulant 8 was found to disrupt the formation of all SSA aggregated structures under those conditions tested, apart from those of SSA 5, which contains the complimentary carboxylic acid functionality. Here, the proportion of SSA anion and cation to become coordinated within these structures was found to double. However, only 4% of the co-formulant was absorbed into these structures as a result.
To better demonstrate the potential for SSA self-associated aggregate species to be developed for real-world application, a second set of experiments were performed under analogous conditions. Here, however, the SSA self-associated aggregates were allowed to form in situ (with the exception of 1, see Table 1), prior to the addition of the co-formulant in a 1:1 ratio. Analogous quantitative 1H NMR experiments were then conducted to confirm the proportion of co-formulant to be absorbed into the SSA aggregate species as a function of time. The results of these studies are summarised in Fig. 3, and compared against the proportions of co-formulant incorporated into the SSA aggregated structure during, rather than after initial aggregate formation.
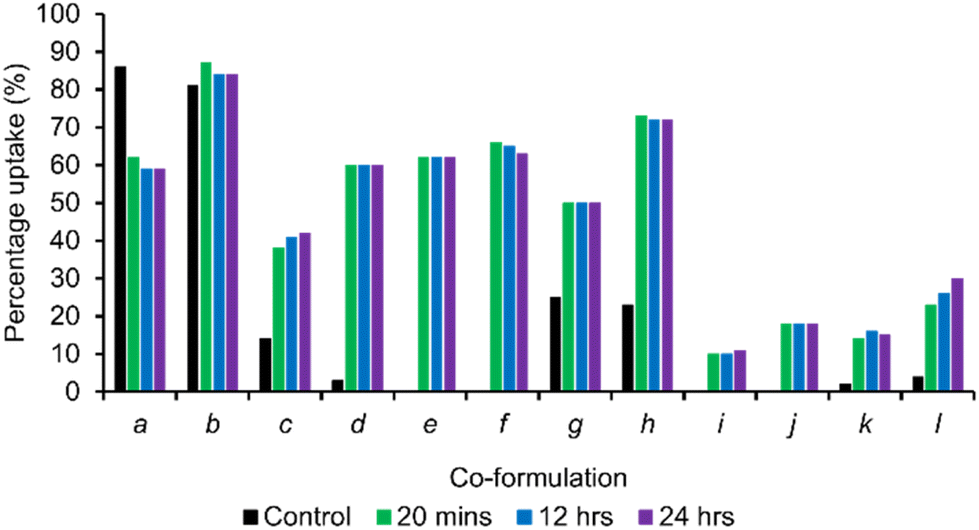 | ||
| Fig. 3 Graph comparing the percentage uptake of co-formulant into preformed SSA self-associated structures of 1–3 and 5 over a 24-hour period at 298 K and a concentration of 5.56 mM. Here the control data set represents the percentage of co-formulant incorporated into SSA self-associated structures as they were formed. Experiments involving co-formulations a–h were undertaken in a D2O/EtOH 19:1 solutions, whereas analogous experiments involving co-formulations i–j were optimised within a D2O/1.0% MeCN solution. Here the EtOH and MeCN signals were used as the internal solution standards as applicable. | ||
The results of these studies show that all co-formulations, except a, increase the proportion of co-formulant absorbed into the SSA aggregate structure, where that aggregated structure is allowed to form in the absence of the co-formulant. In addition, the proportion of co-formulant that is absorbed into the SSA aggregate structure remains fairly constant across the 24 h monitoring period, meaning that these structures, when formed under these experimental conditions, are stable. Only co-formulation c and l show any evidence of time dependant co-formulant uptake, while only co-formulation f shows any evidence of slight aggregate destabilisation and resultant co-formulant release. Interestingly, in the case of co-formulations c and l, this involves pairing a basic/acidic substituted SSA with the complimentary acidic/basic co-formulant.
Also, worthy of note are the enhanced levels of co-formulant adsorption observed for c and d, compared to the control value. As shown in Table 1, under these experimental conditions SSA 1 fails to form stable higher order aggregates. However, in comparison to the control, adding the co-formulant to a solution of ‘pre-formed’ SSA aggregates of 1 dramatically increases co-formulant uptake. Therefore, we suggest the presence of lower-order SSA pre-organisation events to be undertaken by 1 under these experimental conditions, that are not detectable by our quantitative 1H NMR experiments, predisposing this system towards co-formulant uptake.
From these data we conclude that preformed aggregates of 2 are the most effective for the broad-spectrum adsorption of co-formulates 6–8. We believe this is because of the increased hydrophobic nature of the SSA anion basic substituent, which also makes this SSA the most effective at absorbing the carboxylic acid substituted coumarin derivative (6). Furthermore, SSA 5, substituted with the carboxylic acid functionality, was also found to be the most effective towards the adsorption of basic co-formulant 8. In addition, the aggregates formed by SSA 5 were found to be the most effective towards the adsorption of anionic co-formulant 7. Here we believe that this preference maybe due to complimentary hydrogen bond formation between the carboxylic acid group (acting as the hydrogen bond donor) of this SSA anion and the sulfonate moieties, contained within the structure of the co-formulant acting as complimentary hydrogen bond accepting groups.
Fig. 4 provides some insight into the physical properties of higher-order SSA:co-formulant aggregate structures. In this instance all experiments were performed in a H2O/EtOH 19:1 solution to enable direct comparison of all values. DLS analysis (Fig. 4a) showed the size of those structures produced through either SSA co-formulant addition method to remain constant for co-formulations c, g and k, which are all aggregated species produced from SSA 3. This makes 3 the only SSA to retain aggregate size independent of co-formulant addition method. Where SSA aggregate formation was followed by co-formulant addition, SSAs 2 and 5 both showed an increase in the size of co-formulated aggregate produced. However, subsequent comparative zeta potential analysis showed those self-associated aggregates of a, b and h, produced through direct co-formulation, to form structures which exhibit greater stability.
 | ||
| Fig. 4 Summary of (a) DLS (bars) and associated polydispersity index values (crosses) and (b) zeta potential measurements for co-formulations a–l conducted produced through institute formation (black/grey) or after 20 minutes post addition to pre-formed SSA aggregated species (blue) at a concentration of 5.56 mM in a H2O/EtOH 19:1 solution at 298 K. | ||
Conclusions
We present a series of five SSAs (1–5) and have characterised the self-associative properties of these compounds in a H2O(D2O)/EtOH 19:1 solution. We confirm SSAs 2–5 to form self-associated aggregate species at a concentration of 5.56 mM under these experimental conditions, even though in some instances this is below the CAC. Further to this, we confirm our hypothesis that complementary acidic/basic functionalities appended to the SSA anionic component can be used to drive the uptake of (polar) micropollutants from largely aqueous environments. We also report initial evidence that suggests that complementary hydrogen bonding events may also be used to drive polar micropollutant uptake into SSA aggregate species. In summary, we believe the results of this study support our hypothesis that this SSA technology may be adapted to produce rapid (polar) micropollutant adsorption technologies, enabling the effective clean-up of aqueous environments. As a result, further work is ongoing in this area to explore the effects of altering micropollutant:SSA ratio, temperature, SSA structure and solvent environment on the efficacy of these systems.
Author contributions
RJE: Investigation; validation; writing – original draft, review & editing. JEB, LJW and SO: Investigation; validation; writing – review & editing. JRH: Conceptualization; funding acquisition; project administration; supervision; writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JEB would like to thank UKHSA and the University of Kent for PhD studentship funding. RJE and SO would like to thank the University of Kent for funding. JRH and LJW would like to thank the UKRI for JRH's Future Leaders Fellowship (MR/T020415/1).References
- J. Yeston, R. Coontz, J. Smith and C. Ash, Science, 2006, 313, 1067 CrossRef CAS.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. Von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 CrossRef CAS PubMed.
- G. T. Williams, C. J. E. Haynes, M. Fares, C. Caltagirone, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2021, 50, 2737–2763 RSC.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS.
- S. D. Richardson and S. Y. Kimura, Anal. Chem., 2020, 92, 473–505 CrossRef CAS PubMed.
- E. K. Putra, R. Pranowo, J. Sunarso, N. Indraswati and S. Ismadji, Water Res., 2009, 43, 2419–2430 CrossRef CAS PubMed.
- J. J. M. Órfão, A. I. M. Silva, J. C. V. Pereira, S. A. Barata, I. M. Fonseca, P. C. C. Faria and M. F. R. Pereira, J. Colloid Interface Sci., 2006, 296, 480–489 CrossRef PubMed.
- L. Kovalova, D. R. U. Knappe, K. Lehnberg, C. Kazner and J. Hollender, Environ. Sci. Pollut. Res., 2013, 20, 3607–3615 CrossRef CAS PubMed.
- G. San Miguel, S. D. Lambert and N. J. D. Graham, Water Res., 2001, 35, 2740–2748 CrossRef CAS.
- A. M. Vibhute and K. M. Sureshan, ChemSusChem, 2020, 13, 5343–5360 CrossRef CAS PubMed.
- B. Doshi, M. Sillanpää and S. Kalliola, Water Res., 2018, 135, 262–277 CrossRef CAS PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- B. Zhang, S. Chen, H. Luo, B. Zhang, F. Wang and J. Song, J. Hazard. Mater., 2020, 387, 121460 CrossRef PubMed.
- A. Fernando, T. L. Mako, A. M. Levenson, P. T. Cesana, A. M. Mendieta, J. M. Racicot, B. DeBoef and M. Levine, Supramol. Chem., 2019, 31, 545–557 CrossRef CAS.
- A. Detsi, C. Kontogiorgis and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat., 2017, 27, 1201–1226 CrossRef CAS PubMed.
- A.-M. Katsori and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat., 2014, 24, 1323–1347 CrossRef CAS.
- R. Kumar, R. Yadav, M. A. Kolhe, R. S. Bhosale and R. Narayan, Polymer, 2018, 136, 157–165 CrossRef CAS.
- A. Chandra, S. Prasad, H. Iuele, F. Colella, R. Rizzo, E. D'Amone, G. Gigli and L. L. del Mercato, Chem. – Eur. J., 2021, 27, 13318–13324 CrossRef.
- M. Brown and A. A. Hebert, J. Am. Acad. Dermatol., 1997, 36, 243–249 CrossRef CAS PubMed.
- L. R. Blackholly, H. J. Shepherd and J. R. Hiscock, CrystEngComm, 2016, 18, 7021–7028 RSC.
- L. J. White, S. N. Tyuleva, B. Wilson, H. J. Shepherd, K. K. L. Ng, S. J. Holder, E. R. Clark and J. R. Hiscock, Chem. – Eur. J., 2018, 24, 7761–7773 CrossRef CAS PubMed.
- L. J. White, N. J. Wells, L. R. Blackholly, H. J. Shepherd, B. Wilson, G. P. Bustone, T. J. Runacres and J. R. Hiscock, Chem. Sci., 2017, 8, 7620–7630 RSC.
- L. J. White, J. E. Boles, N. Allen, L. S. Alesbrook, J. M. Sutton, C. K. Hind, K. L. F. Hilton, L. R. Blackholly, R. J. Ellaby, G. T. Williams, D. P. Mulvihill and J. R. Hiscock, J. Mater. Chem. B, 2020, 8, 4694–4700 RSC.
- L. J. White, J. E. Boles, K. L. F. Hilton, R. J. Ellaby and J. R. Hiscock, Molecules, 2020, 25, 4126 CrossRef CAS PubMed.
- S. Romand, J. Schappler, J. L. Veuthey, P. A. Carrupt and S. Martel, Eur. J. Pharm. Sci., 2014, 63, 14–21 CrossRef CAS PubMed.
- S. H. Unger, J. R. Cook and J. S. Hollenberg, J. Pharm. Sci., 1978, 67, 1364–1367 CrossRef CAS PubMed.
- Po Y. Lu and R. L. Metcalf, Environ. Health Perspect., 1975, 10, 269–284 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and DLS, zeta potential, tensiometry, mass spectrometry, NMR spectroscopy and crystallography data. CCDC 2108071. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01365g |
| ‡ A suitable crystal was selected and mounted on a Rigaku Oxford Diffraction Supernova diffractometer. Data were collected using Cu Kα radiation at 100 K. Structures were solved with the ShelXT28 or ShelXS structure solution programs via Direct Methods and refined with ShelXL29 on Least Squares minimisation. Olex230 was used as an interface to all ShelX programs. CCDC deposition number for the structure shown in Fig. 2 = 2108071. |
| This journal is © The Royal Society of Chemistry 2022 |