
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclopropylmethyldiphosphates are substrates for sesquiterpene synthases: experimental and theoretical results†
Cong Duc
Tran
a,
Gerald
Dräger
 a,
Henry Frederik
Struwe
a,
Lukas
Siedenberg
a,
Somi
Vasisth
b,
Jörg
Grunenberg
b and
Andreas
Kirschning
*a
a,
Henry Frederik
Struwe
a,
Lukas
Siedenberg
a,
Somi
Vasisth
b,
Jörg
Grunenberg
b and
Andreas
Kirschning
*a
aInstitute of Organic Chemistry, Leibniz University Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: andreas.kirschning@oci.uni-hannover.de
bInstitute of Organic Chemistry, TU Braunschweig, Hagenring 30, 38106 Braunschweig, Germany
First published on 15th September 2022
Abstract
New homo-sesquiterpenes are accessible after conversion of presilphiperfolan-8β-ol synthase (BcBOT2) with cyclopropylmethyl analogs of farnesyl diphosphate, and this biotransformation is dependent on subtle structural refinements. Two of the three cyclisation products are homo variants of germacrene D and germacrene D-4-ol while the third product reported contains a new bicyclic backbone for which no analogue in nature has been described so far. The findings on diphosphate activation are discussed and rationalised by relaxed force constants and dissociation energies computed at the DFT level of theory.
Introduction
Terpene cyclases (TCs) play an important role in the biosynthesis of terpenes.1 They use linear, unsaturated, methyl-branched precursors activated as terminal diphosphate esters to initiate cascades of cation reactions. In these processes, sesquiterpene synthases, such as presilphiperfolan-8β-ol synthase (BcBOT2), a fungal sesquiterpene cyclase from Botrytis cinerea, use farnesyl diphosphate (FPP, 1) as a precursor equipped with a total of 15 carbon atoms.2In the case of BcBOT2, the product of such a cation cascade is the tricyclic sesquiterpene presilphiperfolan-8β-ol (2).3 Interestingly, our recent studies4 show that BcBOT2 is well suited to investigate the scope of substrate promiscuity of sesquiterpene cyclases (STCs).5 In particular, we demonstrated that linear ether and thioether derivatives of FPP 1, as well as isomers in which the methyl groups are shifted by one position on the alkenes toward the diphosphate group, are transformed by BcBOT2, leading to new unnatural sesquiterpene ring systems and backbones.4 The formation of 2 is initiated by a ring closure between carbon atoms 1 and 11.
Chemically, the cyclopropane ring can be considered a homoalkene group, showing similar but reduced π-reactivity compared to alkenes.6 The formation of a carbocation in the neighboring of the cyclopropyl ring leads to a rapid equilibrium between the cyclopropylmethyl and the cyclobutyl cation. (3 → 4).7 Given these features and chemical properties of the cyclopropyl ring, we wondered at the outset of the present study whether TCs and specifically BcBOT2 is also capable of activating cyclopropylmethyl diphosphate and initiating a cascade reaction arising from this situation. And if this particularly promiscuous sesquiterpene cyclase is capable of such an activation, the question arises whether the equilibrium between cations 3 and 4 initially plays a role before the first cyclisation step occurs. This, in turn, is clearly a theoretical issue and can be solved by determining relaxed force constants and dissociation energies calculated, for example, at the DFT level of theory.8 Noteworthy, while the expected transformation products from cyclopropanes 5 and 7 should have the same molecular formula as the typical products from natural FPP 1, diphosphates 6 and 8 should yield “homo-sesquiterpenes” (Scheme 1).
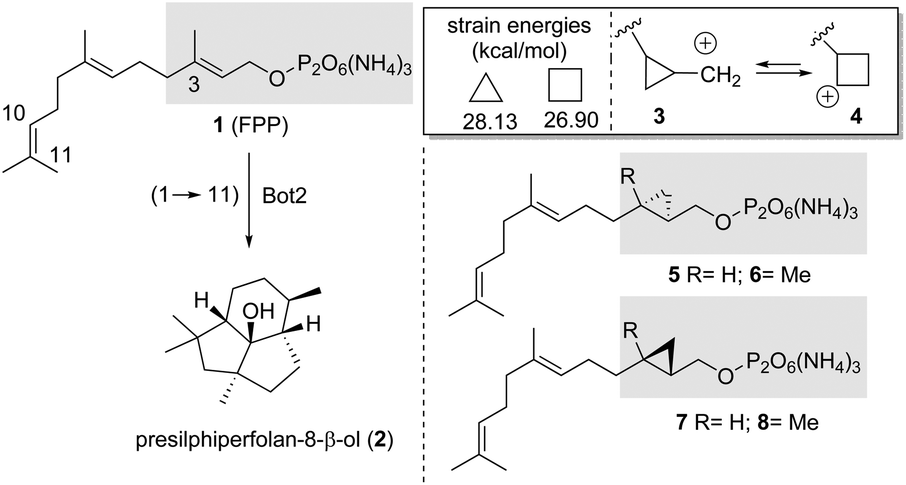 | ||
| Scheme 1 Left: transformation of FPP 1 by BcBOT2 and formation of presilphiperfolan-8β-ol (2). Top right: estimated strain energies (kcal mol−1) of cyclopropane and cyclobutane based on single conformation increments (ref. 9) and rearrangement reaction of the cyclopropylmethyl cation 3. Bottom right: structures of FPP-analogues 5–8 bearing a cyclopropylmethyl diphosphate moiety. | ||
Results and discussion
Synthesis of FPP derivatives
The four diphosphates 5–8 were not known at the beginning of the project, so we had to develop enantioselective syntheses for them. Briefly, the syntheses of the cyclopropane derivatives 13 and 14 devoid of the methyl group began with geraniol (9), which was converted to alcohol 10 after bromination, elongation of the carbon chain using a copper enolate derived from ethyl acetate, followed by ester reduction (Scheme 2, top).9,10 From there, Swern oxidation, olefination, and reduction of the ethyl ester provided the allyl alcohol 11. At this point, we employed the method of Charette11 using the enantiomeric boronates 12a and 12b so that at this point the synthesis split toward the two enantiomeric cyclopropylmethyl alcohols 5 and 7. We found that the common method of converting the secondary allylic alcohol to the corresponding diphosphates, in which the corresponding allylic chlorides or bromides are first formed as intermediates,12 did not work for the cyclopropylmethyl alcohols 13–16. Therefore, we switched to an alternative method that yields diphosphates in one step from the corresponding alcohols.13 However, this method does not provide very high yields because, above all, the corresponding mono- and triphosphates are also formed, which have to be separated from the diphosphates. Consequently, the synthesis to diphosphates 5 and 7 were completed by direct phosphorylation of alcohols 13 and 14 with bis-triethylammonium phosphate. | ||
| Scheme 2 Synthesis of cyclopropylmethyl diphosphates 5–8 (DIPA = diisopropanolamine, EtOAc = ethyl acetate, DIBAL-H = diisobutylaluminium hydride, DMSO = dimethylsulfoxide, DME = dimethoxy ethane, TEAP = bis-triethylammonium phosphate). | ||
Likewise, cyclopropylmethyl diphosphates 6 and 8 were prepared from farnesol (15) after asymmetric cyclopropanation, again using the two chiral ligands 12a and 12b and subsequent phosphorylation (Scheme 2, bottom). The enantiomeric excess of the newly formed cyclopropanes was determined by chiral GC, and the hydrodex-β-6-tbdm column (l = 25 m, inner θ = 0.25 mm, film thickness = 0.25 μm) proved to be the most suitable for separating the two enantiomers in each case (5: >90% ee; 6: 94% ee; 7: >90% ee; 8: >90% ee).14
Biotransformations
Next, BcBOT2 was cloned and expressed in E. coli. as reported before.4In vitro enzyme tests for determining enzyme activity and for optimizing substrate tolerance were conducted on small scales (150 μM substrate, 0.1 mg mL−1 enzyme) using natural precursor FPP 1 (see ESI†). The outcome of biotransformations with FPP derivatives 5–8 with BcBOT2 is summarised in Table 1.| Diphosphate | Products m/z | Retention index (RI) |
|---|---|---|
| a The structures of these products are reported in Scheme 3. b both products are supposedly enantiomers. c the product is formed in about 0.3% at 35–40 °C. | ||
| 5 | 204 | 1771 |
| 6 | Major:a 218, 2× 236 | 1599, 1773, 1782 |
| Minor: 3× 218 | 1517,b 1548, 1560 | |
| 7 | 204, 222 | 1771, 1750 |
| 8 | 218 | 1516b,c |
Biotransformations with cyclopropane derivatives 5 and 7, which lack the methyl group at C3, yielded several products, however, these exhibited very low signal intensities (<1% compared to biotransformation using FPP). The same was true for (R,R)-derivative 8. Cyclopropylmethyl diphosphate 6, however, was readily accepted by BcBOT2. GC/MS analysis identified three major products (one product: m/z = 218 and two products: m/z = 236) that had formed in sufficient quantities to repeat the experiment on a larger scale (batch size: 75 μmol 6).
The constitution of the three new homo-sesquiterpenes 17–19 (Scheme 3) was elucidated by NMR spectroscopy, using in particular the COSY and HMBC correlations. Selected NOE data provided information on the preferred conformation of the macrocycles and hence stereochemical information on the relative stereochemistry of the substituents on the macrocycles in 18 and 19 (Fig. 1). The configuration of the 1,2-disubstituted olefinic double bonds in macrocycles 17 and 18 is proposed based on the recorded coupling constants (J = 15.2 Hz in 17 and 15.4 Hz in 18) (Table 2). The absolute stereochemistry, assuming a common mechanism for the formation of the three biotransformation products, is derived from the intact cyclopropane moiety in bicyclic biotransformation product 19.
 | ||
| Scheme 3 Formation of cyclisation products 17–19 and structures of sesquiterpene analogues 20–22 (numbering of carbon atoms refers to IUPAC nomenclature). Biotransformations of diphosphates 5, 7, 8 and 23. | ||
 | ||
| Fig. 1 COSY and HMBC (2J correlations are not shown) correlations and selected NOEs. | ||
| 17 | J H5,H6 | J H6,H7 | J H7,H8a | J H7,H8b | J H8a,H8b |
| 9.4 | 15.2 | 9.4 | 5.5 | 12.6 | |
| 18 | J H2a,H2b | J H2a,H3 | J H2b,H3 | J H3,H4 | J H4,H5 |
| 13.0 | 10.0 | 5.4 | 15.4 | 10.0 | |
| 19 | J H1,H2 | J H2,H3 | J H1,CHa | J H1,CHb | J CHa,CHb |
| 9.8 | 1.6 | 5.6 | 8.4 | 4.2 |
The crude product obtained was subjected to column chromatographic purification (silica gel; n-pentane/Et2O = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 4:1 → 2:1), which first allowed separation of the triene 17 (24%) from the two alcohols 18 (11%) and 19 (17%) these were present in a ratio of 2:3, as indicated by the integrals in the 1H NMR spectrum. Triene 17 is the homo-analogue of germacrene D (20) and alcohol 18 corresponds to germacrene D-4-ol (21),15 while the backbone of alcohol 19 is closely related to the sesquiterpene kunzeaol (22).16
:1 → 4:1 → 2:1), which first allowed separation of the triene 17 (24%) from the two alcohols 18 (11%) and 19 (17%) these were present in a ratio of 2:3, as indicated by the integrals in the 1H NMR spectrum. Triene 17 is the homo-analogue of germacrene D (20) and alcohol 18 corresponds to germacrene D-4-ol (21),15 while the backbone of alcohol 19 is closely related to the sesquiterpene kunzeaol (22).16
Our previously collected findings on the pronounced promiscuity of presilphiperfolan-8β-ol synthase (BcBOT2) are further confirmed in this study. Among a number of other STCs assayed, only bacterial caryolan-1-ol synthase (GcoA) from Streptomyces griseus, cubebol synthase (Cop4) from Coprinus cinereus, and epi-isozizaene synthase (Cyc1) from Streptomyces coelicolor convert diphosphate 6 to both alcohols 18 and 19, but only to a small extent (1–10% compared to biotransformations with FPP 1) while in no case the formation of triene 17 was detected. Cop4 also accepted the other enantiomeric cyclopropane 8, but again the overall yields were too small for collecting sufficient amounts of the newly formed products (see ESI†). In selected cases optimisation of the conditions were carried out that included temperature dependence of the biotransformation, elongation of reaction times, the addition of PPase to remove the byproduct diphosphate which might act as an inhibitory and the addition of a second portion of enzyme after 30 minutes. However, only the incubation temperature had an effect on yields. When the olefinic double bond is absent, as in the case of the dihydro-FPP derivative rac-23, GC-MS analysis of the crude product revealed no formation of new products upon biotransformation with BcBOT2.
Mechanistic considerations and theoretical calculations
Mechanistically, the formation of the three products 17–19 is based on the same macrocyclisation pathway followed by hydride shifts.17 Interestingly, the first step is a 1 → 10 cyclisation (Scheme 4, top), in contrast to the macrocyclation proposed for the initial cyclisation of FPP 1 by BcBOT2 (1 → 11 to intermediate 27; Scheme 4, bottom). Activation of the diphosphate group is accompanied by formation of the macrocycle, and it appears that cyclopropylmethyl–cyclobutyl rearrangement is suppressed in this process. On the one hand, the different behavior of BcBOT2 with respect to the diphosphates studied here may be associated with the conformational matching of the substrates in the active pocket of STC, especially valid for the pair of enantiomers 5/7 and 6/8, respectively. On the other hand, the electronic properties and in particular the dissociation energies of the C–O (phosphate) bonds of the diphosphate groups must be taken into account. These are influenced by the different chemical environments and can be considered as a driving factor for the inhibition or initiation of carbocationic cascade reactions. To elucidate the different reactivities of the diphosphate group, we performed static and dynamic calculations at the DFT theory level (see Calculation methods in the ESI†). Cystallographic data (pdb: 4OKZ) of selina-4(15),7(11)-diene synthase (SdS) with 1 served as guideline,18 to create five diphosphate model structures A–E, each containing three Mg2+ ions and two significant water molecules. Therefore, the crystallographic data that included the magnesium cofactors served as a common starting structure to obtain a valid electrostatic environment. As a first step, the kinetic stability of the activated C–O bond was investigated by calculating the relevant relaxed force constants,19,20 | ||
| Scheme 4 Top: proposed mechanisms for the BcBOT2-promoted formation of macrocyclic terpenoids 17–19 from cyclopropylmethyl diphosphate 6. The configuration of the newly formed double bonds was elucidated by determining the coupling constants (JH,H); bottom: first proposed step to presilphiperfolan-8β-ol (2) by BcBOT2. | ||
After optimisation of each of the models A–E (Table 3), starting from the ligand-bound conformation fitted to the pdb structure 4OKZ, a def2tzvp basis set, and a continuum solvent, the relaxed force constants were calculated using our freely available COMPLIANCE code (see ESI†). The values obtained show significant differences for the model systems B–E compared to natural (oligo)prenyl diphosphates as precursors in class I terpene cyclases. For the natural farnesol derivative A a weak relaxed force constant of 3.22 N m−1 was calculated. In contrast, the cyclopropane ring has a significant stabilizing kinetic effect on the corresponding C–O bond for case C (E = 3.73 N m−1) and a moderate effect for case E (E = 3.44 N m−1). Removal of the olefinic double bond as in B or shifting the olefin by one position as in D leads to an increased, kinetic stability of the C–O bond by about 20% (3.84 and 3.74 N m−1, respectively).
| Models A–E (→iphosphate numbers) and cation formation | K ij (N m−1) | E (Diss) [kcal mol−1] | |
|---|---|---|---|
| A → 1 |

|
3.22 | 24.11 |
| B → 23 |

|
3.84 | 23.47 |
| C → 6/8 |

|
3.73 | 24.84 |
| D |

|
3.74 | 26.53 |
| A → 5/7 |
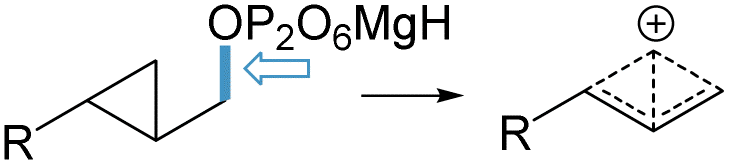
|
3.44 | 30.27 |
However, it is quite possible that the kinetic hindrance of the cyclopropyl-bearing ring in C and E is largely cancelled or overridden by thermodynamic factors. Therefore, the dissociation energies of the relevant C–O (phosphate) bonds were calculated. These are in the range between 24 and 26 kcal mol−1 for models A–D (Table 3), reflecting the comparable relative stability of the resulting carbocations.21 FPP 1 provides a stabilised allyl cation (A). Aliphatic B forms a tertiary carbocation after a rapid and barrier-free hydrogen shift from C3 (see file B.gif in the ESI†). After diphosphate dissociation, the cyclopropylmethyl substructure (C) is the source of a tertiary carbocation that is stabilised by a pronounced hyperconjugation (see ESI†; C.gif). In case D, stabilisation occurs by cyclopropylcarbinyl cation formation (see ESI†; D.gif). Noteworthy, such cations have been discussed in the context of the mechanism of pentalenene formation.22 Finally, in the case of di-instead of a trisubstituted cyclopropyl ring (model system E), a secondary homoallyl carbocation is formed and the cation cascade is about 5 kcal mol−1 more endothermic (30.27 kcal mol−1; Table 3) compared to the model systems A–D.
Finally, we investigated the cyclopropylmethyl/cyclobutyl rearrangement, which is of importance in initiating the cascade reaction. While the reduced strain energy in four-membered ring systems should facilitate cyclopropylmethyl/cyclobutyl ring expansion, it is nevertheless unclear how reliable previous force field calculations are with respect to this equilibrium, particularly because the potential energy surface of C4H7(+) is dominated by strong electron correlation. For example, MP2/cc-pVTZ calculations by Olah and coworkers23 give the bisected cyclopropylcarbinyl cation an energy lower by only 0.4 kcal mol−1 compared to the nonclassical bicyclobutonium ion, while solvent effects seem to complicate the whole situation.24 And indeed, an intrinsic reaction path (IRC) calculation, starting from the transition state of initial macrocyclisation (6 → 24) with the inclusion of continuum solvent effects, suggests a complex situation with CH2 migration without the formation of a cyclobutyl intermediate. This inherent dynamic behavior can also occur in the presence of an enzyme, an important aspect in the context of this work.25
Our calculations demonstrate that cyclopropylmethyl diphosphate precursors such as 6 are capable of entering a cationic reaction cascade via the stabilised cations 28 and without changing the regiochemistry of the first step (Scheme 5), despite their slight kinetic hindrance. When the bicyclobutonium ion 28b is the first relevant cation, the regiochemistry is determined solely by the enzyme-induced folding of the linear substrate. The formation of the newly formed homosesquiterpenes support the theoretical studies by Tantillo et al., on a cyclopropane-to-cyclopropane rearrangement of sterols who concluded that a carbocationic pathway can be preferred over a radical pathway and they propose a bicyclobutonium intermediate in their studies.26
 | ||
| Scheme 5 First step of cyclisation via the cyclopropylmethyl cation 28a or the bicyclobutonium ion 28b. | ||
The results thus also address questions about cyclopropylmethyl cations and activation barriers for cyclopropylmethyl rearrangement. And they extend insights into the remarkable substrate flexibility of STCs, because FPP derivatives in which the C3 methyl group is shifted to C2 are also accepted and activated.
Conclusions
In summary, our results show that small structural changes in the substrate can initiate different and new cyclisation cascades of STCs, recognisable by the fact that in the present case a switch from an initial 1–11 to a 1–10 macrocyclisation occurs. In combination with targeted and computer assisted mutagenesis, there is thus a high potential for terpene synthases to make new and unknown terpenoids synthetically accessible.Experimental
Semipreparative biotransformation of FPP derivative 6 with BcBot2
A solution (50 mL; pH = 7.5) composed of HEPES (50 mM), DTT (5 mM) Tween 20 (0.01%) in a round bottom flask were added the sesquiterpene cyclase (0.1 g L−1) and the FPP derivative 6 (33.5 mg, 75 μmol) continuously using a syringe pump (1 mL h−1) at room temperature. The resulting colorless suspension was incubated for 6 h at 34 °C and 150 rpm. Afterwards, a second portion of BcBot2 (0.05 g L−1) was added and incubation was continued for another 16 h. After this time, a final portion of enzyme solution (0.05 g L−1) was added and incubation was continued for 4 h. The reaction mixture was extracted with Et2O and the phases were separated. The combined organic phases were washed with a sat. aq. NaCl solution, dried over MgSO4, filtered, and concentrated under reduced pressure (≥800 mbar, 40 °C). The crude mixture was purified by column chromatography (SiO2, n-pentane:Et2O) to yield three biotransformation products. Column chromatography (silica, n-pentane:Et2O = 10:1 → 4:1 → 2:1) allowed to separate the triene 17 from the more polar alcohols 18 and 19, which were obtained as a 2:3 mixture.
(R,1E,6E)-5-Isopropyl-2-methyl-9-methylenecycloundeca-1,6-diene (17) (4 mg, 18 μmol, 24%)
R f = 0.90 (PE:EtOAc = 4:1); 1H-NMR (400 MHz, CDCl3): δ = 5.12–5.15 (m, 1 H, H-6), 5.11 (ddd, J = 15.1 Hz, 9.4 Hz, 5.5 Hz, 1 H, H-2), 5.02 (dd, J = 15.3 Hz, 9.5 Hz, 1 H, H-1), 4.81 (d, J = 8.2 Hz, 2 H, H-14), 2.73 (dd, J = 12.5 Hz, 5.4 Hz, 1 H, H-13), 2.48 (dd, J = 12.7 Hz, 9.4 Hz, 1 H, H-13), 2.28–2.32 (m, 1 H, H-5), 2.22–2.25 (m, 1 H, H-8), 2.12–2.15 (m, 1 H, H-4), 2.06–2.10 (m, 1 H, H-8), 1.94–1.96 (m, 1 H, H-4), 1.90–1.92 (m, 1 H, H-5), 1.84–1.87 (m, 1 H, H-10), 1.40–1.43 (m, 2 H, H-9), 1.49 (m, 4 H, H-11, H-15), 0.83 (d, J = 6.8 Hz, 3 H, H-12/16), 0.80 (d, J = 6.9 Hz, 3 H, H-12/16) ppm; 13C-NMR (100 MHz, CDCl3): δ = 150.1 (C-3) 135.2 (C-7), 133.5 (C-1), 128.9 (C-2), 127.3 (C-6), 111.2 (C-14), 53.1 (C-10), 43.4 (C-13), 41.6 (C-8), 33.6 (C-11), 34.2 (C-4), 29.6 (C-5), 20.8 (C-12), 28.2 (C-9), 15.9 (C-15), 19.0 (C-16) ppm.
(3E,5R,8E)-5-Isopropyl-1,8-dimethylcycloundeca-3,8-dien-1-ol (18) (2 mg, 8.5 μmol, 11%)
R f = 0.20 (PE:EtOAc = 4:1); 1H-NMR (400 MHz, CDCl3): δ = 5.28 (ddd, J = 15.4 Hz, 10.0 Hz, 5.2 Hz, 1 H, H-2) 5.13 (dd, J = 15.4 Hz, 9.8 Hz, 1 H, H-1), 5.06–5.09 (m, 1 H, H-6), 2.26–2.33 (m, 1 H, H-5), 2.23–2.25 (m, 1 H, H-8), 2.18–2.19 (m, 1 H, H-13), 2.15–2.17 (m, 1 H, H-8), 2.01 (dd, J = 13.0 Hz, 10.1 Hz, 1 H, H-13), 1.89–1.95 (m, 1 H, H-10), 1.82 (m, 1 H, H-5), 1.80 (m, 1 H, H-4), 1.67–1.69 (m, 1 H, H-4), 1.57 (s, 3 H, H-15), 1.55 (br, 1 H, OH), 1.49–1.52 (m, 1 H, H-11), 1.36–1.40 (m, 2 H, H-9), 1.24 (m, 3 H, H-14), 0.83 (d, J = 6.8 Hz, 3 H, H-12/16), 0.80 (d, J = 6.9 Hz, 3 H, H-12/16) ppm; 13C-NMR (100 MHz, CDCl3): δ = 135.9 (C-1) 133.2 (C-7), 128.8 (C-2), 130.1 (C-6), 72.8 (C-3), 53.5 (C-10), 48.0 (C-13), 33.8 (C-11), 42.2 (C-8), 30.1 (C-14), 40.0 (C-4), 27.2 (C-9), 23.9 (C-5), 16.0 (C-15), 18.8 (C-12/16), 20.7 (C-12/16) ppm.
(1R,2S,3R,10R,E)-3-Isopropyl-6,10-dimethyl-bicyclo[8.1.0]undec-6-en-2-ol (19) (3 mg, 13 μmol, 17%)
R f = 0.20 (PE:EtOAc = 4:1); 1H-NMR (400 MHz, CDCl3): δ = 5.32 (t, J = 7.6 Hz, 1 H, H-6) 3.36 (dd, J = 9.9 Hz, 1.7 Hz, 1 H, H-1), 2.20–2.25 (m, 2 H, H-5, H-8), 2.13–2.17 (m, 1 H, H-5), 1.84–1.88 (m, 1 H, H-4), 1.80 (m, 1 H, H-8), 1.73–1.76 (m, 1 H, H-9), 1.67–1.69 (m, 1 H, H-11), 1.64 (s, 3 H, H-15), 1.55 (br, 1 H, OH), 1.32–1.35 (m, 1 H, H-9), 1.05–1.08 (m, 1 H, H-10), 1.00 (d, J = 6.7 Hz, 3 H, H-12/16), 0.98 (s, 3 H, H-14), 0.93 (d, J = 6.6 Hz, 3 H, H-12/16), 0.85–0.87 (m, 1 H, H-4), 0.62 (ddd, J = 9.7 Hz, 8.3 Hz, 5.6 Hz, 1 H, H-2), 0.40 (dd, J = 8.3 Hz, 4.3 Hz, 1 H, H-13), 0.12 (dd, J = 5.5 Hz, 4.3 Hz, 1 H, H-13) ppm; 13C-NMR (100 MHz, CDCl3): δ = 133.2 (C-7) 124.0 (C-6), 74.7 (C-1), 48.4 (C-10), 39.2 (C-4), 38.8 (C-8), 32.2 (C-11), 30.0 (C-2), 25.6 (C-9), 24.2 (C-5), 21.7 (C-12/16), 21.3 (C-12/16), 18.8 (C-15), 18.5 (C-3), 17.3 (C-14), 17.2 (C-13) ppm.
Further details are found in the ESI.†
Author contributions
C. D. Tran and H. Struwe performed the chemical synthesis, protein expressions and biotransformations. G. Dräger provided analytical support. L. Siedenberg was involved in the optimisation of biontransformations. S. Vasisth and J. Grunenberg conducted the theoretical calculations and simulations. A. Kirschning and J. Grunenberg supervised the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr. Sascha Beutel (LUH) for support in protein production and Dr. Linn Müggenburg (LUH) for NMR support.References
- (a) D. W. Christianson, Structural and chemical biology of terpenoid cyclases, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS; (b) J. S. Dickschat, Bacterial terpene cyclases, Nat. Prod. Rep., 2016, 33, 87–110 RSC; (c) M. Baunach, J. Franke and C. Hertweck, Terpenoid biosynthesis off the beaten track: unconventional cyclases and their impact on biomimetic synthesis, Angew. Chem., Int. Ed., 2015, 54, 2604–2626 CrossRef CAS PubMed.
- R. A. Yoder and J. N. Johnston, A case study in biomimetic total synthesis: polyolefin carbocyclizations to terpenes and steroids, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS PubMed.
- C.-M. Wang, R. Hopson, X. Lin and D. E. Cane, Biosynthesis of the Sesquiterpene Botrydial in Botrytis cinerea. Mechanism and Stereochemistry of the Enzymatic Formation of Presilphiperfolan-8β-ol, J. Am. Chem. Soc., 2009, 131, 8360–8361 CrossRef CAS PubMed.
- (a) C. Oberhauser, V. Harms, K. Seidel, B. Schröder, K. Ekramzadeh, S. Beutel, S. Winkler, L. Lauterbach, J. S. Dickschat and A. Kirschning, Exploiting the synthetic potential of sesquiterpene cyclases for generating unnatural terpenoids, Angew. Chem., Int. Ed., 2018, 57, 11802–11806 CrossRef CAS PubMed; (b) V. Harms, B. Schröder, C. Oberhauser, C. D. Tran, S. Winkler, G. Dräger and A. Kirschning, Methyl-Shifted Farnesyldiphosphate Derivatives Are Substrates for Sesquiterpene Cyclases, Org. Lett., 2020, 22, 4360–4365 CrossRef CAS PubMed; (c) V. Harms, V. Ravkina and A. Kirschning, Mechanistic Similarities of Sesquiterpene Cyclases PenA, Omp6/7, and BcBOT2 Are Unraveled by an Unnatural “FPP-Ether” Derivative, Org. Lett., 2021, 23, 3162–3166 CrossRef CAS PubMed.
- V. Harms, A. Kirschning and J. Dickschat, Nature-driven approaches to non-natural terpene analogues, Nat. Prod. Rep., 2020, 37, 1080–1097 RSC.
- W. A. Bernett, J. Chem. Educ., 1967, 44, 17–24 CrossRef CAS.
- G. A. Olah, V. P. Prakash and G. K. A. Prakas, Long-lived cyclopropylcarbinyl cations, Chem. Rev., 1992, 92, 69–95 CrossRef CAS.
- P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, Evaluation of strain in hydrocarbons. The strain in adamantane and its origin, J. Am. Chem. Soc., 1970, 92, 2377–2386 CrossRef CAS.
- P. Rabe, L. Barra, J. Rinkel, R. Riclea, C. A. Citron, T. A. Klapschinski, A. Janusko and J. S. Dickschat, Conformational Analysis, Thermal Rearrangement, and EI-MS Fragmentation Mechanism of (1(10)E, 4E, 6S, 7R)-Germacradien-6-ol by 13C-Labeling Experiments, Angew. Chem., Int. Ed., 2015, 54, 13448–13451 CrossRef CAS PubMed.
- D. M. Cermak, D. F. Wiemer, K. Lewis and R. J. Hohl, 2-(Acyloxy) ethylphosphonate analogues of prenyl pyrophosphates: synthesis and biological characterization, Bioorg. Med. Chem., 2000, 8, 2729–2737 CrossRef CAS PubMed.
- A. B. Charette, H. Juteau, H. Lebel and C. Molinaro, Enantioselective cyclopropanation of allylic alcohols with dioxaborolane ligands: Scope and synthetic applications, J. Am. Chem. Soc., 1998, 120, 11943–11952 CrossRef CAS.
- A. B. Woodside, Z. Huang and C. D. Poulter, Org. Synth., 1987, 66, 211 Search PubMed.
- R. K. Keller and R. Thompson, Rapid synthesis of isoprenoid diphosphates and their isolation in one step using either thin layer or flash chromatography, J. Chromatogr., 1993, 645, 161–167 CrossRef CAS.
- Due to tailoring, the signal of the minor enantiomer could not be integrated precisely (see ESI†).
- R. Lauchli, J. Pitzer, R. Z. Kitto, K. Z. Kalbarczyk and K. S. Rabe, Improved selectivity of an engineered multi-product terpene synthase, Org. Biomol. Chem., 2014, 12, 4013–4020 RSC.
- B. Pickel, D. P. Drew, T. Manczak, C. Weitzel, H. T. Simonsen and D.-K. Ro, Identification and characterization of a kunzeaol synthase from Thapsia garganica: implications for the biosynthesis of the pharmaceutical thapsigargin, Biochem. J., 2012, 448, 261–271 CrossRef CAS PubMed.
- K. Ekramzadeh, C. Brämer, T. Frister, J. Fohrer, A. Kirschning, T. Scheper and S. Beutel, Optimization of factors influencing enzyme activity and product selectivity and the role of proton transfer in the catalytic mechanism of patchoulol synthase, Biotechnol. Prog., 2020, 36, e2935 CrossRef CAS PubMed.
- P. Baer, P. Rabe, K. Fischer, C. A. Citron, T. A. Klapschinski, M. Groll and J. S. Dickschat, Induced–fit mechanism in class I terpene cyclases, Angew. Chem., Int. Ed., 2014, 53, 7652–7656 CrossRef CAS.
- G. Markopoulos and J. Grunenberg, Predicting Kinetically Unstable C-C Bonds from the Ground–State Properties of a Molecule, Angew. Chem., Int. Ed., 2013, 52, 10648–10651 CrossRef CAS PubMed.
- A. Kreft, A. Lücht, J. Grunenberg, P. G. Jones and D. B. Werz, Kinetic studies of donor–acceptor cyclopropanes: the influence of structural and electronic properties on the reactivity, Angew. Chem., Int. Ed., 2019, 58, 1955–1959 CrossRef CAS PubMed.
- D. J. Tantillo, The carbocation continuum in terpene biosynthesis—where are the secondary cations?, Chem. Soc. Rev., 2010, 39, 2847–2854 RSC.
- P. Gutta and D. J. Tantillo, Theoretical studies on farnesyl cation cyclization: pathways to pentalenene, J. Am. Chem. Soc., 2006, 128, 6172–6179 CrossRef CAS PubMed.
- A. Olah, G. K. S. Prakash and G. Rasu, Ab Initio/GIAO-CCSD(T) Study of Structures, Energies, and 13C NMR Chemical Shifts of C4H7+ and C5H9+ Ions: Relative Stability and Dynamic Aspects of the Cyclopropylcarbinyl vs Bicyclobutonium Ions, J. Am. Chem. Soc., 2008, 130, 9168–9172 CrossRef PubMed.
- J. Casanova, D. R. Kent IV, W. A. Goddard III and J. D. Roberts, Quantum-mechanical calculations of the stabilities of fluxional isomers of C4H7+ in solution, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15–19 CrossRef CAS PubMed.
- S. R. Hare and D. J. Tantillo, Dynamic behavior of rearranging carbocations–implications for terpene biosynthesis, Beilstein J. Org. Chem., 2016, 12, 377–390 CrossRef CAS PubMed.
- Y. J. Hong, J. –L. Giner and D. J. Tantillo, Bicyclobutonium Ions in Biosynthesis – Interconversion of Cyclopropyl-Containing Sterols from Orchids, J. Am. Chem. Soc., 2015, 137, 2085–2088 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01279k |
| This journal is © The Royal Society of Chemistry 2022 |