
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thioarylation of anilines using dual catalysis: two-step synthesis of phenothiazines†
Amy C.
Dodds
,
Sabrina
Puddu
and
Andrew
Sutherland
 *
*
School of Chemistry, The Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Andrew.Sutherland@glasgow.ac.uk
First published on 6th July 2022
Abstract
A two-step synthesis of phenothiazines has been developed using a dual-catalytic ortho-thioarylation reaction of anilines as the key step. Activation of N-(2-bromophenylthio)succinimide was achieved using the super Lewis acid, iron(III) triflimide and the Lewis base, diphenyl selenide, resulting in an accelerated and efficient ortho-thioarylation reaction of various protected aniline derivatives and less reactive, unprotected analogues. The thioarylated adducts were then cyclised to the desired phenothiazines using either an Ullmann–Goldberg or Buchwald–Hartwig coupling reaction. The dual catalytic thioarylation and copper(I)-catalysed cyclisation approach was used for the four-step synthesis of methopromazine, a neuroleptic agent with antipsychotic activity.
Introduction
Phenothiazine heterocycles are an important structural motif, found as the core component of a wide range of pharmaceutically active agents.1,2 Early analogues such as the parent compound, phenothiazine (Fig. 1) was used as an anthelmintic agent in livestock and humans,3 while the dye, methylene blue was used for the treatment of malaria.4 More recently, dimethylaminopropyl analogues such as chlorpromazine have been widely used as antipsychotic drugs for the treatment of mental illnesses such as schizophrenia.1,5 Thioridazine, a first generation antipsychotic drug, withdrawn due to cardiac arrhythmias, has been shown to be an anti-glioblastoma and anticancer stem cell agent.6 As well as displaying other medicinal properties,7 phenothiazine derivatives have also been utilised in applications such as molecular wires,8 chemiluminescence emitters,9 sensors for the detection of flavins10 and as photoredox catalysts.11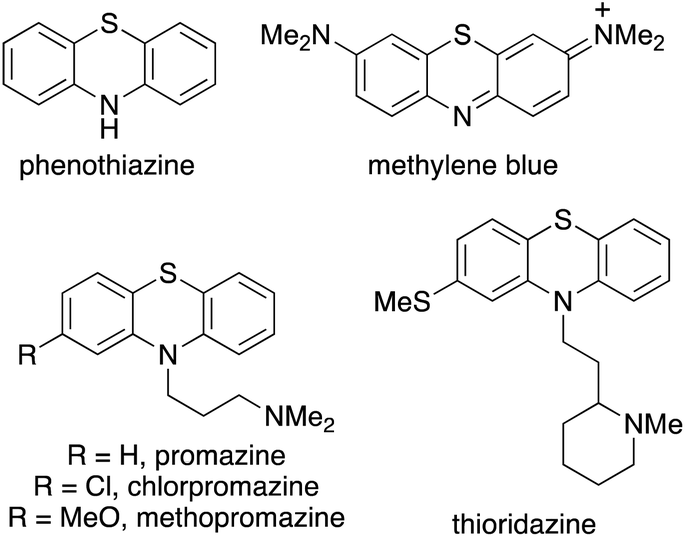 | ||
| Fig. 1 Structures of medicinally important phenothiazines. | ||
Due to their importance, particularly as medicinal agents, there have been significant efforts in developing efficient syntheses of phenothiazines. Historically, phenothiazines were prepared by heating diphenylamines with sulfur at high temperatures (250–260 °C).12 This approach has been improved using microwave heating.13 For example, a range of phenothiazines with antitubercular activity were prepared in a two-step approach involving a Buchwald–Hartwig reaction for the synthesis of diphenylamines, followed by iodine-catalysed cyclisation with sulfur, using microwave heating (Scheme 1a).14 Traditionally, phenothiazines have also been prepared via a four-step approach, using a based-mediated Smiles rearrangement as the key step.15 To overcome the regioselectivity issues associated with both approaches, recent strategies have focused on transition metal-catalysed coupling of pre-functionalised anilines and arene partners.16 Jørgensen and co-workers reported a palladium-catalysed three-component synthesis of phenothiazines using 1-bromo-2-iodobenzenes, primary amines and 2-bromobenzenethiol,17 while the Ma group developed a sequential copper iodide and L-proline-catalysed process from 2-iodoanilines and 2-bromobenzenethiols (Scheme 1b).18 A one-pot, rhodium(III)-catalysed C–H thioarylation reaction and copper-catalysed C–N amination of acetanilides and 2-bromobenzenethiols has also been reported (Scheme 1c).19 In addition to transition metal-catalysed approaches, various base-, enzyme- and iodine-mediated couplings have also recently been described for phenothiazine synthesis.20
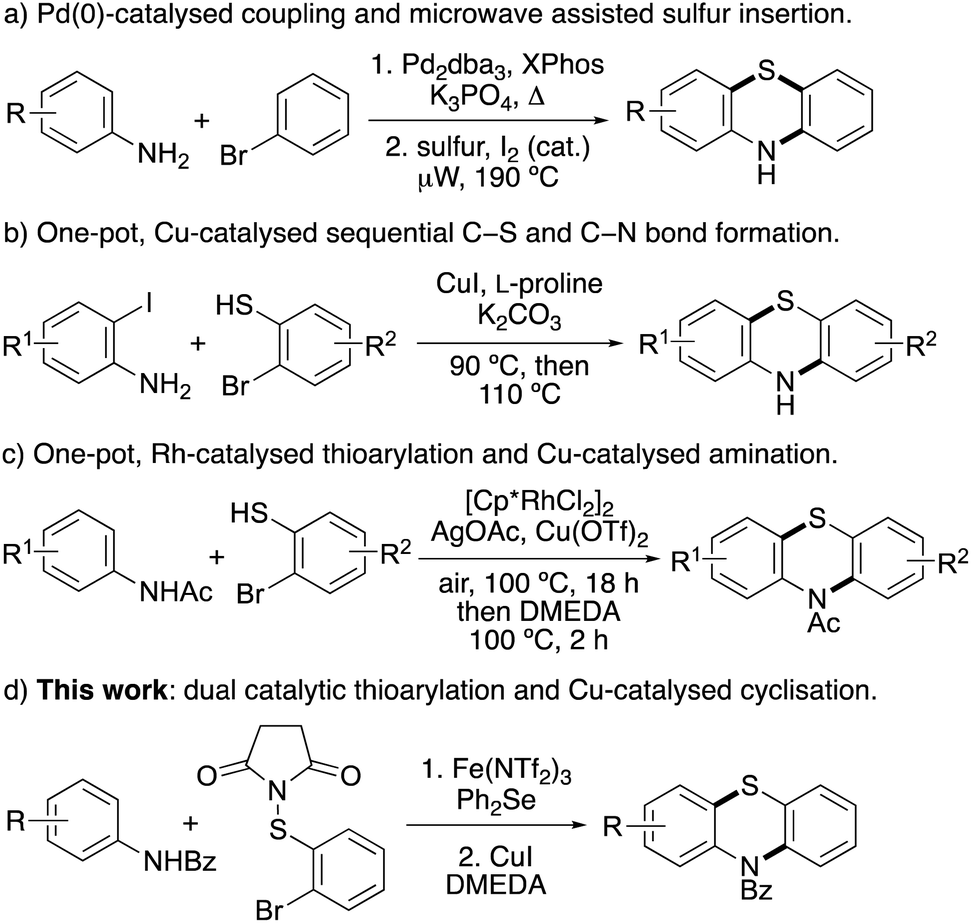 | ||
| Scheme 1 Methods for the synthesis of phenothiazines. | ||
Several of these methods are elegant and efficient but rely on highly functionalised arenes or the use of precious transition metal catalysis. We have developed catalytic methods for the regioselective halogenation of arenes using the super Lewis acid, iron(III) triflimide to activate N-halosuccinimides.21 More recently, this approach has been extended for aryl C–S bond formation with N-thiosuccinimides and used as the key step for the synthesis of phenoxathiins.22 To address the challenge of phenothiazine synthesis using simple aniline starting materials and non-precious transition metal catalysts, it was proposed that iron(III)-catalysed thioarylation of benzoyl-protected anilines with N-(2-bromophenylthio)succinimide would generate intermediates, that following copper-catalysed cyclisation would allow access to phenothiazines (Scheme 1d). Herein, we now report the two-step synthesis of phenothiazines using an accelerated, dual-catalysed thioarylation reaction involving iron(III) triflimide and the Lewis base, diphenyl selenide, followed by copper(I)-catalysed cyclisation. We also describe the use of this two-step approach for the synthesis of the neuroleptic agent, methopromazine.
Results and discussion
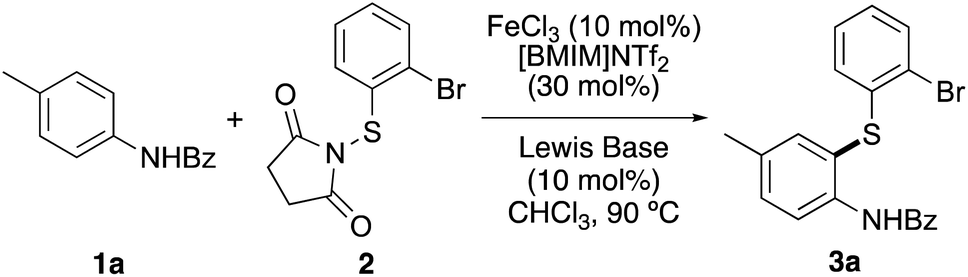
Our studies began with the thioarylation of a suitably protected derivative of p-toluidine with N-(2-bromophenylthio)succinimide (2). Although Fu and co-workers showed that uncatalysed thioarylation of unprotected anilines with N-(arylthio)succinimides could be achieved using high temperatures (100 °C) and extended reaction times (21–30 h),23 examples of catalysed methods with activated N-thiolation reagents are relatively rare and require the use of protected derivatives.22a,24,25 Preliminary work in our group demonstrated that iron-catalysed thioarylation of unprotected anilines gave a mixture of thiolated products via reaction with either the arene or amine moiety. For this reason, N-benzoyl protected p-toluidine 1a was chosen as the model substrate. Using reactions parameters previously optimised (catalyst loading, solvent and temperature),22 an initial reaction was attempted at 90 °C, using iron(III) triflimide (10 mol%), which was generated in situ from iron(III) chloride and the commercially available ionic liquid, [BMIM]NTf2. The transformation was found to be incomplete after 48 h and returned mainly starting material (Table 1, entry 1). This was not surprising as the ortho-position of 1a is more sterically hindered and less activated compared to the parent aniline. Previous work by the Gustafson group has shown that Lewis acid catalysed thioarylations can be accelerated using Lewis bases.24c Therefore, a series of commercially available Lewis base catalysts (10 mol%) were screened to determine whether these could improve both the rate and yield of iron(III)-catalysed thioarylation of 1a. Addition of bis(4-methoxyphenyl)sulfane to the thioarylation reaction of 1a with thiosuccinimide 2 resulted in significant improvement (entry 2). The reaction was complete after 18 h and gave 3a in 88% yield. Similar results were observed using 1-methoxy-4-(methylthio)benzene (entry 3). Despite being used as Lewis bases for aryl chlorination,26 both diaryl thioureas and triphenylphosphine sulfide showed little activity for thioarylation (entries 4 and 5). The best results were observed using the commercially available Lewis base, diphenyl selenide, which allowed complete thioarylation of 1a after 6 hours and gave 3a in 91% yield (entry 6). To demonstrate that both iron triflimide and diphenyl selenide were required for the accelerated reaction, the transformation was repeated in the absence of iron(III) triflimide (entry 7). After 20 h, this gave no product, confirming the dual role of both iron(III) triflimide and diphenyl selenide in accelerating the reaction.|
|
|||
|---|---|---|---|
| Entry | Lewis base | Time (h) | Yieldb (%) |
a Reaction conditions: 1a (0.29 mmol), 2 (1.2 equiv.), 0.6 M in arene.
b Isolated yields.
c Reaction gave a 3.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1a:3a after 48 h.
d Reaction performed in the absence of FeCl3 and [BMIM]NTf2. :1 mixture of 1a:3a after 48 h.
d Reaction performed in the absence of FeCl3 and [BMIM]NTf2.
|
|||
| 1c | — | 48 | — |
| 2 | (4-MeOPh)2S | 18 | 88 |
| 3 | (4-MeOPh)SMe | 18 | 73 |
| 4 | (PhNH)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S S |
24 | — |
| 5 | Ph3PS |
24 | — |
| 6 | Ph2Se | 6 | 91 |
| 7d | Ph2Se | 20 | — |
Using these results, a mechanism for dual-catalysed thioarylation involving both iron(III) triflimide and diphenyl selenide has been proposed (Scheme 2). On activation of N-(2-bromophenylthio)succinimide (2) with the strongly Lewis acidic iron(III) cation, the resulting intermediate undergoes rapid reaction with diphenyl selenide to form a cationic intermediate.27 Due to the charged nature of this intermediate, this undergoes a significantly faster reaction with N-benzoyl protected p-toluidine 1a to give thioarylated product 3a, than using iron(III) triflimide as the only activating agent.
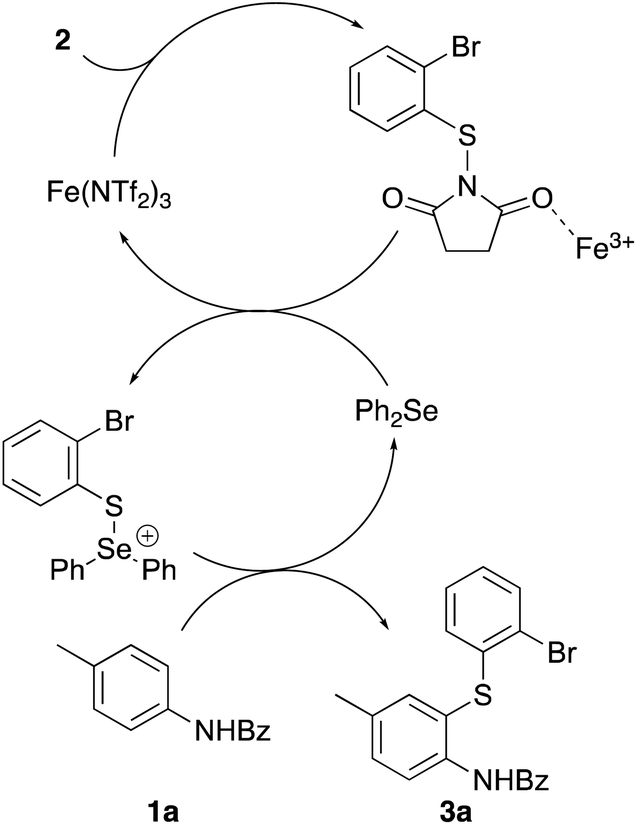 | ||
| Scheme 2 Proposed catalytic pathway for dual catalytic thioarylation. | ||
The scope of the dual catalytic thioarylation method was then explored with various N-benzoyl protected anilines 1a–1g, bearing a range of substituent patterns (Scheme 3). The process allowed fast ortho-thioarylation of most substrates and gave products 3a–3g in 80–95% yields. The transformation was also effective for thioarylation at hindered ortho, ortho-substituted positions, producing 3g after 3 h, in 81% yield. A larger scale reaction was also performed using N-benzoyl protected p-toluidine 1a. At 1.5 mmol scale, this gave thioarylated product 3a in 84% yield, in a similar manner to that of the small-scale reaction (91%). Using the optimised reaction conditions with diphenyl selenide, the scope of the protecting group was further explored. Successful ortho-thioarylation reactions were observed with Cbz-, tosyl- and alkyl-protected p-toluidines (1h–1k), although longer reaction times were required. Attempted thioarylation with less reactive N-benzoyl protected anilines bearing conjugated or deactivating aryl substituents under the optimised conditions showed no reaction or gave low yields (<35%). These results were partly attributed to the low solubility of some of the N-benzoyl protected anilines in chloroform. To overcome this issue, it was proposed that a more efficient transformation with these substrates may be possible with the unprotected anilines, which were readily soluble in chloroform. Despite the aforementioned issues associated with reaction of unprotected anilines with activated N-thiosuccinimides, treatment of N-(2-bromophenylthio)succinimide (2) with p-toluidine (1l), 4-phenylaniline (1m), 2-aminonaphthalene (1n) and 4-chloroaniline (1o) resulted in clean reactions and gave the products in moderate to high yields (42–75%). We believe that a combination of the relatively bulky N-thiosuccinimide reagent 2 and the less nucleophilic nature of the amine moiety of substrates 1m–1o allowed a chemoselective, thioarylation reaction of these anilines. A limitation of this transformation is that anilines with strong electron-withdrawing groups are not substrates for this transformation. For example, 4-aminoacetophenone and 4-aminobenzonitrile showed no reaction under the optimised conditions after 24 h.
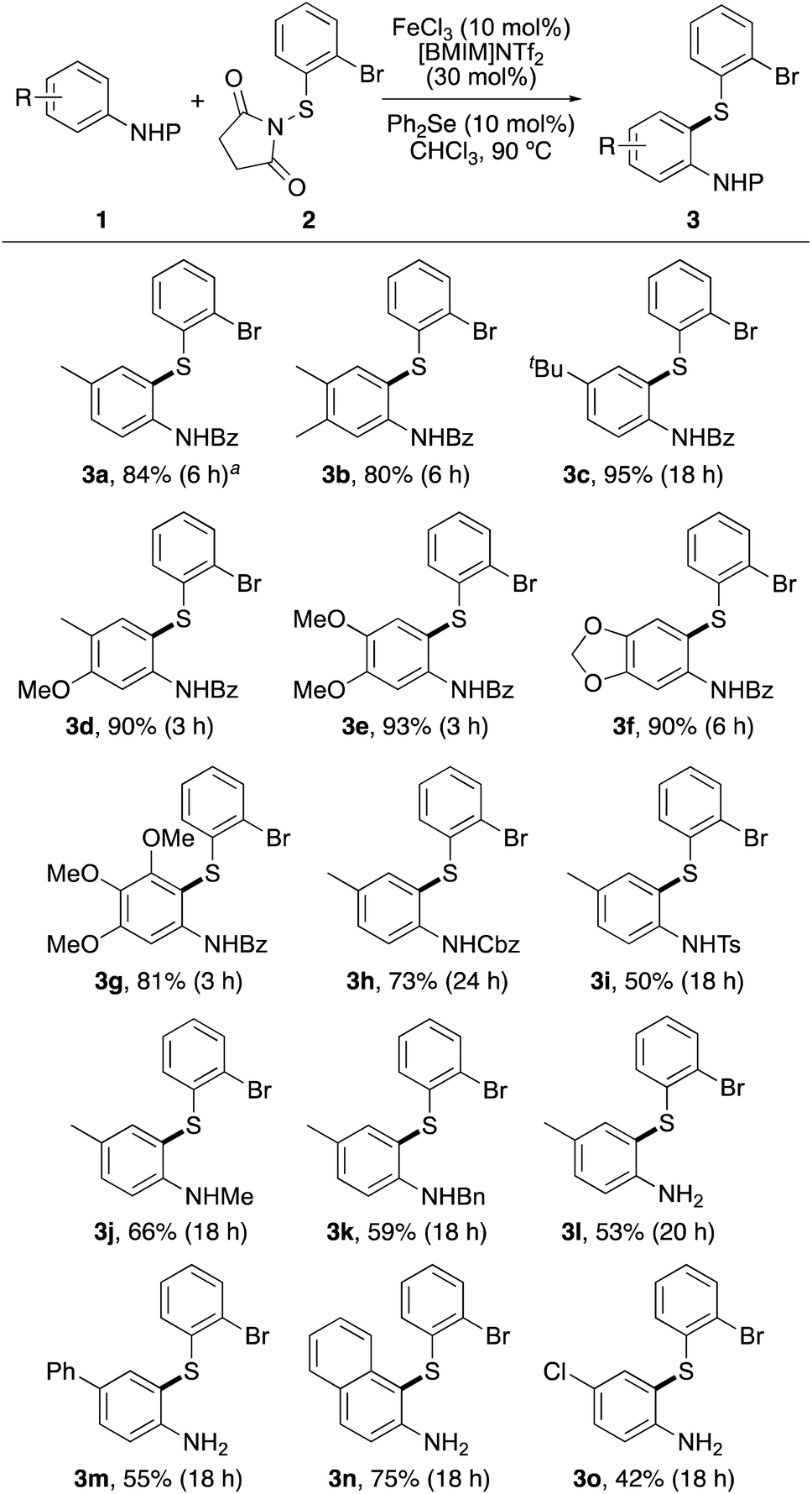 | ||
| Scheme 3 Reaction scope of anilines. General reaction conditions: 1 (0.29 or 0.58 mmol), 2 (1.2 equiv.), 0.6 M in arene. aReaction performed on a 1.5 mmol scale. | ||
Having optimised a dual catalytic thioarylation process to access a range of biaryl sulfides (3), cyclisation to the desired phenothiazine targets was then investigated (Scheme 4). For N-benzoyl-protected derivatives (3a–3g), copper-catalysed Ullmann–Goldberg type cyclisation using a combination of copper(I) iodide and DMEDA under basic conditions proved effective.28 While in some cases, long reaction times were required, this gave the majority of phenothiazine products in good yields. These conditions were found to be non-compatible with Cbz- (3h) and tosyl-protected derivatives (3i), with debrominated and deprotected by-products identified from the crude reaction mixtures. Instead, the use of racemic trans-N,N′-dimethylcyclohexane-1,2-diamine,29 to facilitate a ligand-assisted, copper-catalysed cyclisation was more effective. This permitted faster access to phenothiazines 4h and 4i in 76% and 75% yields, respectively.
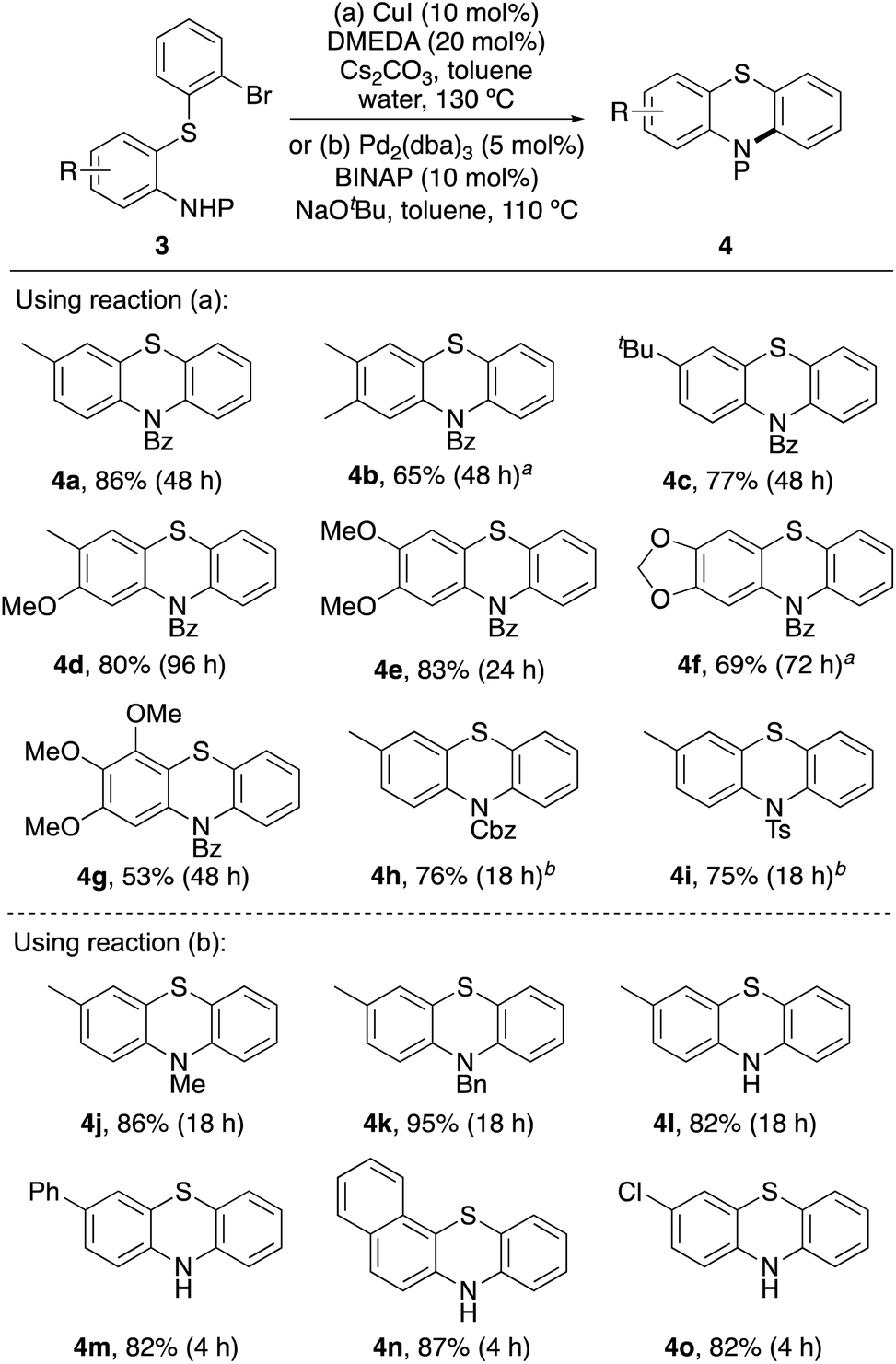 | ||
| Scheme 4 Synthesis of phenothiazines. Reaction conditions for copper-catalysed cyclisation: 3 (0.07 to 0.18 mmol), Cs2CO3 (2.0 equiv.), 0.4 M in arene. Reaction conditions for palladium-catalysed cyclisation: 3 (0.10 to 0.39 mmol), NaOtBu (2.0 equiv.), 0.33 M in arene. aReaction was performed at 150 °C. bReaction was done using trans-N,N′-dimethylcyclohexane-1,2-diamine (10 mol%) instead of DMEDA. | ||
Attempted copper-catalysed cyclisation of N-alkyl or unprotected ortho-thiolated anilines 3j–3o returned only starting material. The inability of these compounds to cyclise under these conditions is likely due to strong coordination with the copper catalyst. Instead, substrates 3j–3o were cyclised to the corresponding phenothiazines using a palladium(0)-catalysed Buchwald–Hartwig reaction (Scheme 4).30 Under standard conditions,31 using Pd2(dba)3 (5 mol%) and BINAP as a ligand, fast and efficient cyclisation to phenothiazines 4j–4o was observed. In particular, cyclisation of 3m–3o required reaction times of 4 h and gave phenothiazines 4m–4o in 82–87% yield.
Finally, the synthetic utility of this two-step approach was demonstrated with a short synthesis of methopromazine, a neuroleptic agent with antipsychotic activity.32N-Benzoyl protected 3-methoxyaniline 5 was chosen as the starting material for this synthesis (Scheme 5). While both substituents are activating, it was proposed that the stronger electron donating methoxy group would promote reaction at the C6-position.33,34 Thioarylation of 5 using N-(2-bromophenylthio)succinimide (2) at 90 °C and a reaction time of 3 h gave 6 in 54% yield. However, the 4,6-dithioarylated product was also observed from the 1H NMR spectrum of the crude reaction mixture and therefore a lower temperature reaction was investigated to minimise side-product formation. At 75 °C, the reaction was completed in a similar reaction time of 4 h and gave 6 in an improved 64% yield. Ullmann–Goldberg cyclisation of 6 using copper(I) iodide and DMEDA gave phenothiazine 7 in 80% yield. The benzoyl protecting was then removed using hydrazine hydrate.35 Finally, alkylation with the commercially available hydrochloride salt of 3-dimethylaminopropyl chloride using sodium hydride as base completed the four-step synthesis of methopromazine.
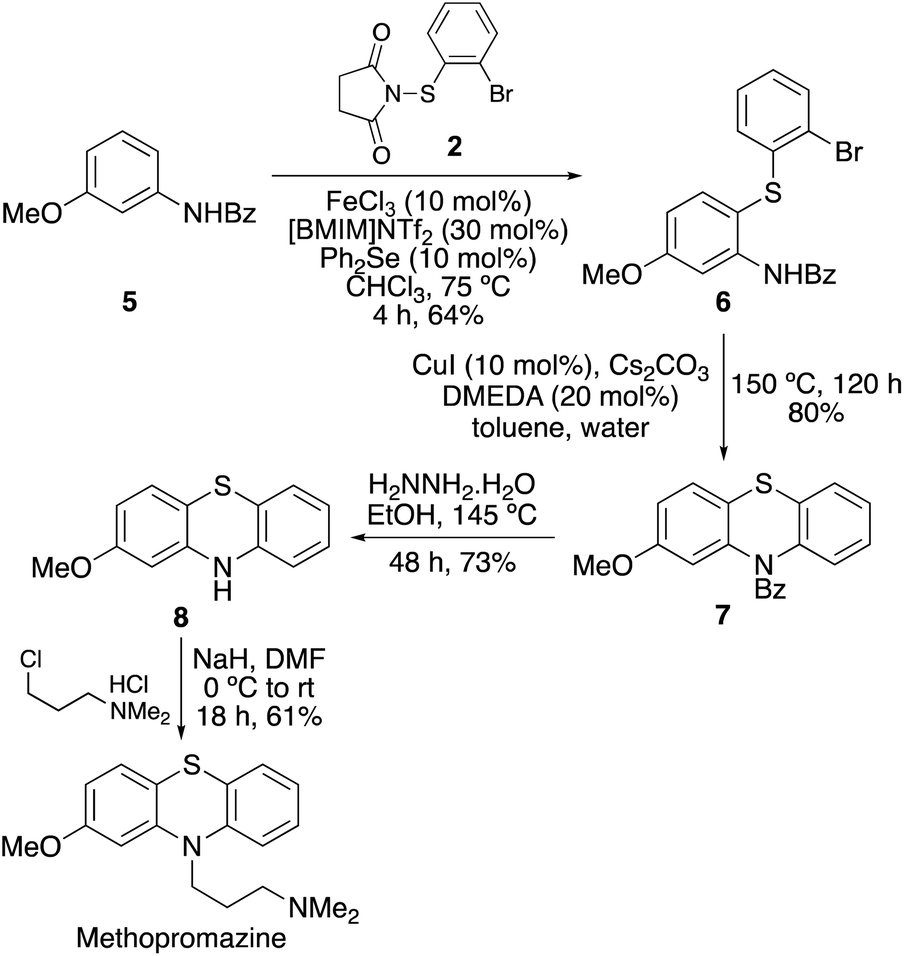 | ||
| Scheme 5 Synthesis of methopromazine. | ||
Conclusions
In summary, a new approach for the synthesis of phenothiazines has been developed using a dual catalytic thioarylation of anilines as the key step. A combination of the Lewis acid, iron(III) triflimide and the Lewis base, diphenyl selenide permitted rapid and efficient thioarylation of N-benzoyl anilines. Ullmann–Goldberg cyclisation of the resulting diaryl sulfides completed the synthesis of a small library of phenothiazines. This straightforward approach was used for a four-step synthesis of the neuroleptic agent, methopromazine. Unprotected anilines and N-alkyl derivatives were also substrates for the dual catalytic process and underwent chemoselective thioarylation, rather than N-sulfenylation. The products of these reactions were readily cyclised to the corresponding phenothiazines using a Buchwald–Hartwig coupling reaction. Overall, this simple, dual catalytic thioarylation process allowed efficient ortho-C–S bond formation of hindered arenes and current work is focused on new applications of this approach for other arene functionalisation reactions.Experimental
All reagents and starting materials were obtained from commercial sources and used as received. N-(2-Bromophenylthio)succinimide (2)22a and starting materials 1a–1g and 5,361h,371i,381j and 1k,39 and 1n40 were prepared as previously described. Reactions were performed open to air unless otherwise mentioned. Brine refers to a saturated aqueous solution of sodium chloride. Flash column chromatography was performed using silica gel 60 (35–70 μm). Aluminium-backed plates pre-coated with silica gel 60F254 were used for thin layer chromatography and were visualised with a UV lamp or by staining with potassium permanganate. 1H NMR spectra were recorded on a NMR spectrometer at either 400 or 500 MHz and data are reported as follows: chemical shift in ppm relative to the solvent as internal standard (CHCl3, δ 7.26 ppm; CH3OH, δ 3.31 ppm; DMSO, δ 2.50), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of non-equivalent resonances, integration). 13C NMR spectra were recorded on a NMR spectrometer at either 101 or 126 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as internal standard (CDCl3, δ 77.16 ppm; CD3OD, δ 49.00 ppm; DMSO-d6, δ 39.52), multiplicity with respect to hydrogen (deduced from DEPT experiments, C, CH, CH2 or CH3). Assignment of 1H and 13C NMR spectra are based on two-dimensional COSY, HSQC, HMBC and DEPT experiments. Infrared spectra were recorded on a FTIR spectrometer; wavenumbers are indicated in cm−1. Mass spectra were recorded using electron impact or electrospray techniques. HRMS spectra were recorded using dual-focusing magnetic analyser or quadrupole time of flight (Q-TOF) mass spectrometers. Melting points are uncorrected.
General procedure A: preparation of sulfenylated products
Iron(III) trichloride (10 mol%) was dissolved in [BMIM]NTf2 (30 mol%) and left to stir for 0.5 h at room temperature before being added to a solution of N-(2-bromophenylthio)succinimide (2) (1.2 equiv.) in chloroform (0.6 M in arene). The arene (1.0 equiv.) and diphenyl selenide (10 mol%) was then added and the mixture was left to stir at 90 °C, until the reaction was deemed complete. The reaction mixture was concentrated in vacuo and purified using flash column chromatography.(2-Benzoylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3a)
The reaction was performed as described in general procedure A using N-(4-methylphenyl)benzamide (1a) (61 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 6 h. Purification by flash column chromatography (hexane/dichloromethane, 1:1) gave (2-benzoylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3a) (103 mg, 89%) as a white solid. Mp 105–107 °C; νmax/cm−1 (neat) 3379 (NH), 2920 (CH), 1678 (CO), 1516, 1443, 1304, 1246, 1018, 822; δH (400 MHz, CDCl3) 2.38 (3H, s, CH3), 6.65 (1H, dd, J 7.9, 1.6 Hz, 6′-H), 6.98 (1H, ddd, J 7.9, 7.4, 1.6 Hz, 4′-H), 7.10 (1H, ddd, J 7.9, 7.4, 1.4 Hz, 5′-H), 7.36–7.44 (3H, m, 4-H, 3′′-H and 5′′-H), 7.47–7.52 (2H, m, 6-H and 4′′-H), 7.54 (1H, dd, J 7.9, 1.4 Hz, 3′-H), 7.65–7.69 (2H, m, 2′′-H and 6′′-H), 8.61 (1H, d, J 8.4 Hz, 3-H), 8.95 (1H, br s, NH); δC (101 MHz, CDCl3) 20.8 (CH3), 119.1 (C), 120.9 (CH), 121.2 (C), 127.1 (2 × CH), 127.3 (CH), 127.4 (CH), 128.4 (CH), 128.9 (2 × CH), 132.0 (CH), 132.6 (CH), 133.1 (CH), 134.7 (C), 134.7 (C), 137.3 (C), 137.5 (CH), 138.0 (C), 165.2 (C); m/z (ESI) 398.0218 (MH+. C20H1779BrNOS requires 398.0209).
(2-Benzoylamino-4,5-dimethylphenyl)(2′-bromophenyl)sulfane (3b)
The reaction was performed as described in general procedure A using N-(3,4-dimethylphenyl)benzamide (1b) (66 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 6 h. Purification by flash column chromatography (hexane/dichloromethane, 1:1) gave (2-benzoylamino-4,5-dimethylphenyl)(2′-bromophenyl)sulfane (3b) (96 mg, 80%) as a white solid. Mp 135–140 °C; νmax/cm−1 (neat) 3375 (NH), 2916 (CH), 1678 (CO), 1519, 1442, 1249, 1199, 1018, 752; δH (400 MHz, CDCl3) 2.28 (3H, s, CH3), 2.38 (3H, s, CH3), 6.64 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.97–7.00 (1H, m, 4′-H), 7.08 (1H, ddd, J 8.0, 7.4, 1.4 Hz, 5′-H), 7.38–7.43 (3H, m, 6-H, 3′′-H and 5′′-H), 7.46–7.54 (2H, m, 3′-H and 4′′-H), 7.64–7.69 (2H, m, 2′′-H and 6′′-H), 8.54 (1H, s, 3-H), 8.92 (1H, br s, NH); δC (101 MHz, CDCl3) 19.3 (CH3), 20.4 (CH3), 115.8 (C), 121.0 (C), 122.1 (CH), 127.1 (2 × CH), 127.1 (CH), 127.1 (CH), 128.3 (CH), 128.9 (2 × CH), 132.0 (CH), 133.0 (CH), 133.6 (C), 134.8 (C), 137.7 (C), 137.9 (CH), 138.2 (C), 141.3 (C), 165.1 (C); m/z (ESI) 412.0366 (MH+. C21H1979BrNOS requires 412.0365).
(2-benzoylamino-5-tert-butylphenyl)(2′-bromophenyl)sulfane (3c)
The reaction was performed as described in general procedure A using N-(4-tert-butylphenyl)benzamide (1c) (74 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave (2-benzoylamino-5-tert-butylphenyl)(2′-bromophenyl)sulfane (3c) (124 mg, 97%) as a white solid. Mp 109–111 °C; νmax/cm−1 (neat) 3352 (NH), 2955 (CH), 1678 (CO), 1505, 1427, 1308, 1022, 841, 752; δH (400 MHz, CDCl3) 1.35 (9H, s, 3 × CH3), 6.62 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 6.98 (1H, ddd, J 7.9, 7.5, 1.5 Hz, 4′-H), 7.10 (1H, ddd, J 8.0, 7.5, 1.4 Hz, 5′-H), 7.38–7.44 (2H, m, 3′′-H and 5′′-H), 7.47–7.52 (1H, m, 4′′-H), 7.54 (1H, dd, J 7.9, 1.4 Hz, 3′-H), 7.60 (1H, dd, J 8.7, 2.3 Hz, 4-H), 7.63–7.70 (3H, m, 6-H, 2′′-H and 6′′-H), 8.63 (1H, d, J 8.7 Hz, 3-H), 8.92 (1H, br s, NH); δC (101 MHz, CDCl3) 31.4 (3 × CH3), 34.7 (C), 118.8 (C), 120.7 (CH), 121.1 (C), 127.1 (2 × CH), 127.1 (CH), 127.2 (CH), 128.4 (CH), 128.9 (2 × CH), 129.1 (CH), 132.0 (CH), 133.1 (CH), 134.1 (CH), 134.8 (C), 137.4 (C), 137.9 (C), 148.2 (C), 165.2 (C); m/z (ESI) 440.0686 (MH+. C23H2379BrNOS requires 440.0678).
(2-Benzoylamino-4-methoxy-5-methylphenyl)(2′-bromophenyl)sulfane (3d)
The reaction was performed as described in general procedure B using N-(3-methoxy-4-methylphenyl)benzamide (1d) (70 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 3 h. Purification by flash column chromatography (hexane/dichloromethane, 2:3) gave (2-benzoylamino-4-methoxy-5-methylphenyl)(2′-bromophenyl)sulfane (3d) (112 mg, 90%) as a white solid. Mp 142–144 °C; νmax/cm−1 (neat) 3379 (NH), 2916 (CH), 1678 (CO), 1581 (CC), 1392, 1249, 1168, 1053, 887, 882, 790; δH (400 MHz, CDCl3) 2.22 (3H, s, CH3), 3.97 (3H, s, OCH3), 6.61 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.97 (1H, ddd, J 7.9, 7.4, 1.6 Hz, 4′-H), 7.09 (1H, ddd, J 8.0, 7.4, 1.4 Hz, 5′-H), 7.39–7.45 (3H, m, 3-H, 3′′-H and 5′′-H), 7.48–7.54 (2H, m, 3′-H and 4′′-H), 7.67–7.71 (2H, m, 2′′-H and 6′′-H), 8.44 (1H, s, 6-H), 9.09 (1H, br s, NH); δC (101 MHz, CDCl3) 15.9 (CH3), 55.8 (CH3), 103.0 (CH), 108.3 (C), 120.7 (C), 123.5 (C), 126.9 (CH), 127.0 (CH), 127.1 (2 × CH), 128.3 (CH), 129.0 (2 × CH), 132.1 (CH), 133.0 (CH), 134.7 (C), 138.1 (C), 138.6 (CH), 140.0 (C), 160.5 (C), 165.3 (C); m/z (ESI) 428.0317 (MH+. C21H1979BrNO2S requires 428.0314).
(2-Benzoylamino-4,5-dimethoxyphenyl)(2′-bromophenyl)sulfane (3e)
The reaction was performed as described in general procedure A using N-(3,4-dimethoxyphenyl)benzamide (1e) (75 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 3 h. Purification by flash column chromatography (hexane/ethyl acetate, 4:1) gave (2-benzoylamino-4,5-dimethoxyphenyl)(2′-bromophenyl)sulfane (3e) (120 mg, 93%) as a pale-yellow solid. Mp 119–120 °C; νmax/cm−1 (neat) 3329 (NH), 2943 (CH), 1659 (CO), 1586 (CC), 1516, 1439, 1250, 1204, 1045, 868; δH (400 MHz, CDCl3) 3.89 (3H, s, OCH3), 4.03 (3H, s, OCH3), 6.61 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 6.98 (1H, td, J 7.7, 1.5 Hz, 4′-H), 7.08–7.13 (2H, m, 6-H and 5′-H), 7.40–7.45 (2H, m, 3′′-H and 5′′-H), 7.48–7.52 (1H, m, 4′′-H), 7.54 (1H, dd, J 7.7, 1.3 Hz, 3′-H), 7.66–7.70 (2H, m, 2′′-H and 6′′-H), 8.52 (1H, s, 3-H), 8.97 (1H, br s, NH); δC (101 MHz, CDCl3) 56.3 (CH3), 56.4 (CH3), 104.7 (CH), 108.6 (C), 118.9 (CH), 120.7 (C), 126.8 (CH), 127.0 (2 × CH), 127.2 (CH), 128.4 (CH), 129.0 (2 × CH), 132.1 (CH), 133.1 (CH), 134.7 (C), 135.6 (C), 137.8 (C) 145.8 (C), 151.7 (C), 165.2 (C); m/z (ESI) 444.0273 (MH+. C21H1979BrNO3S requires 444.0264).
(2-Benzoylamino-4,5-methylenedioxyphenyl)(2′-bromophenyl)sulfane (3f)
The reaction was performed as described in general procedure A using N-(3,4-methylenedioxyphenyl)benzamide (1f) (70 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 6 h. Purification by flash column chromatography (hexane/dichloromethane, 3:7) gave (2-benzoylamino-4,5-methylenedioxyphenyl)(2′-bromophenyl)sulfane (3f) (99 mg, 79%) as a white solid. Mp 160–161 °C; νmax/cm−1 (neat) 3375 (NH), 1670 (CO), 1516 (CC), 1465 (CC), 1238, 1176, 1030, 933, 879; δH (400 MHz, CDCl3) 6.07 (2H, s, OCH2O), 6.69 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.99 (1H, ddd, J 8.0, 7.5, 1.6 Hz, 4′-H), 7.09 (1H, s, 6-H), 7.12 (1H, ddd, J 8.0, 7.5, 1.4 Hz, 5′-H), 7.39–7.44 (2H, m, 3′′-H and 5′′-H), 7.48–7.55 (2H, m, 3′-H and 4′′-H), 7.65–7.70 (2H, m, 2′′-H and 6′′-H), 8.36 (1H, s, 3-H), 9.00 (1H, br s, NH); δC (101 MHz, CDCl3) 102.2 (CH2), 102.9 (CH), 110.2 (C), 115.8 (CH), 121.0 (C), 127.0 (CH), 127.1 (2 × CH), 127.3 (CH), 128.4 (CH), 128.9 (2 × CH), 132.1 (CH), 133.1 (CH), 134.6 (C), 136.3 (C), 137.5 (C), 144.3 (C), 150.6 (C), 165.0 (C); m/z (ESI) 427.9950 (MH+. C20H1579BrNO3S requires 427.9951).
(2-Benzoylamino-4,5,6-trimethoxyphenyl)(2′-bromophenyl)sulfane (3g)
The reaction was performed as described in general procedure A using N-(3,4,5-trimethoxyphenyl)benzamide (1g) (84 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 3 h. Purification by flash column chromatography (hexane/ethyl acetate, 4:1) gave (2-benzoylamino-4,5,6-trimethoxyphenyl)(2′-bromophenyl)sulfane (3g) (112 mg, 81%) as a white solid. Mp 134–137 °C; νmax/cm−1 (neat) 3352 (NH), 2936 (CH), 1663 (CO), 1582 (CC), 1512, 1442, 1288, 1111, 1015, 930; δH (400 MHz, CDCl3) 3.88 (3H, s, OCH3), 3.90 (3H, s, OCH3), 4.01 (3H, s, OCH3), 6.65 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 6.98 (1H, ddd, J 7.9, 7.4, 1.5 Hz, 4′-H), 7.10 (1H, ddd, J 8.0, 7.4, 1.4 Hz, 5′-H), 7.40–7.46 (2H, m, 3′′-H and 5′′-H), 7.49–7.55 (2H, m, 3′-H and 4′′-H), 7.70–7.75 (2H, m, 2′′-H and 6′′-H), 8.36 (1H, s, 3-H), 9.28 (1H, br s, NH); δC (101 MHz, CDCl3) 56.3 (CH3), 61.3 (CH3), 62.0 (CH3), 100.3 (CH), 104.5 (C), 121.1 (C), 126.8 (CH), 127.1 (2 × CH), 127.1 (CH), 128.3 (CH), 129.0 (2 × CH), 132.2 (CH), 133.1 (CH), 134.6 (C), 137.6 (C), 137.6 (C), 139.1 (C), 155.8 (C), 156.4 (C), 165.4 (C); m/z (ESI) 474.0377 (MH+. C22H2079BrNO4S requires 474.0369).
[2-(Benzyloxycarbonyl)amino-5-methylphenyl](2′-bromophenyl)sulfane (3h)
The reaction was performed as described in general procedure A using benzyl-p-tolylcarbamate (1h) (70 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 24 h. Purification by flash column chromatography (hexane/dichloromethane, 7:3) gave [2-(benzyloxycarbonyl)amino-5-methylphenyl](2′-bromophenyl)sulfane (3h) (91 mg, 73%) as a yellow solid. Mp 84–86 °C; νmax/cm−1 (neat) 3381 (NH), 2945 (CH), 1732 (CO), 1519 (CC), 1443, 1219, 1049, 824, 740; δH (400 MHz, CDCl3) 2.24 (3H, s, CH3), 5.07 (2H, s, PhCH2), 6.45 (1H, dd, J 7.7, 1.5 Hz, 6′-H), 6.91 (1H, td, J 7.7, 1.5 Hz, 4′-H), 7.00 (1H, td, J 7.7, 1.4 Hz, 5′-H), 7.17–7.29 (6H, m, 4-H and Ph), 7.31 (1H, d, J 1.6 Hz, 6-H), 7.44 (1H, dd, J 7.7, 1.4 Hz, 3′-H), 7.56 (1H, br s, NH), 8.10 (1H, d, J 8.4 Hz, 3-H); δC (101 MHz, CDCl3) 20.6 (CH3), 67.1 (CH2), 118.1 (C), 119.6 (CH), 121.2 (C), 126.9 (CH), 127.0 (CH), 128.2 (CH), 128.3 (2 × CH), 128.4 (CH), 128.7 (2 × CH), 132.6 (CH), 133.1 (CH), 133.9 (C), 136.1 (C), 137.6 (CH), 137.8 (C), 138.3 (C), 153.4 (C); m/z (ESI) 428.0312 (MH+. C21H1979BrNO2S requires 428.0314).
[2(4′′-Methylphenylsulfonyl)amino-5-methylphenyl](2′-bromophenyl)sulfane (3i)
The reaction was performed as described in general procedure A using N-(4-methylphenyl)-p-toluenesulfonamide (1i) (76 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave [2-(4-methylphenylsulfonyl)amino-5-methylphenyl](2′-bromophenyl)sulfane (3i) (65 mg, 50%) as a white solid. Mp 108–110 °C; νmax/cm−1 (neat) 3212 (NH), 2918 (CH), 1486, 1340, 1185, 1019, 806, 677; δH (400 MHz, CDCl3) 2.27 (3H, s, CH3), 2.35 (3H, s, CH3), 6.23 (1H, dd, J 7.8, 1.6 Hz, 6′-H), 6.92 (1H, ddd, J 7.8, 7.4, 1.5 Hz, 5′-H), 6.98 (1H, dd, J 7.8, 7.4, 1.6 Hz, 4′-H), 7.09–7.13 (2H, m, 3′′-H and 5′′-H), 7.20–7.26 (2H, m, 4-H and 6-H), 7.49 (1H, br s, NH), 7.51 (1H, dd, J 7.8, 1.5 Hz, 3′-H), 7.55–7.60 (2H, m, 2′′-H and 6′′-H), 7.70 (1H, d, J 8.3 Hz, 3-H); δC (101 MHz, CDCl3) 20.7 (CH3), 21.7 (CH3), 120.4 (CH), 121.4 (C), 127.0 (CH), 127.2 (CH), 127.2 (C), 127.4 (2 × CH), 128.1 (CH), 129.7 (2 × CH), 132.6 (CH), 133.1 (CH), 135.4 (C), 136.0 (C), 137.3 (C), 137.4 (C), 137.9 (CH), 144.0 (C); m/z (ESI) 469.9852 (MNa+. C20H1879BrNNaO2S2 requires 469.9855).
(2-Methylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3j)
The reaction was performed as described in general procedure A using N-methyl-p-toluidine (1j) (35 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 9:1) gave (2-methylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3j) (59 mg, 66%) as a yellow oil. νmax/cm−1 (neat) 3395 (NH), 2909 (CH), 1605 (CC), 1512, 1443, 1312, 1169, 1018, 745; δH (400 MHz, CDCl3) 2.26 (3H, s, CH3), 2.81 (3H, d, J 4.9 Hz, NHCH3), 4.73 (1H, q, J 4.9 Hz, NHCH3), 6.56 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.63 (1H, d, J 8.3 Hz, 3-H), 6.95 (1H, ddd, J 7.9, 7.4, 1.6 Hz, 4′-H), 7.08 (1H, ddd, J 8.0, 7.4, 1.4 Hz, 5′-H), 7.21 (1H, dd, J 8.3, 2.1 Hz, 4-H), 7.30 (1H, d, J 2.1 Hz, 6-H), 7.51 (1H, dd, J 7.9, 1.4 Hz, 3′-H); δC (101 MHz, CDCl3) 20.2 (CH3), 30.8 (CH3), 110.5 (CH), 112.4 (C), 120.7 (C), 126.2 (CH), 126.2 (CH), 126.4 (C), 127.9 (CH), 132.8 (CH), 132.9 (CH), 138.3 (CH), 138.5 (C), 148.9 (C); m/z (ESI) 329.9929 (MNa+. C14H1479BrNNaS requires 329.9923).
(2-Benzylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3k)
The reaction was performed as described in general procedure A using N-benzyl-p-toluidine (1k) (57 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 9:1) gave (2-benzylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3k) (66 mg, 59%) as a yellow solid. Mp 72–75 °C; νmax/cm−1 (neat) 3403 (NH), 2897 (CH), 1605 (CC), 1508, 1443, 1316, 1015, 806, 748; δH (400 MHz, CDCl3) 2.23 (3H, s, CH3), 4.34 (2H, d, J 5.8 Hz, PhCH2), 5.24 (1H, t, J 5.8 Hz, NH), 6.56 (1H, d, J 8.3 Hz, 3-H), 6.64 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.98 (1H, ddd, J 7.8, 7.4, 1.6 Hz, 4′-H), 7.07–7.29 (7H, m, 4-H, 5′-H and Ph), 7.32 (1H, d, J 1.8 Hz, 6-H), 7.51 (1H, dd, J 7.8, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 20.2 (CH3), 47.8 (CH2), 111.4 (CH), 112.9 (C), 121.0 (C), 126.4 (CH), 126.7 (CH), 126.8 (C), 127.0 (2 × CH), 127.2 (CH), 127.8 (CH), 128.7 (2 × CH), 132.8 (CH), 132.9 (CH), 138.3 (CH), 138.4 (C), 139.3 (C), 147.4 (C); m/z (ESI) 406.0239 (MNa+. C20H1879BrNNaS requires 406.0236).
(2-Amino-5-methylphenyl)(2′-bromophenyl)sulfane (3l).25f
The reaction was performed as described in general procedure A using p-toluidine (1l) (31 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 20 h. Purification by flash column chromatography (hexane/dichloromethane, 7:3) gave (2-amino-5-methylphenyl)(2′-bromophenyl)sulfane (3l) (46 mg, 53%) as a white solid. Mp 83–86 °C (lit.25f 82–84 °C); δH (400 MHz, CDCl3) 2.26 (3H, s, CH3), 4.16 (2H, br s, NH2), 6.63 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.75 (1H, d, J 8.2 Hz, 3-H), 6.97 (1H, ddd, J 7.9, 7.4, 1.6 Hz, 4′-H), 7.08–7.12 (2H, m, 4-H and 5′-H), 7.29 (1H, d, J 2.0 Hz, 6-H), 7.52 (1H, dd, J 7.9, 1.4 Hz, 3′-H); δC (101 MHz, CDCl3) 20.3 (CH3), 113.2 (C), 115.7 (CH), 120.8 (C), 126.3 (CH), 126.4 (CH), 127.9 (CH), 128.5 (C), 132.7 (CH), 132.9 (CH), 138.0 (CH), 138.3 (C), 146.9 (C); m/z (ESI) 294 (MH+. 100%).
(2-Amino-5-biphenyl)(2′-bromophenyl)sulfane (3m)
The reaction was performed as described in general procedure A using 4-aminobiphenyl (1m) (99 mg, 0.58 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave (2-amino-5-biphenyl)(2′-bromophenyl)sulfane (3m) (113 mg, 55%) as a yellow oil. νmax/cm−1 (neat) 3371 (NH), 3024 (CH), 1613 (CC), 1477, 1443, 1157, 1018, 745; δH (400 MHz, CDCl3) 4.37 (2H, br s, NH2), 6.72 (1H, dd, J 8.0, 1.6 Hz, 6′-H), 6.91 (1H, d, J 8.4 Hz, 3-H), 6.99 (1H, ddd, J 7.9, 7.4, 1.6 Hz, 4′-H), 7.12 (1H, ddd, J 8.0, 7.4, 1.4 Hz, 5′-H), 7.27–7.32 (1H, m, 4′′-H), 7.39–7.44 (2H, m, 3′′-H and 5′′-H), 7.53–7.59 (4H, m, 4-H, 3′-H, 2′′-H and 6′′-H), 7.77 (1H, d, J 2.2 Hz, 6-H); δC (101 MHz, CDCl3) 113.7 (C), 115.9 (CH), 120.9 (C), 126.4 (2 × CH), 126.4 (CH), 126.5 (CH), 126.8 (CH), 128.0 (CH), 128.9 (2 × CH), 130.5 (CH), 132.2 (C), 133.0 (CH), 136.3 (CH), 138.0 (C), 140.1 (C), 148.6 (C); m/z (ESI) 356.0109 (MH+. C18H1579BrNS requires 356.0103).
1-(2′-Bromophenylthio)-2-aminonaphthalene (3n)41
The reaction was performed as described in general procedure A using 2-aminonaphthalene (1n) (83 mg, 0.58 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 7:3) gave 1-(2′-bromophenylthio)-2-aminonaphthalene (3n) (142 mg, 75%) as an orange solid. Mp 125–127 °C; Spectroscopic data were consistent with the literature.41δH (400 MHz, CDCl3) 4.72 (2H, br s, NH2), 6.37–6.46 (1H, m, 8-H), 6.90–6.98 (2H, m, 6-H and 7-H), 7.07 (1H, d, J 8.7 Hz, 3-H), 7.29 (1H, ddd, J 8.0, 6.9, 1.2 Hz, 5′-H), 7.45 (1H, ddd, J 8.4, 6.9, 1.3 Hz, 4′-H), 7.50–7.57 (1H, m, 5-H), 7.74 (1H, dd, J 8.0, 1.3 Hz, 6′-H), 7.80 (1H, d, J 8.7 Hz, 4-H), 8.19 (1H, dd, J 8.4, 1.2 Hz, 3′-H); δC (101 MHz, CDCl3) 103.7 (C), 117.8 (CH), 121.1 (C), 122.9 (CH), 124.1 (CH), 126.1 (CH), 126.2 (CH), 127.9 (CH), 128.1 (CH), 128.6 (CH), 128.6 (C), 132.4 (CH), 132.9 (CH), 136.6 (C), 137.7 (C), 148.8 (C); m/z (ESI) 330 (MH+. 100%).
(2-Amino-5-chloro)(2′-bromophenyl)sulfane (3o)
The reaction was performed as described in general procedure A using 4-chloroaniline (1o) (37 mg, 0.29 mmol). The reaction mixture was stirred at 90 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 7:3) gave (2-amino-5-chloro)(2′-bromophenyl)sulfane (3o) (38 mg, 42%) as a colourless oil. νmax/cm−1 (neat) 3372 (NH), 3055 (CH), 1609 (CC), 1474, 1442, 1296, 1103, 1018, 745; δH (400 MHz, CDCl3) 4.30 (2H, br s, NH2), 6.65 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 6.75 (1H, d, J 8.6 Hz, 3-H), 7.00 (1H, ddd, J 7.9, 7.5, 1.5 Hz, 4′-H), 7.13 (1H, ddd, J 8.0, 7.5, 1.3 Hz, 5′-H), 7.23 (1H, dd, J 8.6, 2.5 Hz, 4-H), 7.45 (1H, d, J 2.5 Hz, 6-H), 7.53 (1H, dd, J 7.9, 1.3 Hz, 3′-H); δC (101 MHz, CDCl3) 114.7 (C), 116.5 (CH), 121.1 (C), 122.8 (C), 126.6 (CH), 126.8 (CH), 128.0 (CH), 131.8 (CH), 133.1 (CH), 136.8 (CH), 137.2 (C), 147.9 (C); m/z (ESI) 313.9401 (MH+. C12H1079BrClNS requires 313.9400).
General procedure B: copper catalysed synthesis of phenothiazines
To a solution of biaryl sulfide (1.0 equiv.) in toluene (0.4 M in arene) was added copper(I) iodide (10 mol%), cesium carbonate (2.0 equiv.), N,N′-dimethylethylenediamine (20 mol%) and water (1 M in arene). The mixture was purged with nitrogen and then heated to 130 °C or 150 °C, until the reaction was deemed complete. The reaction mixture was then cooled to room temperature, diluted with ethyl acetate (20 mL) and washed with a 1 M aqueous solution of sodium thiosulfate solution (20 mL). The aqueous layer was extracted with ethyl acetate (2 × 20 mL) and the combined organic layers were washed with brine (20 mL). The organic phase was dried (MgSO4), filtered and concentrated in vacuo. The resulting material was purified by flash column chromatography.N-Benzoyl-3-methylphenothiazine (4a)
The reaction was performed as described in general procedure B using (2-benzoylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3a) (32 mg, 0.080 mmol). The reaction mixture was stirred at 130 °C for 48 h. Purification by flash column chromatography (hexane/dichloromethane, 2:3) gave N-benzoyl-3-methylphenothiazine (4a) (22 mg, 86%) as a white solid. Mp 123–125 °C; νmax/cm−1 (neat) 2916 (CH), 1667 (CO), 1462, 1319, 1258, 1111, 810, 760; δH (400 MHz, CDCl3) 2.30 (3H, s, 3-CH3), 6.95 (1H, dd, J 8.2, 1.9 Hz, 2-H), 7.11–7.17 (2H, m, 6-H and 8-H), 7.19–7.33 (5H, m, 1-H, 4-H, 3′-H, 4′-H and 5′-H), 7.35–7.46 (4H, m, 7-H, 9-H, 2′-H and 6′-H); δC (101 MHz, CDCl3) 21.0 (CH3), 126.5 (CH), 126.8 (CH), 126.9 (CH), 127.2 (CH), 127.8 (CH), 127.8 (CH), 128.1 (2 × CH), 128.2 (CH), 129.0 (2 × CH), 130.4 (CH), 132.0 (C), 132.4 (C), 135.5 (C), 136.6 (C), 137.1 (C), 139.8 (C), 169.0 (C); m/z (ESI) 318.0954 (MH+. C20H16NOS requires 318.0947).
N-Benzoyl-2,3-dimethylphenothiazine (4b)
The reaction was performed as described in general procedure B using (2-benzoylamino-4,5-dimethylphenyl)(2′-bromophenyl)sulfane (3b) (30 mg, 0.072 mmol). The reaction mixture was stirred at 150 °C for 48 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave N-benzoyl-2,3-dimethylphenothiazine (4b) (15 mg, 65%) as a white solid. Mp 170–171 °C; νmax/cm−1 (neat) 2916 (CH), 1678 (CO), 1492, 1392, 1246, 1014, 883, 794; δH (400 MHz, CDCl3) 2.14 (3H, s, CH3), 2.21 (3H, s, CH3), 7.05–7.13 (2H, m, 6-H and 8-H), 7.19–7.22 (3H, m, 4-H, 3′-H and 5′-H), 7.26–7.31 (3H, m, 1-H, 7-H and 4′-H), 7.35–7.37 (2H, m, 2′-H and 6′-H), 7.42 (1H, dd, J 7.6, 1.8 Hz, 9-H); δC (101 MHz, CDCl3) 19.5 (CH3), 19.6 (CH3), 126.4 (CH), 126.8 (CH), 127.2 (CH), 127.8 (CH), 128.0 (2 × CH), 128.1 (CH), 128.4 (CH), 128.8 (C), 128.9 (2 × CH), 130.3 (CH), 132.9 (C), 135.3 (C), 135.6 (C), 135.8 (C), 137.1 (C), 140.0 (C), 169.0 (C); m/z (ESI) 332.1111 (MH+. C21H18NOS requires 332.1104).
N-Benzoyl-3-tert-butylphenothiazine (4c)
The reaction was performed as described in general procedure B using (2-benzoylamino-5-tert-butylphenyl)(2′-bromophenyl)sulfane (3c) (80 mg, 0.18 mmol). The reaction mixture was stirred at 130 °C for 48 h. Purification by flash column chromatography (hexane/dichloromethane, 1:1) gave N-benzoyl-3-tert-butylphenothiazine (4c) (50 mg, 77%) as a white solid. Mp 123–124 °C; νmax/cm−1 (neat) 2963 (CH), 1651 (CO), 1462, 1323, 1258, 1115, 880, 756; δH (400 MHz, CDCl3) 1.30 (9H, s, 3 × CH3), 7.07–7.16 (2H, m, 7-H and 8-H), 7.18–7.24 (3H, m, 2-H, 3′-H and 5′-H), 7.26–7.33 (2H, m, 6-H and 4′-H), 7.36–7.40 (2H, m, 2′-H and 6′-H), 7.42–7.47 (3H, m, 1-H, 4-H and 9-H); δC (101 MHz, CDCl3) 31.4 (3 × CH3), 34.8 (C), 124.3 (CH), 124.6 (CH), 126.4 (CH), 126.6 (CH), 126.9 (CH), 127.2 (CH), 127.8 (CH), 128.0 (2 × CH), 129.0 (2 × CH), 130.4 (CH), 131.6 (C), 132.6 (C), 135.5 (C), 136.8 (C), 139.9 (C), 150.0 (C), 169.0 (C); m/z (ESI) 360.1425 (MH+. C23H22NOS requires 360.1417).
N-Benzoyl-2-methoxy-3-methylphenothiazine (4d)
The reaction was performed as described in general procedure B using (2-benzoylamino-4-methoxy-5-methylphenyl)(2′-bromophenyl)sulfane (3d) (60 mg, 0.14 mmol). The reaction mixture was stirred at 130 °C for 96 h. Purification by flash column chromatography (hexane/dichloromethane, 3:7) gave N-benzoyl-2-methoxy-3-methylphenothiazine (4d) (52 mg, 80%) as a white solid. Mp 180–181 °C; νmax/cm−1 (neat) 3050 (CH), 1654 (CO), 1462, 1338, 1249, 1057, 756, 706; δH (400 MHz, CDCl3) 2.16 (3H, s, 3-CH3), 3.62 (3H, s, 2-OCH3), 6.91 (1H, s, 4-H), 7.10–7.17 (3H, m, 1-H, 7-H and 8-H), 7.21–7.25 (2H, m, 3′-H and 5′-H), 7.29–7.34 (1H, m, 4′-H), 7.37–7.44 (4H, m, 6-H, 9-H, 2′-H and 6′-H); δC (101 MHz, CDCl3) 16.0 (CH3), 55.7 (CH3), 109.9 (CH), 122.1 (C), 125.7 (C), 126.4 (CH), 126.7 (CH), 127.1 (CH), 127.7 (CH), 128.1 (2 × CH), 128.8 (CH) 128.8 (2 × CH), 130.4 (CH), 133.2 (C), 135.6 (C), 138.3 (C), 139.7 (C), 156.9 (C), 169.1 (C); m/z (ESI) 348.1060 (MH+. C21H18NO2S requires 348.1053).
N-Benzoyl-2,3-dimethoxyphenothiazine (4e)
The reaction was performed as described in general procedure B using (2-benzoylamino-4,5-dimethoxyphenyl)(2′-bromophenyl)sulfane (3e) (50 mg, 0.11 mmol). The reaction mixture was stirred at 130 °C for 24 h. Purification by flash column chromatography (hexane/diethyl ether, 3:2) gave N-benzoyl-2,3-dimethoxyphenothiazine (4e) (34 mg, 83%) as a white solid. Mp 112–114 °C; νmax/cm−1 (neat) 2936 (CH), 1663 (CO), 1501, 1443, 1308, 1261, 1026, 849; δH (400 MHz, CDCl3) 3.66 (3H, s, OCH3), 3.86 (3H, s, OCH3), 6.89 (1H, s, 4-H), 6.92 (1H, s, 1-H), 7.12–7.18 (2H, m, 6-H and 8-H), 7.20–7.25 (2H, m, 3′-H and 5′-H), 7.29–7.33 (1H, m, 4′-H), 7.35–7.46 (4H, m, 7-H, 9-H, 2′-H and 6′-H); δC (101 MHz, CDCl3) 56.2 (CH3), 56.3 (CH3), 109.9 (CH), 111.1 (CH), 123.0 (C), 126.4 (CH), 126.9 (CH), 127.1 (CH), 127.7 (CH), 128.2 (2 × CH), 128.8 (2 × CH), 130.4 (CH), 132.7 (C), 132.8 (C), 135.6 (C), 139.8 (C), 147.6 (C), 148.1 (C), 169.1 (C); m/z (ESI) 364.1011 (MH+. C21H18NO3S requires 364.1002).
N-Benzoyl-2,3-methylenedioxyphenothiazine (4f)
The reaction was performed as described in general procedure B using (2-benzoylamino-4,5-methylenedioxyphenyl)(2′-bromophenyl)sulfane (3f) (79 mg, 0.18 mmol). The reaction mixture was stirred at 150 °C for 72 h. Purification by flash column chromatography (hexane/dichloromethane, 3:7) gave N-benzoyl-2,3-methylenedioxyphenothiazine (4f) (44 mg, 69%) as a white solid. Mp 185–187 °C; νmax/cm−1 (neat) 3055 (CH), 1647 (CO), 1462, 1327, 1238, 1030, 922, 860, 806; δH (400 MHz, CDCl3) 5.95 (2H, s, OCH2O), 6.87 (1H, s, 4-H), 7.03 (1H, s, 1-H), 7.09 (1H, td, J 7.6, 1.4 Hz, 8-H), 7.14 (1H, td, J 7.6, 1.4 Hz, 7-H), 7.21–7.25 (3H, m, 6-H, 3′-H and 5′-H), 7.32 (1H, tt, J 7.4, 1.2 Hz, 4′-H), 7.35–7.38 (2H, m, 2′-H and 6′-H), 7.43 (1H, dd, J 7.6, 1.4 Hz, 9-H); δC (101 MHz, CDCl3) 102.1 (CH2), 107.4 (CH), 108.6 (CH), 124.8 (C), 126.5 (CH), 127.0 (CH), 127.2 (CH), 127.7 (CH), 128.2 (2 × CH), 128.9 (2 × CH), 130.5 (CH), 133.2 (C), 133.6 (C), 135.3 (C), 140.1 (C), 146.3 (C), 147.3 (C), 169.0 (C); m/z (ESI) 348.0691 (MH+. C20H14NO3S requires 348.0689).
N-Benzoyl-2,3,4-trimethoxyphenothiazine (4g)
The reaction was performed as described in general procedure B using (2-benzoylamino-4,5,6-trimethoxyphenyl)(2′-bromophenyl)sulfane (3g) (50 mg, 0.11 mmol). The reaction mixture was stirred at 130 °C for 48 h. Purification by flash column chromatography (hexane/ethyl acetate, 4:1) gave N-benzoyl-2,3,4-trimethoxyphenothiazine (4g) (22 mg, 53%) as a white solid. Mp 111–113 °C; νmax/cm−1 (neat) 2936 (CH), 1659 (CO), 1447, 1308, 1242, 1107, 926, 745; δH (400 MHz, CDCl3) 3.69 (3H, s, OCH3), 3.85 (3H, s, OCH3), 3.98 (3H, s, OCH3), 6.86 (1H, s, 1-H), 7.08–7.16 (2H, m, 7-H and 8-H), 7.20–7.39 (6H, m, 6-H and Ph), 7.46 (1H, dd, J 7.5, 1.6 Hz, 9-H); δC (101 MHz, CDCl3) 56.4 (CH3), 61.3 (CH3), 61.4 (CH3), 107.5 (CH), 118.0 (C), 126.5 (CH), 126.9 (CH), 127.1 (CH), 128.0 (CH), 128.1 (2 × CH), 128.8 (2 × CH), 130.5 (CH), 132.5 (C), 134.7 (C) 135.5 (C), 139.7 (C), 140.8 (C), 149.6 (C), 152.5 (C), 169.2 (C); m/z (ESI) 394.1117 (MH+. C22H20NO4S requires 394.1108).
N-Benzyloxycarbonyl-3-methylphenothiazine (4h)
The reaction was performed as described in general procedure B using ([2-(benzyloxycarbonyl)amino-5-methylphenyl](2′-bromophenyl)sulfane (3h) (50 mg, 0.12 mmol) and trans-N,N′-dimethylcyclohexane-1,2-diamine (3.7 μL, 23 mol%) instead of N,N′-dimethylethylenediamine. The reaction mixture was stirred at 130 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave N-benzyloxycarbonyl-3-methylphenothiazine (4h) (31 mg, 76%) as a colourless oil. νmax/cm−1 (neat) 2952 (CH), 1711 (CO), 1469, 1318, 1216, 1093, 748; δH (400 MHz, CDCl3) 2.33 (3H, s, 3-CH3), 5.26 (2H, s, PhCH2), 7.08 (1H, dd, J 8.2, 1.3 Hz, 2-H), 7.14–7.20 (2H, m, 4-H and 7-H), 7.27 (1H, ddd, J 8.0, 7.5, 1.5 Hz, 8-H), 7.30–7.39 (6H, m, 6-H and Ph), 7.42 (1H, d, J 8.2 Hz, 1-H) 7.54 (1H, dd, J 8.0, 1.0 Hz, 9-H); δC (101 MHz, CDCl3) 21.0 (CH3), 68.1 (CH2), 126.5 (CH), 126.8 (CH), 126.9 (CH), 127.1 (CH), 127.7 (CH), 127.8 (CH), 127.9 (2 × CH), 128.0 (CH), 128.2 (CH), 128.6 (2 × CH), 132.0 (C), 132.4 (C), 135.8 (C), 136.1 (C), 136.6 (C), 138.6 (C), 153.8 (C); m/z (ESI) 348.1052 (MH+. C21H18NO2S requires 348.1053).
N-(4′-Methylphenylsulfonyl)-3-methylphenothiazine (4i)
The reaction was performed as described in general procedure B using [2-(4-methylphenylsulfonyl)amino-5-methylphenyl](2′-bromophenyl)sulfane (3i) (39 mg, 0.087 mmol) and trans-N,N′-dimethylcyclohexane-1,2-diamine (2.7 μL, 20 mol%) instead of N,N′-dimethylethylenediamine. The reaction mixture was stirred at 130 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 3:2) gave N-(4′-methylphenylsulfonyl)-3-methylphenothiazine (4i) (24 mg, 75%) as a yellow solid. Mp 146–148 °C; νmax/cm−1 (neat) 2920 (CH), 1597 (CC), 1447, 1354, 1165, 922, 760; δH (400 MHz, CDCl3) 2.32 (3H, s, 3-CH3), 2.37 (3H, s, 4′-CH3), 6.92 (1H, d, J 1.1 Hz, 4-H), 7.05 (2H, d, J 8.1 Hz, 3′-H and 5′-H), 7.08–7.16 (4H, m, 2-H, 6-H, 2′-H and 6′-H), 7.20 (1H, td, J 7.5, 1.3 Hz, 7-H), 7.32 (1H, ddd, J 7.9, 7.5, 1.5 Hz, 8-H), 7.61 (1H, d, J 8.2 Hz, 1-H), 7.73 (1H, dd, J 7.9, 1.3 Hz, 9-H); δC (101 MHz, CDCl3) 21.1 (CH3), 21.8 (CH3), 127.0 (CH), 127.2 (CH), 127.3 (CH), 127.7 (CH), 127.8 (2 × CH), 128.2 (CH), 129.4 (2 × CH), 129.8 (CH), 130.1 (CH), 132.8 (C), 133.3 (C), 133.5 (C), 136.2 (C), 136.3 (C), 138.0 (C), 144.1 (C); m/z (ESI) 402.0395 (MCl−. C20H1735ClNO2S2 requires 402.0395).
General procedure C: palladium catalysed synthesis of phenothiazines
To a solution of biaryl sulfide (1.0 equiv.) in toluene (0.33 M in arene) was added Pd2(dba)3 (5 mol%) and (S)-BINAP (10 mol%). The reaction mixture was purged with nitrogen and sodium tert-butoxide (2.0 equiv.) was added. The mixture was then heated at 110 °C, until the reaction was deemed complete. The reaction mixture was allowed to cool to room temperature and water (10 mL) was added before being filtered through Celite and washed with dichloromethane (10 mL). The layers were separated and the aqueous layer was further extracted with dichloromethane (2 × 10 mL). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo. The resulting material was purified by flash column chromatography.3,10-Dimethylphenothiazine (4j).20c
The reaction was performed as described in general procedure C using (2-methylamino-5-methylphenyl)(2′-bromophenyl) (3j) (48 mg, 0.16 mmol). The reaction mixture was stirred at 110 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 9:1) gave 3,10-dimethylphenothiazine (4j) (30 mg, 86%) as a white solid. Mp 143–145 °C (lit.20c 145–146 °C); δH (400 MHz, CDCl3) 2.26 (3H, s, 3-CH3), 3.35 (3H, s, NCH3), 6.71 (1H, d, J 8.1 Hz, 1-H), 6.80 (1H, dd, J 8.0, 1.2 Hz, 9-H), 6.92 (1H, td, J 7.6, 1.2 Hz, 7-H), 6.95–7.01 (2H, m, 2-H and 4-H), 7.11–7.21 (2H, m, 6-H and 8-H); δC (101 MHz, CDCl3) 20.4 (CH3), 35.4 (CH3), 114.0 (CH), 114.0 (CH), 122.3 (CH), 123.3 (C), 123.5 (C), 127.3 (CH), 127.5 (CH), 127.8 (CH), 128.0 (CH), 132.1 (C), 143.5 (C), 146.2 (C); m/z (ESI) 228 (MNa+. 100%).
N-Benzyl-3-methylphenothiazine (4k)
The reaction was performed as described in general procedure C using (2-benzylamino-5-methylphenyl)(2′-bromophenyl)sulfane (3k) (46 mg, 0.12 mmol). The reaction mixture was stirred at 110 °C for 18 h. Purification by flash column chromatography (hexane/dichloromethane, 1:1) gave N-benzyl-3-methylphenothiazine (4k) (34 mg, 95%) as a colourless oil. νmax/cm−1 (neat) 2916 (CH), 1578 (CC), 1466, 1358, 1254, 871, 729; δH (400 MHz, CDCl3) 2.21 (3H, s, 3-CH3), 5.07 (2H, s, PhCH2), 6.53 (1H, d, J 8.3 Hz, 1-H), 6.62 (1H, dd, J 8.2, 1.1 Hz, 9-H), 6.77 (1H, dd, J 8.3, 1.6 Hz, 2-H), 6.85 (1H, td, J 7.5, 1.1 Hz, 7-H), 6.92 (1H, d, J 1.6 Hz, 4-H), 6.97 (1H, ddd, J 8.2, 7.5, 1.6 Hz, 8-H), 7.09 (1H, dd, J 7.5, 1.6 Hz, 6-H), 7.22–7.38 (5H, m, Ph); δC (101 MHz, CDCl3) 20.4 (CH3), 52.7 (CH2), 115.3 (CH), 115.4 (CH), 122.3 (CH), 123.0 (C), 123.1 (C), 126.7 (2 × CH), 126.9 (CH), 127.1 (CH), 127.3 (CH), 127.4 (CH), 127.8 (CH), 128.8 (2 × CH), 132.2 (C), 137.0 (C), 142.1 (C), 144.8 (C); m/z (ESI) 326.0982 (MNa+. C20H17NNaS requires 326.0974).
3-Methyl-10H-phenothiazine (4l).42
The reaction was performed as described in general procedure C using (2-amino-5-methylphenyl)(2′-bromophenyl)sulfane (3l) (30 mg, 0.10 mmol). The reaction mixture was stirred at 110 °C for 18 h. Purification by flash column chromatography (hexane/ethyl acetate, 9:1) gave 3-methyl-10H-phenothiazine (4l) (18 mg, 82%) as a white solid. Mp 166–169 °C (lit.42 169–170 °C); δH (400 MHz, DMSO-d6) 2.12 (3H, s, 3-CH3), 6.58 (1H, d, J 8.0 Hz, 1-H), 6.66 (1H, dd, J 7.9, 1.1 Hz, 9-H), 6.68–6.75 (2H, m, 4-H and 7-H), 6.79 (1H, dd, J 8.0, 1.8 Hz, 2-H), 6.89 (1H, dd, J 7.7, 1.3 Hz, 6-H), 6.97 (1H, td, J 7.9, 1.3 Hz 8-H), 8.45 (1H, br s, NH); δC (101 MHz, DMSO-d6) 19.9 (CH3), 114.3 (CH), 114.3 (CH), 116.2 (C), 116.2 (C), 121.4 (CH), 126.2 (CH), 126.5 (CH), 127.5 (CH), 128.0 (CH), 130.7 (C), 139.5 (C), 142.4 (C); m/z (ESI) 214 (MH+. 100%).
3-Phenyl-10H-phenothiazine (4m).43
The reaction was performed as described in general procedure C using (2-amino-5-biphenyl)(2′-bromophenyl)sulfane (3m) (100 mg, 0.28 mmol). The reaction mixture was stirred at 110 °C for 4 h. Purification by flash column chromatography (hexane/dichloromethane, 5:1) gave 3-phenyl-10H-phenothiazine (4m) (63 mg, 82%) as a yellow solid. Mp 220–222 °C (lit.43 216–218 °C); δH (400 MHz, DMSO-d6) 6.69 (1H, dd, J 7.9, 1.1 Hz, 9-H), 6.73–6.79 (2H, m, 1-H and 7-H), 6.93 (1H, dd, J 7.7, 1.4 Hz, 6-H), 7.00 (1H, td, J 7.9, 1.4 Hz, 8-H), 7.22 (1H, d, J 2.1 Hz, 4-H), 7.25–7.32 (2H, m, 2-H and 4′-H), 7.37–7.43 (2H, m, 3′-H and 5′-H), 7.54–7.59 (2H, m, 2′-H and 6′-H), 8.70 (1H, br s, NH); δC (101 MHz, DMSO-d6) 114.4 (CH), 114.7 (CH), 116.1 (C), 117.0 (C), 121.8 (CH), 124.1 (CH), 125.7 (2 × CH), 125.8 (CH), 126.2 (CH), 126.8 (CH), 127.6 (CH), 128.8 (2 × CH), 133.7 (C), 139.1 (C), 141.3 (C), 141.7 (C); m/z (ESI) 299 (MNa+. 100%).
Benzo[a]phenothiazine (4n).44
The reaction was performed as described in general procedure C using 1-(2′-bromophenylthio)-2-aminonaphthalene (3n) (100 mg, 0.39 mmol). The reaction mixture was stirred at 110 °C for 4 h. Purification by flash column chromatography (hexane/dichloromethane, 5:1) gave benzo[a]phenothiazine (4n) (85 mg, 87%) as a yellow solid. Mp 184–186 °C (lit.44 185 °C); δH (400 MHz, DMSO-d6) 6.74 (1H, dd, J 8.3, 1.3 Hz, 13-H), 6.80 (1H, td, J 7.5, 1.3 Hz, 11-H), 7.00–7.05 (3H, m, 1-H, 10-H and 12-H), 7.31 (1H, ddd, J 8.0, 6.9, 1.1 Hz, 4-H), 7.49 (1H, ddd, J 8.3, 6.9, 1.3 Hz, 5-H), 7.63 (1H, d, J 8.6 Hz, 2-H), 7.70 (1H, dd, J 8.3, 1.1 Hz, 6-H), 7.76 (1H, dd, J 8.0, 1.3 Hz, 3-H), 8.80 (1H, br s, NH); δC (101 MHz, DMSO-d6) 107.0 (C), 114.5 (CH), 116.0 (C), 116.8 (CH), 121.4 (CH), 122.3 (CH), 123.3 (CH), 126.6 (CH), 127.0 (CH), 127.4 (CH), 127.8 (CH), 128.4 (CH), 129.6 (C), 129.9 (C), 139.8 (C), 142.4 (C); m/z (ESI) 288 (MK+. 100%).
3-Chloro-10H-phenothiazine (4o).16b
The reaction was performed as described in general procedure C using (2-amino-5-chloro)(2′-bromophenyl)sulfane (3o) (60 mg, 0.19 mmol). The reaction mixture was stirred at 110 °C for 4 h. Purification by flash column chromatography (hexane/dichloromethane, 5:1) gave 3-chloro-10H-phenothiazine (4o) (85 mg, 87%) as a white solid. Mp 201–203 °C (lit.16b 200–201 °C); δH (400 MHz, DMSO-d6) 6.63–6.68 (2H, m, 1-H and 9-H), 6.77 (1H, td, J 7.7, 1.3 Hz, 7-H), 6.91 (1H, dd, J 7.7, 1.4 Hz, 6-H), 6.97–7.05 (3H, m, 2-H, 4-H and 8-H), 8.71 (1H, br s, NH); δC (101 MHz, DMSO-d6) 114.6 (CH), 115.4 (CH), 115.5 (C), 118.6 (C), 122.1 (CH), 124.9 (C), 125.4 (CH), 126.3 (CH), 127.2 (CH), 127.8 (CH), 141.1 (C), 141.6 (C); m/z (EI) 233 (M+. 100%).
(2-Benzoylamino-4-methoxyphenyl)(2′-bromophenyl)sulfane (6)
The reaction was performed as described in general procedure A using N-(3-methoxyphenyl)benzamide (5) (66 mg, 0.29 mmol). The reaction mixture was stirred at 75 °C for 4 h. Purification by flash column chromatography (hexane/ethyl acetate, 9:1) gave (2-benzoylamino-4-methoxyphenyl)(2′-bromophenyl)sulfane (6) (78 mg, 66%) as a white solid. Mp 78–81 °C; νmax/cm−1 (neat) 3360 (NH), 2936 (CH), 1678 (CO), 1574 (CC), 1443, 1250, 1169, 1018, 748; δH (400 MHz, CDCl3) 3.93 (3H, s, OCH3), 6.61 (1H, dd, J 8.0, 1.5 Hz, 6′-H), 6.77 (1H, dd, J 8.6, 2.8 Hz, 5-H), 6.98 (1H, td, J 7.7, 1.5 Hz, 4′-H), 7.10 (1H, ddd, J 8.0, 7.7, 1.4 Hz, 5′-H), 7.40–7.45 (2H, m, 3′′-H and 5′′-H), 7.48–7.55 (2H, m, 3′-H and 4′′-H), 7.56 (1H, d, J 8.6 Hz, 6-H), 7.67–7.71 (2H, m, 2′′-H and 6′′-H), 8.47 (1H, d, J 2.8 Hz, 3-H), 9.13 (1H, br s, NH); δC (101 MHz, CDCl3) 55.8 (CH3), 105.6 (CH), 109.4 (C), 111.7 (CH), 120.8 (C), 126.8 (CH), 127.1 (2 × CH), 127.1 (CH), 128.3 (CH), 129.0 (2 × CH), 132.2 (CH), 133.1 (CH), 134.6 (C), 137.9 (C), 138.4 (CH), 141.9 (C), 162.7 (C), 165.4 (C); m/z (ESI) 414.0160 (MH+. C20H1779BrNO2S requires 414.0158).
N-Benzoyl-2-methoxyphenothiazine (7)
The reaction was performed as described in general procedure B using (2-benzoylamino-4-methoxyphenyl)(2′-bromophenyl)sulfane (6) (250 mg, 0.60 mmol). The reaction mixture was stirred at 150 °C for 120 h. Purification by flash column chromatography (hexane/ethyl acetate, 9:1) gave N-benzoyl-2-methoxyphenothiazine (7) (160 mg, 80%) as a white solid. Mp 156–157 °C; νmax/cm−1 (neat) 2920 (CH), 1655 (CO), 1578 (CC), 1442, 1335, 1246, 1022, 756; δH (400 MHz, CDCl3) 3.65 (3H, s, OCH3), 6.74 (1H, dd, J 8.6, 2.7 Hz, 3-H), 7.01 (1H, d, J 2.7 Hz, 1-H), 7.10–7.17 (2H, m, 7-H and 8-H), 7.21–7.25 (2H, m, 3′-H and 5′-H), 7.29–7.44 (6H, m, 4-H, 6-H, 9-H, 2′-H, 4′-H and 6′-H); δC (101 MHz, CDCl3) 55.7 (CH3), 112.8 (CH), 113.6 (CH), 123.0 (C), 126.5 (CH), 126.9 (CH), 127.2 (CH), 127.8 (CHCH), 128.2 (CH), 128.2 (2 × CH), 128.9 (2 × CH), 130.5 (CH), 133.0 (C), 135.4 (C), 139.7 (C), 140.8 (C), 159.1 (C), 169.1 (C); m/z (ESI) 334.0902 (MH+. C20H16NO2S requires 334.0896).
2-Methoxyphenothiazine (8).18
To a solution of N-benzoyl-2-methoxyphenothiazine (7) (100 mg, 0.30 mmol) in ethanol (5.3 mL) was added hydrazine monohydrate (11 mL) under an atmosphere of argon. The reaction mixture was stirred at 145 °C for 48 h. The resulting residue was cooled and poured onto ice-water (30 mL). The aqueous layer was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. Purification by flash column chromatography (hexane/ethyl acetate, 4:1) gave 2-methoxyphenothiazine (8) (50 mg, 73%) as a white solid. Mp 183–185 °C (lit.18 184–185 °C); δH (400 MHz, DMSO-d6) 3.68 (3H, s, OCH3), 6.32 (1H, d, J 2.7 Hz, 1-H), 6.38 (1H, dd, J 8.4, 2.7 Hz, 3-H), 6.67 (1H, dd, J 8.0, 1.3 Hz, 6-H), 6.75 (1H, ddd, J 7.7, 7.4, 1.3 Hz, 8-H), 6.82 (1H, d, J 8.4 Hz, 4-H), 6.91 (1H, dd, J 7.7, 1.5 Hz, 9-H), 6.98 (1H, ddd, J 8.0, 7.4, 1.5 Hz, 7-H), 8.59 (1H, br s, NH); δC (101 MHz, DMSO-d6) 55.0 (CH3), 100.7 (CH), 107.1 (C), 107.2 (CH), 114.4 (CH), 117.0 (C), 121.8 (CH), 126.2 (CH), 126.8 (CHCH), 127.4 (CH), 141.8 (C), 143.3 (C), 159.3 (C); m/z (EI) 229 (M+. 100%).
Methopromazine45
Sodium hydride (7.5 mg, 0.43 mmol) was slowly added to DMF (0.4 mL) under an atmosphere of argon and the resulting suspension was cooled to 0 °C. 3-Dimethylamino-1-propyl chloride hydrochloride (41 mg, 0.26 mmol) and 2-methoxyphenothiazine (8) (30 mg, 0.13 mmol) were subsequently added. The reaction mixture was stirred at room temperature for 18 h. The resulting residue was diluted with dichloromethane (20 mL) and washed with 5% aqueous lithium chloride (30 mL). The aqueous layer was further extracted with dichloromethane (2 × 20 mL) and the combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. Purification by flash column chromatography (dichloromethane/methanol, 19:1) gave methopromazine (25 mg, 61%) as a brown oil. Spectroscopic data were consistent with the literature.45δH (400 MHz, CDCl3) 1.98 (2H, pent, J 7.1 Hz, 2′-H2), 2.23 (6H, s, 2 × CH3), 2.44 (2H, t, J 7.1 Hz, 3′-H2), 3.78 (3H, s, OCH3), 3.90 (2H, t, J 7.1 Hz, 1′-H2), 6.43–6.52 (2H, m, 1-H and 3-H), 6.86–6.94 (2H, m, 6-H and 8-H), 7.03 (1H, d, J 8.4 Hz, 4-H), 7.09–7.17 (2H, m, 7-H and 9-H); δC (101 MHz, CDCl3) 24.6 (CH2), 45.1 (2 × CH3), 45.3 (CH2), 55.7 (CH3), 57.0 (CH2), 103.6 (CH), 107.3 (CH), 115.9 (CH), 116.5 (C), 122.8 (CH), 126.1 (C), 127.3 (CH), 127.6 (CH), 127.9 (CH), 145.0 (C), 146.7 (C), 160.0 (C); m/z (ESI) 315 (MH+. 100%).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from The Carnegie Trust for the Universities of Scotland (Ph.D. studentship to A. C. D.) and the University of Glasgow is gratefully acknowledged.Notes and references
- M. J. Ohlow and B. Moosmann, Drug Discovery Today, 2021, 16, 119–131 CrossRef PubMed.
- K. Laxmikeshav, P. Kumari and N. Shankaraiah, Med. Res. Rev., 2021, 42, 513–575 CrossRef PubMed.
- (a) S. C. Mitchell, Curr. Drug Targets, 2009, 7, 1181–1189 CrossRef PubMed; (b) W. E. Swales, Can. J. Comp. Med. Vet. Sci., 1939, 3, 188–194 CAS.
- A. Bernthsen, Ber. Dtsch. Chem. Ges., 1883, 16, 2896–2904 CrossRef.
- (a) J. H. Burn, Proc. R. Soc. Med., 1954, 47, 617–621 CAS; (b) D. Boyd-Kimball, K. Gonczy, B. Lewis, T. Mason, N. Siliko and J. Wolfe, ACS Chem. Neurosci., 2019, 10, 79–88 CrossRef CAS PubMed.
- H.-W. Cheng, Y.-H. Liang, Y.-L. Kuo, C.-P. Chuu, C.-Y. Lin, M.-H. Lee, A. T. H. Wu, C.-T. Yeh, E.-T. Chen, J. Whang-Peng, C.-L. Su and C.-Y. F. Huang, Cell Death Dis., 2015, 6, e1753 CrossRef CAS PubMed.
- (a) A. González-González, L. K. Vazquez-Jimenez, A. D. Paz-González, M. L. Bolognesi and G. Rivera, Curr. Med. Chem., 2021, 28, 7910–7936 CrossRef PubMed; (b) E. Uliassi, L. Piazzi, F. Belluti, A. Mazzanti, M. Kaiser, R. Brun, C. B. Moraes, L. H. Freitas-Junior, S. Gul, M. Kuzikov, B. Ellinger, C. Borsari, M. P. Costi and M. L. Bolognesi, ChemMedChem, 2018, 13, 678–683 CrossRef CAS PubMed; (c) K. Vögerl, N. Ong, J. Senger, D. Herp, K. Schmidtkunz, M. Marek, M. Müller, K. Bartel, T. B. Shaik, N. J. Porter, D. Robaa, D. W. Christianson, C. Romier, W. Sippl, M. Jung and F. Bracher, J. Med. Chem., 2019, 62, 1138–1166 CrossRef PubMed; (d) Y. Gao, T. Y. Sun, W. F. Bai and C. G. Bai, Eur. J. Med. Chem., 2019, 183, 111692 CrossRef PubMed.
- E. A. Weiss, M. J. Tauber, R. F. Kelley, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2005, 127, 11842–11850 CrossRef CAS PubMed.
- R. Y. Lai, X. Kong, S. A. Jenekhe and A. J. Bard, J. Am. Chem. Soc., 2003, 125, 12631–12639 CrossRef CAS PubMed.
- H.-W. Rhee, S. J. Choi, S. H. Yoo, Y. O. Jang, H. H. Park, R. M. Pinto, J. C. Cameselle, F. J. Sandoval, S. Roje, K. Han, D. S. Chung, J. Suh and J.-I. Hong, J. Am. Chem. Soc., 2009, 131, 10107–10112 CrossRef CAS PubMed.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- N. L. Smith, J. Org. Chem., 1950, 15, 1125–1130 CrossRef CAS.
- S. V. Filip, I. A. Silberg, E. Surducan, M. Vlassa and V. Surducan, Synth. Commun., 1998, 28, 337–345 CrossRef CAS.
- P. B. Madrid, W. E. Polgar, L. Toll and M. J. Tanga, Bioorg. Med. Chem. Lett., 2007, 17, 3014–3017 CrossRef CAS PubMed.
- (a) H. L. Yale, J. Am. Chem. Soc., 1955, 77, 2270–2272 CrossRef CAS; (b) N. Sharma, R. Gupta, M. Kumar and R. R. Gupta, J. Fluor. Chem., 1999, 98, 153–157 CrossRef CAS; (c) K. Pluta, B. Morak-Mlodawska and M. Jelen, J. Mol. Struct., 2020, 1216, 128320 CrossRef CAS.
- (a) C. Dai, X. Sun, X. Tu, L. Wu, D. Zhan and Q. Zeng, Chem. Commun., 2012, 48, 5367–5369 RSC; (b) W. Hu and S. Zhang, J. Org. Chem., 2015, 80, 6128–6132 CrossRef CAS PubMed.
- T. Dahl, C. W. Tornøe, B. Bang-Andersen, P. Nielsen and M. Jørgensen, Angew. Chem., Int. Ed., 2008, 47, 1726–1728 CrossRef CAS PubMed.
- D. Ma, Q. Geng, H. Zhang and Y. Jiang, Angew. Chem., Int. Ed., 2010, 49, 1291–1294 CrossRef CAS PubMed.
- X. Rui, C. Wang, D. Si, X. Hui, K. Li, H. Wen, W. Li and J. Liu, J. Org. Chem., 2021, 86, 6622–6632 CrossRef CAS PubMed.
- (a) S. Wu, W.-Y. Hu and S.-L. Zhang, RSC Adv., 2016, 6, 24257–24260 RSC; (b) K. Kanemoto, Y. Sakata, T. Hosoya and S. Yoshida, Chem. Lett., 2020, 49, 593–596 CrossRef CAS; (c) Q. Chen, R. Xie, H. Jia, J. Sun, G. Lu, H. Jiang and M. Zhang, J. Org. Chem., 2020, 85, 5629–5637 CrossRef CAS PubMed; (d) V. Hahn, A. Mikolasch, J. Weitemeyer, S. Petters, T. Davids, M. Lalk, J.-W. Lackmann and F. Schauer, ACS Omega, 2020, 5, 14234–14339 CrossRef PubMed; (e) S. Wang, R. Li, S. Jiang, H. Huang, W. Shao and G.-J. Deng, Adv. Synth. Catal., 2022, 364, 1481–1487 CrossRef CAS.
- (a) D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Org. Lett., 2015, 17, 4782–4785 CrossRef CAS; (b) M. A. B. Mostafa, R. M. Bowley, D. T. Racys, M. C. Henry and A. Sutherland, J. Org. Chem., 2017, 82, 7529–7537 CrossRef CAS; (c) M. A. B. Mostafa, E. D. D. Calder, D. T. Racys and A. Sutherland, Chem. – Eur. J., 2017, 23, 1044–1047 CrossRef CAS PubMed.
- (a) A. C. Dodds and A. Sutherland, J. Org. Chem., 2021, 86, 5922–5932 CrossRef CAS PubMed; (b) A. C. Dodds and A. Sutherland, Org. Biomol. Chem., 2022, 20, 1738–1748 RSC.
- H. Tian, H. Yang, C. Zhu and H. Fu, Adv. Synth. Catal., 2015, 357, 481–488 CrossRef CAS.
- (a) P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848–851 CrossRef CAS PubMed; (b) T. Hostier, V. Ferey, R. Ricci, D. G. Pardo and J. Cossy, Org. Lett., 2015, 17, 3898–3901 CrossRef CAS PubMed; (c) C. J. Nalbandian, Z. E. Brown, E. Alvarez and J. L. Gustafson, Org. Lett., 2018, 20, 3211–3214 CrossRef CAS PubMed.
- Methods of aniline thiolation are known using reagents such as diaryl disulfides or thiols with oxidizing agents such as iodine: (a) D. Yang, K. Yan, W. Wei, J. Zhao, M. Zhang, X. Sheng, G. Li, S. Lu and H. Wang, J. Org. Chem., 2015, 80, 6083–6092 CrossRef CAS PubMed; (b) Y.-m. Lin, G.-p. Lu, G.-x. Wang and W.-b. Yi, Adv. Synth. Catal., 2016, 358, 4100–4105 CrossRef CAS; (c) T. Müller and L. Ackermann, Chem. – Eur. J., 2016, 22, 14151–14154 CrossRef PubMed; (d) S. Yang, B. Feng and Y. Yang, J. Org. Chem., 2017, 82, 12430–12438 CrossRef CAS PubMed; (e) X. Jiang, Z. Shen, C. Zheng, L. Fang, K. Chen and C. Yu, Tetrahedron Lett., 2020, 61, 152141 CrossRef CAS; (f) W. Zhao, F. Zhang and G.-J. Deng, J. Org. Chem., 2021, 86, 291–301 CrossRef CAS PubMed.
- (a) S. M. Maddox, C. J. Nalbandian, D. E. Smith and J. L. Gustafson, Org. Lett., 2015, 17, 1042–1045 CrossRef CAS PubMed; (b) S. M. Maddox, A. N. Dinh, F. Armenta, J. Um and J. L. Gustafson, Org. Lett., 2016, 18, 5476–5479 CrossRef CAS PubMed.
- A similar cationic intermediate was proposed by Gustafson and co-workers in their triflic acid and diaryl selenide activated thioarylation reaction. See ref. 24c.
- For reviews of copper-catalysed aryl amination, see: (a) K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428–2439 CrossRef CAS; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed; (c) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC; (d) K. Okano, H. Tokuyama and T. Fukuyama, Chem. Commun., 2014, 50, 13650–13663 RSC.
- (a) A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727–7729 CrossRef CAS PubMed; (b) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS PubMed; (c) M. C. Henry, R. McGrory, R. J. Faggyas, M. A. B. Mostafa and A. Sutherland, Org. Biomol. Chem., 2019, 17, 4629–4639 RSC.
- (a) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2047 CrossRef CAS; (b) B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125–146 CrossRef CAS.
- J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 7215–7216 CrossRef CAS.
- (a) S. Courvoisier, R. Ducrot, J. Fournel and L. C. Julou, C. R. Seances Soc. Biol. Fil. Int., 1957, 151, 689–692 CAS; (b) F. Jourdan, P. Duchene-Marullaz, G. Faucon and P. Bouverot, C. R. Seances Soc. Biol. Fil. Int., 1958, 152, 91–93 CAS; (c) M. Apfeldorf, H. G. Bauer and T. H. McGavack, Am. J. Psychiatry, 1960, 117, 72–73 CrossRef CAS PubMed.
- M. C. Henry, H. M. Senn and A. Sutherland, J. Org. Chem., 2019, 84, 346–364 CrossRef CAS PubMed.
- A limitation of this general approach for ortho-thioarylation of monosubstituted arenes is that the para-position is required to be blocked. However, the iron-catalysed thioarylation of compound 6 is an example where a more activating substituent can facilitate thioarylation at the ortho-position to the N-benzoyl protected amino group, without having the para-position blocked.
- D. L. Boger, K. Machiya, D. L. Hertzog, P. A. Kitos and D. Homes, J. Am. Chem. Soc., 1993, 115, 9025–9036 CrossRef CAS.
- M. C. Henry, V. M. Abbinante and A. Sutherland, Eur. J. Org. Chem., 2020, 2819–2826 CrossRef CAS.
- P. Wipf and J. P. Maciejewski, Org. Lett., 2008, 10, 4383–4386 CrossRef CAS PubMed.
- C. Alp, Ş. Özsoy, N. A. Alp, D. Erdem, M. S. Gültekin, Ö. İ. Küfrevioğlu, M. Şentürk and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2012, 27, 818–824 CrossRef CAS PubMed.
- S. Choi, J. Park, E. Yu, J. Sim and C. M. Park, Angew. Chem., Int. Ed., 2020, 59, 11886–11891 CrossRef CAS PubMed.
- S. Voth, J. W. Hollett and J. A. McCubbin, J. Org. Chem., 2015, 80, 2545–2553 CrossRef CAS PubMed.
- X. Huang, Y. Chen, S. Zhen, L. Song, M. Gao, P. Zhang, H. Li, B. Yuan and G. Yang, J. Org. Chem., 2018, 83, 7331–7340 CrossRef CAS PubMed.
- M. Huang, D. Huang, X. Zhu and Y. Wan, Eur. J. Org. Chem., 2015, 4835–4839 CrossRef CAS.
- Y.-m. Lin, G.-p. Lu, R.-k. Wang and W.-b. Yi, Org. Lett., 2016, 18, 6424–6427 CrossRef CAS PubMed.
- T. Matsumoto and Y. Matsunaga, Bull. Chem. Soc. Jpn., 1981, 54, 648–653 CrossRef CAS.
- V. Quesneau, K. Renault, M. Laly, S. Jenni, F. Ponsot and A. Romieu, Tetrahedron Lett., 2020, 61, 152582 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. See DOI: https://doi.org/10.1039/d2ob01082h |
| This journal is © The Royal Society of Chemistry 2022 |