
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new approach to 10-arylated 5H-dibenzo[b,f]azepines using syn-selective hydrohalogenation of ethynylaniline†
Kento
Iwai
 ab,
Yusuke
Mukaijo
a,
Haruyasu
Asahara
abc and
Nagatoshi
Nishiwaki
*ab
ab,
Yusuke
Mukaijo
a,
Haruyasu
Asahara
abc and
Nagatoshi
Nishiwaki
*ab
aSchool of Environmental Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: nishiwaki.nagatoshi@kochi-tech.ac.jp
bResearch Center for Molecular Design, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
cGraduate School of Pharmaceutical Sciences, Osaka University, Yamadaoka 1-6, Suita, Osaka 565-0871, Japan
First published on 29th June 2022
Abstract
A new synthetic method for 10-arylated dibenzo[b,f]azepines was developed. The pseudo-intramolecular hydrohalogenation of 2-(2′-bromophenyl)ethynylaniline, which proceeded in a syn-selective manner without forming any detectable over-addition product, was a crucial step. All attempts of subsequent arylation via Suzuki–Miyaura cross coupling and construction of a seven membered ring via Ullmann-type intramolecular coupling were unsuccessful because of dehydrohalogenation or other side reactions. This problem was overcome by the N-acetylation of the amino group, which facilitated the abovementioned coupling reactions to afford the desired 10-arylated dibenzoazepines.
Introduction
Although alkynes are good electrophiles for nucleophilic addition, they are an inappropriate substrate for electrophilic addition because the generation of carbocations with highly electronegative sp carbon atoms is difficult. Furthermore, issues such as overaddition to form dihaloalkanes1 and the difficulty in handling gaseous hydrogen chloride must be addressed.2 Consequently, several efficient transition metal-catalyzed systems have been developed recently3–5 for realizing the anti-4 and syn-selective5 hydrochlorination of alkynes.We previously demonstrated an effective hydrohalogenation method that used 2-ethynylaniline as a substrate and involved a pseudo-intramolecular process.6 The salt formation between an amino group and an acidic reagent was a crucial step to increase the proximity between the reactants, and this facilitated hydrochlorination even in the absence of a metal catalyst or special reagent, without the formation of any detectable overaddition products (Fig. 1). This reaction furnished the corresponding haloalkenes in a syn-selective and regioselective manner, with an amino group in the product. These structural features prompted us to investigate the synthesis of 10-aryldibenzo[b,f]azepines (10Ar-DBA).
 | ||
| Fig. 1 Selected dibenzapine derivatives showing biological activity. | ||
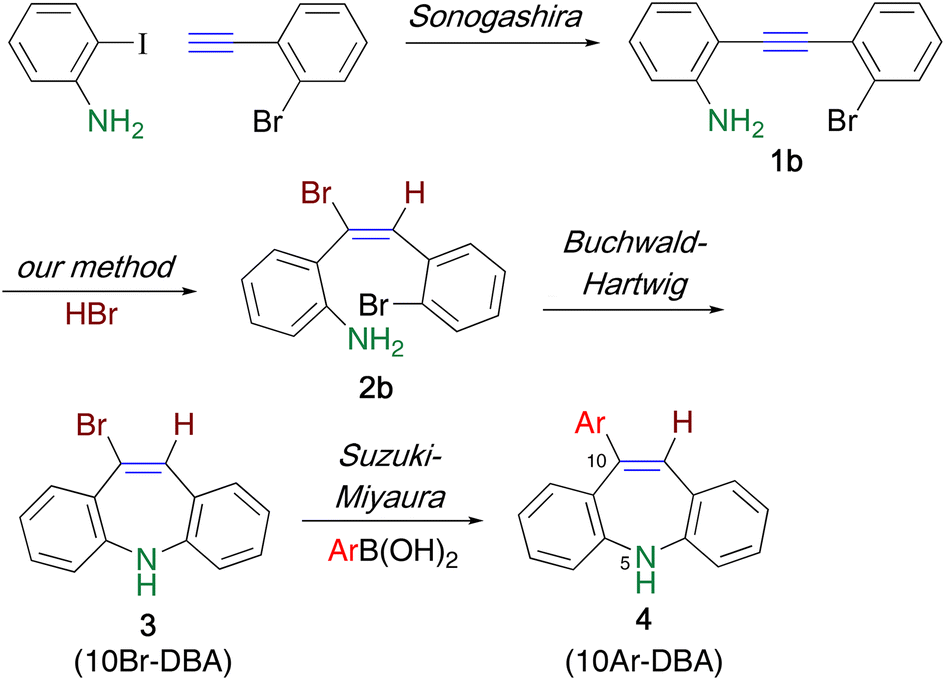
Similar to carbamazepine, the dibenzoazepine (DBA) framework is often found in pharmaceuticals and agrochemicals, and numerous methods have been developed for their synthesis.7 Despite the abundant synthetic methods, there are only a few reports on the synthesis of 10Ar-DBAs. This framework was first synthesized by Bergmann et al. via ring expansion and rearrangement using 9-(α-hydroxybenzyl)acridine.8 Wang et al. constructed an azepine ring and a fused cyclohexane ring in a single step through Pd-catalyzed cyclization using o-ethynylaniline and cyclohexanone in the presence of trimethylsilyl cyanide; the subsequent oxidation reaction afforded the target 10Ar-DBA framework.9 Recently, Dorta et al. used this framework as a planar chiral ligand by introducing phosphorous10 or sulfur11 atom on the ring nitrogen. They introduced a phenyl group at the 10-position through the Suzuki–Miyaura cross-coupling reaction using 10-bromo-5-(trifluoroacetyl)dibenzoazepine.10 In 2019, Doucet et al. reported the Pd-catalyzed arylation at the 10-position; the bromo-substituted DBA was not necessary as the substrate for this reaction.12 Although these methods are excellent for the construction of the DBA framework, the development of a facile synthetic method for 10Ar-DBAs is highly demanded. Herein, we report another synthetic method that involves our previously developed pseudo-intramolecular hydrohalogenation as a key step. The synthetic route (Scheme 1) consists of four steps: (1) preparation of an ethynylaniline by Sonogashira coupling, (2) pseudo-intramolecular hydrohalogenation, (3) construction of a dibenzazepine ring by Buchwald–Hartwig coupling, and (4) arylation at the 10-position by Suzuki–Miyaura coupling.
 | ||
| Scheme 1 Synthetic plan for 10Ar-DBA. | ||
Results and discussion
2-Bromo-1-ethynylbenzene13 was prepared in 86% yield by the Sonogashira cross-coupling14 of 2-bromo-1-iodobenzene with (trimethylsilyl)ethyne, followed by desilylation upon treatment under basic conditions.14 When the prepared ethynylbenzene was subjected to another Sonogashira coupling reaction with 2-iodoaniline, ethynylaniline 1b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 was furnished in 91% yield.
15 was furnished in 91% yield.
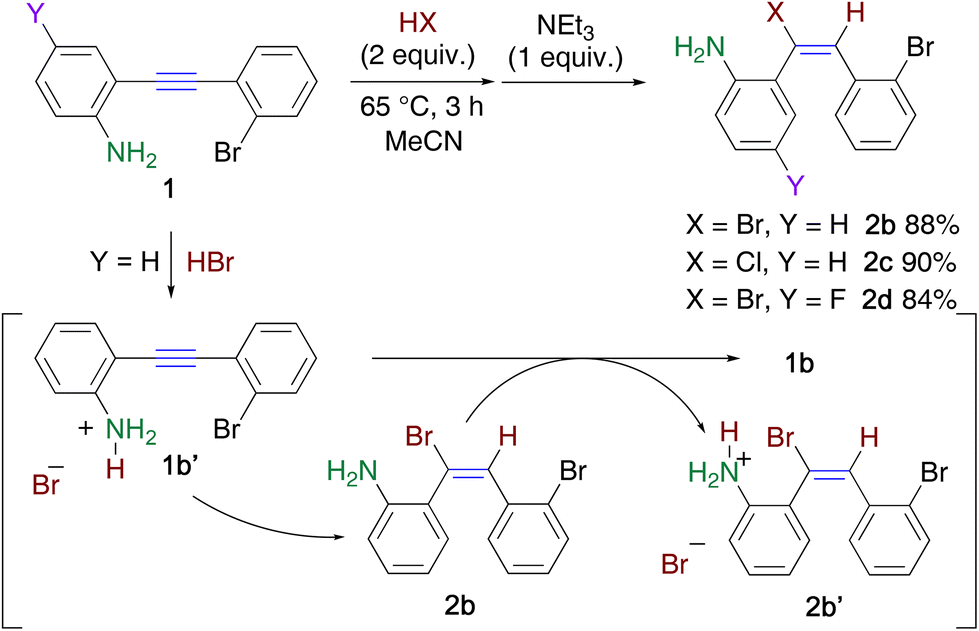
Ethynylaniline 1b (Y = H) immediately precipitated as a yellow salt 1b′ in acetonitrile upon treatment with an equimolar amount of hydrobromic acid. When the suspension was heated at 65 °C for 3 h, the precipitate gradually disappeared. However, almost half of 1b was recovered, along with the formation of hydrobromide 2b′. This is because the less electronegative ethenyl group increased the basicity of the amino group in 2b, which was converted to the deprotonated product 2b′ during the reaction (Scheme 2).16 Hence, 2 equiv. of hydrobromic acid was necessary for the efficient hydrobromination of 1b, and adduct 2b was readily obtained in 88% yield upon the addition of triethylamine to the reaction mixture. Similarly, the hydrochlorination of 1b using 2 equiv. of hydrochloric acid proceeded to afford 2c in 90% yield. An electron-withdrawing fluoro group did not disturb the reaction, which leading to 2d in 84% yield.
 | ||
| Scheme 2 Hydrohalogenation of ethynylaniline 1. | ||
Next, the hydrobrominated product 2b was subjected to the Buchwald–Hartwig coupling reaction according to a method reported in the literature;17 however, a substantial amount of 2b was recovered (Table S1†). Although 2b was completely consumed upon heating at 90 °C or upon treatment with stronger base, it underwent dehydrobromination rather than ring closure to afford 1b (Scheme 3). Dehydrohalogenation is seldom observed in the usual Pd-catalyzed reactions using haloalkenes. Indeed, the rection did not proceed when α-bromostilbene18 was used under the same conditions. Hence, the ortho-amino group in 2b possibly serves as a directing group to attract Pd0 species and facilitate the oxidative addition and the subsequent base-induced dehydrobromination, which affords a triple bond.
 | ||
| Scheme 3 Dehydrobromination of 2b. | ||
To avoid undesirable dehydrohalogenation assisted by the ortho-amino group prevents the cross-coupling reaction, N-acetylation was conducted to decrease the coordinating ability of the amino group. When bromoalkene 5a was subjected to the Buchwald–Hartwig coupling reaction under the conditions corresponding to Scheme 4, a complex mixture was obtained, with 61% recovery of 5; 10Br-DBA 6 was not detected in the mixture (Scheme 4). However, it is worth noting that this reaction pathway did not involve dehydrobromination leading to 7. Therefore, Suzuki–Miyaura coupling using N-acetylated bromoalkene 5 was effective.
 | ||
| Scheme 4 Acetylation of 2b and Pd-catalyzed reaction of 5. | ||
Bromoalkene 5a reacted with 4-methylphenylboronic acid 8a to afford differently triarylated alkene 9a in 92% yield, without any E/Z isomerization (Table 1, entry 1). When boronic acids 8b and 8c were used, the yields of coupling products 9b and 9c decreased considerably, presumably because of the formation of a trimer upon the dehydration of 8b and 8c (entries 2 and 3). This drawback was overcome by adding 10 equiv. of water to hydrolyze the trimer, which afforded 9b and 9c in high yields (entries 4 and 5). In the case of less reactive 8d, heating the reaction mixture was more effective than adding water, and 9d was obtained (entries 6 and 7). The Suzuki–Miyaura coupling efficiently proceeded even in the presence of a fluoro group to afford triarylalkene 9e in 89% yield (entry 8).

Finally, the construction of an azepine ring was investigated (Table 2). When triarylalkene 9 was treated with a Pd catalyst,19 cyclization proceeded to afford 10a in 22% yield, along with 64% recovery of 9 (entry 1). Increasing the catalyst loading resulted in the complete consumption of 9, and the yield of 10a increased up to 53% (entry 2). However, the yield could not be improved further under the other reaction conditions tested. We next studied the intramolecular cross-coupling reaction using CuI, instead of a Pd catalyst.20 Although it was necessary to use stoichiometric amounts of CuI (3 equiv.), 10Ar-DBA 10a was obtained in higher yields than that obtained using a Pd catalyst (entries 3–5). Heating in a sealed tube for a longer time, instead of microwave heating, was also effective, and the reaction proceeded to furnish 10a with 87% yield (entry 6).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Y | X | Metal reagent (equiv.) | Ligand (equiv.) | Base | Solv. | Temp./°C | Time/h | Yield/% | |
| a Microwave heating was used. | ||||||||||
| 1 | H | Me | a | Pd(OAc)2 (0.05) | Xantphos (0.05) | Cs2CO3 | 1,4-Dioxane | 100 | 24 | 22 |
| 2 | H | Me | a | Pd(OAc)2 (0.2) | Xantphos (0.2) | Cs2CO3 | 1,4-Dioxane | 100 | 24 | 53 |
| 3 | H | Me | a | CuI (1) | — | K2CO3 | DMF | 150a | 3 | 13 |
| 4 | H | Me | a | CuI (3) | — | K2CO3 | DMF | 150a | 3 | 64 |
| 5 | H | Me | a | CuI (3) | — | K2CO3 | 1,4-Dioxane | 150 | 24 | 72 |
| 6 | H | Me | a | CuI (3) | — | K2CO3 | 1,4-Dioxane | 150 | 24 | 87 |
| 7 | H | MeO | b | CuI (3) | — | K2CO3 | 1,4-Dioxane | 150 | 24 | 33 |
| 8 | H | Cl | c | CuI (3) | — | K2CO3 | 1,4-Dioxane | 150 | 24 | 51 |
| 9 | H | COOMe | d | CuI (3) | — | K2CO3 | 1,4-Dioxane | 150 | 24 | 39 |
| 10 | F | Me | E | CuI (3) | — | K2CO3 | 1,4-Dioxane | 150 | 24 | 33 |

Other triarylalkenes 9b–e were converted to the corresponding products 10b–e, respectively, in moderate yields, under the same conditions (entries 7–9). When the CuI-mediated ring closure reaction was employed for acetylated bromoalkene 5, a complex reaction mixture was obtained, presumably because of the higher reactivity of the bromoalkene moiety than the bromobenzene moiety. Hence, the sequence of the Suzuki–Miyaura and Ullmann-type intramolecular coupling reactions was crucial for the synthesis of 10Ar-DBAs 10.
In the 1H NMR spectrum of 10a, two types of signals were observed for each proton, indicating that 10a existed in two isomeric forms. In the temperature-variable NMR spectra of 10a acquired in DMSO-d6, each type of signal coalesced (Fig. 2). X-ray crystallographic analysis of 10a revealed that the N5–C1′ bond length (1.38 Å) was shorter than the usual N–C single bond length (1.47 Å), indicating double bond characteristics in the former. Consequently, rotation around this bond was prevented, facilitating the existence of stable atropisomers.21
 | ||
| Fig. 2 Changes of methyl groups in the temperature-variable NMR spectra of 10a (DMSO-d6). | ||
Conclusions
5-Acetylated 10Ar-DBAs 10a–e were successfully synthesized from the corresponding ethynylaniline 1b and 1c (Scheme 5). The developed syn-selective hydrobromination of 1b efficiently afforded substrate 2b for the successive Pd-catalyzed cross-coupling reactions. However, the elimination of hydrogen bromide, which was promoted by the presence of the ortho-amino group, proceeded predominantly. This undesirable reaction was suppressed by N-acetylation, and the subsequent Suzuki–Miyaura and Ullmann-type intramolecular coupling reactions were facilitated to afford 10Ar-DBAs 10. This method will be useful for synthesizing new biologically active compounds because the 10-position of the DBA framework can be easily modified. Although 10Ar-DBA possessing an electron-withdrawing group at the 5-position could not be obtained by direct arylation,11 it was possible to obtain this using our protocol. We believe that the modification of the substituents on 10Ar-DBAs will trigger further investigations on the butterfly-like dynamics of these compounds.21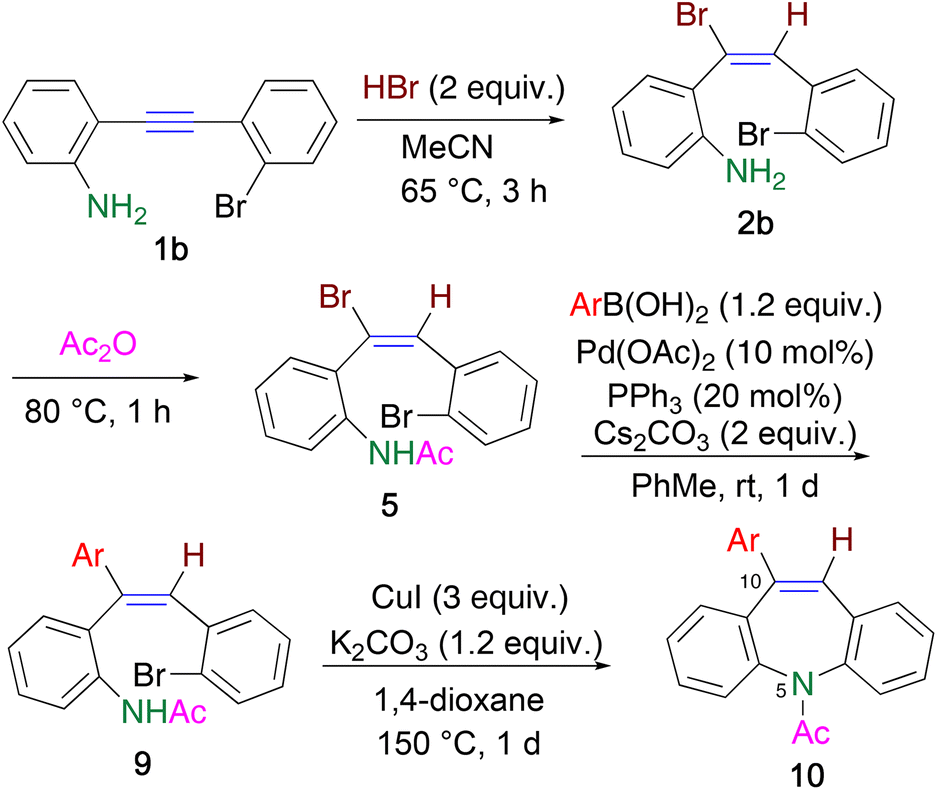 | ||
| Scheme 5 Summary of the synthetic method for 10Ar-DBAs 10. | ||
Experimental
General
All reagents and dry solvents were purchased from commercial sources and used as received. 1H, 13C{1H} and 19F{1H} NMR spectra were recorded on a Bruker DPX-400 spectrometer (400 MHz, 100 MHz, and 367.5 MHz, respectively) and a JEOL JMN-ECZ400S spectrometer (400 MHz and 100 MHz, respectively) in a deuterated solvent using TMS as an internal standard. For 19F{1H} NMR, the chemical shift of hexafluorobenzene (δ = −163.0 ppm in CDCl3) was used as an external reference. The assignments of the 13C{1H} NMR were performed by DEPT experiments. In cases of 10Ar-DBAs 10, NMR spectra were measured using a mixture of diastereomer A and B (A/B = 51/49–56/44). Hence, integral values of 1H NMR were shown using HA and HB, which were belonged to isomer A and B, respectively. Assignments of 13C NMR were also shown using CA and CB, which were belonged to isomer A and B, respectively. IR spectra were recorded on a JASCO FT/IR-4200 spectrometer equipped with an ATM detector. High-resolution mass spectra were obtained on an AB SCEIX Triplet TOF 4600 mass spectrometer. Melting points were recorded on an SRS-Optimelt automated melting point system and were uncorrected. Microwave heating was performed by Anton-Paar Microwave 300 (850 W, 2455 MHz) using 10 mL glass vessel.X-ray crystallographic analysis
Diffraction data were collected at 93 K under a cold N2-gas stream on a Rigaku XtaLAB Synergy-S/Mo system (λ = 0.71073 Å (Mo-Kα)). The integrated data were analyzed by using an Olex2 crystallographic software package.22 The structures were solved with the ShelXT structure solution program23 using Intrinsic Phasing and refined with the ShelXL refinement package24 using the least-squares minimization. Anisotropic refinement was performed for all non-hydrogen atoms, and all the hydrogen atoms were put at calculated positions.Hydrobromination of 1b
In a screw capped test tube, HBr aq. (d = 1.49, 48% (w/w), 116 μL, 1.0 mmol) was added to a solution of ethynylaniline 1b (134 mg, 0.5 mmol) in MeCN (5 mL). Yellow solid immediately precipitated. When the mixture was heated at 65 °C, the yellow solid gradually decreased and completely disappeared after 3 h. After addition of NEt3 (70 μL, 0.5 mmol), solvent was removed under vacuo, and the residue was dissolved in CHCl3 (10 mL). The CHCl3 solution was washed with H2O (10 mL × 3), dried over MgSO4, and evaporated to afford adduct 2b (154 mg, 0.44 mmol, 88%) as a yellow solid. When HCl was used instead of HBr, reaction was conducted in a same way.Pd-Mediated dehydrobromination
To a mixture of bromoalkene 2b (70.2 mg, 0.2 mmol), Pd2(dba)3 (10.7 mg, 0.01 mmol), PBut3 (3.4 mg, 0.01 mmol), and NaOBut (19.4 mg, 0.2 mmol), dry PhMe (2 mL) was added under Ar atmosphere. After stirring the resultant mixture at room temperature for 3 h, filtered through celite. The filtrate was evaporated to afford 2-[(2-bromophenyl)ethynyl]aniline (1b) (48.2 mg, 0.18 mmol, 90%) as a yellow oil.N-Acetylation of bromoalkene 2b
A solution of bromoalkene 2b (70.2 mg, 0.2 mmol) in Ac2O (2 mL) was heated at 80 °C for 1 h. After concentration under reduced pressure, the residue was subjected to column chromatography on SiO2 to afford N-acetylated product 5 (eluted with hexane/EtOAc = 7/3, 60.5 mg, 0.15 mmol, 77%) as a yellow solid. Single crystal was obtained by recrystallization from hexane.Synthesis of triarylalkene 9
To a mixture of bromoalkene 5 (79.0 mg, 0.2 mmol), 4-MeC6H4B(OH)28a (32.6 mg, 0.24 mmol), PPh3 (10.7 mg, 0.04 mmol), Pd(OAc)2 (5.3 mg, 0.02 mmol), and Cs2CO3 (134.3 mg, 0.4 mmol), dry PhMe (1 mL) was added under Ar atmosphere. After stirring the resultant mixture at room temperature for 1 d, H2O (10 mL) was added and extracted with CHCl3 (10 mL × 3). The organic layer was dried over MgSO4, and evaporated to afford yellow oil. The residue was treated by column chromatography on SiO2 to furnish 9a (eluted with hexane/EtOAc = 8/2, 78.2 mg, 0.184 mmol, 92%) as a yellow solid.When other boronic acids 8b–d were used, reactions were conducted in a similar way. In cases of 8b and 8c, 10 equiv. of H2O were added to hydrolyze a trimer of boronic acid. In the case of 8d, the reaction mixture was heated at 80 °C.
Synthesis of dibenzoazepine 10
In a screw capped test tube, to a solution of triarylalkene 9a (81.5 mg, 0.2 mmol) in 1,4-dioxane (1 mL), CuI (114.3 mg, 0.6 mmol) and K2CO3 (27.6 mg, 0.2 mmol) were added and sealed. The resultant mixture was heated at 150 °C for 1 d. When microwave heating was used, the reaction mixture was added in 10 mL glass vessel, and put in the equipment and heated at 150 °C for 3 h. After cooling to room temperature, the reaction mixture was filtered through celite with washing by EtOAc. The filtrate was evaporated to afford yellow solid as a residue, in which production of 10ArDBA 10a with 87% yield was confirmed by 1H NMR. Further purification was performed by column chromatography on SiO2 to furnish 10a (eluted with hexane/EtOAc = 7/3, 47.1 mg, 0.145 mmol, 72%) as a pale yellow solid.Author contributions
Dr Iwai mainly prepared the manuscript and performed experiments. Mr Mukaijo performed experiments. Dr Asahara and Prof. Nishiwaki participated in discussion about this project.Conflicts of interest
There are no conflicts to declare.Notes and references
- S.-i. Kawaguchi, Y. Gonda, H. Masuno, H. T. Vu, K. Yamaguchi, H. Shinohara, M. Sonoda and A. Ogawa, Tetrahedron Lett., 2014, 55, 6779 CrossRef CAS; P. J. Kropp and S. D. Crawford, J. Org. Chem., 1994, 59, 3102 CrossRef; K. Griesbaum, R. Rao and G. Leifker, J. Org. Chem., 1982, 47, 4975 CrossRef.
- C.-X. Xu, C.-H. Ma, F.-R. Xiao, H.-W. Chen and B. Dai, Chin. Chem. Lett., 2016, 27, 1683 CrossRef CAS; M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231 CrossRef; J. T. Hutchings, J. Catal., 1985, 96, 292 CrossRef.
- P. Yu, A. Bismuto and B. Morandi, Angew. Chem., Int. Ed., 2020, 59, 2904 CrossRef CAS PubMed; S. Liang, R. Ebule, G. B. Hammond and B. A. Xu, Org. Lett., 2017, 19, 4524 CrossRef PubMed; S. Dérien, H. Klein and C. Bruneau, Angew. Chem., 2015, 127, 12280 CrossRef.
- J. Derosa, A. L. Cantu, M. N. Boulous, M. L. O'Duill, J. L. Turnbull, Z. Liu, D. M. deLa Torre and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 5183 CrossRef CAS PubMed; R. Ebule, S. Liang, G. B. Hammond and B. Xu, ACS Catal., 2017, 7, 6798 CrossRef PubMed.
- J. Oliver-Meseguer, A. Doménech-Carbó, M. Boronat, A. Leyve-Pérez and A. Corma, Angew. Chem., Int. Ed., 2017, 56, 6435 CrossRef CAS PubMed.
- H. Asahara, Y. Mukaijo, K. Muragishi, K. Iwai, A. Ito and N. Nishiwaki, Eur. J. Org. Chem., 2021, 5747 CrossRef CAS.
- D. W. Nelson, Dibenzaepine-based sodium channel blockers for the treatment of neuropathic pain, in Bioactive Heterocyclic Compound Classes, ed. J. Dinges and C. Lamberth, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012, pp. 115–133 CrossRef CAS PubMed; A. Cassano, A. Manganaro, T. Martin, D. Young, N. Piclin, M. Pintore, D. Bigoni and E. Benfenati, Chem. Cent. J., 2010, 4, S1 CrossRef CAS PubMed; A. Kotali, Curr. Org. Chem., 2002, 6, 965 CrossRef; H. Blattner and A. Storni, Eur. Pat, EP11603, 1980 CrossRef; L. J. Kricka and A. Ledwith, Chem. Rev., 1974, 74, 101 CrossRef.
- E. D. Bergmann, M. Rabinovitz and A. Bromberg, Tetrahedron, 1968, 24, 1289 CrossRef CAS.
- G. K. Dhandabani, M. R. Murta and J.-J. Wang, Adv. Synth. Catal., 2018, 360, 4754 CrossRef CAS.
- A. Herrera, A. Grasruck, F. W. Heinemann, A. Shueurer, A. Chelouan, S. Frieß, F. Seidel and R. Dorta, Organometallics, 2017, 36, 714 CrossRef CAS.
- A. Chelouan, S. Bao, S. Frieß, A. Herrera, F. W. Heinemann, A. Escalona, A. Grasruck and R. Dorta, Organometallics, 2018, 37, 3983 CrossRef CAS.
- H. Li, T. Roisnel, J.-F. Soulé and H. Doucet, Adv. Synth. Catal., 2019, 361, 791 CrossRef CAS.
- I. V. Alabugin, K. Gilmore, S. Patil, M. Manoharan, S. V. Kovalenko, R. J. Clark and I. Ghiviriga, J. Am. Chem. Soc., 2008, 130, 11535 CrossRef CAS PubMed.
- L. Alonso-Marañón, L. A. Sarandeses, M. M. Martínez and J. P. Sestelo, Org. Chem. Front., 2018, 5, 2308 RSC.
- L. Yang, Y. Ma, F. Song and J. You, Chem. Commun., 2014, 50, 3024 RSC.
- K. Muragishi, H. Asahara and N. Nishiwaki, ACS Omega, 2017, 2, 1265 CrossRef CAS PubMed.
- J. A. Sirvent, F. Foubelo and M. Yus, J. Org. Chem., 2014, 79, 1356 CrossRef CAS PubMed.
- T. Hirose, Y. Miyazaki, M. Watabe, S. Akimoto, T. Tachikawa, K. Kodama and M. Yasutake, Tetrahedron, 2015, 71, 4714 CrossRef CAS; Y. Li, L. Cao, X. Luo and W.-P. Deng, Tetrahedron, 2014, 70, 5974 CrossRef.
- J. Yin and S. L. Buchwald, Org. Lett., 2000, 2, 1101 CrossRef CAS.
- A. S. Gajare, K. Toyota, M. Yoshifuji and F. Ozawa, Chem. Commun., 2004, 1994 RSC.
- Y. Kanase, M. Kuniyoshi, H. Tabata, Y. Takahashi, S. Kayama, S. Wakamatsu, T. Oshitari, H. Natsugari and H. Takahashi, Synthesis, 2015, 47, 3907 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra of compounds 2, 5, 9 and 10, temperature-variable NMR spectra of 10a, crystallographic data of 5, 9a and 10a, and optimization of reaction conditions. CCDC 2150669–2150671. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob00950a |
| This journal is © The Royal Society of Chemistry 2022 |