
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of glycopeptides and glycopeptide conjugates
Ward
Doelman
 and
Sander I.
van Kasteren
*
and
Sander I.
van Kasteren
*
Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: s.i.van.kasteren@chem.leidenuniv.nl
First published on 29th July 2022
Abstract
Protein glycosylation is a key post-translational modification important to many facets of biology. Glycosylation can have critical effects on protein conformation, uptake and intracellular routing. In immunology, glycosylation of antigens has been shown to play a role in self/non-self distinction and the effective uptake of antigens. Improperly glycosylated proteins and peptide fragments, for instance those produced by cancerous cells, are also prime candidates for vaccine design. To study these processes, access to peptides bearing well-defined glycans is of critical importance. In this review, the key approaches towards synthetic, well-defined glycopeptides, are described, with a focus on peptides useful for and used in immunological studies. Special attention is given to the glycoconjugation approaches that have been developed in recent years, as these enable rapid synthesis of various (unnatural) glycopeptides, enabling powerful carbohydrate structure/activity studies. These techniques, combined with more traditional total synthesis and chemoenzymatic methods for the production of glycopeptides, should help unravel some of the complexities of glycobiology in the near future.
Introduction
Glycosylation is one of the key post-translational modifications (PTMs) of proteins.1 Like all PTMs, glycans are not directly encoded by the genome. Genetic techniques can therefore not be readily used to study the effects of glycosylation. Furthermore, the glycans introduced onto proteins using the cellular glycosylation machinery are heterogenous in nature. This means that isolated glycoproteins exist as mixtures modified with different glycans; so-called glycoforms.2 Determining the relationship between glycoprotein functions and specific oligosaccharide structures is therefore challenging.This is particularly poignant in immunology, where protein glycosylation plays a major role in many key processes, like regulation of immunotolerance,3,4 uptake of antigens,5,6 the creation of neo-epitopes,7,8 and even the protection of the foetus.9 The precise study of these processes has been hampered by a dearth of well-defined reagents. In some cases, well-defined glycans, in the absence of any protein scaffold, can be used to study these functions. For instance, the carbohydrate binding specificity of many lectins has been determined using synthetic oligosaccharides in the absence of a protein scaffold.10,11 However, for many processes, the combination of carbohydrate and protein is key. For example, antigen (cross-)presentation is thought to be enhanced by uptake of the antigen via lectins12,13 and therefore requires a covalent attachment of the protein and the (specific) oligosaccharide.
Entire glycoproteins, bearing a well-defied glycan, are formidable synthetic targets. Their use is therefore often not feasible. Synthetic, glycosylated peptides can offer a useful alternative; a compromise between reality and synthetic accessibility for biochemical studies. Additionally, synthetic glycosylated peptides in themselves are being actively developed as vaccine candidates.14 These have been shown to induce glycosylation-specific immune responses.15,16 Examples of antigens that have been tried as targets for glycopeptide vaccines are the envelope glycoprotein of human immunodeficiency virus (HIV),17 as well as certain tumor associated antigens.18
From a synthetic perspective, the synthesis of glycosylated peptides is still a challenge in itself, as it involves the combination of two types of bio-organic chemistry: peptide synthesis and carbohydrate chemistry. While these two fields are not inherently incompatible, there are challenges to overcome for successful glycopeptide synthesis. This review will focus on the synthesis of glycopeptides and glycopeptide conjugates useful for immunological studies. To give a good overview of the field both early and recent developments will be highlighted.
Overview of protein glycosylation
Before discussing the synthetic challenges involved, it is important to understand the structure of naturally glycosylated peptides. Native glycosylation of peptides and proteins generally happens in two forms: O-glycosylation, were the reducing end of an oligosaccharide is attached to the hydroxy-function of a serine or threonine (or sometimes tyrosine) residue,1,19 and N-glycosylation, where the oligosaccharide is attached to the carboxamide function of an asparagine residue.1,20 Not only the nature of the glycosidic bond to the peptide is inherently different between these two types of glycosylation; the general structure of the N- and O-linked glycans is also fundamentally different and each presents their own unique synthetic (and biological) challenges.The simplest form of O-glycosylation is the introduction of β-GlcNAc onto serine or threonine (Fig. 1, 1). This is a regulatory modification that is, unlike the more complex forms of O-glycosylation, reversable.21 Despite this, it seems to be an immunologically relevant peptide modification, as O-GlcNAcylated peptides have been shown to be presented by major histocompatibility complexes (MHC) in vivo.22 The non-dynamic types of O-glycosylation start with the introduction of an α-linked N-acetyl galactosamine (GalNAc) residue (2), also called the Tn-antigen, onto serine or threonine. This single carbohydrate can then be enzymatically elaborated towards more complicated structures. For instance, the introduction of galactose onto the 3-OH creates the T-antigen (3), while 2,6-sialylation creates the STn-antigen (4). More elaborate structures are produced by enzymatic extension of different core structures. The four O-glycan cores most important in humans are also shown in Fig. 1 (3, 5–7). The biosynthesis and biochemical functions of these more complex O-glycosylation patterns are, as of yet, poorly understood.23 It cannot, for example, be predicted from the structure where on the protein this modification will be introduced, nor is the glycosylation machinery well defined. Besides these common modifications, glycosylation of serine and threonine with mannose, fucose, galactose and xylose have also been shown to exist within the human glycoproteome.1
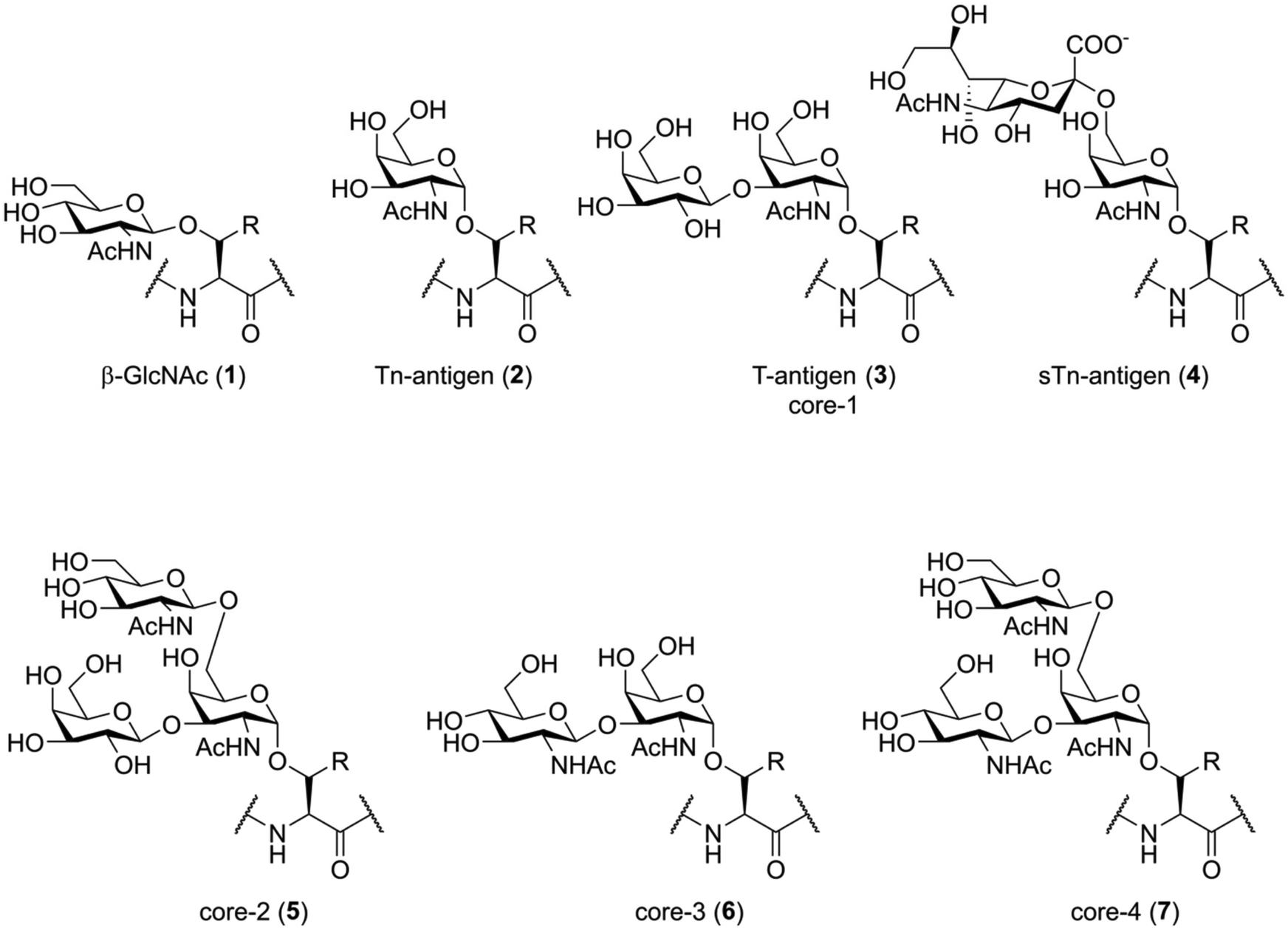 | ||
| Fig. 1 Some examples of O-glycosylation. β-GlcNAcylation of serine or threonine (1) is a commons regulatory PTM that competes with phosphorylation. Minimal O-glycosylation (2–4) patterns known to be relevant in cancer. More complex O-glycosylation is branched of different O-glycan cores represented by compounds 2, 5, 6, 7. All of these modifications occur both on serine (R = H) or threonine (R = CH3). | ||
One of the most well-studied examples of protein O-glycosylation is the mucin family of glycoproteins. The members of this family are produced primarily by epithelial cells and are either cell-membrane bound or secreted into the extracellular matrix. While elaborate O-glycosylation is suspected to exist on many proteins, the total amount and structural diversity found within this protein family is striking. Aberrant glycosylation of mucins, resulting in the proteins being decorated with the more simplistic forms of O-glycosylation (2–4), is found in several types of cancer.24 Targeting of these hypoglycosylated proteins by the immune system is a very active area of research for the development of onco-immunology based cancer treatments.25
N-Glycosylation is biochemically distinct from O-glycosylation. Unlike O-glycosylation, the location of N-glycosylation can be easily predicted: introduction of N-glycans occurs at asparagine residues that are part of the consensus sequence Asn-Xxx-Ser/Thr, with Xxx representing any amino acid except proline.1 Furthermore, the structure of the glycan is very different from those seen for O-glycans. Every N-glycan has the same core structure, that is Manα1-6(Manα1-3)Manβ1-4GlcNAcβ1-4GlcNac (8), containing a rare β-mannoside linkage (Fig. 2).26 Generally, this core structure is further elaborated with additional glycosylation. One commonly seen N-glycan structure is the so-called “high mannose”, or Man9, N-glycan (9), a modification that is introduced early in the biosynthesis of N-glycosylated proteins.26 Remodeling and elaboration of these immature N-glycans by the cellular glycosylation machinery leads to more elaborate N-glycans, the so-called “complex” type, such as a so-called biantennary and sialylated structure 10.
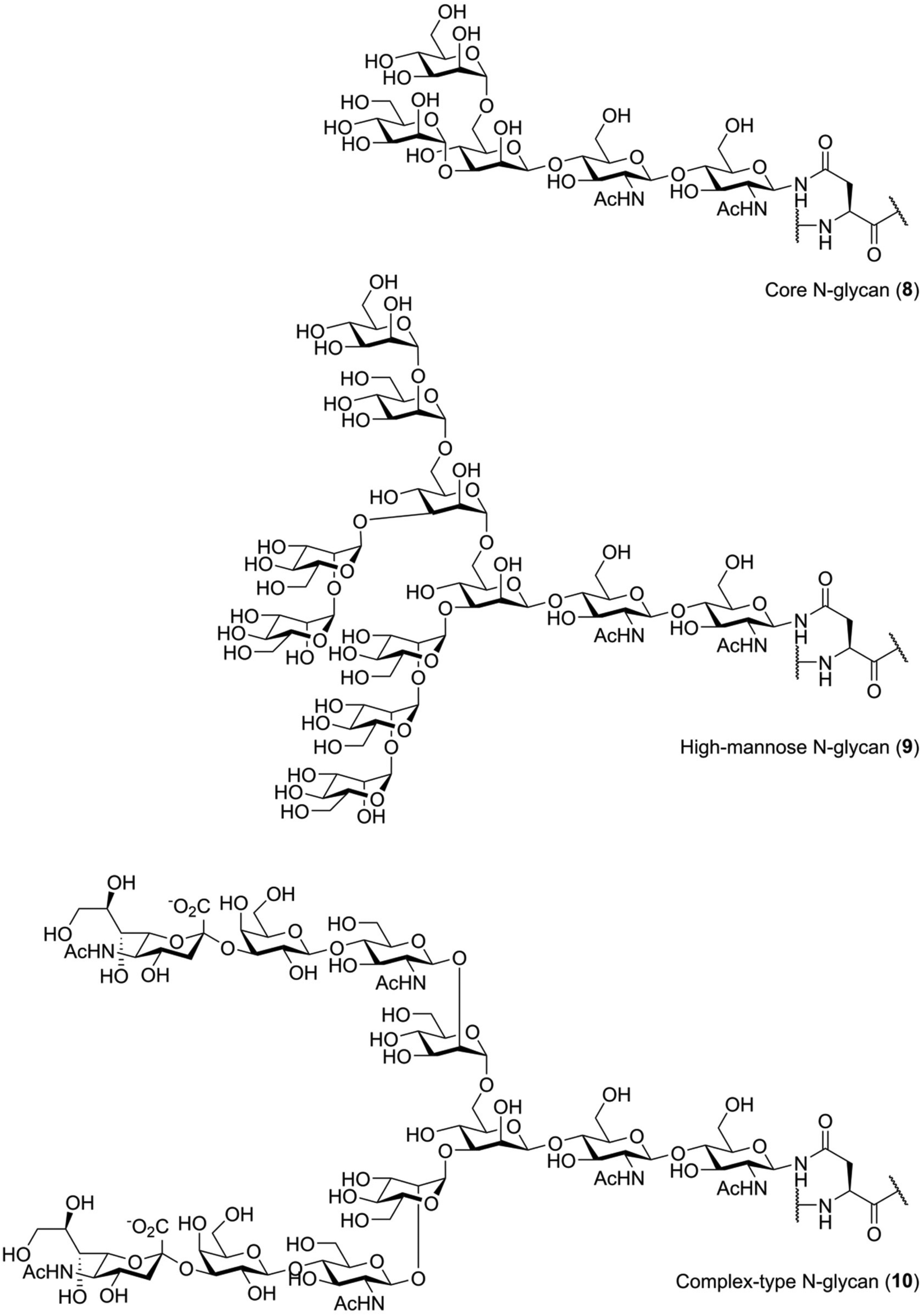 | ||
| Fig. 2 Examples of asparagine residues decorated with N-glycans. Structure 8 represents the core pentasaccharide seen in all natural N-glycans. Structure 9 is an example of the high-mannose or immature N-glycan, while 10 is an example of a sialylated complex N-glycan. | ||
Chemical approaches towards synthetic glycopeptides
Synthetic strategies towards the production of well-defined glycopeptides can take one of three approaches:1. Total chemical synthesis, where the entire molecule is produced using organic transformations;
2. Chemoenzymatic synthesis, where parts of the synthesis are accomplished using enzymatic transformations;
3. Conjugation based approaches, where the covalent linkage of peptide and oligosaccharide is achieved using one of various chemical approaches. These approaches usually yield a glycoconjugate containing one or more unnatural linkages.
While the first two approaches produce glycopeptides identical to naturally glycosylated peptides, this last approach will produce neoglycopeptides that, while not identical to native molecules, can be used to study the biochemical functions of glycosylation in some instances.14,27–32
For some applications, like the development of vaccines against glycosylated epitopes, having the entire, natively glycosylated amino acid in the peptide sequence is of crucial importance. This is necessary to produce antibodies and T-cell responses against a specific antigen in vivo without producing antibodies against unnatural linker-structures.33 However, the majority of lectin interactions only require certain oligosaccharide substructures derived from larger O- or N-glycans.10,29,30,34 For these areas of research, where the interactions under study only depend on a substructure of the glycan, simplified glycopeptides or glycoconjugates can be used. These can mimic some of the effects of natural glycosylation and can be used in place of natively glycosylated peptides. Specific areas of (immunological) interest these conjugates have seen use are the study of lectin mediated peptide uptake,13,28 modulation of cytokine production29,35 or targeting of specific cell-types.27,36 The increased synthetic accessibility of these glycoconjugates enables the synthesis and evaluation of more variants, in turn enabling more in-depth studies of glycan function. Therefore, conjugation methods to synthesize non-natively glycosylated peptides in a facile (compared to synthesis of native structures) manner will be described in more detail.
Total chemical synthesis of glycopeptides
Total synthesis of O-glycosylated peptides
The total chemical synthesis of O-glycosylated peptides is usually carried out using a building block-based approach, where the entire glycosylated serine or threonine residue is first synthesized as an Fmoc-protected building block. These amino acids are then incorporated into the desired peptide sequence using standard Fmoc-SPPS conditions. However, unlike the synthesis of peptides containing only standard, non-glycosylated amino acids, some considerations need to be made. The typical TFA mediated global deprotection of the synthesized peptides can lead to hydrolysis of certain glycosidic bonds, with α-fucosides and sialic acids being the most sensitive.37–39 This was famously demonstrated by Unverzagt and Kunz during the synthesis of a peptide containing an asparagine residue glycosylated with fucosylated chitobiose.37 During deprotection, they found that the acid lability of the α-fucoside linkage was protecting-group dependent. Compound 11, bearing benzyl protection groups on the fucose hydroxy-groups, was not resistant to acidic treatment, while acetylated compound 13 was (Fig. 3A). Additionally, alkaline conditions, like the Fmoc-deprotection step or the coupling step itself, can cause β-elimination of glycosylated threonine (Fig. 3B) or epimerization of glycosylated serine.40,41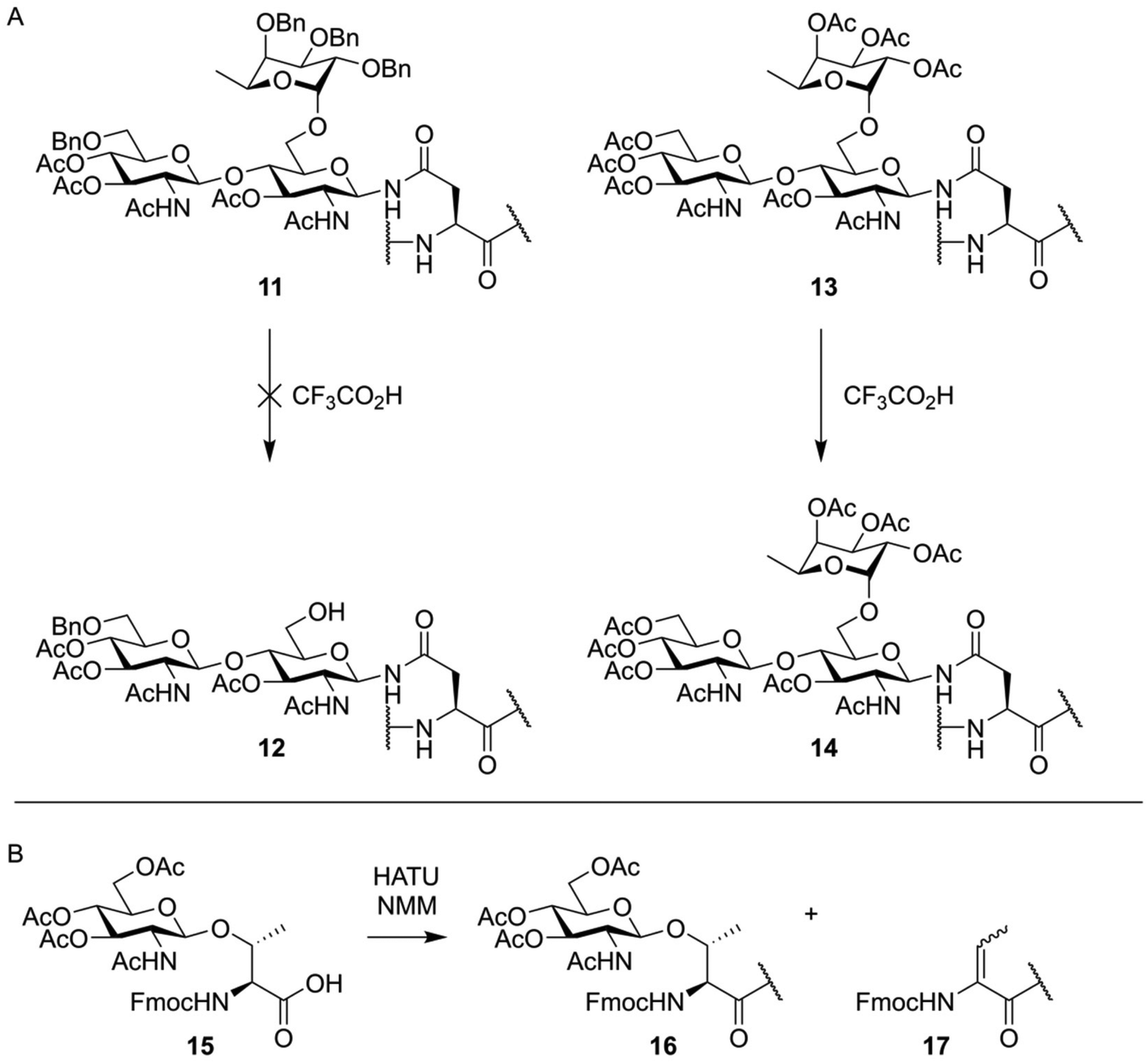 | ||
| Fig. 3 (A) TFA mediated deglycosylation observed by Unverzagt and Kunz.37 During TFA mediated deprotection of a peptide containing a trisaccahride (11) cleavage of the α-fucoside bond was observed yielding partially deprotected glycopeptide 12. A peptide containing an acetyl protected trisaccharide (13), when treated under similar conditions, did fully retain the fucose residue. (B) Base mediated β-elimination of an O-glycosylated threonine residue as observed by Zhang et al.40 | ||
The use of particular protecting groups on the sugar part of the glycopeptide can help alleviate these problems, but require careful consideration during synthesis of the glycosylated amino acid building blocks. O-Acetyls are most often employed, as they improve the acid stability of the molecule, while enabling removal under mild enough conditions to avoid the aforementioned base-mediated side reactions. Treatment with dilute solutions of NaOMe,42 NaOH43 or hydrazine44 can often be used to selectively liberate the protected glycoside without affecting the peptide or glycosidic bonds.
These days, peptides containing (simple) O-glycosylation patterns are synthesized as a matter of course, with many useful building blocks readily available from commercial sources. This, of course, is made possible by decades of development, that are briefly highlighted here. The initial development of synthetic methods to produce O-glycosylated peptides started in the 1980s.45–47 Kunz introduced the building block approach described above, synthesizing the first of Fmoc-protected O-glycosylated amino acids, using esters to protect the sugar hydroxyl groups (Fig. 4A). The common approach to introduce the α-glycosidic bond between the amino acid and the galactosamine, that is glycosylation with a 2-azido donor followed by reduction and acetylation, was used. These amino acids were used to produce several tripeptides decorated with both Tn- and T-antigen structures.47 Paulsen then applied a similar building block to Fmoc-SPPS, producing an octapeptide bearing five threonine residues modified with α-GalNAc,42 opening up the way towards more complicated glycopeptides.
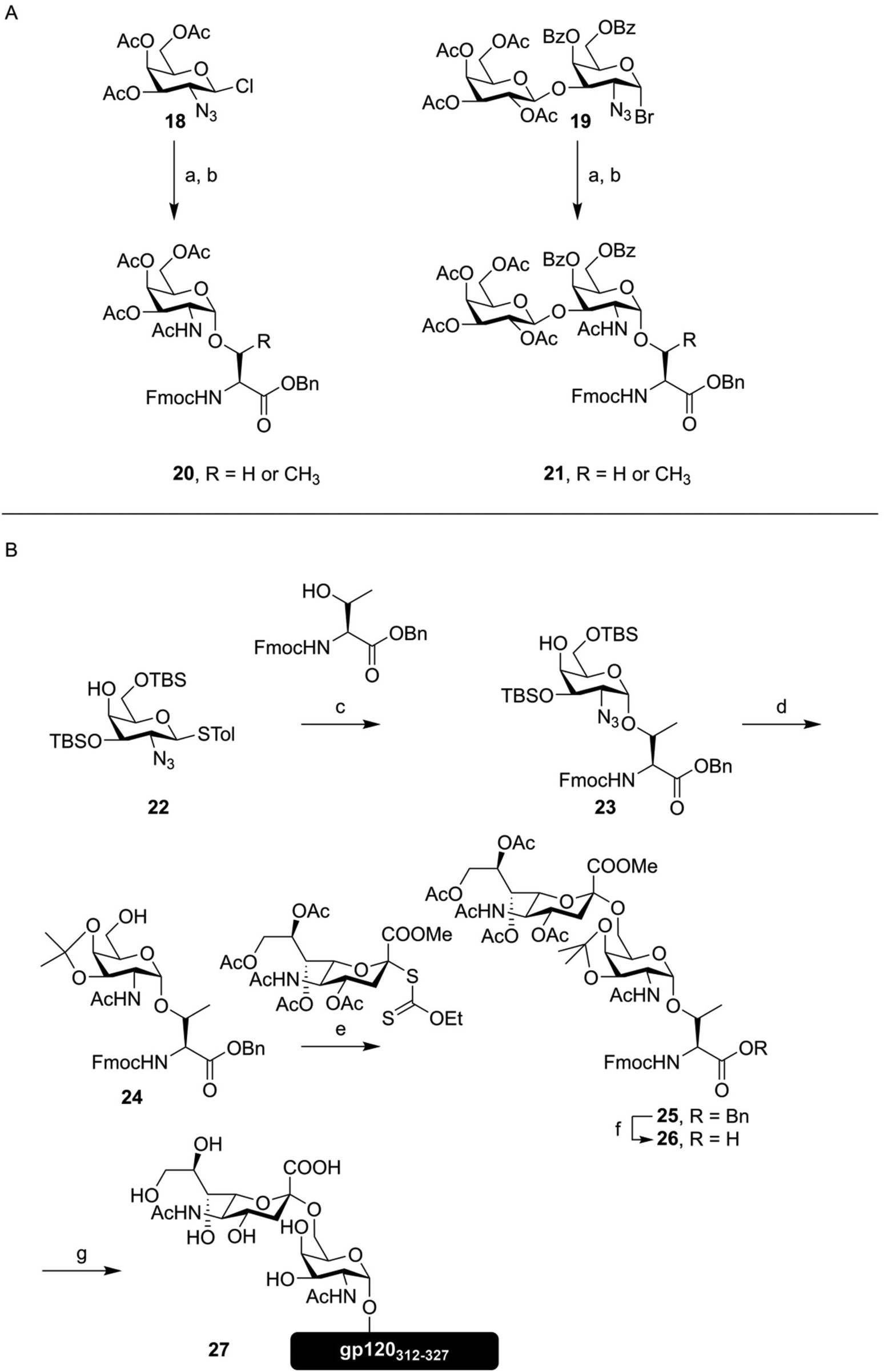 | ||
| Fig. 4 (A) First synthesis of the Tn- (20) and T-antigen (21) as Fmoc-protected amino acids, as described by Kunz.47 (B) Synthesis of a Sialo-Tn antigen building block (26) and use of this building block in Fmoc-SPPS, as described by Kihlberg and coworkers.49 Reagents and conditions: (a) Fmoc-Ser-OBn or Fmoc-Thr-OBn, Ag+, 30–65% (b) i) NaBH4, NiCl2 (ii) Ac2O, pyridine, 60–86% (c) NBS, Bu4NOTf, DCM, −28 °C, 71% (α/β mixture) (d) (i) AcSH, pyridine, 67% (ii) AcOH, MeOH, THF, 60 °C, 86% (iii) DMP, TsOH, 85% (e) MeSBr, AgOTf, DCM, MeCN, −78 °C, 49% (f) H2, Pd/C, EtOAc, 88% (g) (i) Fmoc-SPPS (ii) TFA, PhSMe, HSCH2CH2SH (iii) NaOMe, MeOH (iv) NaOH, H2O, (v) RP-HPLC, 12%. | ||
The synthesis of glycopeptides bearing sialic acid containing O-glycans, a key modification highly abundant on certain tumors,48 would take another decade.43,49 As an example, the synthesis of a sialyl Tn-antigen building block (26) and incorporation into a peptide fragment from the HIV glycopeptide gp12050 (27) by Kihlberg is given in Fig. 4B.49 This synthesis, like the one outlined above, used an azide function on the galactose C-2 to mediate formation of the α-glycosidic bond, followed by transformation to the desired acetamide function. This synthesis also exemplifies an early use of an acid labile protecting group, in the form of the isopropylidene, on a glycosylated amino acid building block. Since the sialic acid containing amino acid is introduced as the methyl ester, final deprotection has to be carried out using a hydroxide solution, in order to obtain the free acid.
With the synthesis of the glycopeptides via Fmoc-SPPS reaching greater maturity, the complexity, as well as the diversity, of the oligosaccharide structures has increased.51–55 For instance, the group of Westerlind developed a microarray containing a wide variety of mucin-protein derived glycopeptides, enabling the profiling of the antibody response of mice vaccinated with different MUC-glycopeptide based vaccine candidates.56
Besides mucin-derived peptides, other O-glycosylated peptides have been the object of study. As an example, glycopeptide ligands for the P- and E-selectin receptors on white blood cells are an important class of O-glycosylated peptides.57 P-selectin binds to the sialyl-LewisX (sLeX) tetrasaccharide (Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAcβ), a synthetically challenging target on its own. Total synthesis of a 12-mer peptide, derived from the protein P-selectin glycoprotein ligand-1 (PSGL-1), containing a threonine residue glycosylated with a hexasaccharide containing the sLeX structure was achieved by Baumann et al.58
Recently, renewed interest in the development of O-glycosylated amino acid building blocks has been seen. Several novel acid-labile protecting group strategies have been developed, enabling a one-step deprotection of the resin-bound glycopeptide. These approaches use electron-rich benzyl ethers59,60 or silyl ethers.49,61,62 While these are promising developments simplifying the synthesis of O-glycosylated peptides, they have only been applied to peptides bearing mono- or disaccharides, and have yet to be proven in the synthesis of glycopeptides bearing larger protected glycans.
Total synthesis of N-glycosylated peptides
While generally more challenging than the synthesis of O-glycosylated peptides, due to the high complexity of even “simple” N-glycans, some total syntheses of N-glycosylated peptides have also been carried out. In contrast to the synthesis of O-glycosylated peptides, where the glycosidic linkage between amino acid and oligosaccharide is often introduced in an early stage, most syntheses of N-glycosylated amino-acids or peptides produce the crucial glycoside-asparagine linkages at a late stage in the synthesis. Most commonly, the entire glycan, either with or without protecting groups, is produced as a glycosyl amine, that is then used in an amide-bond forming reaction with an aspartic acid sidechain.Early work on N-glycosylated peptides used a similar building block approach as was used for the synthesis of O-glycosylated peptides. The group of Ogawa managed the first synthesis of a tripeptide carrying the core-pentasaccharide.63 This synthesis, outlined in Fig. 5, starts with the complete synthesis of the pentasaccharide azide 37. In this synthesis, no particular care was taken to selectively produce the β-mannoside linkage in compound 30, preferring instead to separate the two diastereomers. After allyl ether deprotection (34) and introduction of the final two mannose moieties (36), the phthalimide functions were converted to the acetamides (37). To introduce the (orthogonally protected) asparagine residue, the anomeric azide was selectively reduced by hydrogenation over Lindlar catalyst to obtain the glycosyl amine. This intermediate was then condensed with anhydride 38 in the same pot, to limit hydrolysis of the sensitive anomeric amine. TFA mediated deprotection of the tert-butyl ester produced Fmoc-amino acid 40, usable for Fmoc-SPPS of glycopeptides. In this case, however, solution phase peptide synthesis was employed, with EDC mediated coupling to dipeptide 41 yielding the protected tripeptide 42. Fmoc-deprotection followed by hydrogenolysis yielded the desired deprotected glycopeptide 43.
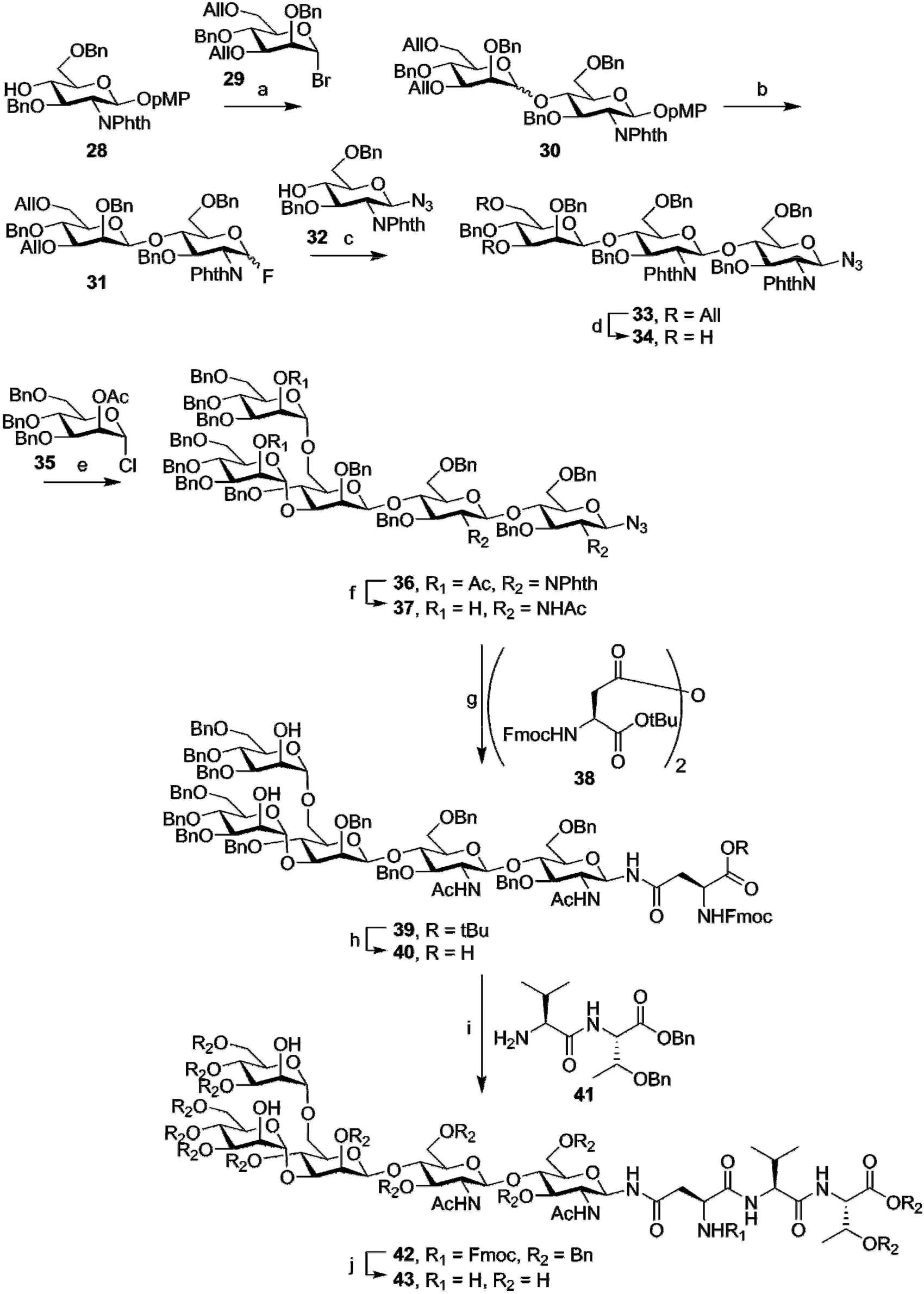 | ||
| Fig. 5 Ogawa's synthesis of an Fmoc-asparagine building block carrying the core N-glycan structure (40), and solution phase synthesis of a simple tripeptide bearing this glycan (43). In contrast to the synthesis of O-glycosylated building blocks for peptide synthesis, the introduction of the amino acid happens after production of the full glycan. Reagents and conditions: (a) silver silica-alumina, DCM, 72% (α/β = 7/8) (b) i) CAN, H2O, MeCN, MePh, 70% (ii) DAST, DCM, quant. (c) AgClO4, hafnocene dichloride, DCM, 79% (d) {Ir(COD)[PCH3(C6H5)2]2}PF6, THF, 79% (e) AgOTf, DCM, −40 °C, 80% (f) (i) NH2CH2CH2NH2, nBuOH (ii) Ac2O, MeOH, 0 °C, 80% (g) Lindlar, H2, MeOH, 79% (h) TFA, DCM, 89% (i) EDC, HOBt, DCM, 90% (j) (i) morpholine (ii) Pd/C, H2, AcOH, H2O, 58%. | ||
Further development of this building block approach by Unverzagt produced a pentapeptide decorated with a heptasaccharide N-glycan, again by in solution peptide synthesis.64,65 This glycosylated amino acid, bearing no protective groups on the sugar hydroxy functions, was later also used in Fmoc-SPPS, where the choice of solid support turned out to be critical to mediate effective introduction of this extremely polar amino acid.66 When using glycosylated amino acids without hydroxyl-group protection, side reactions, in the form of unwanted acylation, limit the maximum length of the peptide that can be synthesized.67 However, when the glycoside was protected with acetyl groups, via on-resin acetylation, another set of unspecified side-reactions still prevented the synthesis of N-glycosylated peptides longer than 20 amino acids.68
To avoid these problems, the total synthesis of N-glycosylated peptides is usually carried out via an convergent approach. This is most often accomplished using the so-called Lansbury aspartylation (Fig. 6).69,70 In this reaction, a glycosylamine derivative of the oligosaccharide is condensed with a partially protected peptide, containing an activated aspartic acid residue. This approach is not free of side reactions, with aspartimide formation and the associated formation of iso-aspartate and loss of stereochemistry of the aspartic acid residue being key problems.71
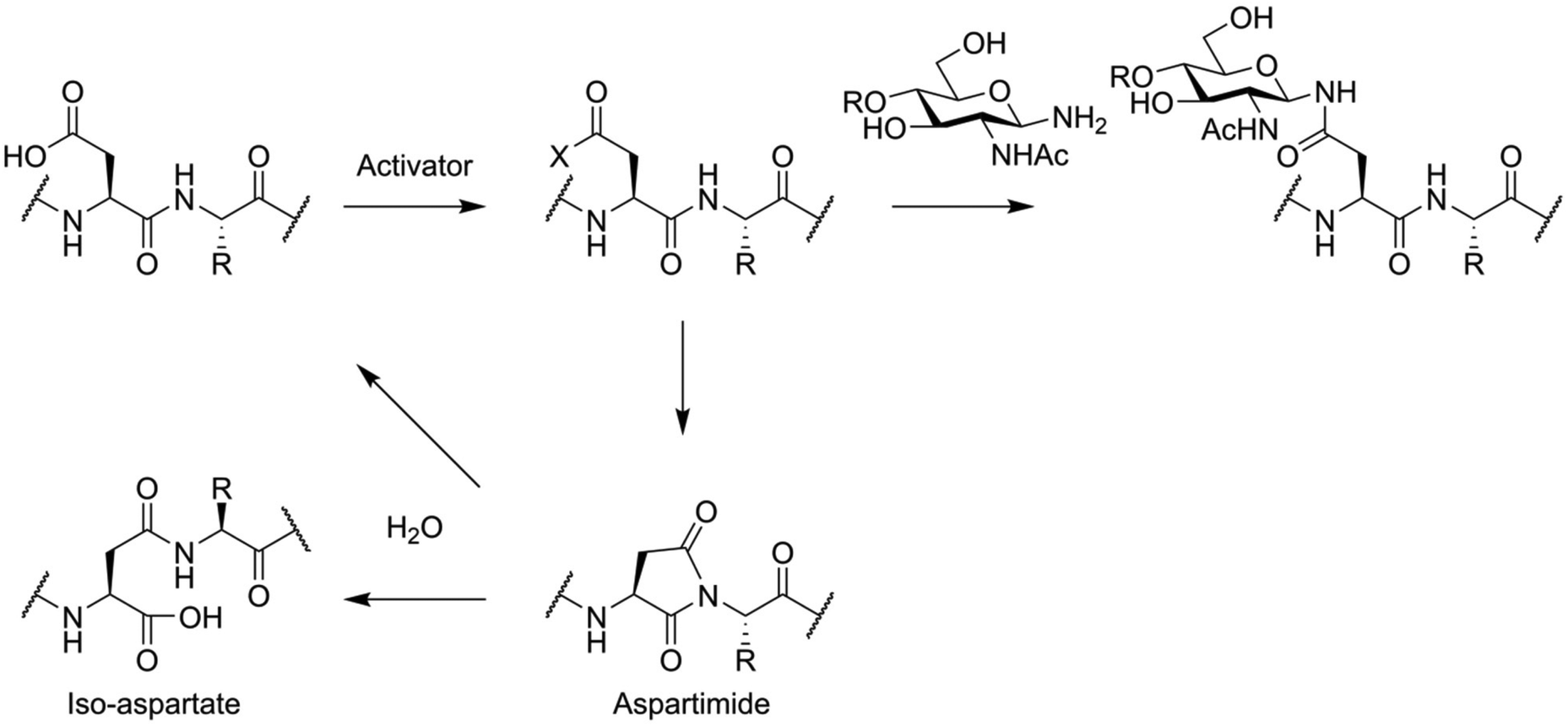 | ||
| Fig. 6 Lansbury aspartylation and the associated aspartimide formation side reactions. The acid function of an unprotected aspartic acid sidechain is activated using a coupling reagent, and the active ester is reacted with a free glycosyl amine. As a sidereaction, the activated ester of aspartic acid can react with the C-terminal amide, forming an aspartimide. Hydrolysis at the Cα-side leads to the formation of an iso-aspartate residue. X = active ester. | ||
Still, in this manner the total synthesis of highly complex N-glycosylated peptides was achieved, with the chemical synthesis of homogeneously glycosylated Erythropoietin by Danishefsky and coworkers the most famous example.72 Also, a glycopeptide derived from the HIV envelope protein, a potential vaccine candidate, was produced by total synthesis. This peptide was used to isolate a so-called broadly neutralizing antibody (BnAb) from the plasma of a HIV patient, as well as induce production of antibodies in a Rhesus Macaque model.73 However, to further avoid the side reactions associated with chemical introduction of N-glycans or N-glycosylated amino acids, as well as the laborious work of producing said glycan by synthesis, many examples of N-glycopeptide synthesis have instead been carried out using chemoenzymatic and semisynthetic means.
Chemoenzymatic (semi)synthesis of glycopeptides
Since N-glycans all have the same pentasaccharide core, the enzymes that deal with this part of the glycan are often somewhat promiscuous. This is especially true for the family of endo-β-N-acetylglucosaminidases (ENGases), which catalyze the cleavage of the majority of an N-glycan from a peptide, by hydrolyzing the glycosidic bond between the first and second GlcNAc at the reducing and of the oligosaccharide.74 It was soon realized that these enzymes could, under the right conditions, be made to perform transglycosylation reactions between an asparagine residue containing a full N-glycan and another bearing only an N-acetyl glucosamyl.75 The discovery that these enzymes were capable of using oxazoline donors76 instead of glycosylated asparagine derivatives helped to further develop this effective enzymatic glycosylation method. Directed mutagenesis of several ENGase enzymes lead to the development of specific mutants that had limited hydrolytic capacity, removing the most pronounced side reaction and increasing the yield of these enzymatic glycosylations.77,78 In Fig. 7A, the general method of ENGase mediated peptide glycosylation, using an oxazoline donor glycoside, is shown.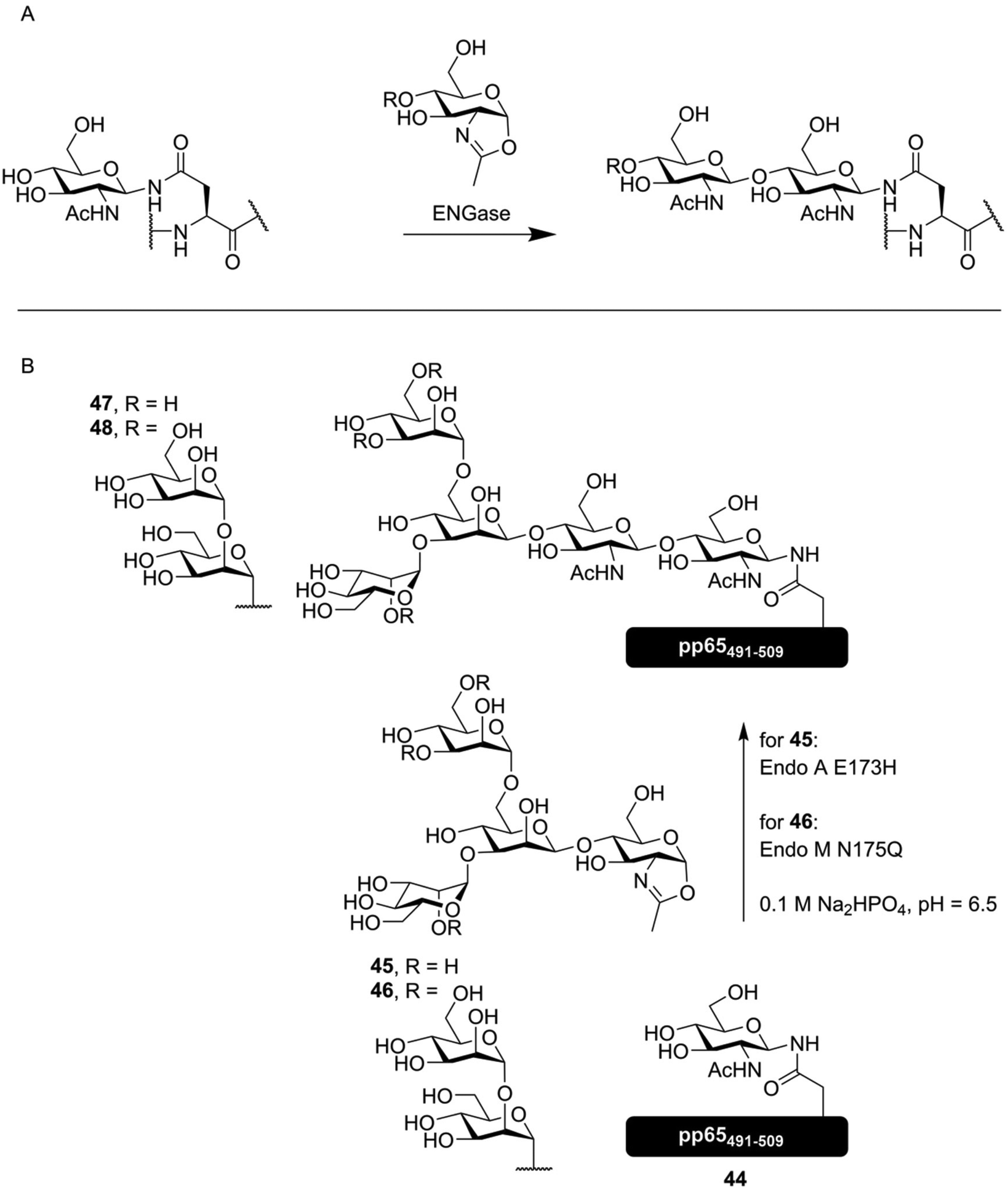 | ||
| Fig. 7 (A) general overview of ENGase mediated glycosylation of a peptide containing an N-GlcNAc asparagine residue with an oxazoline donor. (B) ENGase mediated synthesis of several N-glycosylated derivatives of the cytomegalovirus derived peptide pp65491–509 by McIntosh et al.79 Both synthetic oxazoline donor 45 as well as semisynthetic oxazoline donor 46, produced from an N-glycopeptide obtained from soy flour, were enzymatically introduced onto glycopeptide 44, producing vaccine candidates 47 and 48. | ||
ENGase mediated synthesis has been used to synthesize a peptide derived from the HIV glycoprotein gp120, containing two core pentasaccharides,80 and a vaccine candidate glycopeptide against cytomegalovirus (CMV), decorated with either a core-pentasaccharide (47, Fig. 7B) or a full high mannose-type glycan (48). As the peptide sequence under investigation contains two distinct N-glycosylation sites, the effect of glycosylating one or both sites was evaluated. The found that while glycosylation of Asn507 had negligible inhibitory effect on T-cell activation, introduction of the N-glycans onto both Asn495 and Asn507 completely inhibited T-cell activation.79
ENGase mediated glycopeptide synthesis has also used to synthesize lectin binding peptides; Yamaguchi et al. produced, by total chemical synthesis, an oxazoline donor of a bis-phosphorylated high-mannose type N-glycan (50), and used this to produce a cyclic peptide containing two phosphorylated N-glycans (51, Fig. 8). This molecule had a nanomolar affinity for the cation-independent mannose-6-phophate receptor (CI-MPR). The cyclic nature of the peptide was found to be critical, as linear glycopeptide 52 had a 20-fold lower affinity for the CI-MPR.81 Besides the synthesis of N-glycosylated peptides, ENGase catalyzed glycosylation has also been extensively applied to the production of single glycoform N-glycosylated proteins, including remodeling of the N-glycans on the Fc-domain of antibodies.82–85
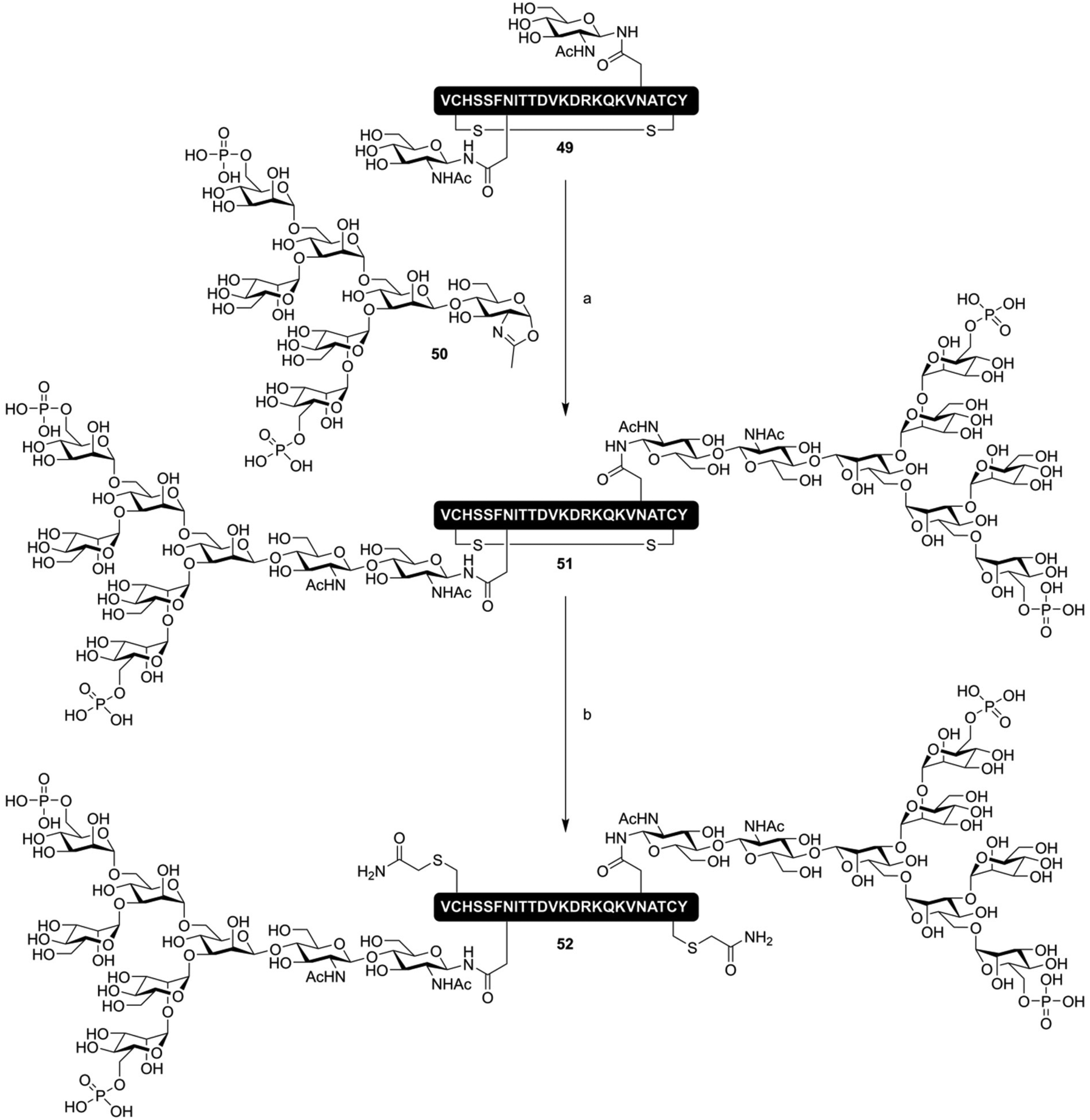 | ||
| Fig. 8 ENGase mediated synthesis of a CI-MPR binding cyclic phosphorylated glycopeptide 51 by Yamaguchi et al.81 Reagents and conditions: (a) Endo A N171A, Tris buffer (pH 7.1) (b) (i) DTT (ii) Iodoacetamide. | ||
Another area where biochemical methods have improved the study of N-glycosylated peptides is in the production of the N-glycosylated asparagine building blocks and oxazoline donors required for the above syntheses. Methods have been developed to isolate N-glycopeptide carrying specific N-glycans from biological sources. This was first demonstrated by the isolation of sialoglycopeptide (SGP) from egg yolk, a hexapeptide bearing a biantennary sialoglycan.86 This peptide was used directly with Endo M (one of the ENGases) to produce a glycosylated form of the CD52 peptide,87 as well as being chemically transformed into an oxazoline donor for other ENGase mediated reactions.88,89 Furthermore, after peptidase mediated liberation of the glycosylated asparagine followed by selective introduction of an Fmoc-group and protection of the sialic acid residues as benzyl esters67 or phenacyl (Pac) esters,90 this semisynthetic glycosylated amino acid has been used in SPPS for the facile production of N-glycosylated peptides.91,92 These protecting groups inhibited the acid mediated hydrolysis of the sensitive glycosidic bond of sialic acid, enabling both Fmoc- and Boc-SPPS approaches. A high mannose type N-glycan (Man9GlcNAc2-Asn) can, in a similar manner, be isolated from soybean flour93,94 or egg yolks,95 and has also been used in ENGase mediated glycotransfer reactions79,96 as well as SPPS.97
Another enzymatic approach that has furthered the synthesis of N-glycopeptides is enzyme-based glycan remodeling. This is the enzymatically removal and/or addition of specific glycosides to an N-glycan. Enzymes have most commonly been employed to install sialic acids onto peptides bearing synthetic complex-type N-glycans,65 or for introduction of the so-called core-fucose moiety (α-fucosylation of the 6-OH of the reducing-end GlcNAc in the core pentasaccharide).98 More extensive remodeling of N-glycosylated asparagine has also been reported. The group of Kajihara used partial acidic hydrolysis of glycosylated asparagine obtained from egg-yolk SGP, followed by further enzymatic hydrolysis to produce various N-glycosylated asparagine residues. These were subsequently used in peptide synthesis.99 More recently, the group of Boons showed the production of various N-glycosylated asparagine structures by enzymatic remodeling of the biantennary N-glycan isolated from egg yolk.100,101 This method has, however, not yet been employed for the synthesis of N-glycosylated peptides.
Chemo-enzymatic approaches have also been applied to the synthesis of O-glycans, by enzymatically elaborating synthetic O-glycosylated peptides. These approaches start by the chemical synthesis of a peptide containing an α-GalNAc-Ser/Thr as a starting point for enzymatic introduction of additional saccharides. Sialic acids have been introduced in a stereoselective manner using this approach,102 a feat that can be challenging using organic synthesis techniques.103 More complex oligosaccharides have also been made chemoenzymatically; for instance, a glycosulfopeptide derived from PGSL-1 has been produced by enzymatic elaboration of an α-GalNac containing synthetic peptide.104 Producing O-glycosylated peptides from non-glycosylated starting material has also been accomplished, as shown by the group of Clausen, who synthesized various glycoforms of a 60-mer MUC1 repeat peptide.105 Two of the peptides produced in this manner were then used to immunize mice, where production of specific antibodies was observed. Using the various differently glycosylated MUC1 peptides produced by the authors, they were able to precisely determine the specificity of these antibodies.105,106
Unnatural glycoconjugate peptides
The glycans of many glycoproteins are very complex (as described above). However, the core of the glycan often does not partake in shaping its biological role.10 In order to study the role glycosylation plays in immunological processes without having the perform highly complex glycopeptide total synthesis, conjugation strategies to produce non-natural glycosylated peptides have been developed. These non-natural structures enable the rapid production of glycopeptide conjugates. This in turn facilitates the study of the role of glycosylation in certain (immune)cellular processes. It also enables more rapid synthesis of libraries of glycopeptides bearing various oligosaccharides, enabling powerful studies in carbohydrate structure/activity relationships.28–31Simplified and stabilized glycosylated amino acid building blocks
Peptides decorated with simplified glycans – either attached via a native or non-native linkage – can provide useful tools for studying the effect of glycosylation on immune reactions. Various approaches have been developed based on this premise. These primarily differ from each other by how close they mimic the chemical structure of natively glycosylated peptides. They can be viewed to be on a spectrum from nearly-native down to glycopeptide conjugates using completely non-native linkages. On the nearly-native side of the spectrum, there are the simplified N-glycans. Here asparagine is, via a natural N-glycosidic linkage, decorated with a truncated glycan. Next are the stabilized linkages, described below, where labile parts of the glycosylated amino acid are replaced with more stable bioisosteres. For example, replacing the oxygen in a glycosidic bond for a sulfur atom renders the glycoside resistant to enzymatic hydrolysis.107 While these are not equivalent to the linkages found in nature, these still have a high degree of similarity to the native structures. On the fully non-native end of the spectrum, completely non-amino acid based building blocks are used to attach a glycoside to a peptide, usually by amide bond formation with the N-terminal or lysine-sidechain amine group(s).The simplified N-glycan mimics, as the most natural-like of the simplified glycans, still retain some key properties of the glycans they are mimicking, i.e. the presence of a glycosidic bond with asparagine. Peptides produced via the simplified N-glycan approach are capable of mimicking certain functional aspects of N-glycans, while circumventing production of the entire, synthetically complex natural N-glycan. The group of Kunz has, for example, developed glycopeptide ligands for the lectin E-selectin, containing asparagine linked Sialyl-LeX determinants (53, Fig. 9A).108,109 Simplified N-glycans, bearing other, non-sialylated and immunologically important Lewis-type sugars, like LeX![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29,110 and LeA111 have also been described. These kinds of simplified N-glycans can be used for functional studies. For instance, LeX is known to bind to the lectin Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), inducing a tolerogenic response from the targeted dendritic cell.112 We produced a peptide, derived from the protein myelin oligodendrocyte glycoprotein (MOG), decorated with a simplified LeXN-glycan on the native N-glycosylation site (54).29 This molecule was able to bind to DC-SIGN and elicit a tolerogenic response in vitro.
29,110 and LeA111 have also been described. These kinds of simplified N-glycans can be used for functional studies. For instance, LeX is known to bind to the lectin Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), inducing a tolerogenic response from the targeted dendritic cell.112 We produced a peptide, derived from the protein myelin oligodendrocyte glycoprotein (MOG), decorated with a simplified LeXN-glycan on the native N-glycosylation site (54).29 This molecule was able to bind to DC-SIGN and elicit a tolerogenic response in vitro.
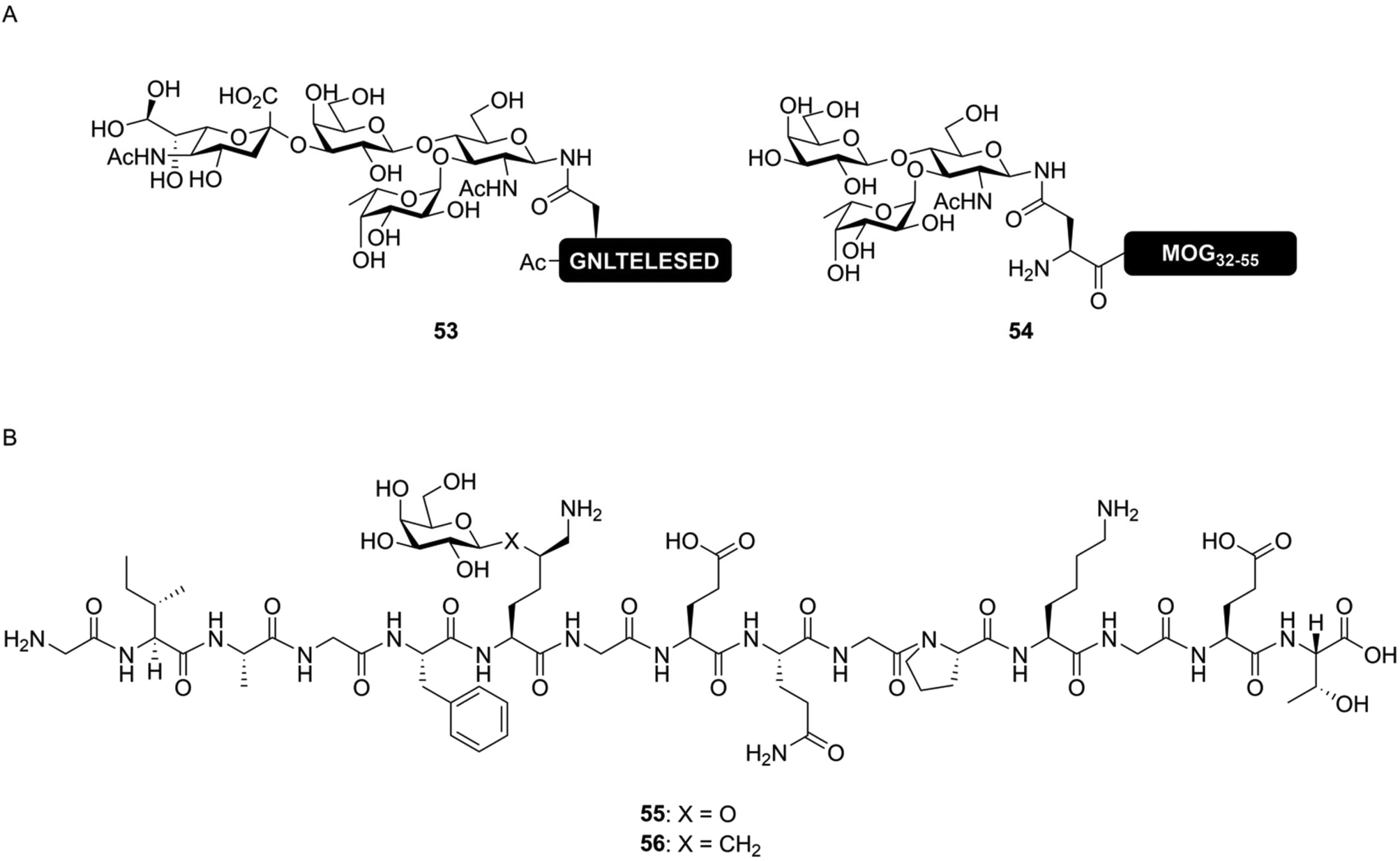 | ||
| Fig. 9 Examples of simplified N-glycopeptides. (A) Synthetic N-glycopeptide bearing simplified N-glycans produced by Filser et al.108 (53) and us (54).29 By including only the part of the N-glycan required for lectin binding, the synthetic complexity of glycopeptides can be limited without compromising on functional activity. (B) Structure of the native (55) and stabilized (56) variants of a neoglycoepitope derived from collagen type II synthesized by Gustafsson et al.113 Peptide 56 was still able to induce a T-cell response, despite the non-native glycosidic linkage. | ||
Stabilized building blocks were developed not to simplify chemical synthesis, but to increase (bio)chemical stability of the glycopeptide. These approaches are occasionally used to suppress the chemical side reactions described earlier. More commonly, stabilized building blocks are applied in glycopeptide synthesis to prevent the enzymatic hydrolysis of natural linkages in biological systems.21,114 These actions of cellular enzymes can be a complication when studying the effect of peptide glycosylation in biological systems. To suppress enzymatic degradation, stabilized glycosylated amino acid building blocks have been developed, based on S- or C-glycosidic linkages. Cys(β-GlcNAc), as mentioned before, is a good functional mimic of Ser(β-GlcNAc), that is resistant to hydrolysis by the glycosidase O-GlcNAcase.107 An S-glycoside analogue of a Tn-antigen containing MUC1-peptide has also been developed and tested as a vaccine candidate. The antibodies obtained from mice immunized with this peptide were able to recognize a human tumor cell line, indicating that the unnatural linkage does not necessarily impede the ability of a synthetic glycopeptide to induce an immune response to the native antigen.115 This result further exemplifies the ability of S-glycopeptides to mimic the functions of O-glycopeptides. C-Glycosides have been similarly used in immunological studies. For instance, the group of Kihlberg produced a glycopeptide epitope, derived from type II collagen containing a galactosylated δ-hydroxylysine (55, Fig. 9B), as well as a C-glycoside mimic of this moiety (56).113 This peptide may play a key role in the pathogenesis of rheumatoid arthritis (RA).116,117 The C-glycoside analogue 56 was able to activated T-cell hybridomas, although at a lower level compared to the natural peptide. Stabilized glycopeptides can also bind lectins; for instance, C-mannoside decorated peptides have been successfully used as DC-SIGN ligands.118
Finally, sometimes glycosylated building blocks bearing very little resemblance to the natively glycosylated amino acids are used. Here, a glycoside is coupled, usually via the anomeric lactol function, to a linker bearing a carboxylic acid. These can be coupled on-resin or in solution to the N-terminus of the peptide or to the sidechain amine of lysine residues. These linkers can take the form of a simple hydroxylated carboxylic acid, as used by Kantchev et al.119 These molecules were used to decorate vaccine peptides with glycans and study the effect of lectin mediated uptake on antigen (cross-)presentation. An alternative glycoside derivatization strategy was developed by the group of Roche (Fig. 10). Here, the free reducing-end lactol of a glycoside (57) is reacted with the amine group of glutamic acid (58), forming the hemiaminal. Cyclization with the sidechain carboxylic acid formed the pyroglutamate derivative, stabilizing the N-glycosidic bond (60).120 After hydrogenolysis of the benzyl ester protecting the carboxylic ester (61) the glycans were coupled in solution phase to various oligolysine peptides (producing, for example, 62). Various fluorescently labeled glycoclusters were build using this method. These were then used to study the binding specificity of the mannose receptor.28 The same group later further expanded on this approach, using it to target antigenic peptides to DC-SIGN in order to improve antigen cross-presentation. Here, they showed a glycan-dependent effect on T-cell activation.121
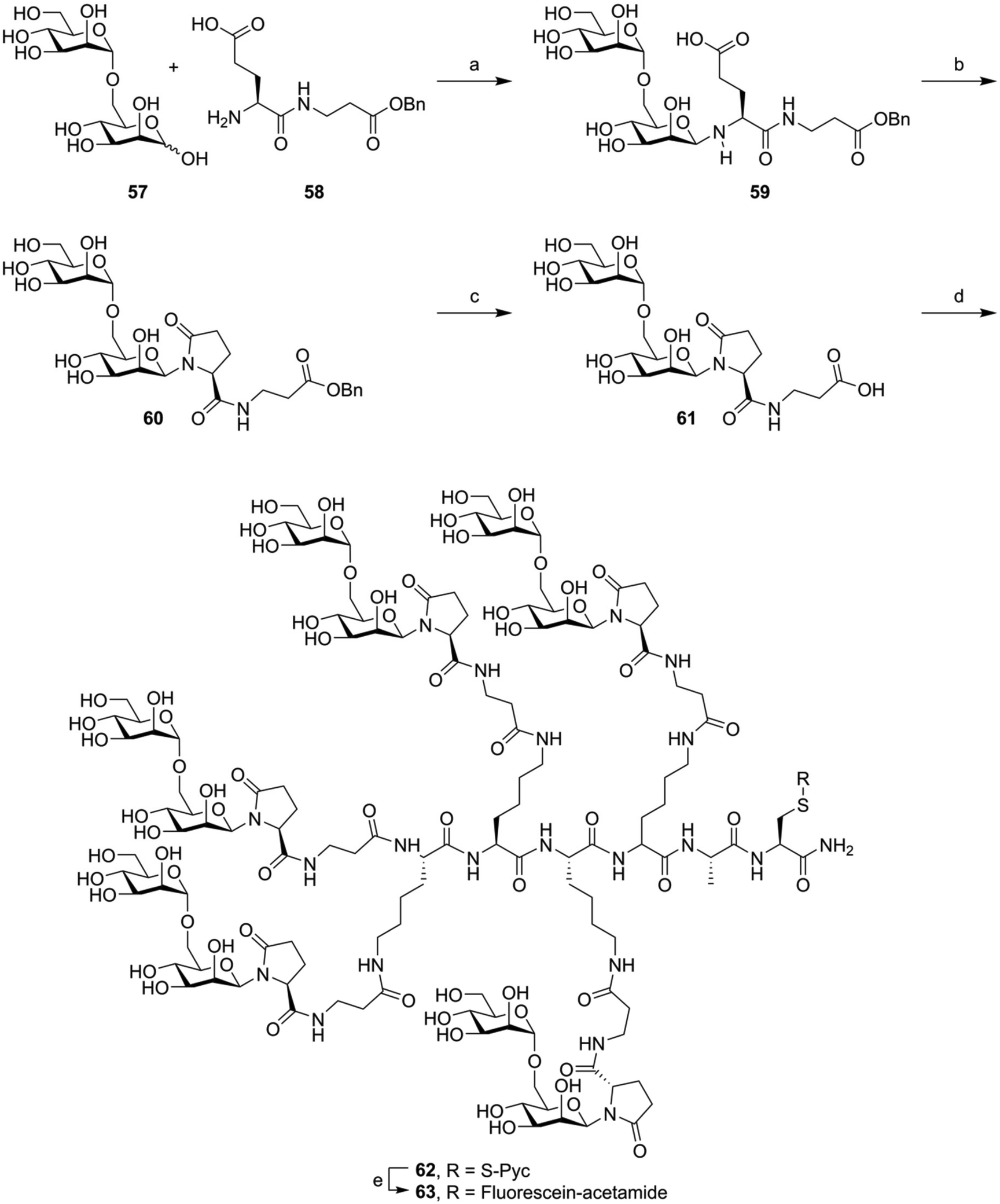 | ||
| Fig. 10 Example synthesis of the fluorophore labeled oligomannose clusters synthesized by Frison et al.28 Reagents and conditions: (a) imidazole, NMP, 50 °C (b) BOP, imidazole (c) H2, Pd/C (d) Lys-Lys-Lys-Lys-Ala-Cys(S-Pyc)-NH2, HBTU, HOBt, DiPEA, NMP (e) (i) TCEP, NMP, NaPi pH = 7.2 (ii) Fluorescien-iodoacetamide. | ||
All of these strategies still rely on introduction of glycosyl building blocks into the peptide on the solid support or in solution. This often requires large amounts of the glyco-building block under investigation and are often limited by low peptide yields. In order to limit the need for large amounts of precious oligosaccharide, conjugation methods were developed, where the carbohydrate part of the molecule can be conjugated to the peptide after SPPS and HPLC purification. This requires far less of the precious oligosaccharide, enabling the use of more complex sugars.
Late-stage glycoconjugation using organic transformations
Many glycopeptide conjugation strategies are based around the unique reactivity of the amino acid cysteine.122 The thiol function of this amino acid represents unique reactivity compared to the 19 other amino acids, enabling some form of chemoselectivity. Various groups have exploited the fact that thiolates are soft nucleophiles to introduce oligosaccharide modifications to peptides (Fig. 11). The group of Stenvall used a bromoethyl linker on the reducing end of several oligosaccharides (for instance 65) to produce O-glycan mimicking neoglycopeptides.123 In a similar manner, maleimidosugars (68) have been used as well.124 Alternatively, the ability of thiols to form disulfides under mild conditions was exploited to produce disulfide linked oligosaccharides (73) by the groups of Davis,125 and Boons,126 using thioglycoside 71 and 72 respectively.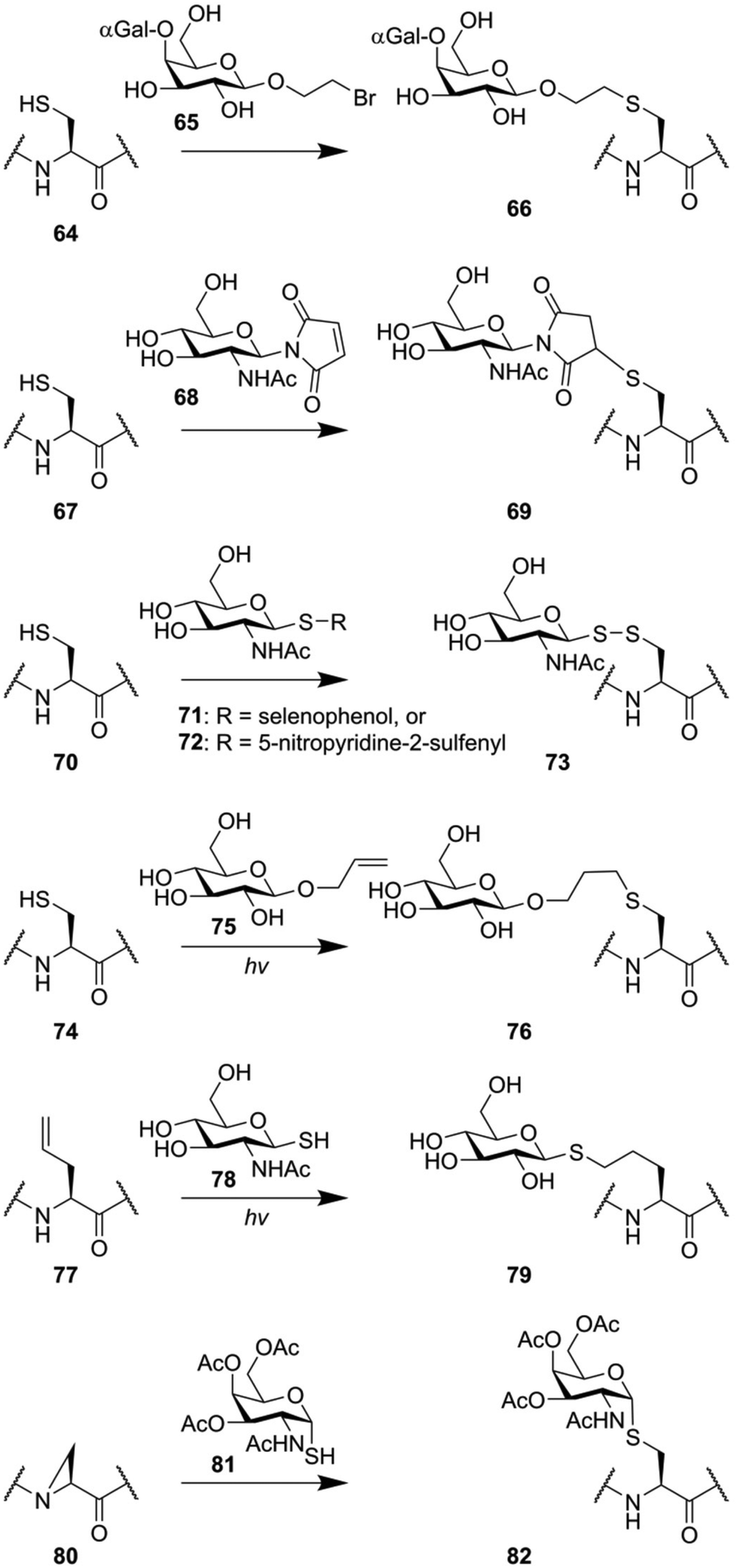 | ||
| Fig. 11 Examples of thiol-based glycoconjugate synthesis. | ||
Another class of thiol selective chemistry that can be applied to cysteine is the photoinduced thiole–ene reaction,127 which has been used to produce glycoconjugate 76 using allyl glycoside 75.128,129 Alternatively, the reactivity of thiols can also be used in the opposite direction. The thiol–ene reaction can also be performed by reacting a thiol glycoside (78) to an alkene containing peptide (76), producing neoglycopeptide 79.130 The nucleophilicity of thioglycosides was also exploited to synthesize peptides containing S-glycosylated cysteine residues in an unconventional manner. The nucleophilic opening of an aziridine amino acid (80) on solid support was used to produce an S-glycoside mimic of the Tn-antigen (82).131
However, the above-described approaches all rely on thiolate functions to mediate chemoselective ligation, making these approaches incompatible with peptides containing natural cysteine residues. Earlier attempts at making glycoconjugation reactions orthogonal to common peptide functionalities hinged on the reactivity between the aldehyde function present in carbohydrates and oxy-amine functionalities (Fig. 12). As a simple example, the oxy-amine derivative of serine (83) can be used to chemoselectively ligate a free lactol.132 The downside of this ligation approach is that it primarily forms the ring-opened configuration of the reducing end glycoside (85).133 When the nitrogen in this oxy-amine is alkylated (86), the cyclic form of the glycoside is conserved after ligation (87).134,135 Another approach, that more closely mimics the structure of N-glycans, uses the hydrazide derivative of aspartic acid (88). Conjugation with the free lactol of GlcNAc (84) leads to the formation of an anomeric β-hydrazide (89), a close mimic of a native N-glycan, with an additional nitrogen in between the glycoside and the asparagine residue.132 The condensation of oxy-amines with carbonyl functionalities has also been used in the form of a chemoselective ligation between a peptide containing a ketone containing amino acid (90) and a hydroxylamine-glycoside (91).136
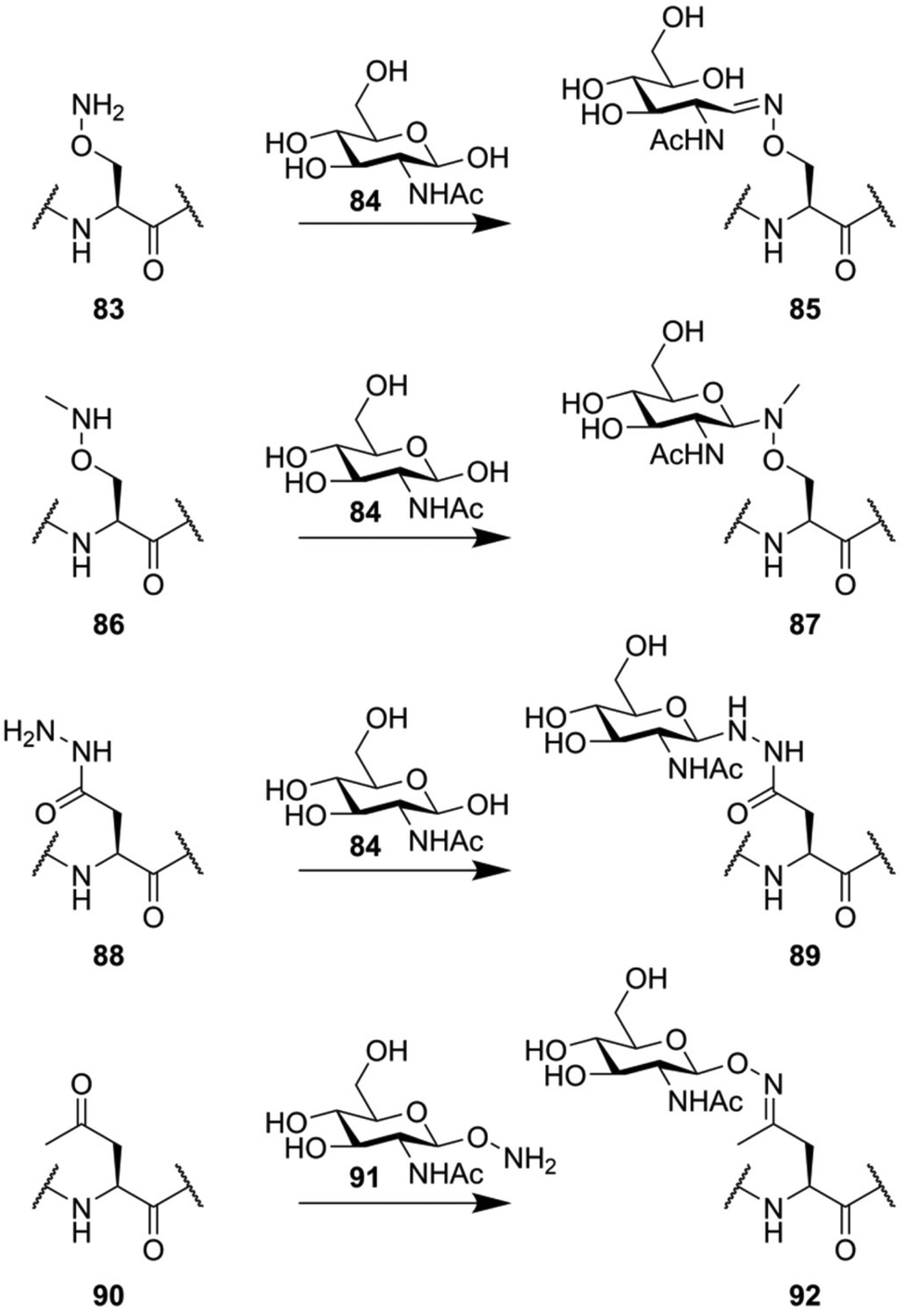 | ||
| Fig. 12 Various glycoconjugation approaches used for the synthesis of neoglycopeptides, based on the unique reactivity of oxy-amine and hydrazide derivatives. | ||
Metal-catalyzed late stage conjugations
One downside to the aforementioned conjugation methods is that they are quite limited in scope, often not producing the desired results when dealing with complex oligosaccharides or peptides. Recently, metal-catalyzed ligation reactions have shown their ability to produce glycoconjugate peptides by true bio-orthogonal means, while simultaneously producing high yields using complex substrates. The earliest work on metal catalyzed synthesis of glycoconjugates made use of the olefin-crossmetathesis reaction. McGarvey et al. used protected peptides containing an allylglycine residue, together with a C-glycoside bearing an anomeric allyl modification.137 Later, the utility of cross-metathesis to produce larger glycoconjugate peptides in aqueous medium was demonstrated by the group of Davis, as they showed effective cross-metathesis between an S-allyl cysteine containing protein and β-allyl glucose or α-allyl mannose.138However, the most popular metal-catalyzed cross-coupling reaction used for the production of glycopeptide conjugates is the copper-catalyzed azide–alkyne cycloaddition (CuAAC). In this reaction, an alkylic azide reacts with an alkyne group to form a triazole structure. The reaction is mediated by Cu(I) catalysis and compatible with a wide variety of solvents including water and other polar media usable for the dissolution of unprotected peptides. Soon after the reactions’ discovery,139,140 the group of Rutjes produced the first triazole-linked glycopeptide by reacting protected dipeptides, containing the unnatural amino acid propargylglycine, with acetyl-protected 1-azido-glucose and -cellobiose.141 The utility of the reaction for the modification of unprotected peptides was discovered soon after. Walsh produced several variants of the peptide antibiotic tyrocidine containing one or more propargylglycine residues. These were, in unprotected form, conjugated with several unprotected sugars, modified with an anomeric azidoethanol function, displaying for the first time the utility of the CuAAC reaction in the late-stage introduction of glycosylation onto a peptide scaffold.142 This was further exemplified by the work of Davis34 and Brimble,143,144 producing various unnatural glycopeptides.
To further streamline the synthesis of glycoconjugate peptides via CuAAC, novel methods to produce glycosyl azides from reducing sugars were developed. Shoda introduced the reagent 2-chloro-1,3-dimethylimidazolinium chloride (DMC) for the activation of anomeric lactol towards nucleophiles like sodium azide, producing exclusively the β-azide on glucose-configured reducing sugars (Fig. 13A).145 This method enables the use of oligosaccharides isolated from natural sources, like the biantennary N-glycan isolated from egg yolk described in section 2, to be conveniently conjugated to peptides containing one or more alkyne handles. Fairbanks further improved on this approach by utilizing a more efficient reagent for the introduction of the azide, 2-azido-1,3-dimethylimidazolinium hexafluorophosphate (ADMP), and using this approach to synthesize several non-natively glycosylated MUC1 derivatives, among them compound 95, bearing a biantennary sialoglycan (Fig. 13B).146
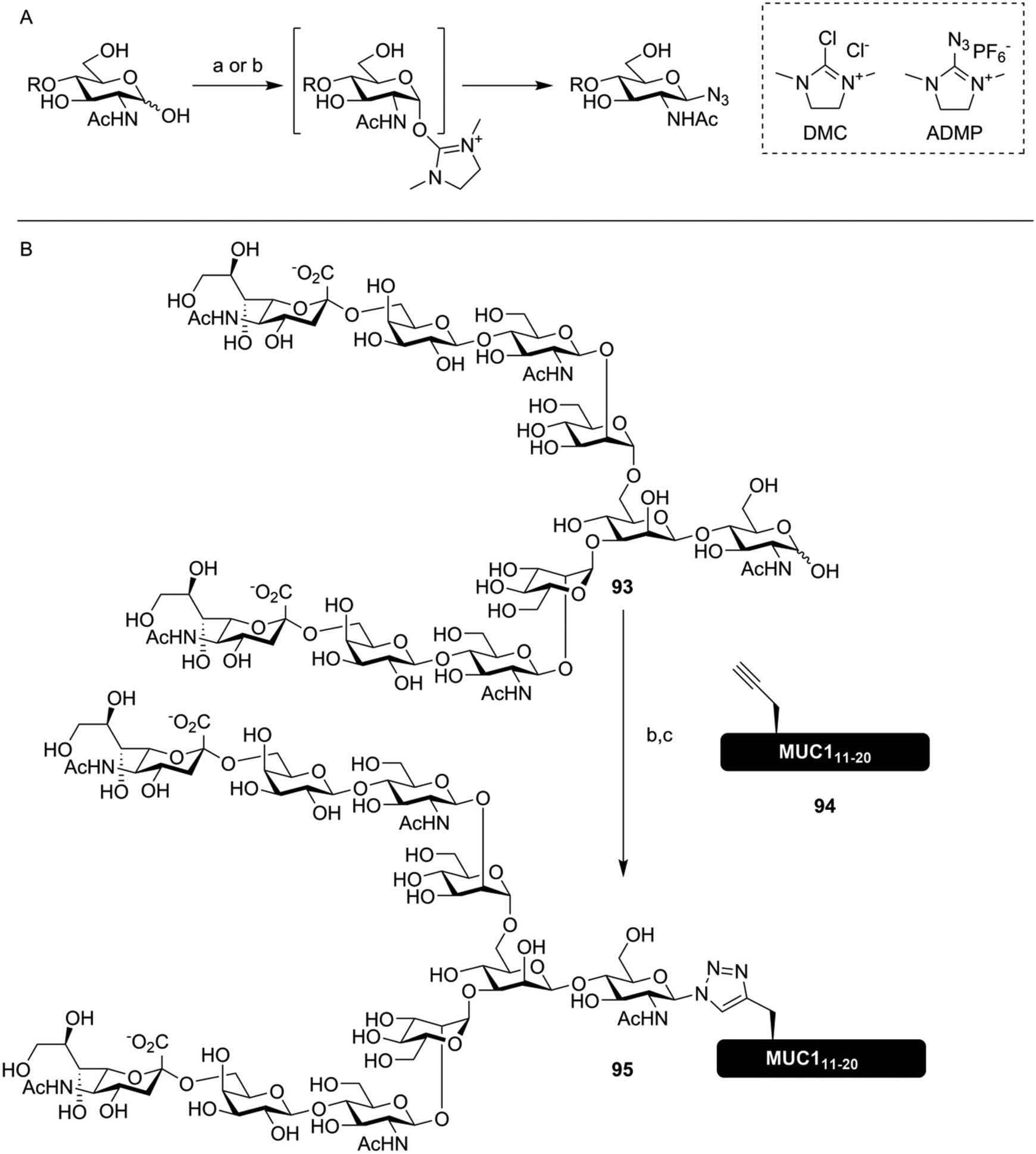 | ||
| Fig. 13 (A) DMC145 or ADMP146 mediated synthesis of azidoglycosides from unprotected sugars in water. (B) One pot synthesis of a glycoconjugate MUC1-peptide (95) using ADMP mediated conversion of lactol 93 into the glycosyl azide. CuAAC conjugation in the same pot with alkyne-containing peptide 94, where Thr16 was replaced with propargylglycine, yielded the desired glycoconjugate.146 Reagents and conditions: (a) DMC, 2,6-lutidine, NaN3, H2O (b) ADMP, Et3N, D2O, MeCN (c) 94, CuSO4, ascorbic acid. | ||
The CuAAC mediated synthesis of glycoconjugates was soon utilized in the synthesis of more glycosylated antigenic peptides. The group of Brimble used CuAAC to synthesize six different MUC1 derived peptides containing 1–5 Tn-antigen mimics, introduced using a late-stage click reaction between unprotected peptides and glycosides (Fig. 14A).147 More recently, CuAAC-based approaches have been used to rapidly produce large libraries of antigenic peptides containing various oligosaccharides, in order to study the role peptide glycosylation has in antigen (cross)-presentation. The group of Codée investigated the binding of mannosylated gp100 antigen, an often studied cancer epitope,148 to the lectins DC-SIGN31 and Langerin.149 To study the effects of both multivalency and carbohydrate configuration, large libraries of glycopeptides bearing 1,2,3 or 6 azide handles, usable for the introduction of alkyne-modified oligomannosides, were produced. While the higher valency conjugates showed an increase in affinity for the targeted receptors, this did not translate into improved presentation of the antigens to T-cells.31,149
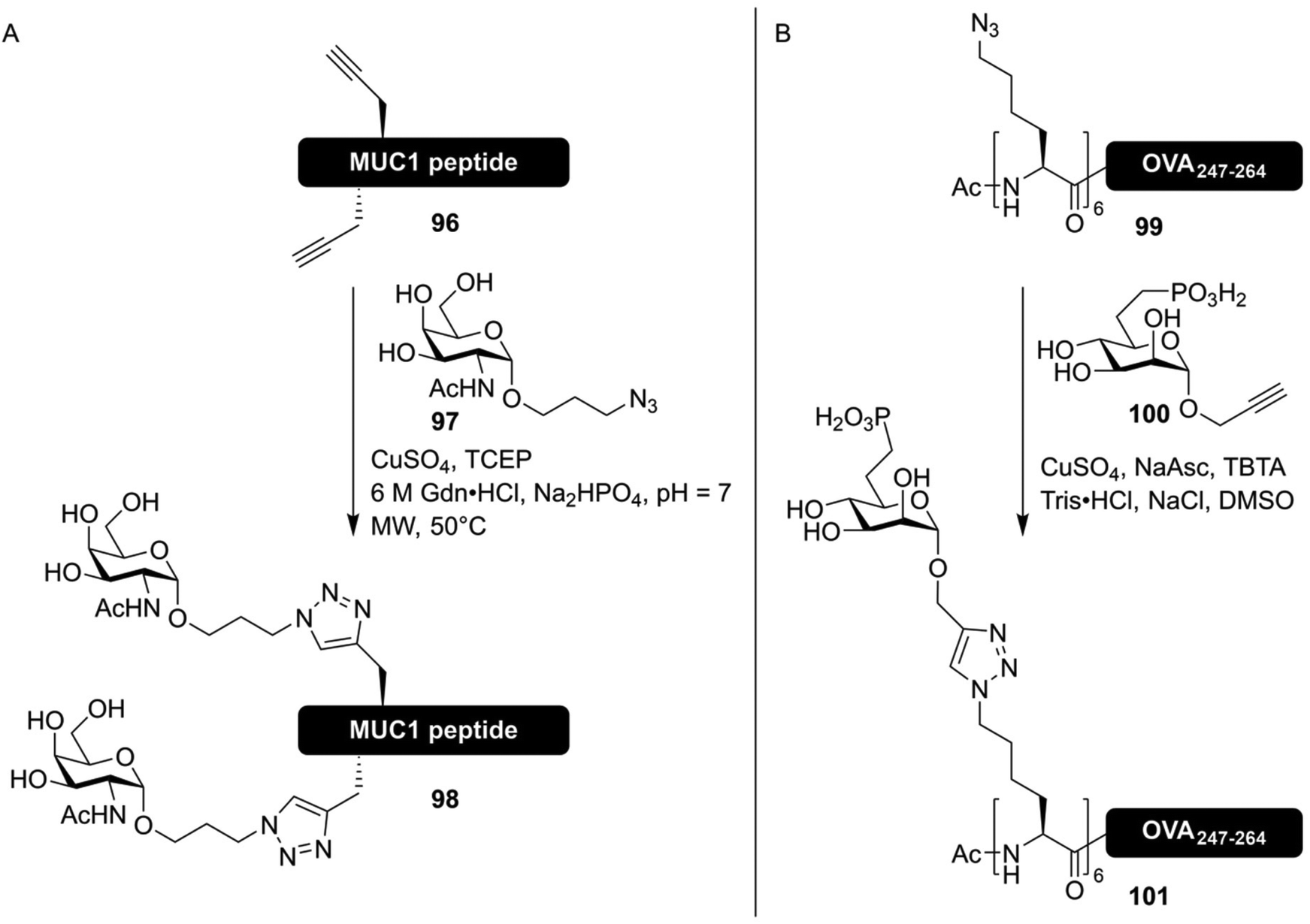 | ||
| Fig. 14 (A) Synthesis of MUC1 derived neo-glycopeptides bearing click-based Tn-antigen mimics by Lee et al.147 Selected threonine and/or serine residues were replaced with propargyl-glycine in the peptide sequence, and a CuAAC reaction with the Tn-antigen mimic 97 facilitated the synthesis of neoglycopeptides. (B) Synthesis of an antigenic peptide derived from chicken ovalbumin bearing a glycocluster of mannose-6-phosphate receptor binding glycans by Reintjes et al.32 By synthesizing a peptide bearing multiple azidonorleucine residues on the N-terminus, followed by CuAAC conjugation using propargyl glycoside 100, the effect of multivalent glycan presentation on antigen uptake could be studied. | ||
In a similar manner, stabilized ligands for the mannose-6-phosphate receptor, in the form of mannose-6-phophonate (100), conjugated to epitopes derived from chicken ovalbumin (99), were produced (Fig. 14B).32 All of these works were based on the synthesis of peptides bearing several azidonorleucine residues, conjugated to different carbohydrates all bearing an anomeric propargyl group.
CuAAC-based neoglycopeptide synthesis strategies have now shown to be able to quickly deliver various unnatural but functional glycoconjugate peptides. It is expected that these methods will be further exploited in the future to elucidate the complex roles of lectins in the immune system.
Conclusions
Over the last few decades, the development of glycopeptides and glycopeptide conjugates has aided our understanding of glycobiology and glyco-immunology. Various glycosylated peptides are being actively studied as potential peptide vaccines, the hypoglycosylated mucin peptides and mannosylated HIV-gp120 derivatives as the most important examples. Glycosylation is also used to enhance peptide uptake and to shape the response of immune cells, by targeting immunolectins with various oligosaccharides. The contemporary development of efficient and rapid glyco-conjugation methods, key among which are the methods utilizing the copper catalyzed click reaction, creates the opportunity to generate many unnatural but functionally interesting glycoconjugates. These in turn enable an ever increasing amount of studies in carbohydrate structure/activity relationships. In the future, these developments could lead to better vaccines and other immune-response shaping synthetic molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. D. was financially supported by a NWO BBoL grant.Notes and references
- K. T. Schjoldager, Y. Narimatsu, H. J. Joshi and H. Clausen, Nat. Rev. Mol. Cell Biol., 2020, 21, 729–749 CrossRef CAS PubMed.
- P. M. Rudd and R. A. Dwek, Crit. Rev. Biochem. Mol. Biol., 1997, 32, 1–100 CrossRef CAS PubMed.
- S. I. Gringhuis, J. den Dunnen, M. Litjens, M. van der Vlist and T. B. H. Geijtenbeek, Nat. Immunol., 2009, 10, 1081–1088 CrossRef CAS PubMed.
- J. Lübbers, E. Rodríguez and Y. van Kooyk, Front. Immunol., 2018, 9, 2807 CrossRef PubMed.
- S. Burgdorf, V. Lukacs-Kornek and C. Kurts, J. Immunol., 2006, 176, 6770–6776 CrossRef CAS PubMed.
- P.-J. Royer, M. Emara, C. Yang, A. Al-Ghouleh, P. Tighe, N. Jones, H. F. Sewell, F. Shakib, L. Martinez-Pomares and A. M. Ghaemmaghami, J. Immunol., 2010, 185, 1522–1531 CrossRef CAS PubMed.
- I. Hafstrand, D. Badia-Martinez, B. J. Josey, M. Norström, J. Buratto, S. Pellegrino, A. D. Duru, T. Sandalova and A. Achour, PLoS One, 2017, 12, e0189584 CrossRef PubMed.
- V. Apostolopoulos, E. Yuriev, P. A. Ramsland, J. Halton, C. Osinski, W. Li, M. Plebanski, H. Paulsen and I. F. C. McKenzie, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15029–15034 CrossRef CAS PubMed.
- G. Rizzuto, J. F. Brooks, S. T. Tuomivaara, T. I. McIntyre, S. Ma, D. Rideaux, J. Zikherman, S. J. Fisher and A. Erlebacher, Nature, 2022, 603, 497–502 CrossRef CAS PubMed.
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. Van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C. H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17033–17038 CrossRef CAS PubMed.
- E. van Liempt, C. M. C. Bank, P. Mehta, J. J. García-Vallejo, Z. S. Kawar, R. Geyer, R. A. Alvarez, R. D. Cummings, Y. van Kooyk and I. van Die, FEBS Lett., 2006, 580, 6123–6131 CrossRef CAS PubMed.
- S. Burgdorf, A. Kautz, V. Böhnert, P. A. Knolle and C. Kurts, Science, 2007, 316, 612–616 CrossRef CAS PubMed.
- J. Rauen, C. Kreer, A. Paillard, S. Van Duikeren, W. E. Benckhuijsen, M. G. Camps, A. R. P. M. Valentijn, F. Ossendorp, J. W. Drijfhout, R. Arens and S. Burgdorf, PLoS One, 2014, 9, e103755 CrossRef PubMed.
- M. Anderluh, F. Berti, A. Bzducha-Wróbel, F. Chiodo, C. Colombo, F. Compostella, K. Durlik, X. Ferhati, R. Holmdahl, D. Jovanovic, W. Kaca, L. Lay, M. Marinovic-Cincovic, M. Marradi, M. Ozil, L. Polito, J. J. Reina, C. A. Reis, R. Sackstein, A. Silipo, U. Švajger, O. Vaněk, F. Yamamoto, B. Richichi and S. J. van Vliet, FEBS J. DOI:10.1111/febs.15909.
- L. Galli-Stampino, E. Meinjohanns, K. Frische, M. Meldal, T. Jensen, O. Werdelin and S. Mouritsen, Cancer Res., 1997, 57, 3214–3222 CAS.
- C. Soliman, E. Yuriev and P. A. Ramsland, Curr. Opin. Struct. Biol., 2017, 44, 1–8 CrossRef CAS PubMed.
- A. Fernández-Tejada, B. F. Haynes and S. J. Danishefsky, Expert Rev. Vaccines, 2015, 14, 815–831 CrossRef PubMed.
- I. Brockhausen and J. Melamed, Glycoconjugate J., 2021, 38, 459–474 CrossRef CAS PubMed.
- D. Dutta, C. Mandal and C. Mandal, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3096–3108 CrossRef CAS PubMed.
- R. G. Spiro, Glycobiology, 2002, 12, 43R–56R CrossRef CAS PubMed.
- X. Yang and K. Qian, Nat. Rev. Mol. Cell Biol., 2017, 18, 452–465 CrossRef CAS PubMed.
- J. S. Haurum, I. B. Høier, G. Arsequell, A. Neisig, G. Valencia, J. Zeuthen, J. Neefjes and T. Elliott, J. Exp. Med., 1999, 190, 145–150 CrossRef CAS PubMed.
- H. H. Wandall, M. A. I. Nielsen, S. King-Smith, N. de Haan and I. Bagdonaite, FEBS J., 2021, 288, 7183–7212 CrossRef CAS PubMed.
- S. Chugh, V. S. Gnanapragassam, M. Jain, S. Rachagani, M. P. Ponnusamy and S. K. Batra, Biochim. Biophys. Acta, Rev. Cancer, 2015, 1856, 211–225 CrossRef CAS PubMed.
- A. R. Kumar, A. R. Devan, B. Nair, B. S. Vinod and L. R. Nath, Mol. Biol. Rep., 2021, 48, 8075–8095 CrossRef CAS PubMed.
- M. Aebi, R. Bernasconi, S. Clerc and M. Molinari, Trends Biochem. Sci., 2010, 35, 74–82 CrossRef CAS PubMed.
- C. M. Fehres, H. Kalay, S. C. M. Bruijns, S. A. M. Musaafir, M. Ambrosini, L. Van Bloois, S. J. Van Vliet, G. Storm, J. J. Garcia-Vallejo and Y. Van Kooyk, J. Controlled Release, 2015, 203, 67–76 CrossRef CAS PubMed.
- N. Frison, M. E. Taylor, E. Soilleux, M.-T. Bousser, R. Mayer, M. Monsigny, K. Drickamer and A.-C. Roche, J. Biol. Chem., 2003, 278, 23922–23929 CrossRef CAS PubMed.
- W. Doelman, M. H. S. Marqvorsen, F. Chiodo, S. C. M. Bruijns, G. A. van der Marel, Y. van Kooyk, S. I. van Kasteren and C. Araman, Chem. – Eur. J., 2021, 27, 2742–2752 CrossRef CAS PubMed.
- R. Riera, T. P. Hogervorst, W. Doelman, Y. Ni, S. Pujals, E. Bolli, J. D. C. Codée, S. I. van Kasteren and L. Albertazzi, Nat. Chem. Biol., 2021, 17, 1281–1288 CrossRef CAS PubMed.
- R. J. E. Li, T. P. Hogervorst, S. Achilli, S. C. Bruijns, T. Arnoldus, C. Vivès, C. C. Wong, M. Thépaut, N. J. Meeuwenoord, H. van den Elst, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov, S. J. van Vliet, F. Fieschi, J. D. C. Codée and Y. van Kooyk, Front. Chem., 2019, 7, 650 CrossRef CAS PubMed.
- N. R. M. Reintjens, E. Tondini, C. Vis, T. McGlinn, N. J. Meeuwenoord, T. P. Hogervorst, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel, F. Ossendorp, G. A. van der Marel, F. Ossendorp and J. D. C. Codée, ChemBioChem, 2021, 22, 434–440 CrossRef CAS PubMed.
- D. M. Beckwith and M. Cudic, Semin. Immunol., 2020, 47, 101389 CrossRef CAS PubMed.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105–1109 CrossRef CAS PubMed.
- J. J. García-Vallejo, M. Ambrosini, A. Overbeek, W. E. van Riel, K. Bloem, W. W. J. Unger, F. Chiodo, J. G. Bolscher, K. Nazmi, H. Kalay and Y. van Kooyk, Mol. Immunol., 2013, 53, 387–397 CrossRef PubMed.
- M. Rentzsch, R. Wawrzinek, C. Zelle-Rieser, H. Strandt, L. Bellmann, F. F. Fuchsberger, J. Schulze, J. Busmann, J. Rademacher, S. Sigl, B. Del Frari, P. Stoitzner and C. Rademacher, Front. Immunol., 2021, 12, 4338 Search PubMed.
- H. Kunz and C. Unverzagt, Angew. Chem., Int. Ed. Engl., 1988, 27, 1697–1699 CrossRef.
- L. Urge, L. Otvos, E. Lang, K. Wroblewski, I. Laczko and M. Hollosi, Carbohydr. Res., 1992, 235, 83–93 CrossRef CAS PubMed.
- S. Ito, Y. Asahina and H. Hojo, Tetrahedron, 2021, 97, 132423 CrossRef CAS.
- Y. Zhang, S. M. Muthana, J. J. Barchi and J. C. Gildersleeve, Org. Lett., 2012, 14, 3958–3961 CrossRef CAS PubMed.
- Y. Zhang, S. M. Muthana, D. Farnsworth, O. Ludek, K. Adams, J. J. Barchi and J. C. Gildersleeve, J. Am. Chem. Soc., 2012, 134, 6316–6325 CrossRef CAS PubMed.
- H. Paulsen, G. Merz and U. Weichert, Angew. Chem., Int. Ed. Engl., 1988, 27, 1365–1367 CrossRef.
- B. Liebe and H. Kunz, Angew. Chem., Int. Ed. Engl., 1997, 36, 618–621 CrossRef CAS.
- T. Bielfeldt, S. Peters, M. Meldal, K. Bock and H. Paulsen, Angew. Chem., Int. Ed. Engl., 1992, 31, 857–859 CrossRef.
- H. Kauth and H. Kunz, Liebigs Ann. Chem., 1983, 1983, 360–366 CrossRef.
- H. Paulsen, M. Paal and M. Schultz, Tetrahedron Lett., 1983, 24, 1759–1762 CrossRef CAS.
- H. Kunz and S. Birnbach, Angew. Chem., Int. Ed. Engl., 1986, 25, 360–362 CrossRef.
- R. Bhatia, S. K. Gautam, A. Cannon, C. Thompson, B. R. Hall, A. Aithal, K. Banerjee, M. Jain, J. C. Solheim, S. Kumar and S. K. Batra, Cancer Metastasis Rev., 2019, 38, 223–236 CrossRef CAS PubMed.
- M. Elofsson, L. A. Salvador and J. Kihlberg, Tetrahedron, 1997, 53, 369–390 CrossRef CAS.
- V. Yoon, M. Fridkis-Hareli, S. Munisamy, J. Lee, D. Anastasiades and L. Stevceva, Curr. Med. Chem., 2010, 17, 741–749 CrossRef CAS PubMed.
- J. B. Schwarz, S. D. Kuduk, X. T. Chen, D. Sames, P. W. Glunz and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 2662–2673 CrossRef CAS.
- P. W. Glunz, S. Hintermann, L. J. Williams, J. B. Schwarz, S. D. Kuduk, V. Kudryashov, K. O. Lloyd and S. J. Danishefsky, J. Am. Chem. Soc., 2000, 122, 7273–7279 CrossRef CAS.
- C. Brocke and H. Kunz, Synlett, 2003, 2052–2056 CAS.
- C. Pett, M. Schorlemer and U. Westerlind, Chem. – Eur. J., 2013, 19, 17001–17010 CrossRef CAS PubMed.
- C. Pett and U. Westerlind, Chem. – Eur. J., 2014, 20, 7287–7299 CrossRef CAS PubMed.
- C. Pett, H. Cai, J. Liu, B. Palitzsch, M. Schorlemer, S. Hartmann, N. Stergiou, M. Lu, H. Kunz, E. Schmitt and U. Westerlind, Chem. – Eur. J., 2017, 23, 3875–3884 CrossRef CAS PubMed.
- W. S. Somers, J. Tang, G. D. Shaw and R. T. Camphausen, Cell, 2000, 103, 467–479 CrossRef CAS PubMed.
- K. Baumann, D. Kowalczyk and H. Kunz, Angew. Chem., Int. Ed., 2008, 47, 3445–3449 CrossRef CAS PubMed.
- N. Takeda, T. Takei, Y. Asahina and H. Hojo, Chem. – Eur. J., 2018, 24, 2593–2597 CrossRef CAS PubMed.
- Y. Asahina, T. Kawakami and H. Hojo, Eur. J. Org. Chem., 2019, 2019, 1915–1920 CrossRef CAS.
- C. Toonstra, M. N. Amin and L. X. Wang, J. Org. Chem., 2016, 81, 6176–6185 CrossRef CAS PubMed.
- B. Yan, W. Li and C. P. R. Hackenberger, Org. Biomol. Chem., 2021, 19, 8014–8017 RSC.
- I. Matsuo, Y. Nakahara, Y. Ito, T. Nukada, Y. Nakahara and T. Ogawa, Bioorg. Med. Chem., 1995, 3, 1455–1463 CrossRef CAS PubMed.
- C. Unverzagt, Carbohydr. Res., 1997, 305, 423–431 CrossRef CAS PubMed.
- C. Unverzagt, Tetrahedron Lett., 1997, 38, 5627–5630 CrossRef CAS.
- S. Mezzato, M. Schaffrath and C. Unverzagt, Angew. Chem., Int. Ed., 2005, 44, 1650–1654 CrossRef CAS PubMed.
- N. Yamamoto, Y. Ohmori, T. Sakakibara, K. Sasaki, L. R. Juneja and Y. Kajihara, Angew. Chem., Int. Ed., 2003, 42, 2537–2540 CrossRef CAS PubMed.
- C. Piontek, D. Varón Silva, C. Heinlein, C. Pöhner, S. Mezzato, P. Ring, A. Martin, F. X. Schmid and C. Unverzagt, Angew. Chem., Int. Ed., 2009, 48, 1941–1945 CrossRef CAS PubMed.
- S. T. Anisfeld and P. T. Lansbury, J. Org. Chem., 2002, 55, 5560–5562 CrossRef.
- S. T. Cohen-Anisfeld and P. T. Lansbury, J. Am. Chem. Soc., 2002, 115, 10531–10537 CrossRef.
- P. Wang, B. Aussedat, Y. Vohra and S. J. Danishefsky, Angew. Chem., Int. Ed., 2012, 51, 11571–11575 CrossRef CAS PubMed.
- P. Wang, S. Dong, J. H. Shieh, E. Peguero, R. Hendrickson, M. A. S. Moore and S. J. Danishefsky, Science, 2013, 342, 1357–1360 CrossRef CAS PubMed.
- S. M. Alam, B. Aussedat, Y. Vohra, R. R. Meyerhoff, E. M. Cale, W. E. Walkowicz, N. A. Radakovich, K. Anasti, L. Armand, R. Parks, L. Sutherland, R. Scearce, M. G. Joyce, M. Pancera, A. Druz, I. S. Georgiev, T. Von Holle, A. Eaton, C. Fox, S. G. Reed, M. Louder, R. T. Bailer, L. Morris, S. S. Abdool-Karim, M. Cohen, H. X. Liao, D. C. Montefiori, P. K. Park, A. Fernández-Tejada, K. Wiehe, S. Santra, T. B. Kepler, K. O. Saunders, J. Sodroski, P. D. Kwong, J. R. Mascola, M. Bonsignori, M. A. Moody, S. Danishefsky and B. F. Haynes, Sci. Transl. Med., 2017, 19(381) DOI:10.1126/scitranslmed.aai7521.
- A. J. Fairbanks, Chem. Soc. Rev., 2017, 46, 5128–5146 RSC.
- K. Yamamoto, S. Kadowaki, J. Watanabe and H. Kumagai, Biochem. Biophys. Res. Commun., 1994, 203, 244–252 CrossRef CAS PubMed.
- M. Fujita, S. Shoda, K. Haneda, T. Inazu, K. Takegawa and K. Yamamoto, Biochim. Biophys. Acta, Gen. Subj., 2001, 1528, 9–14 CrossRef CAS.
- M. Umekawa, W. Huang, B. Li, K. Fujita, H. Ashida, L. X. Wang and K. Yamamoto, J. Biol. Chem., 2008, 283, 4469–4479 CrossRef CAS PubMed.
- M. Umekawa, C. Li, T. Higashiyama, W. Huang, H. Ashida, K. Yamamoto and L. X. Wang, J. Biol. Chem., 2010, 285, 511–521 CrossRef CAS PubMed.
- J. D. McIntosh, M. A. Brimble, A. E. S. Brooks, P. R. Dunbar, R. Kowalczyk, Y. Tomabechi and A. J. Fairbanks, Chem. Sci., 2015, 6, 4636–4642 RSC.
- H. Li, B. Li, H. Song, L. Breydo, I. V. Baskakov and L. X. Wang, J. Org. Chem., 2005, 70, 9990–9996 CrossRef CAS PubMed.
- T. Yamaguchi, M. N. Amin, C. Toonstra and L. X. Wang, J. Am. Chem. Soc., 2016, 138, 12472–12485 CrossRef PubMed.
- P. Priyanka, T. B. Parsons, A. Miller, F. M. Platt and A. J. Fairbanks, Angew. Chem., 2016, 128, 5142–5145 CrossRef.
- H. Ochiai, W. Huang and L. X. Wang, J. Am. Chem. Soc., 2008, 130, 13790–13803 CrossRef CAS PubMed.
- M. Kurogochi, M. Mori, K. Osumi, M. Tojino, S. I. Sugawara, S. Takashima, Y. Hirose, W. Tsukimura, M. Mizuno, J. Amano, A. Matsuda, M. Tomita, A. Takayanagi, S. I. Shoda and T. Shirai, PLoS One, 2015, 10, e0132848 CrossRef PubMed.
- C. W. Lin, M. H. Tsai, S. T. Li, T. I. Tsai, K. C. Chu, Y. C. Liu, M. Y. Lai, C. Y. Wu, Y. C. Tseng, S. S. Shivatare, C. H. Wang, P. Chao, S. Y. Wang, H. W. Shih, Y. F. Zeng, T. H. You, J. Y. Liao, Y. C. Tu, Y. S. Lin, H. Y. Chuang, C. L. Chen, C. S. Tsai, C. C. Huang, N. H. Lin, C. Ma, C. Y. Wu and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10611–10616 CrossRef CAS PubMed.
- A. Seko, M. Koketsu, M. Nishizono, Y. Enoki, H. R. Ibrahim, L. R. Juneja, M. Kim and T. Yamamoto, Biochim. Biophys. Acta, Gen. Subj., 1997, 1335, 23–32 CrossRef CAS.
- H. Li, S. Singh, Y. Zeng, H. Song and L. X. Wang, Bioorg. Med. Chem. Lett., 2005, 15, 895–898 CrossRef CAS PubMed.
- W. Huang, C. Li, B. Li, M. Umekawa, K. Yamamoto, X. Zhang and L. X. Wang, J. Am. Chem. Soc., 2009, 131, 2214–2223 CrossRef CAS PubMed.
- W. Huang, Q. Yang, M. Umekawa, K. Yamamoto and L. X. Wang, ChemBioChem, 2010, 11, 1350–1355 CrossRef CAS PubMed.
- M. Murakami, R. Okamoto, M. Izumi, Y. Kajihara, M. Murakami, R. Okamoto, M. Izumi and Y. Kajihara, Angew. Chem., Int. Ed., 2012, 51, 3567–3572 CrossRef CAS PubMed.
- N. Yamamoto, A. Takayanagi, T. Sakakibara, P. E. Dawson and Y. Kajihara, Tetrahedron Lett., 2006, 47, 1341–1346 CrossRef CAS.
- Y. Kajihara, A. Yoshihara, K. Hirano and N. Yamamoto, Carbohydr. Res., 2006, 341, 1333–1340 CrossRef CAS PubMed.
- D. L. Evers, R. L. Hung, V. H. Thomas and K. G. Rice, Anal. Biochem., 1998, 265, 313–316 CrossRef CAS PubMed.
- L.-X. Wang, J. Ni, S. Singh and H. Li, Chem. Biol., 2004, 11, 127–134 CAS.
- Y. Makimura, T. Kiuchi, M. Izumi, S. Dedola, Y. Ito and Y. Kajihara, Carbohydr. Res., 2012, 364, 41–48 CrossRef CAS PubMed.
- L. X. Wang, H. Song, S. Liu, H. Lu, S. Jiang, J. Ni and H. Li, ChemBioChem, 2005, 6, 1068–1074 CrossRef CAS PubMed.
- T. Kiuchi, M. Izumi, Y. Mukogawa, A. Shimada, R. Okamoto, A. Seko, M. Sakono, Y. Takeda, Y. Ito and Y. Kajihara, J. Am. Chem. Soc., 2018, 140, 17499–17507 CrossRef CAS PubMed.
- C. Li, S. Zhu, C. Ma and L. X. Wang, J. Am. Chem. Soc., 2017, 139, 15074–15087 CrossRef CAS PubMed.
- Y. Kajihara, Y. Suzuki, N. Yamamoto, K. Sasaki, T. Sakakibara and L. R. Juneja, Chem. – Eur. J., 2004, 10, 971–985 CrossRef CAS PubMed.
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. De Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379–383 CrossRef CAS PubMed.
- L. Liu, A. R. Prudden, C. J. Capicciotti, G. P. Bosman, J. Y. Yang, D. G. Chapla, K. W. Moremen and G. J. Boons, Nat. Chem., 2019, 11, 161–169 CrossRef CAS PubMed.
- H. Malekan, G. Fung, V. Thon, Z. Khedri, H. Yu, J. Qu, Y. Li, L. Ding, K. S. Lam and X. Chen, Bioorg. Med. Chem., 2013, 21, 4778–4785 CrossRef CAS PubMed.
- A. M. Vibhute, N. Komura, H. N. Tanaka, A. Imamura and H. Ando, Chem. Rec., 2021, 21, 3194–3223 CrossRef CAS PubMed.
- A. Leppänen, P. Mehta, Y. Ouyang, T. Ju, J. Helin, K. L. Moore, I. Van Die, W. M. Canfield, R. P. McEver and R. D. Cummings, J. Biol. Chem., 1999, 274, 24838–24848 CrossRef PubMed.
- A. L. Sørensen, C. A. Reis, M. A. Tarp, U. Mandel, K. Ramachandran, V. Sankaranarayanan, T. Schwientek, R. Graham, J. Taylor-Papadimitriou, M. A. Hollingsworth, J. Burchell and H. Clausen, Glycobiology, 2006, 16, 96–107 CrossRef PubMed.
- M. A. Tarp, A. L. Sørensen, U. Mandel, H. Paulsen, J. Burchell, J. Taylor-Papadimitriou and H. Clausen, Glycobiology, 2007, 17, 197–209 CrossRef CAS PubMed.
- C. A. De Leon, P. M. Levine, T. W. Craven and M. R. Pratt, Biochemistry, 2017, 56, 3507–3517 CrossRef CAS PubMed.
- C. Filser, D. Kowalczyk, C. Jones, M. K. Wild, U. Ipe, D. Vestweber and H. Kunz, Angew. Chem., Int. Ed., 2007, 46, 2108–2111 CrossRef CAS PubMed.
- U. Sprengard, M. Schudok, W. Schmidt, G. Kretzschmar and H. Kunz, Angew. Chem., Int. Ed. Engl., 1996, 35, 321–324 CrossRef CAS.
- K. von dem Bruch and H. Kunz, Angew. Chem., Int. Ed. Engl., 1994, 33, 101–103 CrossRef.
- J. März and H. Kunz, Synlett, 1992, 589–590 CrossRef.
- J. J. García-Vallejo, J. M. Ilarregui, H. Kalay, S. Chamorro, N. Koning, W. W. Unger, M. Ambrosini, V. Montserrat, R. J. Fernandes, S. C. M. Bruijns, J. R. T. van Weering, N. J. Paauw, T. O'Toole, J. van Horssen, P. van der Valk, K. Nazmi, J. G. M. Bolscher, J. Bajramovic, C. D. Dijkstra, B. A. ‘t Hart and Y. van Kooyk, J. Exp. Med., 2014, 211, 1465–1483 CrossRef PubMed.
- T. Gustafsson, M. Hedenström and J. Kihlberg, J. Org. Chem., 2006, 71, 1911–1919 CrossRef CAS PubMed.
- M. J. Gramer, C. F. Goochee, V. Y. Chock, D. T. Brousseau and M. B. Sliwkowski, Bio/Technology, 1995, 13, 692–698 CAS.
- I. Companión, A. Guerreiro, V. Mangini, J. Castro-López, M. Escudero-Casao, A. Avenoza, J. H. Busto, S. Castillón, J. Jiménez-Barbero, J. L. Asensio, G. Jiménez-Osés, O. Boutureira, J. M. Peregrina, R. Hurtado-Guerrero, R. Fiammengo, G. J. L. Bernardes and F. Corzana, J. Am. Chem. Soc., 2019, 141, 4063–4072 CrossRef PubMed.
- E. Michaëlsson, M. Andersson, R. Holmdahl and Å. Engström, Eur. J. Immunol., 1992, 22, 1819–1825 CrossRef PubMed.
- J. Broddefalk, J. Bäcklund, F. Almqvist, M. Johansson, R. Hoimdahl and J. Kihlberg, J. Am. Chem. Soc., 1998, 120, 7676–7683 CrossRef CAS.
- T. P. Hogervorst, R. J. E. Li, L. Marino, S. C. M. Bruijns, N. J. Meeuwenoord, D. V. Filippov, H. S. Overkleeft, G. A. Van Der Marel, S. J. Van Vliet, Y. Van Kooyk and J. D. C. Codeé, ACS Chem. Biol., 2020, 15, 728–739 CrossRef CAS PubMed.
- E. A. B. Kantchev, C. C. Chang and D. K. Chang, Biopolymers (Peptide Science), 2006, 84, 232–240 CrossRef CAS PubMed.
- N. Frison, P. Marceau, A. C. Roche, M. Monsigny and R. Mayer, Biochem. J., 2002, 368, 111 CrossRef CAS PubMed.
- O. Srinivas, P. Larrieu, E. Duverger, C. Boccaccio, M. T. Bousser, M. Monsigny, J. F. Fonteneau, F. Jotereau and A. C. Roche, Bioconjugate Chem., 2007, 18, 1547–1554 CrossRef CAS PubMed.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- M. Bengtsson, J. Broddefalk, J. Dahmén, K. Henriksson, J. Kihlberg, H. Lönn, B. R. Srinivasa and K. Stenvall, Glycoconjugate J., 1998, 15, 223–231 CrossRef CAS PubMed.
- I. Shin, H. J. Jung and M. R. Lee, Tetrahedron Lett., 2001, 42, 1325–1328 CrossRef CAS.
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., 2004, 116, 846–851 CrossRef.
- W. M. Macindoe, A. H. Van Oijen and G. J. Boons, Chem. Commun., 1998, 847–848 RSC.
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 1060 Search PubMed.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem. – Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- G. Piccirillo, A. Pepe, E. Bedini and B. Bochicchio, Chem. – Eur. J., 2017, 23, 2648–2659 CrossRef CAS PubMed.
- S. R. Alexander, D. Lim, Z. Amso, M. A. Brimble and A. J. Fairbanks, Org. Biomol. Chem., 2017, 15, 2152–2156 RSC.
- D. P. Galonić, W. A. Van Der Donk and D. Y. Gin, J. Am. Chem. Soc., 2004, 126, 12712–12713 CrossRef PubMed.
- S. Peluso and B. Imperiali, Tetrahedron Lett., 2001, 42, 2085–2087 CrossRef CAS.
- P. Finch and Z. Merchant, J. Chem. Soc., Perkin Trans. 1, 1975, 1682–1686 RSC.
- M. R. Carrasco, M. J. Nguyen, D. R. Burnell, M. D. MacLaren and S. M. Hengel, Tetrahedron Lett., 2002, 43, 5727–5729 CrossRef CAS.
- F. Peri, P. Dumy and M. Mutter, Tetrahedron, 1998, 54, 12269–12278 CrossRef CAS.
- E. C. Rodriguez, L. A. Marcaurelle and C. R. Bertozzi, J. Org. Chem., 1998, 63, 7134–7135 CrossRef CAS PubMed.
- G. J. McGarvey, T. E. Benedum and F. W. Schmidtmann, Org. Lett., 2002, 4, 3591–3594 CrossRef CAS PubMed.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- B. H. M. Kuijpers, S. Groothuys, A. R. Keereweer, P. J. L. M. Quaedflieg, R. H. Blaauw, F. L. Van Delft and F. P. J. T. Rutjes, Org. Lett., 2004, 6, 3123–3126 CrossRef CAS PubMed.
- H. Lin and C. T. Walsh, J. Am. Chem. Soc., 2004, 126, 13998–14003 CrossRef CAS PubMed.
- D. J. Lee, K. Mandai, P. W. R. Harris, M. A. Brimble and S. B. H. Kent, Org. Lett., 2009, 11, 5270–5273 CrossRef CAS PubMed.
- N. Miller, G. M. Williams and M. A. Brimble, Org. Lett., 2009, 11, 2409–2412 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S. I. Shoda, Chem. Commun., 2009, 3378–3379 RSC.
- D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., 2014, 126, 12101–12105 CrossRef.
- D. J. Lee, P. W. R. Harris and M. A. Brimble, Org. Biomol. Chem., 2011, 9, 1621–1626 RSC.
- Y. Kawakami and S. A. Rosenberg, Int. Rev. Immunol., 1997, 14, 173–192 CrossRef CAS PubMed.
- R. J. E. Li, T. P. Hogervorst, S. Achilli, S. C. M. Bruijns, S. Spiekstra, C. Vivès, M. Thépaut, D. V. Filippov, G. A. van der Marel, S. J. van Vliet, F. Fieschi, J. D. C. Codée and Y. van Kooyk, Front. Cell Dev. Biol., 2020, 8, 556 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |