
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic total synthesis of (−)-galanthamine via intramolecular anodic aryl–phenol coupling†
Ziyue
Xiong
,
Frauke
Weidlich
,
Camille
Sanchez
and
Thomas
Wirth
 *
*
School of Chemistry, Cardiff University, Park Place, Main Building, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: wirth@cf.ac.uk
First published on 5th May 2022
Abstract
(−)-Galanthamine as a drug for the treatment of Alzheimer's disease has attracted synthetic chemists for decades. However, previous total synthetic and biomimetic approaches often use stoichiometric oxidants (metal oxidants or hypervalent iodine) to prepare the target product. Anodic oxidative coupling offers a sustainable alternative method which is, for the first time, successfully applied to the total synthesis of (−)-galanthamine. We report a new asymmetric total synthesis of (−)-galanthamine by using an anodic aryl–phenol coupling as the key synthetic step.
Introduction
(−)-Galanthamine 1 (Fig. 1), an Amaryllidaceae alkaloid, has attracted the focus of synthetic chemists since 1952 when it was first isolated from the plant Caucasian snowdrop.1 (−)-Galanthamine 1 has an interesting and synthetically challenging structure which contains a strained tetracyclic framework with an all-carbon quaternary stereocentre and has a unique activity as an acetylcholine esterase (AChE) inhibitor.2 In 2001, (−)-galanthamine was approved by the FDA as a drug for the symptomatic treatment of Alzheimer's disease.3 | ||
| Fig. 1 (−)-Galanthamine 1 and (−)-Narwedine 2. | ||
Numerous synthetic studies of (−)-galanthamine and its analogues have been performed and a number of total syntheses of 1 have been reported.4–11 In the early stages of (−)-galanthamine syntheses, the biomimetic intramolecular oxidative coupling was the key step to build up the tetracyclic skeleton.4 These methods typically require stoichiometric metal or hypervalent iodine reagents as oxidants and provide the products in only low to moderate yields. The stereoselectivity of these methods mainly rely on a resolution process where the precursor (−)-Narwedine 2 is crystallized in 70–80% yield (Fig. 1).5 Aside from the biomimetic intramolecular coupling protocol, Trost and co-workers reported an intramolecular Heck reaction to assemble a tricyclic precursor and the associated quaternary carbon centre of galanthamine.6a Inspired by his work, several related approaches have been published later.6 These approaches focus on forming the ABC ring system first before assembling the D ring.5,7 Two recent examples achieved a final construction of the C ring after installing the ABD ring skeleton.8 A recent report by Xu and co-workers uses a Rh-catalyzed C–C activation to form the tetracyclic carbon framework directly,9 while Brown10 and Zhao11 have used other transition-metal mediated approaches. Although a variety of synthetic strategies have been reported so far, the commercial supplies of 1 are still using the Fröhlich–Jordis route4f in which the key phenolic oxidation coupling reaction proceeds in 40–54% yield by using 2 equivalents K3Fe(CN)6 as the oxidant. A similar approach has been reported by electrochemically regenerating the ferrocyanide oxidant.12
In 1984, Vlahov and co-workers reported an electrochemical method for oxidative phenol couplings to synthesise products with a galanthamine skeleton.13 Recently Opatz and co-workers have published the total syntheses of (−)-Thebaine14 and (−)-Oxycodone15 by using an anodic aryl–aryl coupling as the key step. However, more than 3 decades have passed and an electrochemical coupling reaction has still not been successfully applied to the total synthesis of (−)-galanthamine. Herein, we report the optimisation of an anodic aryl–aryl coupling reaction using an effective electrochemical approach with a recently developed electrochemical flow reactor16 and its application to the asymmetric synthesis of (−)-galanthamine 1.
Electrochemical synthesis offers a tunable, cost effective and environmentally friendly alternative to carry out redox reactions using only electrons as traceless reagents, thus obviating the need for dangerous and toxic stoichiometric oxidants.17 Building on the advantages of flow electrochemical approaches,18 we have initially developed a simple and short synthetic plan (Scheme 1).
 | ||
| Scheme 1 Retrosynthetic strategy to access (−)-galanthamine 1 using an anodic aryl–aryl coupling reaction. | ||
It is based around the key norbelladine intermediate 4 which has a methylester19 substituent designed to set up the required stereochemistry by the intramolecular anodic aryl–aryl coupling reaction leading to compound 3. We sought to access 1 from the (−)-Narwedine derivative 3 through removal of the methylester, reduction of the enone and amide moieties of 3 followed by an N-methylation. The norbelladine intermediate 4 could be obtained by the reductive amination of commercially available methyl D-tyrosine 5 and isovanillin 6.
Results and discussion
Initial synthetic investigations began with the much cheaper L-tyrosine and compound ent-4 was obtained by following Yamada's work.19 However, the electrolysis of ent-4 under flow conditions generated only the para–para′-coupled product 7 while the desired para–ortho′-coupled product ent-8 was not observed (Scheme 2a). In order to block the apparently favored para position, the electrolysis substrate was modified to 9 inspired by Vlahov's substrate.13 Unfortunately, all attempted electrolysis reactions of 9 failed to give any products (Scheme 2b). Probably due to the difference in equipment, even Vlahov's work could not be repeated in our hands. Considering the Br group may deactivate the aromatic ring for the coupling reaction, we then redesigned the substrate to ent-11 which contains a 3′,4′,5′-trioxygenated aromatic moiety (Scheme 2c). This was inspired by Node's approach,4g where the symmetrical configuration makes both coupling positions identical to generate a single product.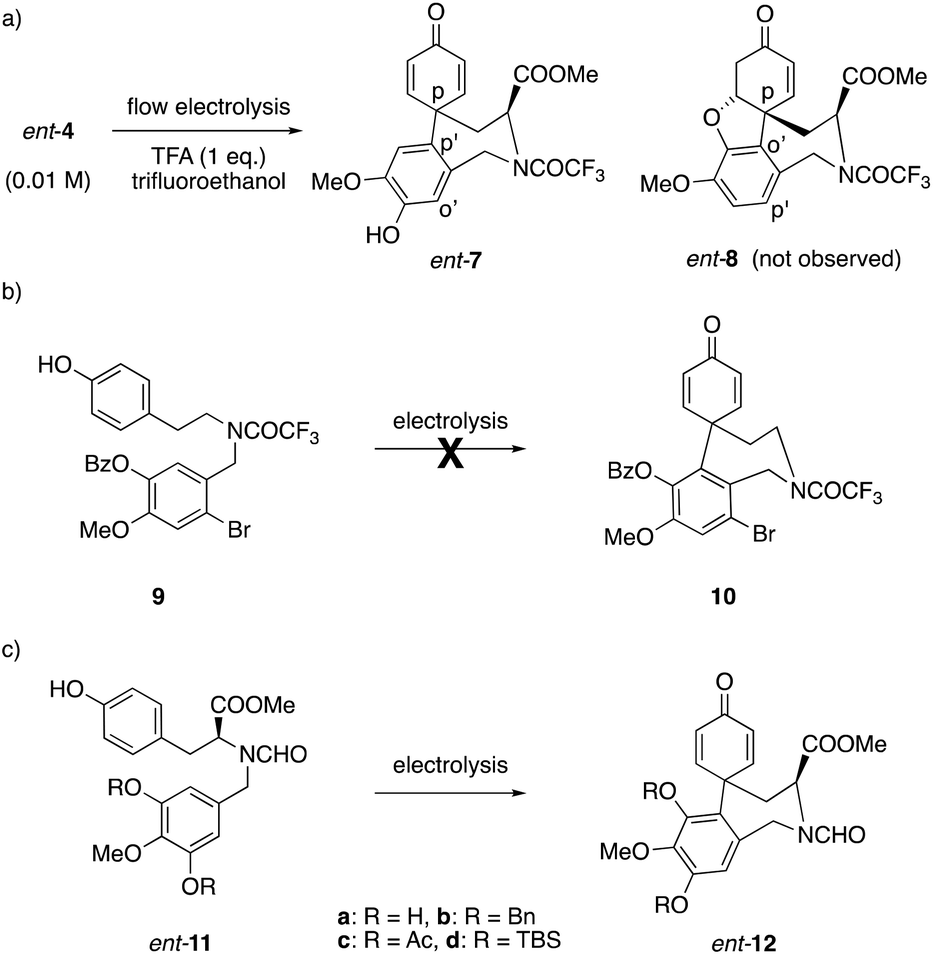 | ||
| Scheme 2 (a) Initial anodic coupling of norbelladine derivative ent-4; (b) unsuccessful anodic coupling attempts of substrate 9; (c) coupling of 3′,4′,5′-trioxygenated norbelladine derivatives 11. | ||
For the first attempt, a batch electrolysis of ent-11b (c = 0.02 M) in MeCN with 0.1 M nBu4NClO4 as supporting electrolyte (graphite anode and platinum cathode) was performed and 8% of product ent-12b was obtained. Encouraged by this result, different parameters of the electrolysis such as electrode material, solvent, temperature, current density and reaction time were scanned (see ESI†). It was found that trifluoroethanol is a very good solvent for this anodic oxidation and that a RVC anode and platinum cathode is the best electrode material combination. Compared to room temperature, higher or lower temperatures gave worse results. Other electrolytes have only minor effects on the yield. Lower concentrations usually lead to cleaner reactions, but the yield was limited to only 20%. When more than 1.6 F was applied under these conditions, only decomposition of the starting material was observed. To increase the yield, different additives were investigated. The addition of an acidic additive such as trifluoroacetic acid successfully increased the yield of the electrolysis to 40% in batch mode (Scheme 3).
 | ||
| Scheme 3 Anodic coupling of 3′,4′,5′-trioxygenated norbelladine derivative ent-11b. | ||
This reaction was then examined using a flow electrochemical reactor,20 where typically the addition of supporting electrolytes is not necessary due to the close distance of the electrodes.13 Although the addition of supporting electrolyte decreases the potential, surprisingly even the addition of only small amounts of supporting electrolyte (0.5% mol) reduced the yield of the product ent-12b under flow conditions. After further optimization of the flow system, the yield was improved to 55% (80% based on recovered starting material) as shown in Scheme 3. The difference is due to chromatographic purification as both, starting material and product are not completely recovered. There are no other products observed in the electrochemical step. The unprotected substrate ent-11a, acetate ent-11c and silylether ent-11d were also examined in this anodic coupling. However, due to the very low solubility of ent-11a in TFE, MeCN or AcOH, no product was formed. Substrates ent-11c and ent-11d were unreactive and did not cyclise under the electrolytic reaction conditions. The mechanism of the anodic coupling reaction has already been investigated and discussed.21
After optimisation of the electrolysis conditions, 11b was then synthesized from methyl D-tyrosine 5 and methyl gallate through 6 steps (see ESI† and Scheme 4). The cyclisation of 11b gave similar results as ent-11b under the electrolysis conditions. With gram amounts of the key intermediate 12b in hand, the deprotection of the benzyl groups was then attempted. The oxa-Michael addition has been shown to proceed spontaneously upon deprotection of 12b of related substrates, and with high diastereoselectivity. Due to the steric hinderance through the methyl ester group, the oxa-Michael addition will be more favoured from the opposite side. However, after investigating different reaction conditions (Scheme 4),4g it was found that the reported combination of dimethyl sulfide with trifluoroacetic acid or methanesulfonic acid led to decomposition. Only the use of BCl3 at −78 °C gave clean results, but irrespective of the reaction time, an inseparable mixture of a mono-debenzylated product and the desired product 13 was obtained (Table S6†). At temperatures above −20 °C the ester is being hydrolysed while at temperatures below −40 °C the reaction does not go to completion. An improved yield of 68% with a ratio of formamide rotamers of 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was obtained by a mono-debenzylation at −78 °C, followed by one additional equivalent of BCl3 at −20 °C (see ESI†).
:1 was obtained by a mono-debenzylation at −78 °C, followed by one additional equivalent of BCl3 at −20 °C (see ESI†).
 | ||
| Scheme 4 Synthetic route to compounds 14. | ||
The free hydroxyl moiety in 13 was then efficiently transformed into a triflate for a subsequent Pd-catalysed deoxygenation by using HCOOH/Et3N as the transfer hydrogen reductant to give 14a in 82% yield over two steps. The hydrolysis of the methyl ester in 14a with LiOH also proceed in high yield (90%) to generate 14b. Converting 14b to the narwedine derivative 14c was then attempted by the Barton–McCombie decarboxylation.22 However, this reaction failed to provide the desired product 14c even after many attempts under a variety of reaction conditions. The nickel-catalyzed Barton decarboxylation reported by Baran's group23 and a photocatalytic decarboxylation24 were also trialed multiple times without success.
It was then decided to convert the ester group into a cyanide group for a reductive decyanation to remove the undesired functional group in analogy to a reported natural product synthesis.19 To achieve this reductive decyanation, an initial reduction of the formamide group to the methylamine is necessary to provide an α-aminonitrile as the decyanation substrate. However, direct methods for decyanations of α-aminonitriles require usually strong reductants such as alkali metals or LiAlH4 as they proceed after cyanide loss via the corresponding iminium ions.25 Such reagents would also affect the enone moiety and the ester group, especially as it was found that the enone moiety is a very sensitive and therefore problematic functional group at a later stage. The L-selectride reduction of the enone was carried out initially to give the alcohol 15 in up to 70% yield. In order to convert the formamide group to a methyl substituent, it was hydrolysed to the amine by methanolic HCl followed by reductive amination with formaldehyde and NaBH(OAc)3 and 16 was obtained in 65% yield over these two steps. The subsequent amidation of 16 with 7 N ammonia in MeOH was very clean but took a long time. After an increase of the reaction time from 2 to 10 days, the yield of 17 was improved from 25% to 45% and 20% starting material was recovered. The following dehydration of the amide by using trifluoroacetic anhydride (TFAA) and Et3N gave the desired α-aminonitrile 18 in 57% yield. The final reduction was initially attempted with LiAlH4 at −20 °C, but this reaction did lead to many side products. NaBH4 gave a much cleaner reaction to provide 41% of (−)-galanthamine 1, while 43% starting material was recovered (Scheme 5).
 | ||
| Scheme 5 Final stages in the synthesis of (−)-galanthamine 1. | ||
Conclusions
In summary, a new biomimetic total synthesis of (−)-galanthamine 1 was achieved in 16 steps with a 0.7% overall yield by using an anodic aryl–aryl coupling as the key synthetic step which was optimised in a flow electrochemical setup. To our knowledge, this demonstrates the first successful application of electrochemistry in the total synthesis of (−)-Galanthamine.Author contributions
T.W. conceptualized the work. Z.X. developed the methodology. Z.X., F.W. and C.S. performed all the experiments and analyzed the data. Z.X. and T.W. prepared the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge financial support from the School of Chemistry, Cardiff University. We thank Dr Md. N. Khan for help with analysis.Notes and references
- (a) N. F. Proskurnina and A. P. Yakovleva, J. Gen. Chem. USSR, 1952, 22, 1899–1902 CAS; (b) H. Kondo, K. Tomimura and S. Ishiwata, J. Pharm. Soc. Jpn., 1932, 52, 51 Search PubMed.
- (a) M. D. Mashkovskii, Pharmacol. Toxicol., 1955, 18, 21 CAS; (b) R. L. Irwin and H. J. Smith, Biochem. Pharmacol., 1960, 3, 147–148 CrossRef CAS PubMed; (c) A. Caricati-Neto, L. C. A. D'angelo, H. Reuter, N. H. Jurkiewicz, A. G. Garcia and A. Jurkiewicz, Eur. J. Pharmacol., 2004, 503, 191–201 CrossRef CAS PubMed.
- L. J. Scott and K. L. Goa, Drugs, 2000, 60, 1095–1122 CrossRef CAS PubMed.
- For the biomimetic oxidative coupling strategy towards the total synthesis of galanthamine, see: (a) D. H. R. Barton and G. W. Kirby, J. Chem. Soc., 1962, 806–817 RSC; (b) T. Kametani, K. Yamaki, H. Yagi and K. Fukumoto, J. Chem. Soc. C, 1969, 2602–2605 RSC; (c) T. Kametani, K. Yamaki, H. Yagi and K. Fukumoto, J. Chem. Soc. D, 1969, 425–426 RSC; (d) K. Shimizu, K. Tomioka, S.-i. Yamada and K. Koga, Heterocycles, 1977, 8, 277–282 CrossRef CAS; (e) Y. Kita, M. Arisawa, M. Gyoten, M. Nakajima, R. Hamada, H. Tohma and T. Takada, J. Org. Chem., 1998, 63, 6625–6633 CrossRef CAS; (f) B. Küenburg, L. Czollner, J. Fröhlich and U. Jordis, Org. Process Res. Dev., 1999, 3, 425–431 CrossRef; (g) M. Node, S. Kodama, Y. Hamashima, T. Katoh, K. Nishide and T. Kajimoto, Chem. Pharm. Bull., 2006, 54, 1662–1679 CrossRef CAS PubMed; (h) J. Marco-Contelles, M. do Carmo Carreiras, C. Rodriguez, M. Villarroya and A. G. García, Chem. Rev., 2006, 106, 116–133 CrossRef CAS PubMed.
- W.-C. Shieh and J. A. Carlson, J. Org. Chem., 1994, 59, 5463–5465 CrossRef CAS.
- For the synthetic studies of (−)-galanthamine using a Heck reaction as a key strategy, see: (a) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 11262–11263 CrossRef CAS; (b) B. M. Trost and W. P. Tang, Angew. Chem., Int. Ed., 2002, 41, 2795–2797 CrossRef CAS; (c) B. M. Trost, W. P. Tang and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 14785–14803 CrossRef CAS PubMed; (d) V. Satcharoen, N. J. McLean, S. C. Kemp, N. P. Camp and R. C. D. Brown, Org. Lett., 2007, 9, 1867–1869 CrossRef CAS PubMed; (e) J.-Q. Chen, J.-H. Xie, D.-H. Bao, S. Liu and Q.-L. Zhou, Org. Lett., 2012, 14, 2714–2717 CrossRef CAS PubMed; (f) Y. Zang and I. Ojima, J. Org. Chem., 2013, 78, 4013 CrossRef CAS PubMed; (g) J. Choi, H. Kim, S. Park and J. Tae, Synlett, 2013, 24, 379 CrossRef CAS.
- For the initial ABC-ring construction towards the total synthesis of galanthamine, see: (a) C. Guillou, J.-L. Beunard, E. Gras and C. Thal, Angew. Chem., Int. Ed., 2001, 40, 4745–4746 CrossRef CAS; (b) X.-D. Hu, Y.-Q. Tu, E. Zhang, S. Gao, S. Wang, A. Wang, C.-A. Fan and M. Wang, Org. Lett., 2006, 8, 1823–1825 CrossRef CAS PubMed; (c) H. Tanimoto, T. Kato and N. Chida, Tetrahedron Lett., 2007, 48, 6267–6270 CrossRef CAS; (d) T. Ishikawa, K. Kudo, K. Kuroyabu, S. Uchida, T. Kudoh and S. Saito, J. Org. Chem., 2008, 73, 7498–7508 CrossRef CAS PubMed; (e) J. H. Chang, H.-U. Kang, I.-H. Jung and C.-G. Cho, Org. Lett., 2010, 12, 2016–2018 CrossRef CAS PubMed; (f) M. G. Banwell, X. Ma, O. P. Karunaratne and A. C. Willis, Aust. J. Chem., 2010, 63, 1437–1447 CrossRef CAS; (g) J. Nugent, E. Matoušová and M. G. Banwell, Eur. J. Org. Chem., 2015, 3771–3778 CrossRef CAS; (h) M. A. Endoma-Arias and T. Hudlicky, Chem. – Eur. J., 2016, 22, 14540–14543 CrossRef PubMed; (i) J. Nugent and M. G. Banwell, Eur. J. Org. Chem., 2016, 5862–5867 CrossRef CAS; (j) Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS PubMed.
- For the initial ABD-ring construction towards the total synthesis of galanthamine, see: (a) L. Li, Q. Yang, Y. Wang and Y. Jia, Angew. Chem., Int. Ed., 2015, 54, 6255–6259 CrossRef CAS PubMed; (b) T. Venkatesh, P. S. Mainkar and S. Chandrasekhar, Org. Biomol. Chem., 2019, 17, 2192–2198 RSC.
- Y. Zhang, S. Shen, H. Fang and T. Xu, Org. Lett., 2020, 22, 1244–1248 CrossRef CAS PubMed.
- I. R. Miller, N. J. McLean, G. A. I. Moustafa, V. Ajavakom, S. C. Kemp, R. K. Bellingham, N. P. Camp and R. C. D. Brown, J. Org. Chem., 2022, 87, 1325–1334 CrossRef CAS PubMed.
- Y.-P. Chang, X. Ma, H. Shao and Y.-M. Zhao, Org. Lett., 2021, 23, 9659–9663 CrossRef CAS PubMed.
- A. Horvath, J. A. Verbraeken, K. A. G. De Maria and M. Novak, EP1560947A1, 2002.
- D. Krikorian, R. Vlahov, S. Parushev, M. Chinova, I. Vlahov, H.-J. Schäfer, H. Duddeck and G. Snatzke, Tetrahedron Lett., 1984, 25, 2969–2972 CrossRef.
- A. Lipp, D. Ferenc, C. Gütz, M. Geffe, N. Vierengel, D. Schollmeyer, H. J. Schäfer, S. R. Waldvogel and T. Opatz, Angew. Chem., Int. Ed., 2018, 57, 11055–11059 CrossRef CAS PubMed.
- A. Lipp, M. Selt, D. Ferenc, D. Schollmeyer, S. R. Waldvogel and T. Opatz, Org. Lett., 2019, 21, 1828–1831 CrossRef CAS PubMed.
- (a) A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef CAS PubMed; (b) M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Génot and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 9811–9815 CrossRef CAS PubMed; (c) M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287–3296 CrossRef CAS PubMed.
- (a) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC; (b) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed; (c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed; (d) G. Hilt, ChemElectroChem, 2020, 7, 395–405 CrossRef CAS; (e) Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed; (f) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- (a) M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287–3296 CrossRef CAS PubMed; (b) T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef PubMed; (c) N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS PubMed.
- K. Tomioka, K. Koga and S. Yamada, Chem. Pharm. Bull., 1977, 25, 2681–2688 CrossRef CAS.
- Ion electrochemical reactor, https://www.vapourtec.com/products/flow-reactors/ion-electrochemical-reactor-features/, (accessed May 2022).
- A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef CAS PubMed.
- (a) D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585 RSC; (b) M. F. Saraiva, M. R. C. Couri, M. Le Hyaric and M. V. de Almeida, Tetrahedron, 2009, 65, 3563–3572 CrossRef CAS.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS PubMed.
- C. Cassani, G. Bergonzini and C.-J. Wallentin, Org. Lett., 2014, 16, 4228–4231 CrossRef CAS PubMed.
- J.-M. R. Mattalia, Beilstein J. Org. Chem., 2017, 13, 267–284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob00669c |
| This journal is © The Royal Society of Chemistry 2022 |