
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borane promoted aryl transfer reaction for the synthesis of α-aryl functionalised β-hydroxy and β-keto esters†
Tanja
Kaehler‡
 ,
Jonas
Lorenz‡
,
Darren M. C.
Ould§
,
Dorothea
Engl
,
Micol
Santi¶
,
Lukas
Gierlichs
,
Thomas
Wirth
and
Rebecca L.
Melen
*
,
Jonas
Lorenz‡
,
Darren M. C.
Ould§
,
Dorothea
Engl
,
Micol
Santi¶
,
Lukas
Gierlichs
,
Thomas
Wirth
and
Rebecca L.
Melen
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, Cymru/Wales, UK. E-mail: MelenR@cardiff.ac.uk
First published on 9th May 2022
Abstract
The synthesis of a series of α-aryl or α-alkyl functionalised β-hydroxy and β-keto esters has been achieved by reacting α-diazoesters with boranes, and aldehydes, ketones, anhydrides, nitriles, esters or isocyanates. In a mild reaction protocol, 26 examples are presented in yields up to 73%.
Diazo compounds are useful building blocks in organic synthesis.1 They are typically activated by transition metals,2 however, in recent times, the use of catalytic amounts of a borane as an alternative to transition metal mediated carbene generation has become more popular.3 The reaction of diazo compounds with equimolar amounts of trialkyl or triaryl boranes has been shown to lead to the insertion of the carbene into the C–B bond and is generally well explored.4 For example, Stephan et al.5 reported in 2013 the generation of boron enolates by reacting ethyl α-diazomethylacetate and B(C6F5)3 in CH2Cl2 at −78 °C (cf.Scheme 1A). The aryl group migration from boron to carbon was confirmed by NMR spectroscopy analysis, revealing the formation of the E- and Z-boron enolate products in a 4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in 42% yield. This reactivity led to the synthesis of new bulky, secondary and tertiary substituted Lewis acidic boranes.5 Already, as early as in the 1970s, Hooz et al. conducted the first studies by trapping boron enolates generated from diazo precursors with aldehydes,6 ketones,9 dimethylethylenammonium iodide,7N-bromo- or N-chlorosuccinimide8 and nitriles (Scheme 1B–E).9 Later, Miranda et al.10 prepared 1,3-diketones and β-keto esters from reacting α-diazocarbonyl compounds with BR3 (R = ethyl, n-propyl, phenyl) and aldehydes followed by oxidation (Scheme 1E).13 To explore further the utility of this approach, we developed the scope for the use of electrophilic aryl boranes as arylation reagents with diazo compounds.11 Initially, the treatment of various α-diazoesters with different triaryl boranes gave similar boron enolates as mentioned above. Basic aqueous work-up of these enolates gave α-aryl functionalised esters in up to 99% yield (Scheme 1F) which we also demonstrated could be applied towards an intermediate in the synthesis of antidepressant diclofensine which had previously been reported using rhodium-based catalysts.6,12 It was also found that the more Lewis acidic boranes are able to transfer more than one aryl group.5,6,13 However, using 2-benzyloxy-substituted diazo compounds as starting materials, biologically important 3,3-disubstituted benzofuranone derivatives could be synthesised (Scheme 1F). Here, the boron enolate formed after the aryl transfer from B(ArF)3 (ArF = fluorinated aryl) intramolecularly attacks the benzyl group generating a quaternary carbon centre. Subsequent cyclisation leads to the benzofuranone heterocycle.6 In this study we investigate alternative synthetic applications of the boron enolate formed after aryl transfer from B(ArF)3 to the diazo precursor, since the previous studies concentrated mostly on diazo carbonyl compounds and alkylboranes, and a wider scope was not investigated (see Scheme 1 for comparison). Therefore, we decided to explore other electrophiles such as aldehydes, ketones, imines, alkylhalides, nitriles, isocyanates, esters and acid chlorides in a three-component reaction with α-diazoesters and fluorinated triaryl boranes, as the incorporation of fluorinated aromatic rings is important for drug synthesis or for medicinal applications.14 For a complete picture, we also included BEt3 in our studies.
:1 ratio in 42% yield. This reactivity led to the synthesis of new bulky, secondary and tertiary substituted Lewis acidic boranes.5 Already, as early as in the 1970s, Hooz et al. conducted the first studies by trapping boron enolates generated from diazo precursors with aldehydes,6 ketones,9 dimethylethylenammonium iodide,7N-bromo- or N-chlorosuccinimide8 and nitriles (Scheme 1B–E).9 Later, Miranda et al.10 prepared 1,3-diketones and β-keto esters from reacting α-diazocarbonyl compounds with BR3 (R = ethyl, n-propyl, phenyl) and aldehydes followed by oxidation (Scheme 1E).13 To explore further the utility of this approach, we developed the scope for the use of electrophilic aryl boranes as arylation reagents with diazo compounds.11 Initially, the treatment of various α-diazoesters with different triaryl boranes gave similar boron enolates as mentioned above. Basic aqueous work-up of these enolates gave α-aryl functionalised esters in up to 99% yield (Scheme 1F) which we also demonstrated could be applied towards an intermediate in the synthesis of antidepressant diclofensine which had previously been reported using rhodium-based catalysts.6,12 It was also found that the more Lewis acidic boranes are able to transfer more than one aryl group.5,6,13 However, using 2-benzyloxy-substituted diazo compounds as starting materials, biologically important 3,3-disubstituted benzofuranone derivatives could be synthesised (Scheme 1F). Here, the boron enolate formed after the aryl transfer from B(ArF)3 (ArF = fluorinated aryl) intramolecularly attacks the benzyl group generating a quaternary carbon centre. Subsequent cyclisation leads to the benzofuranone heterocycle.6 In this study we investigate alternative synthetic applications of the boron enolate formed after aryl transfer from B(ArF)3 to the diazo precursor, since the previous studies concentrated mostly on diazo carbonyl compounds and alkylboranes, and a wider scope was not investigated (see Scheme 1 for comparison). Therefore, we decided to explore other electrophiles such as aldehydes, ketones, imines, alkylhalides, nitriles, isocyanates, esters and acid chlorides in a three-component reaction with α-diazoesters and fluorinated triaryl boranes, as the incorporation of fluorinated aromatic rings is important for drug synthesis or for medicinal applications.14 For a complete picture, we also included BEt3 in our studies.
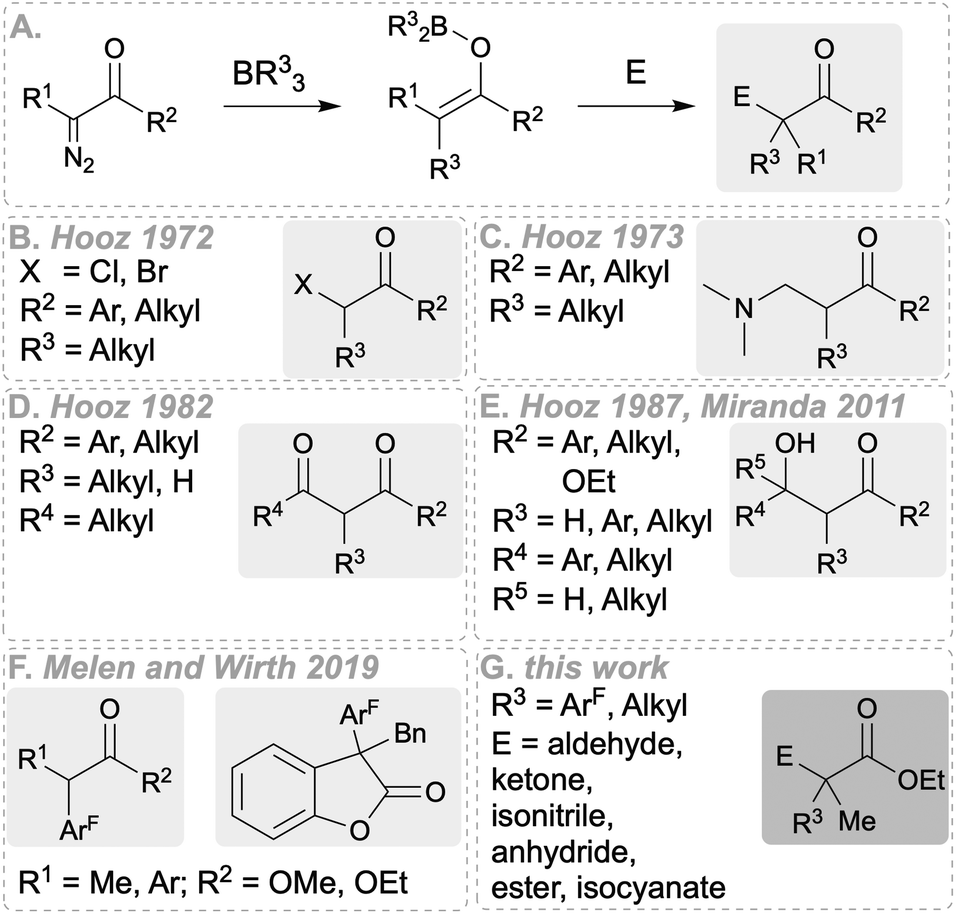 | ||
| Scheme 1 Previous and current work. | ||
Our initial studies investigated the reaction of commercially available ethyl diazoacetate 1a with B(C6F5)3 in the presence of aldehydes. Reaction of 1a with 4-fluorobenzaldehyde and 1 equiv. B(C6F5)3 for 20 h at 45 °C gave a mixture of products including α-aryl substituted β-hydroxy ester 2a, β-keto ester 4 and an unidentified compound (Scheme 2). A small crop of crystals of this latter compound was isolated from the solution which was identified as compound 6 by single crystal X-ray diffraction (Scheme 2, bottom right). We propose that 6 is formed en route to product 4. When using catalytic amounts of B(C6F5)3 (0.1 equiv.) under the same reaction conditions 4, 5 and 6 could be identified in the 1H NMR spectrum of the crude reaction mixture (see the ESI for details†). The formation of compound 3a was not observed. The unselective reaction of diazo compound 1a with other Lewis acids has already been described in the literature.15 However, with 1 equiv. of B(C6F5)3 in CH2Cl2 for 20 h at room temperature, 2a could be isolated in 74% yield as a diastereomeric mixture (dr: 1:0.07). The solid-state structure of 2S,3R-2a could be verified via X-ray diffraction analysis (Scheme 2, bottom left).
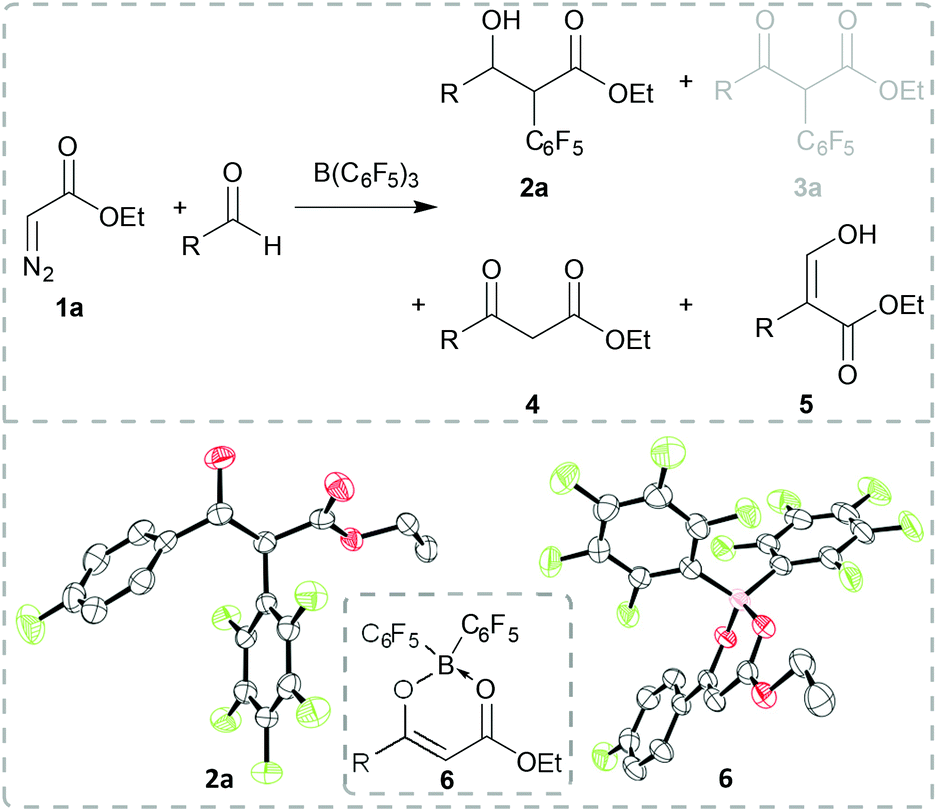 | ||
| Scheme 2 Reaction of ethyl diazoacetate 1a, with 4-fluorobenzaldehyde and B(C6F5)3 (R = 4-fluorophenyl; top). Solid-state structure of compound 2a (bottom left) and 6 (bottom right). Thermal ellipsoids drawn at 50% probability. Carbon: black; oxygen: red; boron: pink; fluorine: green. H atoms omitted for clarity. | ||
Considering the difficulties in the selective synthesis of α-aryl substituted β-hydroxy esters 2 with 1a as a starting material, we investigated ethyl α-diazomethylacetate 1b as an alternative. Indeed, a more selective reaction is observed with 4-fluorobenzaldehyde, B(C6F5)3, and diazo ester 1b as model substrates (Table 1). Under the same reaction conditions as above, the 1:1:1 reaction of 1b with 4-fluorobenzaldehyde and B(C6F5)3 gave the product 2b in 62% yield and a 1:0.10 diastereomeric ratio (Table 1, entry 1). Screening of the solvents showed that CH2Cl2 gave the best isolated yields and diastereomeric ratio, with less polar toluene and hexane giving 41% and 40% yields respectively (Table 1, entries 2–3). Variation of the temperature to room temperature (20 °C) in CH2Cl2 and 60 °C in 1,2-dichloroethane (C2H4Cl2) (Table 1, entries 4 and 5) both showed significantly lower yields of 43% and 31% yield, respectively. When the reaction time was extended from 20 h to 24 h and 30 h (Table 1, entries 6 and 7), a better conversion of the aldehyde starting material was detected in the 1H NMR spectrum of the crude reaction mixture (see ESI for details†). However, the isolated yields from the product mixture were similar. Consequently, a reaction time of 24 h was chosen. Lastly, the amount of B(C6F5)3 was changed since we have previously shown that B(C6F5)3 can transfer more than one of its aryl rings.6 Substoichiometric amounts of borane (0.8 and 0.6 equiv.) however lead to poorer diastereomeric ratios and lower yields (Table 1, entries 8 and 9). Using more than 1 equiv. of B(C6F5)3 showed no advantage (Table 1, entry 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | B(C6F5)3 (equiv.) | Solvent | Temp. (°C) | Time (h) | dra | Yield 2bb (%) |
| a dr determined by 19F NMR analysis of the crude reaction mixture. b Reported yields are isolated yields of both diastereoisomers. All the reactions were carried out on a 0.16 mmol scale. 1b (1 equiv.), 4-fluorobenzaldehyde (1 equiv.), and B(C6F5)3, and 2.0 mL of solvent were used. | ||||||
| 1 | 1.0 | CH2Cl2 | 45 | 20 | 1:0.10 |
62 |
| 2 | 1.0 | Toluene | 45 | 20 | 1:0.14 |
41 |
| 3 | 1.0 | Hexane | 45 | 20 | 1:0.12 |
40 |
| 4 | 1.0 | CH2Cl2 | 20 | 20 | 1:0.10 |
43 |
| 5 | 1.0 | C2H4Cl2 | 60 | 20 | 1:0.26 |
31 |
| 6 | 1.0 | CH2Cl2 | 45 | 24 | 1:0.10 |
72 |
| 7 | 1.0 | CH2Cl2 | 45 | 30 | 1:0.10 |
70 |
| 8 | 0.8 | CH2Cl2 | 45 | 24 | 1:1.15 |
56 |
| 9 | 0.6 | CH2Cl2 | 45 | 24 | 1:1.15 |
45 |
| 10 | 1.2 | CH2Cl2 | 45 | 24 | 1:1.15 |
52 |

With the optimised reaction conditions in hand, we explored the substrate scope for the synthesis of α-substituted β-hydroxy esters 2. Firstly, 1b, B(R3)3 (R3 = C6F5, Et) and aldehydes were used bearing electron-withdrawing, neutral, and electron-releasing groups giving β-hydroxy esters 2b–2h, 2r,s in yields up to 73% (Scheme 3). The conversion of aldehydes with electron withdrawing substituents was found to be better than electron-releasing groups (cf.2b in 72% vs.2e in 32% yield). The highest yield could be achieved for 2f (73%) using the ortho-methyl substituted benzaldehyde as a substrate. The diastereomeric selectivity for the arylated compounds 2b–2h was quite good (e.g. 1:0.09 for 2g) and the diastereoisomers could be separated via preparative thin layer chromatography (TLC) giving a major and a minor product. Crystals of the minor compound of 2c and the major compound of 2i could be formed by slow evaporation of a saturated CHCl3 solution (Fig. 1). Based on this we find that the minor compound of 2c is racemic 2S*,3R*-alcohol and the major compound of 2i is racemic 2S*,3S*-alcohol. From this we propose that the major isomer formed are the racemic 2S*,3S*-alcohols through a 6-membered Zimmerman–Traxler transition state. We then investigated other boranes (B(ArF)3; ArF = 3,4,5-F3C6H2; 2,4,6-F3C6H2; 4-FC6H4) in the reaction giving β-hydroxy esters 2i–2m in yields up to 72%. However, we observed lower diastereoselectivities when using boranes which are devoid of ortho-fluorine atoms or bearing an ethyl group (compounds 2r, 2s, and 2j–2m, Scheme 3). Interestingly, the less Lewis acidic BPh3 did not work for these reactions.6 To further expand the scope of β-hydroxy esters, we investigated other electrophiles such as imines, alkyl halides, and ketones. While reactions with imines and alkyl halides were unsuccessful, treatment of 1b and B(C6F5)3 or BEt3 with various ketones yielded compounds 2n–2q, and 2t (Scheme 3).
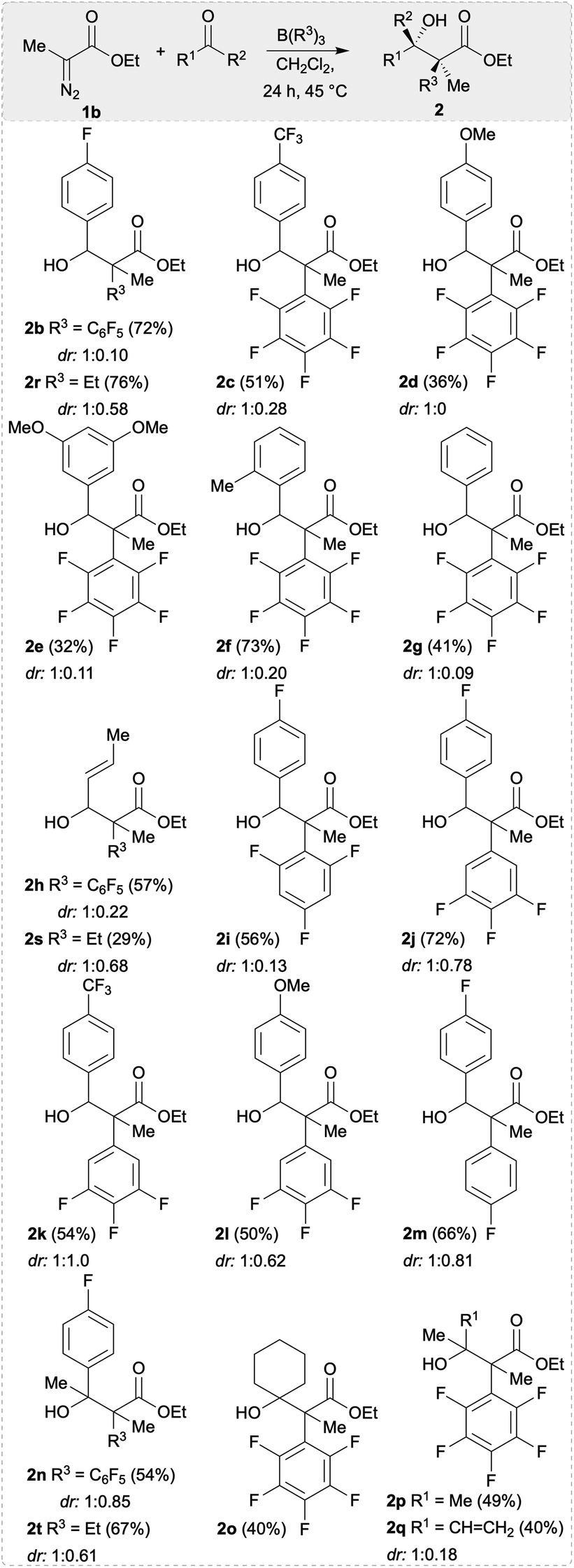 | ||
| Scheme 3 Substrate scope of the synthesised β-hydroxy esters. Yields reported are isolated. Reactions were carried out on a 0.16 mmol scale. 1b (1 equiv.), aldehyde/ketone (1 equiv.), B(R3)3 (1 equiv.; R3 = C6F5, Et), and 2.0 mL of CH2Cl2 were used. | ||
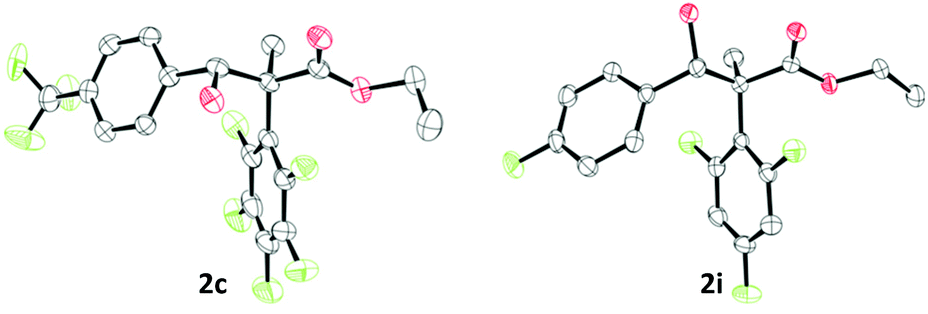 | ||
| Fig. 1 Solid-state structure of compound 2c (left) and 2i (right). Thermal ellipsoids drawn at 50% probability. Carbon: black; oxygen: red; fluorine: green. H atoms omitted for clarity. | ||
This concept was then applied to the synthesis of β-keto esters 3b–3h (Scheme 4). Compound 3b could be synthesised using benzoic anhydride, benzonitrile or benzoyl chloride as substrates, however when using benzoic anhydride the best yields were obtained (60%). In case of benzonitrile, an additional acidic work up was performed to hydrolyse the initially formed β-imine into 3b. With benzoyl chloride, 3b only formed as a minor product with 7d isolated as the main product from an otherwise complex crude reaction mixture (see below, Schemes 4 and 5). When using an ester such as methyl propiolate as electrophile, 3c was isolated in 36% as the main product. No reaction at the unsaturated carbon–carbon bond occurred as was already observed in the synthesis of compounds 2h and 2q. The amino functionalised α-aryl substituted β-keto esters 3d–3f and 3h were synthesised using isocyanates in the three-component reaction (Scheme 4). In case of the C6F5 transfer, electron rich aryl isocyanates bearing a p-OMe gave the highest yield (3e, 54%) whereas p-CF3 isocyanate gave 3d in just 13% yield. Using BEt3 in this reaction, compound 3h could only be isolated in 14% yield which is a significantly lower product formation compared to 3e.
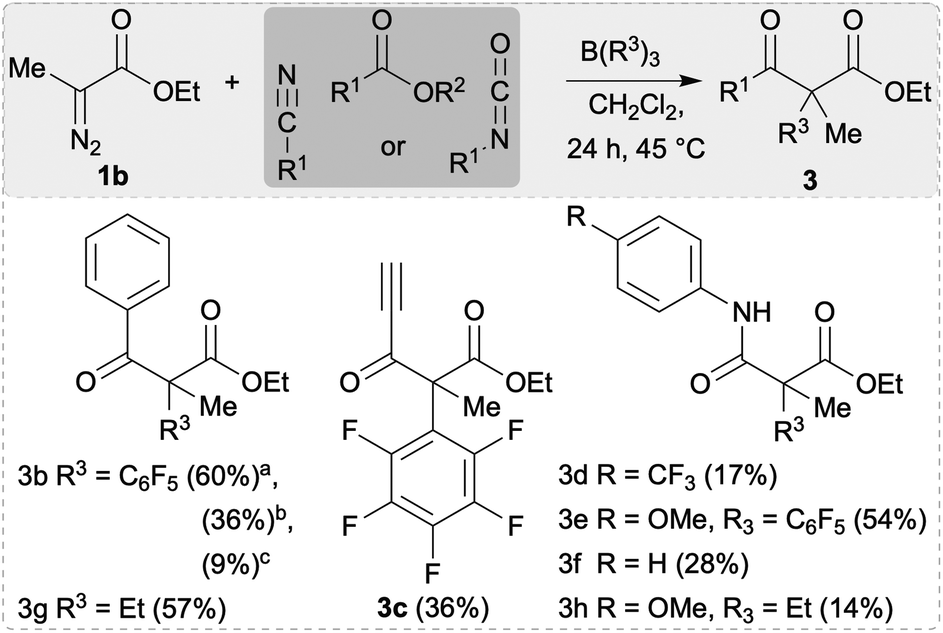 | ||
| Scheme 4 Substrate scope of the synthesised β-keto esters. Yields reported are isolated. All the reactions were carried out on a 0.16 mmol scale. 1b (1 equiv.), electrophile (1 equiv.), B(R3)3 (1 equiv.; R3 = C6F5, Et), and 2.0 mL of CH2Cl2 were used. aYield using benzoic anhydride. bYield using benzonitrile. cYield using benzoyl chloride. | ||
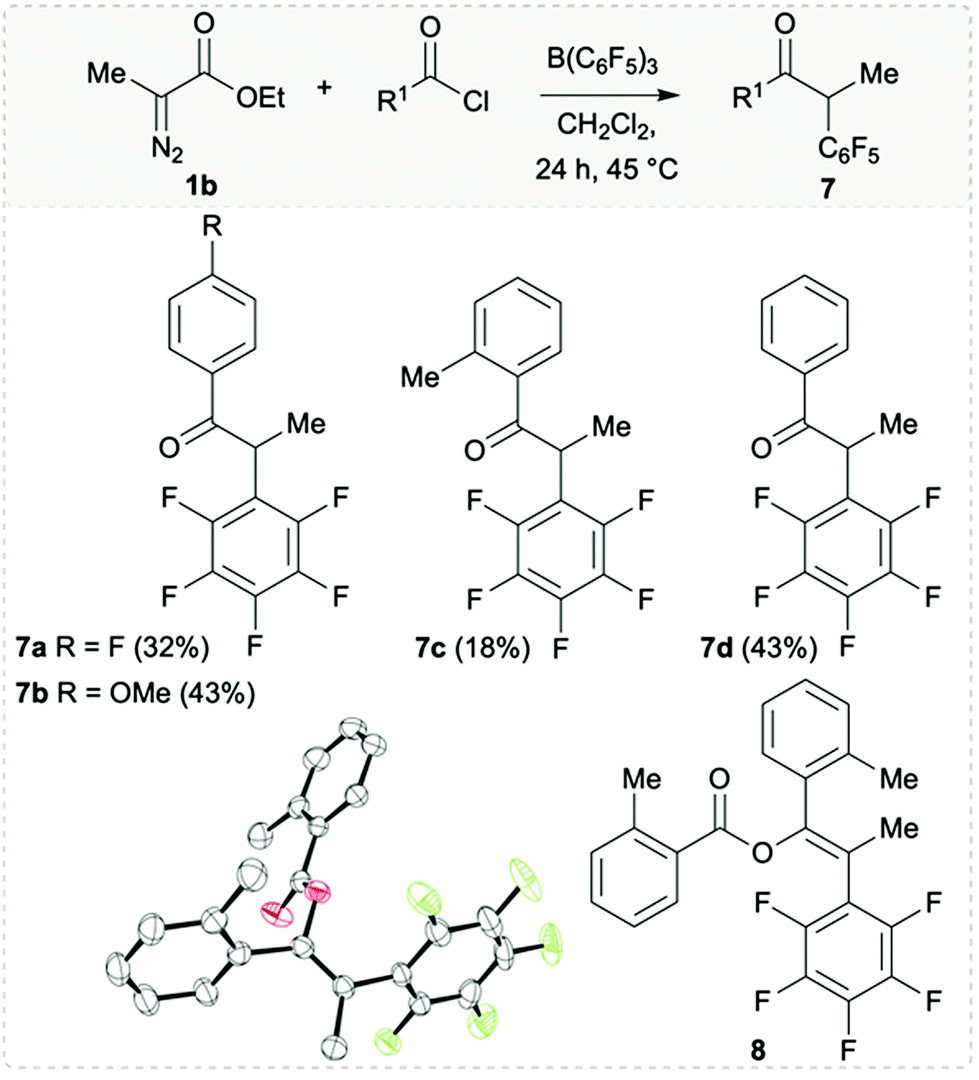 | ||
| Scheme 5 Substrate scope of the synthesised ketones. Yields reported are isolated. All the reactions were carried out on a 0.16 mmol scale. 1b (1 equiv.), acid chloride (1 equiv.), B(C6F5)3 (1 equiv.), and 2.0 mL of CH2Cl2 were used. Solid-state structure of compound 8 (bottom). Thermal ellipsoids drawn at 50% probability. Carbon: black; oxygen: red; fluorine: green. H atoms omitted for clarity. | ||
As alluded earlier, treatment of 1b and B(C6F5)3 with acid chlorides does not give the desired β-keto esters. Instead, we observe the formation of compounds 7a–d isolated in 18–43% yield (Scheme 5). In these reactions, the nucleophilic attack of the boron enolate to the acid chloride took place with a formal decarboxylation step. Conditions for the release of CO2 from β-keto esters usually involves strong acidic/basic conditions,16 metal salts,17 or elevated temperatures.18 A few examples where boric acid promotes decarboxylation of β-keto esters have also been reported.19 From the reaction that yielded compound 7c, a small crop of crystals of another compound was isolated from the solution which was identified as compound 8 by single crystal X-ray diffraction (Scheme 5, bottom). Here, reaction with two equivalents of the acid chloride had taken place indicating that acid chlorides are too reactive for these reactions under the applied conditions.
In conclusion, we synthesised 21 examples of α-aryl and 5 examples of α-alkyl functionalised β-hydroxy and β-keto esters. This is a three-component reaction with ethyl α-diazomethylacetate 1b, boranes B(R3)3 (R3 = C6F5, Et), and electrophiles such as aldehydes, ketones, anhydrides, nitriles, esters and isocyanates. On the other hand, the reaction of ethyl α-diazomethylacetate 1b, B(C6F5)3 and acid chlorides resulted in the elimination of the ester functionality and yielded ketones 7a–7d.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TK and RLM are grateful to the EPSRC for funding (EP/R026912/1). Information about the data that underpins the results presented in this article, including how to access them, can be found at http://doi.org/10.17035/d.2022.0177945377.Notes and references
- (a) K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233 CrossRef CAS PubMed; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
- B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864 CrossRef CAS PubMed.
- (a) A. Dasgupta, E. Richards and R. L. Melen, ACS Catal., 2022, 12, 442 CrossRef CAS PubMed; (b) X.-Y. Wu, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Adv. Synth. Catal., 2022, 364, 750 CrossRef CAS.
- H. Li, Y. Zhang and J. Wang, Synthesis, 2013, 3090 CAS.
- R. C. Neu, C. Jiang and D. W. Stephan, Dalton Trans., 2013, 42, 726 RSC.
- J. Hooz, J. Oudenes, J. L. Roberts and A. Benderly, J. Org. Chem., 1987, 52, 1347 CrossRef CAS.
- J. N. Bridson and J. Hooz, J. Am. Chem. Soc., 1973, 95, 602 CrossRef.
- J. N. Bridson and J. Hooz, Can. J. Chem., 1972, 50, 2387 CrossRef.
- J. Oudenes and J. Hooz, Synth. Commun., 1982, 12, 189 CrossRef.
- M. A. Sanchez-Carmona, D. A. Contreras-Cruz and L. D. Miranda, Org. Biomol. Chem., 2011, 9, 6506 RSC.
- M. Santi, D. M. C. Ould, J. Wenz, Y. Soltani, R. L. Melen and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 7861 CrossRef CAS PubMed.
- J. Ghorai and P. Anbarasan, J. Org. Chem., 2015, 80, 3455 CrossRef CAS PubMed.
- R. C. Neu and D. W. Stephan, Organometallics, 2012, 31, 46 CrossRef CAS.
- D. Barnes-Seeman, J. Beck and C. Springer, Curr. Top. Med. Chem., 2014, 14, 855 CrossRef CAS PubMed.
- (a) A. K. Gupta, M. Mukherjee, G. Hu and W. D. Wulff, J. Org. Chem., 2012, 77, 7932 CrossRef CAS PubMed; (b) M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Chem. Commun., 2009, 5153 RSC; (c) D. Benito-Garagorri, J. Wiedermann, M. Pollak, K. Mereiter and K. Kirchner, Organometallics, 2007, 26, 217 CrossRef CAS.
- (a) K. Kundu and S. K. Nayak, J. Nat. Prod., 2017, 80, 1176 CrossRef PubMed; (b) D. Landini and F. Rolla, J. Org. Chem., 1982, 47, 154 CrossRef CAS; (c) C. R. Schmid, Tetrahedron Lett., 1992, 33, 757 CrossRef CAS; (d) K. L. Kaestle, M. K. Anwer, T. K. Audhya and G. Goldstein, Tetrahedron Lett., 1991, 32, 327 CrossRef CAS; (e) Y. Yamamoto, T. Furuta, J. Matsuo and T. Kurata, J. Org. Chem., 1991, 56, 5737 CrossRef CAS.
- Z.-W. Chen, D.-N. Ye, Y.-P. Qian, M. Ye and L.-X. Liu, Tetrahedron, 2013, 69, 6116 CrossRef CAS.
- Q. Zhang and D. P. Curran, Adv. Synth. Catal., 2003, 345, 329 CrossRef.
- (a) P. Delbecq, J.-P. Celerier and G. Lhommet, Tetrahedron Lett., 1990, 31, 4873 CrossRef CAS; (b) J. M. Lalancette and A. Lachance, Tetrahedron Lett., 1970, 45, 3903 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR data, X-ray data. CCDC 2154538–2154540, 2154542, 2154543 and 2154565. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob00643j |
| ‡ Authors contributed equally to this work. |
| § Current Address: Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK |
| ¶ Current Address: Faculty of Chemistry/Industrial Organic Chemistry and Biothechnology, Universität Bielefeld, Postfach 100131, D-33501 Bielefeld, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |