
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Egg yolk sialylglycopeptide: purification, isolation and characterization of N-glycans from minor glycopeptide species†
Javier Mauricio
Melo Diaz
 *ab,
Alan B.
Moran
ac,
Simon R.
Peel
a,
Jenifer L.
Hendel
a and
Daniel I. R.
Spencer
a
*ab,
Alan B.
Moran
ac,
Simon R.
Peel
a,
Jenifer L.
Hendel
a and
Daniel I. R.
Spencer
a
aLudger Ltd, Culham Science Centre, Oxfordshire, Abingdon, UK. E-mail: javier.melodiaz@ludger.com
bDepartment of Chemistry Royal College of Surgeons in Ireland, 123 St Stephen's Green, Dublin, Ireland
cLeiden University Medical Center, Center for Proteomics and Metabolomics, 2300 RC Leiden, the Netherlands
First published on 16th May 2022
Abstract
Sialylglycopeptide (SGP) is a readily available naturally occurring glycopeptide obtained from hen egg yolk which is now commercially available. During SGP extraction, other minor glycopeptide species are identified, bearing N-glycan structures that might be of interest, such as asymmetrically branched and triantennary glycans. As the scale of SGP production increases, recovery of minor glycopeptides and their N-glycans can become more feasible. In this paper, we aim to provide structural characterization of the N-glycans derived from these minor glycopeptides.
1. Introduction
Sialylglycopeptide (SGP) (Fig. 1) is a naturally occurring glycopeptide that can be extracted from hen egg yolks. It originates from the proteolytic cleavage of vitellogenin during yolk formation.1 SGP has wide applications. Synthetically, SGP offers convenient access to N-glycan oxazolines, needed for glycoprotein remodelling by ENGase mediated transglycosylation.2,3 Furthermore, SGP has been used as a precursor for the synthesis of constructs such as sialyl Lewis-X structures (S-Lex),4 antennary polylactosamine repeats5 and triantennary glycans,6,7 or a combination of all the above,8 providing access to N-glycan structures for which there is no natural source. Analytically, it has been used for testing new matrices for MALDI-MS,9 for sialic acid derivatization strategies10–12 and as a control for N-glycan release and labelling by reductive amination.13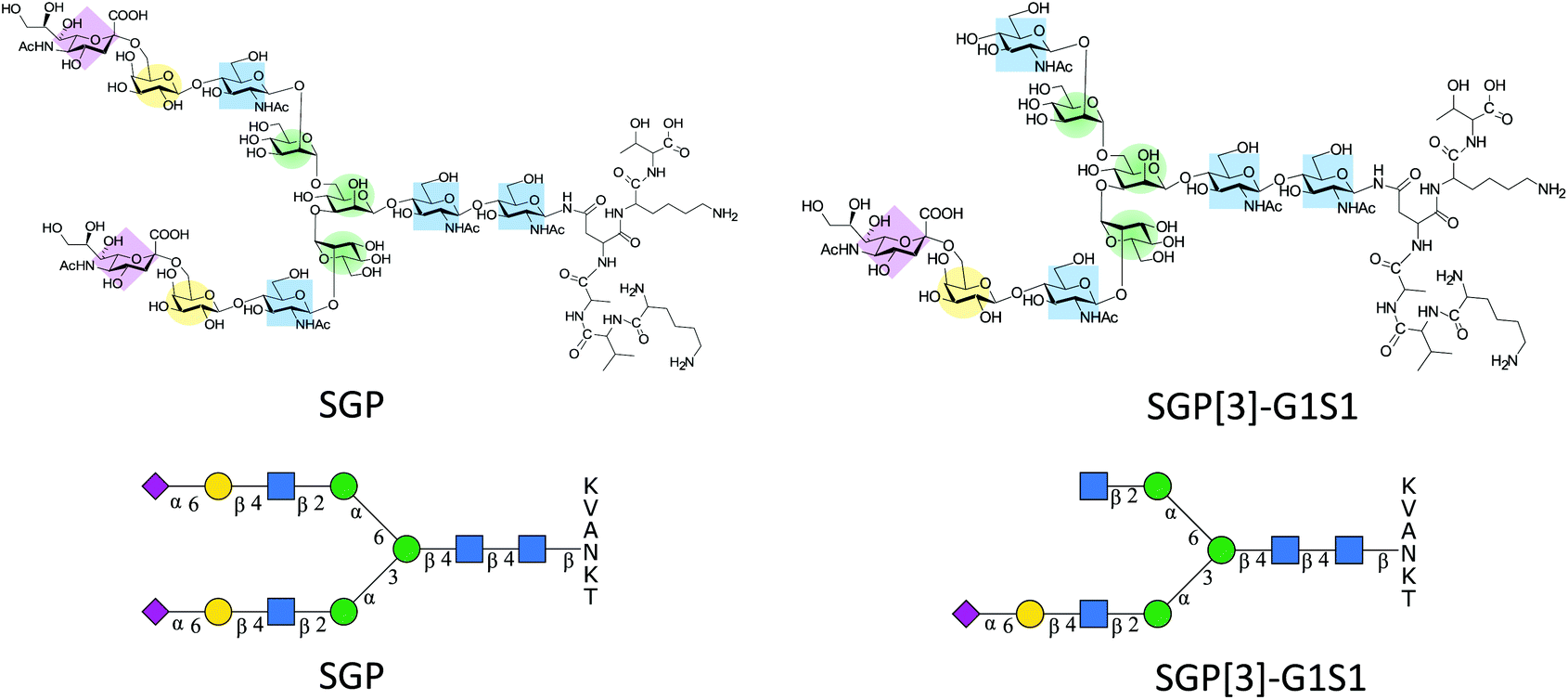 | ||
| Fig. 1 Structural representation of SGP and SGP-A2[3]G1S1* for N-glycan abbreviations see section 2.9 of ESI.† | ||
Even though the amount of glycoconjugates present in egg yolk is only about 0.3% (w/w),14 the usefulness of SGP has led to the development of different isolation protocols that have enabled to increase the purity at which SGP can be obtained.15–18 However, SGP might not be the only relevant glycopeptide target that can be extracted from egg yolks. Early reports of the isolation of SGP indicated the presence of other glycopeptides bearing monosialylated glycans,19,20 some of which allegedly corresponded exclusively to the antennary extension of the α-1,3 mannose arm of the N-glycan core SGP-A2[3]G1S1 (Fig. 1). This glycan asymmetry has been attributed to different processing in the Golgi.21 Normally during SGP purification the monoantennary glycopeptide is removed by size exclusion chromatography22 or digested enzymatically to afford a homogenous albeit de-galactosylated glycan.23,24
Despite previous evidence for the presence of the monosialylated asymmetric glycan in egg yolk, recent publications identify it as a mixture.25 Having an alternative source of asymmetric glycans would be of interest since they are difficult to synthesize and there is growing awareness of the biological implications of N-glycan asymmetry such as the complement-dependent cytotoxicity of monoclonal antibodies,26 and influenza haemagglutinin binding.27 Furthermore, asymmetric structures can be useful for the validation of mass spectrometry-based techniques.28,29 Each of these examples is indicative of potential applications for these compounds.
This paper aims to present a simplified purification strategy for both A2G2S2 and A2[3]G1S1 starting from egg yolk, along with supporting evidence that confirms this structural assignment. Unexpectedly, during method development, it was possible to identify an additional glycopeptide species that corresponded to a triantennary analogue of SGP. The separation and characterization of the triantennary glycans obtained from this glycopeptide will also be presented.
2. Results and discussion
2.1 SGP extraction
Both egg yolk powder and fresh egg yolks were used to produce crude SGP preparations, the details of purification processes can be found in the materials and methods. However, as previously reported25 glycated versions of SGP are also present when using egg yolk powder (Fig S1†), with these species most likely originating during spray drying.30 SGP from the dry yolk powder was used for initial N-glycan analysis, but to provide a better estimation of the overall purification yield based on the weight of the crude SGP, fresh yolks were used.2.2 Preliminary procainamide analysis-sialic acid derivatization
The N-glycans present in the crude SGP (obtained from dry egg yolk) were released with PNGase-F and then procainamide labelled producing a mixture that contains about 56% of A2G2S2 glycan (Fig. 2A), with other species identified summarized in Table 1. Sialic acid linkage characterisation was carried out on this initial mixture by subjecting the procainamide labelled glycans to ethyl esterification-amidation (EEA)31 and were re-analysed by HPLC-MS to determine the mass increments due to derivatization. In brief, for each α-2,6-linked sialic acid, a glycan will exhibit a mass shift of +28.03 Da whereas one α-2,3-linked sialic acid will produce a mass shift of −0.95 Da (Scheme S1†).32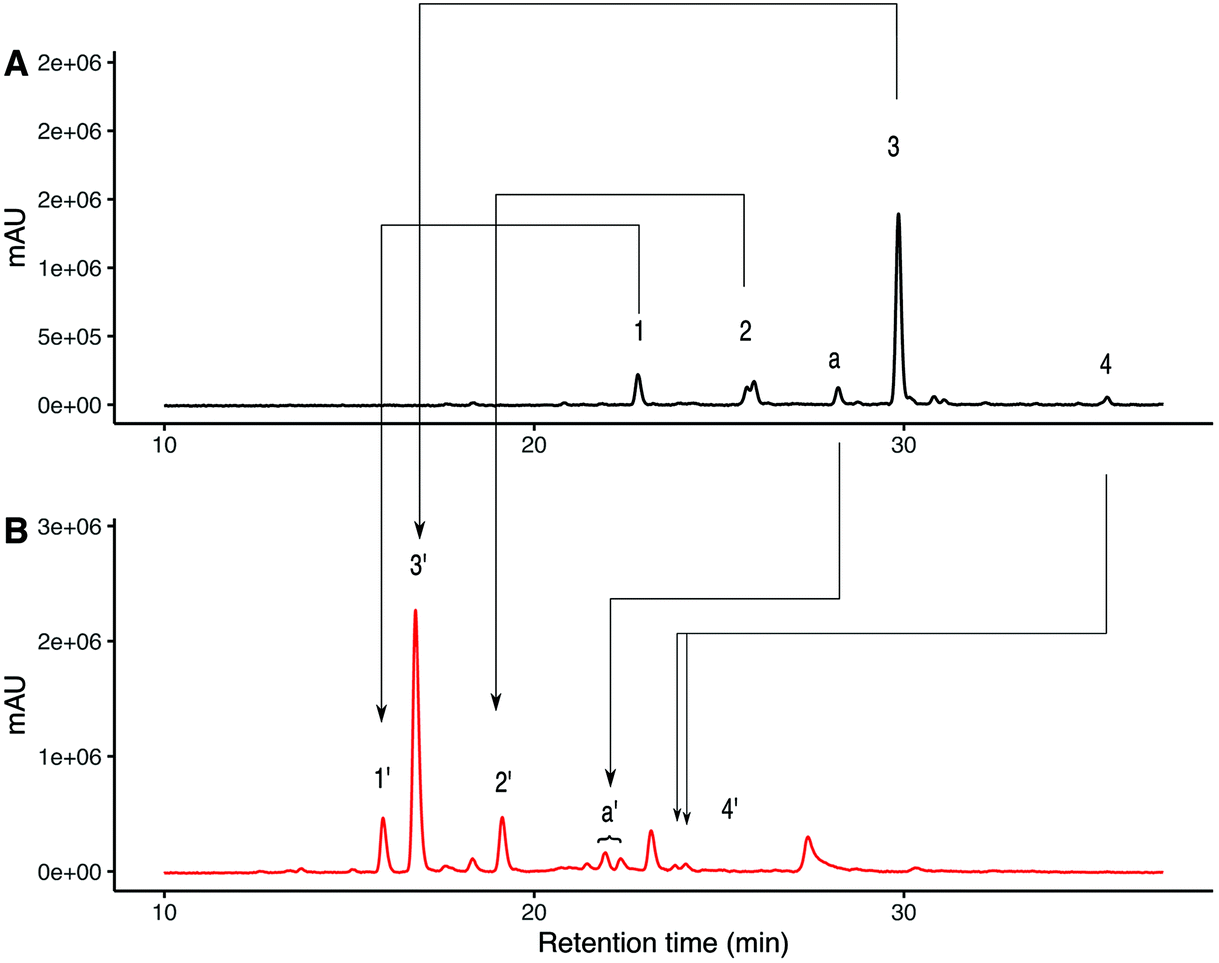 | ||
| Fig. 2 UHPLC analysis for procainamide labelled N-glycans released from crude SGP using PNGase-F. (A) UHPLC-FLD analysis for procainamide labelled glycans species identified 1–4 are described in Table 1. (B) UHPLC-FLD analysis for ethyl esterified and amidated procainamide labelled glycans species identified 1′–4′ are described in Table 2. | ||
| Peak | Name | SNFG representation | Predicted m/z | Observed m/z | ppma |
|---|---|---|---|---|---|
| a Amazon Bruker instrument accuracy quoted as ±0.5 Da. | |||||
| 1 | A2[3]G1S1 |
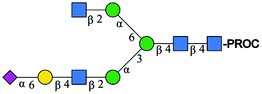
|
995.41 [M + 2H]2+ | 995.57 [M + 2H]2+ | −161 |
| 2 | A2G2S1 |
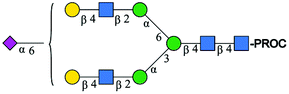
|
1076.44 [M + 2H]2+ | 1076.54 [M + 2H]2+ | −93 |
| 3 | A2G2S2 |
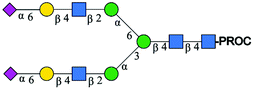
|
1221.98 [M + 2H]2+ | 1221.46 [M + 2H]2+ | 426 |
| a | A2G2S2 |
|
1221.98 [M + 2H]2+ | 1222.34 [M + 2H]2+ | 524 |
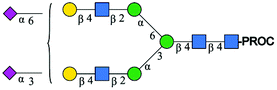
| 4 | A3G3S2(6)S(3) |
|
1033.7 [M + 3H]3+ | 1033.7[M + 3H]3+ | −34 |
| -PROC |
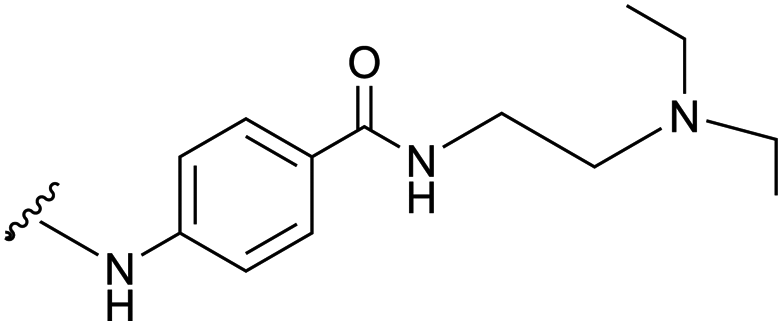
|
||||
The selectivity of the mass changes due to EEA was verified using glycan standards of known sialylation (Fig S2†). ESI-MS/MS fragmentation of α-2,6 linked sialic acid after EEA produced a fragment of 686.35 m/z whereas α-2,3 linked sialic acid produced a fragment of 656.33 which is consistent with ethyl esterification and amidation respectively33 (Fig S5†). Fig. 2B shows the UHPLC separation of procainamide labelled glycans after EEA. Here, the glycans corresponding to peaks 1, 2 and 3 are converted to the EEA products giving peaks 1′,2′ and 3′, (Fig S6†) with mass change consistent with α-2,6 linked sialic acid. However, for the triantennary glycan (peak 4) the EEA results suggest that at least one of the three sialic acids present is α-2,3 linked (Table 2). Interestingly, on the underivatized UHPLC trace, there was an earlier eluting peak of the same m/z as A2G2S2 (Fig. 2A peak a). EEA allowed us to identify this species as an A2G2S2 isomer with one α-2,3 linked sialic acid (Fig. 2B peak a′). The derivatized species was detected as two peaks, suggesting α-2,3 linked sialic acid can be present at either arm.
| Composition | Initial peak | Peak after EEA | Expected monoisotopic m/z after EEA [M + 2H]2+ | Observed monoisotopic m/z after EEA [M + 2H]2+ | ppma | Expected monoisotopic m/z after EEA [M + 3H]3+ | Observed monoisotopic m/z after EEA [M + 3H]3+ | ppm |
|---|---|---|---|---|---|---|---|---|
| a Amazon Bruker instrument accuracy quoted as ±0.5 Da. | ||||||||
| A2G1S1(6) | 1 | 1′ | 1009.43 | 1009.53 | −99 | 673.29 | 673.36 | −104 |
| A2G2S1(6) | 2 | 2′ | 1090.45 | 1090.57 | −110 | 727.30 | 727.38 | −110 |
| A2G2S1(6)S1(3) | a | a’ | 1235.51 | 1236.08 | −461 | 824.01 | 824.09 | −97 |
| A2G2S2(6) | 3 | 3′ | 1250.01 | 1250.57 | −448 | 833.68 | 833.75 | −84 |
| A3G3S2(6)S(3) | 4 | 4′ | 1052.09 | 1052.42 | −314 | |||
2.3 Large scale release and anion exchange chromatography
PNGaseF release of ∼30 mg of crude SGP obtained from fresh yolks was carried out over 2 days at 37 °C and monitored using HPAE-PAD and MALDI-MS analysis. Procainamide labelling of the released glycan mixture indicates it was 54% A2G2S2 (Fig. 3A). The released glycan mixture was separated by anion exchange chromatography producing fractions S1, S2 and S3 (Fig. 3B) which were also labelled with procainamide for HPLC-MS analysis (panels S1–S3 in Fig. 3A). ESI-MS and MS/MS of these fractions (Fig S7†) allowed for glycan composition identification, which was consistent with the preliminary N-glycan analysis. Even though in the original trace peak 2, A2G2S1, had a similar abundance to peak 1 A2[3]G1S1, the most abundant peak in the S1 fraction corresponded to A2[3]G1S1.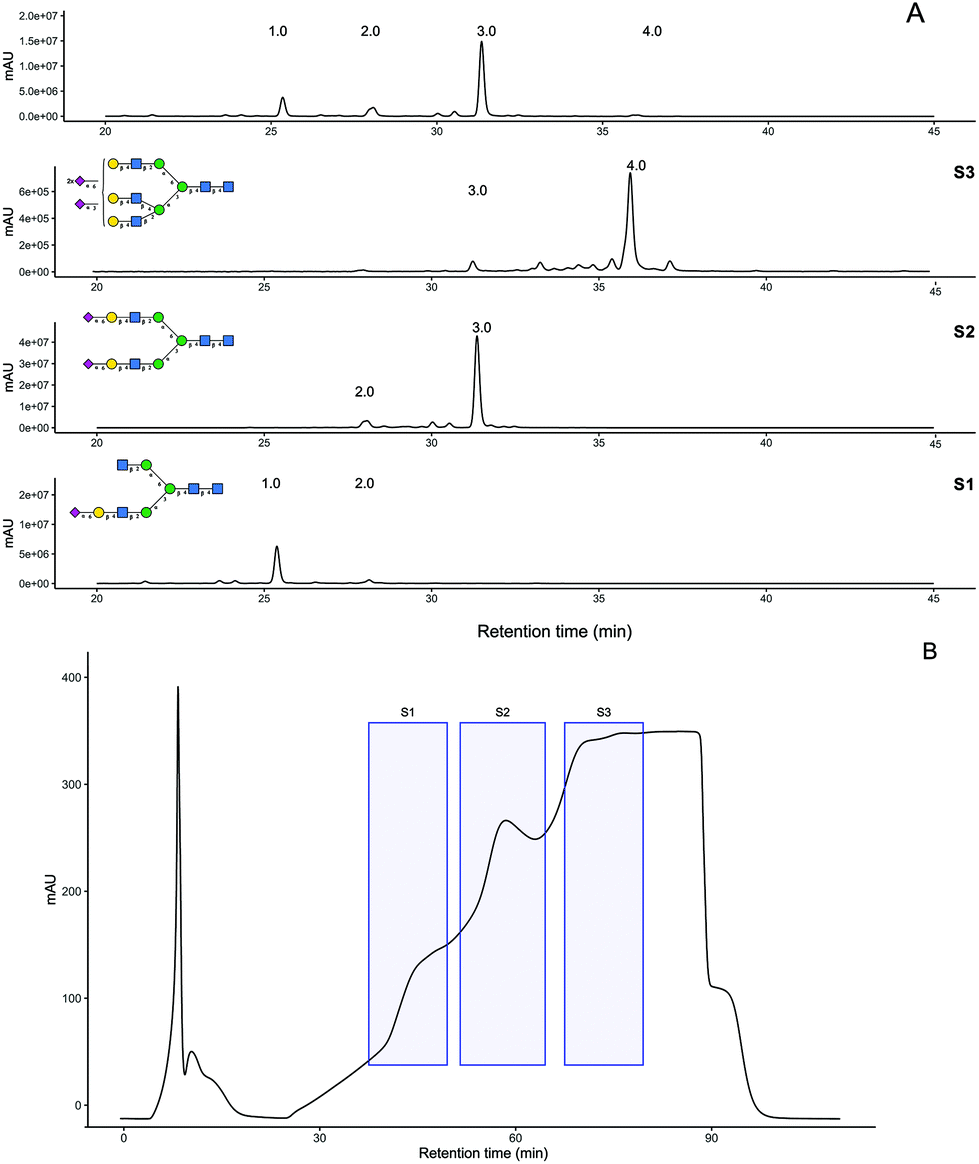 | ||
| Fig. 3 UHPLC-HILIC analysis of procainamide labelled N-glycans at different stages of purification (A) crude SGP before anion exchange separation. S1, S2, S3 correspond to the three regions collected during anion exchange chromatography. (B) UV-214 nm trace for anion exchange separation of released N-glycans from crude SGP, shaded region corresponds to the different pools of mono-sialylated S1, di-sialylated S2, and tri-sialylated S3, N-glycans. | ||
HPLC-MS analysis of the glycopeptide mixture indicates that the recovered N-glycans most likely originate from SPG glycoforms, with a minor contribution of naturally present free N-glycans (Fig S8†). Triantennary N-glycans have been found on egg yolk proteins such as phosvitin.34–36 However, recent glycopeptide analysis of the egg yolk did not identify any glycopeptides with triantennary glycans.37,38 In contrast, our UHPLC-MS analysis indicates that triantennary glycans are indeed present and they are most likely to be SGP analogues i.e., KVANKT glycopeptides (Fig. S9†). The detection of these glycopeptides and related N-glycans might be due to the larger scale of the extraction in comparison to the previously reported analytical approaches.
S1 and S2 fractions were further purified by porous graphitized chromatography (PGC), (Fig S10A†) whereas S3 was desalted by gel filtration. The resulting purified N-glycans were quantified using 2-amino benzamide (2AB) labelling (Fig S10B†) followed by comparison to a quantitative labelled chitotriose standard (Ludger BioQuant chitotriose). From this quantitative method, the purification process produced 8.6 mg of A2G2S2 at 94% purity and 1.3 mg A2[3]G1S1 at 93% purity (Fig S10C†).
2.4 Characterization of the purified N-glycans
2.4.1.1 H1-NMR. After PGC purification of the S1 fraction, 1H-NMR of the isolated glycan A2[3]G1S1 was recorded and compared to the A2G2S2 glycan that was produced in parallel (Fig S11†). The chemical shifts corresponding to the H3ax and H3eq of Neu5Ac indicate that both compounds have the same type of sialic acid linkage. Comparing the chemical shifts against literature data39 suggests that it is an α-2,6 linked sialic acid, which is also in agreement with EEA experiments. Inspection of the region corresponding to the anomeric protons provides information on the different galactosylation between structures (Fig S12A†). The A2[3]G1S1 glycan has only one doublet at 4.38 ppm corresponding to a single galactose whereas the A2G2S2 glycan (Fig S12B†) has 2 doublets at 4.36 ppm, one for each galactose present. Furthermore, the chemical shift for H1-GlcNAcs is also indicative of galactose substitution40 as the anomeric signals for the antennary GlcNAcs for A2[3]G1S1 at 4.50 ppm are resolved from the anomeric proton of GlcNAc-2 at 4.55 pm. On the other hand, for the fully galactosylated A2G2S2 glycan, the H1-GlcNAc of the antennary and the H1-GlcNAc-2 signals appear overlapped in a single multiplet at 4.51–4.52 ppm (Fig S12B†).
2.4.1.2 Elucidation of antennary galactose position in A2G1 isomer. To assist in the identification of which isomers of A2G1 were present in the isolated mixture, sialic acids were removed with HCl 50 mM at 65 °C before positive ion MALDI-MS/MS and UHPLC analysis. MALDI-MS was used to confirm that the sialic acid had been removed. Indeed, a peak at 1501.17 m/z (242 ppm) was observed, which corresponds to the [M + Na]+ for an A2G1 glycan (Fig. 4A). The position of the antennary galactose can be obtained from MS/MS fragmentation; if the [6]-antenna is not galactosylated a 2,5A4 fragmentation event will produce a 462.16 m/z ion. This is the case for the A2G1 glycan obtained from egg (Fig. 4B) for which a fragment of 462.08 m/z (171 ppm) is observed suggesting the galactose is on the [3]-antenna and the isomer is an A2[3]G1 glycan. As further confirmation, if a galactosylated [6]-antenna is present the same 2,5A4 fragmentation will produce a 624.21 m/z fragment (Fig S13†),41 this fragment was not detected in the MS/MS experiment for this isomer. As a positive control, the A2G2 glycan (Fig. 4C) was used as a reference compound containing a galactosylated [6]-antenna, as its MS/MS produces the diagnostic 624.25 m/z fragment (58 ppm) (Fig. 4D). Furthermore, the fragmentation pattern of the A2[3]G1 was preserved even after the enzymatic fucosylation using Helicobacter pyllori fucosyltransferase (Hp α-1,3FT) which will only transfer a fucose unit to the 3-OH of the galactosylated antennary GlcNAc. MS/MS analysis of the produced A2[3]G1F1 still produces a 462.10 m/z ion (125 ppm), thus confirming the vacancy of the GlcNAc at the [6]-antenna. (Fig S14†). Further evidence of the placement of the galactose in the [3]-antenna position was obtained by comparing the retention times for 2AB labelled samples of A2[3]G1 against a mixture of containing both A2[6]G1 and A2[3]G1 as seen in Fig. 5. For the mixture, the later eluting peak has been assigned as A2[3]G1 according to retention time databases (https://www.glycostore.com) (Fig. 5A). The glycan extracted from the egg produced only a single peak and its GU value corresponded with that of the later eluting peak (Fig. 5B). Thus, providing orthogonal confirmation to the MS/MS experiments. This data supports the original report19 that the A2G1S1 glycan isolated from egg corresponds to A2[3]G1S1 and contrasts with recent publications which identified it as a mixture.25 2AB retention times can also provide confirmation of the sialic acid linkage of the original glycan by comparing it against the chemoenzymatically synthetized α-2,3 linked isomers. (Fig S1†) The retention time of the precursor glycan is consistent with that of α-2,6 linked sialic acid,42 which correlates with both the NMR and EEA findings.
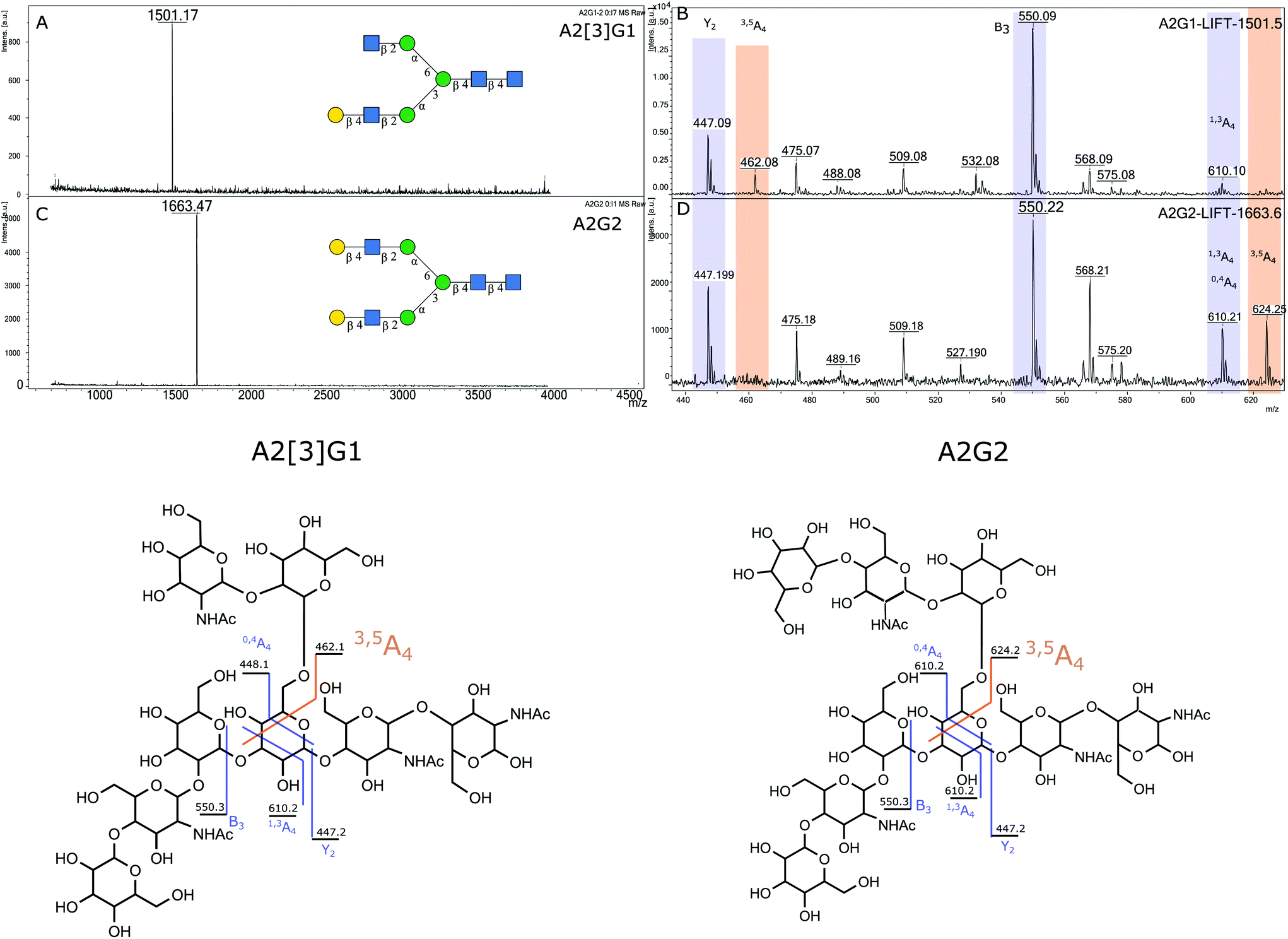 | ||
| Fig. 4 Positive ion MALDI MS and MS/MS analysis for A2[3]G1 and A2G2 (A) MALDI MS for A2G1 (B) MS/MS for A2G1 (C) MALDI MS for A2G2, (D) MS/MS of A2G2. | ||
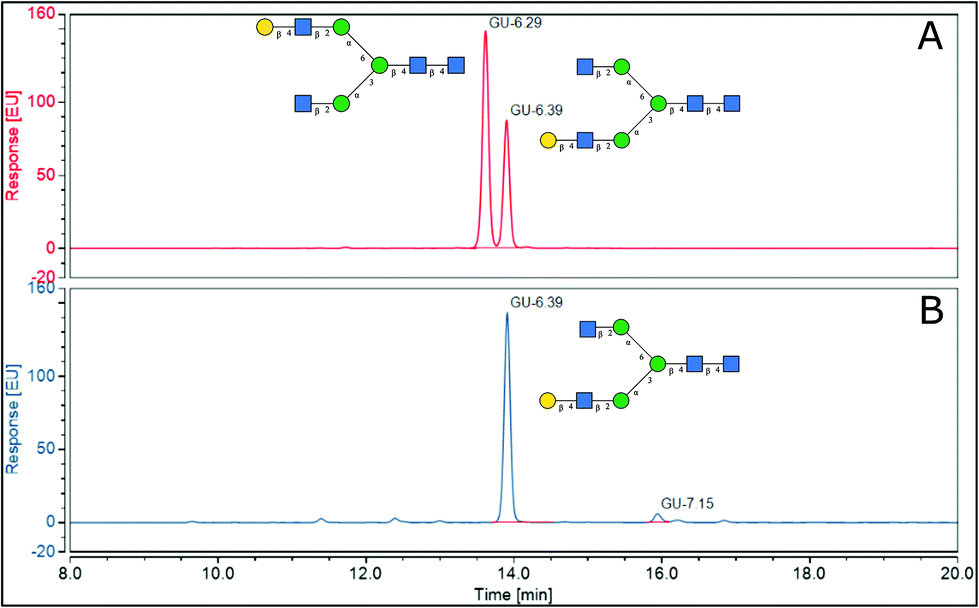 | ||
| Fig. 5 UHPLC analysis of 2AB labelled N-glycans (A) A2[6]G1 and A2[3]G1 glycan mixture (B) A2[3]G1 glycan from egg glycopeptides. | ||
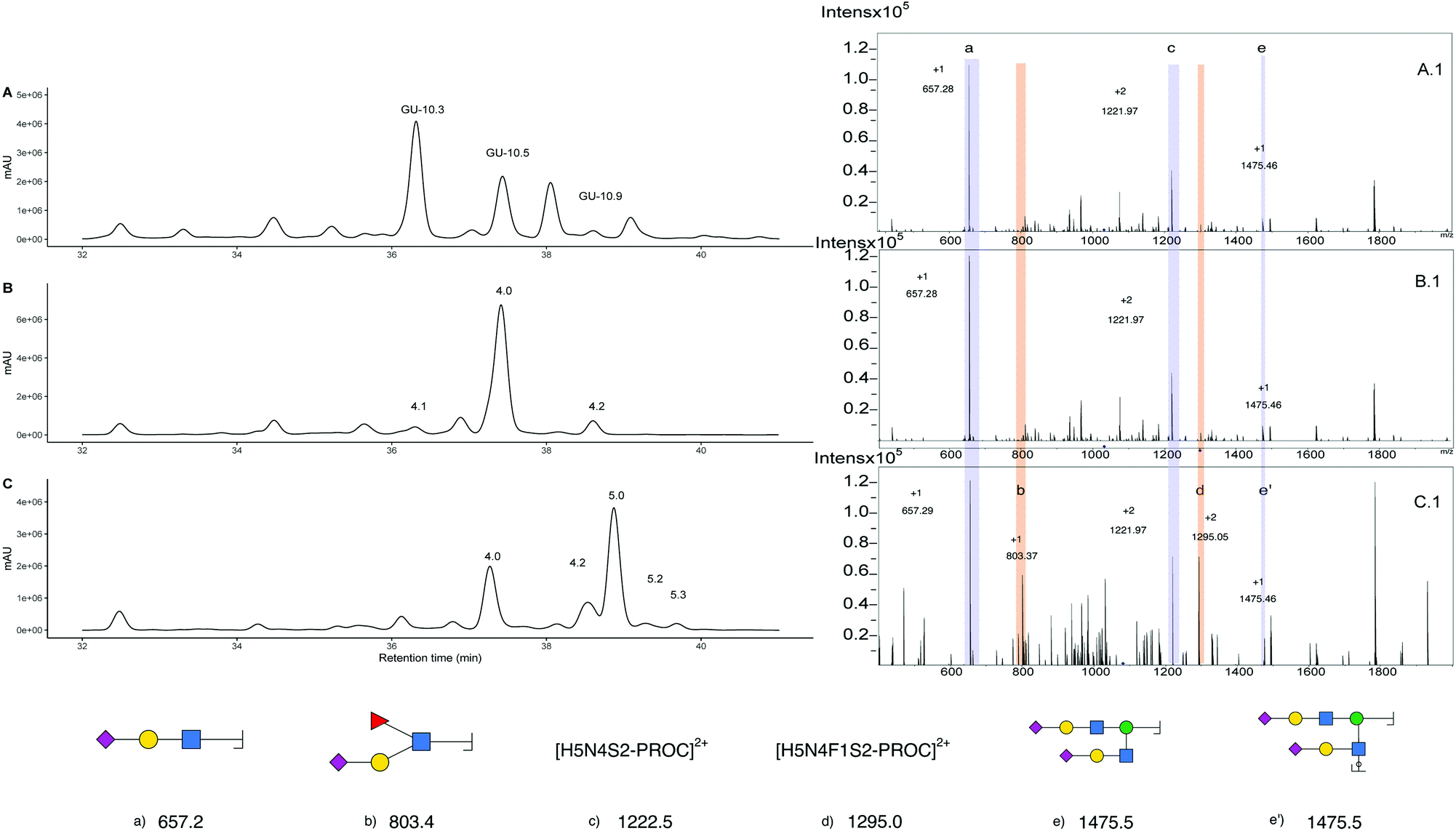 | ||
| Fig. 6 UHPLC analysis of procainamide labelled triantennary N-glycans. (A) Procainamide labelled N-glycans from bovine fetuin (A.1) MS/MS m/z 1033.70 procainamide labelled glycan corresponding to fetuin. (B) UHPLC of procainamide labelled N-glycans corresponding to the S3 fraction. (B.1) ESI-MS/MS of peak 4, m/z 1033.70 for procainamide labelled N-glycans from S3 fractions. (C) UHPLC for procainamide labelled N-glycans from S3, after antennary fucosylation. (C.1) ESI-MS/MS m/z 1082.40, obtained from peak 5.0 after antennary fucosylation. | ||
Further evidence of the presence of α-2,3 linked sialic acid is provided by chemoenzymatic reaction with Helicobacter pyllori Fucosyltransferase (Hp α1,3FT) and GDP-Fucose. The reaction was performed on unlabelled glycans and monitored by MALDI-MS. After one day, an aliquot of the reaction was procainamide labelled and the UHPLC-MS acquired (Fig. 6C). The newly formed peak (5.0) produces a [M + 3H]3+ ion of 1082.80 m/z that corresponds to the addition of only one fucose relative to the triantennary precursor (peak 4.0). Furthermore, MS/MS analysis (Fig. 6C.1) shows the formation of an 803.3 m/z fragment previously absent which is consistent with the addition of fucose to a sialylated antenna (original sialylated antenna mass 657.3 m/z), thus confirming the formation of the S-Lex tetrasaccharide. This enzymatic reaction is helpful for structural confirmation because the enzyme has strict requirements for the addition of fucose. For a GlcNAc to be an acceptor, it must be galactosylated at position 3 or 4. Additionally, the galactose needs to have the C6-OH vacant.44 This way, structures containing the α-2,3 linked sialic acids are acceptors whereas α-2,6 are not. In this case, this is observed as peak 4.2, which corresponds to a glycan containing three antennary α-2,6 sialic acids, was still detected after the enzymatic reaction. Since the main glycan structure present in the egg only has one α-2,3 linked sialic acid, this could allow for the control of the number of fucose units added when compared to N-glycans obtained from bovine fetuin which have more than one α-2,3 linked sialic acid. However, even though only one fucose can be added, there is no control over which antenna is fucosylated and three products (5.1, 5.0 and 5.2) produce ions corresponding to the addition of one fucose. (Fig S17†) that cannot be distinguished from their MS/MS data (Fig S17.B†). Furthermore, the EIC at 1131.10 m/z helps to identify peak 5.3 as the product corresponding to the addition of two fucoses, which likely corresponds to the fucosylation of the glycan at peak 4.1. (Fig S18B and C†). Additionally, other fucosylated structures were detected after the EIC scan for the peak at 803.3 m/z (Fig S19A and B†) suggesting there were additional glycan structures with α-2,3 linked sialic acids capable of acting as fucose acceptor (Fig S19C and D†). Purification of the triantennary acceptors is the next step in obtaining an increased yield and a purer product. Even though bovine fetuin is the most common and abundant source of triantennary glycans45 there is an interest in finding alternative sources such as plasma of other vertebrates.46 What makes the triantennary glycan from the egg special is the preference toward one isomer and the possibility to be isolated alongside the more abundant SGP.
2.5 Comparison with other methods
Although there have been different total synthesis strategies to produce asymmetric glycans,47,48 it is still a challenging process. Different semi-synthetic strategies to produce asymmetric glycans are compared in Fig. 7. Starting from SGP Kajihara et al.,20,49 produced a library of oligosaccharides by performing partial acid hydrolysis on crude pronase digested Asn-Fmoc glycans, from which A2[3]G1S1 and A2[3]G1 can be separated. A2[6]G1S1, the non-naturally occurring isomer, was also accessible by separating out the monosialylated A2G2[6]S1, which is a hydrolytic product of the main species A2G2S2. Even though this process realises the production of a wide number of targets simultaneously, the separation of the different species generated by acid hydrolysis is not trivial. Starting with a higher purity SGP Aoyama et al.26 produced a mixture of asymmetric monosialylated versions of SGP by partial acid hydrolysis. The monosialylated glycopeptides were separated from the unreacted precursor by reverse phase chromatography and were treated with exoglycosidases to produce A2[3]G1 and A2[6]G1 bearing glycopeptides. Although it is an improvement on the previous method, it relies on access to large amounts of pure SGP. Enzymatic methods to introduce asymmetry have also been explored, for example, taking advantage of the branch preference of E. coli Beta-galactosidase and50 starting from fully galactosylated A2G2, Van den Eijnden et al. were able to produce asymmetric N-glycans having galactose only on the [6]-antenna A2[6]G1. Different routes have also been explored to produce A2G2. Starting from bovine fetuin Calderon et al.51 obtained a mixture of triantennary and bi-antennary glycans that were separated by HILIC. The A2G2 product was subjected to partial exoglycosidase digestion producing A2[6]G1 as the main product after HILIC purification. This strategy has also been used to produce asymmetric core fucosylated glycans. Starting from SGP, Asn-Glycans were produced by pronase digestion8 followed by Cbz-amino group protection. Sequential exoglycosidase digestion was then used to produce a completely de-galactosylated Cbz-glycan. The removal of galactose was necessary for the introduction of α-1,6 core fucose.52 Galactose was then re-installed enzymatically, to produce the symmetric fully galactosylated glycan, that when digested with the E. coli beta-galactosidase afforded the FA2[6]G1 as the main product after HILIC purification.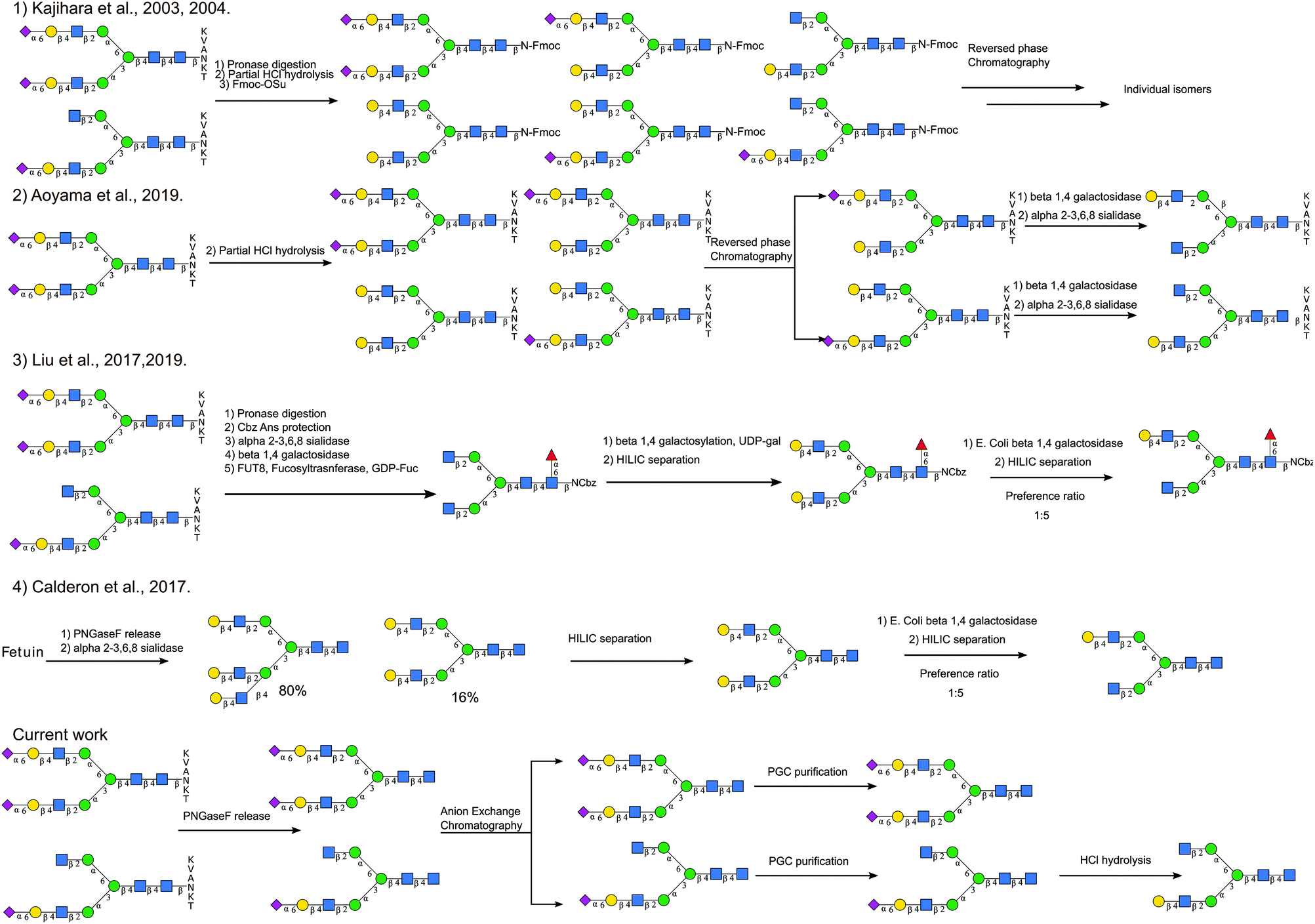 | ||
| Fig. 7 Comparison of different semisynthetic approaches to produce asymmetric N-glycans. | ||
The approach we present here does not require the introduction of branch asymmetry by chemical or enzymatic means, it relies on the separation of the different glycans species occurring natively according to the number of sialic acids present, and then additional HPLC purification. It allowed us, not only to recover asymmetric monosialylated glycans, namely A2[3]G1S1, but also to obtain triantennary glycans that have not been associated previously with the SGP purification process.
3. Conclusions
The isolation of SGP has been extensively reported in the past, and it has contributed significantly to the process of production of homogenous glycoproteins. However, we want to emphasize the additional opportunity to obtain naturally occurring asymmetric and triantennary N-glycans that can be concomitantly isolated alongside the most abundant glycan species. Isolating these N-glycans can complement existing chemoenzymatic approaches or serve as standards to verify the products of total synthetic strategies.Author contributions
JMMD: Conceptualisation, investigation, writing–original draft, writing–review and editing, visualization. AM: Investigation (sialic acid derivatization experiments), writing–review and editing. SRP: Resources, writing–review and editing, supervision. JLH: Writing–review and editing, visualization, supervision, funding acquisition. DIRS: Conceptualisation, writing–review and editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors are based at Ludger Ltd which specialises in the production of commercial glycan standards for use in the field of glycomics and the analysis of biopharmaceuticals.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under a Marie Sklodowska Curie ITN (GlySign, Grant No. 722095 and Nanocarb, Grant No. 814236). Glycosyltransferase enzymes were obtained thanks to the BBSRC/Innovate UK IB catalyst award ‘Glycoenzymes for Bioindustries’(BB/M029018/1) grant.References
- H. Retzek, E. Steyrer, E. J. Sanders, J. Nimpf and W. J. Schneider, Molecular Cloning and Functional Characterization of Chicken Cathepsin D, a Key Enzyme for Yolk Formation, DNA Cell Biol., 1992, 11, 661–672 CrossRef CAS PubMed.
- A. J. Fairbanks, Synthetic and semi-synthetic approaches to unprotected N-glycan oxazolines, Beilstein J. Org. Chem., 2018, 14, 416–429 CrossRef CAS PubMed.
- C. Ou, C. Li, R. Zhang, Q. Yang, G. Zong, Y. Dai, R. L. Francis, S. Bournazos, J. V. Ravetch and L. X. Wang, One-Pot Conversion of Free Sialoglycans to Functionalized Glycan Oxazolines and Efficient Synthesis of Homogeneous Antibody-Drug Conjugates through Site-Specific Chemoenzymatic Glycan Remodeling, Bioconjugate Chem., 2021, 32, 1888–1897 CrossRef CAS PubMed.
- C. H. Lin, M. Shimazaki, C. H. Wong, M. Koketsu, L. R. Juneja and M. Kim, Enzymatic synthesis of a sialyl Lewis X dimer from egg yolk as an inhibitor of E-selectin, Bioorg. Med. Chem., 1995, 3, 1625–1630 CrossRef CAS PubMed.
- Y. Maki, T. Mima, R. Okamoto, M. Izumi and Y. Kajihara, Semisynthesis of Complex-Type Biantennary Oligosaccharides Containing Lactosamine Repeating Units from a Biantennary Oligosaccharide Isolated from a Natural Source, J. Org. Chem., 2018, 83, 443–451 CrossRef CAS PubMed.
- Y. Maki, R. Okamoto, M. Izumi, T. Murase and Y. Kajihara, Semisynthesis of Intact Complex-Type Triantennary Oligosaccharides from a Biantennary Oligosaccharide Isolated from a Natural Source by Selective Chemical and Enzymatic Glycosylation, J. Am. Chem. Soc., 2016, 138, 3461–3468 CrossRef CAS PubMed.
- Y. Maki, R. Okamoto, M. Izumi and Y. Kajihara, Chemical Synthesis of an Erythropoietin Glycoform Having a Triantennary N-Glycan: Significant Change of Biological Activity of Glycoprotein by Addition of a Small Molecular Weight Trisaccharide, J. Am. Chem. Soc., 2020, 142, 20671–20679 CrossRef CAS PubMed.
- L. Liu, A. R. Prudden, C. J. Capicciotti, G. P. Bosman, J. Y. Yang, D. G. Chapla, K. W. Moremen and G. J. Boons, Streamlining the chemoenzymatic synthesis of complex N-glycans by a stop and go strategy, Nat. Chem., 2019, 11, 161–169 CrossRef CAS PubMed.
- H. Hinou, Aniline derivative/DHB/alkali metal matrices for reflectron mode MALDI-TOF and TOF/TOF MS analysis of unmodified sialylated oligosaccharides and glycopeptides, Int. J. Mass Spectrom., 2019, 443, 109–115 CrossRef CAS.
- T. Nishikaze, S. I. Kawabata and K. Tanaka, In-depth structural characterization of N -Linked glycopeptides using complete derivatization for carboxyl groups followed by positive- and negative-ion tandem mass spectrometry, Anal. Chem., 2014, 86, 5360–5369 CrossRef CAS PubMed.
- T. Nishikaze, Sensitive and Structure-Informative N-Glycosylation Analysis by MALDI-MS; Ionization, Fragmentation, and Derivatization, Mass Spectrom., 2017, 6, A0060 CrossRef PubMed.
- S. Yang, L. Zhang, S. Thomas, Y. Hu, S. Li, J. Cipollo and H. Zhang, Modification of sialic acids on solid-phase: accurate characterization of protein sialylation, Anal. Chem., 2017, 89, 6330–6335 CrossRef CAS PubMed.
- C. Ma, H. Zhu, C. Xiao, D. Liu, G. Edmunds, L. Wen, C. Ma, J. Li and P. G. Wang, Solid-phase reductive amination for glycomic analysis, Anal. Chim. Acta, 2017, 962, 32–40 CrossRef PubMed.
- M. Koketsu, in Hen Eggs Their basic and applied Science, ed. T. Yamamoto, Y. Lekn Raj, H. Hatta and M. Kim, 1997, pp. 99–133 Search PubMed.
- A. Seko, M. Koketsu, M. Nishizono, Y. Enoki, H. R. Ibrahim, L. R. Juneja, M. Kim and T. Yamamoto, Occurrence of a sialylglycopeptide and free sialylglycans in hen's egg yolk, Biochim. Biophys. Acta, 1997, 1335, 23–32 CrossRef CAS.
- Y. Zou, Z. Wu, L. Chen, X. Liu, G. Gu, M. Xue, P. G. Wang and M. Chen, An efficient approach for large-scale production of sialyglycopeptides from egg yolks, J. Carbohydr. Chem., 2012, 31, 436–446 CrossRef CAS.
- B. Sun, W. Bao, X. Tian, M. Li, H. Liu, J. Dong and W. Huang, A simplified procedure for gram-scale production of sialylglycopeptide (SGP) from egg yolks and subsequent semi-synthesis of Man3GlcNAc oxazoline, Carbohydr. Res., 2014, 396, 62–69 CrossRef CAS PubMed.
- K. Alagesan and D. Kolarich, Improved strategy for large scale isolation of sialylglycopeptide (SGP) from egg yolk powder, MethodsX, 2019, 6, 773–778 CrossRef PubMed.
- M. Koketsu, L. R. Juneja, M. Kim, M. Ohta, F. Matsuura and T. Yamamoto, Sialyoligosaccharides of Delipidated Egg Yolk Fraction, J. Food Sci., 1993, 58, 743–747 CrossRef CAS.
- Y. Kajihara, Y. Suzuki, K. Sasaki and L. R. Juneja, Chemoenzymatic synthesis of diverse asparagine-linked oligosaccharides, Methods Enzymol., 2003, 362, 44–64 CAS.
- W. Sumiyoshi, S. Nakakkita, N. Miyanishi and J. Hirabayashi, Strategic Glycan Elution Map for the Production of Human-Type N -Linked Oligosaccharides: The Case of Hen Egg Yolk and White, Biosci., Biotechnol., Biochem., 2009, 73, 543–551 CrossRef CAS PubMed.
- F. Tang, L. X. Wang and W. Huang, Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody-drug conjugates, Nat. Protoc., 2017, 12, 1702–1721 CrossRef CAS PubMed.
- A. García-García, L. Ceballos-Laita, S. Serna, R. Artschwager, N. C. Reichardt, F. Corzana and R. Hurtado-Guerrero, Structural basis for substrate specificity and catalysis of α1,6-fucosyltransferase, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- A. Garcia-Garcia, S. Serna, Z. Yang, I. Delso, V. Taleb, T. Hicks, R. Artschwager, S. Y. Vakhrushev, H. Clausen, J. Angulo, F. Corzana, N. C. Reichardt and R. Hurtado-guerrero, FUT8-Directed Core Fucosylation of N - glycans Is Regulated by the Glycan Structure and Protein Environment, ACS Catal., 2021, 11(15), 9052–9065 CrossRef CAS.
- L. Liu, A. R. Prudden, G. P. Bosman and G. J. Boons, Improved isolation and characterization procedure of sialylglycopeptide from egg yolk powder, Carbohydr. Res., 2017, 452, 122–128 CrossRef CAS PubMed.
- M. Aoyama, N. Hashii, W. Tsukimura, K. Osumi, A. Harazono, M. Tada, M. Kiyoshi, A. Matsuda and A. Ishii-Watabe, Effects of terminal galactose residues in mannose α1-6 arm of Fc-glycan on the effector functions of therapeutic monoclonal antibodies, MAbs, 2019, 11, 826–836 CrossRef CAS PubMed.
- S. Pawar, L. Hsu, T. Narendar Reddy, M. Ravinder, C. T. Ren, Y. W. Lin, Y. Y. Cheng, T. W. Lin, T. L. Hsu, S. K. Wang, C. H. Wong and C. Y. Wu, Synthesis of Asymmetric N-Glycans as Common Core Substrates for Structural Diversification through Selective Enzymatic Glycosylation, ACS Chem. Biol., 2020, 15, 2382–2394 CrossRef CAS PubMed.
- C. Y. Liew, C. C. Yen, J. L. Chen, S. T. Tsai, S. Pawar, C. Y. Wu and C. K. Ni, Structural identification of N-glycan isomers using logically derived sequence tandem mass spectrometry, Commun. Chem., 2021, 4, 1–11 CrossRef.
- P. Bansal, A. Ben Faleh, S. Warnke and T. R. Rizzo, Identification of: N -glycan positional isomers by combining IMS and vibrational fingerprinting of structurally determinant CID fragments, Analyst, 2022, 147, 704–711 RSC.
- Q. Rao, M. C. Fisher, M. Guo and T. P. Labuza, Storage stability of a commercial hen egg yolk powder in dry and intermediate-moisture food matrices, J. Agric. Food Chem., 2013, 61, 8676–8686 CrossRef CAS PubMed.
- G. S. M. Lageveen-Kammeijer, N. de Haan, P. Mohaupt, S. Wagt, M. Filius, J. Nouta, D. Falck and M. Wuhrer, Highly sensitive CE-ESI-MS analysis of N-glycans from complex biological samples, Nat. Commun., 2019, 10, 2137 CrossRef PubMed.
- T. Pongracz, A. Verhoeven, M. Wuhrer and N. De Haan, The structure and role of lactone intermediates in linkage-specific sialic acid derivatization reactions, Glycoconj. J., 2021, 38, 157–166 CrossRef CAS PubMed.
- A. B. Moran, R. A. Gardner, M. Wuhrer, G. S. M. Lageveen-Kammeijer and D. I. R. Spencer, Sialic Acid Derivatization of Fluorescently Labeled N -Glycans Allows Linkage Differentiation by Reversed-Phase Liquid Chromatography–Fluorescence Detection–Mass Spectrometry, Anal. Chem., 2022, 94, 6639–6648 CrossRef CAS PubMed.
- R. L. Brockbank and H. J. Vogel, Structure of the oligosaccharide of hen phosvitin as determined by two-dimensional proton NMR of the intact glycoprotein, Biochemistry, 1990, 29(23), 5574–5583 CrossRef CAS PubMed.
- R. Shainkin and G. E. Perlmann, Phosvitin, a phosphoglycoprotein: Composition and partial structure of carbohydrate moiety, Arch. Biochem. Biophys., 1971, 145, 693–700 CrossRef CAS PubMed.
- R. Shainkin and G. E. Perlmann, Phosvitin a Phosphoglycoprotein: isolation and characterization of a glycopeptide from phosvitin, J. Biol. Chem., 1971, 246, 2278–2284 CrossRef CAS PubMed.
- S. Arena and A. Scaloni, An Extensive Description of the Peptidomic Repertoire of the Hen Egg Yolk Plasma, J. Agric. Food Chem., 2018, 66, 3239–3255 CrossRef CAS PubMed.
- F. Geng, Y. Xie, J. Wang, K. Majumder, N. Qiu and M. Ma, N-Glycoproteomic Analysis of Chicken Egg Yolk, J. Agric. Food Chem., 2018, 66, 11510–11516 CrossRef CAS PubMed.
- J. F. G. Vliegenthart and J. P. Kamerling, 1H NMR Structural-Reporter-Group Concepts in Carbohydrate Analysis, Compr. Glycosci., 2007, 2–4, 133–191 Search PubMed.
- J. F. G. Vliegenthart, L. Dorland and H. van Halbeek, High-Resolution, 1H- Nuclear Magnatic Resonance spectroscopy as a tool in the strucutral analysis of carbohydrates related to glycoproteins, Academic Press, 1983, vol. 41 Search PubMed.
- D. J. Harvey, R. H. Bateman and M. R. Green, High-energy collision-induced fragmentation of complex oligosaccharides ionized by matrix-assisted laser desorption/ionization mass spectrometry, J. Mass Spectrom., 1997, 32, 167–187 CrossRef CAS.
- Y. Huang, Y. Nie, B. Boyes and R. Orlando, Resolving isomeric glycopeptide glycoforms with hydrophilic interaction chromatography (HILIC), J. Biomol. Tech., 2016, 27, 98–104 CrossRef PubMed.
- E. D. Green, G. Adelt, J. U. Baenziger, S. Wilson and H. van Halbeek, The asparagine-linked oligosaccharides on bovine fetuin. Structural analysis of N-glycanase-released oligosaccharides by 500-megahertz1H NMR spectroscopy, J. Biol. Chem., 1988, 263, 18253–18268 CrossRef CAS PubMed.
- J. Ye, H. Xia, N. Sun, C. C. Liu, A. Sheng, L. Chi, X. W. Liu, G. Gu, S. Q. Wang, J. Zhao, P. Wang, M. Xiao, F. Wang and H. Cao, Reprogramming the enzymatic assembly line for site-specific fucosylation, Nat. Catal., 2019, 2, 514–522 CrossRef CAS.
- R. L. Valk-Weeber, L. Dijkhuizen and S. S. van Leeuwen, Large-scale quantitative isolation of pure protein N-linked glycans, Carbohydr. Res., 2019, 479, 13–22 CrossRef CAS PubMed.
- J. Seino, H. Fujihira, S. I. Nakakita, Y. Masahara-Negishi, E. Miyoshi, J. Hirabayashi and T. Suzuki, Occurrence of free sialyl oligosaccharides related to N-glycans (sialyl free N-glycans) in animal sera, Glycobiology, 2016, 26, 1072–1085 CrossRef CAS PubMed.
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. Mcbride, R. P. de Vries, J. Glushka, J. C. Paulson and G. Boons, A General Strategy for the Chemoenzymatic Synthesis of Asymmetrically Branched N-Glycans, Science, 2014, 341, 40–47 Search PubMed.
- I. A. Gagarinov, T. Li, J. S. Toraño, T. Caval, A. D. Srivastava, J. A. W. Kruijtzer, A. J. R. Heck and G. J. Boons, Chemoenzymatic approach for the preparation of asymmetric bi-, tri-, and tetra-antennary N-glycans from a common precursor, J. Am. Chem. Soc., 2017, 139, 1011–1018 CrossRef CAS PubMed.
- Y. Kajihara, Y. Suzuki, N. Yamamoto, K. Sasaki, T. Sakakibara and L. R. Juneja, Prompt Chemoenzymatic Synthesis of Diverse Complex-Type Oligosaccharides and Its Application to the Solid-Phase Synthesis of a Glycopeptide with Asn-Linked Sialyl-undeca- and Asialo-nonasaccharides, Chem. – Eur. J., 2004, 10, 971–985 CrossRef CAS PubMed.
- D. H. van den Eijnden, W. M. Blanken and A. van Vliet, Branch specificity of B-Galactosidase from Escherichia coli, Carbohydr. Res., 1986, 151, 329–335 CrossRef CAS PubMed.
- A. D. Calderon, J. Zhou, W. Guan, Z. Wu, Y. Guo, J. Bai, Q. Li, P. G. Wang, J. Fang and L. Li, An enzymatic strategy to asymmetrically branched: N -glycans, Org. Biomol. Chem., 2017, 15, 7258–7262 RSC.
- A. D. Calderon, Y. Liu, X. Li, X. Wang, X. Chen, L. Li and P. G. Wang, Substrate specificity of FUT8 and chemoenzymatic synthesis of core-fucosylated asymmetric: N -glycans, Org. Biomol. Chem., 2016, 14, 4027–4031 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of analytical method, isolation, purification, characterization of N-glycans and N-glycan nomenclature. See DOI: https://doi.org/10.1039/d2ob00615d |
| This journal is © The Royal Society of Chemistry 2022 |