
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scale-up and optimization of the synthesis of dual CBP/BRD4 inhibitor ISOX-DUAL†
Anthony K.
Edmonds
a,
Catherine S.
Oakes
b,
Storm
Hassell-Hart
 a,
Didier
Bruyère
b,
Graham J.
Tizzard
c,
Simon J.
Coles
c,
Robert
Felix
b,
Hannah J.
Maple
b,
Graham P.
Marsh
b and
John
Spencer
*ad
a,
Didier
Bruyère
b,
Graham J.
Tizzard
c,
Simon J.
Coles
c,
Robert
Felix
b,
Hannah J.
Maple
b,
Graham P.
Marsh
b and
John
Spencer
*ad
aChemistry Department, School of Life Sciences, University of Sussex, Falmer, Brighton BN1 9QJ, UK. E-mail: j.spencer@sussex.ac.uk; Tel: +44(0) 1273 872722
bBio-Techne (Tocris), The Watkins Building, Atlantic Road, Bristol, BS11 9QD, UK
cNational Crystallography Service, School of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
dSussex Drug Discovery Centre, School of Life Sciences, University of Sussex, Falmer, Brighton BN1 9QJ, UK
First published on 27th April 2022
Abstract
ISOX-DUAL is a dual inhibitor of CBP/p300 (IC50 = 0.65 μM) and BRD4 (IC50 = 1.5 μM) bromodomains, and a useful chemical probe for epigenetic research. Aspects of the published synthetic route to this compound and its analogues are small-scale, poor-yielding or simply unamenable to scale-up without optimization. Herein we describe the development of a refined synthesis that circumvents the challenges of the original report, with notable improvements to several of the key synthetic transformations. Moreover, a general Suzuki Miyaura protocol for the late stage installation of alternative dimethyl-isoxazole acetyl-lysine (KAc) binding motifs is presented.
Introduction
ISOX-DUAL ([3-[4-[2-[5-(dimethyl-1,2-oxazol-4-yl)-1-[2-(morpholin-4-yl)ethyl]-1H-1,3-benzodiazol-2-yl]ethyl]phenoxy]propyl]dimethylamine) 1 is a dual inhibitor of BRD4 and CBP/p300 bromodomains.1 It is structurally related to a series of CBP/p300 and BRD4 bromodomain inhibitors through a benzimidazole central scaffold with an adjoining 3,5-dimethylisoxazole group, which acts as an acetylated lysine (KAc) mimic.2,3 In previous work, differential activity and overall potency were tuned through structure-based design, yielding compounds with selectivity for CBP/p300 over BRD4, together with a probe (ISOX DUAL) possessing balanced potency against both targets (Fig. 1). ISOX-DUAL is a useful tool compound for exploring transcriptional regulation.1,2,4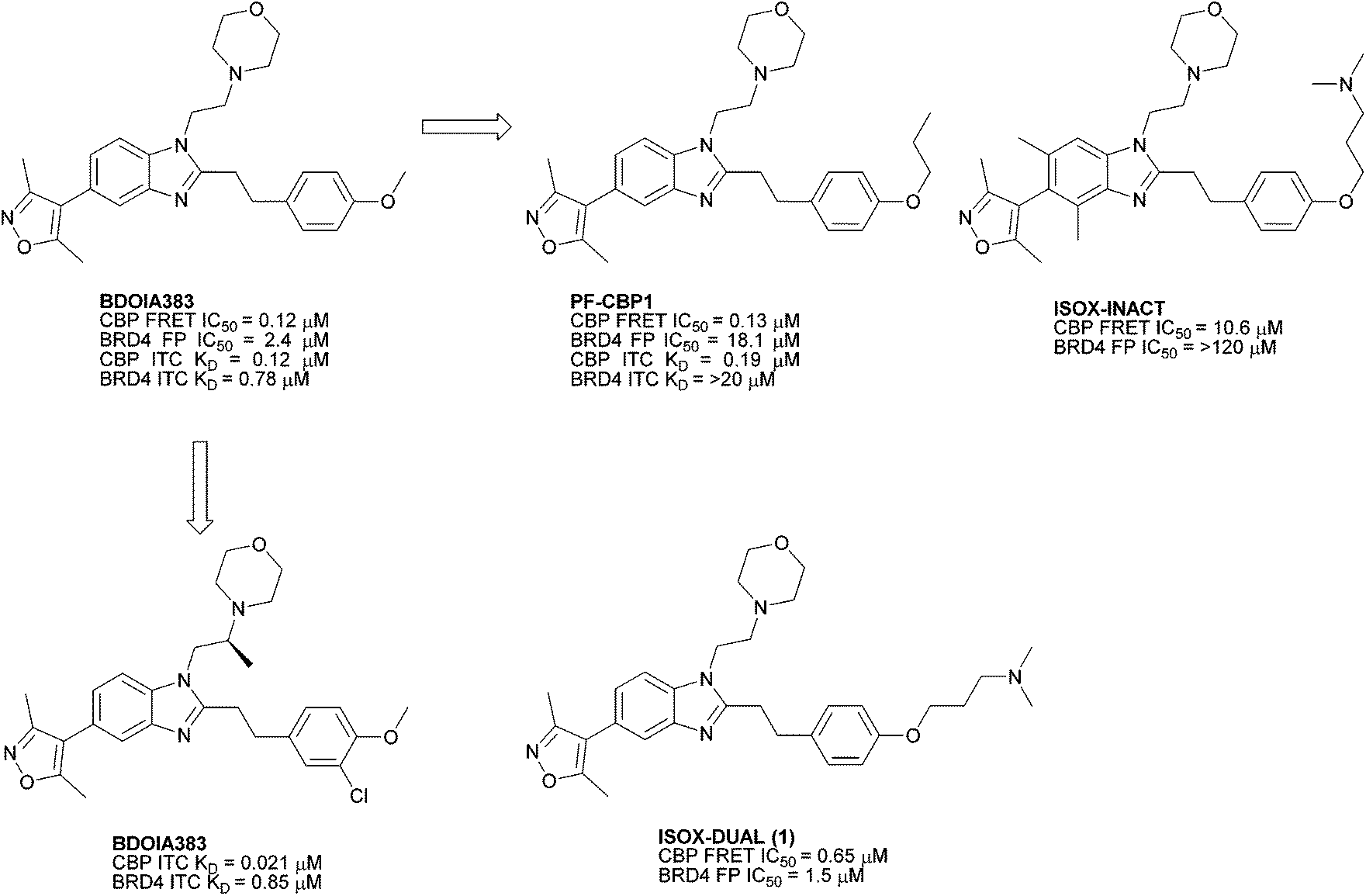 | ||
| Fig. 1 Reported CBP BRD inhibitors including dual binding ISOX-DUAL (1).1 | ||
Our interest in functionalised bromodomain inhibitors necessitated a feasible, scalable route to 1 and its analogues. However, the reported synthetic strategy (Scheme 1) is, in places, both small-scale in nature and inviting of optimisation.1 The key transformation is an elegant, albeit low-yielding, one-pot nitro-reduction and in situ cyclisation sequence, in which exposure of nitroaniline 4 to sodium dithionite in the presence of aldehyde 5 facilitated the installation of the benzimidazole framework in one synthetic operation. Of additional note, the late-stage alkylation of phenol 7 to furnish 1 was also of modest yield: these two crucial stages thereby being significant contributors to an overall isolated yield of just 1%.
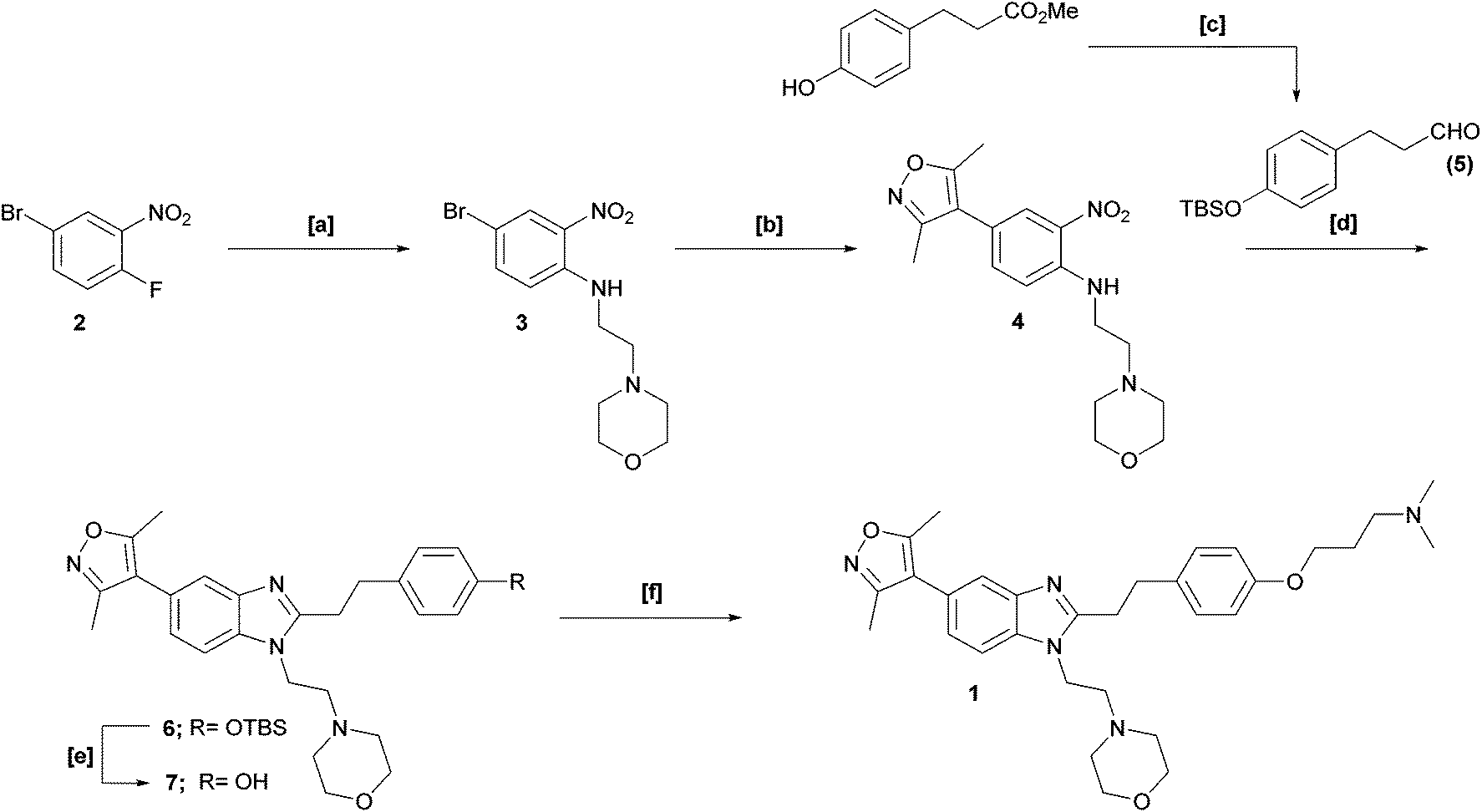 | ||
| Scheme 1 Published synthetic strategy for the preparation of ISOX-DUAL 1. Reagents & Conditions: [a] 4-(2-aminoethyl)morpholine, TEA, DMSO, 80 °C, 1 h (80%); [b] 3,5-dimethylisoxazole-4-boronic acid, PdCl2(dppf)·DCM, Cs2CO3, dioxane/H2O, 100 °C, 16 h (62%); [c] (i) TBSCl, imidazole, DCM, rt; (ii) LiAlH4, THF, 30 °C; (iii) DMP, DCM, rt, 18 h (72% over 3 steps); [d] Na2S2O4 (aq), EtOH, 80 °C, 12 h (33%); [e] TBAF, THF, rt, 3 h (62%); [f] Br(CH2)3NMe2.HCl, KOtBu, DMA, 60 °C, 2 h (11%). | ||
Our modified retrosynthetic analysis identified that independent reduction of nitroaniline 4 to bis-aniline 8, followed by an amide coupling with subsequent acid-catalysed benzimidazole formation, might furnish the target 1 in higher yield (Scheme 2). Employing carboxylic acid 9 instead of aldehyde 5 offers the prospect of shortening the synthetic route by two chemical stages in comparison to the original, and further provides the distinct advantage of enabling an alternative early-stage phenolic alkylation, the comparative transformation having been a significant limitation to the output of the published synthesis.
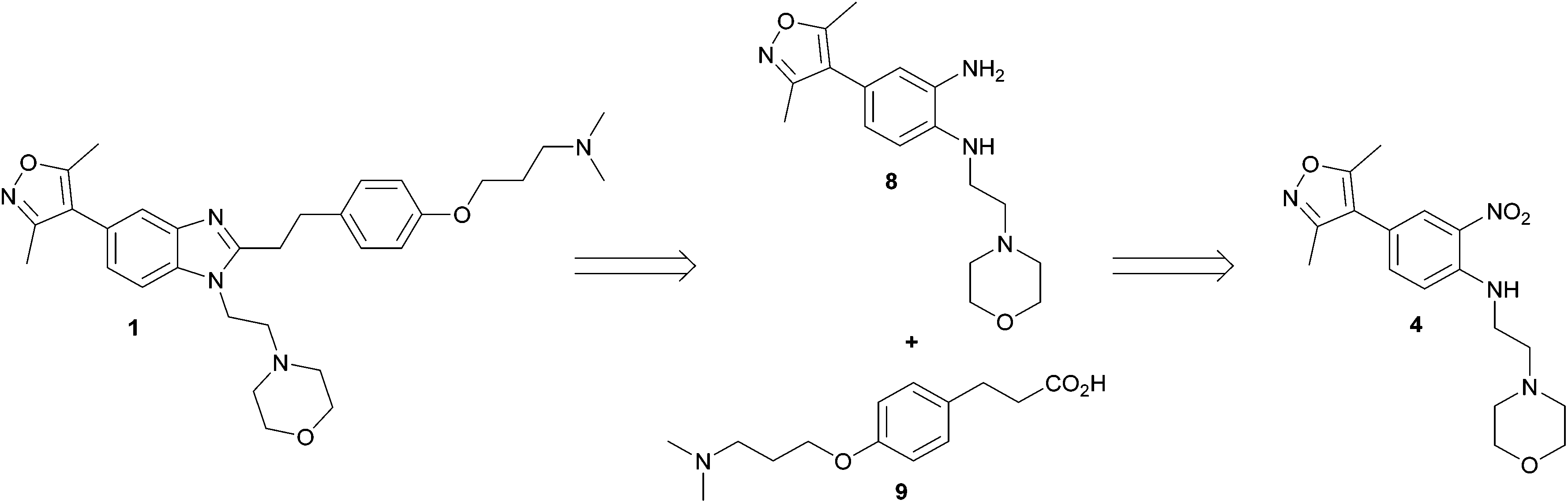 | ||
| Scheme 2 Retrosynthetic approach to ISOX-DUAL 1. | ||
The initial stage in both synthetic approaches involves an SNAr reaction between commercially available 4-bromo-1-fluoro-nitrobenzene 2 and 4-(2-aminoethyl)morpholine. In our hands, this transformation, to prepare bromide 3, was readily replicated on large scale in near-quantitative yield, and further, was demonstrated to proceed rapidly under microwave (MW) conditions with commensurate output.
Next, in sequence, we turned to elaboration of 3 to install the requisite pendant isoxazole functionality. This necessitated examination of the Suzuki–Miyaura cross-coupling reaction, where we hypothesised that competing hydrodehalogenation and protodeboronation could be potential contributory factors to a drop-off in expected product yield in the original report.5–8 A screen for substrate scope was performed under microwave conditions using a range of palladium catalysts and bases, exploring their effectiveness as boronic acid, boronate ester and MIDA boronate coupling partners, with selected results displayed in Table 1. From this screen, we found that employment of a boronate ester bearing the isoxazole moiety performed the best, with potassium phosphate and PdCl2(dppf)·DCM acting as base and catalyst, respectively. This reaction could be conducted on small scale in a microwave and was successfully demonstrated thermally on a larger scale to give almost 40 g of product, in near quantitative yield.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Methoda | (3) (mmol) | Boronate Species | Catalyst | Base | Yieldb,c (%) |
| a Typical method: 5 mol% Pd-catalyst, 2.6 eq. base (1 M solution in H2O), 1,4-dioxane, MW, 150 °C, 3 h, or reflux, 16 h. b Isolated yield after purification. c Crude yield assessed by 1H NMR. d 7.6 eq. base used.9 e 1 mol% catalyst. | ||||||
| 1 | Thermal | 30 | Boronic acid | PdCl2(dppf)·DCM | Cs2CO3 | 53b |
| 2 | MW | 0.5 | Boronic acid | PdCl2(dppf)·DCM | Cs2CO3 | 54c |
| 3 | MW | 0.5 | Boronate ester | PdCl2(dppf)·DCM | Cs2CO3 | 74c |
| 4 | MW | 0.5 | Boronate ester | PdCl2(dtbpf) | Cs2CO3 | 71c |
| 5 | MW | 0.5 | Boronate ester | PdCl2(dppf)·DCM | CsF | 72c |
| 6 | MW | 0.5 | Boronate ester | PdCl2(dtbpf) | CsF | 47c |
| 7 | MW | 0.5 | Boronate ester | PdCl2(dppf)·DCM | K3PO4 | 72c |
| 8 | MW | 0.5 |
|
PdCl2(dppf)·DCM | K3PO4 | 60c |
| 9 | MW | 0.5 | MIDA ester | PdCl2(dppf)·DCM | K3PO4d | 51c |
| 10 | MW | 0.5 | Boronate ester | PdCl2(dppf)·DCM | CsF | 74c |
| 11 | MW | 1 | Boronate ester | PdCl2(dppf)·DCM | Cs2CO3 | 79b |
| 12 | MW | 1 | Boronate ester | PdCl2(dppf)·DCM | CsF | 95b |
| 13 | MW | 1 | Boronate ester | PdCl2(dppf)·DCM | K3PO4 | 95b |
| 14 | MW | 1 | Boronate ester | PdCl2(dppf)·DCMe | K3PO4 | 66b |
| 15 | Thermal | 115 | Boronate ester | PdCl2(dppf)·DCM | K3PO4 | 94b |
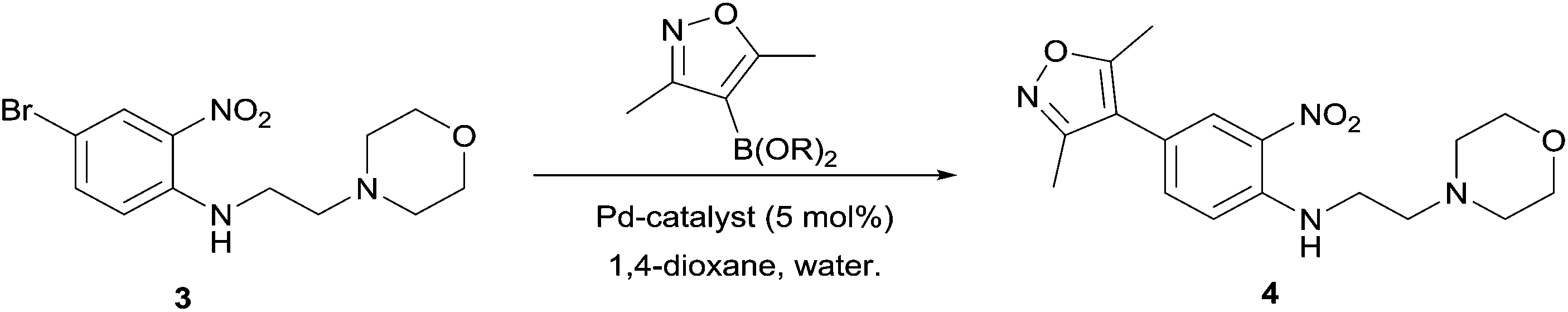
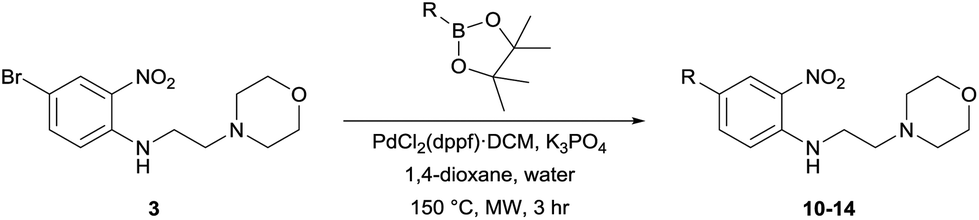
To demonstrate the synthetic utility and reliability of this now optimised procedure, a small series of analogues (10–14) were synthesised employing a variety of heterocyclic boronate esters (Table 2). This demonstrates the potential for late-stage KAc bio-isostere modification to these and similar scaffolds, which is of interest to ongoing work in our laboratory. Yields tend to be good to excellent and all analogues were further characterised in the solid state by X-ray studies.
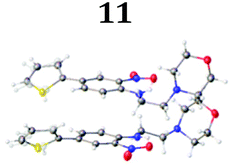
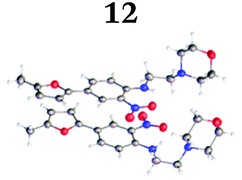
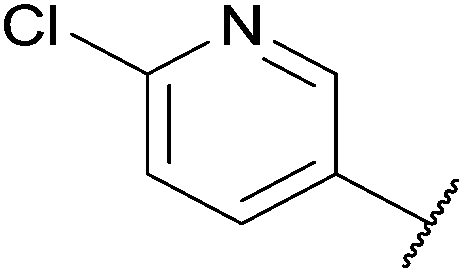
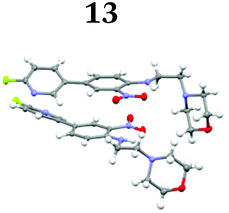
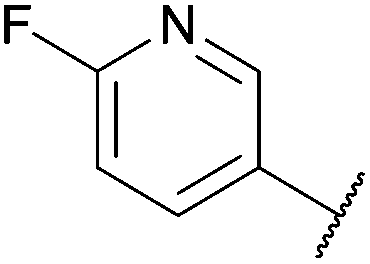
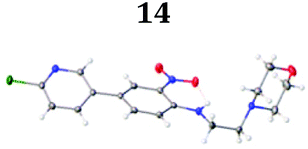
With key precursor nitro-aniline 4 in-hand, we turned our attention to the reduction of the nitro group, whereupon standard exposure to palladium on carbon under a balloon of hydrogen, the desired product 8 was readily obtained in reasonably high isolated yield. However, analysis of the profile of the reaction indicated the formation of a minor by-product, presumed to be generated by cleavage of the labile isoxazole N–O bond.10,11 This was established by repetition of the hydrogenation reaction under pressure, which, solely delivered the ring-opened product. Indicative confirmation was obtained by the changes observed in the 1H NMR spectrum where the signals corresponding to the two methyl groups moved upfield from δ = 2.26 and 2.40 ppm to δ = 1.72 and 1.87 ppm respectively. The crude product was then carried forward through an amide coupling and cyclisation sequence to afford benzimidazole 15, whose structure was solved by single crystal X-ray analysis (Scheme 3).
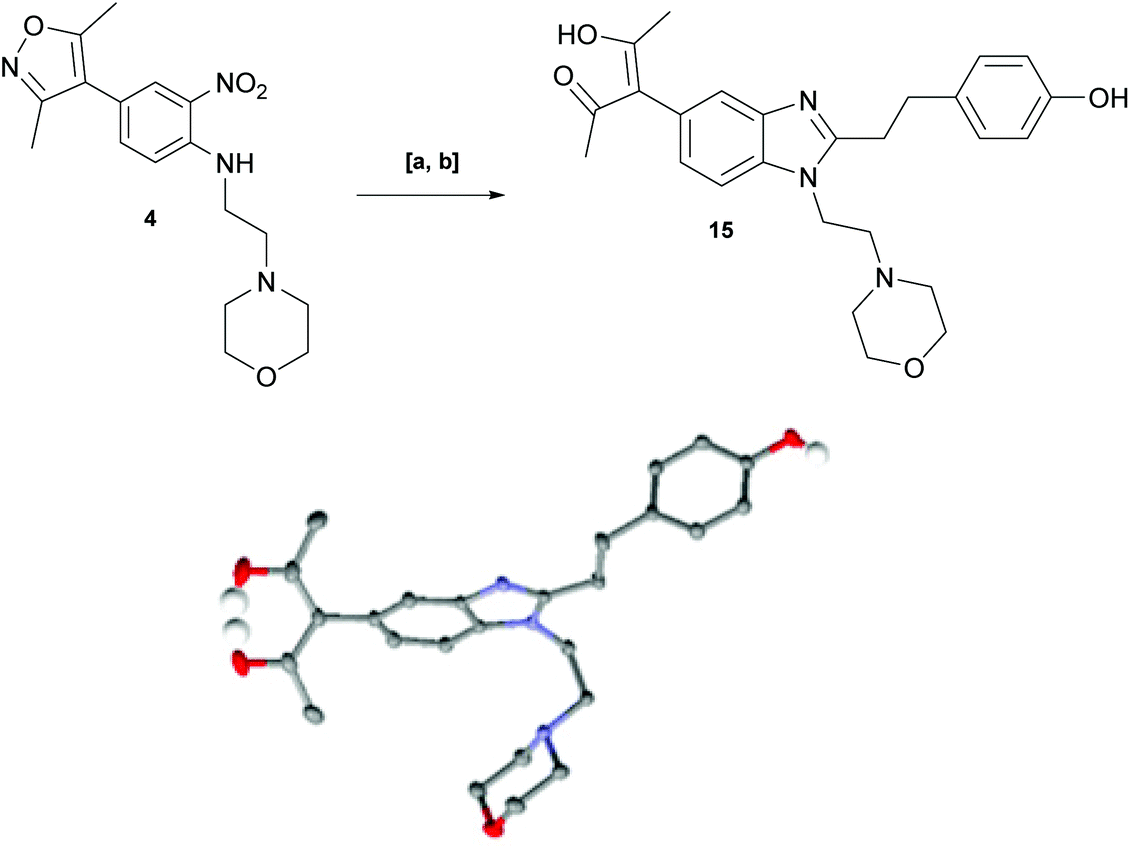 | ||
Scheme 3 Forced generation and structural confirmation of open chain side-product. Reagents & Conditions: [a] 10% Pd/C, EtOAc/MeOH (5 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), COware (with Zn and 7 M HCl in 2nd chamber) (quant).; [b] 3-(4-hydroxyphenyl)propanoic acid, HATU, TEA, DMF, rt, 16 h, then AcOH, reflux, 2 h (51%). :1), COware (with Zn and 7 M HCl in 2nd chamber) (quant).; [b] 3-(4-hydroxyphenyl)propanoic acid, HATU, TEA, DMF, rt, 16 h, then AcOH, reflux, 2 h (51%). | ||
We opted to replicate and scale-up the reported reduction which can be induced by employment of sodium dithionite as the reducing agent2 affording aniline 8 in high yield (81%) and purity (Scheme 4).
 | ||
| Scheme 4 Scaled-up and optimised ISOX-DUAL (1) synthesis. Reagents & Conditions: [a] 4-(2-aminoethyl)morpholine, TEA, DMSO, 80 °C, 2 h (quant.); or 4-(2-aminoethyl)morpholine, TEA, 120 °C, MW, 10 mins (quant.); [b] 3,5-dimethylisoxazole-4-boronic acid pinacol ester, PdCl2(dppf)·DCM, K3PO4, 1,4-dioxane/H2O, reflux, 16 h (94%); [c] (i) 1 M Na2S2O4 (aq.), EtOH, 80 °C, 1 h; (ii) 10% aq. NH3 solution (81%); [d] Cl(CH2)3NMe2·HCl, Cs2CO3, NaI, MeCN, reflux, 16 h (87%); [e] LiOH·H2O, THF/H2O, rt, 16 h (quant.); [f] (i) HATU, DMF, rt, 16 h; (ii) AcOH, reflux, 2 h (55%). | ||
Independent construction of the requisite amide coupling partner 9 successfully allowed circumvention of the low yielding alkylation step from the original synthesis. As such, after conventional phenolic alkylation to give methyl ester 16 in high yield (87%), a subsequent ester hydrolysis gave rise to carboxylate 9 in quantitative yield, which proved most convenient to isolate and manipulate as the lithium salt (Scheme 4). To complete the synthesis, a HATU-mediated amide coupling between 8 and 9, followed by acetic acid treatment of the crude product induced cyclisation to 1 in a one-pot procedure with good yield (55%) and high purity.
Conclusions
In summary, we detail an optimised synthetic route to ISOX-DUAL (1) and related analogues, with improved yields at every stage. By repositioning the phenolic alkylation to earlier in the plan, and by implementing a redesigned stepwise construction approach to the benzimidazole core, we now report an improved synthesis, shortened from eight stages to six, with a significant overall isolated yield increase from 1% to 42%. Additionally, development of the Suzuki coupling invoking a boronate ester establishes prospective in-roads to structural modification and opportunity to employ late stage Pd coupling process in the installation of alternative KAc bioisosteres in bioactive scaffolds.Experimental
General methods
All reagents and solvents were purchased from commercial sources and used without further purification. Nuclear magnetic resonance spectra were recorded on a Bruker Avance III HD spectrometer operating at 400 MHz or a Varian VNMRS 500 or VNMRS 600 spectrometer operating at 500 MHz or 600 MHz for 1H NMR and 126 MHz or 151 MHz for 13C NMR, respectively. 19F NMR spectra were recorded on a Varian VRMS 400 spectrometer operating at 376 MHz. 1H NMR and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) and are referenced to residual protium in solvent and to the carbon resonances of the residual solvent peak, respectively. DEPT and correlation spectra were run in conjunction to aid assignment. 19F NMR chemical shifts are reported in ppm and are uncorrected. Coupling constants (J) are quoted in Hertz (Hz), and the following abbreviations were used to report multiplicity: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet.Purification by flash column chromatography was carried out using Fisher Scientific silica gel 60 Å (35–70 μm), or with silica gel or C18 columns using a Combi flash RF 75 PSI or 150 PSI, Teledyne ISCO unit. Analytical thin layer chromatography was performed on glass plates pre-coated with silica gel (250 μm/UV254), with visualization being achieved using UV light (254 nm) and/or by staining with alkaline potassium permanganate dip. Reaction monitoring LC-MS analyses were conducted using a Shimadzu LC-MS 2020, on a Gemini 5 m C18 110 Å column. High resolution mass spectral (HRMS) data was collected in the laboratories of the University of Sussex Chemistry Department using a Bruker Daltonics Apex III (Apollo ESI ion source). Single crystal X-ray measurements were recorded in the laboratories of the UK National Crystallography Service at the University of Southampton.
Microwave method. A CEM Explorer microwave vial was equipped with a magnetic flea and 4-bromo-1-fluoro-2-nitrobenzene 2 (3.10 mL, 25 mmol, 1 eq.). Triethylamine (15 mL, 100 mmol, 4 eq.) and 4-(2-aminoethyl)morpholine (3.34 mL, 25.25 mmol, 1.01 eq.) were added successively and the vessel was sealed and heated using the dynamic heating method, with max power set to 300 W, max pressure 300 psi, max temperature 120 °C, high stirring throughout and power max turned off. This method was used to hold the method at 120 °C for 10 min. After cooling, the reaction mixture was partitioned between EtOAc (250 mL) and water (300 mL). The organic layer was collected and the aqueous was extracted with EtOAc (4 × 150 mL). The combined organic extracts were washed with NaHCO3 (sat. aq.) and brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound as an orange solid (7.95 g, 24.07 mmol, quant.).
Thermal method. To a stirred solution of 4-bromo-1-fluoro-2-nitrobenzene (2) (29.9 g, 136 mmol, 1 eq.) in DMSO (300 mL) at ambient temperature was added triethylamine (56 mL, 408 mmol, 3 eq.) followed by 4-(2-aminoethyl)morpholine (18.7 mL, 143 mmol, 1.05 eq.) in a dropwise fashion. The reaction mixture was then heated to 80 °C for 2 hours. Upon completion of the reaction, the mixture was cooled to ambient temperature and partitioned between EtOAc (500 mL) and water (500 mL). The organic layer was collected and the aqueous was extracted with EtOAc (3 × 750 mL). The combined organic extracts were washed successively with NaHCO3 (sat. aq.) and brine (1 L), dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound as an orange solid (40.6 g, 123 mmol, quant.). δH (400 MHz, CDCl3) 2.52 (s, 4H), 2.72 (t, J = 6.3 Hz, 2H), 3.34 (q, J = 5.8 Hz, 2H), 3.76 (s, 4H), 6.73 (d, J = 9.2 Hz, 1H), 7.49 (dd, J = 8.6, 1.6 Hz, 1H), 8.33 (d, J = 2.2 Hz, 1H), 8.53 (s, 1H). LC-MS (5-95 MeCN in 5 min) tR = 3.178, purity = >99%, m/z = 332.1 [M + H+]+. Data in accordance with literature.1
General Suzuki reaction microwave method
A 35 mL microwave vessel was charged with 3 (1 eq,), 3,5-dimethylisoxazole-4-boronate species (1.12 eq.), and palladium catalyst (5 mol%) before dissolution in 1,4-dioxane (12 mL mmol−1). The solution was sealed, degassed with argon (×3) before the addition of base (2.6 eq.) as a 1 M solution in water. The vessel was then heated using the dynamic heating method, with max power set to 300 W, max pressure 300 psi, max temperature 150 °C, high stirring throughout and power max turned off. This method was used to hold the method at 150 °C for 3 hours. The reaction mixture was cooled and filtered through Celite™. The filter cake was washed with EtOAc (3 × 50 mL), and the filtrates were concentrated under reduced pressure.Thermal method. A stirred solution of 4-bromo-N-(2-morpholinoethyl)-2-nitroaniline (3) (38 g, 115 mmol, 1 eq.), potassium phosphate (63.5 g, 299 mmol, 2.6 eq.) and 3,5-dimethylisoxazole boronic acid pinacol ester (25.6 g, 115 mmol, 1 eq.) in 1,4-dioxane (1.2 L) and water (120 mL) was degassed with argon (×3) before the addition of PdCl2(dppf)·DCM (4.7 g, 5.75 mmol, 0.05 eq.). The reaction mixture was then degassed and refilled with argon once further, heated to reflux and stirred overnight under a stream of N2. The reaction mixture was then cooled to ambient temperature and filtered through a pad of Celite™ before concentrating under reduced pressure to approximately 300 mL. The residue was then partitioned between water (600 mL) and EtOAc (600 mL), the organic phase was collected, and the aqueous phase extracted with EtOAc (3 × 250 mL). The combined organic extracts were washed with brine (3 × 400 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 0–80% EtOAc in hexane, afforded the title compound as an orange solid (37.4 g, 108 mmol, 94%). δH (400 MHz, CDCl3) 2.26 (s, 3H), 2.40 (s, 3H), 2.55 (d, J = 4.6 Hz, 3H), 2.75 (t, J = 6.1 Hz, 2H), 3.38–3.44 (m, 2H), 3.78 (d, J = 4.6 Hz, 4H), 6.91 (d, J = 8.8 Hz, 1H), 7.34 (dd, J = 8.8, 2.2 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 8.58 (s, 1H). LC-MS (5-95 MeCN in 5 min) tR = 3.266, purity = >99%, m/z = 347.2 [M + H+]+. Data in accordance with literature.1
:MeOH (5:1, 20 mL). Zinc (10 g) was added to the other chamber and the vessel was flushed with argon for a further 5 mins before sealing. 7 M HCl (aq.) (14.22 mL, 99.5 mmol) was then added to the zinc chamber. The reaction mixture was left to stir at room temperature overnight behind a blast shield, with the addition of more 7 M HCl (aq.) after 16 h (14.2 mL, 99.5 mmol). The reaction vessel was carefully vented, and the organic component was filtered over Celite, washing with EtOAc (3 × 50 mL) and the filtrate was concentrated under reduced pressure to afford the crude product as a brown oil (7.5 g, 23.6 mmol, 99%) which was used directly in the next stage without further purification. δH (600 MHz, CDCl3) 1.72 (s, 3H), 1.87 (s, 3H), 2.51 (s, 4H), 2.70 (t, J = 5.7 Hz, 2H), 3.19 (t, J = 5.7 Hz, 2H), 6.51 (s, 1H), 3.73 (s, 4H), 6.62–6.53 (m, 2H), 10.45 (s, 1H). LC-MS (30-95 MeCN in 20 min) tR = 3.05 min, purity = 90%, m/z = 319.00 [M + H+]+.
Part B: To a stirred solution of 4-hydroxyphenyl propionic acid (914 mg, 5.05 mmol, 1.01 eq.) and HATU (2.465 g, 6.5 mmol, 1.3 eq.) in DMF (15 mL) at ambient temperature was added triethylamine (1.4 mL, 10 mmol, 2 eq.) followed by a solution of the crude di-aniline (1.732 g, 5 mmol, 1 eq.) in DMF (15 mL). After stirring overnight at ambient temperature, the reaction mixture was partitioned between DCM (50 mL) and water (50 mL) and the organic phase was separated. The aqueous phase was extracted with DCM (3 × 25 mL). The combined organic extracts were washed with saturated aqueous NaHCO3 (100 mL) and brine (100 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was dissolved in AcOH (50 mL) and heated to reflux for 2 hours. The reaction mixture was then cooled to ambient temperature and concentrated under reduced pressure. The residue was treated with DCM (50 mL) followed by neutralisation with saturated aqueous NaHCO3. The organic phase was collected, and the aqueous phase was extracted with DCM (4 × 50 mL). The combined organic extracts were washed with brine (250 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 0–15% MeOH in DCM with 0.5% NH4OH, afforded the title compound as a yellow oil (1.14 g, 2.50 mmol, 51%). δH (600 MHz, CDCl3) 1.87 (s, 6H). 2.50 (s, 4H), 2.67 (t, J = 7.1 Hz, 2H), 3.21−3.11 (m, 5H), 3.69 (s, 4H), 4.18 (t, J = 7.1 Hz, 2H), 6.74 (d, J = 8.0 Hz, 2H), 7.00 (d, J = 8.1 Hz, 2H), 7.05 (d, J = 8.2 Hz, 1H), 7.32 (d, J = 8.2 Hz, 1H), 7.51 (s, 1H). δC (151 MHz, CDCl3) 24.3, 29.7, 33.1, 41.5, 54.0, 57.6, 66.7, 109.5, 115.3, 115.8, 121.4, 125.5, 129.3, 131.0, 131.5, 134.0, 142.4, 155.3, 155.4, 191.20. LC-MS (30–95 MeCN in 20 min) tR = 3.36 min, purity = 99%, m/z = 450.10. [M + H+]+. HR-MS (m/z): [M + H+]+ calculated for C26H32N3O4, 450.2393; found, 450.2397.
Abbreviations
| CBP | CREB-binding protein |
| HATU | Hexafluorophosphate azabenzotriazole tetramethyl uronium |
| MW | Microwave |
| PdCl2(dppf)·DCM | [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane |
| PdCl2(dtbpf) | 1,1′-Bis(di-tert-butylphosphino)ferrocene] palladium(II) |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest in this study.Acknowledgements
We are grateful for an EPSRC iCASE award (to AKE) from Bio-Techne (Tocris) and EPSRC as well as EPSRC funding (SHH; grant number: EP/P026990/1) and for the National Crystallography Service for structural studies.12References
- E. L. P. Chekler, J. A. Pellegrino, T. A. Lanz, R. A. Denny, A. C. Flick, J. Coe, J. Langille, A. Basak, S. Liu, I. A. Stock, P. Sahasrabudhe, P. D. Bonin, K. Lee, M. T. Pletcher and L. H. Jones, Chem. Biol., 2015, 22, 1588–1596 CrossRef CAS PubMed.
- D. A. Hay, O. Fedorov, S. Martin, D. C. Singleton, C. Tallant, C. Wells, S. Picaud, M. Philpott, O. P. Monteiro, C. M. Rogers, S. J. Conway, T. P. C. Rooney, A. Tumber, C. Yapp, P. Filippakopoulos, M. E. Bunnage, S. Müller, S. Knapp, C. J. Schofield and P. E. Brennan, J. Am. Chem. Soc., 2014, 136, 9308–9319 CrossRef CAS PubMed.
- D. S. Hewings, O. Fedorov, P. Filippakopoulos, S. Martin, S. Picaud, A. Tumber, C. Wells, M. M. Olcina, K. Freeman, A. Gill, A. J. Ritchie, D. W. Sheppard, A. J. Russell, E. M. Hammond, S. Knapp, P. E. Brennan and S. J. Conway, J. Med. Chem., 2013, 56, 3217–3227 CrossRef CAS PubMed.
- D. S. Hewings, M. Wang, M. Philpott, O. Fedorov, S. Uttarkar, P. Filippakopoulos, S. Picaud, C. Vuppusetty, B. Marsden, T. D. Heightman, S. Knapp, P. Brennan and S. J. Conway, RSC Advances in Synthesis and Medicinal Chemistry, 2012, vol. 243 Search PubMed.
- N. A. Isley, M. S. Hageman and B. H. Lipshutz, Green Chem., 2015, 17, 893–897 RSC.
- Z. Ahmadi and J. S. McIndoe, Chem. Commun., 2013, 49, 11488–11490 RSC.
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2016, 138, 9145–9157 CrossRef CAS PubMed.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC.
- A. J. Close, P. Kemmitt, M. K. Emmerson and J. Spencer, Tetrahedron, 2014, 70(47), 9125–9131 CrossRef CAS.
- P. Vitale and A. Scilimati, Recent Developments in the Chemistry of 3-Arylisoxazoles and 3-Aryl-2-isoxazolines,Elsevier Ltd, 2017, vol. 122 Search PubMed.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, 5th edn., 2010 Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2110085 (10), 2110086 (11), 2110087 (12), 2110088 (14), 2110089 (13) and 2110090 (15). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob00609j |
| This journal is © The Royal Society of Chemistry 2022 |