
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA stepwise one-pot synthesis of aliphatic thiols and their derivatives from acrylamides and sulfur†
András Gy.
Németh
ab,
Renáta
Szabó
ab,
Krisztina
Németh
c,
György M.
Keserű
 *ab and
Péter
Ábrányi-Balogh
*ab
*ab and
Péter
Ábrányi-Balogh
*ab
aDepartment of Organic Chemistry and Technology, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3, H-1111 Budapest, Hungary
bMedicinal Chemistry Research Group, Research Centre for Natural Sciences, Magyar tudósok krt. 2, H-1117 Budapest, Hungary. E-mail: keseru.gyorgy@ttk.hu; abranyi-balogh.peter@ttk.hu
cMS Metabolomics Research Laboratory, Research Centre for Natural Sciences, Magyar tudósok krt. 2, H-1117 Budapest, Hungary
First published on 7th May 2022
Abstract
Elemental sulfur enables the convenient formation of C–S bonds and the direct incoporation of S–S bonds. The reactivity of easily accessible electron deficient alkenes towards sulfur, however, is barely disclosed. Herein, we investigated the reactivity of acrylamides with sulfur and eventually developed a new pseudo-multicomponent reaction for the preparation of polysulfides. Sequential one-pot reduction led to diversely substituted thiols. Additional third stage one-pot modifications provided thioethers, unsymmetric disulfide and thioester.
Introduction
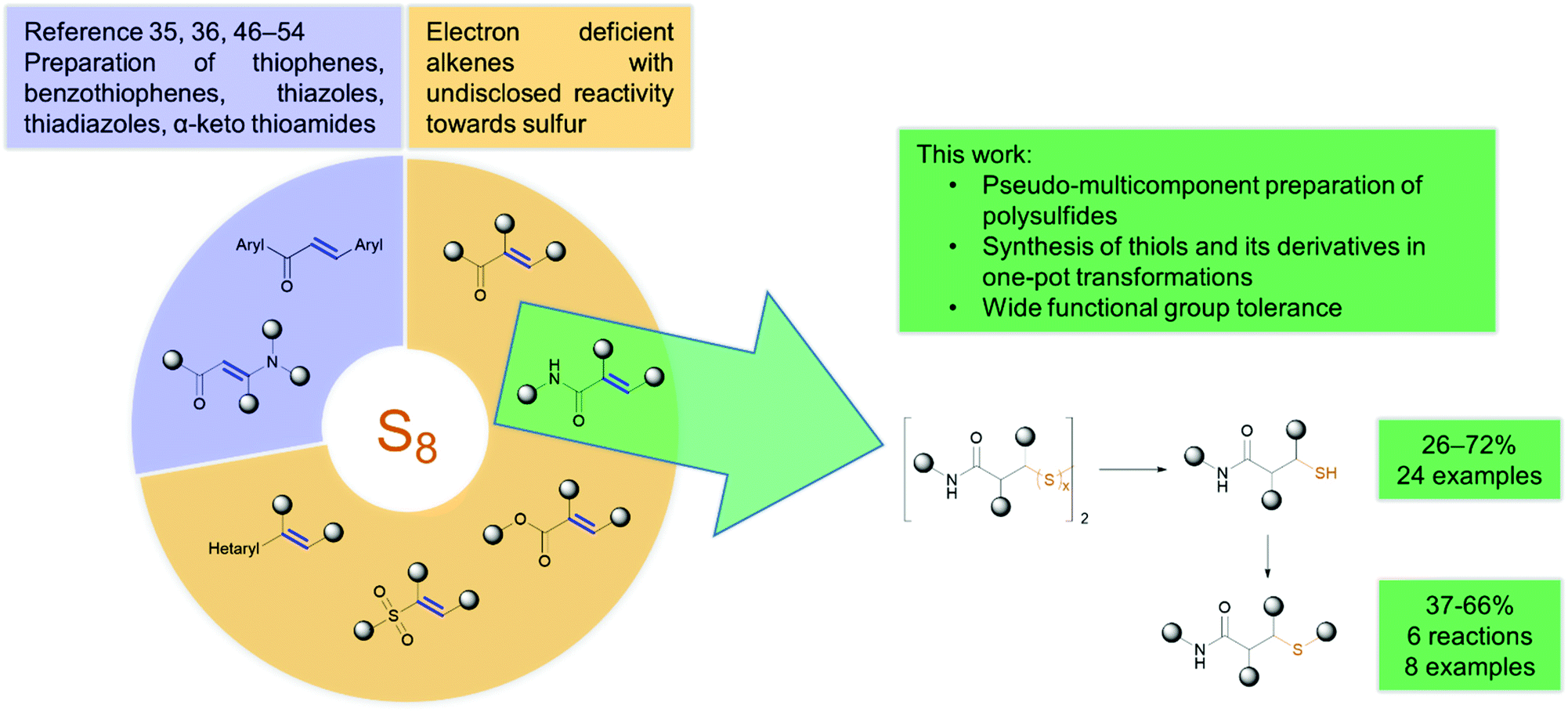
C–S and S–S bonds appear in important natural products, pharmaceuticals and functional materials.1,2 In particular, organic di-, tri and tetrasulfides (–Sx–) are considered potent sources of H2S, being an important gasotransmitter.3–5 Di- and trisulfide bridges provide higher stability and potency to peptide based therapeutics, while tetrasulfides find application in rechargeable lithium–sulfur batteries.6,7 Therapeutic applications of sulfides are also significant. A number of di- and trisulfides are high potency antitumor agents. In fact, calicheamicin and shishijimicin A derivatives are used as payloads of ADCs.8–10 In addition, polysulfides might act as precursors to thiols, which are biologically and synthetically important compounds. Mucolytic N-acetylcysteine and carbocisteine are used to loosen thick mucus in bronchitis or COPD.11,12 Mycothiol serves as an intracellular detoxification agent, similar to GSH in functionality, scavenging reactive oxygen and nitrogen species (Fig. 1).13 Captopril, derived from L-proline is a potent ACE inhibitor prescribed for hypertension and to treat heart failure.14 | ||
| Fig. 1 Selected examples of biologically active thiols and thioethers. | ||
Due to their nucleophilicity thiols are often applied in late-stage appendage reactions for the formation of C–S bonds. For example, the last steps in the synthesis of antifungal butoconazole and immunosuppressive azathioprine are S-alkylation and nucleophilic aromatic substitution (SNAr) reactions, respectively.15,16 Michael addition or thiol–ene click reaction is popularly applied in the preparation of linear, cross-linked polymers and dendrimers or in medicinal chemistry and chemical biology settings for protein labelling. One might mention covalent drugs having an acrylamide function for labelling cysteine residues (e.g. ibrutinib or osimertinib) and the synthesis of the highly potential antibody–drug conjugate Kadcyla from the cytotoxic payload mertansine and the antibody trastuzumab both used in oncology indications.17–20
Traditional approaches toward the formation of S–S bonds consists of reacting thiols or disulfides with sulfenyl halides21,22 or other disulfides equipped with leaving groups (Scheme 1).23 Recently, Jiang and co-workers developed multifaceted bilateral sulfurating reagents giving access to di-, tri and tetrasulfides starting from thiols (S–S bonds) or boronic acids (C–S bonds).24,25 Bhabak and co-workers evaluated a selective method to generate symmetric trisulfides from Bunte salts and sodium sulfide.3 Nonetheless, these methods generate halogen waste and the special reagents have to be prepared in advance. The readily available and cheap elemental sulfur, however, enables the direct and convenient incorporation of S–S bonds and the formation of C–S bonds.26–29 Yamaguchi and co-workers reported a Rh-catalyzed method for the insertion of sulfur atoms into disulfides and trisulfides leading to higher polysulfides.30 Most recently, Hilt and Fährmann investigated the electrochemical induced insertion of sulfur atoms to bridge thiols and turn disulfides into higher polysulfides.31 One should note that literature known methods focus on increasing the number of sulfur atoms in sulfur containing compounds. Herein we present a method for inserting sulfur atoms and S–S bonds into acrylamides using elemental sulfur.
 | ||
| Scheme 1 Methods for the formation of S–S bonds and polysulfides and this work. | ||
Certain nucleophiles, such as aliphatic amines, sulfide anions, carbonates and hydroxides are able to activate sulfur under mild conditions, generating open-chain ionic polysulfide anions.32–38 The in situ generated active species may be used under metal-free conditions to create new C–S bonds by the atom efficient sulfuration of electrophiles, e.g. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds,39,40 isocyanides,29,37,41–43 isothiocyanates44 and iodonium salts.45 Recently, a handful of new methods exploited the divergent reactivity of chalcones36,46–49 and β-oxo-enamines35,50–54 towards sulfur giving access to the selective formation of sulfur containing heterocycles and other structures (Scheme 2). In fact, Retailleau and Nguyen demonstrated that starting from chalcones, the careful selection of the base may enable the selective formation of either sultams or benzothiophenes.46,47 Although the great synthetic potential of these reactions have been demonstrated, we have noted that the reactivity of other easily accessible electron-deficient alkenes towards sulfur remained undisclosed.55 As a continuous effort towards the development of new sulfur based reactions,37,41–43 we designed the generation of polysulfides starting from N-phenylacrylamide and sulfur in the presence of tertiary amines. Our objective was to investigate the formation and application of polysulfides in a multistep one-pot process to synthesize thiols and their derivatives.
N double bonds,39,40 isocyanides,29,37,41–43 isothiocyanates44 and iodonium salts.45 Recently, a handful of new methods exploited the divergent reactivity of chalcones36,46–49 and β-oxo-enamines35,50–54 towards sulfur giving access to the selective formation of sulfur containing heterocycles and other structures (Scheme 2). In fact, Retailleau and Nguyen demonstrated that starting from chalcones, the careful selection of the base may enable the selective formation of either sultams or benzothiophenes.46,47 Although the great synthetic potential of these reactions have been demonstrated, we have noted that the reactivity of other easily accessible electron-deficient alkenes towards sulfur remained undisclosed.55 As a continuous effort towards the development of new sulfur based reactions,37,41–43 we designed the generation of polysulfides starting from N-phenylacrylamide and sulfur in the presence of tertiary amines. Our objective was to investigate the formation and application of polysulfides in a multistep one-pot process to synthesize thiols and their derivatives.
 | ||
| Scheme 2 Chemical space of easily accessible electron deficient alkenes and their reactivity towards elemental sulfur and this work. | ||
Results and discussion
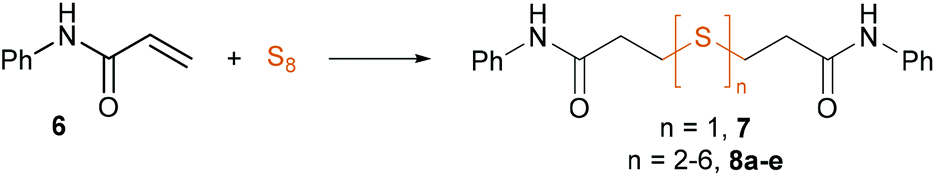
First, we probed the reactivity of heteroaromatic alkenes, vinyl ketones, vinyl sulfones, acrylesters and acrylamides towards sulfur in aqueous mixtures in the presence of PMDTA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine, 1) at room temperature to 100 °C, being reaction conditions regularly used in our lab for sulfur-based reactions (Scheme 3).37,41–43 We investigated the reaction mixtures with HPLC-MS and noted that 4-vinylpyiridne (2), phenyl vinyl ketone (3) and phenyl vinyl sulfone (4) led to a complex mixture of products while phenyl acrylate (5) instantly hydrolyzed to phenol. To our delight, we noticed the direct sulfuration of N-phenylacrylamide (6) with sulfur leading to a mixture of symmetric thioether 7 and polysulfides 8a–e.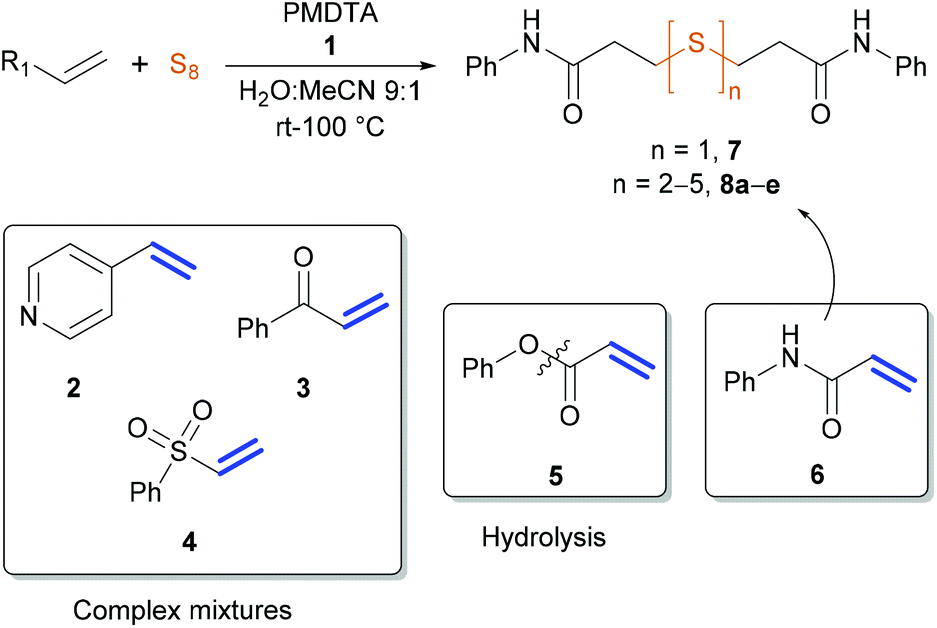 | ||
| Scheme 3 Preliminary experiments with selected electron deficient alkenes and sulfur. | ||
We started the optimization of this reaction employing PMDTA in a mixture of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN 9:1 at 80 °C. We obtained full conversion of acrylamide 6 in 2 hours and the mixture contained the thioether 7 and polysulfides 8a–e in a ratio of 31:69 respectively (Table 1, entry 1). We aimed to shift the selectivity of the reaction towards the formation of polysulfides as we figured they might serve as precursors to thiols. We determined the selectivity by comparing the relative MS intensity of 7 and 8a–e based on their tight structural similarity and similar ionization profile. Application of various tertiary amines highlighted the fundamental effect of the base on the selectivity of the reaction. DIPEA (N,N-diisopropylethylamine), Et3N and N-ethylpiperidine all favored the formation of 8a–e, the latter being the most efficient providing an excellent 2:98 ratio of 7:8a–e (Table 1, entries 2–4). Application of NaOH or Na2S together with sulfur switched the selectivity of the reaction completely favoring the generation of thioether 7 (Table 1, entries 5 and 6). Other tertiary amines or inorganic bases did not promote the reaction at all (Table 1, entry 7). Reducing the excess of the base or elevating the temperature to 100 °C resulted in slightly lower selectivity (Table 1, entries 8–11). Employing 60 °C or reducing the excess of sulfur to two equivalents enabled only low conversion in 2 hours (Table 1, entries 12 and 13). Larger excess of sulfur did not have any positive effect on the rate of the reaction or on the selectivity (Table 1, entry 14). Replacement of the reaction medium to aprotic apolar solvents such as toluene, dioxane or tetrahydrofuran inhibited the reaction (Table 1, entry 15), while in polar aprotic solvents, such as DMF, DMSO or NMP, we only acquired complex reaction mixtures (Table 1, entry 16).
:MeCN 9:1 at 80 °C. We obtained full conversion of acrylamide 6 in 2 hours and the mixture contained the thioether 7 and polysulfides 8a–e in a ratio of 31:69 respectively (Table 1, entry 1). We aimed to shift the selectivity of the reaction towards the formation of polysulfides as we figured they might serve as precursors to thiols. We determined the selectivity by comparing the relative MS intensity of 7 and 8a–e based on their tight structural similarity and similar ionization profile. Application of various tertiary amines highlighted the fundamental effect of the base on the selectivity of the reaction. DIPEA (N,N-diisopropylethylamine), Et3N and N-ethylpiperidine all favored the formation of 8a–e, the latter being the most efficient providing an excellent 2:98 ratio of 7:8a–e (Table 1, entries 2–4). Application of NaOH or Na2S together with sulfur switched the selectivity of the reaction completely favoring the generation of thioether 7 (Table 1, entries 5 and 6). Other tertiary amines or inorganic bases did not promote the reaction at all (Table 1, entry 7). Reducing the excess of the base or elevating the temperature to 100 °C resulted in slightly lower selectivity (Table 1, entries 8–11). Employing 60 °C or reducing the excess of sulfur to two equivalents enabled only low conversion in 2 hours (Table 1, entries 12 and 13). Larger excess of sulfur did not have any positive effect on the rate of the reaction or on the selectivity (Table 1, entry 14). Replacement of the reaction medium to aprotic apolar solvents such as toluene, dioxane or tetrahydrofuran inhibited the reaction (Table 1, entry 15), while in polar aprotic solvents, such as DMF, DMSO or NMP, we only acquired complex reaction mixtures (Table 1, entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Base (eq.) | S8 eq. | T [°C] | Relative MS intensity of 7:8a–ea,b |
|
a Ratio of 7:8a–e calculated by extracted MS peak intensities.
b Reaction conditions: 6 (0.2 mmol), S8 (3 eq.), base (7.5 eq.) in H2O:MeCN 9:1 (1 mL) at 80 °C for 2 h.
c NMM, TMEDA, DABCO, DMAP, pyridine, Na2CO3.
d Solvent: toluene, dioxane, THF.
e Solvent: DMF, NMP or DMSO.
|
||||
| 1 | PMDTA (7.5) | 3 | 80 | 31:69 |
| 2 | DIPEA (7.5) | 3 | 80 | 9:91 |
| 3 | Et3N (7.5) | 3 | 80 | 5:95 |
| 4 | N-Ethylpiperidine (7.5) | 3 | 80 |
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :98 :98
|
| 5 | NaOH (7.5) | 3 | 80 | 91:9 |
| 6 | Na2S (7.5) | 3 | 80 | 99:1 |
| 7c | Other bases (7.5) | 3 | 80 | No reaction |
| 8 | N-Ethylpiperidine (5) | 3 | 80 | 4:96 |
| 9 | N-Ethylpiperidine (2.5) | 3 | 80 | 6:94 |
| 10 | N-Ethylpiperidine (1.5) | 3 | 80 | 12:88 |
| 11 | N-Ethylpiperidine (7.5) | 3 | 100 | 5:95 |
| 12 | N-Ethylpiperidine (7.5) | 3 | 60 | Traces |
| 13 | N-Ethylpiperidine (7.5) | 2 | 80 | Low conversion |
| 14 | N-Ethylpiperidine (7.5) | 5 | 80 | 2:98 |
| 15d | N-Ethylpiperidine (7.5) | 3 | 80 | No reaction |
| 16e | N-Ethylpiperidine (7.5) | 3 | 80 | Complex mixture |
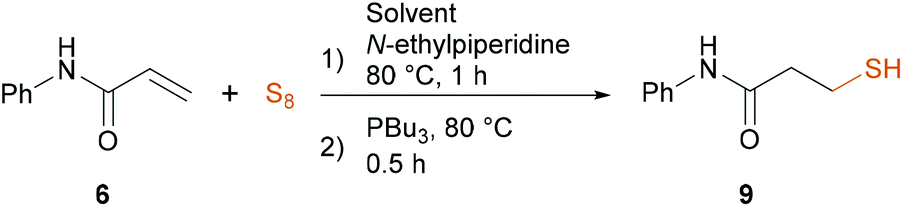
Next, we aimed the reduction of polysulfides to synthetically more versatile thiols that could be used in further transformations. Also, Hirsch and co-workers recently observed the efficient inhibitory effect of N-aryl mercaptopropionamides against metallo-β-lactamases, being responsible for the hydrolysis of β-lactam drugs in bacteria.56,57 We investigated the reaction using several reducing agents, particularly a mixture of NaBH4 and methanol, tricyclohexylphosphine, tris(2-carboxyethyl)phosphine (TCEP), triphenylphosphine and tributylphosphine (PBu3) under usual reducing conditions, and finally selected tributylphosphine as the most efficient reducing agent turning the polysulfides into the thiol 9 (for details see ESI†). The reduction was smooth and efficient below 80 °C, but still, in later work-up procedures, we observed the trisulfide 8b by the reaction of the thiol 9 with the remaining excess of sulfur. This could be avoided by keeping the reaction temperature at 80 °C. Finally, with the optimized synthesis protocol we performed the sequence one-pot, and isolated thiol 9. Applying the solvent mixture of H2O:MeCN 9:1, we observed the full conversion of 6 in 1 hour, then added PBu3 and continued the reaction for 30 minutes. After work-up, we isolated 9 in 57% yield (Table 2, entry 1). In MeCN or under solvent-free conditions the yield changed to 62% and 49%, respectively (Table 2, entries 2 and 3). Addition of 5 eq. water to the solvent-free mixture, however, had an advantageous effect on the reaction resulting in 63% yield (Table 2, entry 4). Thus, we have combined these observations and used MeCN with 5 eq. H2O that further enhanced the yield to 72% (Table 2, entry 5). Next, we applied dioxane or isopropyl alcohol (IPA) in the presence of 5 eq. H2O but encountered significantly longer reaction times in both cases and decreased yield in dioxane (Table 2, entries 6 and 7). Despite the slightly higher yield acquired in IPA, we selected MeCN as the main solvent considering the shorter reaction time (1 hour compared to overnight). Eventually, the optimal reaction conditions were 80 °C in a mixture of MeCN and 5 eq. water, 3 eq. sulfur, 7.5 eq. N-ethylpiperidine, followed by a reduction step using 3 eq. PBu3 at 80 °C for further 30 minutes.
|
|
||
|---|---|---|
| Entry | Solvent | Yielda [%] |
| a Reaction conditions: 6 (0.5 mmol), S8 (1.5 mmol), N-ethylpiperidine (3.75 mmol), solvent (2 mL), 80 °C, 1 h; PBu3 (1.5 mmol), 80 °C, 0.5 h. b Overnight reaction time. | ||
| 1 | H2O:MeCN 9:1 |
57 |
| 2 | MeCN | 62 |
| 3 | — | 49 |
| 4 | 5 eq. H2O | 63 |
| 5 | MeCN + 5 eq. H 2 O | 72 |
| 6b | Dioxane + 5 eq. H2O | 61 |
| 7b | iPrOH + 5 eq. H2O | 75 |
We investigated the distribution of the polysulfides under different reaction conditions (for more details see ESI†). Generally, disulfide 8a and trisulfide 8b dominated the mixtures, followed by tetrasulfide 8c. We also observed the formation of pentasulfide 8d and hexasulfide 8e and in a few cases we also detected the presence of heptasulfide 8f. The average ratio of the polysulfides 8a–e were 19:16:10:4:1 respectively. The careful selection of the base and the reaction medium mainly favored the formation of di- and trisulfides and efficiently suppressed the formation of the synthetically irrelevant thioether 7.
Conducting the reaction under the optimized conditions (N-ethylpiperidine at 80 °C in MeCN and 5 eq. H2O), we investigated the scope of acrylamides 10 (Scheme 4). The developed methodology provided a set of diversely substituted 3-mercapto-N-arylpropanamides, including 4-NO2 (11), 4-CN (12), 4-COMe (13), 4-COOEt (14), 4-COOH (15) and halogen atoms (16–18) in good yields. Although the 2-NO2 derivative 19 is known to participate in intramolecular cyclization reactions and photolysis, we did not observe the formation of any side-products by HPLC-MS.58 Repeating the experiment in dark, we obtained 19 in similar, 24% yield. In the case of N-phenylacrylamides equipped with electron-donating groups the reaction went smoothly, resulting in the formation of 20–22 in 54–65% yield. The 58% yield of ortho-phenyl substituted 23 showed that no steric effect compromises the reaction. We isolated the α-naphthalene derivative 24 in 53% yield. N-Phenylmethacrylamide and N-phenylbut-2-enamide reacted significantly slower at 80 °C with sulfur. Thus, we performed these reactions at 100 °C, obtaining 25 and 26 in 33% and 45% yield, respectively, after overnight stirring. N,N-Disubstituted thiol 27 was obtained in 51% yield, showing that the NH group does not participate in the reaction. Heteroaromatic thiols 28 and 29 were isolated in 36% and 56% yield, respectively. We observed the partial and complete cleavage of the acrylamidic N–C(O) bond in the case of the Boc-protected L-tryptophan methyl ester analog 30 and the benzimidazole analogue 31, respectively, which eventually enabled the synthesis of 30 in 31% yield. Aliphatic acrylamides generally reacted slower than the less electron rich aromatic and electron poor heteroaromatic analogs, therefore we conducted these reactions at 100 °C, obtaining 32–34 in 40%, 36% and 55% yield respectively.
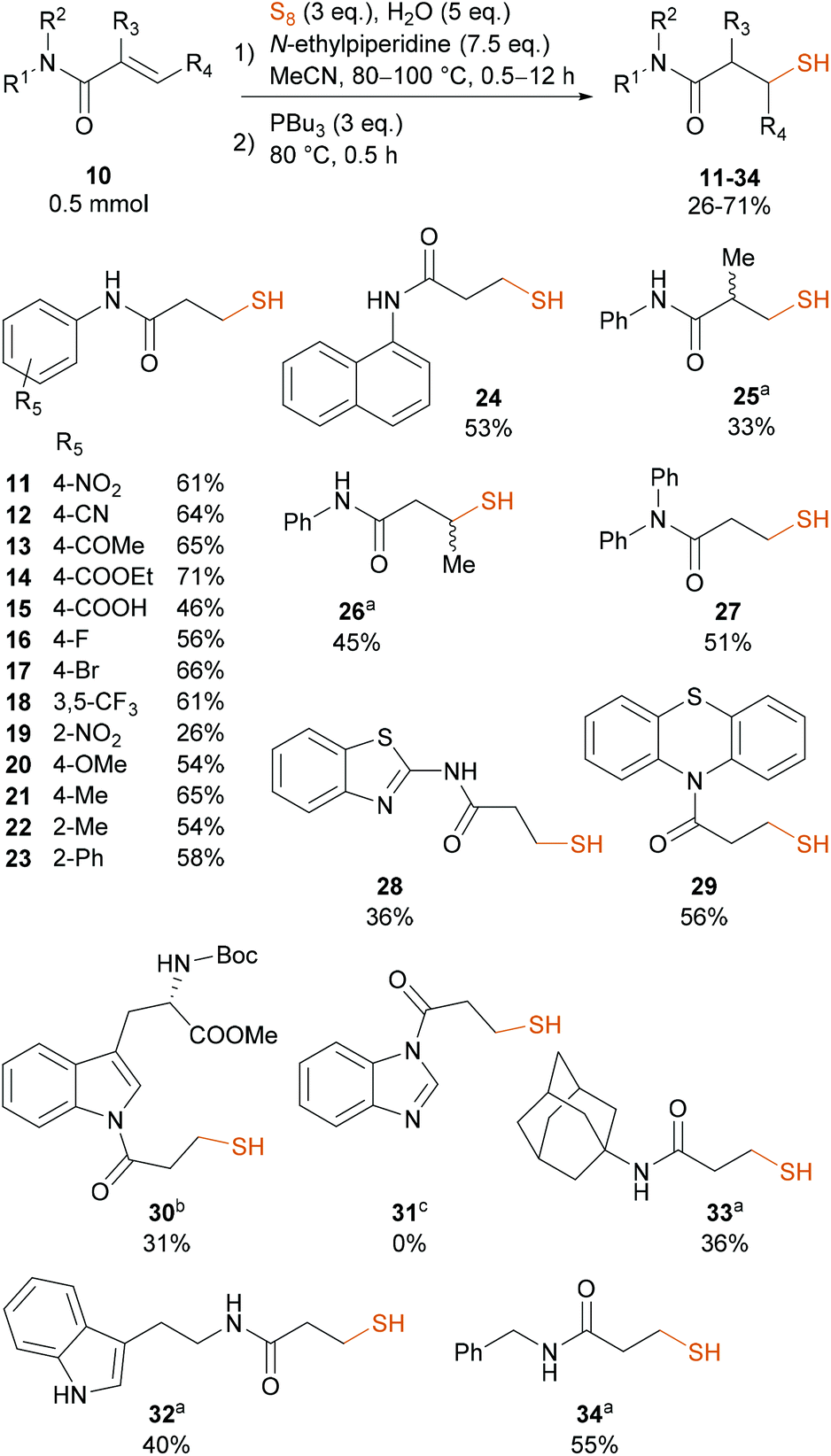 | ||
| Scheme 4 Scope of acrylamides in the pseudo-multicomponent one-pot synthesis of aliphatic thiols. aThe pseudo-multicomponent reaction was conducted at 100 °C; bwe observed the partial decomposition of the starting material to the free N-Boc-tryptophan methyl ester under the applied reaction conditions; cwe observed the instant and complete cleavage of the amide bond to form benzimidazole under the applied reactions conditions. | ||
To demonstrate the robustness of the reaction we performed the 20-fold scale-up synthesis of 9 (10 mmol, Scheme 5) in a sealed tube. To our delight, the pseudo-multicomponent reaction was ready in 1 hour, followed by the reduction in 30 minutes providing 9 in 61% yield (1.17 g).
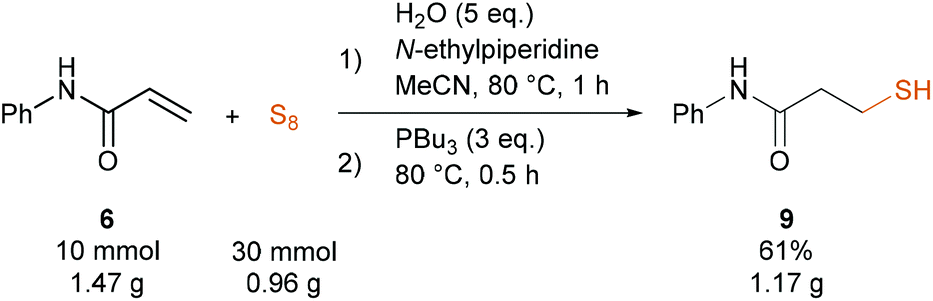 | ||
| Scheme 5 Scale-up synthesis of 9. | ||
Next, we turned our attention to one-pot secondary transformations of the thiols leading to more complex sulfur containing structures such as thioethers, unsymmetric disulfide and thioester. We have applied the optimized reaction conditions leading to thiol 9 and added N-phenylmaleimide (35) to the reaction mixture at room temperature without isolating the thiol intermediate. The Michael addition led to the thioether 36 in an overall 52% yield respect to the acrylamide 6 after three consecutive reactions (i.e. formation of polysulfides, reduction and Michael addition, Scheme 6). Application of N-phenylpropiolamide (37) enabled the isolation of E-38 and Z-38 by flash column chromatography in 25% and 26% yield, respectively. Alkylation with benzyl bromide (39) at room temperature and SNAr with 2-chloro-5-nitropyridine (40) at 60 °C and 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (41) at 100 °C led to the corresponding thioethers 42–44 in 37%, 66% and 54%, respectively. Thioester 45 was obtained in 60% yield via Schotten–Baumann acylation at room temperature using benzoyl chloride (46). Oxidation with benzylmercaptan (47) in the presence of excess iodine led to the unsymmetrical disulfide 48 in 63% yield. These successful reactions showed that after a pseudo-multicomponent reaction and the subsequent reduction, even third stage functionalization is feasible in a one pot sequential manner.
 | ||
| Scheme 6 One-pot secondary transformations of aliphatic thiol 9 synthesized from N-phenylacrylamide (6). Reaction conditions: art, 0.5 h, 1.5 eq. electrophile; b60 °C, 0.5 h, 1.5 eq. electrophile; c100 °C, overnight, 1.5 eq. electrophile; drt, 0.5 h, 3 eq. I2, 5 eq. benzylmercaptan. | ||
In order to gain mechanistic insights on the reaction, we isolated thiol 49 in 66% yield by switching from H2O to D2O (Scheme 7). Characterization by 1H NMR revealed deuteration on the α-carbon atom, thus water might serve as a proton source in the reaction. No reaction happened in the absence of sulfur or N-ethylpiperidine, and HPLC-MS proved the stability of the acrylamide under the conditions applied. Next, small samples of the polysulfide intermediates 8a–e were isolated and characterized by 1H NMR, 13C NMR and HRMS measurements confirming the structure of the intermediates.
 | ||
| Scheme 7 Control experiments and isolation of polysulfide intermediates 8a–e. | ||
Based on literature data and our control experiments, we propose a mechanism for the formation of polysulfides 8a–e. The first step is the cleavage of the octasulfur ring by the base resulting in polysulfide anions of different chain length (50) or disulfide anions (51), which may further react with sulfur to provide 50 (Scheme 8).38,59 The crucial effect of the base on the selectivity between thioether 7 and polysulfides 8a–e may depend on its behavior against sulfur during the generation of polysulfide anions. Conjugate addition of the polysulfide anions on the acrylamide (10), followed by recombination with another acrylamide and proton transfer from water leads to the intermediates 8a–e. Also, the last steps might be interchangeable, the proton transfer might precede the recombination of 52 with the second acrylamide.
 | ||
| Scheme 8 Proposed mechanism for the formation of polysulfides 8a–e. | ||
Conclusion
This study reports a new pseudo-multicomponent reaction for the preparation of polysulfides from acrylamides and sulfur. Taking advantage of this transformation we designed a multistep one-pot process leading to a wide range of thiols and after a one-pot third stage modification thioethers, unsymmetric disulfide and thioester. Our methodology features excellent functional group tolerance and operational simplicity in gram scale, enabling the direct and fast synthesis of thiols and various derivatives.Author contributions
Conceptualization, András Gy. Németh and Péter Ábrányi-Balogh; investigation András Gy. Németh and Renáta Szabó; HR-MS measurements Krisztina Németh; supervision, György M. Keserű, Péter Ábrányi-Balogh; writing—original draft preparation András Gy. Németh; writing—review and editing, György M. Keserű, Péter Ábrányi-Balogh. All authors have read and agreed to the published version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Á-B. is grateful for the support of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. R. Sz. is grateful for the support of ÚNKP-21-2-II-BME-276 grant.References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- D. Bhattacherjee, A. Sufian, S. K. Mahato, S. Begum, K. Banerjee, S. De, H. K. Srivastava and K. P. Bhabak, Chem. Commun., 2019, 55, 13534–13537 RSC.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS.
- M. M. Gadalla and S. H. Snyder, J. Neurochem., 2010, 113, 14–26 CrossRef CAS PubMed.
- D. Y. Wang, W. Guo and Y. Fu, Acc. Chem. Res., 2019, 52, 2290–2300 CrossRef CAS PubMed.
- M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef PubMed.
- K. C. Nicolaou, Z. Lu, R. Li, J. R. Woods and T. I. Sohn, J. Am. Chem. Soc., 2015, 137, 8716–8719 CrossRef CAS PubMed.
- N. Oku, S. Matsunaga and N. Fusetani, J. Am. Chem. Soc., 2003, 125, 2044–2045 CrossRef CAS PubMed.
- W. M. Maiese, J. Korshalla, N. Kuck, A. A. Fantini, M. J. Wildey, J. Thomas, M. Greenstein and D. P. Labeda, J. Antibiot., 1990, 43, 253–258 CrossRef CAS PubMed.
- P. N. Dekhuijzen and W. J. van Beurden, Int. J. Chronic Obstruct. Pulm. Dis., 2006, 1, 99–106 CAS.
- J. Zheng, J. Kang, S. Huang, P. Chen, W. Yao, L. Yang, C. Bai, C. Wang and C. Wang, Lancet, 2008, 371, 2013–2018 CrossRef CAS.
- C. E. Hand and J. F. Honek, J. Nat. Prod., 2005, 68, 293–308 CrossRef CAS PubMed.
- L. Køber, A. P. Maggioni, S. D. Solomon, K. Swedberg, D. Ph, F. Van De Werf, D. Ph, H. White, D. Sc, J. D. Leimberger, D. Ph, M. Henis, S. Edwards, S. Zelenkofske, M. A. Sellers and R. M. Califf, N. Engl. J. Med., 2003, 349, 1893–1906 CrossRef PubMed.
- R. S. Vardanyan and V. J. Hruby, in Synthesis of Essential Drugs, 2006, pp. 419–424 Search PubMed.
- R. S. Vardanyan and V. J. Hruby, in Synthesis of Essential Drugs, 2006, pp. 535–547 Search PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- G. von Minckwitz, C.-S. Huang, M. S. Mano, S. Loibl, E. P. Mamounas, M. Untch, N. Wolmark, P. Rastogi, A. Schneeweiss, A. Redondo, H. H. Fischer, W. Jacot, A. K. Conlin, C. Arce-Salinas, I. L. Wapnir, C. Jackisch, M. P. DiGiovanna, P. A. Fasching, J. P. Crown, P. Wülfing, Z. Shao, E. Rota Caremoli, H. Wu, L. H. Lam, D. Tesarowski, M. Smitt, H. Douthwaite, S. M. Singel and C. E. Geyer, N. Engl. J. Med., 2019, 380, 617–628 CrossRef CAS PubMed.
- M. Santarpia, A. Liguori, N. Karachaliou, M. Gonzalez-Cao, M. G. Daffinà, A. D'Aveni, G. Marabello, G. Altavilla and R. Rosell, Lung Cancer: Targets Ther., 2017, 8, 109–125 CAS.
- Z. Pan, H. Scheerens, S. J. Li, B. E. Schultz, P. A. Sprengeler, L. C. Burrill, R. V. Mendonca, M. D. Sweeney, K. C. K. Scott, P. G. Grothaus, D. A. Jeffery, J. M. Spoerke, L. A. Honigberg, P. R. Young, S. A. Dalrymple and J. T. Palmer, ChemMedChem, 2007, 2, 58–61 CrossRef CAS PubMed.
- Y. Hou, I. A. Abu-Yousef, Y. Doung and D. N. Harpp, Tetrahedron Lett., 2001, 42, 8607–8610 CrossRef CAS.
- G. Derbesy and D. N. Harpp, Tetrahedron Lett., 1994, 35, 5381–5384 CrossRef CAS.
- S. Xu, Y. Wang, M. N. Radford, A. J. Ferrell and M. Xian, Org. Lett., 2018, 20, 465–468 CrossRef CAS PubMed.
- J. Xue and X. Jiang, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- J. Xue and X. Jiang, Org. Lett., 2020, 22, 8044–8048 CrossRef CAS PubMed.
- T. B. Nguyen, Adv. Synth. Catal., 2017, 359, 1066–1130 CrossRef CAS.
- T. B. Nguyen, Adv. Synth. Catal., 2020, 362, 3448–3484 CrossRef CAS.
- S. Liu, G. J. Deng and H. Huang, Synlett, 2021, 142–158 Search PubMed.
- A. G. Németh and P. Ábrányi-Balogh, Catalysts, 2021, 11, 1081 CrossRef.
- M. Arisawa, K. Tanaka and M. Yamaguchi, Tetrahedron Lett., 2005, 46, 4797–4800 CrossRef CAS.
- J. Fährmann and G. Hilt, Chem. – Eur. J., 2021, 27, 11141–11149 CrossRef PubMed.
- M. Saito, S. Murakami, T. Nanjo, Y. Kobayashi and Y. Takemoto, J. Am. Chem. Soc., 2020, 142, 8130–8135 CrossRef CAS.
- J. Ma, J. Luo, K. Jiang, G. Zhang, S. Liu and B. Yin, Org. Lett., 2021, 23, 8033–8038 CrossRef CAS PubMed.
- Y. Liao and X. Jiang, Org. Lett., 2021, 23, 8862–8866 CrossRef CAS.
- R. G. Fu, Y. Wang, F. Xia, H. L. Zhang, Y. Sun, D. W. Yang, Y. W. Wang and P. Yin, J. Org. Chem., 2019, 84, 12237–12245 CrossRef CAS PubMed.
- T. B. Nguyen, D. H. Mac and P. Retailleau, J. Org. Chem., 2020, 85, 13508–13516 CrossRef CAS.
- A. G. Németh, G. M. Keserű and P. Ábrányi-Balogh, Beilstein J. Org. Chem., 2019, 15, 1523–1533 CrossRef.
- Y. Jiang, Y. Qin, S. Xie, X. Zhang, J. Dong and D. Ma, Org. Lett., 2009, 11, 5250–5253 CrossRef CAS PubMed.
- X. Ma, X. Yu, H. Huang, Y. Zhou and Q. Song, Org. Lett., 2020, 22, 5284–5288 CrossRef CAS PubMed.
- R. Mei, C. Yang, F. Xiong and J. Zhao, Adv. Synth. Catal., 2021, 1–7 Search PubMed.
- A. G. Németh, R. Szabó, A. Domján, G. M. Keserű and P. Ábrányi-Balogh, ChemistryOpen, 2021, 10, 16–27 CrossRef.
- A. G. Németh, R. Szabó, G. Orsy, I. M. Mándity, G. M. Keserű and P. Ábrányi-Balogh, Molecules, 2021, 26, 303 CrossRef PubMed.
- A. G. Németh, B. Marlok, A. Domján, Q. Gao, X. Han, G. M. Keserű and P. Ábrányi-Balogh, Eur. J. Org. Chem., 2021, 28–33 Search PubMed.
- T. B. Nguyen and P. Retailleau, Org. Lett., 2021, 23, 5344–5348 CrossRef CAS PubMed.
- N. S. Antonkin, Y. A. Vlasenko, A. Yoshimura, V. I. Smirnov, T. N. Borodina, V. V. Zhdankin, M. S. Yusubov, A. Shafir and P. S. Postnikov, J. Org. Chem., 2021, 86, 7163–7178 CrossRef CAS PubMed.
- T. B. Nguyen and P. Retailleau, Org. Lett., 2017, 19, 3879–3882 CrossRef CAS PubMed.
- T. B. Nguyen and P. Retailleau, Org. Lett., 2017, 19, 4858–4860 CrossRef CAS PubMed.
- T. B. Nguyen and P. Retailleau, Org. Lett., 2018, 20, 186–189 CrossRef CAS PubMed.
- K. X. Nguyen, P. H. Pham, T. T. Nguyen, C. H. Yang, H. T. B. Pham, T. T. Nguyen, H. Wang and N. T. S. Phan, Org. Lett., 2020, 22, 9751–9756 CrossRef CAS PubMed.
- M. Wu, Y. Jiang, Z. An, Z. Qi and R. Yan, Adv. Synth. Catal., 2018, 360, 4236–4240 CrossRef CAS.
- B. Zhang, D. Liu, Y. Sun, Y. Zhang, J. Feng and F. Yu, Org. Lett., 2021, 23, 3076–3082 CrossRef CAS PubMed.
- Z. Yang, Y. Liang, A. Li, K. Liu, L. Li, T. Yang and C. Zhou, J. Org. Chem., 2019, 84, 16262–16267 CrossRef CAS PubMed.
- L. Gan, Y. Gao, L. Wei and J. P. Wan, J. Org. Chem., 2019, 84, 1064–1069 CrossRef CAS.
- L. F. Yang, C. G. Liu, X. P. Xu and S. J. Ji, Org. Biomol. Chem., 2016, 14, 2993–2999 RSC.
- J. S. Martin, C. J. MacKenzie, D. Fletcher and I. H. Gilbert, Bioorg. Med. Chem., 2019, 27, 2066–2074 CrossRef CAS PubMed.
- C. Kaya, J. Konstantinović, A. M. Kany, A. Andreas, J. S. Kramer, S. Brunst, L. Weizel, M. J. Rotter, D. Frank, S. Yahiaoui, R. Müller, R. W. Hartmann, J. Haupenthal, E. Proschak, T. A. Wichelhaus and A. K. H. Hirsch, J. Med. Chem., 2022, 65, 3913–3922 CrossRef CAS PubMed.
- R. W. Hartmann, J. Konstantinović, J. Haupenthal, A. K. H. Hirsch, A. Kany, C. Kaya, S. Yahiaoui, T. Wichelhaus and E. Proschak, 2021191219A1, WO Pat., 2021.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- L. Li, Q. Chen, H. H. Xu, X. H. Zhang and X. G. Zhang, J. Org. Chem., 2020, 85, 10083–10090 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob00512c |
| This journal is © The Royal Society of Chemistry 2022 |