
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a Lewis b hexasaccharide thioglycoside donor and its use towards an extended mucin core Tn heptasaccharide structure and a photoreactive biotinylated serine linked hexasaccharide†
Martin
Hollinger
,
Filippo
Bonaccorsi
,
Aisling Ní
Cheallaigh
and
Stefan
Oscarson
 *
*
Centre for Synthesis and Chemical Biology, UCD School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: stefan.oscarson@ucd.ie
First published on 16th May 2022
Abstract
Investigation into Heliobacter pylori binding to Lewis b (Leb) antigens through the blood group antigen binding adhesion protein (BabA) requires structurally well-defined tools. A Leb hexasaccharide thioglycoside donor was chemically prepared through a linear approach starting from D-lactose. This donor can be used to attach reducing end linkers providing a range of options for conjugation techniques or to further extend the oligosaccharide structure. To evaluate its efficiency as a donor, it was coupled to a 6-OH GalNAc acceptor, producing an extended Leb-containing Tn mucin core structure in 84% yield, and to L-serine in 72% yield. The latter compound was subsequently functionalized with a photolabile diazirine linker and biotin, creating a Leb hexasaccharide structure–function tool suitable for lectin tagging interaction studies. This donor opens a wide range of possibilities for conjugation of Leb structures to produce a variety of chemical biology tools to assist in the study of these interactions.
Introduction
Heliobacter pylori infections of the gastric epithelium can lead to gastric inflammation and may progress to gastroduodenal diseases such as peptic ulceration and gastric adenocarcinoma.1–5H. pylori binds to Lewis b-antigen (Leb) structures present in the gastric epithelium. This binding is mediated by its blood group antigen binding adhesion protein (BabA).6–8 Chemically defined structures have proved to be an invaluable tool for probing the binding of H. pylori BabA to Leb antigens.8–11 In our first synthesis of the Leb hexasaccharide, we constructed and utilized a Leb tetrasaccharide thioglycoside donor that was coupled to an azide-propyl lactose acceptor to afford the hexasaccharide.12 However, the donor was prone to elimination rather than glycosylation reaction. More effective routes to the hexasaccharide were developed using a linear synthesis of the Leb hexasaccharide starting from the same lactose acceptor;13–15 these syntheses are high-yielding and can be performed on a large scale but give little flexibility for changes at the reducing end of the molecule. Therefore, a more adaptable approach involving a Leb hexasaccharide donor was investigated, allowing the construction of a variety of Leb-containing chemical biology tools. Two earlier examples of Leb hexasaccharide donors have been reported, a fluoride donor by Sato et al.16 and a 1,2-epoxide donor by Danishefsky et al.17 However, a very low yield16 and no stereoselectivity17 were experienced in initial glycosylations with these two donors limiting their use in further applications. Considering our earlier good experiences with large block thioglycoside donors,18–22 a Leb hexasaccharide thioglycoside 1 (Fig. 1) was designed as the target donor.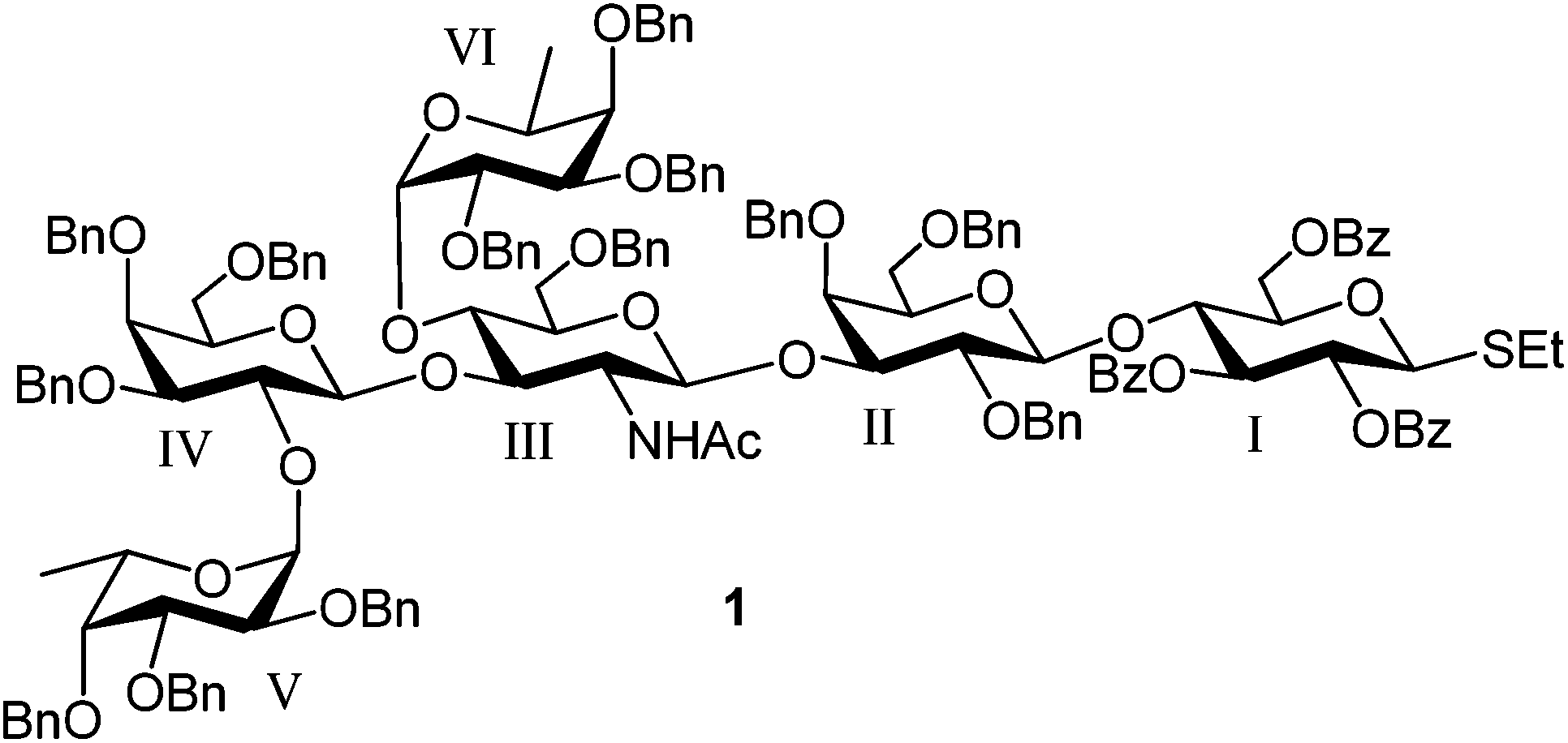 | ||
| Fig. 1 Target Leb hexasaccharide thioglycoside donor. | ||
Results and discussion
Apart from the orthogonal glycosylations required, the major difference from our earlier syntheses was the design and synthesis of the lactose acceptor, in particular, its protecting group pattern. Benzyl protecting groups were used on the galactose residue to ensure good reactivity of the 3′-OH acceptor, and acyl protecting groups on the glucose residue, to ensure β-selectivity in the glycosylations with the hexasaccharide donor 1. This protecting group pattern could be introduced starting from known compound 2 (Scheme 1).23,24 The Catelani group published procedure to 2 can effectively be performed on a large scale, the triisopropylidenelactose intermediate has been synthesised on a 250 kg scale25 and we synthesized compound 2 on a 30 g scale. Regioselective introduction of a naphtylmethyl ether (Nap) in the 3′-position using tin activation (→3) followed by benzylation afforded compound 4, with the desired protecting group pattern in the galactose moiety. Acidic acetal hydrolysis followed by acetylation gave compound 5 with an acylated glucose. Due to the presence of the benzyl protecting groups, the Lewis acid-promoted formation of the ethyl thiodisaccharide 6, had to be monitored very carefully by TLC (Rf 0.44 in toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc 4:1, v/v) to avoid decomposition. Attempts to use TMSOTf for the introduction of the thioethyl group were not successful, but with BF3-etherate as promoter compound 6 was obtained in an 81% yield based on recovered starting material predominantly consisting of the less reactive α-acetate. The Nap group of disaccharide 6 was then cleaved using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give lactose acceptor 7 in a 79% yield.
:EtOAc 4:1, v/v) to avoid decomposition. Attempts to use TMSOTf for the introduction of the thioethyl group were not successful, but with BF3-etherate as promoter compound 6 was obtained in an 81% yield based on recovered starting material predominantly consisting of the less reactive α-acetate. The Nap group of disaccharide 6 was then cleaved using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give lactose acceptor 7 in a 79% yield.
 | ||
| Scheme 1 Reagents and conditions: (a) dibutyltin oxide, tetra-butyl ammonium bromide, 2-(bromomethyl)naphthalene, toluene, reflux, 81%; (b) NaH, BnBr, 0 °C → rt, 89%; (c) 80% AcOH, 70 °C; (d) sodium acetate, Ac2O, reflux, 83% (over 2 steps, α/β mixture (3:1)); (e) EtSH, BF3·OEt2, ClCH2CH2Cl, 0 °C, 81% (based on recovered starting material); (f) DDQ, CH2Cl2–MeOH 4:1, rt, 79%. | ||
An orthogonal glycosylation strategy was needed to build the hexasaccharide without activating the reducing end thioglycoside donor. It was decided to use trichloroacetimidate (TCA) donors for the introduction of the glucosamine and galactose residues and glycosyl bromides with halide-assisted glycosylation conditions for the introduction of the two fucose moieties, the latter being reproducibly high yielding in earlier synthesis.13–15 When performing orthogonal glycosylations with thioglycoside acceptors, especially with non-bulky thio aglycons, there is always the risk of intramolecular aglycon transfer, the extent is hard to predict and very much dependent on the acceptor structure and of the thioaglycone used.26 In this synthesis we found that acceptor 7, although being an ethyl thiosaccharide, showed no tendency at all for aglycon transfer reactions.
For the introduction of the glucosamine moiety a suitable donor with a β-directing participating group and orthogonal protecting groups at the 3- and 4-position was needed to be able to selectively glycosylate these positions at a later stage, N-phthalimido derivative 827,28 (Scheme 2) was selected. Compound 8 possesses an orthogonal para-methoxybenzyl group (PMB) at the 3-position, and a benzylidene acetal in the 4,6-positions, which can be selectively opened to free the 4-position at a later stage. The thioethyl donor 8 was converted into its trichloroacetimidate 9 by hydrolysis of the thioethyl group using N-iodosuccinimide (NIS), followed by formation of the imidate with trichloroacetonitrile and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) (92% over 2 steps after silica gel column chromatography).
 | ||
| Scheme 2 Reagents and conditions: (a) NIS, CH3CN–H2O 10:1, rt; (b) Cl3CCN, DBU, ClCH2CH2Cl, 0 °C, 92% (over 2 steps); (c) TMSOTf, CH2Cl2, −20 °C, 78%; (d) sodium methoxide, MeOH, rt; (e) EtOH, hydrazine hydrate, reflux; (f) Ac2O, MeOH, rt.; (g) BzCl, pyridine, 0 °C, 83% (over 4 steps); (h) DDQ, CH2Cl2/iso-butanol (4:1), 78%. | ||
Donor 9 was employed in a TMSOTf-catalyzed glycosylation reaction with acceptor 7 affording the desired trisaccharide 10 in a 78% yield. Due to earlier findings, in which the removal of the phthalimido group in a fully assembled hexasaccharide was problematic and low yielding,13 we decided to remove this protecting group and introduce the acetamide moiety prior to further glycosylation steps, despite being aware of problematic reports on the use of acetamido-containing acceptors.29–31 The acetyl groups in trisaccharide 10 were removed using Zemplén conditions and the phthalimido group cleaved using hydrazine hydrate in ethanol. The amino group was then chemoselectively acetylated with acetic anhydride in methanol followed by benzoylation of the hydroxyl groups to give compound 11 in a yield of 83% over 4 steps. The benzoyl groups were preferred over acetyl protecting groups to enhance lipophilicity and therefore solubility of subsequent intermediates but also to improve glycosylation properties of the target hexasaccharide donor.32 In contrast to the removal of the Nap group in compound 6, the removal of the PMB group to obtain derivative 12 was challenging due to persistent side reactions resulting in poor yields when carrying out the reaction with DDQ in CH2Cl2–MeOH 4:1. By varying the solvent mixture to CH2Cl2–isobutanol, yields were significantly improved and trisaccharide acceptor 12 obtained in 78% yield.33
For the introduction of the galactose moiety, a donor with an orthogonal β-selective participating group at the 2-position was needed. Since benzoyl groups were present in trisaccharide 12, and it was known that removal of a 2′′′-O-acetyl group in a lacto-N-tetraose (LNT) tetrasaccharide requires unusually strong basic conditions,15 we chose a chloroacetyl group to protect this position (Scheme 3). The acetyl group of the known galactose derivative 1334 was removed using Zemplén conditions and replaced with a chloroacetyl group (→14, 88%) followed by conversion into the corresponding trichloroacetimidate donor 15. This time, NBS was used to hydrolyse the thioethyl group whereafter treatment with DBU and trichloroacetonitrile afforded compound 15 as a α/β mixture (10:1). It was possible to separate the anomers using silica gel chromatography. However, the high reactivity of the compound35 resulting in a rather low yield of 47% after chromatography.
 | ||
| Scheme 3 Reagents and conditions: (a) sodium methoxide, MeOH, rt, 99%; (b) chloroacetyl chloride, CH2Cl2–pyridine 14:1, 0 °C → rt, 89%; (c) NBS, CH3CN–H2O 10:1, rt; (d) trichloroacetonitrile, DBU, ClCH2CH2Cl, 0 °C, 47% (over 2 steps, α/β mixture (10:1)). (e) TMSOTf, CH2Cl2, −30 °C → −5 °C; (f) lutidine, thiourea, MeOH, 70 °C, 47% (over 2 steps); (g) NaCNBH3, HCl (1 M in Et2O), 71%; (h) Br2, CH2Cl2, rt; (i) tetraethylammonium bromide, CH2Cl2–DMF 3.5:1, rt, 89%. | ||
In the next step, donor 15 was employed in a TMSOTf-catalyzed glycosylation reaction with trisaccharide acceptor 12 (Scheme 3). The reaction had to be monitored carefully by TLC (toluene:EtOAc 1:1) as an intermediate (possibly the orthoester) was initially formed which was slowly converted into the desired tetrasaccharide. Due to difficulties in the isolation of the product, the crudely worked-up reaction mixture was directly treated with thiourea and lutidine in methanol to remove the chloroacetyl group affording the 2′′′-OH tetrasaccharide 16 (47% over two steps). The benzylidene group of the GlcNAc moiety of compound 16 was then selectively opened to free the 4′′-position using NaCNBH3, affording tetrasaccharide 2′′′,4′′-diol acceptor 17. In the final step towards the hexasaccharide donor, fucose thioglycoside 1836 was converted into its bromide 19 and then directly employed in a completely α-selective halide-assisted glycosylation with tetrasaccharide acceptor 17 to give the target Lewis b donor 1 in an 89% yield. In summary, hexasaccharide donor 1 (600 mg) was synthesized from compound 2 in a ten step sequence and in a 15% overall yield.
The donor properties of hexasaccharide donor 1 were then investigated. We were interested in Lewis b extended mucin core structures so GalNAc derivative 21 was chosen as a trial acceptor. Acceptor 21 was prepared from compound 2037via introduction of a benzylidene group at the 4,6-positions, followed by subsequent benzoylation and final removal of the acetal (Scheme 4). Glycosylation of the 4,6-diol acceptor 21 with hexasaccharide donor 1, using NIS and AgOTf as promoters, afforded exclusively the β-(1 → 6)-linked heptasaccharide 22 in an 84% yield. Heptasaccharide 22 was then deprotected, the benzoyl groups were removed under Zemplén conditions, followed by hydrogenolysis of the benzyl and azido groups to afford globally deprotected heptasaccharide 23 in a 92% yield over the 2 steps.
 | ||
| Scheme 4 Reagents and conditions: (a) benzaldehyde dimethylacetal, p-TsOH·H2O, DMF, rt; (b) benzoyl chloride, pyridine, rt; (c) AcOH (80% in H2O), 85 °C, 34% (over 3 steps); (d) AgOTf, NIS, CH2Cl2, −40 °C → 0 °C, 84%; (e) sodium methoxide, MeOH, rt; (f) H2 (5 bar), Pd/C, HCl (1 N in H2O), THF–EtOH–H2O, 92% (over 2 steps). | ||
We were further interested in photo-active tools to enable cross-linking of Lewis b-structures to neighbouring interacting structures. Hexasaccharide donor 1 was therefore glycosylated with Boc-Ser-OBn using the NIS/AgOTf promotor system producing the serine linked hexasaccharide 24 in 72% yield (Scheme 5). Hydrogenolysis with Pd/C and H2 followed by saponification using Zemplén conditions produced the globally deprotected compound (88% over 2 steps) which was carried through as crude material to the following two-step bifunctionalising process. A photolabile diazirine linker was introduced to the free amine of the serine residue using an activated N-hydroxysuccinimide ester of the corresponding diazirine linker, subsequently the carboxylic acid group of the serine was coupled to biotin hydrazate, using HOBt and EDCI as coupling reagents, producing the photoreactive biotinylated hexasaccharide 25.
 | ||
| Scheme 5 Reagents and conditions (a) NIS, AgOTf, −40 °C, 72%; (b) Pd/C, H2; (c) NaOMe, MeOH, 88% (over 2 steps); (d) N-succinimidyl-diazirine (SDA), PBS buffer; (e) HOBt, EDCl, biotin hydrazate. | ||
Conclusions
We have successfully synthesized a novel Leb block donor via a linear approach using a lactose acceptor that can be produced on a large scale and two new selectively protected trichloroacetimidate donors. The synthesized Leb hexasaccharide block donor gave high yields and complete stereoselectivity in a glycosylation reaction to obtain a Leb-extended Tn mucin core structure. Coupling of the donor to serine and additional extension to a bifunctionalised diazirine-biotin linked hexasaccharide further showed the versatility of the donor and its potential use in carbohydrate–protein structure–function interaction elucidation. This donor opens a wide range of possibilities for conjugation of Leb structures to produce alternative targets.Experimental
General methods
Please see ESI† for NMR spectra and experimental for compounds 3–7, 9, 14–15 and 21.General methods
The 1H/13C NMR spectra (δ in ppm, relative to TMS in CDCl3 and relative to solvent peak in D2O) were recorded with Varian spectrometers (400/100 MHz, 500/125 MHz or 600/150 MHz) at 25 °C. Assignments were aided by 1H–1H and 1H–13C correlation experiments. HRMS spectra were recorded on a micromass LCT instrument from Waters. Optical rotations were measured on a PerkinElmer polarimeter with a Na lamp (589 nm) at 20 °C and are not corrected. TLC was carried out on precoated 60 F254 silica gel alumina plates (Merck) using UV-light, H2SO4 (10% in ethanol) and/or ninhydrin-solution (ninhydrin/CH3COOH/ethanol [0.3:3:100 w/v/v]. Flash chromatography was performed on silica gel (Apollo scientific, pore size 60 Å, particle size 40–63 μm) or via pre-packed columns (Biotage AB, particle size 50 μm) on a Biotage SP4 system. Size exclusion chromatography was performed using Biogel P2 (Biorad, <45 μm bead size, 100–1800 MW) with H2O-n-butanol (99:1) as eluent.
:1); [α]20D –1.7° (c 1.33, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.70–7.63 (m, 1H, Ar), 7.56–7.50 (m, 3H, Ar), 7.43–7.26 (m, 14H, Ar), 7.19–7.10 (m, 4H, Ar), 6.90–6.84 (m, 4H, Ar) and 6.36–6.31 (m, 2H, Ar), 5.62 (s, 1H, CHPh), 5.46 (d, J1,2 = 8.4 Hz, 1H, H-1III), 5.00–4.91 (m, 2H), 4.88–4.82 (m, 1H), 4.68 (d, J = 12.1 Hz, 1H), 4.52–4.35 (m, 7H), 4.29–4.20 (m, 3H), 4.07 (d, J1,2 = 7.6 Hz, 1H, H-1II), 4.04–3.98 (m, 2H), 3.88–3.75 (m, 4H), 3.70–3.59 (m, 3H), 3.57 (s, 3H, C6H4OCH3), 3.54–3.44 (m, 3H), 3.37 (dd, J = 9.8, 7.6 Hz, 1H), 3.11 (ddd, J = 9.8, J = 5.4, J = 1.7 Hz, 1H), 2.63–2.50 (m, 2H, CH2CH3), 2.00, 1.88 and 1.78 (9H, COCH3), 1.17 (t, J = 7.4 Hz, 3H, CH2CH3); 13C NMR (125 MHz, CDCl3) δ 170.5, 170.2 and 169.6 (COCH3), 167.37 (NCO(Phth)), 158.8, 138.9, 138.5, 137.8, 137.3, 133.5, 131.2, 130.1, 129.5, 129.1, 128.5, 128.3, 128.2, 127.9, 127.9, 127.7, 127.4, 126.7, 126.3, 126.1, 123.1 and 113.3 (Ar), 102.5 (C-1II), 101.4 (CHPh), 100.1 (C-1III), 83.2 (C-1I), 83.1 (2C, C-4III, C-3II), 79.0 (C-2II), 77.1 (C-5I (HSQC)), 76.1 (C-4II), 74.8 (CH2Ph), 74.2 (2C, CH2Ph, C-3III), 73.8 (2C, CH2PhOCH3, C-4I), 73.5 (2C, C-3I, CH2Ph), 73.4 (C-5II), 69.8 (C-2I), 68.8 (C-6III), 68.6 (C-6II), 65.9 (C-5III), 62.3 (C-6I), 56.4 (C-2III), 54.9 (PhOCH3), 24.1 (CH2CH3), 20.8, 20.7, 20.6 (COCH3), 14.9 (CH2CH3); ES-HRMS calcd for C70H75NO20S [Na]+ 1304.4501 found 1304.4436.
:1) the solution was neutralised with Dowex 50 W+ ion exchange resin, filtered, concentrated under reduced pressure and directly used for the next step. The residue was dissolved in EtOH (150 mL), hydrazine hydrate (5.54 mL, 114 mmol) added and the mixture refluxed for 36 h. After completion of the reaction the solvent was evaporated and the residue co-evaporated with toluene (2 × 30 mL). The crude material was dissolved in EtOAc (150 mL), washed with saturated aq NaHCO3 (100 mL) and the aqueous layer extracted with EtOAc (2 × 50 mL). The combined organic fractions were dried over MgSO4, filtered, concentrated under reduced pressure, and the crude was dried in vacuo. The residue was dissolved in MeOH (150 mL) and the resulting solution cooled to 0 °C before acetic anhydride (0.467 mL, 4.95 mmol) was added. The reaction mixture was stirred for 3 h while allowing to warm to room temperature. Then the mixture was concentrated, co-evaporated with toluene (3 × 20 mL) and dried in vacuo. The crude material was dissolved in pyridine (40 mL), benzoyl chloride (1.55 mL, 13.3 mmol) added at 0 °C and the mixture stirred for 2 h. After completion of the reaction, MeOH (2 mL) was added and the mixture concentrated and co-evapoarted with toluene. The residue was dissolved in CH2Cl2 (200 mL), washed with saturated aq NaHCO3 (100 mL) and the organic layer dried over MgSO4, filtered and concentrated. Purification by flash chromatography on silica gel (toluene–EtOAc) gave 11 (4.36 g, 83%) as a colourless solid. Rf 0.62 (toluene–EtOAc 2:1); [α]20D +2.7° (c 1.79, CHCl3); 1H NMR (500 MHz, CDCl3) 8.01–7.81 (m, 6H, Ar), 7.60–7.08 (m, 31H, Ar), 6.82–6.76 (m, 2H, Ar), 5.68–5.61 (m, 1H), 5.53 (s, 1H, CHPh), 5.46–5.40 (m, 1H), 4.91–4.84 (m, 2H), 4.77 (d, J = 11.8 Hz, 1H), 4.73–4.66 (m, 4H), 4.61 (d, J = 12.3 Hz, 1H), 4.47 (d, J = 11.5 Hz, 1H), 4.42 (dd, J = 12.1, J = 5.2 Hz, 1H), 4.35–4.26 (m, 3H), 4.15–4.06 (m, 3H), 3.80–3.49 (m, 11H), 3.44–3.37 (m, 1H), 3.31–3.26 (m, 1H), 2.91 (dd, J = 9.0, J = 5.1 Hz, 1H), 2.85–2.79 (m, 1H), 2.73–2.59 (m, 2H, CH2CH3), 1.46 (s, 3H, NCOCH3), 1.19 (t, J = 7.4 Hz, 3H, CH2CH3); 13C NMR (125 MHz, CDCl3) δ 169.9 (NCOCH3), 165.9, 165.7 and 165.4 (COPh), 159.2, 139.2, 138.8, 138.0, 137.4, 133.2, 133.1, 132.6, 130.5, 130.3, 129.9, 129.8, 129.6, 129.3, 129.0, 128.5, 128.4, 128.3, 128.0, 127.8, 127.6, 127.5, 127.1, 126.6, 126.1 and 113.7 (Ar), 103.1 (C-1II), 101.9 (C-1III), 101.2 (CHPh), 83.6 (C-1I), 82.4 (C-4III), 80.7 (C-3II), 79.9 (C-2II), 77.6 (C-5I), 77.0 (C-3III (HSQC)) 75.6 (C-4II), 75.3 (C-4I), 74.9 (CH2Ph), 74.4 (CH2Ph), 74.2 (C-3I), 73.6 (CH2PhOCH3), 73.1 (2C, CH2Ph, C-5II), 70.6 (C-2I), 68.8 (C-6II), 67.7 (C-6III), 65.9 (C-5III), 63.0 (C-6I), 56.7 (C-2III), 55.3 (PhOCH3), 24.3 (CH2CH3), 23.1 (NCOCH3), 14.9 (CH2CH3); ES-HRMS calcd for C79H81NO19S [Na]+ 1402.5021 found 1402.4957.
:1); [α]20D –10.5° (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.01–7.92 (m, 4H), 7.90–7.85 (m, 2H, Ar), 7.60–7.54 (m, 1H, Ar), 7.53–7.26 (m, 21H, Ar), 7.23–7.10 (m, 7H, Ar), 5.71–5.63 (m, 1H), 5.53 (s, 1H), 5.49–5.43 (m, 1H), 5.30 (d, J = 5.2 Hz, 1H), 5.08 (d, J = 12.7 Hz, 1H), 4.81–4.74 (m, 1H), 4.68 (m, 5H), 4.45 (dd, J = 12.1, J = 5.1 Hz, 1H), 4.38–4.28 (m, 3H), 4.19–4.09 (m, 3H), 3.84–3.77 (m, 1H), 3.77–3.68 (m, 4H), 3.62–3.49 (m, 3H), 3.40 (m, 1H), 3.36–3.30 (m, 1H), 2.97 (dd, J = 9.0, J = 5.3 Hz, 1H), 2.86–2.80 (m, 1H), 2.75–2.61 (m, 2H, CH2CH3), 1.48 (s, 3H, COCH3), 1.20 (t, J = 7.4 Hz, 3H, CH2CH3) (OH was not observed); 13C NMR (125 MHz, CDCl3) 172.4 (NCOCH3), 165.9, 165.8 and 165.3 (COPh), 138.9, 138.3, 137.8, 137.0, 133.3, 132.6, 130.2, 129.9, 129.7, 129.6, 129.3, 129.1, 128.8, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.3, 126.4 and 126.1 (Ar), 103.0 (C-1II), 102.3 (C-1III), 101.9 (CHPh), 83.6 (C-1I), 81.5 (C-4III), 81.1 (C-3II), 79.6 (C-2II), 77.5 (C-5I), 75.7 (C-4II), 75.0 (C-4I), 74.6 (CH2Ph), 74.5 (CH2Ph), 74.1 (C-3I), 73.2 (2C, CH2Ph, C-5II), 72.7 (C-3III), 70.5 (C-2I), 68.5 (C-6III), 67.5 (C-6II), 66.3 (C-5III), 63.1 (C-6I), 59.1 (C-2III), 24.3 (CH2CH3), 22.9 (NHCOCH3), 14.9 (CH2CH3); ES-HRMS calcd for C71H73NO18S [Na]+ 1282.4446 found 1282.4392.
:1); [α]20D –9.6° (c 1.88, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.01–7.92 (m, 4H, Ar), 7.89–7.83 (m, 2H, Ar), 7.58–7.53 (m, 1H, Ar), 7.52–7.08 (m, 43H, Ar), 5.67–5.61 (m, 1H), 5.50 (s, 1H), 5.48–5.40 (m, 2H), 4.90 (d, J = 11.5 Hz, 1H), 5.67–5.61 (m, 1H), 5.50 (s, 1H), 5.48–5.40 (m, 2H), 4.90 (d, J = 11.5 Hz, 1H), 4.86–4.77 (m, 3H), 4.70 (d, J1,2 = 9.9 Hz, 1H, H-1I), 4.66 (m, 2H), 4.59 (m, 3H), 4.40 (dd, J = 12.0, J = 5.3 Hz, 1H), 4.36 (d, J = 12.0 Hz, 1H), 4.33–4.24 (m, 4H), 4.22 (d, J1,2 = 7.9 Hz, 1H, H-1IV), 4.14–4.04 (m, 3H), 4.01–3.95 (m, 1H), 3.91–3.87 (m, 1H), 3.86–3.84 (m, 1H), 3.74 (m, 1H), 3.71–3.63 (m, 4H), 3.60–3.54 (m, 2H), 3.52 (dd, J = 10.7, J = 5.5 Hz, 1H), 3.49–3.44 (m, 1H), 3.39 (dd, J = 9.7, J = 4.9 Hz, 1H), 3.35 (dd, J = 8.4, J = 4.9 Hz, 1H), 3.31–3.25 (m, 2H), 2.96–2.75 (m, 3H), 2.73–2.59 (m, 2H, SCH2CH3), 1.53 (s, J = 11.6 Hz, 3H, NCOCH3), 1.18 (t, J = 7.4 Hz, 3H, CH2CH3); 13C NMR (125 MHz, CDCl3) δ 170.0 (NCOCH3), 164.8, 164.7 and 164.3 (COPh), 138.2, 137.8, 137.7, 137.5, 137.0, 136.7, 135.8, 132.2, 132.1, 131.5, 129.3, 128.8, 128.6, 128.3, 128.1, 127.4, 127.3, 127.2, 127.0, 126.9, 126.8, 126.7, 126.6, 126.5, 126.4, 126.1, 125.8 and 125.2 (Ar), 102.2 (C-1IV), 102.1 (C-1II), 101.6 (C-1III), 100.6 (CHPh), 82.5 (C-1I), 80.5 (C-3IV), 80.0 (C-3II), 79.2 (C-4III), 78.6 (C-2II), 76.6 (C-5I), 74.8 (C-3III), 74.6 (C-4II), 74.3 (C-4I), 73.9 (CH2Ph), 73.7 (CH2Ph), 73.4 (CH2Ph), 73.2 (C-3I), 72.7 (C-5IV), 72.4 (2C, C-4IV, CH2Ph), 72.1 (2C, CH2Ph, C-5II), 71.6 (CH2Ph), 69.5 (2C, C-2I, C-2IV), 67.7 (C-6III), 67.3 (C-6IV), 66.7 (C-6II), 65.2 (C-5III), 62.0 (C-6I), 55.2 (C-2III), 23.2 (CH2CH3), 22.2 (NCOCH3), 13.9 (CH2CH3); ES-HRMS calcd for C98H101NO23S [Na]+ 1714.6383 found 1714.6360.
:1); [α]20D +4.1° (c 0.38, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.99–7.92 (m, 4H, Ar), 7.89–7.84 (m, 2H, Ar), 7.56–7.52 (m, 1H, Ar), 7.52–7.48 (m, 1H, Ar), 7.41–7.10 (m, 42H, Ar), 5.68–5.63 (m, 1H), 5.46–5.40 (m, 1H), 5.06 (d, J = 8.2 Hz, 1H, NHCOCH3), 4.95 (d, J = 12.4 Hz, 1H), 4.87 (d, J = 11.6 Hz, 1H), 4.81–4.75 (m, 2H), 4.73–4.63 (m, 5H), 4.53 (d, J = 11.7 Hz, 1H), 4.49–4.46 (m, 2H), 4.44–4.39 (m, 2H), 4.37–4.30 (m, 4H), 4.13–4.05 (m, 3H), 4.01 (d, J1,2 = 7.7 Hz, 1H, H-1IV), 3.92–3.88 (m, 2H), 3.83–3.75 (m, 4H), 3.68–3.63 (m, 1H), 3.62–3.53 (m, 4H), 3.48–3.42 (m, 3H), 3.40 (dd, J = 8.8, J = 5.2 Hz, 1H), 3.34 (dd, J = 9.8, J = 2.9 Hz, 1H), 3.32–3.28 (m, 1H), 2.93–2.92 (m, 1H), 2.90 (dd, J = 9.0, J = 5.4 Hz, 1H), 2.80 (dd, J = 8.9, J = 7.8 Hz, 1H), 2.72–2.60 (m, 2H, SCH2CH3), 1.48 (s, 3H, NCOCH3), 1.20–1.16 (m, 3H, SCH2CH3); 13C NMR (150 MHz, CDCl3) δ 171.7 (NHCOCH3), 165.9, 165.8 and 165.3 (COPh), 139.1, 138.9, 138.4, 138.3, 138.2, 138.0, 137.5, 130.2, 129.9, 129.8, 129.6, 129.3, 128.6, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.1 and 126.2 (Ar), 104.8 (C-1IV), 103.0 (C-1II), 101.6 (C-1III), 87.1 (C-3III), 83.5 (C-1I), 81.4 (2C, C-3II, C-3IV), 79.7 (C-2II), 77.6 (C-5I), 75.7 (C-4II), 75.3 (C-4III), 75.2 (C-4I), 74.7 (CH2Ph), 74.6 (CH2Ph), 74.5 (CH2Ph), 74.1 (2C, C-3I, C-5IV), 73.7 (CH2Ph), 73.4 (CH2Ph), 73.3 (2C, C-4IV, C-5II), 73.1 (CH2Ph), 72.9 (CH2Ph), 71.2 (C-2IV), 70.5 (C-2I), 69.8 (C-6III), 69.3 (C-5III), 68.8 (C-6IV), 67.8 (C-6II), 63.0 (C-6I), 55.6 (C-2III), 24.2 (SCH2CH3), 23.1 (NHCOCH3), 14.9 (SCH2CH3); ES-HRMS calcd for C98H103NO23S [Na]+ 1716.6539 found 1716.6548.
:1 to 2:1, column 2: toluene–acetonitrile 7:1 to 4:1) to obtain 1 (0.600 g, 89%) as a colourless solid. Rf 0.72 (toluene–EtOAc 2:1), [α]20D –36.9° (c 0.73, CHCl3); 13C NMR (125 MHz, CD2Cl2) δ 169.63 (NHCOCH3), 166.3, 166.1 and 165.7 (COPh), 140.3, 140.0, 139.8, 139.7, 139.5, 139.3, 139.0, 138.9, 138.8, 138.6, 133.9, 133.7, 133.3, 130.8, 130.5, 130.3, 130.2, 130.1, 129.9, 129.2, 129.1, 129.0, 128.9, 128.8, 128.7, 128.6, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.5, 127.4 and 127.0 (Ar), 104.0 (C-1II), 102.7 (C-1III), 102.0 (C-1IV), 99.2 (C-1VI), 98.2 (C-1V), 84.3 (C-3IV), 84.1 (C-1I), 81.1 (C-2II), 80.8 (C-3V), 80.5 (C-3II), 79.5 (C-3VI), 79.1 (C-4VI), 78.8 (C-4V), 78.1 (C-5I), 76.6 (2C, C-2V, C-4II), 76.5 (2C, C-2VI, C-4I), 76.2 (CH2Ph), 76.0 (C-5III), 75.8 (C-3III), 75.5 and 75.3 (CH2Ph), 75.2 (2C, CH2Ph), 74.8 (2C, C-3I, C-2IV), 74.3 (C-4IV), 74.1 (CH2Ph), 73.8 (C-5II), 73.6 (CH2Ph), 73.5 (2C, C-5IV, CH2Ph) 73.3 (C-4III), 73.2 (CH2Ph), 72.2 (2C, CH2Ph), 71.9 (CH2Ph), 71.3 (C-2I), 69.2 (C-6IV), 68.2 (2C, C-6III, C-6II), 67.3 (C-5V), 66.9 (C-5VI), 63.5 (C-6I), 56.5 (C-2III), 24.9 (CH2CH3), 23.8 (NHCOCH3), 16.5 (2C, C-6V, C-6VI), 15.4 (CH2CH3); ES-HRMS calcd for C152H159NO31S [Na]+ 2549.0515 found 2549.0415.
:1); [α]20D –21.1° (c 2.33, CHCl3); 13C NMR (150 MHz, CDCl3) δ 170.1 and 169.1 (NHCOCH3), 166.4, 166.0, 165.7 and 165.3 (COPh), 139.5, 139.3, 139.2, 138.9, 138.6, 138.4, 138.2, 138.0, 137.7, 133.4, 133.3, 133.2, 132.6, 130.1, 130.0, 129.8, 129.6, 129.4, 128.6, 128.4, 128.3, 128.2, 128.0, 127.8, 127.7, 127.6, 127.5, 127.3, 127.1, 127.0, 126.9, 126.5 and 126.2 (Ar), 103.0 (C-1II), 101.8 (C-1III), 101.6 (C-1IV), 101.2 (C-1I), 98.4 (C-1VI), 97.8 (2C, C-1V, C-1VII), 83.8 (C-3IV), 80.6 (C-2II), 80.3 (C-3V), 79.7 (C-3II), 79.1 (C-3VI), 78.1 (C-4V), 78.1 (C-4VI), 75.8 (C-4II), 75.7 (C-2V), 75.4 (3C, C-2VI, C-5III, CH2Ph), 75.1 (C-3III), 75.0 (C-4I), 74.9 and 74.8 (CH2Ph), 74.7 (2C, CH2Ph), 74.5 (CH2Ph), 74.0 (C-2IV), 73.8 (C-5I), 73.6 (CH2Ph), 73.5 (C-5II), 73.2 (CH2Ph), 73.1 (2C, C-4IV, C-4III), 72.9 (2C, CH2Ph, C-5IV), 72.6 (2C, C-3I, CH2Ph), 71.7 (4C, C-2I, C-3VII, 2 x CH2Ph) 71.2 (CH2Ph), 69.4 (C-5VII), 68.9 (C-6VII), 68.5 (C-6IV), 67.9 (C-6II), 67.3 (C-6III), 67.1 (C-4VII), 67.0 (CH2CH2N3), 66.8 (C-5V), 66.6 (C-5VI), 62.2 (C-6I), 56.4 (C-2III), 50.2 (CH2CH2N3), 47.3 (C-2VII), 23.4 and 23.2 (NHCOCH3), 16.3 and 16.2 (C-6V and C-6VI); ES-HRMS calcd for C167H175N5O38 [Na]+ 2881.1813 found 2881.1875.
:2:2 +0.5% AcOH); [α]20D –12.8° (c 0.67, H2O); 13C NMR (150 MHz, D2O) δ 174.6 and 174.1 (NHCOCH3), 103.2 (C-1III), 102.9 (C-1II), 102.4 (C-1V), 100.6 (C-1IV), 99.5 (C-1VI), 97.7 (C-1I), 97.6 (C-1VII), 81.5, 78.1, 76.4, 75.1, 74.8, 74.7 (2C), 74.4, 74.2, 73.6, 72.7, 71.9 (2C), 71.7, 70.2, 70.1, 69.4, 69.2, 69.0, 68.7, 68.5, 68.4, 68.2, 67.7, 67.4, 67.0, 66.2, 64.5, 61.5, 60.9, 59.9, 59.4, 55.7, 49.5, 39.2 (CH2CH2N3), 22.1, 21.9 (NHCOCH3), 15.3, 15.2 (C-6VI, C-6VII); ES-HRMS calcd for C48H83N3O34 [Na]+ 1268.4756 found 1268.4805.
:10 to 50:50) followed by a Biogel P2 size exclusion column to give 25 (6.5 mg, 0.0045 mmol, 11% over four steps). 1H NMR (600 MHz, D2O) δ 5.16 (d, J = 4.1 Hz, 1H), 5.04 (d, J = 3.9 Hz, 1H), 4.88 (d, J = 6.8 Hz, 1H), 4.75 (d, J = 5.1 Hz,1H), 4.65–4.59 (m, 2H), 4.49–4.40 (m, 2H), 4.35 (q, J = 6.6 Hz, 1H), 4.25 (dd, J = 10.7, 5.3 Hz, 1H), 4.15 (m, 3H), 4.00–3.50 (m, 28H), 3.36 (m, 2H), 3.02 (dd, J = 13.1, 5.0 Hz, 1H, biotin h), 2.80 (d, J = 13.0 Hz, 1H, biotin h), 2.35 (t, J = 7.3 Hz, 2H, biotin a), 2.29 (t, J = 7.4 Hz, 2H, diazirine i), 2.07 (s, 3H, COOCH3), 1.78–1.67 (m, 4H, biotin d′, b and diazirine j), 1.62 (dd, J = 14.1, 7.6 Hz, 1H, biotin d′′), 1.48 (q, J = 7.6 Hz, 2H, biotin c), 1.28 (dd, J = 9.2, 6.6 Hz, 6H, CHV,VI3), 1.04 (s, 3H, diazirine CH3). 13C NMR (151 MHz, D2O) δ 175.5, 174.1, 165.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 103.17, 102.93, 102.08, 100.57, 99.49, 97.71 (C-1I−VI), 81.48, 78.12, 76.42, 75.11, 74.72, 74.40, 74.06, 73.56, 72.62, 71.91, 71.71, 70.07, 69.37, 69.04, 68.67, 68.55 (CH2 Serine), 68.20, 67.73, 66.96, 66.17, 61.90, 61.52, 60.88, 59.9, 59.4 (C6I−IV), 60.19, 55.68, 55.15, 52.33, 39.63 (CH2 biotin h), 33.09 (CH2 biotin a), 29.81 (CH2 diazirine i), 29.42 (CH2 biotin b), 27.59 (CH2 biotin c), 27.45 (CH2 biotin d), 24.59 (CH2 diazirine j), 22.10 (COOCH3), 18.49 (CH3 diazirine k), 15.29 (CHV,VI3). MALDI-TOF MS: C56H92N8NaO33S [M + Na]+ calcd 1459.5385, found 1459.49.
O), 103.17, 102.93, 102.08, 100.57, 99.49, 97.71 (C-1I−VI), 81.48, 78.12, 76.42, 75.11, 74.72, 74.40, 74.06, 73.56, 72.62, 71.91, 71.71, 70.07, 69.37, 69.04, 68.67, 68.55 (CH2 Serine), 68.20, 67.73, 66.96, 66.17, 61.90, 61.52, 60.88, 59.9, 59.4 (C6I−IV), 60.19, 55.68, 55.15, 52.33, 39.63 (CH2 biotin h), 33.09 (CH2 biotin a), 29.81 (CH2 diazirine i), 29.42 (CH2 biotin b), 27.59 (CH2 biotin c), 27.45 (CH2 biotin d), 24.59 (CH2 diazirine j), 22.10 (COOCH3), 18.49 (CH3 diazirine k), 15.29 (CHV,VI3). MALDI-TOF MS: C56H92N8NaO33S [M + Na]+ calcd 1459.5385, found 1459.49.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Science Foundation Ireland (Grants 08/IN.1/B2067, 13/IA/1959, and 16/RC/3889) is gratefully acknowledged.References
- J. Parsonnet, G. D. Friedman, D. P. Vandersteen, Y. Chang, J. H. Vogelman, N. Orentreich and R. K. Sibley, N. Engl. J. Med., 1991, 325, 1127 CrossRef CAS PubMed.
- A. Dubois, Emerging Infect. Dis., 1995, 1, 79 CrossRef CAS PubMed.
- N. Uemura, S. Okamoto, S. Yamamoto, N. Matsumura, S. Yamaguchi, M. Yamakido, K. Taniyama, N. Sasaki and R. J. Schlemper, N. Engl. J. Med., 2001, 345, 784 CrossRef CAS PubMed.
- S. Suerbaum and P. Michetti, N. Engl. J. Med., 2002, 347, 1175 CrossRef CAS PubMed.
- C.-Y. Kao, B.-S. Sheu and J.-J. Wu, Biomed. J., 2016, 39, 14 CrossRef PubMed.
- T. Borén, P. Falk, K. A. Roth, G. Larsson and S. Normark, Science, 1993, 262, 1892 CrossRef PubMed.
- D. Ilver, A. Arnqvist, J. Ögren, I. Frick, D. Kersulyte, E. Incecik, D. Berg, A. Covacci, L. Engstrand and T. Borén, Science, 1998, 279, 373 CrossRef CAS PubMed.
- M. Aspholm-Hurtig, G. Dailide, M. Lahmann, A. Kalia, D. Ilver, N. Roche, S. Vikström, R. Sjöström, S. Lindén, A. Bäckström, C. Lundberg, A. Arnqvist, J. Mahdavi, U. J. Nilsson, B. Velapatiño, R. H. Gilman, M. Gerhard, T. Alarcon, M. López-Brea, T. Nakazawa, J. G. Fox, P. Correa, M. G. Dominguez-Bello, G. I. Perez-Perez, M. J. Blaser, S. Normark, I. Carlstedt, S. Oscarson, S. Teneberg, D. E. Berg and T. Borén, Science, 2004, 305, 519 CAS.
- E. G. Sweeney and K. Guillemin, Cell Host Microbe, 2016, 19(1), 5 CrossRef CAS PubMed.
- K. Moonens, P. Gideonsson, S. Subedi, M. Mendez, J. Bugaytsova, E. Romaõ, M. Lahmann, G. Castaldo, F. Coppens, A. Lo, J. Nordén, A. Shevtsova, K. Brännström, J. Solnick, L. Hammarström, G. Vandenbussche, S. Oscarson, D. E. Berg, A. Arnqvist, S. Muyldermans, T. Borén and H. Remaut, Cell Host Microbe, 2016, 19(1), 55 CrossRef CAS PubMed.
- J. Bugaytsova, S. Vikström, S. Eriksson, O. Björnham, M. Mendez, C. Aisenbrey, G. Bylun, J. Mahdavi, J. Ögren, D. Ilver, R. Sjöström, R. H. Gilman, A. Chowdhury, A. K. Mukhopadhyay, L. Engstrand, S. Oscarson, S. Odenbreit, R. Haas, G. Gröbner, S. Schedin, D. E. Berg, A. Arnqvist and T. Borén, Cell Host Microbe, 2017, 21(3), 376 CrossRef CAS PubMed.
- A. Chernyak, S. Oscarson and D. Turek, Carbohydr. Res., 2000, 329, 309 CrossRef CAS PubMed.
- M. Lahmann, L. Buelow, P. Teodorovic, H. Gybäck and S. Oscarson, Glycoconjugate J., 2004, 21, 251 CrossRef CAS PubMed.
- V. Fournière, L. Skantz, F. Sajtos, S. Oscarson and M. Lahmann, Tetrahedron, 2010, 66, 7850 CrossRef.
- M. Long, A. Ní Cheallaigh, M. Reihill, S. Oscarson and M. Lahmann, Org. Biomol. Chem., 2020, 18, 4452 RSC.
- S. Sato, Y. Ito and T. Ogawa, Carbohydr. Res., 1986, 155, C1 CrossRef CAS PubMed.
- S. J. Danishefsky, V. Behar, J. T. Randolph and K. O. Lloyd, J. Am. Chem. Soc., 1995, 117, 5701 CrossRef CAS.
- K. Ekelöf and S. Oscarson, J. Org. Chem., 1996, 61, 7711 CrossRef PubMed.
- C. Bernlind, S. Bennett and S. Oscarson, Tetrahedron: Asymmetry, 2000, 11, 481 CrossRef CAS.
- A. Sundgren, M. Lahmann and S. Oscarson, J. Carbohydr. Chem., 2005, 24, 379 CrossRef CAS.
- J. D. M. Olsson and S. Oscarson, Tetrahedron: Asymmetry, 2009, 20, 879 CrossRef.
- L. Guazelli, C. J. Crawford, R. Ulc, A. Bowen, O. McCabe, A. Jedlicka, M. P. Wear, A. Casadevall and S. Oscarson, Chem. Sci., 2020, 11, 9209 RSC.
- G. Catelani, F. D'Andrea and L. Puccioni, Carbohydr. Res., 2000, 324, 204 CrossRef CAS PubMed.
- P. L. Barili, G. Catelani, F. D'Andrea, F. De Rensis and P. Falcini, Carbohydr. Res., 1997, 298, 75 CrossRef CAS.
- K. Agoston, M. J. Hederos, I. Bajza and G. Dekany, Carbohydr. Res., 2019, 476, 71 CrossRef CAS PubMed.
- H. M. Christensen, S. Oscarson and H. H. Jensen, Carbohydr. Res., 2015, 408, 48 CrossRef PubMed.
- M. Nilsson and T. Norberg, Carbohydr. Res., 1988, 183, 71 CrossRef CAS PubMed.
- K. Sarkar, I. Mukherjee and N. Roy, J. Carbohydr. Chem., 2003, 22, 95 CrossRef CAS.
- R. J. Pougny, M. Nasser, N. Naulet and P. Sinaÿ, Nouv. J. Chim., 1978, 2, 389 Search PubMed.
- L. Liao and F. I. Auzanneau, Org. Lett., 2003, 5, 2607 CrossRef CAS PubMed.
- A. Sundgren, M. Lahmann and S. Oscarson, Beilstein J. Org. Chem., 2010, 6, 704 CrossRef PubMed.
- P. J. Garegg, P. Konradsson, I. Kvarnström, T. Norberg, S. C. T. Svensson and B. Wigilius, Acta Chem. Scand., Ser. B, 1985, 39, 569 CrossRef.
- D. Crich and Q. Yao, J. Am. Chem. Soc., 2004, 126, 8232 CrossRef CAS PubMed.
- Y. Zhang, D. Dong, H. Qu, M. Sollogoub and Y. Zhang, Eur. J. Org. Chem., 2011, 7133 CrossRef CAS.
- L. K. Mydock and A. V. Demchenko, Org. Lett., 2008, 10, 2103 CrossRef CAS PubMed.
- H. Lönn, Carbohydr. Res., 1985, 139, 105 Search PubMed.
- Q. Wang, S. A. Ekanayaka, J. Wu, J. Zhang and Z. Guo, Bioconjugate Chem., 2008, 19, 2060 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob00477a |
| This journal is © The Royal Society of Chemistry 2022 |