
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile protocol for the preparation of 2-carboxylated thieno [2,3-b] indoles: a de novo access to alkaloid thienodolin†
Giacomo
Mari
 *,
Lucia
De Crescentini
,
Gianfranco
Favi
,
Stefania
Santeusanio
and
Fabio
Mantellini
*
*,
Lucia
De Crescentini
,
Gianfranco
Favi
,
Stefania
Santeusanio
and
Fabio
Mantellini
*
Department of Biomolecular Sciences, Section of Chemistry and Pharmaceutical Technologies, University of Urbino “Carlo Bo”, Via I Maggetti 24, 61029 Urbino, PU, Italy. E-mail: Fabio.mantellini@uniurb.it
First published on 4th May 2022
Abstract
A metal-free strategy, alternative to the known complex cycloaddition reactions, towards 2-carboxylated thieno [2,3-b] indole derivatives has been successfully developed. The novel approach involves as starting materials easy accessible 1,2-diaza-1,3-dienes and indoline 2-thione and requires mild reaction conditions. Furthermore, the easy work-up required makes this method amenable for a one-pot approach as demonstrated in the preparation of thienodolin, a natural product isolated from Streptomyces albogriseolus that exhibits valuable biological properties.
Introduction
The indole core fused with other carbo or hetero rings constitutes a recurrent skeleton in various pharmaceuticals or bioactive derivatives.1 The fusion of several rings originates rigid structures characterized by a well-defined geometry that permits an optimal and predictable orientation in the three-dimensional space of the substituents thus permitting a high functional specialization.2 In this context, an interesting example is represented by the thieno[2,3-b]indoles, a fused polyheterocyclic system that recurs in a variety of compounds able to act on the central nervous system,3 that exhibit antifungal properties,4 or that were employed in the construction of small efficient push–pull chromophores (Fig. 1).5 A natural product worthy of particular attention is the thienodolin, an amido substituted thieno[2,3-b]indole isolated from Streptomyces albogriseolus whose biological properties include plant–growth regulatory capacity and the nitric oxide synthase inhibition (Fig. 1).6 The most common synthetic approaches for this core require the use of indole or indoline 2-thione as starting materials. In the first case, the indole is employed with elemental sulfur and ketones,7 with elemental sulfur and alkenes or alkynes,8 with α,β-unsaturated carboxylic acids via sulfur-mediated decarboxylative cyclization,9via palladium-catalyzed dehydrative–dehydrogenative double C–H sulfuration,10 just to mention some of the more recent examples. | ||
| Fig. 1 Relevant thieno[2,3-b]indoles. | ||
Alternatively, the indoline-2-thione is employed in a base-mediated reaction with Morita–Baylis–Hillman or Rauhut–Currier adducts of nitroalkenes,11 with 1,2-diiodobenzene in a copper- and palladium-catalyzed tandem coupling reaction,12 with α-bromo-substituted ketones or aldehydes in the presence of Et3N,13 or with 1-aryloxy-4-chlorobut-2-ynes.14
Despite this plethora of methods, only few procedures that allow to obtain functionalized heterocycles in position 2 by amide or ester functions are reported in the literature (Fig. 2).5a,15 All the aforementioned syntheses needed highly functionalized indoles, furans, and thiophenes, which required a complicated and tedious route to be prepared, and in some cases, the scope is limited. Furthermore, the reactions often require vigorous conditions, or metal catalysts are necessary which, even if present in traces in the final product, can compromise the human consumption,16 or the efficiency of organic electronic devices.17
 | ||
| Fig. 2 Procedures to obtain 2-carbonyl thieno[2,3-b]indoles. | ||
Then, the development of efficient and metal-free methods for the assembly of 2-carbonyl substituted thieno[2,3-b]indoles from readily available starting materials are highly desirable. A retrosynthetic analysis reveals that this scaffold could be obtained starting from halo-ketosters and indoline-2-thione (Fig. 3, Path A). Initially, we unsuccessfully tested the conditions optimized by Boeini for the reactions between indoline-2-thiones with simple α-bromo-ketones or aldehydes.13 Unfortunately, this synthetic route is not practicable, even using other reaction conditions,18 and we have therefore designed an alternative synthetic way that involves the use of 1,2-diaza-1,3-dienes (DDs) in place of the halo-ketosters (Fig. 3, Path B).
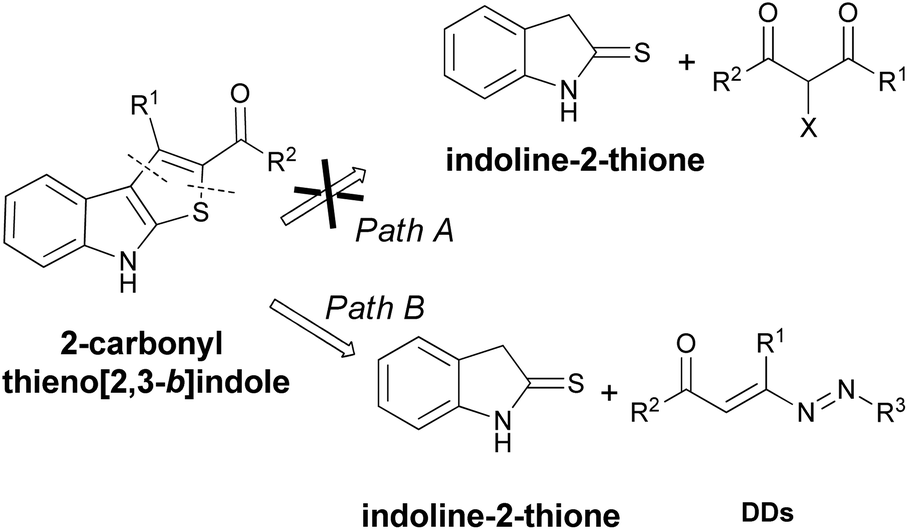 | ||
| Fig. 3 Possible disconnections of 2-carbonyl substituted thieno[2,3-b]indole core. | ||
In this case, the 1,2-diaza-1,3-dienes can represent an effective alternative being excellent Michael acceptors,19 able to regioselectively react with different nucleophiles, including sulfur compounds.20 Then, based on our experience in the thia-Michael reaction onto the azo-ene system of DDs, we have identified the indoline-2-thiones as potential nucleophilic candidates for easily obtaining 2-carbonyl substituted thieno[2,3-b]indole derivatives (Fig. 3, Path B).
Results and discussion
To investigate the capacity of DDs 1 to generate 2-carbonyl substituted thieno[2,3-b]indoles 4, DD 1a and indoline-2-thione 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 were chosen as representative model. The synthesis can be divided into two steps: (i) the initial formation of the α-thiohydrazone 3a; (ii) the internal cyclization process (Scheme 1).
21 were chosen as representative model. The synthesis can be divided into two steps: (i) the initial formation of the α-thiohydrazone 3a; (ii) the internal cyclization process (Scheme 1).
 | ||
| Scheme 1 Reaction between DD 1a and indoline-2-thione 2a to achieve 2-carbonyl thieno[2,3-b]indoles 4a: reaction conditions optimization. | ||
As expected, the indoline-2-thione 2a promptly reacts with DD 1a at room temperature and without any catalysts by providing the corresponding α-thiohydrazone 3a (Scheme 1, step i, Table 1). Different solvents such as acetonitrile (ACN), dichloromethane (DCM), ethanol (EtOH), methanol (MeOH) and tetrahydrofuran (THF) were tested (Table 1, entries 1–5). The reactions conducted in acetonitrile, in methanol or in ethanol, in equimolar ratio of 1a and 2a at room temperature furnished rapidly and in a quantitative manner whitish solid that was identified as the desired α-thiohydrazone 3a (Table 1, entries 1, 3 and 4). In DCM, the reaction proceeded slower and 10 mL of solvent were necessary to fully solubilize 2a (Table 1, entry 2), while, the profile of the reaction conducted in THF was more complicated (monitored by TLC) and the desired hydrazone 3a was obtained in lower yields than the ones gained in the previous cases (Table 1, entry 5).
| Entrya,b | Solvent | Temp. (°C) | Time (h) | Yield of 3ac (%) |
|---|---|---|---|---|
| a The reactions were conducted on 0.5 mmol scale of DD 1a in 3.0 mL of solvent unless differently stated. b 1.0 equiv. of indoline-2-thione 2a was employed. c Isolated yields of 3a calculated on DD 1a. d The reaction was conducted on 0.5 mmol scale of 1a in 10.0 mL of solvent. | ||||
| 1c | ACN | r.t. | 0.1 | Quantitative |
| 2d | DCM | r.t. | 2.5 | 86 |
| 3c | EtOH | r.t. | 0.1 | Quantitative |
| 4c | MeOH | r.t. | 0.1 | Quantitative |
| 5c | THF | r.t. | 1.75 | 67 |
The next step concerned the cyclization optimization of 3a to the thieno[2,3-b]indole derivative 4a (Scheme 1, Step ii, Table 2). This ring closure process occurs following a SEAr reaction in which the C(3)-indole carbon acts as a nucleophile by attacking the carbonyl deriving from the starting DDs. For this reason, we have focused our attention on acid catalysts capable of favouring the cyclization by increasing the electrophilic character of the hydrazone moiety (Scheme 3).
| Entrya | Solvent | Temp. (°C) | Catalyst | Amount of catalyst (eq.)b | Time (h) | Yield of 4ac (%) |
|---|---|---|---|---|---|---|
| a The reactions were conducted on 0.5 mmol scale in 3.0 mL of solvent. b Equivalents added referred to 3a. c Isolated yields of 4a calculated on hydrazone 3a. | ||||||
| 1 | ACN | r.t | CuCl2 | 0.2 | 72.0 | — |
| 2 | ACN | r.t | CuCl2·2H2O | 0.2 | 72.0 | — |
| 3 | ACN | r.t | InBr3 | 0.2 | 72.0 | — |
| 4 | ACN | r.t | ZnCl2 | 0.2 | 72.0 | — |
| 5 | ACN | r.t | Zn(OTf)2 | 0.2 | 72.0 | — |
| 6 | ACN | r.t | AcOH | 1.0 | 48.0 | Traces |
| 7 | ACN | r.t | TFA | 1.0 | 6.5 | 65 |
| 8 | ACN | r.t | PTSA | 1.0 | 4.0 | 74 |
| 9 | ACN | r.t | Amberlyst 15H | 1.0 | 4.0 | 75 |
| 10 | ACN | r.t | Amberlyst 15H | 0.5 | 5.5 | 54 |
| 11 | ACN | r.t | Amberlyst 15H | 2.0 | 4.0 | 74 |
| 12 | MeOH | r.t | Amberlyst 15H | 1.0 | 3.5 | 85 |
| 13 | EtOH | r.t | Amberlyst 15H | 1.0 | 3.5 | 79 |
Then, several Lewis acids (LA) such as CuCl2, CuCl2·2H2O, InBr3, ZnCl2, Zn(OTf)2, or Brønsted-acids as well as acetic- (AcOH), trifluoroacetic- (TFA), p-toluenesulfonic-acid (PTSA) and Amberlyst 15H were tested (Table 2, entries 1–9). Unfortunately, by employing the LA as catalysts the cyclization did not happen and the α-thiohydrazone 3a was recovered unchanged even 72.0 h later (Table 2, entries 1–5). On the contrary, the Brønsted-acids were able to promote the formation of the expected 4a and the PTSA has proved to be the most effective (Table 2, entries 6–8). Encouraged by this result, we then tried Amberlyst 15H, a reticular polystyrene-based ion exchange resin with strongly acidic sulfonic group analogous to PTSA. With our pleasure, we observed that also this heterogeneous catalyst efficiently promoted the formation of 4a (Table 2, entry 9). The Amberlyst's use entails several benefits compared to catalysis in homogeneous phase since this resin is easy to measure, is safe to use, and can be readily removed at the end of the reaction by a simple filtration. Furthermore, an additional advantage of this catalyst is related to its easy regeneration and reuse making it environment friendly.22 Furthermore, all the drawbacks associated with the use of metal catalysts such as contamination of the final products, especially when these are intended for human use or in organic electronics, elevated toxicity, eventual necessity of complex ligands and higher cost are obviated.23 So, different amounts of resin were tested for the cyclization step in ACN: the use of 1.0 equiv. yielded 4a in 75%; by using 0.5 equiv. a decrease was observed while no significant increment was obtained by employing 2.0 equiv. (Table 2, entries 10 and 11). Finally, 1.0 equiv. was checked also in those solvents that provided the best results in the first step observing that in MeOH the yield increased to 85%, while in EtOH reached the 79% (Table 2, entries 12 and 13). These identified optimized conditions were then employed to test the feasibility of conducting a sequential one-pot reaction without the isolation of the α-thiohydrazone 3a. With our great pleasure, we observed that the reaction between 1a (0.5 mmol) and 2a (0.5 mmol) in 3.0 mL of MeOH, at room temperature after 0.1 h furnished the intermediate 3a (TLC monitoring). By adding directly to the solution 1.0 equiv. of Amberlyst 15H, after 4.0 hours, the desired 2-carbonyl thieno[2,3-b]indoles 4a was isolated in 84% yield, in accord with the data obtained in the step-by-step procedure.
The reaction is very easy to monitor: the addition of the resin can be carried out when the typical red color due to the conjugation of the azo-ene system disappears.
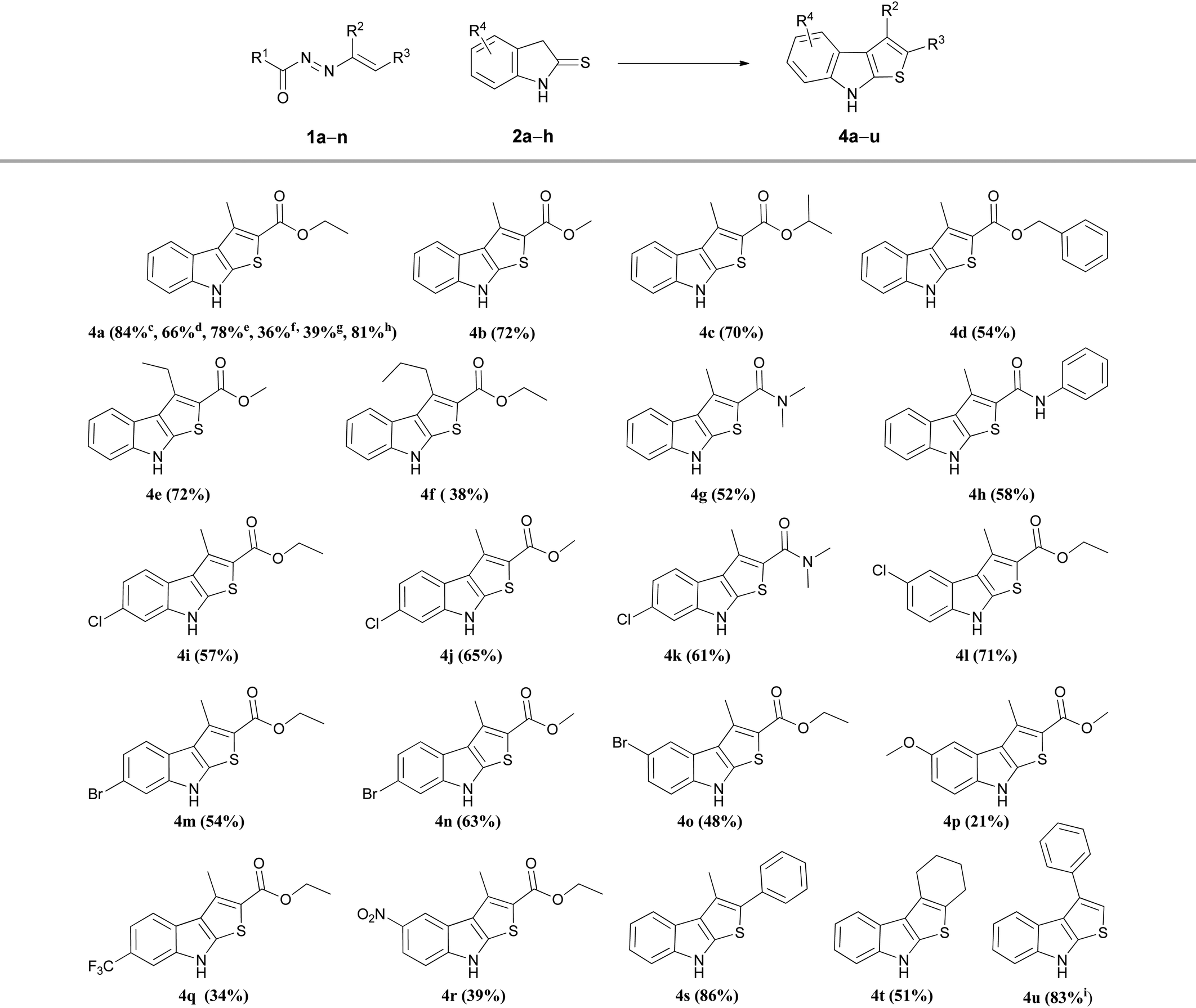
To check the applicability and effectiveness of these optimized sequential one-pot conditions, linear and cyclic DDs with different N-protective groups such as esters (R1 = OBut, OMe, OEt), or amides (R1 = NH2, NHPh), bearing in 3-position (R2) of the azo-ene system alkyl groups of different length such as methyl, ethyl or n-propyl, or substituted in 4-position (R3) with esters (1a–f,i–l) amides (1g,h) (see Table S2, ESI†) were reacted with different indoline-2-thiones 2a–h (Table S3, ESI†) to synthesize the corresponding functionalized 2-carbonyl thieno[2,3-b]indoles 4a–r (Table 3).
|
a Reaction conditions: 1 (1.0 mmol), 2 (1.0 mmol), MeOH (6.0 mL), room temperature (0.1–0.5 h), then to the disappearance of the typical red colour of the solution to the crude Amberlyst 15H (1.0 mmol) was added and the reaction was magnetically stirred for 2.0–5.0 h. The Amberlyst 15H was removed by filtration and the solvent was evaporated under reduced pressure. Thieno[2,3-b]indoles 4 were purified by column chromatography on silica gel (elution mixture: cyclohexane:ethyl acetate, 80:20) and the pure products were precipitated from ethyl ether/petrol ether.
b The isolated yields are shown inside the brackets.
c Isolated yields employing the 1-tert-butyl carboxylate DD 1a.
d Isolated yields employing the 1-methyl carboxylate DD 1i (Table S2, ESI†).
e Isolated yields employing the 1-ethyl carboxylate DD 1j.
f Isolated yields employing the 1-carboxyamide DD 1k.
g Isolated yields employing the 1-(N-phenyl-carboxyamide) DD 1l.
h
4a (1.681 g) was obtained starting from 8.0 mmol of 1-tert-butyl carboxylate DD 1a.
i The 3-phenyl-thieno[2,3-b]indole 4s was obtained from 4-unsubstituted DD generated in situ by basic treatment of the corresponding halogenated hydrazone 5a.25
|
|---|
|
As described in Table 3 for compound 4a, the N-protective group of the azo-ene system is crucial for the success of the synthesis. The best results in terms both of yields and reaction times were obtained with the tert-butyl ester (Boc), that is the most easily removable group in the acid conditions employed. A slight decrease in yields was observed using the methyl and ethyl ester derivatives, while a sharp decrement was checked in the cases when the nitrogen is substituted with amide protecting group (R1 = CONH2, CONHPh) (Table 3). On the basis of these evidences, all the other variously functionalized DDs employed in the subsequent preparations were protected on the nitrogen with a Boc (Table S2, ESI†). From the analysis of the collected data, it is possible to note how the yields are generally good both for 2-amido- and for the 2-ester substituted heterocycles 4, with the exceptions of 3-n-propyl-, 5-methoxy-, 6-trifluoromethyl-, and 5-nitro-substituted-thieno[2,3-b]indoles 4f, 4p, 4q and 4r respectively.
The adaptability to scale-up and the synthetic utility of this method were highlighted conducing a gram-scale synthesis of 4a that was obtained without loss of yield (81%, Table 3).
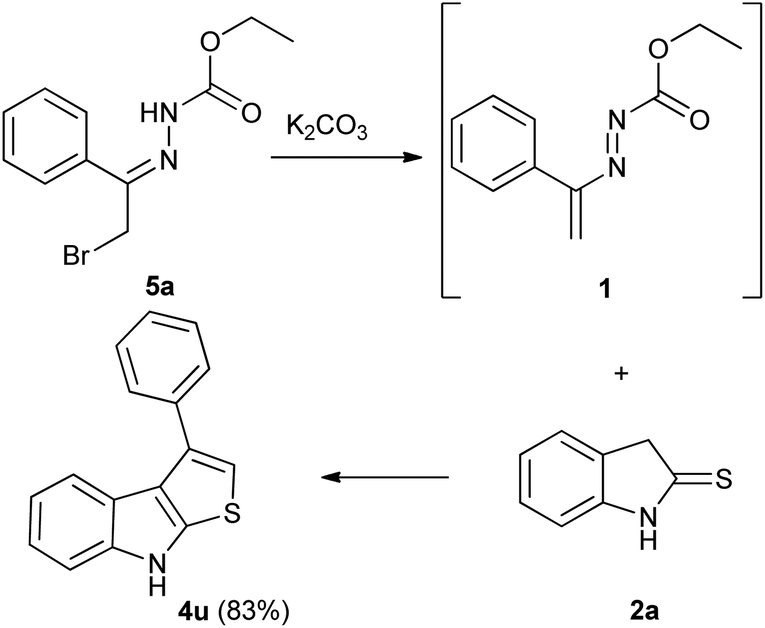
It is noteworthy that this procedure is suitable and effective also for the preparation of differently substituted thieno[2,3-b]indoles. Under the optimized conditions, using the appropriate DDs 1m,n (Table S2, ESI†), the reaction easily furnished in good yields the corresponding 2-aryl (4s) and 2,3-cycloalkyl (4t) derivatives (Table 3). To obtain the 2-unsubstituted compound 4u, the necessary DD was generated in situ by basic treatment of the corresponding α-bromohydrazone 5a with potassium carbonate. This further step is required due to the typical poor stability of this azo-ene derivative which prevents its isolation and conservation.24,25 However, also in this case the desired 3-phenyl thieno[2,3-b]indoles 4u was achieved in good yield (Table 3).
On the contrary, the reaction between the 4,4-disubstituted DDs 1o,p and the indoline-2-thiones 2a,b (Scheme 2) provided exclusively the corresponding hydrazones 3b,c suggesting that the hydrogen atom initially located in position 4 of the azo-ene system of DD 1 is needful to allow the formation of the new thiophene cycle. Moreover, the comparable yields relative to the hydrazones 3b,c indicate that the different nitrogen protecting groups (3b R1 = Boc; 3c R1 = CONHPh) only influence the second step of the synthesis, i.e. the cyclization process.
 | ||
| Scheme 2 Reaction between the 4,4-disubstituted DDs 1o,p and the indoline-2-thiones 2a,b. | ||
On the basis of these evidences, a plausible mechanism foresees an initial tautomerism of the indoline-2-thiones 2 that provides the 1H-indole-2-thiols 2′ that promptly act as nucleophile reacting with the terminal carbon atom of the azo-ene system of the DDs 1 yielding the corresponding α-sulfur hydrazones 3 (Scheme 3). The activation of the sulfur nucleophilicity by means of the thioamide 2 imidothioate 2′′ tautomerism can be excluded as proved by the isolation of hydrazones 3a–c. Clearly, the driving force of the reaction is related to the aromaticity of the formed 1H-indole-2-thiol. In the next step, probably, the addition of Amberlyst 15H activates the hydrazonic function (intermediate A) facilitating the cyclization process which occurs through the nucleophilic attack of the carbon in position 3 of the indole nucleus on the hydrazonic moiety with consequent formation of the intermediate 3,3a-dihydro-2H-thieno[2,3-b]indol-8-ium B.
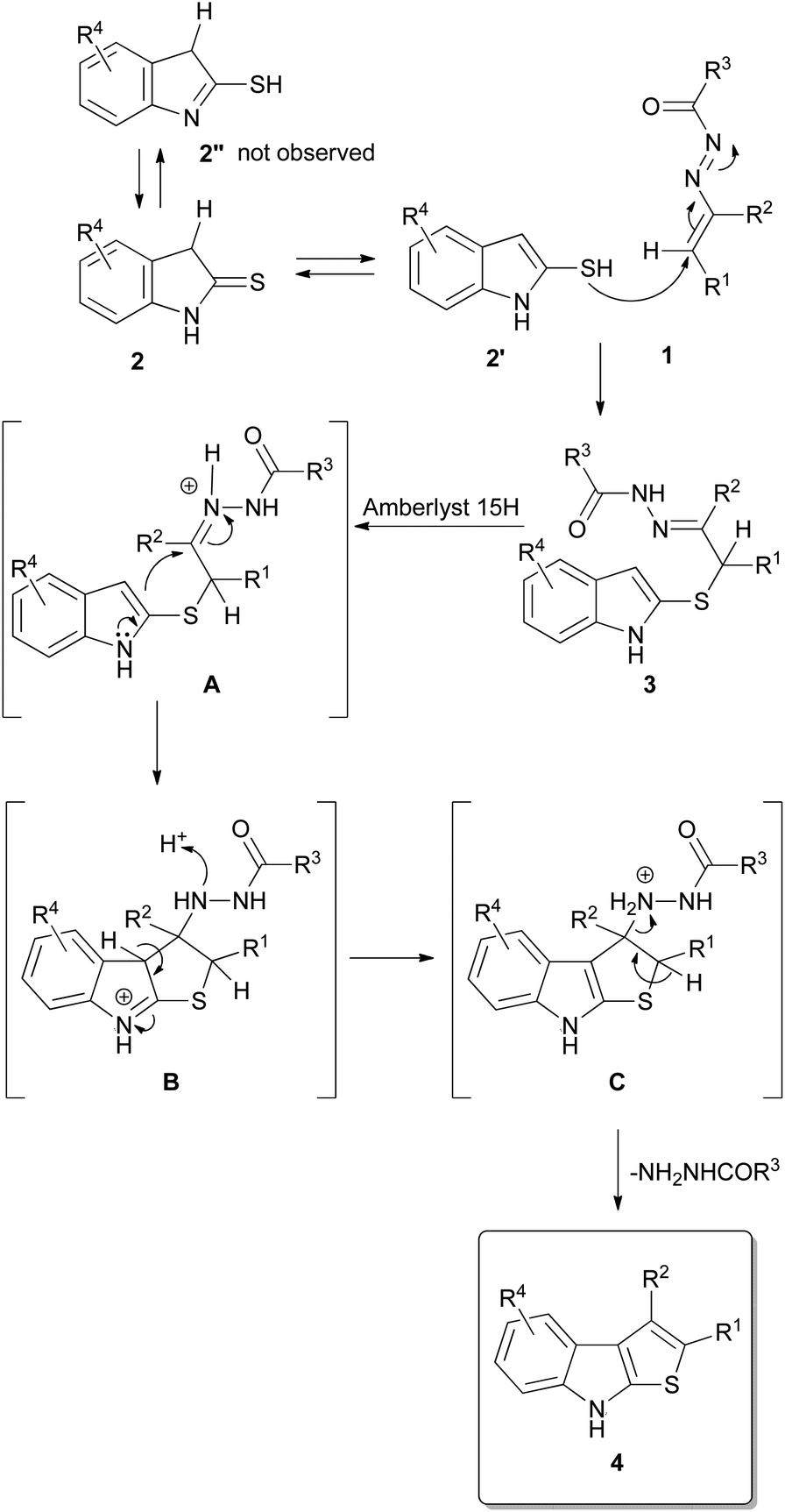 | ||
| Scheme 3 Plausible reaction mechanism. | ||
This aspect can explain the modest yields obtained in the synthesis of compounds 4f,p–r: the plausible protonation of the methoxy group (4p) or the presence of strong electron withdrawing groups (4q trifluoromethyl), (4r nitro) heavily decrease the nucleophilicity of the indolic carbon disfavouring the cyclization process which, in turn, results more difficult also in presence of an increased steric hindrance of the electrophilic site as in the case of intermediate A, precursor of 4f (R2 = n-Propyl). The successive loss of the proton restores the aromaticity of the indolic nucleus (intermediate B, Scheme 3), while the elimination of the hydrazine provides the final thieno[2,3-b]indoles 4. This last step is also favored by the acid reaction environment as confirmed by the best results obtained with the N-Boc protected-DDs.
Given the success obtained with the preparation of 2-carbonyl thieno[2,3-b]indoles 4, we decided to apply this protocol to the synthesis of thienodolin (Scheme 4).6a,b To achieve this target, the 3-unsubstituted DD E is required. This azoalkene E manifests poor stability and cannot be isolated, so a one-pot procedure was adopted. To a commercial ethyl 2-chloro-3-oxopropanoate 6a in methanol at room temperature, one equivalent of tert-butyl hydrazinecarboxylate 7a was added. The reaction furnishes after 4.5 h the corresponding α-chlorohydrazone D that was treated with potassium carbonate to promote the conjugated elimination providing the desired DD E, that, in turn, promptly reacted with indoline-2-thione 2b furnishing the corresponding α-thiohydrazone F intermediate (TLC monitoring).
 | ||
| Scheme 4 Approach to the synthesis of thienodolin. aIsolated yields of 4v calculated on ethyl 2-chloro-3-oxopropanoate 6a. | ||
The direct addition to the crude of two equivalents of Amberlyst 15H furnished the ethyl 6-chloro-8H-thieno[2,3-b]indole-2-carboxylate 4v in 14% overall yield (calculated on the starting 6a) that, in turn, is the direct precursor of the target thienodolin.15b It is noteworthy that, to the best of our knowledge, only the total synthesis proposed by Bergman is reported so far in the literature.15b
Experimental
General
All the commercially available reagents and solvents were used without further purification. 1,2-Diaza-1,3-dienes 1a–p were synthesized as a mixture of E/Z isomers as previously reported.26 Indoline-2-thiones 2a–h were prepared according to the reported method.21 Chromatographic purification of compounds was carried out on silica gel (60–200 μm). TLC analysis was performed on pre-loaded (0.25 mm) glass supported silica gel plates (Kieselgel 60); compounds were visualized by exposure to UV light and by dipping the plates in 1% Ce(SO4)·4H2O, 2.5% (NH4)6Mo7O24·4H2O in 10% sulphuric acid followed by heating on a hot plate. All 1H NMR and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, using [D6]DMSO or CDCl3 as solvent. Chemical shift (δ scale) are reported in parts per million (ppm) relative to the central peak of the solvent and are sorted in ascending order within each group. The following abbreviations are used to describe peak patterns where appropriate: s = singlet, d = doublet, dd = doublet of doublet, dt = doublet of triplet, t = triplet, q = quartet, sex = sextet, sept = septet, m = multiplet and br = broad signal. All coupling constants (J value) are given in Hertz [Hz]. High-and low-resolution mass spectroscopy was performed on a Micromass Q-ToF Micro mass spectrometer (Micromass, Manchester, UK) using an ESI source. Melting points were determined in open capillary tubes and are uncorrected. Elemental analyses were within ± 0.4 of the theoretical values (C, H, N).General procedure for the synthesis of tert-butyl 2-(3-((1H-indol-2-yl)thio)-4-ethoxy-4-oxobutan-2-ylidene)hydrazinecarboxylate 3a, tert-butyl 2-(2-((6-chloro-1H-indol-2-yl)thio)-2-(ethoxycarbonyl)cyclopentylidene)hydrazinecarboxylate 3b and ethyl 1-((1H-indol-2-yl)thio)-2-(2-(phenylcarbamoyl)hydrazono)cyclohexanecarboxylate 3c
To a solution of 1,2-diaza-1,3-dienes 1a,o,p (1.0 mmol) in methanol (6.0 mL) at room temperature indoline-2-thiones 2a,b (1.0 mmol) were added and the reaction mixture was stirred at room temperature until the disappearance of the reagents (TLC monitoring 0.10–0.50 h) as also evidenced by the color change from red, typical of DDs, to pale yellow and the solvent was evaporated under reduced pressure. The α-thio-functionalized hydrazones 3a–c were purified by column chromatography on silica gel (elution mixture: cyclohexane:ethyl acetate, 80:20 for compounds 3a,b and cyclohexane:ethyl acetate, 65:35 for compound 3c) and the pure products were precipitated in ethyl acetate/petroleum ether. In the case of compounds 3b,c, the addition of 1.0 equiv. of Amberlyst 15H (dry form) to the reaction medium does not cause the cyclization process to the corresponding thieno[2,3-b]indoles.
One pot procedure for the synthesis of 2-carbonyl thieno[2,3-b]indoles 4a–r, 3-methyl-2-phenyl-8H-thieno[2,3-b]indole 4s, 2,3,4,6-tetrahydro-1H-benzo[4,5]thieno[2,3-b]indole 4t
To a solution of 1,2-diaza-1,3-dienes 1a–n (1.0 mmol) in methanol (6.0 mL) at room temperature, indoline-2-thiones 2a–h (1.0 mmol) were added and the reaction mixture was stirred at room temperature until the disappearance of the reagents (TLC monitoring 0.10–0.50 h) as also evidenced by the color change from red, typical of DDs, to pale yellow. Directly to the reaction medium, 1.0 equiv. of Amberlyst 15H (dry form) was then added, and the reaction was softly magnetically stirred. After 2.0–5.0 h (TLC monitoring), the corresponding thieno[2,3-b]indoles 4a–t were formed. The Amberlyst 15H was removed by filtration and the solvent was evaporated under reduced pressure. Thieno[2,3-b]indoles 4a–t were purified by column chromatography on silica gel (elution mixture: cyclohexane:ethyl acetate, 80:20) and the pure products were precipitated from ethyl ether/petroleum ether.
One pot procedure for the synthesis of 3-phenyl-8H-thieno[2,3-b]indole 4u
To a solution of ethyl 2-(2-bromo-1-phenylethylidene)hydrazinecarboxylate 5a (1.0 mmol)25 in methanol (6.0 mL) at room temperature, indoline-2-thione 2a (1.0 mmol) and potassium carbonate (2.0 mmol) were added and the reaction mixture was stirred at room temperature until the disappearance of the reagents (TLC monitoring 0.50 h). The crude was filtered to remove the excess of potassium carbonate, and then to the reaction medium 1.0 equiv. of Amberlyst 15H (dry form) was added. The reaction mixture was softly stirred at room temperature until the disappearance of the reagents (TLC monitoring, 3.0 h). The Amberlyst 15H was removed by filtration, and the solvent was evaporated under reduced pressure. Thieno[2,3-b]indole 4u was purified by column chromatography on silica gel (elution mixture: cyclohexane:ethyl acetate, 90:10) and the pure product was precipitated from ethyl ether/petroleum ether.
One pot procedure for the synthesis of ethyl 6-chloro-8H-thieno[2,3-b]indole-2-carboxylate 4v
To a solution of 2-chloro-3-oxopropanoate 6a (1.0 mmol) in methanol (6.0 mL) at room temperature, tert-butyl hydrazinecarboxylate 7a (1.0 mmol) was added. At the disappearance of 6a (TLC monitoring, 4.50 h), to the crude, potassium carbonate (2.0 mmol) was added and the reaction mixture was stirred at room temperature until the disappearance of the reagents (TLC monitoring 0.50 h). The crude was filtered to remove the excess of potassium carbonate, and then to the reaction medium 2.0 equiv. of Amberlyst 15H (dry form) were added and the reaction mixture was softly stirred at room temperature until the disappearance of the reagents (TLC monitoring, 5.0 h). The Amberlyst 15H was removed by filtration, and the solvent was evaporated under reduced pressure. Thieno[2,3-b]indole 4v was purified by column chromatography on silica gel (elution mixture: cyclohexane:ethyl acetate, 80:20) and the pure product was precipitated from ethyl ether/petroleum ether.
Conclusion
In conclusion, here we describe an efficient and easy methodology that gives rapid access to a variety of 2-carbonyl thieno[2,3-b]indoles. The high availability of cheap starting materials, the mild and simple reaction conditions that do not require metal catalysts, or dry solvents, or inert atmosphere, and the potential utilities of these products can increase the synthetic usefulness of this protocol. Furthermore, the easy work-up required makes this method amenable for a sequential multicomponent approach as demonstrated in the preparation of 4t, the direct precursor of the valuable thienodolin.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Erika Palazzetti and Dr Viviana Bartolacci for their precious advices and Dr Anna Maria Gioacchini who competently performed the mass spectra.Notes and references
- (a) Z. Xu, Q. Wang and J. Zhu, Chem. Soc. Rev., 2018, 47, 7882 RSC; (b) Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425 CrossRef CAS PubMed; (c) W. Zi, Z. Zuo and D. Ma, Acc. Chem. Res., 2020, 48, 702 CrossRef PubMed; (d) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (e) A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2019, 48, 4401 RSC; (f) E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390 CrossRef CAS PubMed; (g) G. W. Gribble and J. C. Badenock, Heterocyclic Scaffolds II: Reactions and Applications of Indoles, Springer, Berlin, 2010 CrossRef.
- N. K. Kaushik, N. Kaushik and P. Attri, et al. , Molecules, 2013, 18, 6620 CrossRef CAS PubMed.
- P. Jakobsen, A. Kanstrup, P. Faarup, P. H. Olesen and J. M. Lundbech, U.S, US5783575A19980721, 1998 Search PubMed.
- M. Pedras, C. Soledade and M. Suchy, Bioorg. Med. Chem., 2006, 14, 714 CrossRef CAS PubMed.
- (a) F. Baert, C. Cabanetos, M. Allain, V. Silvestre and P. Leriche, Org. Lett., 2016, 18, 1582 CrossRef CAS PubMed; (b) T. Qi, W. Qiu, Y. Liu, H. Zhang, X. Gao, Y. Liu, K. Lu, C. Du, G. Yu and D. Zhu, J. Org. Chem., 2008, 73, 4638 CrossRef CAS PubMed; (c) R. K. Konidena, K. H. Lee and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 13912 RSC; (d) J. Kim, S. Chae, A. Yi, S. Hong, H. J. Kim and H. Suh, Appl. Polym. Sci., 2019, 136, 47624 CrossRef.
- (a) K. Kanbe, M. Okamura, S. Hattori, H. Naganawa, M. Hamada, Y. Okami and T. Takeuchi, Biosci. Biotechnol. Biochem., 1993, 57, 632 CrossRef CAS; (b) K. Kanbe, H. Naganawa, K. T. Nakamura, Y. Okami and T. Takeuchi, Biosci. Biotechnol. Biochem., 1993, 57, 636 CrossRef CAS.
- (a) T. T. Nguyen and P. H. Tran, RSC Adv., 2020, 10, 9663 RSC; (b) C. Lu, H. Huang, X. Tuo, P. Jiang, F. Zhang and G.-J. Deng, Org. Chem. Front., 2019, 6, 2738 RSC; (c) P. Ni, B. Li, H. Huang, F. Xiao and G.-J. Deng, Green Chem., 2017, 19, 5553 RSC.
- B. Li, P. Ni, H. Huang, F. Xiao and G.-J. Deng, Adv. Synth. Catal., 2017, 359, 4300 CrossRef CAS.
- H.-L. Zhang, F. Wen, W.-B. Sheng, P. Yin, C.-T. Zhang, C.-Y. Peng, D.-M. Peng, D.-F. Liao and R.-G. Fu, Tetrahedron Lett., 2019, 60, 80 CrossRef CAS.
- Y. Liao, Y. Peng, H. Qi, G.-J. Deng, H. Gong and C.-J. Li, Chem. Commun., 2015, 51, 1031 RSC.
- V. Mane, T. V. Baiju and I. N. N. Namboothiri, ACS Omega, 2018, 3, 17617 CrossRef CAS PubMed.
- S. Zhou, G. Xiao and Y. Liang, Tetrahedron Lett., 2017, 58, 338 CrossRef CAS.
- H. Z. Boeini, Helv. Chim. Acta, 2009, 92, 1268 CrossRef CAS.
- K. C. Majumdar and S. Alam, J. Chem. Res., 2006, 5, 289 CrossRef.
- (a) P. H. Olesen, J. B. Hansen and M. Engelstoft, J. Heterocycl. Chem., 1995, 32, 1641 CrossRef CAS; (b) R. Engqvist, A. Javaid and J. Bergman, Eur. J. Org. Chem., 2004, 2589 CrossRef CAS; (c) A. V. Butin, F. A. Tsiunchik, V. T. Abaev and V. E. Zavodnik, Synlett, 2008, 1145 CrossRef CAS; (d) F. M. Moghaddam, H. Saeidian, Z. Mirjafary, S. Taheri and S. Kheirjou, Synlett, 2009, 1047 CrossRef CAS; (e) I. Savych, T. Glaesel, A. Villinger, V. Y. Sosnovskikh, V. O. Iaroshenko and P. Langer, Org. Biomol. Chem., 2015, 13, 729 RSC; (f) J. Liu, Y. Zhang, Y. Yue, Z. Wang, H. Shao, K. Zhuo, Q. Lv and Z. Zhang, J. Org. Chem., 2019, 84, 12946 CrossRef CAS PubMed; (g) L. Yang, S. Zhou, J.-Q. Zhao, Y. You, Z.-H. Wang, M.-Q. Zhou and W.-C. Yuan, Org. Biomol. Chem., 2021, 19, 3678 RSC.
- European Medicines Agency. in ICH Guideline Q3D on Elemental Impurities (London, 2015), https://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180284.pdf.
- Ö. Usluer, M. Abbas, G. Wantz, L. Vignau, L. Hirsch, E. Grana, C. Brochon, E. Cloutet and G. Hadziioannou, ACS Macro Lett., 2014, 3, 1134 CrossRef.
- The reaction between indoline-2-thiones with alkyl 2-halo acetoacetate under different conditions was investigated employing the 6-bromoindoline-2-thione 2d and the ethyl 2-chloro acetoacetate A as representative model. The results obtained are summarized in the Table S1 reported in the ESI.†.
- (a) L. Wei, C. Shen, Y.-Z. Hu, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2019, 55, 6672 RSC; (b) S. M. M. Lopes, A. L. Cardoso, A. Lemos and T. M. V. D. Pinho e Melo, Chem. Rev., 2018, 118, 11324 CrossRef CAS PubMed; (c) O. A. Attanasi, L. De Crescentini, G. Favi, P. Filippone, F. Mantellini, F. R. Perrulli and S. Santeusanio, Eur. J. Org. Chem., 2009, 3109 CrossRef CAS.
- (a) O. A. Attanasi, L. De Crescentini, G. Favi, P. Filippone, S. Lillini, F. Mantellini and S. Santeusanio, Org. Lett., 2005, 7, 2469 CrossRef CAS PubMed; (b) O. A. Attanasi, S. Bartoccini, G. Favi, G. Giorgi, F. R. Perrulli and S. Santeusanio, J. Org. Chem., 2012, 77, 1161 CrossRef CAS PubMed; (c) S. Santeusanio, R. Majer, F. R. Perrulli, L. De Crescentini, G. Favi, G. Giorgi and F. Mantellini, J. Org. Chem., 2017, 82, 9773 CrossRef CAS PubMed; (d) G. Mari, M. Verboni, L. De Crescentini, G. Favi, S. Santeusanio and F. Mantellini, Org. Chem. Front., 2018, 5, 2108 RSC; (e) G. Mari, L. De Crescentini, G. Favi, S. Santeusanio and F. Mantellini, Eur. J. Org. Chem., 2018, 6548 CrossRef CAS.
- Indoline-2-thione was prepared following a general procedure for conversion of the carbonyl group to the thione group: J. W. Scheeren, P. H. J. Ooms and R. J. F. Nivard, Synthesis, 1973, 149 CrossRef CAS.
- For reviews on Amberlyst-15H in organic synthesis, see: (a) R. Pal, T. Sarkar and S. Khasnobis, ARKIVOC, 2012,(i), 570 Search PubMed; (b) T. Okuhara, Chem. Rev., 2002, 102, 3641 CrossRef CAS PubMed.
- (a) European Medicines Agency. in ICH Guideline Q3D on Elemental Impurities (London, 2015), https://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180284.pdf; (b) Ö. Usluer, M. Abbas, G. Wantz, L. Vignau, L. Hirsch, E. Grana, C. Brochon, E. Cloutet and G. Hadziioannou, ACS Macro Lett., 2014, 3, 1134 CrossRef.
- Some recent examples of in situ generated DDs: (a) T. Chen, C. Che, Z. Guo, X.-Q. Dong and C.-J. Wang, Org. Chem. Front., 2021, 8, 4392 RSC; (b) W.-C. Yuan, B.-X. Quan, J.-Q. Zhao, Y. You, Z.-H. Wang and M.-Q. Zhou, J. Org. Chem., 2020, 85, 11812 CrossRef CAS PubMed; (c) J. Feng, T. He, Y. Xie, Y. Yu, J. B. Baell and F. Huang, Org. Biomol. Chem., 2020, 18, 9483 RSC; (d) W. Yin, L. Fang, Z. Wang, F. Gao and Z. Li, Org. Lett., 2019, 21, 7361 CrossRef CAS PubMed; (e) Y. Zhang, Y. Cao, L. Lu, S. Zhang, W. Bao, S. Huang and Y. Rao, J. Org. Chem., 2019, 84, 7711 CrossRef CAS PubMed; (f) B. Cheng, Y. Li, T. Wang, X. Zhang, H. Li, Y. Li and H. Zhai, Chem. Commun., 2019, 55, 14606 RSC; (g) S. Emamian, M. Soleymani and S. S. Moosavi, New J. Chem., 2019, 43, 4765 RSC.
- To synthesize compound 4u the necessary 4,4-unsubstituted DD was prepared in situ by treatment of the α-bromohydrazone 5a with one equivalent of potassium carbonate:
. - L. Preti, O. A. Attanasi, E. Caselli, G. Favi, C. Ori, P. Davoli, F. Felluga and F. Prati, Eur. J. Org. Chem., 2010, 4312 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob00440b |
| This journal is © The Royal Society of Chemistry 2022 |